 Martin Lindmeyer1
Martin Lindmeyer1 Susann Müller
Susann Müller Bruno Bühler
Bruno Bühler- 1Laboratory of Chemical Biotechnology, Department of Biochemical and Chemical Engineering, TU Dortmund University, Dortmund, Germany
- 2Helmholtz Centre for Environmental Research - UFZ, Department for Environmental Microbiology, Leipzig, Germany
- 3Helmholtz Centre for Environmental Research - UFZ, Department of Solar Materials, Leipzig, Germany
Pivotal challenges in industrial biotechnology are the identification and overcoming of cell-to-cell heterogeneity in microbial processes. While the development of subpopulations of isogenic cells in bioprocesses is well described (intra-population variability), a possible variability between genetically identical cultures growing under macroscopically identical conditions (clonal variability) is not. A high such clonal variability has been found for the recombinant expression of the styrene monooxygenase genes styAB from Pseudomonas taiwanensis VLB120 in solvent-tolerant Pseudomonas putida DOT-T1E using the alk-regulatory system from P. putida GPo1. In this study, the oxygenase subunit StyA fused to eGFP was used as readout tool to characterize the population structure in P. putida DOT-T1E regarding recombinant protein content. Flow cytometric analyses revealed that in individual cultures, at least two subpopulations with highly differing recombinant StyA-eGFP protein contents appeared (intra-population variability). Interestingly, subpopulation sizes varied from culture-to-culture correlating with the specific styrene epoxidation activity of cells derived from respective cultures (clonal variability). In addition, flow cytometric cell sorting coupled to plasmid copy number (PCN) determination revealed that detected clonal variations cannot be correlated to the PCN, but depend on the combination of the regulatory system and the host strain employed. This is, to the best of our knowledge, the first work reporting that intra-population variability (with differing protein contents in the presented case study) causes clonal variability of genetically identical cultures. Respective impacts on bioprocess reliability and performance and strategies to overcome respective reliability issues are discussed.
Introduction
The use of whole-cell biocatalysts is a promising approach for applications in industrial biotechnology and is favored over the use of isolated enzymes, when host intrinsic cofactor regeneration, degradation of reactive oxygen species, and/or continuous synthesis of instable enzymes are required (Schmid et al., 2001; Woodley, 2006; Leak et al., 2009; Faber, 2011; Schrewe et al., 2013). The still limited understanding of intracellular regulatory network operation and causes for variability in isogenic microbial cultures often restricts industrial applications (Kuhn et al., 2010a). The overall productivity and stability of a bioprocess strongly relies on the physiology and metabolic performance of the microbial host. A major critical aspect is the occurrence of phenotypically diverse cells or subpopulations (described by the term “phenotypic heterogeneity”), which contribute differently to the production, conversion, or degradation of a desired product or substrate and thus hampering process efficiency (Achilles et al., 2007; Delvigne and Goffin, 2014). Thereby, variations in protein contents or variable expression patterns among different cells in an isogenic population appear (Elowitz et al., 2002; Fraser and Kaern, 2009; Jahn et al., 2012). Phenotypic heterogeneity often arises from stochastic variations in regulatory network operation and can be linked to variations in the amounts of intracellular compounds involved in gene expression such as transcription factors or inhibitors (Ryall et al., 2012). These variations are attributed to two different types of noise: extrinsic and intrinsic noise (Elowitz et al., 2002). Extrinsic noise arises from differing predispositions regarding the availability of molecules with global relevance for regulatory network operation, such as biomolecules involved in the biomass synthesis machinery (ribosomes, DNA-polymerases, RNA-polymerases). In contrast, intrinsic noise does not depend on regulatory predispositions but results from stochastic events, e.g., the frequency of collision/contact between the individual functional units involved in gene expression (inducer, promotor, regulator). Both intrinsic and extrinsic noise jointly contribute to phenotypic heterogeneity, whereas extrinsic noise was shown to make a greater contribution to total gene expression variability than intrinsic noise (Elowitz et al., 2002; Kaern et al., 2005; Avery, 2006).
Bimodality resulting from differential gene expression is a well described phenomenon (Novick and Weiner, 1957; Veening et al., 2006). Simplified, after induction of a certain regulatory system, one subpopulation contains a low level of a distinct protein signaling an OFF-state, whereas another subpopulation surpasses a threshold in the respective protein level signaling an ON-state. Resulting subpopulations and respective dynamics can be detected and characterized via flow cytometry in combination with protein marker molecules (Diaz et al., 2010; Müller et al., 2010). In contrast to bimodality, where cells can fluctuate between two states depending on the threshold signal concentration for activation, bistability describes a binary system in which the state of the regulatory system is maintained even after the trigger input is gone (Nikel et al., 2013). Such bistability can also result from genetic heterogeneities, e.g., caused by instable plasmid proliferation (Bentley and Quiroga, 1993). We recently have developed a method for the accurate quantification of the plasmid copy number (PCN) in subpopulations differently expressing a fluorescence marker gene (Jahn et al., 2014). For Pseudomonas putida KT2440, the bistability in protein production could be directly attributed to plasmid loss of a large fraction of cells resulting in genetic variability.
Regarding phenotypic heterogeneity, two types of variability can be considered: “intra-population variability” characterized by the development of phenotypically diverse subpopulations within a single isogenic culture and “clonal variability” describing the variability between individual isogenic cultures operated under macroscopically identical conditions.
Common examples for intra-population variability in bioprocessing include the appearance of cells with altered physiological properties in large scale fed-batch bioreactors (Enfors et al., 2001) or the development of a “non-producing” subpopulation under production conditions (Alonso et al., 2012). Most studies addressing phenotypic heterogeneity focus on intra-population variability, its characterization, and respective causes. Only few microbiological studies have targeted clonal variability and its relation to intra-population variability. In case of mammalian cells such as Chinese hamster ovary (CHO) cells used for recombinant gene expression, clones tend to differ markedly in expression after transfection due to variable gene copies in the chromosome or random integration. The occurrence of high producer clones after transfection is rare and respective screening and isolation represents a considerable challenge for the development of industrial applications (Pilbrough et al., 2009). However, the emergence of clonal variability is not restricted to mammalian cells, but also occurs in bacteria for plasmid-based heterologous gene expression. In a previous study, a high clonal variability in recombinant oxygenase levels has been detected for P. putida DOT-T1E and S12 but not for P. putida KT2440, Pseudomonas taiwanensis VLB120, and E. coli JM101 (Lindmeyer et al., 2015), when expression was based on the alk-regulatory system from P. putida GPo1 (van Beilen et al., 1994; Staijen et al., 1999). This variability was found not to depend on the type of heterologous enzyme and substrate/product nor on inducer toxicity or antibiotic resistance mechanisms. However, such clonal variability did not occur, when the lac-regulatory system was used. Thus, the interaction between the host-specific regulatory network and the regulatory system used for recombinant gene expression appeared to play a crucial role, giving evidence for an alk-regulatory system-related extrinsic type of noise as the cause for the observed clonal variability (Lindmeyer et al., 2015).
In this study, we investigated if such clonal variability in recombinant gene expression involves a variable subpopulation structure or relies on a random distribution of a large range of expression levels among individual cells, and if and to what extent such variability is related to PCNs. For this purpose, the styrene monooxygenase (StyAB) of P. taiwanensis VLB120 was chosen as model system. StyAB is composed of an oxygenase (StyA) and a reductase (StyB) component. Whereas, StyB catalyzes the transfer of electrons from NADH to freely diffusible FAD, StyA makes use of resulting FADH2 to reductively activate molecular oxygen and catalyze the epoxidation of styrene and derivatives to corresponding (S)-epoxides at a high enantiomeric access (ee) of ≥99% (Otto et al., 2004). Plasmid-based expression of a styA-eGFP gene fusion (Jahn et al., 2014) was studied regarding specific styrene epoxidation activity and specific fluorescence involving flow cytometry. The host strains P. taiwanensis VLB120, E. coli JM101, P. putida DOT-T1E, and P. putida KT2440 and the alk-, tac-, and lac-regulatory systems were used for styA-eGFP tool validation. For analyzing the interrelation of clonal and intra-population variability, the alk- and lac-regulatory systems were compared in P. putida DOT-T1E.
Materials and Methods
Bacterial Strains, Plasmids, Media, and Chemicals
Unless otherwise stated, all chemicals and solutions were purchased from Sigma Aldrich (Steinheim, Germany) in the highest purity available. Bacterial strains and plasmids used in this study are listed in Table 1.
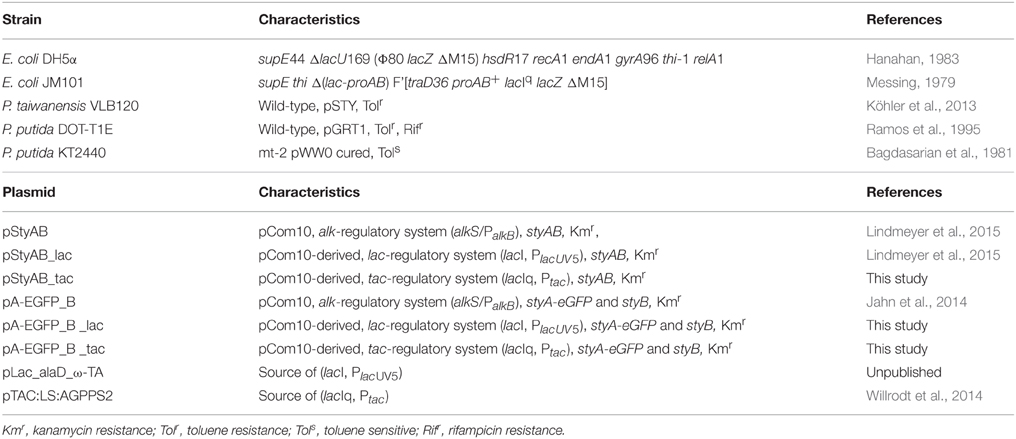
Table 1. Bacterial strains and plasmids used.
Bacteria were either grown on lysogeny broth (Bertani, 1951) composed of 5 g L−1 yeast extract (Difco, Detroit, USA), 5 g L−1 NaCl, and 10 g L−1 Tryptone (Oxoid, Hampshire, United Kingdom) or on minimal M9* medium (25.5g L−1 Na2HPO4•2H2O, 9.0 g L−1 KH2PO4, 1.0 g L−1 NH4Cl, 0.5 g L−1 NaCl) supplemented with 0.2% (v/v) 1M MgSO4 and 0.1% (v/v) US* trace elements solution (4.87 g L−1 FeSO4•7H2O, 4.12 g L−1 CaCl2•7H2O, 1.5 g L−1 MnCl2•4H2O, 1.87g L−1 ZnSO4, 0.84 g L−1 Na2EDTA•2H2O, 0.3 g L−1 H3BO3, 0.25 g L−1 Na2MoO4•2H2O, 0.15 g L−1 CuCl2•2H2O and 82.81 mL 37% fuming HCl). For cultivations with E. coli M9* medium was complemented with 0.5% (w/v) glucose. For the cultivation of Pseudomonas strains in M9* medium, 0.5% (w/v) citrate was used as carbon source to avoid glucose-mediated catabolite repression of the alk-regulatory system (Staijen et al., 1999). M9* medium was adjusted to pH 7.4 using NaOH and, if necessary, supplemented with 50 μg mL−1 kanamycin and 10 mg L−1 thiamine.
DNA Manipulation
Agarose gel-electrophoresis and ligation was performed as described by Sambrook and Russel (2001). Enzymes (Phusion High-Fidelity polymerase, T4 DNA ligase, restriction enzymes) and buffers were purchased from Thermo Scientific (St. Leon-Rot, Germany) and oligonucleotides from Sigma Aldrich (Steinheim, Germany). Plasmids and DNA-fragments were isolated using the peqGOLD plasmid Miniprep Kit I (peqLab, Erlangen, Germany) and purified using NucleoSpin Gel and PCR Clean-up (Macherey-Nagel, Düren, Germany) according to the supplier's protocol. Transformation of E. coli was performed as described (Sambrook and Russel, 2001). Transformation of Pseudomonas strains was performed according to Choi et al. (Choi et al., 2006) by means of electroporation (2500 V, Equibio EasyjecT Prima, Kent, UK).
The plasmid pA-EGFP_B_lac (8802 bp) was based on the pA-EGFP_B backbone, which was isolated via PCR using the primers F5 and B6 (Table 2). The 7161 bp fragment was purified via agarose gel-electrophoresis digested with XbaI and SacI and ligated with purified and identically digested PCR-fragments using F3 and B4 as primers (1655 bp) and pLac_alaD_ω-TA (encoding the lac-regulatory system) as template. pA-EGFP_B_tac (8683 bp) was constructed in analogy to pA-EGFP_B_lac using primers F5 and B6 and pA-EGFP_B as template for the plasmid backbone and primers F1 and B1 and pTAC:LS:AGPPS2 as template for the tac-regulatory system (1546 bp). For the construction of plasmid pStyAB_tac (7948 bp), pStyAB served as PCR template using primers F5 and F6. The resulting 6426 bp fragment was digested with XbaI and SacI and ligated with a purified and identically digested PCR-fragment (1546 bp) isolated via PCR with the primers F1 and B2 and pTAC:LS:AGPPS2 as template. The constructs were verified by digestion with different restriction enzymes and DNA sequencing.
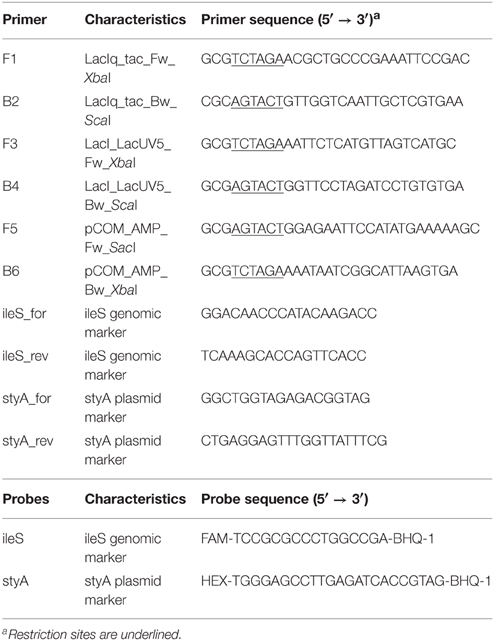
Table 2. Oligonucleotide primers and probes used in this study.
Growth Conditions and Clonal-variability Experiments
Cultivations were carried out in screw-capped, baffled Erlenmeyer shaking flasks at a gas/liquid ratio (v/v) of 9:1, 200 rpm, and 30°C in a Multitron shaker (Infors, Bottmingen, Switzerland). Inoculation and cultivation for clonal variability experiments were carried out as described (Lindmeyer et al., 2015). In short, single colonies (clones) from LB agar plates, which had been incubated for 12–16 h, were picked, grown separately for 6–8 h in liquid LB-precultures, and transferred as a 1% (v/v) inoculum into an overnight M9* preculture, by which the main M9* culture was inoculated to a starting cell concentration of 0.02–0.03 g cell dry weight (CDW) per liter. Recombinant gene expression was induced with 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) or 0.025% (v/v) dicyclopropyl ketone (DCPK) 3.5 h after inoculation for PlacUV5- and Ptac- or PalkB-based expression vectors, respectively. After induction, incubation was continued for 4 h, before cells were harvested by centrifugation and resuspended to 0.5 gCDW L−1 in 50 mM Potassium phosphate (KPi) buffer (pH 7.4) supplemented with 0.5% (w/v) citrate or glucose. Styrene epoxidation assays with nitrogen limited cells (resting-cells) were performed in cell suspension aliquots of 1 mL distributed into 10-mL Teflon sealed Pyrex tubes and incubated for 5 min at 30°C in a rotary shaker (Aquatron, Infors) at 300 rpm. After this equilibration time, the biotransformation was started for 10 min by the addition of 1.5 mM for styrene and stopped by addition of 1 mL ice-cold diethyl ether containing 0.2 mM n-decane as internal standard. After 1 min extraction by mixing and phase separation via centrifugation, the organic phase was transferred into a GC-vial for analysis.
For the quantification of variability in clonal-variability experiments, the following equations were used for the determination of the coefficient of variation (cν) and the experimental error (σexp):
Coefficient of variation:
Experimental Error:
in which σ1, 2, …, n−1, n is the standard deviation of the specific StyAB activity among the tested colonies, the mean specific StyAB activity among the tested colonies, n the amount of tested colonies, the mean specific activity of one colony, and xn, A and xn, B the styrene oxide epoxidation activity of one colony determined in duplicates (A, B). The calculation of the coefficient of variation based on specific eGFP fluorescence (defined as eGFP fluorescence intensity normalized to the cell density in terms of OD450, see below) was carried out using the same equations replacing specific activity with specific fluorescence. Based on the assumption that, for a consistent biocatalyst performance, the coefficient of variation should not exceed 20%, strains were classified to exhibit low clonal variabilities, if the variation did not exceed 20% (Lindmeyer et al., 2015).
Analytical Procedures
Biomass concentrations were determined by monitoring the optical density at 450 nm using a Libra S11 spectrophotometer (Biochrom Ltd., Cambridge, UK). Correlations of the cell dry weight (CDW) concentration with OD450 were reported before and are given in the following per unit OD450: P. putida DOT-T1E: 0.211 gCDW L−1 (Lindmeyer et al., 2015), P. putida KT2440: 0.235 gCDW L−1 (Lindmeyer et al., 2015), E. coli JM101: 0.166 gCDW L−1 (Blank et al., 2008a) and P. taiwanensis VLB120: 0.186 gCDW L−1 (Halan et al., 2011). Styrene and (S)-styrene oxide quantification was performed by GC analysis as described (Kuhn et al., 2010b; Volmer et al., 2014).
For the quantification of the extracellular metabolites glucose, gluconate, and citrate, HPLC analysis was performed using a LaChrome Elite HPLC system (Merck Hitachi, Darmstadt, Germany) equipped with a Trentec 308R-Gel.H column (300x3 mm, Trentec Analysetechnik, Gerlingen, Germany) and coupled to UV (λ = 210 nm) and RI detectors. The following analytic conditions were applied: 40°C, 1.0 mL min−1 isocratic flow rate, 5 mM H2SO4 in demineralized water as mobile phase, 20 μL injection volume.
Specific eGFP fluorescence was measured in microtiter plates using an Infinite M200 reader (Tecan, Mannedorf, Switzerland) at 488 nm and 511 nm for excitation and emission, respectively, with a gain factor of 80. Cells were harvested by centrifugation, washed, resuspended to a cell concentration of ~0.1 gCDW L−1 in PBS Buffer (145 mM NaCl, 1.8 mM NaH2 PO4•H2O, 6 mM Na2HPO4) containing 10% (v/v) glycerol, and stored at −20°C until measurement. Volumes of 200 μL each were transferred to microtiter plates for OD450 measurements (Nunc™ Microwell™ 96-well flat transparent, Nunc, Roskilde, Denmark) and eGFP fluorescence determination (Nunc™ Microwell™ 96-well flat black, Nunc, Roskilde, Denmark). The specific fluorescence [OD] was determined as the ratio of the absolute fluorescence and the respective OD450 value. SDS-PAGE (Laemmli, 1970) was performed to visualize StyA.
Sample Preparation for Flow Cytometry and PCN Determination
Samples for flow cytometry and PCN determination were processed as described above. After resuspension in PBS, cells were fixed in a 1% (v/w) paraformaldehyde (PFA) solution in PBS-buffer for 30 min at room temperature, washed with PBS, and stored in a cryo-protective solution [10% (v/v) glycerol in PBS] at −20°C until analysis. To investigate possible influences of the fixation agent on eGFP fluorescence and PCN, a control experiment with different concentrations of PFA in P. putida DOT-T1E and P. putida KT2440 was performed (Figures S2, S3). Fixation influenced PCN determination, as, upon fixation, generally a lower PCN (25–30%) was detected as compared to unfixed cells (Figure S3).
Flow Cytometry and Cell Sorting
For flow cytometry experiments, fixed cell suspensions were thawed, filtered by a 30 μm CellTrics mesh (Partec-Sysmex, Münster, Germany), and analyzed by a Beckman-Coulter MoFlo cell sorter (Beckman-Coulter, Pasadena, USA) equipped with a 488 nm blue laser (400 mW) for excitation. Forward (FSC) and side scatter (SSC) signals were recorded using a 488/10 nm band pass filter together with a neutral density filter of 2.0, while eGFP fluorescence (FL1) was recorded using a 530/40 nm band pass filter together with a neutral density filter of 0.3. The MoFlo cell sorter was run using the sheath buffer described by Koch et al. (2013) at 2-fold dilution and was calibrated before analysis using fluorescent beads according to Koch et al. (2013) and eGFP-expressing bacterial cells (P. putida KT2440) as a biological standard. Fluorescent (eGFP+) and non-fluorescent (eGFP−) populations were manually gated based on FSC and FL1 intensity, with non-induced P. putida DOT-T1E (pA-EGFP_B) cells as the eGFP-negative reference (see Figure S9, I–IV, 0 h). Cell samples were generally analyzed at a speed of 3000 cells s−1 and sorted into 8-well PCR strips (G003-SF, Kisker Biotech, Steinfurt, Germany) at a lower speed of 200 cells s−1 using the CyCLONE robotic tray (Cytomation, Fort Collins, USA) and the most accurate sorting mode (“single cell,” “one drop”). Wells were prefilled with 7 μL deionized H2O, 1000 cells per well were sorted resulting in a final volume of 8 μL, and sorted samples were immediately deep-frozen at −20°C until digital PCR analysis. As a no-template-control for digital PCR, 1000 beads (Fluoresbrite Bright Blue Microspheres, Ø = 0.5 μm, Polysciences, Warrington, USA) were sorted instead of cells. Data were recorded using Summit v4.3 (Beckman-Coulter) and detailed analysis was performed using the Bioconductor framework for R (Hahne et al., 2009).
PCN Determination by Droplet Digital PCR
PCN determination and calculation were carried out as described by Jahn et al. (2014). Briefly, DNA was extracted from sorted cells by heat treatment at 95°C for 20 min as optimized for P. putida DOT-T1E (Figure S4), followed by cooling on ice. Droplet Digital PCR (ddPCR) was performed using a duplex reaction setup with simultaneous detection of the chromosomal reference gene ileS and the plasmid-located target gene styA using a Taqman assay. Primers as well as FAM- and HEX-labeled probes for ileS and styA are listed in Table 2. A final 20 μL reaction mixture contained 8 μL of template solution, which was completed by adding 12 μL PCR master mix composed of 10 μL 2x ddPCR Supermix (Bio-Rad, Hercules, USA) and 2 μL of a solution containing primers (final concentration of 900 nM) and probes (final concentration of 250 nM for ileS, 125 nM for styA). The reaction mixture was mixed, briefly centrifuged at 500 × g for 3 s and transferred to DG8 cartridges (Bio-Rad) for droplet generation with the QX100 system (Bio-Rad). The resulting 40 μL of droplet emulsion were pipetted into twin.tec 96-well PCR plates (Eppendorf, Hamburg, Germany), sealed, and subjected to PCR using the following program: 95°C for 10 min, 40 cycles of 94°C for 30 s and 58°C for 60 s, and 98°C for 10 min, with a ramp rate of 2.5°C s−1. A Bio-Rad QX100 droplet reader was used for simultaneous detection of FAM and HEX labeled droplets, and PCN was calculated as the ratio of styA and ileS concentration per well.
Results
Characterization of StyA-eGFP Fusion
The fusion construct styA-eGFP (Jahn et al., 2014) was chosen as a tool to investigate clonal variability regarding recombinant gene expression and a possible connectivity with intra-population variability. In this construct, styA, encoding the oxygenase component of the styrene monooxygenase StyAB from P. taiwanensis VLB120, is connected to eGFP via a flexible linker sequence (GGC GGC GGC GGC GGC GCC) integrated between the C-terminus of StyA and the N-terminus of eGFP (Figure S1). The glycine-rich peptide linker (Gly)5Ala thereby functions as a flexible tether minimizing folding interferences between the fluorescence protein and the tagged protein (Tian et al., 2004). As a first step, StyA-eGFP, encoded on the plasmid pA-EGFP_B and expressed under control of the alk-regulatory system, was characterized regarding its catalytic styrene epoxidation performance in whole microbial cells (with the specific styrene epoxidation activity given in U g as readout) by means of resting-cell activity assays (as described in the Materials and Methods section). For this purpose, P. taiwanensis VLB120 was used as host strain, as it is the native host strain of StyAB and was found to show a low clonal variability upon heterologous gene expression under control of the alk-regulatory system (Lindmeyer et al., 2015). Native styAB expressed from an analogous plasmid (pStyAB) served as the control.
P. taiwanensis VLB120 (pA-EGFP_B) and P. taiwanensis VLB120 (pStyAB), cultivated in duplicates starting from two individual colonies of each strain, showed a similar behavior in terms of growth rates and yields on citrate (Figure 1). The specific fluorescence of P. taiwanensis VLB120 (pA-EGFP_B) cultures correlated well with the steadily increasing specific styrene epoxidation activities and SDS-PAGE band intensities. Compared to native StyAB, for which specific activities of 60–80 U g were observed for the time range 4–10 h after induction, a higher activity of 106 ± 3.4 U g was reached with the fusion construct after 10 h of induction. Apparently, expression of the fusion construct was slower as observed by SDS-PAGE band intensities, reaching the maximal level at the beginning of the stationary phase. These results indicate that the styA-eGFP expression rate is reduced compared to styA gene expression, which might be due to the 5 codon repetitions in the linker sequence leading to a slowdown in translation. The fusion, however, appeared to slightly enhance the maximum in vivo oxygenase activity reached after full induction.
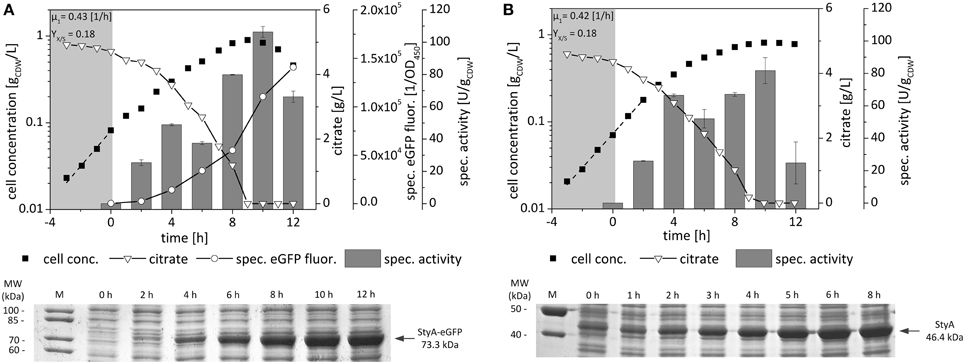
Figure 1. Growth, expression, and specific styrene epoxidation activity profiles of P. taiwanensis VLB120 (pA-EGFP_B) (A) and P. taiwanensis VLB120 (pStyAB) (B) grown on citrate as sole carbon source. Cells were cultivated in M9* medium and induced with 0.025% (v/v) DCPK after 3 h of growth. Samples for resting-cell activity, specific fluorescence, and SDS-Page analyses were taken at regular time intervals (every 2 h) after induction. For determination of the specific styrene epoxidation activity, cells were harvested, resuspended in KPI buffer containing 0.5% (v/v) citrate, and supplied with 1.5 mM styrene to start the reaction. Growth rates [h−1] were determined during exponential phase (dashed line). Yields on citrate were determined after inoculation and at the time point of citrate depletion. Error bars represent standard deviations of two independent assays.
To elucidate if the fusion construct also is stable and active, when different expression systems are used, gene expression and specific styrene epoxidation activity of E. coli JM101 were studied using three different regulatory systems (alk, lac, tac) (Figure S5). Again, specific styrene epoxidation activities maximally achieved with the fusion construct were as high as those obtained with the native StyAB. Thus, the results obtained confirm the applicability of the fusion construct as readout system for recombinant StyAB levels.
Analysis and Quantification of Clonal Variability by Means of Fused StyA-eGFP
As next step, fused StyA-eGFP was tested as a tool to analyze the high clonal variability in specific styrene epoxidation activity observed for P. putida DOT-T1E (pStyAB). For this purpose, specific fluorescence was measured in 21 individual P. putida DOT-T1E (pA-EGFP_B) cultures, originating from individual colonies picked after transformation, and correlated with specific styrene epoxidation activities. For comparison, 8 individual cultures were analyzed of each, P. taiwanensis VLB120 showing high StyAB activities (Figure 1) and low respective clonal variability and P. putida KT2440 showing low StyAB activities and low clonal variability (Lindmeyer et al., 2015).
Using the fusion construct, the high and low specific styrene epoxidation activity variations with P. putida DOT-T1E and P. putida KT2440 could be confirmed (Figure 2). As it was the case for P. taiwanensis VLB120 (Figure 1), specific styrene epoxidation activities of P. putida DOT-T1E and P. putida KT2440 correlated well with the specific eGFP fluorescence (Figure 2). The average specific styrene epoxidation activities of P. putida KT2440 (pA-EGFP_B) (11.3 ± 1.8 U g) were significantly lower as compared to P. taiwanensis VLB120 (pA-EGFP_B) and P. putida DOT-T1E (pA-EGFP_B) (48.9 ± 10.8 U g, respectively) and were in accordance with reported activities (Lindmeyer et al., 2015).
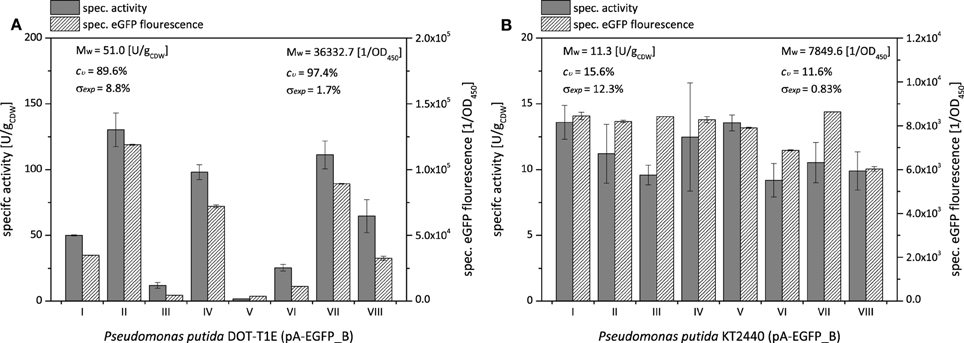
Figure 2. Clonal variability in specific styrene epoxidation activity and styA-eGFP expression levels for P. putida DOT-T1E (A) and P. putida KT2440 (B) both harboring pA-EGFP_B. Specific styrene epoxidation activities and expression levels were determined via resting-cell activity assays and specific eGFP fluorescence, respectively. Exemplary results for 8 independent clones are shown. Cultures were induced with 0.025% (v/v) DCPK for 4 h. SDS-PAGE analyses and induction kinetics of respective clones are shown in Figures S6, S7. Average specific styrene epoxidation activities Mw [U g], average specific fluorescence values [OD], coefficients of variation cν [%], and experimental errors σexp [%] given correspond to 21 and 8 tested cultures of P. putida DOT-T1E (pA-EGFP_B) and P. putida KT2440 (pA-EGFP_B), respectively.
In order to evaluate the fusion construct as a tool to quantify clonal variability, coefficients of variation cν and experimental errors σexp were determined, and respective values obtained via the specific eGFP fluorescence and the specific styrene epoxidation activities 4 h after induction were compared (Figure 3A). With a significantly lower experimental error σexp, the determination of the eGFP fluorescence turned out to be more accurate compared to the specific styrene epoxidation activity. This, in consequence, results in a more accurate coefficient of variation cν.
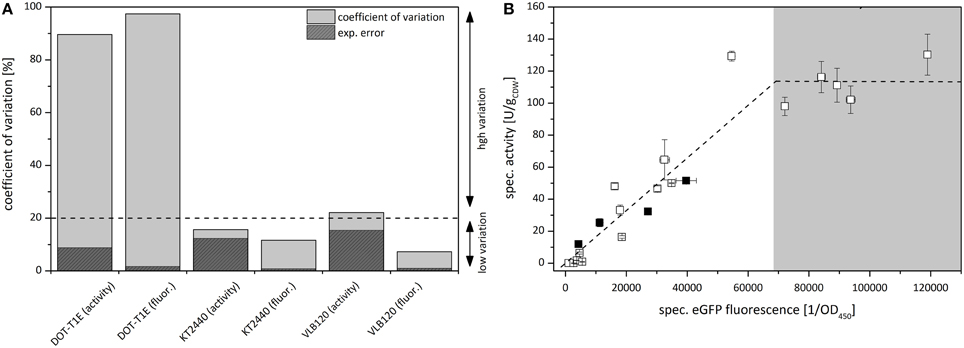
Figure 3. (A) Comparison of clonal variability in specific styrene epoxidation activity and specific eGFP fluorescence with P. putida DOT-T1E (pA-EGFP_B), P. putida KT2440 (pA-EGFP_B), and P. taiwanensis VLB120 (pA-EGFP_B) expressing styA-eGFP under control of the alk-regulatory system. Variations among cultures are displayed via the coefficient of variation cν. Dark gray areas represent calculated experimental errors σexp [%]. For P. putida KT2440 and P. taiwanensis VLB120 harboring pA-EGFP_B, the cν represented by a bar was determined via specific styrene epoxidation activity and specific eGFP fluorescence measurements performed for 8 separate cultures originating from 8 freshly transformed colonies. For P. putida DOT-T1E (pA-EGFP_B), cν determination was based on 21 separate cultures. (B) Correlation of specific activities of 21 independent P. putida DOT-T1E (pA-EGFP_B) cultures with respective specific eGFP fluorescence. The correlation of activity and fluorescence is divided in two sections showing a different interdependence. Black squares represent the 4 colonies which were further analyzed via flow cytometry and digital PCR for PCN determination.
The specific eGFP fluorescence of P. putida DOT-T1E (pA-EGFPB) cultures correlated linearly with respective specific StyAB activities up to a specific eGFP fluorescence of ~4*105 OD (Figure 3B), indicating that below the corresponding StyA-eGFP expression level, the styrene epoxidation rate is limited by the intracellular oxygenase concentration. At higher expression levels, the specific activity remained constant at around 110 U g and obviously was not limited by the intracellular StyAB concentration, but rather by substrate availability (i.e., mass transfer limitation for styrene and/or O2 and/or a limiting NADH regeneration via glucose metabolism). Thus, regarding the analysis of clonal variability, the fusion construct combined with specific fluorescence determination outperformed specific activity determination not only in terms of accuracy but also with respect to the range of expression levels which can be detected.
The Clonal Variability in Gene Expression Observed for P. putida DOT-T1E is Characterized by a Heterogeneous Subpopulation Distribution
To elucidate if and how the clonal variability observed for P. putida DOT-T1E (pA-EGFP_B) depends on subpopulation formation, the subpopulation structure in individual cultures was analyzed via flow cytometry. Since clonal variability did not appear using the lac- instead of the alk-regulatory system in P. putida DOT-T1E (Lindmeyer et al., 2015), pA-EGFP_B_lac was used for comparison. As plasmid backbone and fusion construct were kept identical, emerging differences in gene expression can be attributed to the regulatory system employed. Growth physiology (Figures 4A,B) and styA-eGFP expression dynamics (Figures 4C,D) were investigated in detail for 4 cultures each of P. putida DOT-T1E, harboring pA-EGFP_B (Figures 4A,C) or pA-EGFP_B_lac (Figures 4B,D) including the single cell level (Figure 5). To distinguish between subpopulations and uncover dynamics in recombinant gene expression, flow cytometry analysis was performed before induction as well as 4 and 8 h after induction.
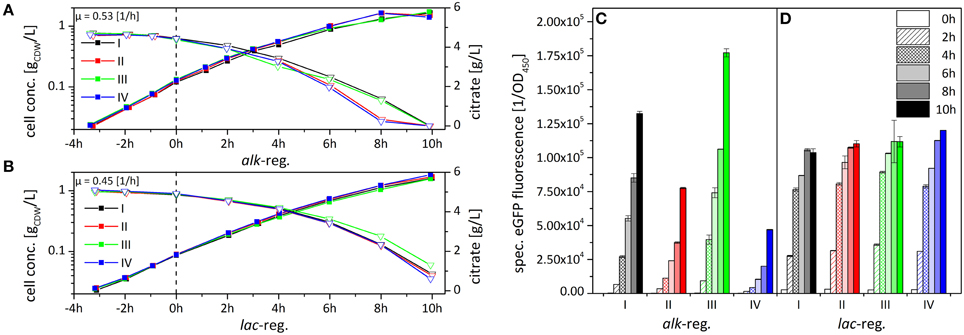
Figure 4. Comparison of growth behavior (A,B) and variability in StyA-eGFP levels (C,D) upon expression under the control of the alk- (A,C) and the lac- (B,D) regulatory systems in P. putida DOT-T1E. Results for 4 representative cultures are shown each for P. putida DOT-T1E (pA-EGFP_B) and P. putida DOT-T1E (pA-EGFP_B_lac) (see text for details on clone selection). Cells were cultivated in M9* medium supplement with 0.5% (w/v) citrate. Induction was initiated at 0 h by the addition of 0.025% (v/v) DCPK or 1 mM IPTG for the alk- or the lac-based expression, respectively. The specific fluorescence was determined via eGFP fluorescence measurement and normalization to the cell concentration. Error bars give the standard deviations of two independent fluorescence measurements.
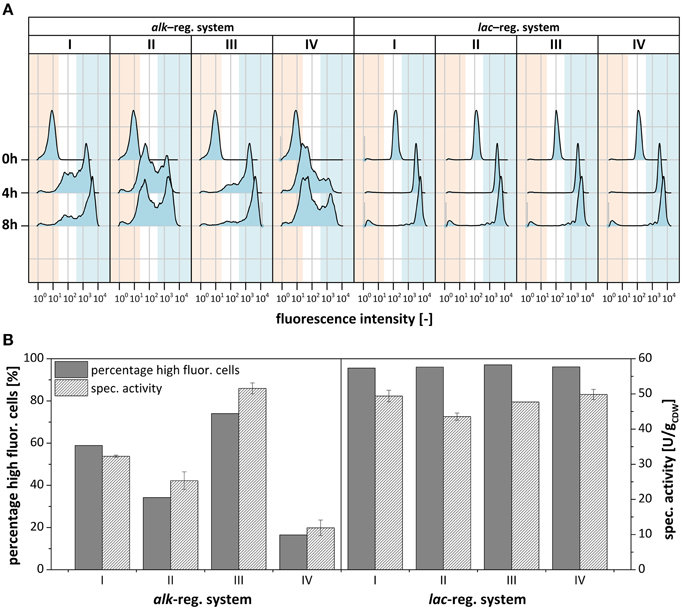
Figure 5. Subpopulation formation of recombinant P. putida DOT-T1E regarding styA-eGFP expression determined via flow cytometry. (A) Flow cytometry analysis of cultures visualized in Figure 4 expressing the fusion construct styA-eGFP under control of the alk- (left) and the lac-regulatory systems (right) before, 4 h after, and 8 h after induction. Populations are split into non-fluorescing (red), low- to medium-fluorescing (white) and high-fluorescing (blue) subpopulations. (B) Bar chart correlating the percentage of cells showing high eGFP fluorescence and respective specific styrene epoxidation activities of the whole population after 4 h of induction.
Only minor variability in growth was observed during cultivation of P. putida DOT-T1E (pA-EGFP_B_lac) clones (Figures 4A,B), whereas P. putida DOT-T1E (pA-EGFP_B) cultures showed higher growth variation after induction with a negative correlation of the average eGFP-fluorescence and the overall growth rate. This can be explained by stress induced by styA-eGFP overexpression (Figures 4A,C), as it has been observed for styAB overexpression in E. coli (Bühler et al., 2008).
In contrast to the high clonal variability in StyA-eGFP content among the 4 chosen P. putida DOT-T1E (pA-EGFP_B) cultures (Figure 4C), P. putida DOT-T1E (pA-EGFP_B_lac) cultures did not vary regarding StyA-eGFP content and respective expression dynamics (Figure 4D).
These clonal cell populations were further investigated by flow cytometry (Figure 5). Before induction, minor leaky gene expression was observed with the lac-regulatory system, whereas no leaky expression was detected with the alk-regulatory system (Figures 4D, 5A). For cells harboring the lac-vector, mainly one population with high fluorescence intensity and thus low intra-population variability was found after 4 h of induction meaning that cells homogenously expressed styA-eGFP at a high-level. After 8 h of induction, when the growth rate decreased, a non-fluorescing subpopulation had increased in size. Such a small non-fluorescing subpopulation (< 10%) also developed upon alk-based expression. In stark contrast to this observation, P. putida KT2440 (pA-EGFP_B_lac) as well as P. putida KT2440 (pA-EGFP_B) showed the formation of large non-fluorescing subpopulations, which, under comparable growth conditions, were very similar in size (63 and 62% of all cells, respectively; Figure S2B). This indicates that P. putida KT2440, in contrast to P. putida DOT-T1E, forms large fluorescence-free subpopulations upon both alk- and the lac-based expression despite the presence of kanamycin. In P. putida KT2440, such fluorescence loss was related to plasmid loss (Jahn et al., 2014).
As it was the case for P. putida DOT-T1E (pA-EGFP_B_lac), almost all cells in all 4 clonal P. putida DOT-T1E (pA-EGFP_B) cultures actively expressed the eGFP construct. However, in contrast to P. putida DOT-T1E (pA-EGFP_B_lac), P. putida DOT-T1E (pA-EGFP_B) cultures developed, beside a small non-fluorescing subpopulation, at least two distinct subpopulations with different recombinant protein contents (high and low) reflecting high intra-population variability (Figure 5A). Considering the different abundancies of highly fluorescing cells among individual cultures of P. putida DOT-T1E (pA-EGFP_B), average eGFP fluorescence variations (Figure 4C) correlated well with the respective distribution of the two subpopulations (Figure 5A). It can be concluded that clonal variability in specific styrene epoxidation activity depends on the sizes of the two subpopulations with high or low oxygenase expression levels: the larger the size of the high fluorescing subpopulation the higher the mean specific StyAB activity of the total population (Figure 5B).
Variation in PCN is Not the Cause for the Intra-population and Clonal Variabilities Observed for alk-based Gene Expression in P. putida DOT-T1E
The intra-population variability in recombinant gene expression observed for P. putida DOT-T1E (pA-EGFP_B) (Figure 5A) can be caused by interactions of expression regulation and the host specific regulatory network or by PCN variation, which might occur due to an interference of the alk-regulatory system with plasmid replication/segregation. In order to test this, cells of different subpopulations were sorted and the PCN was determined in respective high-fluorescing (eGFP+) and non-fluorescing (eGFP−) cells of both P. putida DOT-T1E (pA-EGFP_B) and P. putida DOT-T1E (pA-EGFP_B_lac) (Figure 6). For P. putida DOT-T1E (pA-EGFP_B), the average PCN per cell (3-4) was the same for eGFP+ and eGFP− cells and thus did not correlate with the respective recombinant StyA-eGFP fluorescence. A difference in PCN was found for eGFP+ (PCN = 3-6) and eGFP− (PCN = 1) cells of P. putida DOT-T1E (pA-EGFP_B_lac). Thus, the appearance of the small non-fluorescing subpopulation of P. putida DOT-T1E (pA-EGFP_B_lac) obviously is associated with instable plasmid proliferation. However, the expression variability exhibited by P. putida DOT-T1E (pA-EGFP_B) was not related to plasmid proliferation, indicating that interactions of the alk-regulatory system and the host specific regulatory network finally lead to the observed high clonal variability in expression and biocatalytic activity.
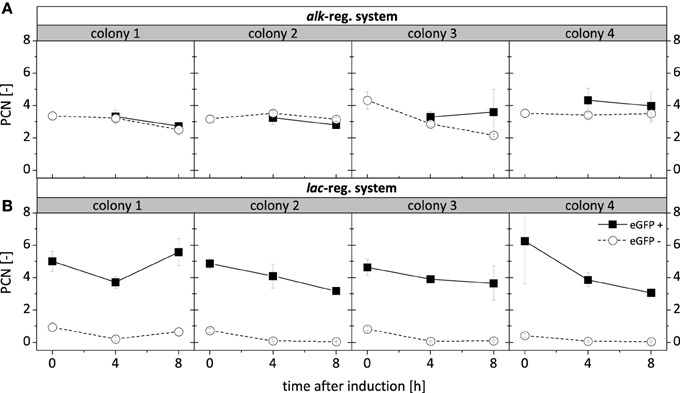
Figure 6. Average PCN per cell after 0 h, 4 h and 8 h of induction of P. putida DOT-T1E recombinants. One thousand cells originating from the 8 respective cultures visualized in Figure 5 expressing the fusion construct styA-eGFP under control of the alk- (A) and the lac- (B) regulatory systems were sorted according to defined gates (Figure S9) for fluorescing (eGFP+) and non-fluorescing (eGFP−) cells. PCNs were determined in quadruplicates for each subpopulation and are presented as an average value with standard deviations.
Discussion
The occurrence of two distinct cell states in an isogenic bacterial culture is a classical phenomenon of intra-population variability, with both cells not acting as catalysts although being alive and cells being highly productive (Müller et al., 2010). For the elucidation of such a binary mechanism in bioprocesses, flow cytometry can provide quantitative measures on cell state variations (Diaz et al., 2010). In this study, flow cytometry analysis of P. putida DOT-T1E (pA-EGFP_B) cultures has revealed the formation of two main subpopulations (high and low styA-eGFP expression levels) varying in size in individual cultures originating from different isogenic colonies. This variability in subpopulation sizes was found to be the cause for the observed clonal variability in biocatalytic performance (in terms of specific styrene epoxidation activity). Furthermore, the observed intra-population variability seems to be caused by interactions of the alk-regulatory system and the host specific regulatory network. This situation differed for expression in P. putida KT2440, for which a regulatory system-independent intra-population variability, but no clonal variability has been found (Table 3; Jahn et al., 2012; Lindmeyer et al., 2015). A main difference between P. putida KT2440 and P. putida DOT-T1E lies in the tolerance against organic solvents. In contrast to solvent-sensitive P. putida KT2440, P. putida DOT-T1E harbors the self-transmissible megaplasmid pGRT1 (133 kbp) encoding genes involved in solvent tolerance such as the most relevant solvent efflux pump TtgGHI. However, on the mechanistic level, solvent tolerance and variability do not seem to be interconnected (Lindmeyer et al., 2015). These observations emphasize the diversity among different P. putida strains. In the following, the utility of eGFP-fusion as a tool to investigate intra-population variability, the connectivity of clonal variability and subpopulation formation, and implications of the results obtained in this study for biocatalyst and bioprocess engineering are discussed.
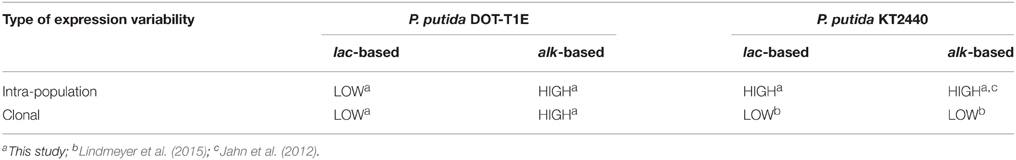
Table 3. Expression variabilities observed for P. putida DOT-T1E and P. putida KT2440.
Fusion to eGFP as Tool to Analyze Clonal Variability and its Connectivity to Intra-population Variability
Transcriptional fusion of GFP or its derivatives (Shaner et al., 2005) to target proteins is a commonly used non-invasive technique to assess recombinant gene expression by flow cytometry (Tracy et al., 2010). Such biosensors are applied to analyze intra-population variability and/or to quantify/optimize recombinant protein production (Burmølle et al., 2005; Sevastsyanovich et al., 2009). Due to its reliability and simplicity, the styA-eGFP fusion construct was shown to enable a highly accurate calculation of the coefficient of variation as a measure for clonal variability. Thereby, the detectable range of expression levels was found not to be biased by substrate availability as it was the case for the specific activity as readout (Figure 3).
In all strains tested, the fusion construct showed a high and even superior styrene epoxidation performance compared to native StyAB. Obviously, in vivo styrene epoxidation activity was not negatively affected by the fusion of StyA to eGFP or the linker in-between. Since protein linkers have effects on the folding stability, oligomeric state, proteolytic resistance, and solubility of fusion proteins (Robinson and Sauer, 1998; Xue et al., 2004), one may speculate that such effects positively influenced the in vivo styrene epoxidation performance of StyA-eGFP StyB.
For P. putida DOT-T1E containing the fusion construct, the variable specific styrene epoxidation activity among cultures originating from different isogenic colonies correlated linearly with the specific eGFP fluorescence up to a threshold level of around 110 U g, which was not exceeded at further increasing fluorescence intensities, i.e., expression levels (Figure 3B). This may be explained by substrate limitation at high expression levels. Considering the three StyAB substrates styrene, O2, and NADH, limitation by the former two can be excluded since cell densities used in resting-cell assays were optimized to avoid respective substrate depletion, as it was reflected by constant activities over 20 min (data not shown) (Panke et al., 1998; Lindmeyer et al., 2015). This points to a competition of the increasing epoxidation-related NADH demand with other intracellular NADH dependent reactions, especially respiration, and/or stress imposed by oxygenase overexpression affecting the metabolic capacity (Bühler et al., 2006, 2008; Blank et al., 2008b).
The Connectivity of Clonal Variability and Subpopulation Formation
Subpopulation formation has already been reported for P. putida KT2440 and was related to variable DNA contents of cells in chemostat cultures or bistability during recombinant gene expression (Figure S2B) (Jahn et al., 2012; Lieder et al., 2014). In contrast to P. putida KT2440 (pA-EGFP_B), which showed the formation of a large non-fluorescing subpopulation associated with plasmid loss (Jahn et al., 2014), the PCN was found not to be reduced in eGFP− cells of P. putida DOT-T1E (pA-EGFP_B), indicating that no or low styrene monooxygenase production cannot be ascribed to the lack of the respective genetic information and plasmid replication/segregation. This together with the finding that the clonal variability observed for P. putida DOT-T1E was found not to depend on the type of heterologous enzyme, antibiotic resistance mechanisms, nor on inducer toxicity (Lindmeyer et al., 2015) indicates that the observed clonal and intra-population variabilities in recombinant gene expression are related to interactions of the alk-regulatory system with the host regulatory network. A similar behavior can be assumed for P. putida S12, for which a very similar clonal variability for recombinant gene expression under control of the alk-regulatory system has been found (Lindmeyer et al., 2015).
The alk-regulatory system, which originates from P. putida GPo1 (van Beilen et al., 1994; Staijen et al., 1999), has been intensively investigated and employed as powerful tool for recombinant gene expression in E. coli and Pseudomonas strains (Yuste et al., 1998; Canosa et al., 1999, 2000; Panke et al., 2007). It features a positive feedback mechanism governed by the regulator AlkS. Regarding alkS expression, a constitutive promoter (PalkS1) establishes a basic AlkS level, whereas, in the presence of an inducer such as alkanes or DCPK, AlkS activates its own formation from a second promoter (PalkS2) and gene expression at the PalkB promotor. In Pseudomonas strains, the alk-regulatory system is subject to catabolite repression, e.g., by glucose, but not by citrate, pyruvate, and glycerol, whereas catabolite repression is absent in E. coli (Staijen et al., 1999; Yuste and Rojo, 2001; Dinamarca et al., 2002). AlkS belongs to the unique group of the Large ATP-binding regulators of the LuxR family (LAL-regulators) (De Schrijver and De Mot, 1999), characterized by their unusually large size (>800 aa) and the presence of an N-terminal ATP/GTP-binding domain and a C-terminal LuxR-like DNA-binding domain. Typically, LAL-regulators act globally controlling various cellular processes via a complex modulation involving the interaction with multiple effectors. They participate in the regulation of diverse types of metabolism in both Gram-negative and Gram-positive bacteria and are proposed to play a role in higher steps of regulatory cascades (Zhao et al., 2010; Guerra et al., 2012). Autoinduction, as described for AlkS, is not a common feature of LAL-regulator proteins (Boos and Shuman, 1998). However, a positive feedback mechanism was proposed by Gerrites et al. for the LAL-regulator protein OrfV from Pseudomonas alcaligenes (Gerritse et al., 1998). In a recent publication, the inherent clonal variability of P. putida DOT-T1E was proposed to rely on phenotypic predispositions of the source-clone (colony), thereby assuming that concentrations of regulatory molecules, such as AlkS or related compounds, differ from source-clone to source-clone and might be bequeathed to the daughter cells (Lindmeyer et al., 2015). This stochasticity in predispositions of the regulatory network was proposed to affect AlkS/PalkB mediated expression. It can be speculated that a certain threshold concentration of AlkS, established via the PalkS1, must be present before induction to efficiently trigger its own formation upon induction via PalkS2. After induction of P. putida DOT-T1E (pA-EGFP_B) cultures, almost all of these cells expressed the styA-eGFP gene at least at a low level. In such a scenario, the AlkS threshold concentration for PalkB promotor activation would be lower than that required to trigger its own formation via PalkS2. Thus, the high intra-population variability observed for P. putida DOT-T1E may be attributable to the architecture of the alk-regulatory system. In general, depending on the sensitivity of autoinduction and the concentration of the transcription factor (TF), a simple positive feedback regulation motif (TF enhances its own production in the presence of an inducer) leads to a broad distribution in gene expression and, in case of strong autoinduction, can result in a bi-modal behavior under the premise that in some cells the TF concentration is low and in others high (Alon, 2007; Silva-Rocha and de Lorenzo, 2010). In contrast, negative regulation and especially feedback regulation are classified as the most “noise-attenuating” regulatory network motifs and generally provide increased stability and homogenous gene expression regardless of inherent extrinsic and intrinsic noises (Patnaik, 2006). The lac-regulatory system applied in this study is not subject to autoregulation, but is under both negative and positive control. Thereby, the low intra-population variability observed may well be attributable to the negative control by LacI.
It is known that, in fully induced cells, the level of recombinant gene expression via PalkB is limited by the intracellular AlkS level (Yuste and Rojo, 2001). Thus, variability in intracellular AlkS concentrations would also affect gene expression at the PalkB promotor. Variability may further be promoted by the fact that AlkS is a highly instable protein (Yuste and Rojo, 2001; Moreno et al., 2007). P. taiwanensis VLB120, E. coli JM101, and P. putida KT2440 did not show any clonal variability, although bimodality and thus intra-population variability was observed for both alk- as well as lac-controlled styA-eGFP expression in P. putida KT2440 (Figure S2B). However, clonal variability characterized by variable subpopulation sizes obviously did not appear in P. putida KT2440 (pA-EGFP_B) (Lindmeyer et al., 2015), where intra-population variability was associated with the appearance of a plasmid-free subpopulation (Jahn et al., 2014). Thus, a possible variability in AlkS levels obviously did not occur or cause expression variability in the three reference strains. In general, the clonal consistency of StyAB-expression levels and activities in these strains indicates that certain stochastic variabilities (“noise”) may either be reduced or not interact with the alk-regulatory system.
Overall, the observed clonal variability in StyA-eGFP levels might be the result of heterogeneous intracellular concentrations of AlkS and/or other regulatory compounds and may be promoted by AlkS features such as its instability, its autoinduction, and its connectivity to the regulatory network of Pseudomonas strains. Further, studies are in progress to investigate such connectivity with intracellular AlkS concentrations with a special emphasis on constitutive alkS overexpression.
Implications for Biocatalyst and Bioprocess Engineering
Recombinant P. putida DOT-T1E harboring the fusion-construct under control of the lac-regulatory system showed stable and reproducible gene expression levels auguring well for application. Before induction, specific eGFP fluorescence measurements revealed a low signal, indicating a slight leakiness of this expression system (Figure 5). For the PlacUV5–lacI system, such leaky expression also was reported in E. coli (Baneyx, 1999; Zhou et al., 2004). After induction, the high styA-eGFP expression levels were accompanied by growth inhibition owing to an increased stress caused by oxygenase overproduction as it also was observed for E. coli (Bühler et al., 2008; Leak et al., 2009). Beyond that, specific styrene epoxidation activities of P. putida DOT-T1E (pA-EGFP_B_lac) decreased after extended induction periods by 67% (8 h, Figure S8), which also can be explained by increasing oxygenase-related stress and/or by inclusion body formation (Yanase et al., 2002; Fahnert et al., 2004), aggregation of misfolded proteins caused by aminoglycoside antibiotics (Ling et al., 2012; Goltermann et al., 2013), or the formation of subpopulations not contributing to the biotransformation. However, the increase in population-wide specific eGFP fluorescence between 4 and 8 h after induction indicates that the development of the small subpopulation lacking the fusion protein (~10% of the cells) did not decisively contribute to the reduced styrene epoxidation performance.
Instable proliferation of the plasmid pA-EGFP_B has been observed for P. putida KT2440 (Jahn et al., 2014), which appeared to develop a mutation- and plasmid-independent adaptive resistance to kanamycin leading to the formation of a large plasmid-free inactive subpopulation. This subpopulation formation was independent of the regulatory system employed as very similar subpopulation sizes upon alk- and lac-controlled styA-eGFP expression in P. putida KT2440 were detected under comparable conditions (Figure S2B). Strain-inherent efflux pumps have been reported to be involved in such adaptive antibiotic resistance (Sánchez-Romero and Casadesús, 2014). However, the small size of the plasmid-free population of P. putida DOT-T1E (pA-EGFP_B_lac) and the time point of its formation, both in contrast to the behavior of P. putida KT2440 (Jahn et al., 2014), indicate that a substantial adaptive kanamycin resistance did not develop.
Although, only 3–5 plasmid copies per cell were detected in recombinant P. putida DOT-T1E, high expression levels and styrene epoxidation activities up to ~130 U g (Figure 2) were achieved, exceeding those of E. coli (up to 100 U g) exhibiting PCNs up to 100 per cell (Jahn, unpublished data). The high gene expression levels achieved from a low gene copy number, as it also has been described for other Pseudomonas strains (Panke et al., 1999), can be considered beneficial for the construction of stable production strains, given that reproducibility issues as described in this study can be avoided. Unequal partitioning of low PCNs (< 3) might cause intra-population variability. Such heterogeneity can be overcome via the use of toxin-antitoxin systems or systems actively controlling equal segregation (Jahn et al., 2015). Alternatively, chromosomal integration can be combined with the careful selection of a tunable expression system, thereby avoiding difficulties related to antibiotic markers, plasmid instability, gene overexpression, and a metabolic burden by high DNA load (Birnbaum and Bailey, 1991; Andersson and Hughes, 2010). Considering the variability issues, which arose using the Pseudomonas-derived alk-regulatory system, the chosen regulatory system should be as orthologous as possible to the host regulatory network to avoid interactions with host specific sigma factors (Canosa et al., 1999), small regulatory RNAs (Moreno et al., 2012), or carbon catabolite repression (Rojo, 2010). In this sense, the combination of well-chosen tunable regulatory systems and stable optimized Pseudomonas chassis strains (Martínez-Garcia et al., 2014; Lieder et al., 2015) augurs well for industrial application with minimal clonal and intra-population variability.
Conclusion
To the best of our knowledge, the observed intra-population variability displayed by heterogeneous subpopulation sizes in individual cultures is reported for the first time as an underlying principle for clonal variability. Furthermore, the presented results show that the detected variations in recombinant gene expression (i.e., alk-based styA-eGFP expression in P. putida DOT-T1E) are not owing to plasmid instability and PCN but rather depend on the regulatory background of the host strain itself and its interaction with the regulatory system employed. In general, clonal variability and expression-related stress are important factors to be considered for the development of production strains and the exploitation of the beneficial characteristics of Pseudomonads such as solvent tolerance, high metabolic capacity, and low gene dosage requirement. Careful selection of tunable expression systems and chromosome-based expression are considered crucial.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Financial support by the German Federal Ministry of Education and Research–BMBF (ERA-IB project “Pseudomonas 2.0,” EIB.10.041) and the German Federal Ministry of Food and Agriculture FNR (ERA-IB project “CONTIbugs,” EIB.12.028) is acknowledged.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2015.01042
References
Achilles, J., Stahl, F., Harms, H., and Müller, S. (2007). Isolation of intact RNA from cytometrically sorted Saccharomyces cerevisiae for the analysis of intrapopulation diversity of gene expression. Nat. Protoc. 2, 2203–2211. doi: 10.1038/nprot.2007.322
Alon, U. (2007). Network motifs: theory and experimental approaches. Nat. Rev. Genet. 8, 450–461. doi: 10.1038/nrg2102
Alonso, S., Rendueles, M., and Díaz, M. (2012). Physiological heterogeneity of Pseudomonas taetrolens during lactobionic acid production. Appl. Microbiol. Biotechnol. 96, 1465–1477. doi: 10.1007/s00253-012-4254-2
Andersson, D. I., and Hughes, D. (2010). Antibiotic resistance and its cost: is it possible to reverse resistance? Nat. Rev. Microbiol. 8, 260–271. doi: 10.1038/nrmicro2319
Avery, S. V. (2006). Microbial cell individuality and the underlying sources of heterogeneity. Nat. Rev. Microbiol. 4, 577–587. doi: 10.1038/nrmicro1460
Bagdasarian, M., Lurz, R., Ruckert, B., Franklin, F. C. H., Bagdasarian, M. M., Frey, J., et al. (1981). Specific-purpose plasmid cloning vectors II. Broad host range, high copy number, Rsf1010-derived vectors, and a host-vector system for gene cloning in Pseudomonas. Gene 16, 237–247. doi: 10.1016/0378-1119(81)90080-9
Baneyx, F. (1999). Recombinant protein expression in Escherichia coli. Curr. Opin. Biotechnol. 10, 411–421. doi: 10.1016/S0958-1669(99)00003-8
Bentley, W. E., and Quiroga, O. E. (1993). Investigation of subpopulation heterogeneity and plasmid stability in recombinant Escherichia coli via a simple segregated model. Biotechnol. Bioeng. 42, 222–234. doi: 10.1002/bit.260420210
Bertani, G. (1951). Studies on lysogenesis. I. The mode of phage liberation by lysogenic Escherichia coli. J. Bacteriol. 62, 293–300.
Birnbaum, S., and Bailey, J. E. (1991). Plasmid presence changes the relative levels of many host cell proteins and ribosome components in recombinant Escherichia coli. Biotechnol. Bioeng. 37, 736–745. doi: 10.1002/bit.260370808
Blank, L. M., Ebert, B. E., Bühler, B., and Schmid, A. (2008a). Metabolic capacity estimation of Escherichia coli as a platform for redox biocatalysis: constraint-based modeling and experimental verification. Biotechnol. Bioeng. 100, 1050–1065. doi: 10.1002/bit.21837
Blank, L. M., Ionidis, G., Ebert, B. E., Bühler, B., and Schmid, A. (2008b). Metabolic response of Pseudomonas putida during redox biocatalysis in the presence of a second octanol phase. FEBS J. 275, 5173–5190. doi: 10.1111/j.1742-4658.2008.06648.x
Boos, W., and Shuman, H. (1998). Maltose/maltodextrin system of Escherichia coli: transport, metabolism, and regulation. Microbiol. Mol. Biol. Rev. 62, 204–229.
Bühler, B., Park, J. B., Blank, L. M., and Schmid, A. (2008). NADH availability limits asymmetric biocatalytic epoxidation in a growing recombinant Escherichia coli strain. Appl. Environ. Microbiol. 74, 1436–1446. doi: 10.1128/AEM.02234-07
Bühler, B., Straathof, A. J. J., Witholt, B., and Schmid, A. (2006). Analysis of two-liquid-phase multistep biooxidation based on a process model: indications for biological energy shortage. Org. Process Res. Dev. 10, 628–643. doi: 10.1021/op060028g
Burmølle, M., Hansen, L. H., and Sørensen, S. J. (2005). Use of a whole-cell biosensor and flow cytometry to detect AHL production by an indigenous soil community during decomposition of litter. Microb. Ecol. 50, 221–229. doi: 10.1007/s00248-004-0113-8
Canosa, I., Sánchez-Romero, J. M., Yuste, L., and Rojo, F. (2000). A positive feedback mechanism controls expression of AlkS, the transcriptional regulator of the Pseudomonas oleovorans alkane degradation pathway. Mol. Microbiol. 35, 791–799. doi: 10.1046/j.1365-2958.2000.01751.x
Canosa, I., Yuste, L., and Rojo, F. (1999). Role of the alternative sigma factor ςs in expression of the AlkS regulator of the Pseudomonas oleovorans alkane degradation pathway. J. Bacteriol. 181, 1748–1754.
Choi, K. H., Kumar, A., and Schweizer, H. P. (2006). A 10-min method for preparation of highly electrocompetent Pseudomonas aeruginosa cells: application for DNA fragment transfer between chromosomes and plasmid transformation. J. Microbiol. Methods 64, 391–397. doi: 10.1016/j.mimet.2005.06.001
Delvigne, F., and Goffin, P. (2014). Microbial heterogeneity affects bioprocess robustness: dynamic single-cell analysis contributes to understanding of microbial populations. Biotechnol. J. 9, 61–72. doi: 10.1002/biot.201300119
De Schrijver, A., and De Mot, R. (1999). A subfamily of MalT-related ATP-dependent regulators in the LuxR family. Microbiology 145, 1287–1288. doi: 10.1099/13500872-145-6-1287
Diaz, M., Herrero, M., Garcia, L. A., and Quiros, C. (2010). Application of flow cytometry to industrial microbial bioprocesses. Biochem. Eng. J. 48, 385–407. doi: 10.1016/j.bej.2009.07.013
Dinamarca, M. A., Ruiz-Manzano, A., and Rojo, F. (2002). Inactivation of cytochrome o ubiquinol oxidase relieves catabolic repression of the Pseudomonas putida GPo1 alkane degradation pathway. J. Bacteriol. 184, 3785–3793. doi: 10.1128/JB.184.14.3785-3793.2002
Elowitz, M. B., Levine, A. J., Siggia, E. D., and Swain, P. S. (2002). Stochastic gene expression in a single cell. Science 297, 1183–1186. doi: 10.1126/science.1070919
Enfors, S. O., Jahic, M., Rozkov, A., Xu, B., Hecker, M., Jürgen, B., et al. (2001). Physiological responses to mixing in large scale bioreactors. J. Biotechnol. 85, 175–185. doi: 10.1016/S0168-1656(00)00365-5
Fahnert, B., Lilie, H., and Neubauer, P. (2004). Inclusion bodies: formation and utilisation. Adv. Biochem. Eng. Biotechnol. 89, 93–142. doi: 10.1007/b93995
Fraser, D., and Kaern, M. (2009). A chance at survival: gene expression noise and phenotypic diversification strategies. Mol. Microbiol. 71, 1333–1340. doi: 10.1111/j.1365-2958.2009.06605.x
Gerritse, G., Ure, R., Bizoullier, F., and Quax, W. J. (1998). The phenotype enhancement method identifies the Xcp outer membrane secretion machinery from Pseudomonas alcaligenes as a bottleneck for lipase production. J. Biotechnol. 64, 23–38. doi: 10.1016/S0168-1656(98)00101-1
Goltermann, L., Good, L., and Bentin, T. (2013). Chaperonins fight aminoglycoside-induced protein misfolding and promote short-term tolerance in Escherichia coli. J. Biol. Chem. 288, 10483–10489. doi: 10.1074/jbc.M112.420380
Guerra, S. M., Rodríguez-Garcia, A., Santos-Aberturas, J., Vicente, C. M., Payero, T. D., Martín, J. F., et al. (2012). LAL regulators SCO0877 and SCO7173 as pleiotropic modulators of phosphate starvation response and actinorhodin biosynthesis in Streptomyces coelicolor. PLoS ONE 7:e31475. doi: 10.1371/journal.pone.0031475
Hahne, F., Lemeur, N., Brinkman, R. R., Ellis, B., Haaland, P., Sarkar, D., et al. (2009). Flowcore: a bioconductor package for high throughput flow cytometry. BMC Bioinformat. 10:106. doi: 10.1186/1471-2105-10-106
Halan, B., Schmid, A., and Buehler, K. (2011). Real-time solvent tolerance analysis of Pseudomonas sp. strain VLB120ΔC catalytic biofilms. Appl. Environ. Microbiol. 77, 1563–1571. doi: 10.1128/AEM.02498-10
Hanahan, D. (1983). Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 166, 557–580. doi: 10.1016/S0022-2836(83)80284-8
Jahn, M., Günther, S., and Müller, S. (2015). Non-random distribution of macromolecules as driving forces for phenotypic variation. Curr. Opin. Microbiol. 25, 49–55. doi: 10.1016/j.mib.2015.04.005
Jahn, M., Seifert, J., Von Bergen, M., Schmid, A., Bühler, B., and Müller, S. (2012). Subpopulation-proteomics in prokaryotic populations. Curr. Opin. Biotechnol. 24, 79–87. doi: 10.1016/j.copbio.2012.10.017
Jahn, M., Vorpahl, C., Türkowsky, D., Lindmeyer, M., Bühler, B., Harms, H., et al. (2014). Accurate determination of plasmid copy number of flow-sorted cells using droplet digital PCR. Anal. Chem. 86, 5969–5976. doi: 10.1021/ac501118v
Kaern, M., Elston, T. C., Blake, W. J., and Collins, J. J. (2005). Stochasticity in gene expression: from theories to phenotypes. Nat. Rev. Genet. 6, 451–464. doi: 10.1038/nrg1615
Koch, C., Günther, S., Desta, A. F., Hübschmann, T., and Müller, S. (2013). Cytometric fingerprinting for analyzing microbial intracommunity structure variation and identifying subcommunity function. Nat. Protoc. 8, 190–202. doi: 10.1038/nprot.2012.149
Köhler, K. A., Rückert, C., Schatschneider, S., Vorhölter, F. J., Szczepanowski, R., and Schmid, A. (2013). Complete genome sequence of Pseudomonas sp. strain VLB120 a solvent tolerant, styrene degrading bacterium, isolated from forest soil. J. Biotechnol. 168, 729–730. doi: 10.1016/j.jbiotec.2013.10.016
Kuhn, D., Bühler, B., Blank, L. M., and Schmid, A. (2010a). Systems biotechnology - rational whole-cell biocatalyst and bioprocess design. Eng. Life Sci. 10, 384–397. doi: 10.1002/elsc.201000009
Kuhn, D., Kholiq, M. A., Heinzle, E., Bühler, B., and Schmid, A. (2010b). Intensification and economic and ecological assessment of a biocatalytic oxyfunctionalization process. Green Chem. 12, 815–827. doi: 10.1039/b921896c
Laemmli, U. K. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685. doi: 10.1038/227680a0
Leak, D. J., Sheldon, R. A., Woodley, J. M., and Adlercreutz, P. (2009). Biocatalysts for selective introduction of oxygen. Biocatal. Biotransform. 27, 1–26. doi: 10.1080/10242420802393519
Lieder, S., Jahn, M., Seifert, J., Von Bergen, M., Müller, S., and Takors, R. (2014). Subpopulation-proteomics reveal growth rate, but not cell cycling, as a major impact on protein composition in Pseudomonas putida KT2440. AMB Express 4:71. doi: 10.1186/s13568-014-0071-6
Lieder, S., Nikel, P. I., de Lorenzo, V., and Takors, R. (2015). Genome reduction boosts heterologous gene expression in Pseudomonas putida. Microb. Cell. Fact. 14:23. doi: 10.1186/s12934-015-0207-7
Lindmeyer, M., Meyer, D., Kuhn, D., Bühler, B., and Schmid, A. (2015). Making variability less variable: matching expression system and host for oxygenase-based biotransformations. J. Ind. Microbiol. Biotechnol. 42, 851–866. doi: 10.1007/s10295-015-1615-8
Ling, J., Cho, C., Guo, L. T., Aerni, H. R., Rinehart, J., and Söll, D. (2012). Protein aggregation caused by aminoglycoside action is prevented by a hydrogen peroxide scavenger. Mol. Cell 48, 713–722. doi: 10.1016/j.molcel.2012.10.001
Martínez-Garcia, E., Nikel, P. I., Aparicio, T., and De Lorenzo, V. (2014). Pseudomonas 2.0: genetic upgrading of P. putida KT2440 as an enhanced host for heterologous gene expression. Microb. Cell. Fact. 13:159. doi: 10.1186/s12934-014-0159-3
Messing, J. (1979). A multipurpose cloning system based on the single-stranded DNA bacteriophage M13. Recomb. DNA Tech. Bull. 3, 43–49.
Moreno, R., Fonseca, P., and Rojo, F. (2012). Two small RNAs, CrcY and CrcZ, act in concert to sequester the Crc global regulator in Pseudomonas putida, modulating catabolite repression. Mol. Microbiol. 83, 24–40. doi: 10.1111/j.1365-2958.2011.07912.x
Moreno, R., Ruiz-Manzano, A., Yuste, L., and Rojo, F. (2007). The Pseudomonas putida Crc global regulator is an RNA binding protein that inhibits translation of the AlkS transcriptional regulator. Mol. Microbiol. 64, 665–675. doi: 10.1111/j.1365-2958.2007.05685.x
Müller, S., Harms, H., and Bley, T. (2010). Origin and analysis of microbial population heterogeneity in bioprocesses. Curr. Opin. Biotechnol. 21, 100–113. doi: 10.1016/j.copbio.2010.01.002
Nikel, P. I., Silva-Rocha, R., Benedetti, I., and de Lorenzo, V. (2013). The private life of environmental bacteria: pollutant biodegradation at the single cell level. Environ. Microbiol. 16, 628–642. doi: 10.1111/1462-2920.12360
Novick, A., and Weiner, M. (1957). Enzyme induction as an all-or-none phenomenon. Proc. Natl. Acad. Sci. U.S.A. 43, 553–566. doi: 10.1073/pnas.43.7.553
Otto, K., Hofstetter, K., Röthlisberger, M., Witholt, B., and Schmid, A. (2004). Biochemical characterization of StyAB from Pseudomonas sp. strain VLB120 as a two-component flavin-diffusible monooxygenase. J. Bacteriol. 186, 5292–5302. doi: 10.1128/JB.186.16.5292-5302.2004
Panke, S., de Lorenzo, V., Kaiser, A., Witholt, B., and Wubbolts, M. G. (1999). Engineering of a stable whole-cell biocatalyst capable of (S)-styrene oxide formation for continuous two-liquid-phase applications. Appl. Environ. Microbiol. 65, 5619–5623.
Panke, S., Makart, S., and Heinemann, M. (2007). Characterization of the AlkS/PalkB-expression system as an efficient tool for the production of recombinant proteins in Escherichia coli fed-batch fermentations. Biotechnol. Bioeng. 96, 326–336. doi: 10.1002/bit.21117
Panke, S., Witholt, B., Schmid, A., and Wubbolts, M. G. (1998). Towards a biocatalyst for (S)-styrene oxide production: characterization of the styrene degradation pathway of Pseudomonas sp. strain VLB120. Appl. Environ. Microbiol. 64, 2032–2043.
Patnaik, P. (2006). External, extrinsic and intrinsic noise in cellular systems: analogies and implications for protein synthesis. Biotechnol. Mol. Biol. Rev. 1, 121–127.
Pilbrough, W., Munro, T. P., and Gray, P. (2009). Intraclonal protein expression heterogeneity in recombinant CHO cells. PLoS ONE 4:e8432. doi: 10.1371/journal.pone.0008432
Ramos, J. L., Duque, E., Huertas, M. J., and Haïdour, A. (1995). Isolation and expansion of the catabolic potential of a Pseudomonas putida strain able to grow in the presence of high-concentrations of aromatic-hydrocarbons. J. Bacteriol. 177, 3911–3916.
Robinson, C. R., and Sauer, R. T. (1998). Optimizing the stability of single-chain proteins by linker length and composition mutagenesis. Proc. Natl. Acad. Sci. U.S.A. 95, 5929–5934. doi: 10.1073/pnas.95.11.5929
Rojo, F. (2010). Carbon catabolite repression in Pseudomonas: optimizing metabolic versatility and interactions with the environment. FEMS Microbiol. Rev. 34, 658–684. doi: 10.1111/j.1574-6976.2010.00218.x
Ryall, B., Eydallin, G., and Ferenci, T. (2012). Culture history and population heterogeneity as determinants of bacterial adaptation: the adaptomics of a single environmental transition. Microbiol. Mol. Biol. Rev. 76, 597–625. doi: 10.1128/MMBR.05028-11
Sambrook, J., and Russel, D. W. (2001). Molecular Cloning: A Laboratory Manual, Vol. 1. NewYork, NY: Cold Spring Harbor Laboratory.
Sánchez-Romero, M. A., and Casadesús, J. (2014). Contribution of phenotypic heterogeneity to adaptive antibiotic resistance. Proc. Natl. Acad. Sci. U.S.A. 111, 355–360. doi: 10.1073/pnas.1316084111
Schmid, A., Dordick, J. S., Hauer, B., Kiener, A., Wubbolts, M., and Witholt, B. (2001). Industrial biocatalysis today and tomorrow. Nature 409, 258–268. doi: 10.1038/35051736
Schrewe, M., Julsing, M. K., Bühler, B., and Schmid, A. (2013). Whole-cell biocatalysis for selective and productive C-O functional group introduction and modification. Chem. Soc. Rev. 42, 6346–6377. doi: 10.1039/c3cs60011d
Sevastsyanovich, Y., Alfasi, S., Overton, T., Hall, R., Jones, J., Hewitt, C., et al. (2009). Exploitation of GFP fusion proteins and stress avoidance as a generic strategy for the production of high-quality recombinant proteins. FEMS Microbiol. Lett. 299, 86–94. doi: 10.1111/j.1574-6968.2009.01738.x
Shaner, N. C., Steinbach, P. A., and Tsien, R. Y. (2005). A guide to choosing fluorescent proteins. Nat. Methods 2, 905–909. doi: 10.1038/nmeth819
Silva-Rocha, R., and de Lorenzo, V. (2010). Noise and robustness in prokaryotic regulatory networks. Annu. Rev. Microbiol. 64, 257–275. doi: 10.1146/annurev.micro.091208.073229
Staijen, I. E., Marcionelli, R., and Witholt, B. (1999). The PalkBFGHJKL promoter is under carbon catabolite repression control in Pseudomonas oleovorans but not in Escherichia coli alk+ recombinants. J. Bacteriol. 181, 1610–1616.
Tian, G. W., Mohanty, A., Chary, S. N., Li, S. J., Paap, B., Drakakaki, G., et al. (2004). High-throughput fluorescent tagging of full-length arabidopsis gene products in planta. Plant Physiol. 135, 25–38. doi: 10.1104/pp.104.040139
Tracy, B. P., Gaida, S. M., and Papoutsakis, E. T. (2010). Flow cytometry for bacteria: enabling metabolic engineering, synthetic biology and the elucidation of complex phenotypes. Curr. Opin. Biotechnol. 21, 85–99. doi: 10.1016/j.copbio.2010.02.006
van Beilen, J. B., Wubbolts, M. G., and Witholt, B. (1994). Genetics of alkane oxidation by Pseudomonas oleovorans. Biodegradation 5, 161–174. doi: 10.1007/BF00696457
Veening, J. W., Smits, W. K., Hamoen, L. W., and Kuipers, O. P. (2006). Single cell analysis of gene expression patterns of competence development and initiation of sporulation in Bacillus subtilis grown on chemically defined media. J. Appl. Microbiol. 101, 531–541. doi: 10.1111/j.1365-2672.2006.02911.x
Volmer, J., Neumann, C., Bühler, B., and Schmid, A. (2014). Engineering of Pseudomonas taiwanensis VLB120 for constitutive solvent tolerance and increased specific styrene epoxidation activity. Appl. Environ. Microbiol. 80, 6539–6548. doi: 10.1128/AEM.01940-14
Willrodt, C., David, C., Cornelissen, S., Bühler, B., Julsing, M. K., and Schmid, A. (2014). Engineering the productivity of recombinant Escherichia coli for limonene formation from glycerol in minimal media. Biotechnol. J. 9, 1000–1012. doi: 10.1002/biot.201400023
Woodley, J. M. (2006). Choice of biocatalyst form for scalable processes. Biochem. Soc. Trans. 34, 301–303. doi: 10.1042/BST0340301
Xue, F., Gu, Z., and Feng, J. A. (2004). LINKER: a web server to generate peptide sequences with extended conformation. Nucleic Acids Res. 32, W562–W565. doi: 10.1093/nar/gkh422
Yanase, H., Moriya, K., Mukai, N., Kawata, Y., Okamoto, K., and Kato, N. (2002). Effects of GroESL coexpression on the folding of nicotinoprotein formaldehyde dismutase from Pseudomonas putida F61. Biosci. Biotechnol. Biochem. 66, 85–91. doi: 10.1271/bbb.66.85
Yuste, L., Canosa, I., and Rojo, F. (1998). Carbon-source-dependent expression of the PalkB promoter from the Pseudomonas oleovorans alkane degradation pathway. J. Bacteriol. 180, 5218–5226.
Yuste, L., and Rojo, F. (2001). Role of the crc gene in catabolic repression of the Pseudomonas putida GPo1 alkane degradation pathway. J. Bacteriol. 183, 6197–6206. doi: 10.1128/JB.183.21.6197-6206.2001
Zhao, K. X., Huang, Y., Chen, X., Wang, N. X., and Liu, S. J. (2010). PcaO positively regulates pcaHG of the beta-ketoadipate pathway in Corynebacterium glutamicum. J. Bacteriol. 192, 1565–1572. doi: 10.1128/JB.01338-09
Keywords: phenotypic heterogeneity, clonal variability, intra-population variability, fluorescent reporter, flow cytometry, plasmid copy number, Pseudomonas putida, alk-regulatory system
Citation: Lindmeyer M, Jahn M, Vorpahl C, Müller S, Schmid A and Bühler B (2015) Variability in subpopulation formation propagates into biocatalytic variability of engineered Pseudomonas putida strains. Front. Microbiol. 6:1042. doi: 10.3389/fmicb.2015.01042
Received: 10 July 2015; Accepted: 14 September 2015;
Published: 01 October 2015.
Edited by:
Robert Kourist, Ruhr-University Bochum, GermanyReviewed by:
Victor De Lorenzo, National Center of Biotechnology of the National Research Council, SpainFrank Delvigne, University of Liège, Belgium
Rebekka Biedendieck, Technische Universität Braunschweig, Germany
Copyright © 2015 Lindmeyer, Jahn, Vorpahl, Müller, Schmid and Bühler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bruno Bühler, Helmholtz Centre for Environmental Research - UFZ, Department of Solar Materials, Permoserstrasse 15, 04318 Leipzig, Germany,YnJ1bm8uYnVlaGxlckB1ZnouZGU=