 Luca Freschi1†
Luca Freschi1† Julie Jeukens1†
Julie Jeukens1† Irena Kukavica-Ibrulj1†
Irena Kukavica-Ibrulj1† Brian Boyle1Marie-Josée Dupont1Jérôme Laroche1Stéphane Larose1
Brian Boyle1Marie-Josée Dupont1Jérôme Laroche1Stéphane Larose1 Halim Maaroufi1Joanne L. Fothergill2
Halim Maaroufi1Joanne L. Fothergill2 Matthew Moore2
Matthew Moore2 Geoffrey L. Winsor3Shawn D. Aaron4
Geoffrey L. Winsor3Shawn D. Aaron4 Jean Barbeau5
Jean Barbeau5 Scott C. Bell6
Scott C. Bell6 Jane L. Burns7Miguel Camara8André Cantin9
Jane L. Burns7Miguel Camara8André Cantin9 Steve J. Charette1,10,11Ken Dewar12
Steve J. Charette1,10,11Ken Dewar12 Éric Déziel13
Éric Déziel13 Keith Grimwood14
Keith Grimwood14 Robert E. W. Hancock15
Robert E. W. Hancock15 Joe J. Harrison16
Joe J. Harrison16 Stephan Heeb8
Stephan Heeb8 Lars Jelsbak17
Lars Jelsbak17 Baofeng Jia18Dervla T. Kenna19
Baofeng Jia18Dervla T. Kenna19 Timothy J. Kidd20,21
Timothy J. Kidd20,21 Jens Klockgether22
Jens Klockgether22 Joseph S. Lam23
Joseph S. Lam23 Iain L. Lamont24
Iain L. Lamont24 Shawn Lewenza16
Shawn Lewenza16 Nick Loman25
Nick Loman25 François Malouin9Jim Manos26
François Malouin9Jim Manos26 Andrew G. McArthur18
Andrew G. McArthur18 Josie McKeown8
Josie McKeown8 Julie Milot27Hardeep Naghra8
Julie Milot27Hardeep Naghra8 Dao Nguyen12,28
Dao Nguyen12,28 Sheldon K. Pereira18Gabriel G. Perron29Jean-Paul Pirnay30Paul B. Rainey31,32
Sheldon K. Pereira18Gabriel G. Perron29Jean-Paul Pirnay30Paul B. Rainey31,32 Simon Rousseau12
Simon Rousseau12 Pedro M. Santos33Anne Stephenson34
Pedro M. Santos33Anne Stephenson34 Véronique Taylor23
Véronique Taylor23 Jane F. Turton19
Jane F. Turton19 Nicholas Waglechner18
Nicholas Waglechner18 Paul Williams8
Paul Williams8 Sandra W. Thrane17
Sandra W. Thrane17 Gerard D. Wright18Fiona S. L. Brinkman3
Gerard D. Wright18Fiona S. L. Brinkman3 Nicholas P. Tucker35
Nicholas P. Tucker35 Burkhard Tümmler22
Burkhard Tümmler22 Craig Winstanley2
Craig Winstanley2 Roger C. Levesque1*
Roger C. Levesque1*- 1Institute for Integrative and Systems Biology, Université Laval, Quebec, QC, Canada
- 2Institute of Infection and Global Health, University of Liverpool, Liverpool, UK
- 3Department of Molecular Biology and Biochemistry, Simon Fraser University, Vancouver, BC, Canada
- 4Ottawa Hospital Research Institute, Ottawa, ON, Canada
- 5Faculté de Médecine Dentaire, Université de Montréal, Montréal, QC, Canada
- 6QIMR Berghofer Medical Research Institute, Brisbane, QLD, Australia
- 7Seattle Children's Research Institute, University of Washington School of Medicine, Seattle, WA, USA
- 8School of Life Sciences, University of Nottingham, Nottingham, UK
- 9Département de Médecine, Université de Sherbrooke, Sherbrooke, QC, Canada
- 10Centre de Recherche de l'Institut Universitaire de Cardiologie et de Pneumologie de Québec, Quebec, QC, Canada
- 11Département de Biochimie, de Microbiologie et de Bio-informatique, Faculté des Sciences et de Génie, Université Laval, Quebec, QC, Canada
- 12Department of Human Genetics, McGill University, Montreal, QC, Canada
- 13INRS Institut Armand Frappier, Laval, QC, Canada
- 14School of Medicine, Griffith University, Gold Coast, QLD, Australia
- 15Department of Microbiology and Immunology, University of British Columbia, Vancouver, BC, Canada
- 16Biological Sciences, University of Calgary, Calgary, AB, Canada
- 17Department of Systems Biology, Technical University of Denmark, Lyngby, Denmark
- 18M.G. DeGroote Institute for Infectious Disease Research, McMaster University, Hamilton, ON, Canada
- 19Antimicrobial Resistance and Healthcare Associated Infections Reference Unit, Public Health England, London, UK
- 20Child Health Research Centre, The University of Queensland, Brisbane, QLD, Australia
- 21Centre for Infection and Immunity, Queen's University Belfast, Belfast, UK
- 22Klinische Forschergruppe, Medizinische Hochschule, Hannover, Germany
- 23Department of Molecular and Cellular Biology, University of Guelph, Guelph, ON, Canada
- 24Department of Biochemistry, University of Otago, Dunedin, New Zealand
- 25Institute for Microbiology and Infection, University of Birmingham, Birmingham, UK
- 26Department of Infectious Diseases and Immunology, The University of Sydney, Sydney, NSW, Australia
- 27Department of Pneumology, Institut Universitaire de Cardiologie et de Pneumologie de Québec, Université Laval, Quebec, QC, Canada
- 28Department of Microbiology and Immunology and Department of Experimental Medicine, McGill University, Montreal, QC, Canada
- 29Department of Biology, Bard College, Annandale-On-Hudson, NY, USA
- 30Laboratory for Molecular and Cellular Technology, Queen Astrid Military Hospital, Brussels, Belgium
- 31New Zealand Institute for Advanced Study, Massey University, Albany, New Zealand
- 32Max Planck Institute for Evolutionary Biology, Plön, Germany
- 33Department of Biology, University of Minho, Braga, Portugal
- 34St. Michael's Hospital, Toronto, ON, Canada
- 35Strathclyde Institute of Pharmacy and Biomedical Sciences, University of Strathclyde, Glasgow, UK
The International Pseudomonas aeruginosa Consortium is sequencing over 1000 genomes and building an analysis pipeline for the study of Pseudomonas genome evolution, antibiotic resistance and virulence genes. Metadata, including genomic and phenotypic data for each isolate of the collection, are available through the International Pseudomonas Consortium Database (http://ipcd.ibis.ulaval.ca/). Here, we present our strategy and the results that emerged from the analysis of the first 389 genomes. With as yet unmatched resolution, our results confirm that P. aeruginosa strains can be divided into three major groups that are further divided into subgroups, some not previously reported in the literature. We also provide the first snapshot of P. aeruginosa strain diversity with respect to antibiotic resistance. Our approach will allow us to draw potential links between environmental strains and those implicated in human and animal infections, understand how patients become infected and how the infection evolves over time as well as identify prognostic markers for better evidence-based decisions on patient care.
Importance of P. aeruginosa as a Model in Large-scale Bacterial Genomics
Studies of the genetic structure of microbial populations are central to understand the evolution, ecology and epidemiology of infectious diseases. However, numerous studies describing the genetic structure of pathogen populations are based on samples drawn mostly and overwhelmingly from clinical collections (Wiehlmann et al., 2007; Pirnay et al., 2009). This approach has resulted in a limited view of bacterial pathogens with respect to the evolutionary history of disease-causing lineages as well as the development and distribution of antibiotic resistance genes via the resistome and mobilome (D'Costa et al., 2006; Perry and Wright, 2013). The environmental bacterium and opportunistic pathogen P. aeruginosa is a model system in large-scale bacterial genomics (Gellatly and Hancock, 2013). It exhibits extensive metabolic adaptability enabling survival in a wide range of niches including soil, water, plants and animals. Genome rearrangements and a varying complement of genes contribute to strain-specific activities but the detailed molecular mechanisms are still poorly understood (Silby et al., 2011). Multiple studies, some of which include environmental isolates, have sought to resolve the population structure of P. aeruginosa using various typing methods and are not in full agreement (Kiewitz and Tümmler, 2000; Pirnay et al., 2002, 2009; Wiehlmann et al., 2007; Fothergill et al., 2010; Lam et al., 2011; Kidd et al., 2012; Martin et al., 2013). Next-generation sequencing (NGS) coupled with whole genome comparison is now becoming the new gold standard for understanding bacterial population structure and offers previously unmatched resolution for phylogenetic analysis (Hilker et al., 2015; Marvig et al., 2015; Williams et al., 2015). The combination of NGS with a more extensive set of strains promises to resolve the population structure of P. aeruginosa and shed light on the genetic basis of its adaptability.
Prominent Role of P. aeruginosa in Cystic Fibrosis Lung Infections
Pseudomonas aeruginosa can cause serious opportunistic infections in humans, in particular among immunocompromised individuals, those having cancer, skin burns and wounds, and most notably, cystic fibrosis (CF) patients (Lyczak et al., 2000). Although transmission routes are difficult to establish, it is generally accepted that the lungs of most CF patients become infected with P. aeruginosa from the environment, and it is difficult to devise strategies to counter such infections (Emerson et al., 2002; Hauser et al., 2011). Even though patient management has increased the median life expectancy of CF patients to about 50 years (Stephenson et al., 2015), most patients eventually develop chronic lung infection, which is the main cause of morbidity and mortality associated with CF. While there has been significant progress in early eradication therapy for P. aeruginosa (Lee, 2009; Heltshe et al., 2015), chronic lung infections remain challenging to eradicate. Indeed, the basic principles on which clinical bacteriology practices are based have altered little over the past 50 years and suffer severe limitations in the context of opportunistic and chronic lung infections. P. aeruginosa populations often exist in biofilms and diversify phenotypically in the CF lung (Mowat et al., 2011), hence antimicrobial susceptibility profiles applied to single isolates are poorly predictive of therapeutic efficacy (Keays et al., 2009). Moreover, despite the availability of a number of completely sequenced and annotated P. aeruginosa genomes (Stover et al., 2000; Lee et al., 2006; Winstanley et al., 2009; Roy et al., 2010; Jeukens et al., 2014), knowledge on genome evolution and the genomic requirements for opportunistic and CF infections is limited. In fact, sequenced strains reported to date (www.pseudomonas.com, Winsor et al., 2011) were randomly selected and are unlikely to reflect population diversity, hence representing an incomplete snapshot of the pathogen, even in the context of CF. Understanding the biology of P. aeruginosa requires the exploration of nontraditional niches in the environment and the widest possible repertoire of opportunistic infections.
The International P. aeruginosa Consortium (IPC)
The science of genomics applied to opportunistic infections via whole bacterial genome sequencing promises to transform the practice of clinical microbiology. With rapidly falling costs and turnaround times, microbial genome sequencing and analysis are becoming a viable strategy to understand CF lung infections as well as other human and animal infections. The objective of the International P. aeruginosa Consortium (IPC) is to sequence a minimum of 1000 P. aeruginosa genomes, link the data to the Pseudomonas Genome Database (www.pseudomonas.com, Winsor et al., 2011), integrate the information with the Canadian CF registry and develop a user-friendly pipeline to study these genomes. Genomics data will support molecular epidemiology for the surveillance of outbreaks and has the potential for future genotypic antimicrobial susceptibility testing as well as the identification of novel therapeutic targets and prognostic markers. This project is supported by an international consortium from five continents; its outcomes will have worldwide dissemination for the benefit of clinical microbiology and especially for CF patients.
Sequencing Over 1000 P. aeruginosa Genomes: Objectives and Strategy
By generating a comprehensive genome sequence database truly representative of the worldwide P. aeruginosa population, we will:
(1) Assemble a large and representative strain collection, with associated genome data, useful for antimicrobial testing, identification of resistance markers, and data mining for new therapeutic targets;
(2) Develop platforms and pipelines to enable synergy between genomic and clinical data, which will allow identification of prognostic markers and stratification of patients, leading to improvements in patient care;
(3) Transform CF diagnostic microbiology through innovations in genomics;
(4) Develop user-friendly tools that will enable CF clinicians to interpret genomic data leading to better informed decisions on issues of cross infection.
Our working hypothesis is that a high-quality, large-scale bacterial genome database available through a user-friendly pipeline will have a major impact on epidemiology, diagnostics and treatment. A major objective is to identify representative isolates from groups of closely related genomes to become reference type isolates and provide reference type genomic data. To this end, genomes are initially sequenced on an Illumina MiSeq instrument with an average median coverage of 40. This first sequencing step will help to determine whether there is already a good reference genome for each new genome by investigating both core and accessory genetic material. First, for conserved regions among isolates, i.e., the core genome, we will perform phylogenetic analysis and identify single nucleotide polymorphisms (SNPs). Second, we will determine whether a new genome significantly contributes to expansion of the full genetic repertoire of P. aeruginosa (i.e., the pan-genome), for instance, with at least 0.1% of its genome (about 6000 bp) representing previously unknown accessory genetic material. It will also be possible to identify indels and structural variations among genomes. We will analyze genomes for the presence of genomic islands using IslandViewer and related genome-comparison and sequence-composition based tools (Langille and Brinkman, 2009; Grant et al., 2012; Dhillon et al., 2015). Finally, each new genome will be characterized based on the presence/absence of regions of genome plasticity (Klockgether et al., 2011), the virulence factor (VF) database (Barken et al., 2008), curated VF data at www.pseudomonas.com, and the Comprehensive Antibiotic Resistance Database (McArthur et al., 2013). In light of this information, a limited set of new reference genomes will eventually be selected for PACBIO RS II sequencing to enable full assembly and detailed annotation.
The International P. aeruginosa Consortium Database (IPCD)
The IPC's strain collection is harbored at the Institute for integrative and systems biology (IBIS), in Quebec City, Canada. It currently contains 1514 entries for P. aeruginosa isolates spanning 135 years back to 1880 and covering 85 locations, in 35 countries, on five continents. It includes previously described collections (Pirnay et al., 2009; Stewart et al., 2011; Kidd et al., 2012) and was assembled with the aim of representing maximal genomic diversity. To this end, various criteria were taken into consideration, including geographic origin, previous genotyping, phenotype, and in vivo behavior. We envisage that the collection can accommodate over 10,000 isolates.
In order to manage phenotypic and genomic data for the growing P. aeruginosa collection in addition to sharing this data, we created the International P. aeruginosa Consortium Database (IPCD), an open source web application available at http://ipcd.ibis.ulaval.ca/. It includes isolate identification, host, researcher, date of isolation, geographical origin, phenotypic data, anonymized patient information, DNA extraction details, NGS information and genome assembly. Its structured vocabulary is being developed further. IPCD currently contains NGS data and unpublished draft genomes from CF patients and from most of the other types of known human infections. For comparative genomics purposes, IPCD also contains animal infection isolates and environmental isolates from plants, soil and water. Researchers who provided strains have priority access to corresponding genome sequences through personal user accounts, but all genome sequences produced by the IPC will become publically available.
The IBIS Bioinformatics Pipeline for Bacterial Genome Assembly
Analysis software for genome assembly and selection of additional reference genomes is required to extract relevant information in a fully automated and reliable fashion without human intervention. Ideally, this software should be platform independent and analyze sequence data directly without being tied to proprietary data formats. This ensures maximal flexibility and reduces lag time to a minimum. We are currently using an integrated pipeline for de novo assembly of microbial genomes based on the A5 pipeline (Tritt et al., 2012) and parallelized on a Silicon Graphics UV 100 to accommodate data from 96 genomes and provide assembly statistics in about 30 h. This automated approach currently results in 20–60 contigs per genome (median N50 = 415 kb) and is anticipated to improve as sequencing technology improves.
Phylogeny of P. aeruginosa
P. aeruginosa is well known to have an adaptable genome (5.5–7 Mbps) that enables it to colonize a wide range of ecological niches; comparative genomics approaches have identified changes in surface antigens, loss of virulence-associated traits, increased antibiotic resistance, inserted genomic islands including phage, and pyocin operons, overproduction of alginate and the modulation of metabolic pathways. Its genome also has many regions that exhibit plasticity (Klockgether et al., 2011). The IPC will provide fine-scale analyses to evaluate these changes, allowing the comparison of VFs, loss/gain of function mutations and antibiotic resistance genes as well as complete core and accessory genomes.
As a proof of concept for this aim, we used the 389 genomes that constitute our current sequence dataset, including the 335 draft genomes produced to date by the IPC, to perform phylogenetic analysis of the core genome using the Harvest suite (Treangen et al., 2014). We found that P. aeruginosa strains can be divided into three major groups (Figure 1A), a result providing concrete support and agreement to previous results with a limited but diverse set of 55 strains (Stewart et al., 2014). The tree presented here also provides novel information since it shows new subgroups and provides unmatched resolution. The high number of strains included in our analysis and the fact that these strains come from a wide array of sources (including environmental, clinical and animal strains, and a wide geographical spread) may suggest that group 1 strains, including PAO1, are naturally more abundant than group 2, which includes PA14. However, it is noteworthy that the opposite conclusion was reached in a previous typing-based study (Wiehlmann et al., 2007), and that our dataset is biased toward clinical isolates. Further, the third major group that includes strain PA7 is now populated with 12 new strains. Given that phylogenetic analysis of 389 genomes has produced at least 20 distinct branches, it is clear that a more extensive survey sequencing approach, as proposed by the IPC, is required to populate these branches. Data analysis with Harvest also revealed that the core genome represents 17.5% of the average P. aeruginosa genome size. This is much less than what previous studies, which typically do not include group 3 strains, have reported (e.g., 79% in Dettman et al., 2015), and is due to a combined effect of diversity and number of strains, as it can be deduced from Figure 1B. Inclusion of sister species P. resinovorans in this analysis resulted in a drop of core genome coverage to 2.4%. Figure 1C presents the number of core genome SNPs.
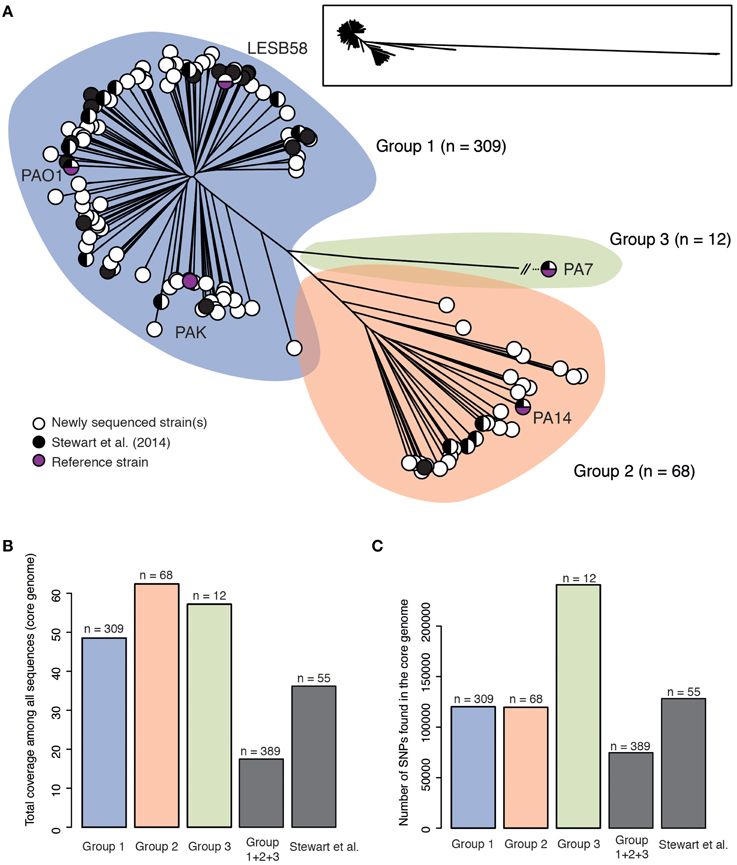
Figure 1. (A) Unrooted maximum likelihood tree of 389 Pseudomonas aeruginosa genomes based on SNPs within the core genome as defined by Harvest (100 bootstraps). Strains are divided into three major groups (group 1: blue, group 2: orange and group 3: green). The number of strains for each group is shown. Black circles represent strains that were already sequenced before this study while white circles represent one or more strains that were sequenced in this study. Group 3 was contracted for visualization purposes; a framed miniature of the true appearance of this tree is presented. The tree in Newick format is available as Supplementary Data Sheet 1 (B) Total coverage of the P. aeruginosa genome by the core genome for each of the three groups shown in (A), all 389 genomes (Group 1+2 + 3) and a diverse set of 55 strains from Stewart et al. (2014). (C) Total number of core genome SNPs for each of the three groups shown in (A), all 389 genomes (Group 1+2 + 3) and a diverse set of 55 strains from Stewart et al. (2014).
Linking IPCD with Pseudomonas.com and the Comprehensive Antibiotic Resistance Database
The Pseudomonas Genome Database (www.pseudomonas.com; Winsor et al., 2011) is an established platform for searching and comparing multiple genome sequences and annotations for Pseudomonas species. This publically available database hosts sequence and annotation data including orthologs, function, expression, cross references, and various predictions for 70 complete Pseudomonas genomes plus draft genomes for more than 561 additional P. aeruginosa isolates. It provides ongoing high-quality curated updates to existing annotations based on community involvement. The IPCD will draw on existing tools such as Sybil that have been developed for both carrying out comparative analyses and presenting data over the web (Riley et al., 2012). Reliable methods for the phylogenetic analysis of our dataset are used including analysis of core genome SNPs using Harvest (Treangen et al., 2014). Close attention to the links between the presence of strain specific genomic islands and patterns of SNPs in the core genome will help identify diagnostic sequences and SNP combinations for the development of new P. aeruginosa typing methods with the highest resolution to date. This will be done using a combination of de novo island prediction using IslandViewer (Langille and Brinkman, 2009; Dhillon et al., 2015) and analysis using the CG View Comparison Tool (Grant et al., 2012).
As an additional feature, we plan to link IPCD with The Comprehensive Antibiotic Resistance Database (CARD; McArthur et al., 2013) available at http://arpcard.mcmaster.ca/, which provides data for ~3000 antibiotic resistance genes and is under continuous curation efforts. Here, for instance, we used the CARD reference data to detect the presence of resistance genes through Resistance Gene Identifer searches (McArthur et al., 2013) on 389 P. aeruginosa strains (Figure 2). Approximately 40% of the 73 detected resistance genes were found in a majority of strains, including genes involved in transport, efflux and permeability as well as genes involved in beta-lactam resistance. Approximately 60% of the resistance genes we detected were found only in a restricted group of strains, particularly for aminoglycoside, macrolide, and sulfonamide resistance, highlighting the great variability of P. aeruginosa strains with respect to resistance genes. This variation is now being unraveled thanks to our extensive sampling. These data will be used to study and understand the pool of resistance genes present in clinical strains with a particular focus on CF strains, and to understand the links between clinical and environmental strains with respect to these genes.
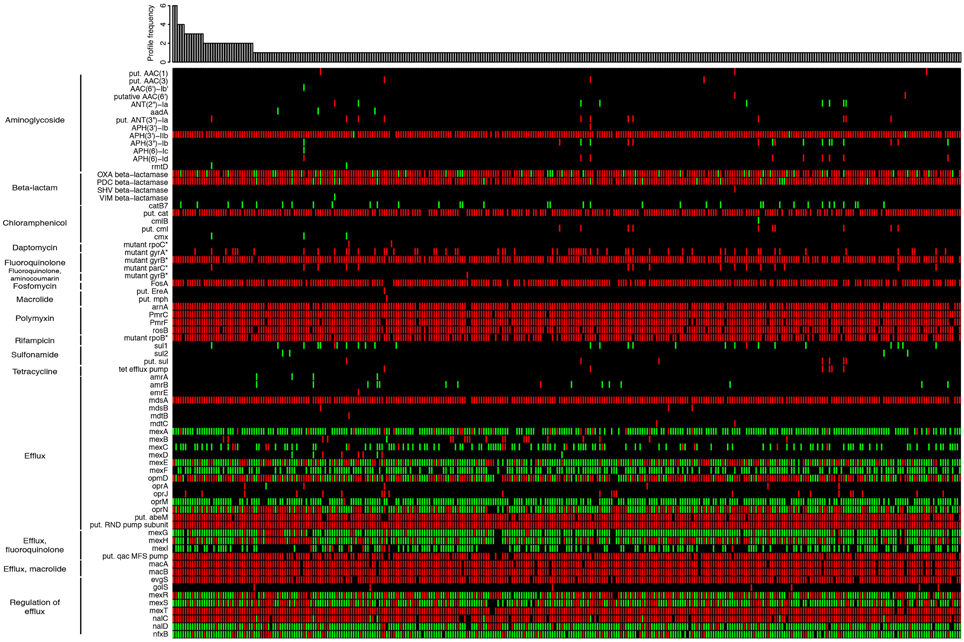
Figure 2. Heat map showing the unique distribution profiles of antibiotic resistance genes for 389 Pseudomonas aeruginosa strains (black: no sequence matching the protein; green: perfect match to known antimicrobial resistance (AMR) gene sequence; red: variant of known AMR gene sequence). The heat map was obtained by performing a Resistance Gene Identifier (RGI) analysis against reference sequences of the Comprehensive Antibiotic Resistance Database (CARD; McArthur et al., 2013). The bar plot shows in how many strains each profile was observed. On the left, proteins are grouped according to their biological function or the resistance they confer. In rare cases, more than a single copy of a resistance gene may be present within an individual strain. For those genes with resistance conferred by mutation (labeled with an asterisk), all detected mutations are known from other pathogens and may require functional verification in P. aeruginosa. Genes labeled as “putative” (“put.” in the figure) are similar to a number of known sequence variants within a family of AMR genes. All perfect matches to OXA β-lactamases are OXA-50. The complete heat map with the full set of P. aeruginosa strains is available in Supplementary Image 1. The raw data used to generate the heat map is available as Supplementary Table 1.
Linking Genomic and Clinical Data
It will be essential to match phenotypic and clinical data (antibiotic resistance, virulence, anonymized clinical observations) to the genomic data produced. We will categorize data within the IPCD so that isolates can be sorted by phenotype, allowing rapid identification of linked genomic signatures and the development of prognostic approaches to treat CF infections. The development of a pipeline to initially map “new” P. aeruginosa genomes will evolve toward becoming a routine clinical tool for the use of genomic data in CF and could represent a very powerful sentinel surveillance system. We will develop tools to rapidly collate data for a given strain type and produce a concise phenotypic and clinical profile that provides clinicians with an evidence based decision making platform. The Canadian CF Registry was created so that data entry could be standardized in CF clinics across Canada. We will use this system to link IPCD with clinical data for CF isolates.
Future Genomic Analyses and Biological Studies of P. aeruginosa
We are committed to continuously improve the IPCD and pseudomonas.com by adding P. aeruginosa genomes from other human, animal, and environmental isolates as well as by enhancing metadata. The consortium has identified a number of research priorities in the international P. aeruginosa community, many of which will be long-term projects shared among members of the consortium as a research working group. The goal of the IPC is to avoid duplication of efforts in P. aeruginosa genomics and enhance interest from researchers having common goals. Additional members are welcome to join in so that the “depth and breadth” of P. aeruginosa biology expands beyond what we outlined for the initial consortium. We also intend to seek collaboration with other groups to connect our database with those developed for other Pseudomonad genomes. Finally, the IPC could become a model for other groups interested in the bacterial genomics of infectious diseases, as the combination of large-scale genomics and evolutionary biology tools may lead to new strategies for countering infections (Little et al., 2012; Casadevall and Pirofski, 2014).
Author Contributions
LF, JJ, IK, and RL collected Pseudomonas strains, performed the analyses and drafted the manuscript. BB provided support for sequencing and analysis. MD, JL, SL, and HM contributed to the genome assembly pipeline and the development of IPCD. AM, BJ, SP, and NW performed the resistome analysis. GW and FB provided database input for links with www.pseudomonas.com. All other authors handled Pseudomonas strains and collected metadata. All authors revised the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We express our gratitude to members of the Plateforme d'analyses génomiques and bioinformatics platform at IBIS. We also thank Tariq Elsayegh (Royal College of Surgeons in Ireland) for his assistance with annotation of polymyxin resistance genes. JJ is supported by a Cystic Fibrosis Canada postdoctoral fellowship. RL is funded by Cystic Fibrosis Canada and by a CIHR-UK team grant. CW, JF, and MM are supported by the UK Cystic Fibrosis Trust. CW and NL would like to acknowledge funding from Fight for Sight. JB was supported by NIH grant P30 DK089507. SB is supported by a Queensland Health Fellowship. SB, TK, PR, and KG were supported by grants by NHMRC (#455919) and the TPCH Foundation. TK is the recipient of an ERS-EU RESPIRE2 Marie Skłodowska-Curie Postdoctoral Research Fellowship—MC RESPIRE2 1st round 4571-2013. IL is supported by grants from CureKids, Cystic Fibrosis New Zealand, and the New Zealand Lotteries Board (Health). AM holds a Cisco Research Chair in Bioinformatics, supported by Cisco Systems Canada, Inc. GD Wright's laboratory is funded by the Canadian Institutes of Health Research (CIHR), the Natural Sciences and Engineering Research Council (NSERC), and a Canada Research Chair. NW is supported by a CIHR Doctoral research award. GW and FB acknowledge Cystic Fibrosis Foundation Therapeutics and Genome Canada for support. ED's laboratory is supported by Canadian Institutes of Health Research (CIHR) operating grant MOP-142466 and a Canada Research Chair.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2015.01036
Supplementary Table 1. Complete antibiotic resistance proteins matrix.
Supplementary Data Sheet 1. Core genome phylogenetic tree (FigTree).
Supplementary Image 1. Complete antibiotic resistance proteins heat map.
References
Barken, K. B., Pamp, S. J., Yang, L., Gjermansen, M., Bertrand, J. J., Klausen, M., et al. (2008). Roles of type IV pili, flagellum-mediated motility and extracellular DNA in the formation of mature multicellular structures in Pseudomonas aeruginosa biofilms. Environ. Microbiol. 10, 2331–2343. doi: 10.1111/j.1462-2920.2008.01658.x
Casadevall, A., and Pirofski, L.-A. (2014). Microbiology: ditch the term pathogen. Nature 516, 165–166. doi: 10.1038/516165a
D'Costa, V. M., McGrann, K. M., Hughes, D. W., and Wright, G. D. (2006). Sampling the antibiotic resistome. Science 311, 374–377. doi: 10.1126/science.1120800
Dettman, J. R., Rodrigue, N., and Kassen, R. (2015). Genome-wide patterns of recombination in the opportunistic human pathogen Pseudomonas aeruginosa. Genome Biol. Evol. 7, 18–34. doi: 10.1093/gbe/evu260
Dhillon, B. K., Laird, M. R., Shay, J. A., Winsor, G. L., Lo, R., Nizam, F., et al. (2015). IslandViewer 3: more flexible, interactive genomic island discovery, visualization and analysis. Nucleic Acids Res. 43, W104–W108. doi: 10.1093/nar/gkv401
Emerson, J., Rosenfeld, M., McNamara, S., Ramsey, B., and Gibson, R. L. (2002). Pseudomonas aeruginosa and other predictors of mortality and morbidity in young children with cystic fibrosis. Pediatr. Pulmonol. 34, 91–100. doi: 10.1002/ppul.10127
Fothergill, J. L., White, J., Foweraker, J. E., Walshaw, M. J., Ledson, M. J., Mahenthiralingam, E., et al. (2010). Impact of Pseudomonas aeruginosa genomic instability on the application of typing methods for chronic cystic fibrosis infections. J. Clin. Microbiol. 48, 2053–2059. doi: 10.1128/JCM.00019-10
Gellatly, S. L., and Hancock, R. E. (2013). Pseudomonas aeruginosa: new insights into pathogenesis and host defenses. Pathog. Dis. 67, 159–173. doi: 10.1111/2049-632X.12033
Grant, J. R., Arantes, A. S., and Stothard, P. (2012). Comparing thousands of circular genomes using the CGView comparison tool. BMC Genomics 13:202. doi: 10.1186/1471-2164-13-202
Hauser, A. R., Jain, M., Bar-Meir, M., and McColley, S. A. (2011). Clinical significance of microbial infection and adaptation in cystic fibrosis. Clin. Microbiol. Rev. 24, 29–70. doi: 10.1128/CMR.00036-10
Heltshe, S. L., Mayer-Hamblett, N., Burns, J. L., Khan, U., Baines, A., Ramsey, B. W., et al. (2015). Pseudomonas aeruginosa in cystic fibrosis patients with G551D-CFTR treated with ivacaftor. Clin. Infect. Dis. 60, 703–712. doi: 10.1093/cid/ciu944
Hilker, R., Munder, A., Klockgether, J., Losada, P. M., Chouvarine, P., Cramer, N., et al. (2015). Interclonal gradient of virulence in the Pseudomonas aeruginosa pangenome from disease and environment. Environ. Microbiol. 17, 29–46. doi: 10.1111/1462-2920.12606
Jeukens, J., Boyle, B., Kukavica-Ibrulj, I., Ouellet, M. M., Aaron, S. D., Charette, S. J., et al. (2014). Comparative genomics of isolates of a Pseudomonas aeruginosa epidemic strain associated with chronic lung infections of cystic fibrosis patients. PLoS ONE 9:e87611. doi: 10.1371/journal.pone.0087611
Keays, T., Ferris, W., Vandemheen, K. L., Chan, F., Yeung, S. W., Mah, T. F., et al. (2009). A retrospective analysis of biofilm antibiotic susceptibility testing: a better predictor of clinical response in cystic fibrosis exacerbations. J. Cyst. Fibros. 8, 122–127. doi: 10.1016/j.jcf.2008.10.005
Kidd, T. J., Ritchie, S. R., Ramsay, K. A., Grimwood, K., Bell, S. C., and Rainey, P. B. (2012). Pseudomonas aeruginosa exhibits frequent recombination, but only a limited association between genotype and ecological setting. PLoS ONE 7:e44199. doi: 10.1371/journal.pone.0044199
Kiewitz, C., and Tümmler, B. (2000). Sequence diversity of Pseudomonas aeruginosa: impact on population structure and genome evolution. J. Bacteriol. 182, 3125–3135. doi: 10.1128/JB.182.11.3125-3135.2000
Klockgether, J., Cramer, N., Wiehlmann, L., Davenport, C. F., and Tümmler, B. (2011). Pseudomonas aeruginosa genomic structure and diversity. Front. Microbiol. 2:150. doi: 10.3389/fmicb.2011.00150
Lam, J. S., Taylor, V. L., Islam, S. T., Hao, Y., and Kocincová, D. (2011). Genetic and functional diversity of Pseudomonas aeruginosa Lipopolysaccharide. Front. Microbiol. 2:118. doi: 10.3389/fmicb.2011.00118
Langille, M. G., and Brinkman, F. S. (2009). IslandViewer: an integrated interface for computational identification and visualization of genomic islands. Bioinformatics 25, 664–665. doi: 10.1093/bioinformatics/btp030
Lee, D. G., Urbach, J. M., Wu, G., Liberati, N. T., Feinbaum, R. L., Miyata, S., et al. (2006). Genomic analysis reveals that Pseudomonas aeruginosa virulence is combinatorial. Genome Biol. 7:R90. doi: 10.1186/gb-2006-7-10-r90
Lee, T. W. (2009). Eradication of early Pseudomonas infection in cystic fibrosis. Chron. Respir. Dis. 6, 99–107. doi: 10.1177/1479972309104661
Little, T. J., Allen, J. E., Babayan, S. A., Matthews, K. R., and Colegrave, N. (2012). Harnessing evolutionary biology to combat infectious disease. Nat. Med. 18, 217–220. doi: 10.1038/nm.2572
Lyczak, J. B., Cannon, C. L., and Pier, G. B. (2000). Establishment of Pseudomonas aeruginosa infection: lessons from a versatile opportunist. Microbes Infect. 2, 1051–1060. doi: 10.1016/S1286-4579(00)01259-4
Martin, K., Baddal, B., Mustafa, N., Perry, C., Underwood, A., Constantidou, C., et al. (2013). Clusters of genetically similar isolates of Pseudomonas aeruginosa from multiple hospitals in the UK. J. Med. Microbiol. 62, 988–1000. doi: 10.1099/jmm.0.054841-0
Marvig, R. L., Sommer, L. M., Jelsbak, L., Molin, S., and Johansen, H. K. (2015). Evolutionary insight from whole-genome sequencing of Pseudomonas aeruginosa from cystic fibrosis patients. Future Microbiol. 10, 599–611. doi: 10.2217/fmb.15.3
McArthur, A. G., Waglechner, N., Nizam, F., Yan, A., Azad, M. A., Baylay, A. J., et al. (2013). The comprehensive antibiotic resistance database. Antimicrob. Agents Chemother. 57, 3348–3357. doi: 10.1128/AAC.00419-13
Mowat, E., Paterson, S., Fothergill, J. L., Wright, E. A., Ledson, M. J., Walshaw, M. J., et al. (2011). Pseudomonas aeruginosa population diversity and turnover in cystic fibrosis chronic infections. Am. J. Respir. Crit. Care Med. 183, 1674–1679. doi: 10.1164/rccm.201009-1430OC
Perry, J. A., and Wright, G. D. (2013). The antibiotic resistance “mobilome”: searching for the link between environment and clinic. Front. Microbiol. 4:138. doi: 10.3389/fmicb.2013.00138
Pirnay, J. P., Bilocq, F., Pot, B., Cornelis, P., Zizi, M., van Eldere, J., et al. (2009). Pseudomonas aeruginosa population structure revisited. PLoS ONE 4:e7740. doi: 10.1371/journal.pone.0007740
Pirnay, J. P., De Vos, D., Cochez, C., Bilocq, F., Vanderkelen, A., Zizi, M., et al. (2002). Pseudomonas aeruginosa displays an epidemic population structure. Environ. Microbiol. 4, 898–911. doi: 10.1046/j.1462-2920.2002.00321.x
Riley, D. R., Angiuoli, S. V., Crabtree, J., Dunning Hotopp, J. C., and Tettelin, H. (2012). Using Sybil for interactive comparative genomics of microbes on the web. Bioinformatics 28, 160–166. doi: 10.1093/bioinformatics/btr652
Roy, P. H., Tetu, S. G., Larouche, A., Elbourne, L., Tremblay, S., Ren, Q., et al. (2010). Complete genome sequence of the multiresistant taxonomic outlier Pseudomonas aeruginosa PA7. PLoS ONE 5:e8842. doi: 10.1371/journal.pone.0008842
Silby, M. W., Winstanley, C., Godfrey, S. A., Levy, S. B., and Jackson, R. W. (2011). Pseudomonas genomes: diverse and adaptable. FEMS Microbiol. Rev. 35, 652–680. doi: 10.1111/j.1574-6976.2011.00269.x
Stephenson, A. L., Tom, M., Berthiaume, Y., Singer, L. G., Aaron, S. D., Whitmore, G. A., et al. (2015). A contemporary survival analysis of individuals with cystic fibrosis: a cohort study. Eur. Respir. J. 45, 670–679. doi: 10.1183/09031936.00119714
Stewart, L., Ford, A., Sangal, V., Jeukens, J., Boyle, B., Kukavica-Ibrulj, I., et al. (2014). Draft genomes of 12 host-adapted and environmental isolates of Pseudomonas aeruginosa and their positions in the core genome phylogeny. Pathog. Dis. 71, 20–25. doi: 10.1111/2049-632X.12107
Stewart, R. M., Wiehlmann, L., Ashelford, K. E., Preston, S. J., Frimmersdorf, E., Campbell, B. J., et al. (2011). Genetic characterization indicates that a specific subpopulation of Pseudomonas aeruginosa is associated with keratitis infections. J. Clin. Microbiol. 49, 993–1003. doi: 10.1128/JCM.02036-10
Stover, C. K., Pham, X. Q., Erwin, A. L., Mizoguchi, S. D., Warrener, P., Hickey, M. J., et al. (2000). Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406, 959–964. doi: 10.1038/35023079
Treangen, T. J., Ondov, B. D., Koren, S., and Phillippy, A. M. (2014). The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 15:524. doi: 10.1186/s13059-014-0524-x
Tritt, A., Eisen, J. A., Facciotti, M. T., and Darling, A. E. (2012). An integrated pipeline for de novo assembly of microbial genomes. PLoS ONE 7:e42304. doi: 10.1371/journal.pone.0042304
Wiehlmann, L., Wagner, G., Cramer, N., Siebert, B., Gudowius, P., Morales, G., et al. (2007). Population structure of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U.S.A. 104, 8101–8106. doi: 10.1073/pnas.0609213104
Williams, D., Evans, B., Haldenby, S., Walshaw, M. J., Brockhurst, M. A., Winstanley, C., et al. (2015). Divergent, coexisting, Pseudomonas aeruginosa lineages in chronic cystic fibrosis lung infections. Am. J. Respir. Crit. Care Med. 191, 775–785. doi: 10.1164/rccm.201409-1646OC
Winsor, G. L., Lam, D. K. W., Fleming, L., Lo, R., Whiteside, M. D., Yu, N. Y., et al. (2011). Pseudomonas genome database: improved comparative analysis and population genomics capability for Pseudomonas genomes. Nucleic Acids Res. 39, D596–D600. doi: 10.1093/nar/gkq869
Winstanley, C., Langille, M. G., Fothergill, J. L., Kukavica-Ibrulj, I., Paradis-Bleau, C., Sanschagrin, F., et al. (2009). Newly introduced genomic prophage islands are critical determinants of in vivo competitiveness in the liverpool epidemic strain of Pseudomonas aeruginosa. Genome Res. 19, 12–23. doi: 10.1101/gr.086082.108
Keywords: Pseudomonas aeruginosa, next-generation sequencing, bacterial genome, phylogeny, database, cystic fibrosis, antibiotic resistance, clinical microbiology
Citation: Freschi L, Jeukens J, Kukavica-Ibrulj I, Boyle B, Dupont M-J, Laroche J, Larose S, Maaroufi H, Fothergill JL, Moore M, Winsor GL, Aaron SD, Barbeau J, Bell SC, Burns JL, Camara M, Cantin A, Charette SJ, Dewar K, Déziel É, Grimwood K, Hancock REW, Harrison JJ, Heeb S, Jelsbak L, Jia B, Kenna DT, Kidd TJ, Klockgether J, Lam JS, Lamont IL, Lewenza S, Loman N, Malouin F, Manos J, McArthur AG, McKeown J, Milot J, Naghra H, Nguyen D, Pereira SK, Perron GG, Pirnay J-P, Rainey PB, Rousseau S, Santos PM, Stephenson A, Taylor V, Turton JF, Waglechner N, Williams P, Thrane SW, Wright GD, Brinkman FSL, Tucker NP, Tümmler B, Winstanley C and Levesque RC (2015) Clinical utilization of genomics data produced by the international Pseudomonas aeruginosa consortium. Front. Microbiol. 6:1036. doi: 10.3389/fmicb.2015.01036
Received: 13 May 2015; Accepted: 11 September 2015;
Published: 29 September 2015.
Edited by:
John R. Battista, Louisiana State University, USAReviewed by:
Awdhesh Kalia, University of Texas MD Anderson Cancer Center, USASuleyman Yildirim, Istanbu Medipol University International School of Medicine, Turkey
Copyright © 2015 Freschi, Jeukens, Kukavica-Ibrulj, Boyle, Dupont, Laroche, Larose, Maaroufi, Fothergill, Moore, Winsor, Aaron, Barbeau, Bell, Burns, Camara, Cantin, Charette, Dewar, Déziel, Grimwood, Hancock, Harrison, Heeb, Jelsbak, Jia, Kenna, Kidd, Klockgether, Lam, Lamont, Lewenza, Loman, Malouin, Manos, McArthur, McKeown, Milot, Naghra, Nguyen, Pereira, Perron, Pirnay, Rainey, Rousseau, Santos, Stephenson, Taylor, Turton, Waglechner, Williams, Thrane, Wright, Brinkman, Tucker, Tümmler, Winstanley and Levesque. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roger C. Levesque, Institute for Integrative and Systems Biology, Université Laval, 1030 Avenue de la Médecine, Quebec, QC G1E 7A9, Canada,cmNsZXZlc3FAaWJpcy51bGF2YWwuY2E=
†These authors have contributed equally to this work.