 Claudia Knief
Claudia Knief- Institute of Crop Science and Resource Conservation – Molecular Biology of the Rhizosphere, University of Bonn, Bonn, Germany
Methane-oxidizing bacteria are characterized by their capability to grow on methane as sole source of carbon and energy. Cultivation-dependent and -independent methods have revealed that this functional guild of bacteria comprises a substantial diversity of organisms. In particular the use of cultivation-independent methods targeting a subunit of the particulate methane monooxygenase (pmoA) as functional marker for the detection of aerobic methanotrophs has resulted in thousands of sequences representing “unknown methanotrophic bacteria.” This limits data interpretation due to restricted information about these uncultured methanotrophs. A few groups of uncultivated methanotrophs are assumed to play important roles in methane oxidation in specific habitats, while the biology behind other sequence clusters remains still largely unknown. The discovery of evolutionary related monooxygenases in non-methanotrophic bacteria and of pmoA paralogs in methanotrophs requires that sequence clusters of uncultivated organisms have to be interpreted with care. This review article describes the present diversity of cultivated and uncultivated aerobic methanotrophic bacteria based on pmoA gene sequence diversity. It summarizes current knowledge about cultivated and major clusters of uncultivated methanotrophic bacteria and evaluates habitat specificity of these bacteria at different levels of taxonomic resolution. Habitat specificity exists for diverse lineages and at different taxonomic levels. Methanotrophic genera such as Methylocystis and Methylocaldum are identified as generalists, but they harbor habitat specific methanotrophs at species level. This finding implies that future studies should consider these diverging preferences at different taxonomic levels when analyzing methanotrophic communities.
Occurrence and Role of Methane-oxidizing Bacteria
The activity of methane-oxidizing bacteria contributes significantly to the global methane budget. Methane is the second most abundant carbon compound in the atmosphere with a current concentration of 1.8 ppmv and a 26-fold stronger radiative efficiency compared to carbon dioxide (IPCC, 2013). The major sink of atmospheric methane is its oxidation by OH radicals, but soils also serve as sink by about 5% due to the activity of methanotrophic bacteria (IPCC, 2013). Moreover, methanotrophs are of particular importance in attenuating net fluxes of this greenhouse gas into the atmosphere in diverse ecosystems that are sources of atmospheric methane (De Visscher et al., 2007; Reeburgh, 2007; Conrad, 2009). Known sources are freshwater and permafrost ecosystems, some animal species and termites, and the release of methane from geological processes, wildfires and hydrates. Another 50–65% of the total emissions are due to anthropogenic activities including ruminant husbandry, fossil fuel extraction and use, rice paddy agriculture and emissions from landfills and waste, resulting in a current elevation of the atmospheric methane concentration by a factor of 2.5 compared to preindustrial times (IPCC, 2013). All these ecosystems with source function for atmospheric methane are typical habitats of methane-oxidizing bacteria. These include freshwater and marine sediments and water columns, aquifers, floodplains, peat bogs, high-arctic, and tundra wetlands, upland soils, rice paddies, landfill covers, and sewage sludge (Hanson and Hanson, 1996; Conrad, 2007; Bowman, 2014).
Besides their importance in the global methane cycle, aerobic methanotrophic bacteria are of biotechnological interest since a long time. They can be used for biodegradation processes of organic pollutants based on the fact that the key enzyme for methanotrophy in these organisms, the methane monooxygenase, catalyzes diverse non-specific oxidation reactions, e.g., of chlorinated solvents such as trichloroethylene (Hanson and Hanson, 1996; Smith and Dalton, 2004; Dalton, 2005; Jiang et al., 2010; Semrau et al., 2010; Strong et al., 2015). Moreover, methanotrophs have been studied in detail with regard to their potential to convert methane to complex organic molecules of higher value. Since the 1970s, methanotrophic bacteria have been studied for single cell protein production (Dalton, 2005). Besides, biopolymers such as polyhydroxybutyrate, metabolic products such as organic acids, vitamins, pigments or lipids (for biodiesel production) may be produced from methane by methanotrophs (Strong et al., 2015). Further possible applications for biosynthesis processes are based on the co-metabolic activities of the methane monooxygenase, e.g., for epoxide production via the conversion of propene to epoxypropane (Hanson and Hanson, 1996; Dalton, 2005). Moreover, researchers address the question to what extent methanotrophic bacteria can be used to increase reduction of methane emissions from anthropogenic sources such as landfills or coal mines (Jiang et al., 2010).
Diversity and Ecophysiology of Cultivated Methanotrophic Bacteria
Brief History About the Cultivation of Aerobic Methanotrophic Bacteria and Current Diversity and Phylogeny of Cultivated Methanotrophs
Methanotrophic bacteria have been studied since the beginning of the last century, initiated by the work of Kaserer (1905) and Söhngen (1906) who reported for the first time the existence of methane-oxidizing bacteria. The first isolates were methanotrophic Gammaproteobacteria, among them Methylomonas methanica, initially referred to as “Bacillus methanicus” (Söhngen, 1906), and Methylococcus capsulatus (Foster and Davis, 1966). Extensive enrichment and isolation work by Whittenbury et al. (1970b) led to isolates of further Gammaproteobacteria and the genera Methylocystis and Methylosinus, i.e., the first methanotrophic Alphaproteobacteria. During the following years and with the availability of molecular methods for the rapid identification and classification of bacteria, several existing strains were reclassified and new genera were described (e.g., Bowman et al., 1993, 1995; Bodrossy et al., 1997). In particular the work of the last 10 years has resulted in a doubling of the number of known genera and species. Currently, 18 genera of cultivated aerobic methanotrophic Gammaproteobacteria and 5 genera of Alphaproteobacteria are known, represented by approx. 60 different species (Table 1). The number of Gammaproteobacteria increases to 20 if “Candidatus Crenothrix polyspora” and “Candidatus Clonothrix fusca” are included. These genera do not contain cultivated representatives but were only studied in natural enrichments so far (Stoecker et al., 2006; Vigliotta et al., 2007). To give an exact number of known methanotrophic taxa at species level is difficult because the taxonomic status of some species, e.g., “Methylomonas rubra,” Methylococcus chroococcus, Methylococcus mobilis or Methylococcus thermophilus is unclear (Table 2). In addition to the species considered in this review, more species have been described in the (early) literature, in particular within the genera Methylomonas and Methylocystis (e.g. Whittenbury et al., 1970b; Gal'chenko et al., 1977), but these were never validated. Several of them will probably be members of species that have been described in the meantime. For an overview of non-validated species with uncertain taxonomic position the reader is referred to Green (1992) or the relevant chapters in taxonomic textbooks (Bowman, 2005a,b, 2014).
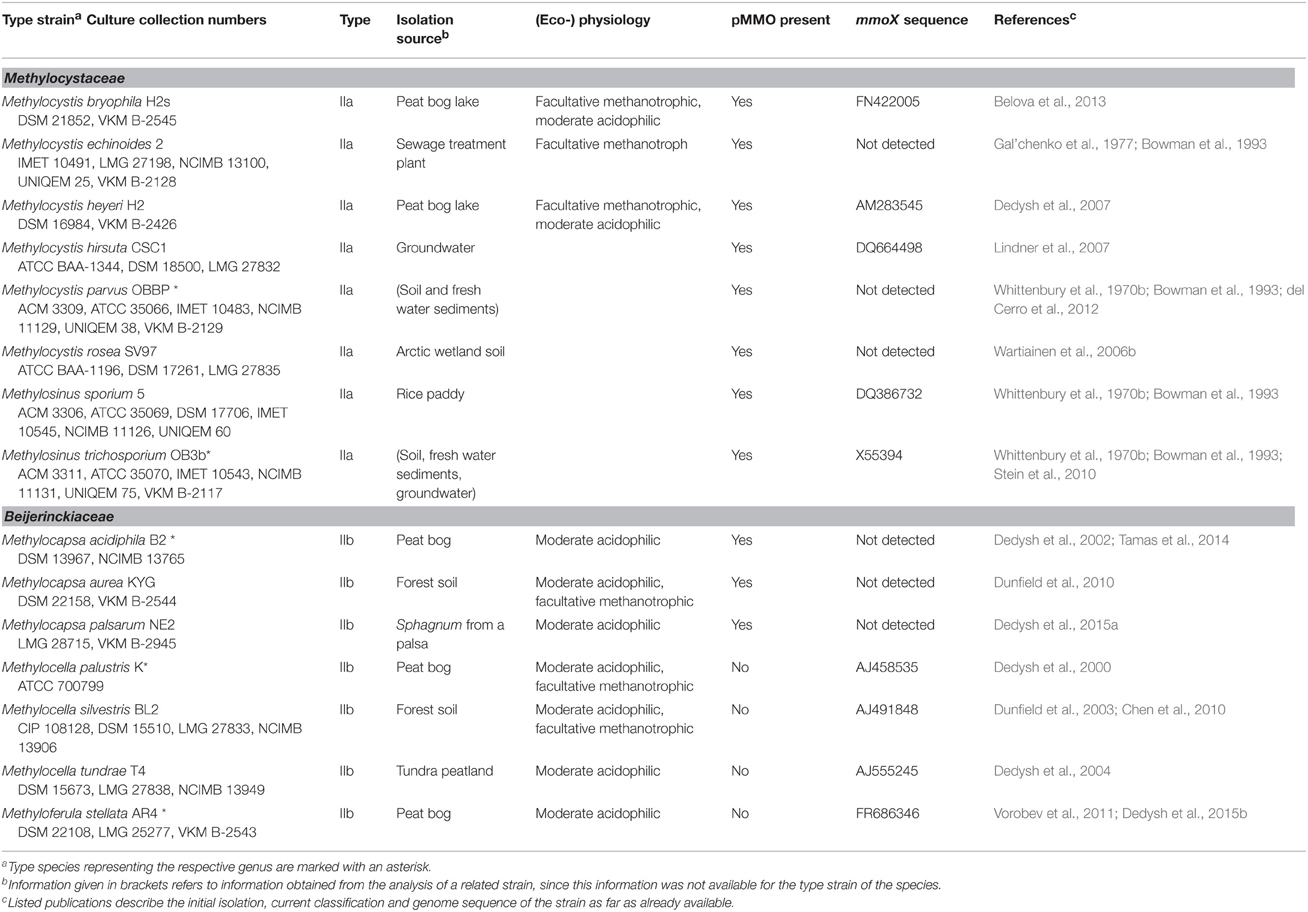
Table 1. Taxonomic and physiological characteristics of aerobic methanotrophic Alphaproteobacteria.
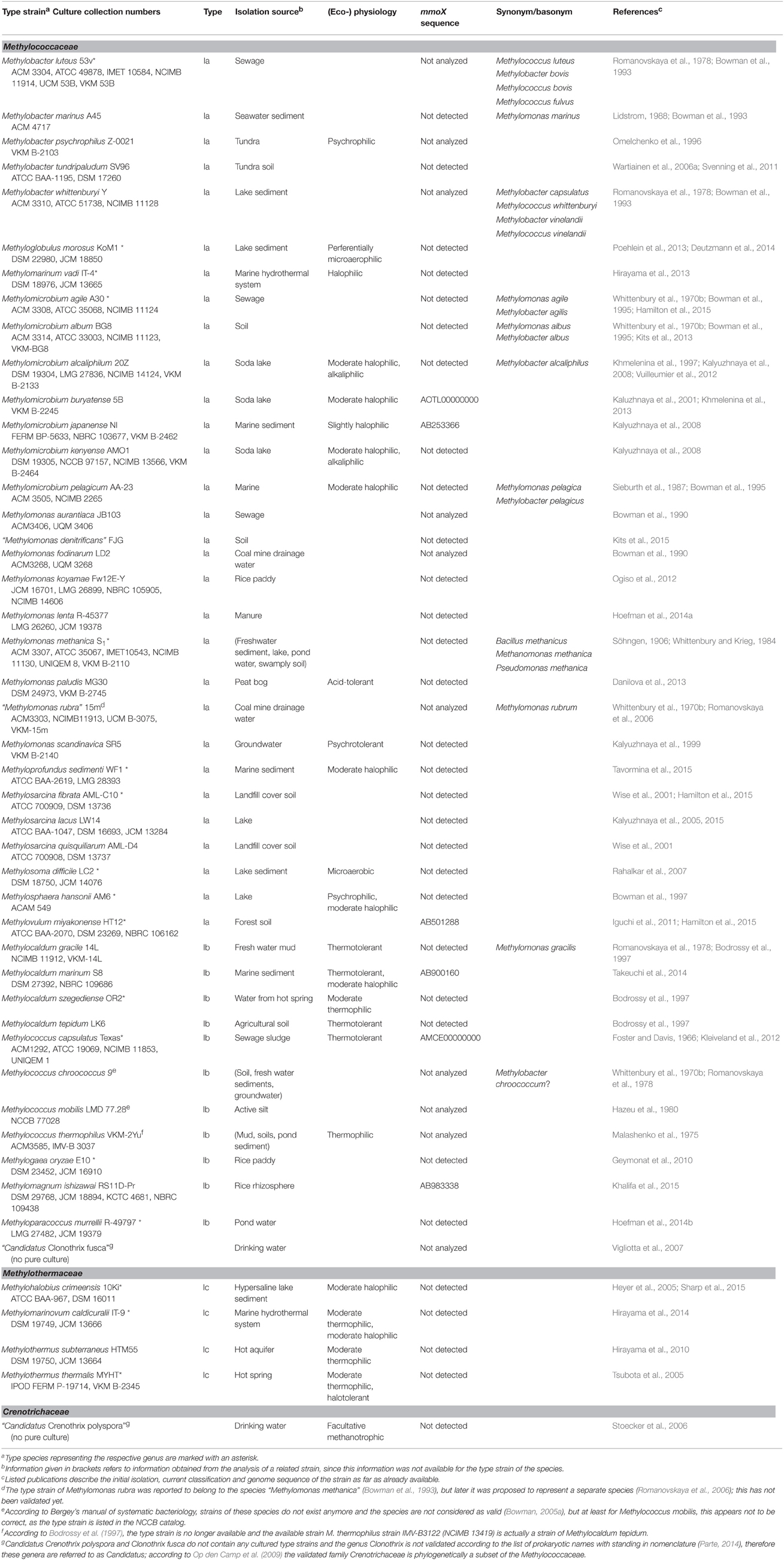
Table 2. Taxonomy, isolation source, and physiological characteristics of aerobic methanotrophic Gammaproteobacteria.
The known diversity of aerobic methanotrophic bacteria was further expanded by the detection of methanotrophic bacteria within the phylum Verrucomicrobia (Table 3). Their existence was described in three independent studies in 2007 and 2008 (Dunfield et al., 2007; Pol et al., 2007; Islam et al., 2008) and they were reported to represent distinct species of the genus “Methylacidiphilum” (Op den Camp et al., 2009). Recently, a second genus within the newly formed methanotrophic family Methylacidiphilaceae was proposed, “Methylacidimicrobium,” also consisting of three species (van Teeseling et al., 2014).
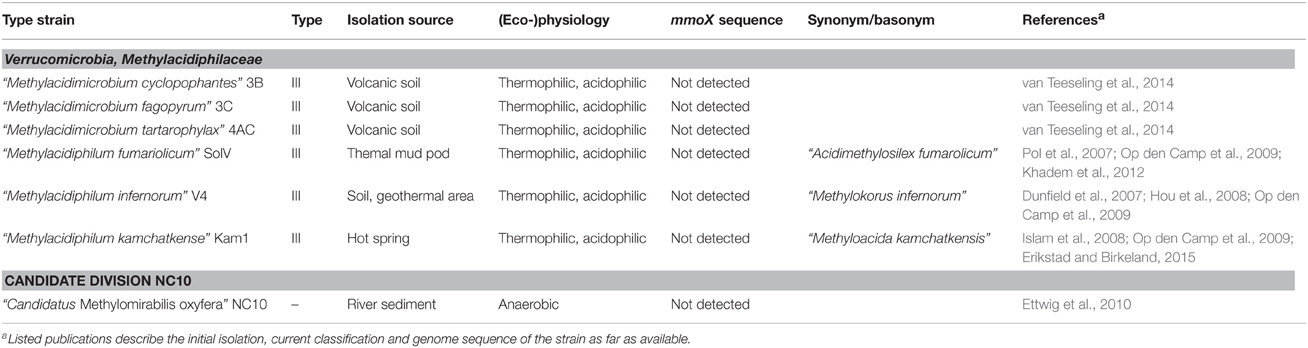
Table 3. Isolation source and physiological characteristics of methanotrophic bacteria harboring pmoA outside the phylum Proteobacteria.
Phylogenetically, the methanotrophic Alphaproteobacteria belong to two families, the Methylocystaceae and Beijerinckiaceae (Figure 1, Table 1). Both families include additional genera of non-methanotrophic bacteria. Nearly all methanotrophic Gammaproteobacteria are classified into the families Methylococcaceae or the recently delineated Methylothermaceae (Hirayama et al., 2014). These families do not contain any non-methanotrophic bacteria. “Candidatus Crenothrix polyspora” is the only exception as it belongs to a distinct family, the Crenotrichaceae, but this classification was put into question by Op den Camp et al. (2009), who proposed that Crenothrix could be a member of the Methylococcaceae, based on its 16S rRNA gene phylogeny (Figure 1).
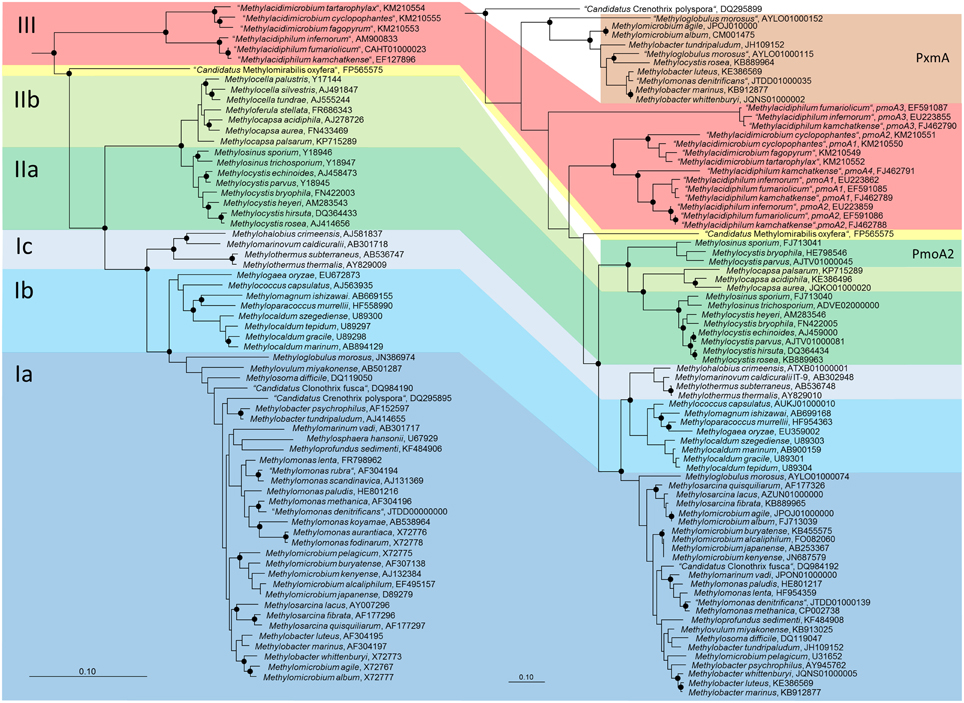
Figure 1. Phylogenetic trees showing the phylogeny of methanotrophic type strains based on 16S rRNA gene sequences (left tree) and PmoA sequences (right tree). The neighbor joining trees were calculated using the ARB software package (Ludwig et al., 2004) based on 1556 nucleotide positions with Jukes Cantor correction or 160 amino acid positions with Kimura correction, respectively. PmoA sequences of Methylobacter luteus, Methylobacter whittenburyi, and Methylomicrobium pelagicum are not available from the type strains, but were taken from a different strain representing the species. The 16S rRNA gene based tree was rooted with sequences of methanogenic Archaea (AB301476, M60880, AB065296, AM114193, AB196288), the PmoA tree with AmoA sequences of ammonia-oxidizing bacteria (NC_004757, X90822). Dots label branch points that were confirmed in maximum likelihood trees. The scale bars display 0.10 changes per nucleotide or amino acid position.
Classification of Cultivated Methanotrophic Bacteria into Type I and Type II Methanotrophs?
The characterization of several new genera of methanotrophs in the 1970s and 1980s resulted in the classification into two major groups, type I and type II methanotrophs based on physiological, morphological, ultrastructural and chemotaxonomic traits (Whittenbury and Dalton, 1981). Major distinctive characteristics between type I and type II methanotrophs were the arrangement of internal membranes as vesicular discs (type I) or paired membranes aligned to the cell periphery (type II), the carbon fixation mechanism via the ribulose monophosphate pathway (type I) or serine cycle (type II), the capability of nitrogen fixation, the formation of resting stages, and the predominance of specific C16 (type I) or C18 (type II) fatty acids (Hanson and Hanson, 1996; Trotsenko and Murrell, 2008). In some studies, type X methanotrophs were further differentiated from type I methanotrophs based on characteristics such as the presence of ribulose-1,5-bisphosphate carboxylase, differences in nitrogen fixation capability or higher optimum growth temperatures (Green, 1992; Hanson and Hanson, 1996; Bowman, 2006). Phylogenetic analyses of 16S rRNA gene sequences confirmed this classification, whereby type I and type X methanotrophs correspond to the Gammaproteobacteria and type II to the Alphaproteobacteria. However, the characterization of several new genera and species during the last years has turned this distinction based on the mentioned criteria largely into question. While the major carbon fixation pathway is still a distinctive feature, several other characteristics are no longer exclusively found in one or the other group:
(1) Methanotrophic Beijerinckia species are not considered as typical type II methanotrophs as most of them lack the characteristic internal membrane system. Some may have vesicles instead, but only Methylocapsa palsarum has a well-developed membrane system. Furthermore, the genera Methylocella and Methyloferula do not possess the particulate methane monooxygenase (Dedysh et al., 2000, 2002, 2004, 2015a; Dunfield et al., 2003, 2010; Vorobev et al., 2011).
(2) All methanotrophic Beijerinckia species lack the classical 18:1ω8c signature fatty acid of type II methanotrophs (Dedysh et al., 2000, 2002, 2004, 2015a; Dunfield et al., 2003, 2010; Vorobev et al., 2011). Similarly, Methylosinus sporium does not possess this signature fatty acid (Bodelier et al., 2009).
(3) Methylocystis heyeri possesses with 16:1ω8c a signature fatty acid of type I methanotrophs (Dedysh et al., 2007).
(4) Most members of the Methylothermaceae have signature fatty acids of type II methanotrophs: Methylohalobius crimeensis, Methylothermus subterraneaus, and Methylomarinovum caldicuralii contain 18:1ω7c among their major fatty acids (Heyer et al., 2005; Hirayama et al., 2010, 2014). The fourth member of this family, Methylothermus thermalis, contains 18:1ω9c, a C18 fatty acid neither abundant in the other Methylothermaceae nor in type II methanotrophs (Tsubota et al., 2005).
(5) Methanotrophic Verrucomicrobia do not fit well into the scheme as most of them lack the typical intracytoplasmic membranes (only exception “Methylacidimicrobium fagopyrum”) and have distinct dominant fatty acids (i14:0, a15:0) (Op den Camp et al., 2009; van Teeseling et al., 2014).
(6) Further differentiation criteria such as nitrogen fixation capability, the formation of resting stages, or the optimum growth temperature, which were initially applied, are not indicative for one or the other type anymore.
Based on these exceptions, the initial concept of type I and II methanotrophs is no longer useful to categorize all known aerobic methanotrophic bacteria and it has been proposed to abandon it (Op den Camp et al., 2009; Semrau et al., 2010). Nevertheless, the terms are still frequently used and adapted to the increasing diversity of methanotrophs, but should only be considered as synonyms for the phylogenetic groups of methanotrophic Alpha- and Gammaproteobacteria. In this way the terms will be used in this review.
The methanotrophic Alphaproteobacteria were recently divided into type IIa (Methylocystaceae) and type IIb (Beijerinckiaceae) methanotrophs (Deng et al., 2013; Dumont et al., 2014). Likewise, the methanotrophic Gammaproteobacteria are frequently differentiated into subgroups. Often they are divided into two groups, whereby the genera Methylococcus, Methylocaldum, Methylogaea and the Methylothermaceae form type 1b methanotrophs, while the remaining gammaproteobacterial genera are grouped as type 1a methanotrophs (Chen et al., 2008; Deutzmann et al., 2011; Dumont et al., 2011; Siljanen et al., 2011; Krause et al., 2014). Some recent studies differentiated the methanotrophic Gammaproteobacteria into three type I subgroups, but this categorization is not consistent among different publications. A taxonomic review referred to the clade consisting of Methylococcus, Methylocaldum, Methylogaea, Methyloparacoccus as type Ia and to members of the family Methylothermaceae as type Ic, while the remaining Gammaproteobacteria represented type Ib methanotrophs (Bowman, 2014). In contrast, in some cultivation independent studies the above mentioned frequent grouping into type Ia and Ib was applied and extended by introducing type Ic, comprising pmoA sequences of uncultivated taxa (USCγ, JR2, JR3, OPU1) and the amoA sequence of Nitrosococcus (Lüke and Frenzel, 2011; Henneberger et al., 2012; Dumont et al., 2014). It is thus referring to a group of uncultivated methanotrophs. Such a further differentiation of the methanotrophic Gammaproteobacteria appears useful to refer to the specific subgroups of cultivated and uncultivated methanotrophs easily. In this review, the nomenclature of type Ia and Ib methanotrophs as applied in diverse cultivation-independent studies is kept, while the Methylothermaceae are referred to as type Ic methanotrophs (Table 2). The clade of Nitrosococcus and related uncultivated clusters represent type Id organisms when discussing diversity based on pmoA phylogeny. Methanotrophic Verrucomicrobia are referred to as type III.
Ecophysiology of Aerobic Methanotrophic Bacteria
Aerobic methanotrophic bacteria occur in terrestrial, aquatic and marine ecosystems, typically at oxic-/anoxic interfaces, where oxygen is available as electron acceptor and methane as carbon and energy source, which is released as end product from the anaerobic degradation of organic matter. They are likewise present in diverse upland soils where they are responsible for atmospheric methane oxidation or become temporarily active when higher concentrations of methane are available (Knief et al., 2006; Dunfield, 2007; Kolb, 2009). The ecology of methanotrophic bacteria has been reviewed in diverse articles and will not be discussed in detail here (e.g., Hanson and Hanson, 1996; Conrad, 2007; Semrau et al., 2010; Chowdhury and Dick, 2013; Bowman, 2014). The focus in this article is on physiological adaptations to particular environmental conditions in relation to phylogeny.
In terms of metabolic adaptations, some methanotrophic bacteria show higher versatility than initially thought. They are capable of growing on carbon compounds with C-C bond, while most methanotrophic bacteria are obligate methanotrophs. The existence of such facultative methanotrophs had been debated for a long time (reviewed in Theisen and Murrell, 2005; Semrau et al., 2011), until it was rigorously proven for Methylocella silvestris BL2 (Dedysh et al., 2005). This strain has the broadest versatility currently known among methanotrophs; besides C1-compounds, it can use a variety of organic acids including acetate, pyruvate, propionate, succinate, malate, and gluconate, alcohols such as ethanol and 2-propanol and the gaseous compounds ethane and propane (Crombie and Murrell, 2014). Growth on acetate is more efficient than on methane and methane monooxygenase expression is down-regulated in the presence of acetate (Dedysh et al., 2005; Theisen et al., 2005). In contrast, methane and propane are consumed simultaneously in this strain (Crombie and Murrell, 2014). A facultative lifestyle with a much narrower substrate range has been reported for other members of the genus Methylocella and for Methylocapsa aurea (Table 1), but it is not a general feature of all methanotrophic Beijerinckiaceae. Moreover, several Methylocystis strains including diverse type strains are able to grow on acetate or ethanol, but with growth rates 3–10-fold lower compared to growth on methane (Belova et al., 2011; Im et al., 2011). Gene expression of methane monooxygenase appears not to be regulated by acetate in these methanotrophs (Belova et al., 2011; Yoon et al., 2011). It remains to be proven whether the capability to grow on acetate is linked to phylogeny within this genus. Crenothrix polyspora is the only methanotrophic gammaproteobacterium for which uptake of acetate and, to lesser extent, glucose, has been reported (Stoecker et al., 2006), but besides evidence from fluorescence in situ hybridization experiments coupled to microautoradiography (FISH-MAR) performed on natural enrichments, this phenomenon has not been further proven. It is obvious that a facultative lifestyle can provide a benefit for methanotrophic bacteria. However, the relevance of facultative methanotrophy in nature remains little understood, and linked to this the question to what extent a facultative life style may influence methane emissions in the environment. Only few studies have analyzed the consumption of methane and alternative substrates under in situ conditions so far. In mire samples, acetate addition resulted in a reduction of methane emission rates and decreased pmoA expression rates of Methylocystis (Wieczorek et al., 2011). Likewise, acetate addition decreased methane oxidation rates and stimulated growth of Methylocystis in paddy soil samples. Stable isotope probing with 13C-labeled acetate under aerobic conditions resulted in a labeling of Methylocystis in these samples, demonstrating that the labeled carbon was somehow metabolized and incorporated by the cells (Leng et al., 2015).
Another aspect that has repeatedly been addressed is the adaptation to low methane concentrations. The observations made in competition experiments with isolates grown in continuous culture and in incubations with rice field soils resulted in the frequently cited conclusion that type I methanotrophs are more competitive under low methane concentrations compared to type II methanotrophs (Graham et al., 1993; Henckel et al., 2000b; Macalady et al., 2002). This seems to apply to ecosystems as long as methane supply remains at a rather high level, but when methane concentrations drop below 1000 or even 100 ppmv for prolonged periods of time, Methylocystaceae have the better potential to remain active (Knief and Dunfield, 2005).
Most methanotrophic bacteria are mesophilic and neutrophilic organisms, but several isolates were obtained from more extreme habitats and are specifically adapted to lower or higher pH, temperature, salt or oxygen concentrations (Trotsenko and Khmelenina, 2002). Methanotrophic bacteria adapted to warmer or colder temperatures are found in a couple of distinct genera of Gammaproteobacteria, often side by side with mesophilic species (Table 2). Among the methanotrophic Alphaproteobacteria adaptations to temperatures outside the mesophilic range appear to be uncommon. Outstanding are the verrucomicrobial methanotrophs, which represent the most thermophilic methanotrophs (optimum temperature 55–60°C) (Op den Camp et al., 2009). These are at the same time acidophiles, with pH optima for growth between 2.0 and 4.3. All isolates were obtained from geothermally influenced environments (Op den Camp et al., 2009; van Teeseling et al., 2014). The occurrence of these thermoacidophilic methanotrophs appears to be largely restricted to such geothermal environments, in particular to acidic conditions, while they seem to have a broader temperature range, as revealed by cultivation-dependent and -independent analyses (Sharp et al., 2014; van Teeseling et al., 2014).
An adaptation to mildly acidic pH values (growth optima between 5.0 and 6.0) is characteristic for methanotrophic Beijerinckiaceae and some Methylocystis strains, which were mostly isolated from acidic peatlands or forest soils (Table 1). Cultivation-independent analyses suggest that the occurrence of Methylocella is not limited to these acidic environments (Rahman et al., 2011). Less common are acidophilic methanotrophs among the Gammaproteobacteria. Members of the species Methylomonas paludis have been described as acid-tolerant and are inhabitants of acidic peatlands (Danilova et al., 2013; Danilova and Dedysh, 2014). Methanotrophs that are adapted to high pH values are found within the Gammaproteobacteria, in particular within the genus Methylomicrobium. The occurrence of alkaliphilic methanotrophs is not restricted to the class of Gammaproteobacteria, the isolation of an alkaliphilic Methylocystis isolate has also been reported (Eshinimaev et al., 2008). Some alkaliphilic Gammaproteobacteria are at the same time halophiles (Methylomicrobium alcaliphium and Methylomicrobium kenyense), isolated from soda lakes (Kalyuzhnaya et al., 2008). Methanotrophic bacteria that were isolated from marine ecosystems are also adapted to higher salt concentrations and are likewise found among methanotrophic Gammaproteobacteria. High salt tolerance is not necessarily a characteristic of all members of a genus, as exemplified by Methylocaldum and Methylomicrobium (Table 2). In the last few years, the first methanotrophic isolates were described that live preferentially under lower oxygen concentrations (Methylosoma difficile and Methyloglobulus morosus). They were enriched in systems with opposing gradients of methane and oxygen, thus mimicking the conditions in sediments (Rahalkar et al., 2007; Deutzmann et al., 2014).
In conclusion, a broad versatility in terms of adaptation to different environmental conditions can be found among the methanotrophic Gammaproteobacteria (low and high temperatures, low and high pH, high salt, low oxygen), which comes along with a high diversity of methanotrophs within this group. Cultivated methanotrophic Alphaproteobacteria are less diverse and show less and different adaptations (low pH, low methane availability) based on current knowledge. At genus level, the occurrence of methanotrophic bacteria that are adapted to a specific environmental condition is not necessarily limited to one phylogenetic lineage, but can often be found within different genera of methanotrophs side by side with species that show different adaptations and habitat preferences. Thus, some genera have a broad ecological niche, though the individual species or strains have smaller niches, while others are less diverse in term of ecophysiological adaptations and have a rather narrow niche. Habitat adaptation and specialization appear to occur at different taxonomic levels. Consequently, the distribution of methanotrophic bacteria in the environment should be evaluated at these different taxonomic levels in order to better understand distribution and community composition. Such a detailed evaluation is undertaken in this review, based on a meta-analysis including the large diversity of uncultivated methanotrophs (see Sections Description of Major Uncultivated Groups of Methanotrophic Bacteria and Their Habitat Specificity and Habitat Specificity of Methanotrophic Taxa Evaluated at Higher Taxonomic Resolution).
Cultivation-independent Detection of Aerobic Methanotrophic Bacteria Based on Molecular Markers
Tools for the cultivation-independent detection of aerobic methanotrophic bacteria exist since 20 years and have been used in diverse studies. The most frequently targeted gene in environmental studies, the 16S rRNA gene, can be used for the detection of methanotrophic bacteria using taxon specific primers and probes that are available for several different groups (compiled by McDonald et al., 2008). While the analysis of this gene provides valuable information about the phylogenetic placement of methanotrophic bacteria detected in environmental samples, it does not allow the identification of methanotrophic bacteria beyond the well-known families.
Functional Marker Genes as Molecular Markers
Such a limitation is of less relevance when functional genes are used as markers, such as the methane monooxygenase encoding genes (McDonald et al., 2008). The methane monooxygenase is the key enzyme responsible for the initial conversion step of methane to methanol. Two forms of this enzyme are known, the soluble methane monooxygenase (sMMO) and a membrane-bound enzyme, the particulate methane monooxygenase (pMMO). The pmoA gene encoding the β-subunit of the particulate methane monooxygenase is the most frequently used marker, as it is present in most aerobic methanotrophic bacteria with exceptions among the Beijerinckiaceae (Table 1). It is also present in anaerobic denitrifying bacteria, represented by an enriched culture of “Candidatus Methylomirabilis,” a bacterium of the NC10 phylum (Ettwig et al., 2010).
To include Beijerinckiaceae and to obtain a more complete picture about the methanotrophs present in a sample, the mmoX gene encoding the α-subunit of the soluble methane monooxygenase hydroxylase component has been used in addition to pmoA in some studies (e.g., Morris et al., 2002; Chen et al., 2008; Deng et al., 2013). However, due to its limited occurrence in methanotrophs (Tables 1–3), mmoX is much less frequently used as marker. It is not uniformly present or absent within the same genus and variation exists even at species level, as evident from studies with Methylocystis, Methylosinus, and Methylomonas strains (Shigematsu et al., 1999; Heyer et al., 2002).
Further gene markers that can be used for the detection of methanotrophs are not unique to this metabolic guild, but shared with other organisms. Among those are the mxaF gene, which encodes the large subunit of the methanol dehydrogenase, and a couple of other markers of the methylotrophic metabolism (reviewed by Kolb and Stacheter, 2013; Dumont, 2014).
pmoA as Molecular Marker
Both, pmoA and mmoX have been shown to produce phylogenies that are largely congruent with those of the 16S rRNA gene (Auman and Lidstrom, 2002; Heyer et al., 2002; Kolb et al., 2003), which allows to draw conclusions about the phylogenetic placement of methanotrophs possessing genes with novel sequence types. Updated trees (Figure 1) show that this is still the case, but research of the last few years has revealed that this congruency includes more and more exceptions. The presence of paralogous gene copies in methanotrophic bacteria as well as the detection of evolutionary related monooxygenases in non-methanotrophic bacteria contribute to sequence diversity in cultivation-independent studies (see next section). Hence, conclusions about the taxonomic identity of bacteria detected based on their pmoA sequences have to be drawn with care, in particular if sequences cluster distantly to those of well-known methanotrophs. This is also exemplified by the pmoA sequence of the gammaproteobacterial “Candidatus Crenothrix polyspora,” which is highly divergent from those of all other methanotrophic Gammaproteobacteria. Besides these issues, inconsistency exists among the type Ia methanotrophs (Figure 1). Tree reconstructions within this group are in general not highly robust, but both, Methylobacter and Methylomicrobium species do not form monophyletic clusters, independent of the applied treeing method and the phylogenetic marker. Methylobacter psychrophilus and Methylobacter tundripaludum appear to be distinct from the other Methylobacter species, likewise as Methylomicrobium album and Methylomicrobium agile cluster with Methylobacter whittenburyi in 16S rRNA gene based trees and with Methylosarcina species in pmoA based trees rather than with the other Methylomicrobium species. Elaborate taxonomic analyses including information derived from whole genome sequencing projects of these and further reference strains will be necessary to ensure the taxonomic placement of these species.
A couple of different primer sets were developed for the amplification of pmoA gene fragments, but remarkably, the first published primer pair (A189/A682) is still most frequently used (Holmes et al., 1995). Only one alternative system (A189/mb661) is often used instead or in addition to the before mentioned system (Costello and Lidstrom, 1999). This second primer combination is more specific for methanotrophic bacteria as it does not amplify the amoA gene of ammonia-oxidizing bacteria (Costello and Lidstrom, 1999). However, it fails to detect some of the clusters that have a phylogenetic position between pmoA and amoA sequences, such as the RA21 or the pxmA cluster, it largely discriminates USCα and amplifies type IIb methanotrophs less efficiently (Bourne et al., 2001; Deng et al., 2013). A third primer, A650 does not show this limitation while excluding amoA, but has less frequently been used (Bourne et al., 2001; Shrestha et al., 2012). Because primer system A189/A682 results in the production of additional unspecific PCR products in some cases, a semi-nested approach was used in these studies. After a first PCR using primers A189/A682 a second PCR with primers A189/mb661 or A189/A650 was applied (Singh et al., 2007; Qiu et al., 2008; Kip et al., 2011; Siljanen et al., 2011; Barbier et al., 2012). Alternatively, a combination of both reverse primers in a multiplex PCR was used in the second PCR to overcome the detection limitations of primer mb661 (Horz et al., 2005). Some further general and several specific primers for the detection of subgroups were developed, as compiled in review articles (McDonald et al., 2008; Dumont, 2014). Many of them were developed for qPCR assays targeting subgroups (Kolb et al., 2003, 2005; Degelmann et al., 2010; Wieczorek et al., 2011; Sharp et al., 2014). Moreover, specific primers are needed to amplify pmoA genes of Verrucomicrobia (Erikstad et al., 2012; Sharp et al., 2012), the homologous pmoA2 gene (Tchawa Yimga et al., 2003), or the pmoA genes of anaerobic methanotrophic bacteria of the NC10 phylum (Luesken et al., 2011).
pmoA Paralogs and Evolutionary Related Monooxygenases
Paralogous copies of the pmoA gene and evolutionary related monooxygenases in non-methanotrophic bacteria are sometimes detected in cultivation-independent studies, depending on the primers used to amplify the target gene. They can thus contribute to the diversity of detected sequence types in environmental studies, but do not represent distinct methanotrophs. A couple of sequence clusters in pmoA based phylogenetic trees have meanwhile been identified as paralogs or alternative monooxygenases.
pmoA Paralogs in Methanotrophic Bacteria
Many methanotrophs have multiple copies of the pmo operon and initially it appeared that these copies are (nearly) identical (Auman et al., 2000; Gilbert et al., 2000), so that they do not affect diversity studies that are based on pmoA gene detection. Methylocystis sp. SC2 was the first methanotrophic strain in which two different pmoA genes were discovered, the conventional and a second copy, referred to as pmoA2, with only 73% identity to the well-known pmoA gene of Methylocystaceae (Dunfield et al., 2002). The application of specific primers for the detection of the pmoA2 gene revealed that this second gene copy is present in diverse though not all Methylocystis and Methylosinus strains (Tchawa Yimga et al., 2003). The pmoA2 gene is localized in the pmoCAB2 operon, which encodes a functional methane monooxygenase, enabling Methylocystis SC2 to oxidize methane at lower mixing ratios compared to the conventional monooxygenase, which is downregulated under these conditions in strain SC2 (Baani and Liesack, 2008). This finding was taken as explanation for the previously described capability of Methylocystis species to oxidize methane at very low mixing ratios down to atmospheric level over a period of several months and their capability to grow at mixing rations as low as 10–100 ppmv (Knief and Dunfield, 2005). Moreover, this corresponds very well to the observation that Methylocystis strains are frequently detected in upland soils and hydromorphic soils, where they face low methane supply almost constantly (Dunfield, 2007). However, the pmoA2 gene of Methylocystis and Methylosinus has not been detected very frequently in upland soils, but rather in different other ecosystems (Tables S1–S4). Either the commonly applied primers are not well suited to amplify pmoA2 genes of those Methylocystaceae that occur in upland soils, or the pmoA2 gene is more important for survival of methanotrophs residing in habitats with fluctuating methane supply at higher concentrations.
In methanotrophic Verrucomicrobia, multiple different pmoA gene copies are present (Figure 1). All genes are highly divergent from those of proteobacterial methanotrophs and quite different to each other (Op den Camp et al., 2009). The strains “Methylacidiphilum fumariolicum” SolV and “Methylacidiphilum infernorum” V4 possess three complete pmoCAB operons, while “Methylacidiphilum kamchatkense” Kam1 has a fourth distinct copy of pmoA, localized in a truncated pmoCA operon. An expression study performed with this strain revealed that the methane monooxygenase encoded by pmoCAB2 is strongly expressed when cells are grown under laboratory conditions (Erikstad et al., 2012). The function of the other copies and regulatory mechanisms that may control the expression of these genes remain currently largely unknown.
The pxmA Gene
In methanotrophic Gammaproteobacteria of the genera Methylomonas, Methylobacter and Methylomicrobium another homolog of pmoA has been detected, the pxmA gene (Tavormina et al., 2011). Recent genome sequencing projects reveal that pxmA genes occur more widespread in methanotrophs. They are present in further Methylococcaceae strains, which are distantly related to the known genera but have so far not been further described in the literature. A pxmA copy is also present in an alphaproteobacterial strain, Methylocystis rosea. In Methyloglobulus morosus an additional pxmA like gene is present besides pmoA and pxmA. All pxmA gene sequences form a monophyletic cluster that is clearly distinct from pmoA sequences of methanotrophic Proteobacteria and Verrucomicrobia (Figure 1). Already before their description by Tavormina et al. (2011), pxmA genes were detected in environmental samples, they were referred to as “pmoA/amoA like” sequences or as Cluster WC306-54 (Nold et al., 2000; Lau et al., 2007; Dörr et al., 2010). The presence of pxmA appears not to be closely linked to phylogeny, similarly to the occurrence of pmoA2 among Methylocystaceae or mmoX among the methanotrophic Proteobacteria. The function of the gene product and regulation of gene expression remain currently largely unknown. So far, it could be shown that the gene, which is localized in the pxmABC operon, is expressed under environmental and in vitro conditions (Tavormina et al., 2011; Kits et al., 2015).
Evolutionary Related Monooxygenases
It is well known that the particulate methane monooxygenase and the ammonia monooxygenase of nitrifying bacteria and archaea are evolutionary related (Holmes et al., 1995). Meanwhile, further monooxygenases of the superfamily of copper-containing membrane-bound monooxygenases have been identified, involved in the oxidation of short chain hydrocarbons, but not methane (Redmond et al., 2010; Sayavedra-Soto et al., 2011; Coleman et al., 2012; Suzuki et al., 2012). In phylogenetic trees, the sequences of these genes form clusters that are distantly related to those of the known pmoA and amoA genes. Due to the high sequence divergence, most of these sequence types have not frequently been detected in cultivation-independent PCR-based studies using current pmoA primers, but some of them have been found in metagenomic or metatranscriptomic datasets, e.g., in hydrocarbon-rich marine ecosystems (Li et al., 2014).
The existence of a butane monooxygenase in Nocardioides sp. CF8 related to the particulate methane monooxygenase was already postulated by Hamamura and Arp (2000), but molecular evidence was provided only recently when the whole genome of the strain was sequenced (Sayavedra-Soto et al., 2011). The butane-oxidizing monooxygenase is encoded by the genes in the bmoCAB operon, which have less than 50% amino acid similarity to the genes of the methane and ammonia monooxygenase. Similar genes were also detected in Mycobacterium smegmatis strains NBB4 and NBB3 (Coleman et al., 2012). The enzyme in strain NBB4 was shown to oxidize ethane, propane, butane and ethylene. Due to the broader substrate spectrum of the enzyme in Mycobacterium, the enzyme was referred to as hydrocarbon monooxygenase, encoded in the hmoCAB operon. Genome sequencing projects suggest that similar monooxygenases exist in Mycobacterium chubuense B4, Nocardioides luteus FB or the uncultured deltaproteobacterial SAR324 clade, which is ubiquitous in the ocean (Sheik et al., 2014).
Redmond et al. (2010) described another cluster of putative hydrocarbon monooxygenases (emoA), detected upon stable isotope probing with 13C-ethane at a hydrocarbon seep. The authors speculate that the labeled organisms are members of the Methylococcaceae, which seem to be incapable of methane oxidation. These assumptions can currently only be confirmed by sequence data from isolates referred to as Methylococcaceae ET-SHO and ET-HIRO, which were deposited in the NCBI database in an independent study, but remain to be published. Based on the entries in the NCBI database it appears that these Methylococcaceae isolates, which were also obtained from a marine habitat, could grow on ethane, but not on methane.
Further types of monooxygenase genes related to pmoA and amoA are found in Gammaproteobacteria of the genus Haliea and in the genome of the alphaproteobacterium Skermanella aerolata KACC 11604 (= 5416T-32T). Strains Haliea ETY-M and ETY-NAG grow on ethylene and oxidize in addition ethane, propane and propylene, but not methane (Suzuki et al., 2012). In case of Skermanella aerolata KACC 11604 growth on hydrocarbons has not yet been studied. The sequence of their monooxygenase is different from the hmoA and emoA genes, but related to the pmoA sequences of type II methanotrophs.
A Comparison of Cultivation-dependent and –Independent Diversity of Methanotrophs Based on pmoA as Phylogenetic Marker
Classification of pmoA Sequences Based on Phylotyping or OTU Clustering
Using pmoA as molecular marker for the detection of methanotrophic bacteria it turned out that there is a huge diversity of methanotrophs present in nature that is not represented by isolates in the laboratory. Approximately 15,000 pmoA and pmoA-like sequences can be found in the Genbank database. To describe and discuss the current diversity of aerobic methanotrophic bacteria based on this data resource, sequences have to be grouped based on similarity. In many studies such groups are defined based on their clustering in phylogenetic trees in relation to known phylotypes, which are represented by sequences of type strains or other well-studied reference strains as well as selected sequences of uncultivated clades. Dumont et al. (2014) recently defined 53 representative sequences for major cultivated and uncultivated phylogenetic clusters.
Another approach is the grouping of similar sequences into operational taxonomic units (OTUs) using a predefined cut-off value. Some studies applied a 3% cut-off without explicitly linking this to a specific phylogenetic resolution (Saidi-Mehrabad et al., 2013; Sharp et al., 2014). Other studies determined and used cut-off values with the aim to reflect genus and species resolution. These values were determined in correspondence to the routinely used cut-off values known from 16S rRNA gene sequence analyses, i.e., 3% sequence difference to distinguish between species and 5% to differentiate genera (Schloss and Handelsman, 2005). For pmoA sequences, Lüke et al. (2010) defined cut-off values at 10 and 17% sequence dissimilarity for species and genus delineation, respectively, based on the fact that the nucleotide substitution rate of pmoA is 3.5 times higher than that of 16S rRNA genes. The factor 3.5 was derived by correlation of 16S rRNA and pmoA gene sequence identities of approx. 75 Methylocystis and Methylosinus strains (Heyer et al., 2002). Degelmann et al. (2010) included Gammaproteobacteria in the comparative analysis and compiled 16S gene sequence identity values of 22 methanotrophs. They correlated 16S rRNA gene to pmoA gene as well as to deduced PmoA protein sequence identity values and defined a cut-off of 13% at DNA level for species delineation, corresponding to 7% cut-off at protein level. When comparing these cut-off values to the sequence differences observed between methanotrophic type strains within the same and of different genera, it is apparent that they reflect the average sequence difference between type strains so that genera and species will not be fully resolved using these values (Figure 2). At the same time the diagrams, which display minimum and maximum sequence difference of each type strain to another type strain within the same genus and family, reveal that it will be impossible to find cut-off values that differentiate perfectly well all genera without already differentiating species within a genus. Similar difficulties in determining cut-off values that correspond to a certain phylogenetic resolution are known from 16S rRNA gene based analyses (Schloss and Westcott, 2011).
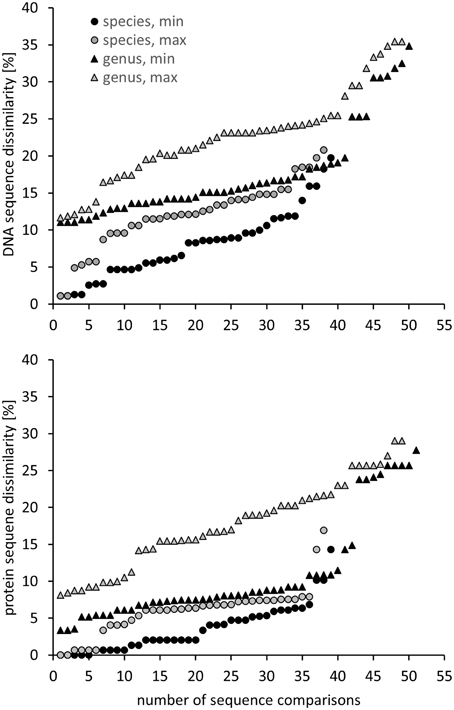
Figure 2. Minimum and maximum pmoA sequence dissimilarity at DNA (upper panel) and protein level (lower panel) between a type strain and its most closely and most distantly related type strain within the same species, as well as to the most closely and distantly related type strain of a different genus within the same family or type (according to Table 1). DNA and protein distance matrices were calculated in ARB based on 480 aligned nucleotide positions or 160 deduced amino acid positions. Methylomicrobium album and Methylomicrobium agile were not included, due to the very distant clustering from the other Methylomicrobium strains (Figure 1), while “Candidatus Crenothrix polyspora” was excluded due to the fact that it contains a highly divergent pmoA sequence compared to all other Gammaproteobacteria.
For the evaluation of the diversity of methanotrophic bacteria in this review article, OTU clustering was performed based on cut-off values that reflect a higher resolution compared to the published values to resolve the distinct genera and species as good as possible. The compilation of minimal DNA sequence differences between genera reveals that a cut-off value of 11% is necessary to differentiate all genera (Figure 2). Indicative for an adequate resolution is the separation of the two most closely related genera, Methylocystis and Methylosinus. To further evaluate the 11% cut-off value, it was applied to cluster all available high quality pmoA sequences using the Mothur classification tool with average neighbor algorithm. Sequences of at least 400 bp length and without accumulation of evident sequencing errors were considered as high quality here and the dataset is referred to as “large pmoA dataset” in the following. When performing OTU clustering using different cut-off values it turned out that not 11% but 12% cut-off is sufficient for nearly full resolution at genus level (Figure 3). At the same time, type strains belonging to the same genus were grouped into distinct clusters in five cases: “Methylacidiphilum,” “Methylacidimicrobium,” Methylocapsa, Methylomicrobium, and Methylobacter. In case of Methylobacter and Methylomicrobium, this finding corresponds to the polyphyletic clustering in pmoA trees (Figure 1). To fully prevent the formation of more than one OTU for these genera, a much higher cut-off value of >20% would be necessary.
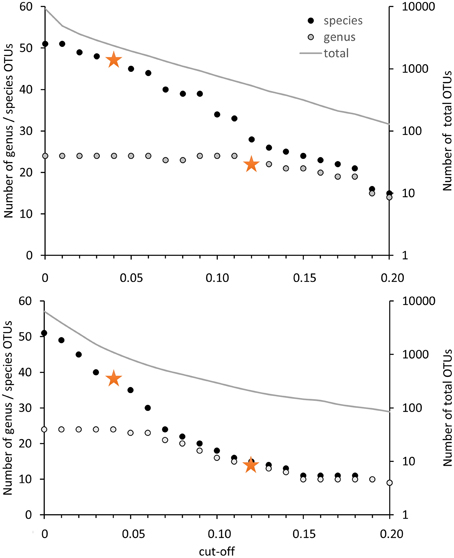
Figure 3. Number of OTUs in dependence on the cut-off value applied for OTU differentiation. The number of OTUs containing type strains of different genera or species are displayed on the left axis, the number of OTUs formed based on all high quality sequences (= total) is presented on the right axis at logarithmic scale. Clustering was performed with 12502 high quality pmoA sequences (upper panel) or the deduced amino acid sequences (lower panel) available from Genbank. Sequences with at least 400 bp sequence length and without accumulation of sequencing errors were included. Distance matrices were calculated in ARB based on 480 aligned nucleotide positions or 160 deduced amino acid positions. OTU clustering was done using Mothur by applying the average neighbor algorithm. Orange stars denote the cut-off values applied in this review.
The differentiation of pmoA sequences at species level is affected by similar difficulties. A cut-off value of 1% is necessary to resolve all species (except Methylomicrobium album and Methylomicrobium agile, which have even more similar pmoA sequences), while such a low value will classify at the same time many strains belonging to the same species into distinct taxonomic units. A higher cut-off value of 3 or 4% leaves only some species unresolved (Figure 2), namely the two Methylothermus species, Methylocystis hirsuta, and Methylocystis rosea, as well as some of the Methylomicrobium species. OTU clustering applied to the “large pmoA dataset” confirmed these findings and shows that a cut-off value of 4% is sufficient to differentiate the majority of species.
Phylogenetic analysis of functional genes is frequently based on protein sequences. This excludes sequence variability at nucleotide positions that are not under evolutionary selection pressure, but provides at the same time less information, so that resolution of closely related taxa becomes more difficult. OTU clustering of sequences with a cut-off that roughly reflects genus level resolution can be achieved at 6% sequence dissimilarity (Figure 2). It only fails to resolve Methylomarinovum from Methylothermus, but a lower value should nevertheless not be selected as the 6% value already provides higher resolution compared to the 12% cut-off value at DNA level when considering the large PmoA dataset including sequences of uncultivated methanotrophs (Figure 3). Differentiation of species based on protein sequences is even more difficult. Full resolution cannot be obtained as Methylomicrobium and Methylothermus species are not even separated at 1% cut-off. A cut-off of 2% already fails to resolve the majority of type strains within the genera Methylocystis, Methylomicrobium, and Methylothermus, although it still gives a higher number of OTUs compared to the 4% cut-off at DNA level when sequences from cultivation-independent studies are included (Figure 3).
Due to the difficulties in finding appropriate cut-off values at protein level, pmoA sequence diversity was evaluated based on DNA sequences but not protein sequences in the present work. The 12% cut-off was applied to differentiate sequences at a level that allows resolution of most methanotrophic genera and a 4% cut-off was used to differentiate species reasonably well. To distinguish in the following OTU classification done with 12% cut-off from classification with 4% cut-off, the OTUs are referred to as OTU12 and OTU4, respectively.
How well do Cultivated Strains Cover the Diversity of Methanotrophic Bacteria as Seen Based on Cultivation-Independent Studies?
Of the 15,000 pmoA sequences that have been deposited in the Genbank database, the vast majority was derived from cultivation-independent studies. Slightly less than 3% were obtained from cultured methanotrophic strains. Most of them belong to the well-known genera Methylocystis, Methylosinus, Methylomonas, Methylobacter, Methylocaldum, or Methylomicrobium (Table 4). Approximately 20 sequences represent isolates that cannot be assigned to a specific known genus; at least some of them may represent new genera. At species level, isolates that are similar to Methylocystis rosea, Methylocystis hirsuta, Methylocystis echinoides, Methylosinus sporium, and Methylosinus trichosporium or “Methylomonas denitrificans” have most frequently been obtained (Table 5). In contrast, more than half of the described species are represented by only one single strain at the moment.
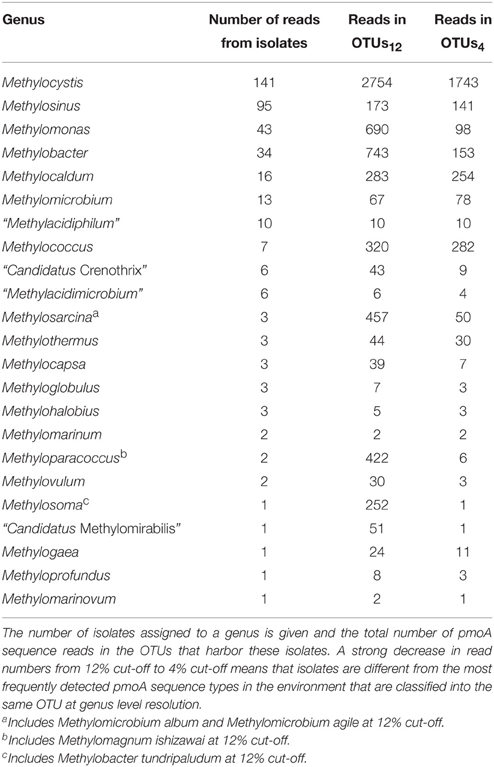
Table 4. Detection frequency of methanotrophic genera in cultivation-dependent and -independent studies.
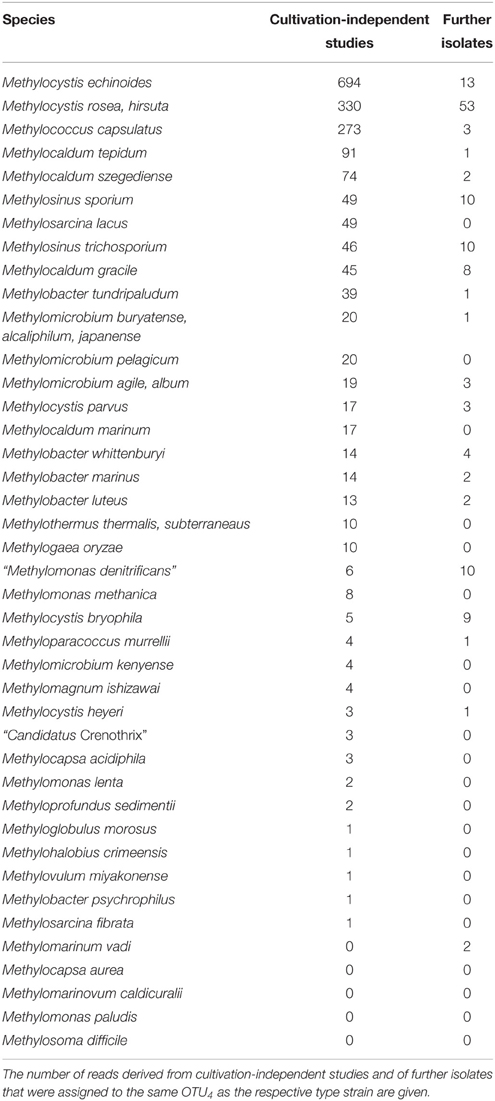
Table 5. Representativeness of methanotrophic type strains at species level resolution.
To evaluate how well cultivated strains cover the diversity of methanotrophic bacteria as seen in cultivation-independent studies, the distribution of their pmoA sequences upon OTU clustering was assessed based on the above mentioned “large pmoA dataset” containing 12,502 high quality sequences. The dataset includes different homologs of pmoA that have been detected in methanotrophs. Clustering of the sequences applying the 12 and 4% cut-off value resulted in 522 and 2287 OTUs, respectively (Table 6). In both cases, there was a rather low number of clusters with high read numbers, while one third of the OTUs12 or even half of the OTUs4 were represented by just one read (singletons). This demonstrates the existence of a very high number of taxa that are rarely detected. The percentage of OTUs that contained sequences of cultivated strains was 12 and 6%, respectively, at the different cut-off levels, which means that only a small fraction of the methanotrophic diversity is represented by cultivated strains. But remarkably, when considering the size of the OTUs12, it turned out that 52% of all available sequences fall into clusters that contain pmoA sequences of isolates. This demonstrates that half of the sequences that have been detected in cultivation-independent studies are closely related to or represented by cultivated genera. At species level, still 24% of all sequences fall into the same OTU4 as a cultivated strain. In conclusion, a surprisingly high number of sequence reads that are detected in environmental studies are closely affiliated to cultivated genera or species, despite the fact that the total diversity of methanotrophs that is present in nature is substantially higher than the cultured diversity.
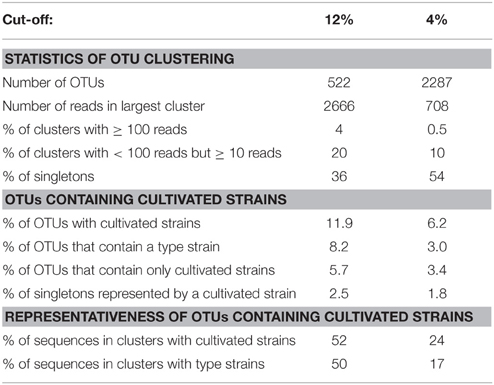
Table 6. Statistics about OTU clustering and distribution of pmoA sequences of cultivated methanotrophic strains within these clusters.
To further evaluate the representativeness of the cultivated genera and species, the size of the OTUs harboring isolates was evaluated. The most frequently detected genera of methanotrophic bacteria in environmental studies are the alphaproteobacterial genera Methylocystis and Methylosinus and the gammaproteobacterial genera Methylomonas, Methylobacter, Methylosarcina, Methylomicrobium, Methylococcus, Methylocaldum, Methylosoma as well as the recently described genus Methyloparacoccus (Table 4). At higher taxonomic resolution, the isolated Methyloparacoccus species remains distinct from the related sequences that have been frequently detected in environmental samples. The same applies to Methylosoma and “Candidatus Methylomirabilis.” Further methanotrophic genera that have very rarely or not yet been detected in environmental samples via cultivation-independent methods comprise Methylomarinovum, Methylomarinum, Methylohalobius, Methyloglobulus and the verrucomicrobial lineages “Methylacidiphilum” and “Methylacidimicrobium” (Table 4). At lower phylogenetic resolution, the genera Methyloglobulus and Methylomarinum do serve as cultivated representatives for major uncultivated clusters (see Section Cluster 2 (CL2) or TUSC). In case of the verrucomicrobial lineages, the limited detection in environmental samples is explained by their highly divergent pmoA sequences, which prevents PCR amplification using the standard pmoA primers. At species level, the frequently detected taxa in cultivation-independent studies are Methylocystis echinoides, Methylocystis rosea, and Methylocystis hirsuta, the two Methylosinus species, Methylococcus capsulatus and most species of the genus Methylocaldum (Table 5). Nearly half of the validly described methanotrophic species have only rarely been detected in environmental samples based on cultivation-independent studies (<10 reads), showing that our culture collections contain many strains of which the ecological relevance in their natural ecosystems remains unknown. Remarkably, Methylosinus strains have very frequently been isolated, but not that frequently been detected by cultivation-independent studies. This is evident from the fact that 54% of all Methylosinus sequences in the database are from isolates, while most other frequently detected genera have only about 5% cultivated representatives (Table 4).
Description of Major Uncultivated Groups of Methanotrophic Bacteria and Their Habitat Specificity
Clusters of pmoA sequences representing uncultivated methanotrophs have been defined in diverse studies mostly at a taxonomic resolution above genus level. They are often named according to the habitat in which they are predominantly found, the sampling site from which they were obtained, or derived from the name of the first described clones of a cluster. The assignment of sequences to a characteristic cluster is usually done in the context of phylogenetic tree reconstruction, guided by a few characteristic reference sequences that are given in the literature as representatives.
The same approach was used here to assign OTUs to described clusters of uncultivated methanotrophic bacteria. Neighbor joining and maximum likelihood trees were constructed using one representative sequence for each OTU12. These representative sequences were selected within each OTU based on the following criteria: OTUs harboring a cultivated strain were represented by the sequence of this strain. For OTUs consisting of sequences from uncultivated bacteria only, the most representative sequence from the first dataset reporting about this sequence type was taken. All representative sequences are listed along with their cluster assignment and accession number in Table S1. Uncultivated clusters were identified in the phylogenetic trees based on the position of published reference sequences. Several OTUs showed inconsistent clustering (in particular among the type I methanotrophs), they were excluded from clusters and are referred to as “incerta sedis” or by their family names and are displayed as “unknown” in Figure 4.
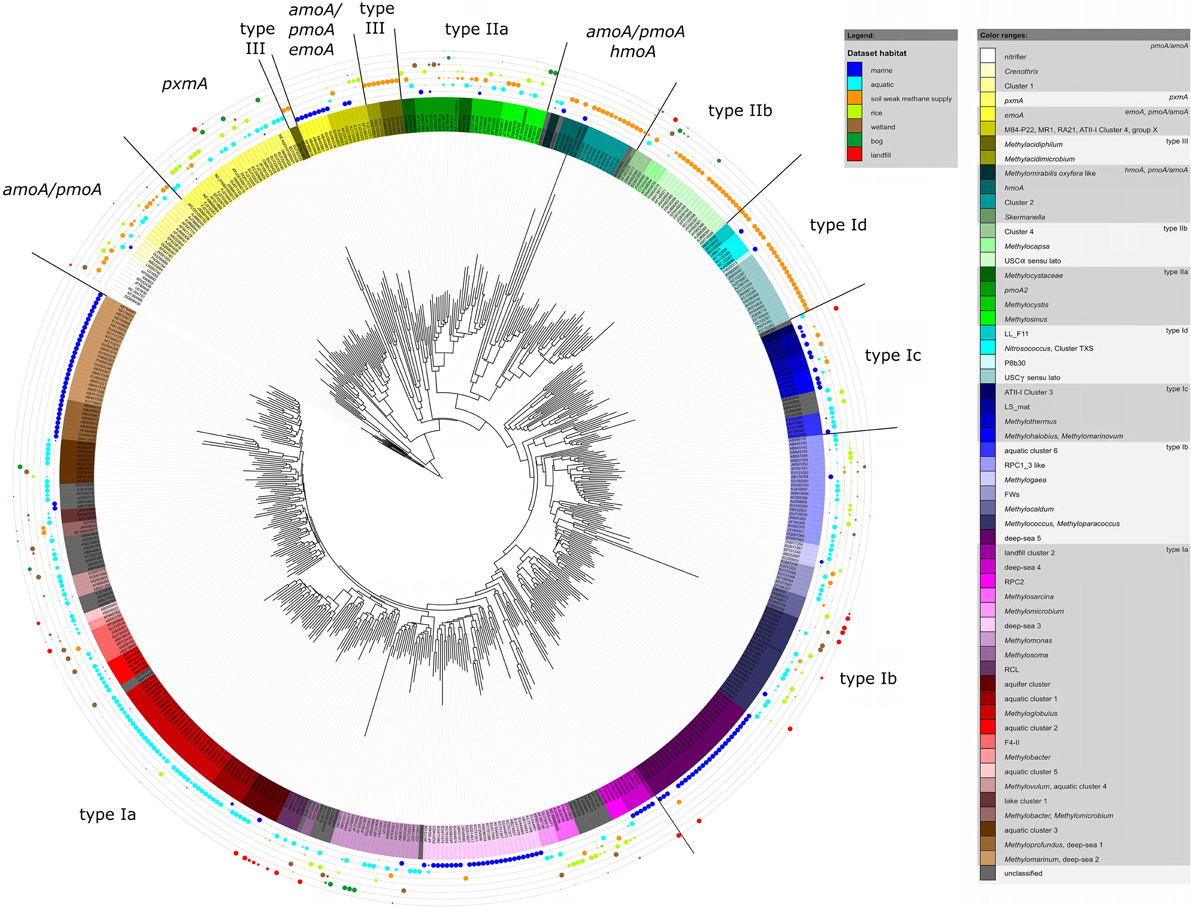
Figure 4. Neighbor joining tree showing the phylogeny of representative pmoA sequences and the habitats in which they were detected. For each OTU12 one representative sequence was included. The dots on the rings around the tree display detection in different habitat types. The size of the dots corresponds to the relative frequency with which sequences of an OTU12 were detected in the seven habitats. The diagram was set up using the iTOL online package (Letunic and Bork, 2011). The phylogenetic tree was calculated in the ARB software package based on 480 nucleotide positions and a Jukes-Cantor correction.
To integrate habitat preferences of methanotrophic lineages into the phylogenetic tree (Figure 4), information about the habitat from which sequences were obtained was collected from the literature and the NCBI database. The definition of categories was largely guided by the terminology used in the literature and the number of sequence reads obtained for each of these categories. The majority of sequences that are currently stored in the public database are from studies that analyzed methanotrophic communities in rice fields, upland soils, aquatic or marine environments (Figure 5). 2.4% of the sequences remained unclassified, either because no information about the habitat was available or they were obtained from studies analyzing rather unusual and thus little studied habitats of methanotrophs (bioreactor, manure, rumen, waste water or plants). Seven major habitat types were defined based on this information and the relative detection frequency of each OTU12 within these habitats calculated. The presentation of these data in combination with phylogeny allows the identification of major clusters with habitat preferences (Figure 4). Habitat specificity of individual OTUs cannot be inferred from this presentation, as a substantial number of OTUs are represented by just one sequence and thus displayed with 100% recovery from one single habitat. To evaluate this aspect, further data analysis is needed as described in Section Habitat Specificity of Methanotrophic Taxa Evaluated at Higher Taxonomic Resolution.
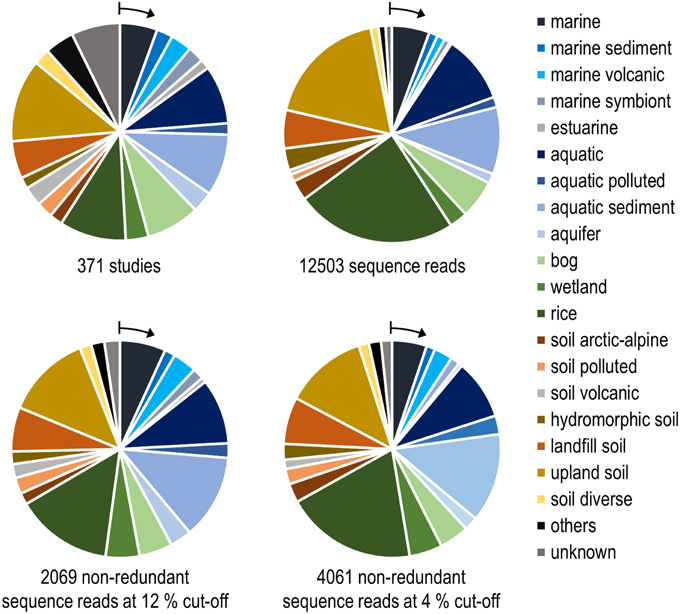
Figure 5. Number of habitats that were analyzed in research studies (upper left) and grouping of pmoA sequences from the NCBI database according to the habitats in which they were detected. The upper right diagram is based on all available high quality sequences, while the lower diagrams include only non-redundant sequence reads. Redundant reads are those that were detected in the same study and fall within the same OTU. Arrows denote the position of the group that is shown as first entry in the legend.
Remarkably, three-fourths of all OTUs12 represent type I methanotrophs in the phylogenetic tree (Figure 4), with nearly 50% belonging to type Ia methanotrophs. This confirms that methanotrophic diversity is highest within the Gammaproteobacteria. Furthermore, it is evident from Figure 4 that the methanotrophs that are found in upland soils, aquatic and marine environments form distinctive and large clusters, while the methanotrophs that are found in other habitats such as rice field soils, wetlands or landfill cover soils are found in smaller clusters that are often detected in different habitats. It is tempting to speculate that colonization of the rather young anthropogenic habitats such as rice field soils or landfill cover soils occurs via methanotrophs that evolved in the much older pristine habitats, so that evolutionary processes leading to diversification and specialization are still in a very early phase in these human made habitats. Moreover, rice field soils and wetlands may represent transitions between terrestrial and aquatic ecosystems and thus share more taxa with other habitats. The absence of specific clusters in wetlands may at least partially be the result of a rather small number of studies in which pmoA sequences were published for this ecosystem (Figure 5) leading to an underrepresentation of sequence reads from this habitat.
In the following, information about the major uncultivated clusters of methanotrophs residing in different habitats is compiled. A condensed phylogenetic tree shows the phylogenetic placement of these clusters in relation to each other and to cultivated type species (Figure 6).
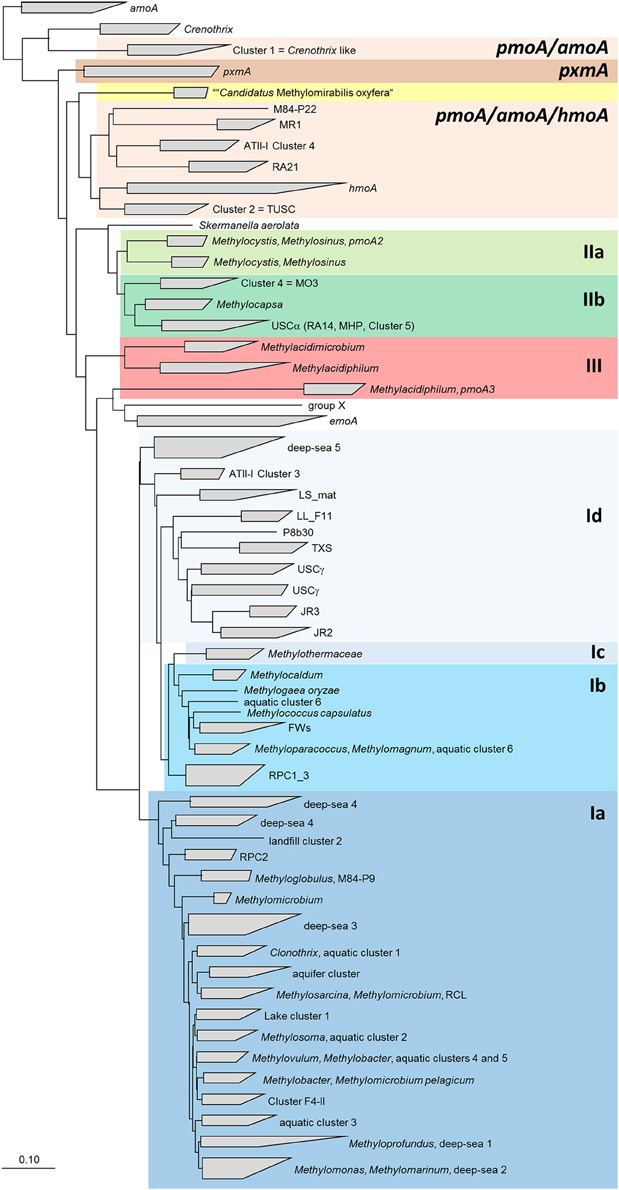
Figure 6. Neighbor joining tree showing the phylogeny of uncultivated clusters in relation to methanotrophic type strains. The tree includes pmoA sequences from all OTUs that were assigned to uncultivated clusters. It was calculated based on 480 nucleotide positions with Jukes Cantor correction. The scale bars display 0.10 changes per nucleotide or amino acid position.
Rice Paddy Clusters (RPC) and Japanese Rice Clusters (JRC), Including the Lake Washington Cluster (LWs), and the Organic Soil Cluster (OSC)
Several different rice paddy clusters and Japanese rice clusters have been defined (Lüke et al., 2010; Stralis-Pavese et al., 2011), but only some of them are regularly detected in diverse studies and implemented in phylogenetic trees. These are RPC1, 2, 3, and JRC3 as well as JRC4, which has meanwhile a cultivated representative, Methylogaea oryzae (Geymonat et al., 2010). JRC3, RPC1, and RPC3 are distantly related to Methylocaldum and Methylococcus and thus part of the type Ib group (Figure 6). RPC2 was reported to show variable clustering either with type Ia or Ib, depending on the algorithm used for tree reconstruction (Lüke and Frenzel, 2011). It is composed of a high number of OTUs at species level resolution, but contains only four OTUs at genus level. RPC1, RPC3, and JRC3 were combined into a larger monophyletic cluster referred to as RPC1_3 in this review, because JRC3 did not form a monophyletic cluster and could not be clearly delineated from RPC1. The RPC1_3 like cluster consists of 25 OTUs12, including in addition the clusters LWs and OSC. Similarly, a large cluster containing the sequences of RPC1, LWs, and OSC but without RPC3 was also formed in some other studies and referred to as freshwater lineage 1 (Lüke and Frenzel, 2011). The major habitat of the methanotrophs belonging to the RPC1_3 like cluster are rice field and aquatic ecosystems (Figure 4). RPC1 and JRC3 were initially exclusively detected in rice paddy associated habitats (Lüke et al., 2010; Lüke and Frenzel, 2011). Exceptional within the RPC1_3 like cluster is OSC, which occurs predominantly in bogs and in some upland soils (Figure 4, Tables S1–S4). Thus, the large RPC1_3 cluster is heterogeneous in terms of habitat preference, with some habitat-specific subgroups. In in-depth studies, biogeographic patterns have been shown for clusters RPC1 and JRC3 (Lüke et al., 2010). Moreover, they respond to the environmental factor rice genotype, either directly or possibly indirectly via altered physicochemical conditions in the plant rhizosphere (Lüke et al., 2011).
Upland Soil Clusters (USCα and USCγ), Jasper Ridge Clusters (JR1, JR2 and JR3), Moor House Peat Cluster (MHP), and Cluster 5
Phylogenetically, the upland soil clusters form two major groups. Sequences of USCα, JR1, and MHP (also referred to as Cluster 5) are related to Methylocapsa (Figure 6). USCα was initially detected by Holmes et al. (1999) and termed RA14. The name USCα was proposed for this sequence type when a second group of sequences with preferential occurrence in upland soils but related to sequences of methanotrophic Gammaproteobacteria, USCγ, was discovered (Knief et al., 2003). USCγ as well as JR2 and JR3 belong to the type Id group (Figure 6). These sequences are related to methanotrophic Gammaproteobacteria and the amoA sequence of Nitrosococcus oceani.
It was proposed to refer to the large group of USCα, JR1/Cluster 5, and MHP sequences as USCα-like sequences or USCα sensu lato, while the initially discovered RA14 clade was defined as USCα sensu stricto (Shrestha et al., 2012). Based on the sequence dataset used in this study, USCα sensu lato consists of 18 OTUs12 and shows an enormous diversity at lower resolution with 133 OTUs4 (Table 7). In particular USCα sensu stricto shows a high diversity at species level resolution. In analogy to this differentiation of USCα sensu lato, sequence clusters USCγ, JR2, and JR3 will be referred to as USCγ sensu lato in this review, while USCγ sensu stricto refers specifically to the USCγ clade. The USCγ sensu lato group is less diverse compared to USCα, consisting of 15 OTUs12 and 98 OTUs4 with USCγ sensu stricto as most diverse group, especially at species level resolution (Table 7).
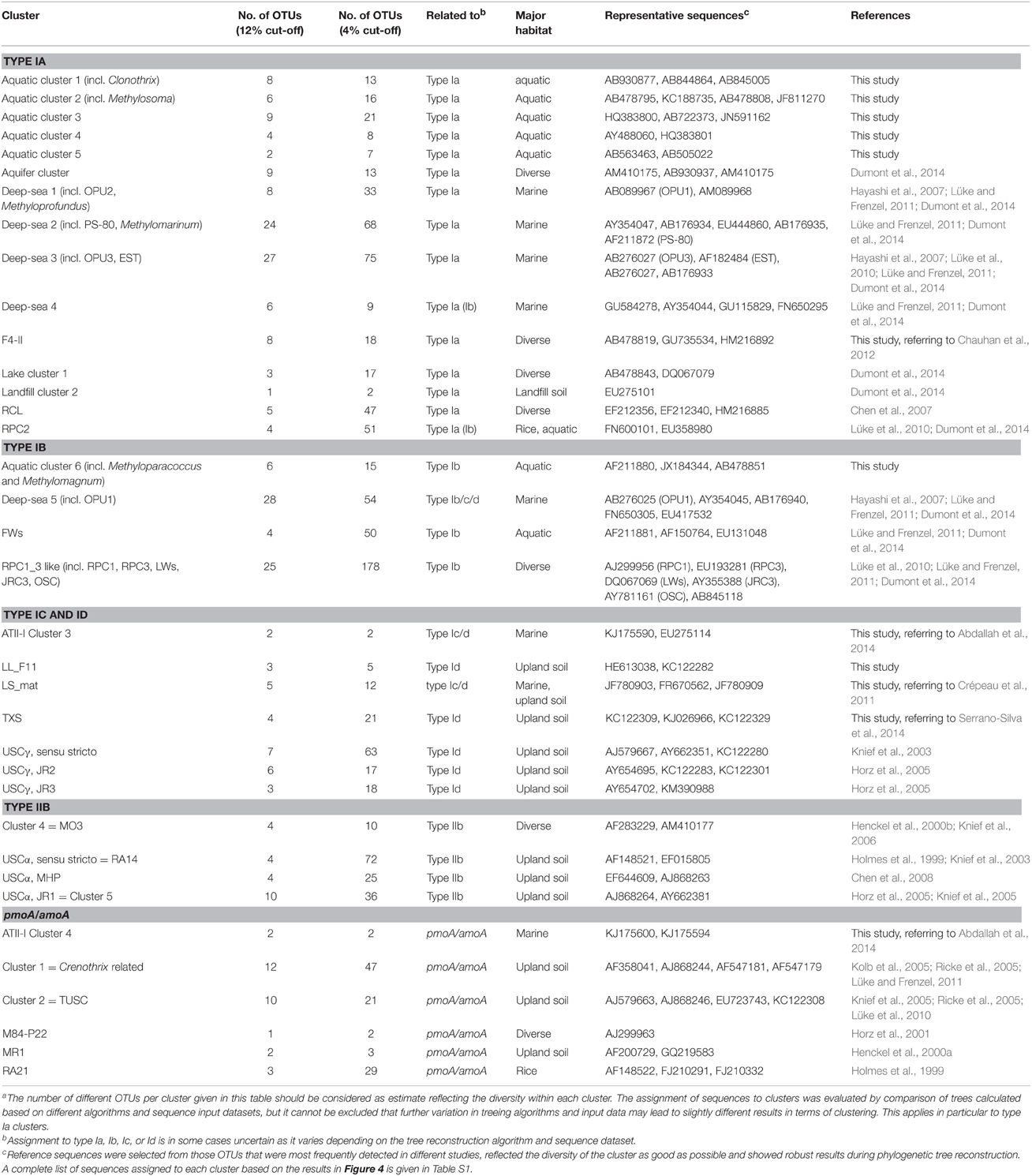
Table 7. Characteristics of uncultivated clusters of pmoA and pmoA-like sequencesa.
All upland soil cluster sequences occur in soils, predominantly in upland soils. USCα sensu lato has been identified as dominant pmoA type in different forest soils (Kolb et al., 2005; Degelmann et al., 2010; Dörr et al., 2010). Some USCα sequence types have additionally been detected in hydromorphic soils (Figure 4, Tables S1–S4) (Knief et al., 2006; Shrestha et al., 2012). USCγ sensu lato occurs in pH neutral and alkaline soils and has been reported to dominate in soils collected from an alpine meadow, an arid desert ecosystem and a former lake (Angel and Conrad, 2009; Zheng et al., 2012; Serrano-Silva et al., 2014). Moreover, USCγ OTUs have been detected sporadically in landfill cover soils (Kumaresan et al., 2009; Henneberger et al., 2012). The occurrence of the two upland soil clusters is clearly pH dependent. USCα sensu lato occurs in acidic to pH neutral soils, while USCγ is only detected in pH neutral and alkaline soils (Knief et al., 2003; Kolb, 2009).
The occurrence of the USC methanotrophs is in most soils reduced the more intensively a soil is agriculturally managed. The clusters are consistently found in forest soils, often as most abundant group, they are quite frequently detected in grassland soils, but rarely detected in intensively managed agricultural soils (Knief et al., 2006; Dunfield, 2007). It has been reported that populations decrease and become inactive when forest soils are converted into agricultural soils, or grasslands are subjected to grazing (Knief et al., 2005; Abell et al., 2009; Dörr et al., 2010; Lima et al., 2014). They recover in afforested or reforested sites and grassland soil in which nitrogen fertilization is reduced (Nazaries et al., 2011; Shrestha et al., 2012). The data of Degelmann et al. (2010) suggest that habitat specificity may exist within USCα sensu lato, as some OTUs occurred in deciduous but not in spruce forest soils.
The USC methanotrophs are assumed to be involved in the oxidation of atmospheric methane (Dunfield, 2007; Kolb, 2009), but this might be different for one specific OTU within USCα sensu lato. OTU 75 (USCα 5, MHP) has more frequently been detected in soils with higher methane supply, i.e., peatlands and wetland, than in typical upland soils (Tables S1–S4) (Chen et al., 2008; Liebner and Svenning, 2013; Yun et al., 2015). Initially it was assumed that the USC methanotrophs may obtain enough energy from atmospheric methane oxidation for cell maintenance and growth (Knief and Dunfield, 2005; Kolb et al., 2005), but later calculations based on methane uptake rates and estimated cell numbers in forest soils indicated that an additional energy source is needed for survival (Degelmann et al., 2010). Indeed, it could be proven that 13C-labeled acetate is incorporated into the biomass of USCα methanotrophs, suggesting that these are facultative methanotrophs (Pratscher et al., 2011).
Cluster 4 (CL4) or MO3
Besides USCα sensu lato, only one further cluster of sequences representing an uncultivated group of methanotrophs is known among the type II group. This is Cluster 4, also known as MO3. It consists of only four OTUs12, is related to Methylocapsa and was initially detected in rice field soil (Henckel et al., 2000b). Upon repeated detection it was defined as cluster 4 (Knief et al., 2006). The cluster has been detected quite frequently in diverse soil habitats including landfill cover, hydromorphic, upland and wetland soils (Figure 4, Tables S1–S4). Its growth was stimulated when rice field soil was incubated under high methane and oxygen concentrations (Henckel et al., 2000b).
Cluster 1 (CL1) or Crenothrix Related Cluster
A sequence cluster related to pmoA of Crenothrix, amoA of nitrifying bacteria and hydrocarbon monooxygenases (hmoA, emoA) was described as cluster 1 (Kolb et al., 2005; Ricke et al., 2005; Knief et al., 2006; Lüke and Frenzel, 2011). It was later also referred to as Crenothrix related cluster (Lüke and Frenzel, 2011). Cluster 1 contains some sequences from methanotrophic isolates that were obtained from Canadian Arctic soils (Pacheco-Oliver et al., 2002). Based on their 16S rRNA gene sequences, these isolates are related to Methylocystis and Methylosinus. Unfortunately, the isolates have been lost and similar isolates could so far not be obtained again, so that the identity and characteristics of the bacteria harboring this pmoA sequence type remain unclear. It has been speculated that cluster 1 organisms are responsible for atmospheric methane uptake, as they were detected as dominant pmoA sequence type in some upland soils, in particular in pH neutral soils (Kolb et al., 2005; Ricke et al., 2005; Kolb, 2009). Experimental proof for this hypothesis is still missing. Further sequences assigned to Cluster 1 were detected in aquatic sediments and aquifers (Figure 4, Tables S1, S3). This corresponds well to the habitat of the related Crenothrix organisms, which were enriched from backwash water of sand filters fed with ground water (Stoecker et al., 2006). Thus, at least some Cluster 1 organisms may be similar to Crenothrix and the whole cluster appears to harbor methanotrophs adapted to different habitats.
Cluster 2 (CL2) or TUSC
Another sequence cluster with pmoA/amoA like sequences was referred to as cluster 2 upon its recurring detection (Knief et al., 2003, 2005; Ricke et al., 2005). In later studies it was named tropical upland soil cluster (TUSC) (Lüke et al., 2010), though its occurrence is not restricted to tropical soils. Instead, it has been detected in diverse upland soils and some hydromorphic soils. It shows similarities in dispersal to USCγ, as it is largely absent in wetlands and acidic soils (Kolb, 2009; Martineau et al., 2014). Moreover, it shows reduced occurrence in intensively managed agricultural soils (Lima et al., 2014) with the exception that it has been found in some agricultural fields that are subjected to organic farming and/or that are characterized by higher carbon content (upon biochar or organic residue application; Dörr et al., 2010; Lima et al., 2014; Ho et al., 2015).
It has been speculated that the organisms harboring genes of this sequence cluster are involved in atmospheric methane oxidation, but this is solely based on the specific detection of this sequence type in upland soils. Further proof for this hypothesis is missing. It can currently not even be excluded that the genes of this sequence cluster encode a non-methane hydrocarbon monooxygenase, which is suggested by the fact that the sequences are related to those of hydrocarbon monooxygenases (Figure 6). The only evidence that supports the assumption that cluster 2 sequences may represent methanotrophic Gammaproteobacteria comes from a study of Kalyuzhnaya et al. (2006), who enriched methanotrophic bacteria from lake Washington sediment by cell sorting using 16S rRNA targeted fluorescent probes. Twenty percent of a pmoA clone library, constructed from a cell suspension enriched with a probe for type I methanotrophs, represented cluster 2 pmoA sequences. Unusual in this context remains the unique detection of this sequence type in a lake sediment.
Deep-Sea Clusters 1 to 5 Including OPU1, OPU3, and PS-80
Sequences retrieved from marine environments can be grouped into five major clusters, referred to as deep-sea clusters 1 to 5 (Lüke and Frenzel, 2011). Deep-sea clusters 1, 2, and 3 belong to the type Ia methanotrophs (Figure 6). Deep-sea cluster 4 is distantly related to known type Ia and Ib methanotrophs. Depending on the subset of sequences and the method used for tree reconstruction this cluster falls within either type Ia or type Ib methanotrophs (Lüke and Frenzel, 2011). Deep-sea cluster 5 is a deeply branching lineage related to type Ib and Ic methanotrophs. The clustering is variable in different phylogenetic trees, so that an unambiguous assignment to one or the other type is difficult. In some studies, this cluster was even assigned to type Id (referred to as type Ic in those studies; Lüke and Frenzel, 2011; Henneberger et al., 2012).
Deep-sea clusters 1 and 2 have meanwhile cultivated representatives. Cluster 1 includes the cultivated genus Methyloprofundus and cluster 2 the genus Methylomarinum. These genera represent one single OTU within the respective clusters, while the clusters contain in total eight and 27 OTUs12. Thus, it appears likely that they consist of more than one genus. Hence, the well-established names deep-sea cluster 1 and 3 are kept for these larger clusters of sequences in this review. Deep-sea cluster 2 includes the uncultivated PS-80 cluster, which is displayed as distinct cluster in some phylogenetic trees or given as alternative name for deep-sea cluster 2 (Deng et al., 2013; Dumont et al., 2014; Li et al., 2014). Likewise, deep-sea cluster 3 includes the sub-clusters OPU3 and EST, which are repeatedly mentioned in the literature and sometimes given as synonym for deep-sea cluster 3 (Lüke et al., 2010; Tavormina et al., 2010, 2013; Crespo-Medina et al., 2014; Li et al., 2014). The same applies to deep-sea cluster 5, which includes or corresponds to OPU1.
Deep-sea clusters 1 and 4 are rather small with only 6 and 8 OTUs12 and have less frequently been detected compared to the other three clusters, which contain between 20 and 30 OTUs12 (Table 7). Most deep-sea clusters consist exclusively of sequences from marine habitats, the only exceptions are found in deep-sea clusters 3 and 5 (Figure 4). They contain one single OTU12, which was retrieved from a terrestrial habitat, i.e., a mud volcano and a landfill cover soil (Henneberger et al., 2012). Furthermore, OTU 271 in cluster 3 contains some sequences from an aquatic habitat. These were detected in an estuarine sediment, which harbored otherwise sequences that are typical for aquatic habitats (McDonald et al., 2005).
Possible habitat preferences of the different deep-sea clusters remain currently largely unknown. In most studies, sequences of two or more deep-sea clusters have been detected in the same sample (Nercessian et al., 2005; Yan et al., 2006; Redmond et al., 2010; Ruff et al., 2013). Nevertheless, methanotrophic communities can differ substantially between sites (Ruff et al., 2013). Clear differences were also seen between sediment and water column within the same site (Tavormina et al., 2008), but overall, all five clusters have been detected in samples from the water column or the sediment with roughly equal frequency. Evidence for habitat specificity is only seen within deep-sea cluster 1, which harbors the majority of sequences that were found in association with marine animals (Zbinden et al., 2008; Wendeberg et al., 2012; Raggi et al., 2013). These methanotrophs live as endosymbionts in mussels, tube worms or shrimps and contribute to the food web of deep-water ecosystems (Petersen and Dubilier, 2009). Deep-sea cluster 2 and 4 sequences have also been detected as endosymbionts or epibionts of marine animals, but less consistently (Zbinden et al., 2008; Rodrigues et al., 2011; Watsuji et al., 2014).
Lake Cluster 1, Aquifer Cluster, and Aquatic Clusters 1 to 6
Sequence types that have predominantly been detected in aquatic habitats are grouped into lake cluster 1 and 2 and the aquifer cluster (Dumont et al., 2014). Lake cluster 1 is a small group of sequences (3 OTUs12) belonging to the type Ia methanotrophs (Figure 6). Most lake cluster 1 sequences were detected in aquatic ecosystems, while few were found in a wetland. Lake cluster 2 sequences represent also type Ia methanotrophs and were grouped by the Mothur classification tool into one single large OTU together with Methyloparacoccus. Thus, it is referred to as Methyloparacoccus here instead of lake cluster 2. This OTU was not only detected in aquatic ecosystems, but also in rice ecosystems and sporadically in other habitats (Figure 4).
The aquifer cluster consists of nine OTUs12 and just a few more OTUs at species level resolution. It is also representing type Ia methanotrophs. The name refers to the initial detection in a petroleum-contaminated aquifer (Urmann et al., 2008), but sequences of this cluster occur in different habitats. Half of the OTUs12 are common in aquatic ecosystems, while others were detected in landfill cover soils (Figure 4, Tables S1, S2). This applies even to the OTU harboring the aquifer sequences; it was also detected in landfill cover soils.
The evaluation of the relationship between phylogeny and habitat revealed the existence of possible further aquatic clusters that were defined in this work (Figure 4). The aquatic clusters 1 to 5 are related to type Ia methanotrophs, while aquatic cluster 6 is a member of the type Ib methanotrophs. Aquatic cluster 1 is related to Clonothrix, aquatic cluster 2 to Methylosoma, and cluster 4 often includes Methylovulum in phylogenetic trees. All clusters are rather small, consisting of two to nine OTUs12 (Table 7). They contain dominantly sequences from aquatic ecosystems plus some sequences from other habitats, often from marine ecosystems (Figure 4). Most aquatic clusters and the lake cluster 1 OTUs were detected in samples from the water column as well as the sediment. Only aquatic cluster 4 shows a much higher detection frequency in studies of sediment samples, while cluster 2 shows a higher detection frequency in samples from the water column (Tables S1–S4). Similarly, the aquifer cluster has not yet been detected in aquatic sediment samples.
Further Clusters of Uncultivated Gammaproteobacterial Methanotrophs
Two further clusters of uncultivated methanotrophs are related to type Ia methanotrophs, represented by cluster RCL and F4-II. Cluster RCL was named after the first clones, obtained during a study analyzing active methanotrophs in landfill cover soil (Chen et al., 2007). It consists of only five OTUs12, but a much higher number of 47 OTUs4 at higher taxonomic resolution (Table 7). It has been detected in different ecosystems, in particular in aquatic sediments and landfill cover soils (Tables S1–S4, Figure 4). Cluster F4-II was defined in this work, referring to the first study in which this sequence type was discovered (Chauhan et al., 2012). It consists of eight OTUs12 and contains sequences from diverse habitats, especially aquatic and wetland ecosystems.
Cluster FWs is present within the type Ib methanotrophs and was defined recently (Dumont et al., 2014). It has a relatively high diversity at species level and has most frequently been detected in aquatic environments.
Two rather small clusters of uncultivated methanotrophs, clusters LS-mat and ATII-I cluster 3 can be assigned to the type Ic or Id methanotrophs, depending on the treeing approach (Figures 4, 6). These clusters were named in this work in accordance with the sample and cluster names given in the studies in which they were first described (Crépeau et al., 2011; Abdallah et al., 2014). They are closely related to each other and were detected in different marine studies and with lower frequency in some terrestrial habitats.
Besides USCγ sensu lato one further cluster of uncultivated sequences is present within the group of type Id methanotrophs. The TXS cluster consists of four OTUs12 and has been exclusively detected in upland soils so far, likewise as the other uncultivated type Id clusters (Serrano-Silva et al., 2014). Whether the organisms of this cluster are also involved in atmospheric methane oxidation is unknown.
Further pmoA/amoA Like Clusters: MR1, RA21, and Others
Several further sequence types form small clusters that are distantly related to the well-known pmoA and amoA sequences as well as to those of pxmA and non-methane hydrocarbon monooxygenase genes. Cluster MR1 is represented by two OTUs12 in this study and has only been detected in some upland soils (Table 7). In contrast, RA21, which has been more frequently retrieved and consists of three OTUs12, is predominantly found in rice field soils. Some further clusters have been defined in this region of the phylogenetic tree, such as the two marine clusters referred to as group X (Tavormina et al., 2010) and ATII-I Cluster 4 (Abdallah et al., 2014), or cluster M84-P22 (Horz et al., 2001). These clusters have until now only been detected very rarely, so that it is too early to draw further conclusions about possible habitat preferences.
Habitat Specificity of Methanotrophic Taxa Evaluated at Higher Taxonomic Resolution
To evaluate habitat specificity for cultivated and uncultivated taxa of methanotrophic bacteria in more detail and at higher taxonomic resolution, 19 different habitat types were defined, which contained at least 30 sequence reads. The assignment of sequences to one of these more specific habitat types was in most cases unambiguous, but for the soil categories an overlap between habitats may exist. This applies for instance to soils collected in arctic-alpine environments, which include samples from glacier forefields as well as alpine meadows and grasslands. Some of these soils may also represent the category “upland soil” or “hydromorphic soil.” Likewise, a polluted soil may at the same time be an “upland soil.” Soils in the category “polluted soils” were collected from areas with hydrocarbon pollution, near coal mines or above oil and gas reservoirs. Four percent of the soil derived sequence reads could not be assigned to a specific soil habitat since no further information about the type of soil habitat was available. These sequences are presented as “soil diverse” in Figure 5, but were excluded from subsequent analyses as they formed a very heterogeneous group. Certain overlap may also exist between wetlands and bog ecosystems, as it cannot be fully excluded that the term wetland was in some cases used by authors for the description of samples from bog ecosystems.
Due to the fact that methanotrophic communities were analyzed at very great depth in some studies, numerous redundant reads are present in the database and a high number of sequence reads assigned to certain OTUs may be the result of just a few studies rather than frequent detection in diverse studies. To correct for this possible artifact, replicate sequence reads, i.e., those that represent the same study and the same OTU, were excluded during the further analysis. This resulted in 2079 non-redundant reads at OTU12 cut-off and 4061 reads at OTU4 cut-off level. In particular at 12% cut-off, this caused a more even distribution of sequence reads across the different habitat types (Figure 5). The recovery of specific sequence types in different habitats was thus evaluated based on their presence or absence in individual studies, while the information from approximately 370 studies was used to estimate the detection frequency of each OTU quantitatively. Non-metric multidimensional scaling plots were calculated to visualize (dis-)similarities between habitats (Figure 7). The major pattern was largely similar, regardless of the applied OTU resolution, demonstrating that major differences between samples are indeed already manifest at genus level. Methanotrophic communities in marine habitats are most distinct from those of all other habitats, as evident from their clear separation along the first axis of the plot. This was also seen when applying other multivariate approaches (principal component analysis, hierarchical cluster analysis) and is in agreement with the existence of the very specific marine clades deep-sea clusters 1 to 5. The second axis separates volcanic soils from all other samples, which can be explained by the unique presence of Verrucomicrobia in several of these soils (Sharp et al., 2014). The high dissimilarities of methanotrophic communities in marine ecosystems and volcanic soil samples compared to all other ecosystems were verified by an analysis of similarity (ANOSIM), which revealed very high values of R = 0.985 (P = 0.001) at OTU12 level and of R = 0.926 (P = 0.001) at OTU4 level. To better evaluate dissimilarities between the remaining aquatic and terrestrial ecosystems, the marine and volcanic soil sample data were excluded from NMDS plots (Figure 7). These reduced datasets reveal that aquatic habitats including the estuarine habitat are again distinct from the other habitats, supported by ANOSIM values of R = 0.444 (P = 0.006) for OTU12 and R = 0.460 (P = 0.004) for OTU4. This agrees with the existence of different aquatic clades (Figure 4). Methanotrophic communities in aquifers appear to be somewhat different from those in aquatic habitats (Figure 7). The terrestrial samples did not show highly consistent patterns in the NMDS plots (or in other multivariate approaches), besides the observation that those soils that are exposed to low methane concentrations, i.e., upland soils, arctic-alpine soils, and hydromorphic soils, are often located close to each other. This is in agreement with the unique occurrence of the upland soil clusters and some other clades in these soils (Figure 4). The limited resolution of differences between the different soil sample types may be related to the fact that these categories may partially overlap, as explained above.
Figure 7. Habitat specificity of methanotrophic bacteria evaluated in non-metric multidimensional scaling (NMDS) plots. Non-redundant sequence reads were used to calculate the relative detection frequency of all OTUs in a habitat. The upper plots show differences between all 18 different habitats, while the lower plots focus on the 13 most similar habitats. OTU clustering was done using 12% (left panels) and 4% dissimilarity cut-off (right panels). The NMDS plots were set up based on Bray-Curtis dissimilarities calculated from Hellinger transformed data using the online tool GUSTA ME (Buttigieg and Ramette, 2014).
Habitat-specific OTUs
To identify common and habitat specific groups at OTU12 and OTU4 level, the relative detection frequency of OTUs across habitats was determined based on non-redundant read counts. OTUs that were detected in at least five studies were included in this evaluation. Otherwise, OTUs may appear erroneously as habitat-specific based on the fact that they have been detected in a limited number of studies. The detection frequency of OTUs across habitats is displayed as heat map and reveals that a rather low number of OTUs is highly habitat specific (Figure 8). The identified habitat specific and common OTUs are listed in Tables 8, 9. The number of specific OTUs increases at species level resolution. This is to some extent the result of splitting a habitat specific genus into several habitat specific species. Furthermore, it is based on the fact that some habitat specific species exist within genera that show a broad distribution, as observed for some Methylocystis species. The genus is commonly found in diverse environments, but some Methylocystis species show habitat specificity and appear to be characteristic for aquatic environments or landfill cover soils (Tables 8, 9). Likewise, the genus Methylocaldum has been detected in diverse habitats, while the species Methylocaldum gracile was found with very high frequency in landfill cover soils.
Figure 8. Relative detection frequency of OTUs across habitats. Non-redundant reads were normalized by the number of studies available for each habitat and the relative frequency with which each OTU was detected across the different habitats was calculated. The upper panel shows the results at genus level resolution (OTU12), the lower panel at species level resolution (OTU4). OTUs displayed in red are highly specific for a certain type of habitat. OTUs that were detected in less then five studies were set to zero. The identity of the most habitat-specific and most common OTUs is given in Tables 8, 9. A list including detailed information about all OTUs is provided as Supplementary Material.
Table 8. Broadly distributed and habitat-specific OTUs12.
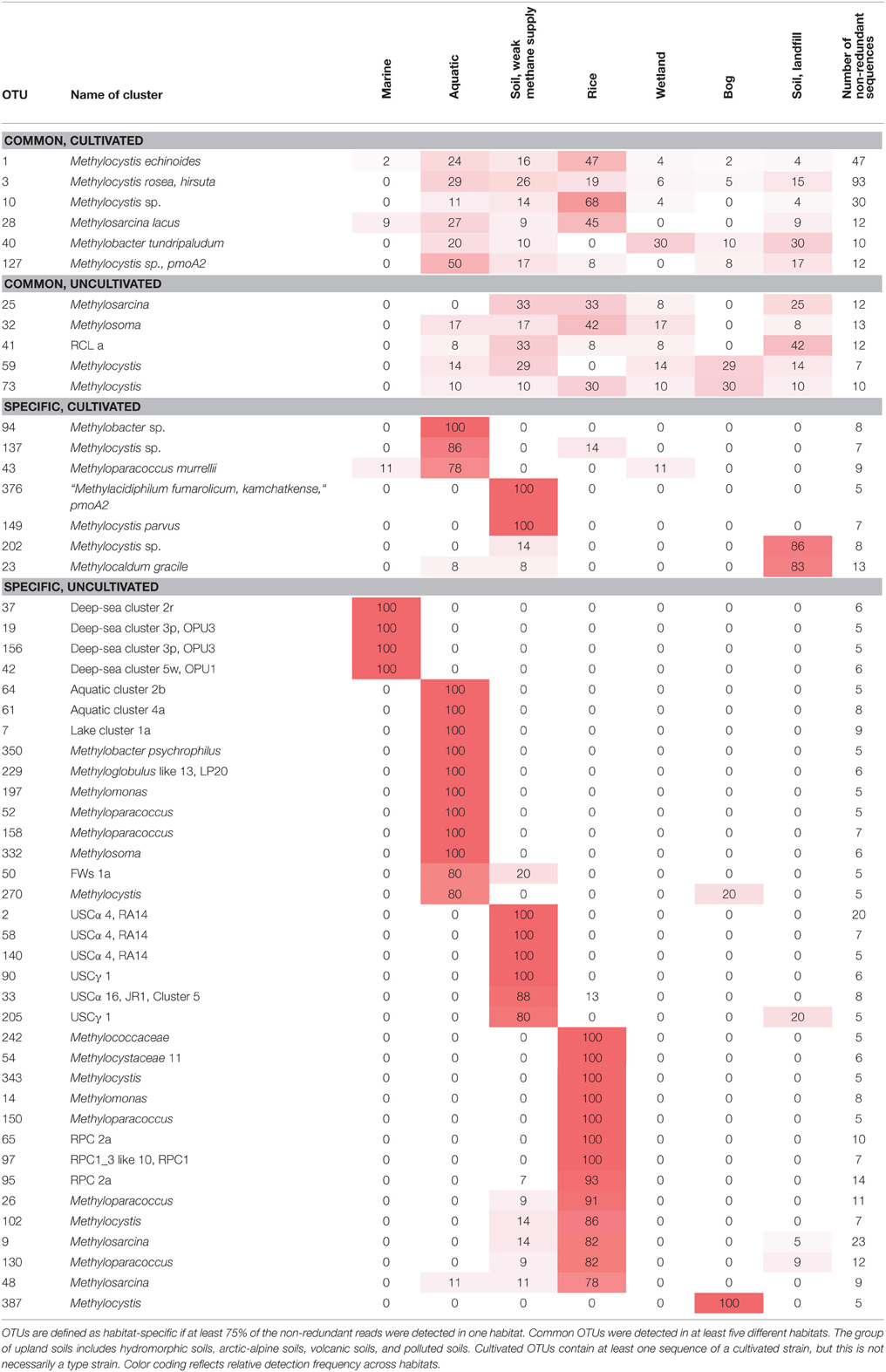
Table 9. Broadly distributed and habitat-specific OTUs4.
The marine habitats, which appeared most distinct in the NMDS plots, are not only characterized by the presence of very unique taxa that are mostly absent from all other ecosystems. Additionally, most taxa with broad distribution in diverse habitats are largely absent in marine ecosystems, in particular at species level resolution (Figure 8). The OTUs that are characteristic for marine habitats belong to the deep-sea clusters 1 to 5 (Table 8). Due to a high phylogenetic diversity within these clusters, most of the individual OTUs have so far only been detected in a few studies, so that many OTUs of these clades were excluded from this kind of analysis. This explains the unexpectedly low number of OTUs that are displayed for the marine samples at species level resolution in the heat map (Figure 8).
The different aquatic habitats have several OTUs at genus and species level in common (Figure 8). This includes the uncultivated clusters FWs, lake cluster 1 and LP20, which have already been described as habitat-specific before (Dumont et al., 2014), and most of the aquatic clusters that were defined in this article. Moreover, the genus Methyloparacoccus murrellii as well as specific OTUs4 of the genera Methylobacter, Methylomonas, Methylosoma, and Methylocystis are characteristic for aquatic habitats (Table 9). Some OTUs are even more habitat specific and occur preferentially either in the water column or the sediment. Specific for the water column are the aquatic clusters 2b and 5a and lake cluster 1, while aquatic cluster 4a, the Methyloglobulus like cluster LP20 and some further OTUs related to Methylobacter psychrophilus, Methyloglobulus morosus, Methyloparacoccus murrellii and Methylosoma difficile are specific for the sediment (Figure 8, Tables S1–S4). In agreement with this preferential occurrence, the cultivated strains of these species were also obtained from aquatic habitats (Table 2).
The terrestrial habitats show a lower number of specific OTUs, in agreement with the weaker resolution in the NMDS plots. Rice associated habitats harbor no characteristic OTUs at genus level resolution, but some specific OTUs related to Methyloparacoccus and Methylocystis or the uncultivated lineages RPC1 and RPC2 at higher taxonomic resolution (Table 9). Characteristic in landfill cover soils are strains of Methylocaldum gracile and of an unclassified Methylocystis species, but no specific clusters of uncultivated methanotrophs were detected. As expected, different lineages of USCα and USCγ are specific for upland soils, while the genus Methylocapsa and a specific uncultivated Methylocystis species are typical inhabitants of bog ecosystems (Tables 8, 9).
Broadly Distributed Methanotrophic Taxa
Several OTUs12 occur in diverse habitats. These include a number of cultivated genera, in particularly those that have been discovered and described quite early and that have been obtained as isolates frequently (Tables 4, 8). Furthermore, some lineages of uncultivated methanotrophs are broadly distributed such as OTUs of the clusters FWs, RCL, RA21, or RPC1_3. At species level resolution, the number of common OTUs is lower (Table 9). This can be explained by habitat specialization with increasing taxonomic resolution, as observed for cultivated and uncultivated members of the genera Methylocystis, Methylocaldum, or Methylobacter (Tables 8, 9).
Understanding the Influence of Environmental Factors on Methanotrophic Community Composition and Activity
It is obvious that the occurrence and activity of methanotrophic bacteria in different ecosystems is largely influenced by abiotic and biotic environmental conditions. Important factors are methane and oxygen concentrations, nutrient availability, pH, temperature, salinity and water availability (Semrau et al., 2010). Additional factors will influence these bacteria indirectly such as soil moisture content, which affects gas diffusion and thus methane and oxygen supply, or plant cover, which alters the water and nutrient status in soil. Among these factors, methane concentration, nitrogen status and the role of copper have been studied in most detail and were identified as very important for shaping methanotrophic communities and for influencing their activity (Conrad, 2007; Semrau et al., 2010; Ho et al., 2013). Future research needs to address the question how the different factors act alone and in combination on the members of methanotrophic communities in different ecosystems. The present study evaluated only the presence or absence of methanotrophic bacteria in the different ecosystems, but this does not implement information about metabolic activity. In particular type IIa methanotrophs are capable of forming resting stages, which enable prolonged survival under unfavorable conditions (Whittenbury et al., 1970a). To link ecosystem function with community composition, activity in dependence on environmental parameters needs to be analyzed in more detail in future studies.
The present review provides a comprehensive overview about habitat preferences of methanotrophic taxa, considering the complete diversity as represented by pmoA as marker and including all major ecosystems in which these bacteria occur. However, habitat preferences do also exist within these ecosystems. The preferential occurrence of USCα in acidic and USCγ in pH neutral upland soils or the plant genotype specific colonization of rice by uncultivated groups of methanotrophs are just two examples (Knief et al., 2003; Lüke et al., 2011). In the latter case, differences can be seen as shifts in the methanotrophic community composition, but not based on pure presence absence data. Likewise, shifts have been observed in aquatic ecosystems, where methanotrophic communities differ in dependence on depth or type of sediment (Pester et al., 2004; Rahalkar and Schink, 2007; Biderre-Petit et al., 2011; Deutzmann et al., 2011). In contrast, almost nothing is known about niche differentiation and habitat preferences among all those OTUs that represent uncultivated genera and species of the marine deep-sea clusters. These methanotrophs appear to coexist in marine habitats, or differentiation occurs at a finer scale. In-depth studies within the different ecosystems are needed to obtain further knowledge about habitat preferences of the individual clusters of methanotrophic bacteria. Such studies need to implement meta-data describing the physicochemical and biological characteristics of the habitat or have to be done under controlled conditions whereby specific parameters are manipulated.
There is a clear need to study the impact of environmental factors at different taxonomic resolution in order to gain comprehensive understanding about mechanisms that lead to niche differentiation among methanotrophs. In initial studies, a simple differentiation between type I and type II methanotrophs was made (e.g., Graham et al., 1993; Amaral et al., 1995; Henckel et al., 2000b), which is certainly appropriate due to some major differences that exist between these groups, e.g., in terms of physiology. Hence, these studies provided valuable insight concerning the differential responses of the studied methanotrophs to high and low methane, oxygen and nitrogen concentrations (Conrad, 2007; Ho et al., 2013). However, the compilation of ecophysiological characteristics from type strains in this study has shown that responses of methanotrophic bacteria to specific environmental factors are often not closely linked to phylogeny, a finding that was recently also reported by Krause et al. (2014), so that other approaches may be necessary to categorize methanotrophs. A concept that has several times been applied considers type I methanotrophs as r-strategists and type II methanotrophs as k-strategists (Steenbergh et al., 2010; Siljanen et al., 2011). A recent proposition is based on a classification of methanotrophic bacteria into more specific ecological response groups based on specific functional traits: methanotrophic genera were classified based on their life strategies as competitors, stress tolerators or ruderals (Bodelier et al., 2012; Ho et al., 2013). The data compiled in this review clearly support the assumption that methanotrophic bacteria have developed different life strategies. Several groups of methanotrophs, among them many uncultivated lineages, appear to be specifically adapted to a certain habitat type and may thus represent good competitors in this specific environment. Some others have been found more widespread in different habitat types and may thus represent stress tolerators and/or ruderals.
Importance to Obtain Further Isolates of Methanotrophic Bacteria
The evaluation of the representativeness of cultured model strains has revealed that they cover already a substantial fraction of the frequently detected methanotrophs in environmental samples. Several of them are common colonizers in diverse habitats. This encompasses in particular those taxa that are easily recovered in enrichment studies, while other isolated species and genera have not (yet) been frequently detected in nature. The fact that major clusters of uncultivated methanotrophs are detected in diverse ecosystems clearly shows the need for further isolation efforts to get hands on these organisms. This applies in particular to the frequently detected methanotrophs belonging to the diverse rice paddy clusters, the marine deep-sea clusters, the upland soil clusters or the different aquatic clusters. It is likely that these organisms are well adapted to their respective habitats, so that specific enrichment strategies may have to be applied, which better mimic the natural conditions of these methanotrophs to stimulate their growth. Several attempts were already made to enrich USCα methanotrophs, but until now, these resulted in the retrieval of well-known methanotrophic genera such as Methylocystis and Methylosinus rather than an enrichment of bacteria harboring USCα gene sequences (Dunfield et al., 1999; Knief and Dunfield, 2005; Kravchenko et al., 2010).
Only the combination of community analyses in natural environments, under controlled conditions in microcosms or mesocosms and of pure cultures or enrichment cultures will allow to understand the physiological and regulatory mechanisms at cellular level that ultimately control activity and affect dispersal of methanotrophs in nature. The fact that many gene functions and regulatory mechanisms in methanotrophic bacteria are until now only little understood, e.g., the role of pxmA, limits also the gain of knowledge from cultivation-independent studies when global analysis approaches such as metagenomics, -transcriptomics or -proteomics are applied. This underlines the need for studying pure cultures under laboratory conditions.
The analysis of dispersal patterns at high taxonomic resolution needs a sufficiently large data basis. Conclusions about habitat preferences can only be drawn for frequently detected genera and species, but not so easily for those methanotrophic genera that are currently represented by a single strain or a very small number of sequences. Their less frequent recovery in cultivation-dependent and -independent approaches might point toward higher specialization. In order to draw further conclusions about habitat preferences for these smaller groups, the detection of similar sequences in cultivation-independent studies and/or the isolation of further representatives are necessary. The application of next generation sequencing techniques will facilitate the detection of such rare methanotrophs due to the higher sequencing depth that can be reached. However, currently the integration of NGS data from studies into existing sequence databases is time consuming, as tools for data mining are still largely lacking. At the moment, this limitation can most conveniently be overcome if authors deposit representative pmoA sequences in the NCBI nucleotide database or provide them as fasta files. NGS sequencing technology is more and more frequently applied to characterize methanotrophic communities and will lead to an enormous amount of data in the next years. If these data are supplemented with detailed information about the sampling sites and the experimental conditions, it may become a very valuable data resource, enabling more detailed meta-analyses, focusing on specific ecosystems, environmental factors, or taxonomic groups.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2015.01346
References
Abdallah, R. Z., Adel, M., Ouf, A., Sayed, A., Ghazy, M. A., Alam, I., et al. (2014). Aerobic methanotrophic communities at the Red Sea brine-seawater interface. Front. Microbiol. 5:487. doi: 10.3389/fmicb.2014.00487
Abell, G. C. J., Stralis-Pavese, N., Sessitsch, A., and Bodrossy, L. (2009). Grazing affects methanotroph activity and diversity in an alpine meadow soil. Environ. Microbiol. Rep. 1, 457–465. doi: 10.1111/j.1758-2229.2009.00078.x
Amaral, J. A., Archambault, C., Richards, S. R., and Knowles, R. (1995). Denitrification associated with Groups I and II methanotrophs in a gradient enrichment system. FEMS Microbiol. Ecol. 18, 289–298. doi: 10.1111/j.1574-6941.1995.tb00185.x
Angel, R., and Conrad, R. (2009). In situ measurement of methane fluxes and analysis of transcribed particulate methane monooxygenase in desert soils. Environ. Microbiol. 11, 2598–2610. doi: 10.1111/j.1462-2920.2009.01984.x
Auman, A. J., and Lidstrom, M. E. (2002). Analysis of sMMO-containing Type I methanotrophs in Lake Washington sediment. Environ. Microbiol. 4, 517–524. doi: 10.1046/j.1462-2920.2002.00323.x
Auman, A. J., Stolyar, S., Costello, A. M., and Lidstrom, M. E. (2000). Molecular characterization of methanotrophic isolates from freshwater lake sediment. Appl. Environ. Microbiol. 66, 5259–5266. doi: 10.1128/AEM.66.12.5259-5266.2000
Baani, M., and Liesack, W. (2008). Two isozymes of particulate methane monooxygenase with different methane oxidation kinetics are found in Methylocystis sp. strain SC2. Proc. Natl. Acad. Sci. U.S.A. 105, 10203–10208. doi: 10.1073/pnas.0702643105
Barbier, B. A., Dziduch, I., Liebner, S., Ganzert, L., Lantuit, H., Pollard, W., et al. (2012). Methane-cycling communities in a permafrost-affected soil on Herschel Island, Western Canadian Arctic: active layer profiling of mcrA and pmoA genes. FEMS Microbiol. Ecol. 82, 287–302. doi: 10.1111/j.1574-6941.2012.01332.x
Belova, S. E., Baani, M., Suzina, N. E., Bodelier, P. L., Liesack, W., and Dedysh, S. N. (2011). Acetate utilization as a survival strategy of peat-inhabiting Methylocystis spp. Environ. Microbiol. Rep. 3, 36–46. doi: 10.1111/j.1758-2229.2010.00180.x
Belova, S. E., Kulichevskaya, I. S., Bodelier, P. L., and Dedysh, S. N. (2013). Methylocystis bryophila sp. nov., a facultatively methanotrophic bacterium from acidic Sphagnum peat, and emended description of the genus Methylocystis (ex Whittenbury et al. 1970) Bowman et al. 1993. Int. J. Syst. Evol. Microbiol. 63, 1096–1104. doi: 10.1099/ijs.0.043505-0
Biderre-Petit, C., Jézéquel, D., Dugat-Bony, E., Lopes, F., Kuever, J., Borrel, G., et al. (2011). Identification of microbial communities involved in the methane cycle of a freshwater meromictic lake. FEMS Microbiol. Ecol. 77, 533–545. doi: 10.1111/j.1574-6941.2011.01134.x
Bodelier, P. L., Gillisen, M. J., Hordijk, K., Damsté, J. S., Rijpstra, W. I., Geenevasen, J. A., et al. (2009). A reanalysis of phospholipid fatty acids as ecological biomarkers for methanotrophic bacteria. ISME J. 3, 606–617. doi: 10.1038/ismej.2009.6
Bodelier, P. L. E., Bär-Gilissen, M. J., Meima-Franke, M., and Hordijk, K. (2012). Structural and functional response of methane-consuming microbial communities to different flooding regimes in riparian soils. Ecol. Evol. 2, 106–127. doi: 10.1002/ece3.34
Bodrossy, L., Holmes, E. M., Holmes, A. J., Kovacs, K. L., and Murrell, J. C. (1997). Analysis of 16S rRNA and methane monooxygenase gene sequences reveals a novel group of thermotolerant and thermophilic methanotrophs, Methylocaldum gen. nov. Arch. Microbiol. 168, 493–503. doi: 10.1007/s002030050527
Bourne, D. G., McDonald, I. R., and Murrell, J. C. (2001). Comparison of pmoA PCR primer sets as tools for investigating methanotroph diversity in three Danish soils. Appl. Environ. Microbiol. 67, 3802–3809. doi: 10.1128/AEM.67.9.3802-3809.2001
Bowman, J. (2006). “The methanotrophs—The families Methylococcaceae and Methylocystaceae,” in The Prokaryotes, eds M. Dworkin, S. Falkow, E. Rosenberg, K. H. Schleifer, and E. Stackebrandt (New York, NY: Springer), 266–289.
Bowman, J. P. (2005a). “Methylococcales,” in Bergey's Manual of Systematic Bacteriology, eds G. Garrity, D. J. Brenner, N. R. Krieg, and J. T. Staley (New York, NY: Springer), 248–271.
Bowman, J. P. (2005b). “Methylocystaceae,” in Bergey's Manual of Systematic Bacteriology, eds G. Garrity, D. J. Brenner, N. R. Krieg, and J. T. Staley (New York, NY: Springer), 411–422.
Bowman, J. P. (2014). “The family Methylococcaceae,” in The Prokaryotes, eds E. Rosenberg, E. F. Delong, S. Lory, E. Stackebrandt, and F. Thompson (Berlin Heidelberg: Springer-Verlag), 411–441.
Bowman, J. P., McCammon, S. A., and Skerratt, J. H. (1997). Methylosphaera hansonii gen. nov., sp. nov., a psychrophilic, group I methanotroph from Antarctic marine-salinity, meromictic lakes. Microbiology 143, 1451–1459. doi: 10.1099/00221287-143-4-1451
Bowman, J. P., Sly, L. I., Cox, J. M., and Hayward, A. C. (1990). Methylomonas fodinarum sp. nov. and Methylomonas aurantiaca sp. nov.—two closely related type I obligate methanotrophs. Syst. Appl. Microbiol. 13, 279–287. doi: 10.1016/S0723-2020(11)80199-2
Bowman, J. P., Sly, L. I., Nichols, P. D., and Hayward, A. C. (1993). Revised taxonomy of the methanotrophs: description of Methylobacter gen. nov., emendation of Methylococcus, validation of Methylosinus and Methylocystis species, and a proposal that the family Methylococcaceae includes only the Group I methanotrophs. Int. J. Syst. Bacteriol. 43, 735–753. doi: 10.1099/00207713-43-4-735
Bowman, J. P., Sly, L. I., and Stackebrandt, E. (1995). The phylogenetic position of the family Methylococcaceae. Int. J. Syst. Bacteriol. 45, 182–185. doi: 10.1099/00207713-45-1-182
Buttigieg, P. L., and Ramette, A. (2014). A guide to statistical analysis in microbial ecology: a community-focused, living review of multivariate data analyses. FEMS Microbiol. Ecol. 90, 543–550. doi: 10.1111/1574-6941.12437
Chauhan, A., Pathak, A., and Ogram, A. (2012). Composition of methane-oxidizing bacterial communities as a function of nutrient loading in the Florida everglades. Microb. Ecol. 64, 750–759. doi: 10.1007/s00248-012-0058-2
Chen, Y., Crombie, A., Rahman, M. T., Dedysh, S. N., Liesack, W., Stott, M. B., et al. (2010). Complete genome sequence of the aerobic facultative methanotroph Methylocella silvestris BL2. J. Bacteriol. 192, 3840–3841. doi: 10.1128/JB.00506-10
Chen, Y., Dumont, M. G., Cébron, A., and Murrell, J. C. (2007). Identification of active methanotrophs in a landfill cover soil through detection of expression of 16S rRNA and functional genes. Environ. Microbiol. 9, 2855–2869. doi: 10.1111/j.1462-2920.2007.01401.x
Chen, Y., Dumont, M. G., McNamara, N. P., Chamberlain, P. M., Bodrossy, L., Stralis-Pavese, N., et al. (2008). Diversity of the active methanotrophic community in acidic peatlands as assessed by mRNA and SIP-PLFA analyses. Environ. Microbiol. 10, 446–459. doi: 10.1111/j.1462-2920.2007.01466.x
Chowdhury, T. R., and Dick, R. P. (2013). Ecology of aerobic methanotrophs in controlling methane fluxes from wetlands. Agric. Ecosyst. Environ. Appl. Soil Ecol. 65, 8–22. doi: 10.1016/j.apsoil.2012.12.014
Coleman, N. V., Le, N. B., Ly, M. A., Ogawa, H. E., McCarl, V., Wilson, N. L., et al. (2012). Hydrocarbon monooxygenase in Mycobacterium: recombinant expression of a member of the ammonia monooxygenase superfamily. ISME J. 6, 171–182. doi: 10.1038/ismej.2011.98
Conrad, R. (2007). Microbial ecology of methanogens and methanotrophs. Adv. Agron. 96, 1–63. doi: 10.1016/S0065-2113(07)96005-8
Conrad, R. (2009). The global methane cycle: recent advances in understanding the microbial processes involved. Environ. Microbiol. Rep. 1, 285–292. doi: 10.1111/j.1758-2229.2009.00038.x
Costello, A. M., and Lidstrom, M. E. (1999). Molecular characterization of functional and phylogenetic genes from natural populations of methanotrophs in lake sediments. Appl. Environ. Microbiol. 65, 5066–5074.
Crépeau, V., Bonavita, M. A. C., Lesongeur, F., Randrianalivelo, H., Sarradin, P. M., Sarrazin, J., et al. (2011). Diversity and function in microbial mats from the Lucky Strike hydrothermal vent field. FEMS Microbiol. Ecol. 76, 524–540. doi: 10.1111/j.1574-6941.2011.01070.x
Crespo-Medina, M., Meile, C. D., Hunter, K. S., Diercks, A. R., Asper, V. L., Orphan, V. J., et al. (2014). The rise and fall of methanotrophy following a deepwater oil-well blowout. Nat. Geosci. 7, 423–427. doi: 10.1038/ngeo2156
Crombie, A. T., and Murrell, J. C. (2014). Trace-gas metabolic versatility of the facultative methanotroph Methylocella silvestris. Nature 510, 148–153. doi: 10.1038/nature13192
Dalton, H. (2005). The Leeuwenhoek Lecture 2000—The natural and unnatural history of methane-oxidizing bacteria. Philos. Trans. R. Soc. B Biol. Sci. 360, 1207–1222. doi: 10.1098/rstb.2005.1657
Danilova, O. V., and Dedysh, S. N. (2014). Abundance and diversity of methanotrophic Gammaproteobacteria in northern wetlands. Microbiology 83, 67–76. doi: 10.1134/S0026261714020040
Danilova, O. V., Kulichevskaya, I. S., Rozova, O. N., Detkova, E. N., Bodelier, P. L. E., Trotsenko, Y. A., et al. (2013). Methylomonas paludis sp. nov., the first acid-tolerant member of the genus Methylomonas, from an acidic wetland. Int. J. Syst. Evol. Microbiol. 63, 2282–2289. doi: 10.1099/ijs.0.045658-0
De Visscher, A., Boeckx, P., and Van Cleemput, O. (2007). “Artificial methane sinks,” in Greenhouse gas Sinks, eds D. S. Reay, C. N. Hewitt, and J. Grace (Wallingford: Oxfordshire), 184–200.
Dedysh, S. N., Belova, S. E., Bodelier, P. L., Smirnova, K. V., Khmelenina, V. N., Chidthaisong, A., et al. (2007). Methylocystis heyeri sp. nov., a novel type II methanotrophic bacterium possessing ‘signature’ fatty acids of type I methanotrophs. Int. J. Syst. Evol. Microbiol. 57, 472–479. doi: 10.1099/ijs.0.64623-0
Dedysh, S. N., Berestovskaya, Y. Y., Vasylieva, L. V., Belova, S. E., Khmelenina, V. N., Suzina, N. E., et al. (2004). Methylocella tundrae sp. nov., a novel methanotrophic bacterium from acidic tundra peatlands. Int. J. Syst. Evol. Microbiol. 54, 151–156. doi: 10.1099/ijs.0.02805-0
Dedysh, S. N., Didriksen, A., Danilova, O. V., Belova, S. E., Liebner, S., and Svenning, M. M. (2015a). Methylocapsa palsarum sp. nov., a methanotrophic bacterium from a Sub-Arctic discontinuous permafrost ecosystem. Int. J. Syst. Evol. Microbiol. 65, 3618–3624. doi: 10.1099/ijsem.0.000465
Dedysh, S. N., Khmelenina, V. N., Suzina, N. E., Trotsenko, Y. A., Semrau, J. D., Liesack, W., et al. (2002). Methylocapsa acidiphila gen. nov., sp. nov., a novel methane-oxidizing and dinitrogen-fixing acidophilic bacterium from Sphagnum bog. Int. J. Syst. Evol. Microbiol. 52, 251–261. doi: 10.1099/00207713-52-1-251
Dedysh, S. N., Knief, C., and Dunfield, P. F. (2005). Methylocella species are facultatively methanotrophic. J. Bacteriol. 187, 4665–4670. doi: 10.1128/JB.187.13.4665-4670.2005
Dedysh, S. N., Liesack, W., Khmelenina, V. N., Suzina, N. E., Trotsenko, Y. A., Semrau, J. D., et al. (2000). Methylocella palustris gen. nov., sp. nov., a new methane-oxidizing acidophilic bacterium from peat bogs, representing a novel subtype of serine-pathway methanotrophs. Int. J. Syst. Evol. Microbiol. 50, 955–969. doi: 10.1099/00207713-50-3-955
Dedysh, S. N., Naumoff, D. G., Vorobev, A. V., Kyrpides, N., Woyke, T., Shapiro, N., et al. (2015b). Draft genome sequence of Methyloferula stellata AR4, an obligate methanotroph possessing only a soluble methane monooxygenase. Genome Announc. 3, e01555–e01514. doi: 10.1128/genomeA.01555-14
Degelmann, D. M., Borken, W., Drake, H. L., and Kolb, S. (2010). Different atmospheric methane-oxidizing communities in European beech and Norway spruce soils. Appl. Environ. Microbiol. 76, 3228–3235. doi: 10.1128/AEM.02730-09
del Cerro, C., García, J. M., Rojas, A., Tortajada, M., Ramon, D., Galan, B., et al. (2012). Genome sequence of the methanotrophic poly-beta-hydroxybutyrate producer Methylocystis parvus OBBP. J. Bacteriol. 194, 5709–5710. doi: 10.1128/JB.01346-12
Deng, Y., Cui, X., Lüke, C., and Dumont, M. G. (2013). Aerobic methanotroph diversity in Riganqiao peatlands on the Qinghai-Tibetan Plateau. Environ. Microbiol. Rep. 5, 566–574. doi: 10.1111/1758-2229.12046
Deutzmann, J. S., Hoppert, M., and Schink, B. (2014). Characterization and phylogeny of a novel methanotroph, Methyloglobulus morosus gen. nov., spec. nov. Syst. Appl. Microbiol. 37, 165–169. doi: 10.1016/j.syapm.2014.02.001
Deutzmann, J. S., Wörner, S., and Schink, B. (2011). Activity and diversity of methanotrophic bacteria at methane seeps in eastern Lake Constance sediments. Appl. Environ. Microbiol. 77, 2573–2581. doi: 10.1128/AEM.02776-10
Dörr, N., Glaser, B., and Kolb, S. (2010). Methanotrophic communities in Brazilian ferralsols from naturally forested, afforested, and agricultural sites. Appl. Environ. Microbiol. 76, 1307–1310. doi: 10.1128/AEM.02282-09
Dumont, M. G. (2014). “Primers: functional marker genes for methylotrophs and methanotrophs,” in Hydrocarbon and Lipid Microbiology Protocols, eds T. J. McGenity, K. N. Timmis, and B. Nogales (Berlin: Springer-Verlag), 1–21.
Dumont, M. G., Lüke, C., Deng, Y. C., and Frenzel, P. (2014). Classification of pmoA amplicon pyrosequences using BLAST and the lowest common ancestor method in MEGAN. Front. Microbiol. 5:e34. doi: 10.3389/fmicb.2014.00034
Dumont, M. G., Pommerenke, B., Casper, P., and Conrad, R. (2011). DNA-, rRNA- and mRNA-based stable isotope probing of aerobic methanotrophs in lake sediment. Environ. Microbiol. 13, 1153–1167. doi: 10.1111/j.1462-2920.2010.02415.x
Dunfield, P. F. (2007). “The soil methane sink,” in Greenhouse Gas Sinks, eds D. S. Reay, C. N. Hewitt, K. A. Smith, and J. Grace (Wallingford: CABI), 152–170.
Dunfield, P. F., Belova, S. E., Vorob'ev, A. V., Cornish, S. L., and Dedysh, S. N. (2010). Methylocapsa aurea sp. nov., a facultative methanotroph possessing a particulate methane monooxygenase, and emended description of the genus Methylocapsa. Int. J. Syst. Evol. Microbiol. 60, 2659–2664. doi: 10.1099/ijs.0.020149-0
Dunfield, P. F., Khmelenina, V. N., Suzina, N. E. A. T. Y., and Dedysh, S. N. (2003). Methylocella silvestris sp. nov. a novel methanotrophic bacterium isolated from an acidic forest cambisol. Int. J. Syst. Evol. Microbiol. 53, 1231–1239. doi: 10.1099/ijs.0.02481-0
Dunfield, P. F., Liesack, W., Henckel, T., Knowles, R., and Conrad, R. (1999). High-affinity methane oxidation by a soil enrichment culture containing a type II methanotroph. Appl. Environ. Microbiol. 65, 1009–1014.
Dunfield, P. F., Tchawa Yimga, M., Dedysh, S. N., Berger, U., Liesack, W., and Heyer, J. (2002). Isolation of a Methylocystis strain containing a novel pmoA-like gene. FEMS Microbiol. Ecol. 41, 17–26. doi: 10.1111/j.1574-6941.2002.tb00962.x
Dunfield, P. F., Yuryev, A., Senin, P., Smirnova, A. V., Stott, M. B., Hou, S., et al. (2007). Methane oxidation by an extremely acidophilic bacterium of the phylum Verrucomicrobia. Nature 450, 879–882. doi: 10.1038/nature06411
Erikstad, H. A., and Birkeland, N. K. (2015). Draft genome sequence of “Candidatus Methylacidiphilum kamchatkense” Strain Kam1, a thermoacidophilic methanotrophic Verrucomicrobium. Genome Announc. 3, e00065–e00015. doi: 10.1128/genomeA.00065-15
Erikstad, H. A., Jensen, S., Keen, T. J., and Birkeland, N. K. (2012). Differential expression of particulate methane monooxygenase genes in the verrucomicrobial methanotroph 'Methylacidiphilum kamchatkense' Kam1. Extremophiles 16, 405–409. doi: 10.1007/s00792-012-0439-y
Eshinimaev, B. T., Khmelenina, V. N., and Trotsenko, Y. A. (2008). First isolation of a type II methanotroph from a soda lake. Microbiology 77, 628–631. doi: 10.1134/S0026261708050196
Ettwig, K. F., Butler, M. K., Le Paslier, D., Pelletier, E., Mangenot, S., Kuypers, M. M., et al. (2010). Nitrite-driven anaerobic methane oxidation by oxygenic bacteria. Nature 464, 543–548. doi: 10.1038/nature08883
Foster, J. W., and Davis, R. H. (1966). A methane-dependent coccus, with notes on classification and nomenclature of obligate, methane-utilizing bacteria. J. Bacteriol. 91, 1924–1931.
Gal'chenko, V. F., Shishkina, V. N., Suzina, N. E., and Trotsenko Iu, A. (1977). Isolation and properties of new strains of obligate methanotrophs. Mikrobiologiya 46, 890–897.
Geymonat, E., Ferrando, L., and Tarlera, S. E. (2010). Methylogaea oryzae gen. nov., sp. nov., a novel mesophilic methanotroph from a rice paddy field in Uruguay. Int. J. Syst. Evol. Microbiol. 61, 2568–2572. doi: 10.1099/ijs.0.028274-0
Gilbert, B., McDonald, I. R., Finch, R., Stafford, G. P., Nielsen, A. K., and Murrell, J. C. (2000). Molecular analysis of the pmo (particulate methane monooxygenase) operons from two type II methanotrophs. Appl. Environ. Microbiol. 66, 966–975. doi: 10.1128/AEM.66.3.966-975.2000
Graham, D. W., Chaudhary, J. A., Hanson, J. S., and Arnold, R. G. (1993). Factors affecting competition between type I and type II methanotrophs in two-organism, continuous-flow reactors. Microb. Ecol. 25, 1–17. doi: 10.1007/BF00182126
Green, P. N. (1992). “Taxonomy of methylotrophic bacteria,” in Microbial Growth on C1-compounds, eds J. C. Murrell and D. P. Kelly (Andover: Intercept Press. Ltd.), 23–84.
Hamamura, N., and Arp, D. J. (2000). Isolation and characterization of alkane-utilizing Nocardioides sp. strain CF8. FEMS Microbiol. Lett. 186, 21–26. doi: 10.1111/j.1574-6968.2000.tb09076.x
Hamilton, R., Kits, K. D., Ramonovskaya, V. A., Rozova, O. N., Yurimoto, H., Iguchi, H., et al. (2015). Draft genomes of gammaproteobacterial methanotrophs isolated from terrestrial ecosystems. Genome Announc. 3, e00515–e00515. doi: 10.1128/genomeA.00515-15
Hayashi, T., Obata, H., Gamo, T., Samo, Y., and Naganuma, T. (2007). Distribution and phylogenetic characteristics of the genes encoding enzymes relevant to methane oxidation in oxygen minimum zones of the eastern pacific ocean. Res. J. Environ. Sci. 1, 275–284. doi: 10.3923/rjes.2007.275.284
Hazeu, W., Batenburg-Van Der Vegte, W. H., and De Bruyn, J. C. (1980). Some characteristics of Methylococcus mobilis sp. nov. Arch. Microbiol. 124, 211–220. doi: 10.1007/BF00427729
Henckel, T., Jäckel, U., Schnell, S., and Conrad, R. (2000a). Molecular analyses of novel methanotrophic communities in forest soil that oxidize atmospheric methane. Appl. Environ. Microbiol. 66, 1801–1808. doi: 10.1128/AEM.66.5.1801-1808.2000
Henckel, T., Roslev, P., and Conrad, R. (2000b). Effects of O2 and CH4 on presence and activity of the indigenous methanotrophic community in rice field soil. Environ. Microbiol. 2, 666–679. doi: 10.1046/j.1462-2920.2000.00149.x
Henneberger, R., Lüke, C., Mosberger, L., and Schroth, M. H. (2012). Structure and function of methanotrophic communities in a landfill-cover soil. FEMS Microbiol. Ecol. 81, 52–65. doi: 10.1111/j.1574-6941.2011.01278.x
Heyer, J., Berger, U., Hardt, M., and Dunfield, P. F. (2005). Methylohalobius crimeensis gen. nov., sp. nov., a moderately halophilic, methanotrophic bacterium isolated from hypersaline lakes of Crimea. Int. J. Syst. Evol. Microbiol. 55, 1817–1826. doi: 10.1099/ijs.0.63213-0
Heyer, J., Galchenko, V. F., and Dunfield, P. F. (2002). Molecular phylogeny of type II methane-oxidizing bacteria isolated from various environments. Microbiology 148, 2831–2846. doi: 10.1099/00221287-148-9-2831
Hirayama, H., Abe, M., Miyazaki, M., Nunoura, T., Furushima, Y., Yamamoto, H., et al. (2014). Methylomarinovum caldicuralii gen. nov., sp nov., a moderately thermophilic methanotroph isolated from a shallow submarine hydrothermal system, and proposal of the family Methylothermaceae fam. nov. Int. J. Syst. Evol. Microbiol. 64, 989–999. doi: 10.1099/ijs.0.058172-0
Hirayama, H., Fuse, H., Abe, M., Miyazaki, M., Nakamura, T., Nunoura, T., et al. (2013). Methylomarinum vadi gen. nov., sp. nov., a methanotroph isolated from two distinct marine environments. Int. J. Syst. Evol. Microbiol. 63, 1073–1082. doi: 10.1099/ijs.0.040568-0
Hirayama, H., Suzuki, Y., Abe, M., Miyazaki, M., Makita, H., Inagaki, F., et al. (2010). Methylothermus subterraneus sp. nov., a moderately thermophilic methanotrophic bacterium from a terrestrial subsurface hot aquifer in Japan. Int. J. Syst. Evol. Microbiol. 61, 2646–2653. doi: 10.1099/ijs.0.028092-0
Ho, A., El-Hawwary, A., Kim, S. Y., Meima-Franke, M., and Bodelier, P. (2015). Manure-associated stimulation of soil-borne methanogenic activity in agricultural soils. Biol Fertil Soils 51, 511–516. doi: 10.1007/s00374-015-0995-2
Ho, A., Kerckhof, F. M., Luke, C., Reim, A., Krause, S., Boon, N., et al. (2013). Conceptualizing functional traits and ecological characteristics of methane-oxidizing bacteria as life strategies. Environ. Microbiol. Rep. 5, 335–345. doi: 10.1111/j.1758-2229.2012.00370.x
Hoefman, S., Heylen, K., and De Vos, P. (2014a). Methylomonas lenta sp nov., a methanotroph isolated from manure and a denitrification tank. Int. J. Syst. Evol. Microbiol. 64, 1210–1217. doi: 10.1099/ijs.0.057794-0
Hoefman, S., Van Der Ha, D., Iguchi, H., Yurimoto, H., Sakai, Y., Boon, N., et al. (2014b). Methyloparacoccus murrellii gen. nov., sp. nov., a methanotroph isolated from pond water. Int. J. Syst. Evol. Microbiol. 64, 2100–2107. doi: 10.1099/ijs.0.057760-0
Holmes, A. J., Costello, A., Lidstrom, M. E., and Murrell, J. C. (1995). Evidence that particulate methane monooxygenase and ammonia monooxygenase may be evolutionarily related. FEMS Microbiol. Lett. 132, 203–208. doi: 10.1111/j.1574-6968.1995.tb07834.x
Holmes, A. J., Roslev, P., McDonald, I. R., Iversen, N., Henriksen, K., and Murrell, J. C. (1999). Characterization of methanotrophic bacterial populations in soils showing atmospheric methane uptake. Appl. Environ. Microbiol. 65, 3312–3318.
Horz, H.-P., Yimga, M. T., and Liesack, W. (2001). Detection of methanotroph diversity on roots of submerged rice plants by molecular retrieval of pmoA, mmoX, mxaF, and 16S rRNA and ribosomal DNA, including pmoA-based terminal restriction fragment length polymorphism profiling. Appl. Environ. Microbiol. 67, 4177–4185. doi: 10.1128/AEM.67.9.4177-4185.2001
Horz, H. P., Rich, V., Avrahami, S., and Bohannan, B. J. (2005). Methane-oxidizing bacteria in a California upland grassland soil: diversity and response to simulated global change. Appl. Environ. Microbiol. 71, 2642–2652. doi: 10.1128/AEM.71.5.2642-2652.2005
Hou, S. B., Makarova, K. S., Saw, J. H. W., Senin, P., Ly, B. V., Zhou, Z. M., et al. (2008). Complete genome sequence of the extremely acidophilic methanotroph isolate V4, Methylacidiphilum infernorum, a representative of the bacterial phylum Verrucomicrobia. Biol. Direct 3:26. doi: 10.1186/1745-6150-3-26
Iguchi, H., Yurimoto, H., and Sakai, Y. (2011). Methylovulum miyakonense gen. nov., sp. nov., a type I methanotroph isolated from forest soil. Int. J. Syst. Evol. Microbiol. 61, 810–815. doi: 10.1099/ijs.0.019604-0
Im, J., Lee, S. W., Yoon, S., Dispirito, A. A., and Semrau, J. D. (2011). Characterization of a novel facultative Methylocystis species capable of growth on methane, acetate and ethanol. Environ. Microbiol. Rep. 3, 174–181. doi: 10.1111/j.1758-2229.2010.00204.x
IPCC. (2013). “Climate change 2013: the physical science basis,” in Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, eds T. F. Stocker, D. Qin, G.-K. Plattner, M. Tignor, S. K. Allen, J. Boschung, et al. (Cambridge; New York, NY: Cambridge University Press), 1–1535.
Islam, T., Jensen, S., Reigstad, L. J., Larsen, O., and Birkeland, N. K. (2008). Methane oxidation at 55 degrees C and pH 2 by a thermoacidophilic bacterium belonging to the Verrucomicrobia phylum. Proc. Natl. Acad. Sci. U.S.A. 105, 300–304. doi: 10.1073/pnas.0704162105
Jiang, H., Chen, Y., Jiang, P. X., Zhang, C., Smith, T. J., Murrell, J. C., et al. (2010). Methanotrophs: multifunctional bacteria with promising applications in environmental bioengineering. Biochem. Eng. J. 49, 277–288. doi: 10.1016/j.bej.2010.01.003
Kaluzhnaya, M., Khmelenina, V. N., Eshinimaev, B., Suzina, N. E., Nikitin, D., Solonin, A., et al. (2001). Taxonomic characterization of new alkaliphilic and alkalitolerant methanotrophs from soda lakes of the Southeastern Transbaikal region and description of Methylomicrobium buryatense sp. nov. Syst. Appl. Microbiol. 24, 166–176. doi: 10.1078/0723-2020-00028
Kalyuzhnaya, M. G., Khmelenina, V., Eshinimaev, B., Sorokin, D., Fuse, H., Lidstrom, M., et al. (2008). Classification of halo(alkali)philic and halo(alkali)tolerant methanotrophs provisionally assigned to the genera Methylomicrobium and Methylobacter and emended description of the genus Methylomicrobium. Int. J. Syst. Evol. Microbiol. 58, 591–596. doi: 10.1099/ijs.0.65317-0
Kalyuzhnaya, M. G., Khmelenina, V. N., Kotelnikova, S., Holmquist, L., Pedersen, K., and Trotsenko, Y. A. (1999). Methylomonas scandinavica sp. nov., a new methanotrophic psychrotrophic bacterium isolated from deep igneous rock ground water of Sweden. Syst. Appl. Microbiol. 22, 565–572. doi: 10.1016/S0723-2020(99)80010-1
Kalyuzhnaya, M. G., Lamb, A. E., McTaggart, T. L., Oshkin, I. Y., Shapiro, N., Woyke, T., et al. (2015). Draft genome sequences of gammaproteobacterial methanotrophs isolated from lake washington sediment. Genome Announc. 3, e00103–e00115. doi: 10.1128/genomeA.00103-15
Kalyuzhnaya, M. G., Stolyar, S. M., Auman, A. J., Lara, J. C., Lidstrom, M. E., and Chistoserdova, L. (2005). Methylosarcina lacus sp. nov., a methanotroph from Lake Washington, Seattle, USA, and emended description of the genus Methylosarcina. Int. J. Syst. Evol. Microbiol. 55, 2345–2350. doi: 10.1099/ijs.0.63405-0
Kalyuzhnaya, M. G., Zabinsky, R., Bowerman, S., Baker, D. R., Lidstrom, M. E., and Chistoserdova, L. (2006). Fluorescence in situ hybridization-flow cytometry-cell sorting-based method for separation and enrichment of type I and type II methanotroph populations. Appl. Environ. Microbiol. 72, 4293–4301. doi: 10.1128/AEM.00161-06
Kaserer, H. (1905). Über die Oxidation des Wasserstoffes und des Methans durch Mikroorganismen. Z. landw. Versuchsw. in Österreich 8, 789–792.
Khadem, A. F., Wieczorek, A. S., Pol, A., Vuilleumier, S., Harhangi, H. R., Dunfield, P. F., et al. (2012). Draft genome sequence of the volcano-inhabiting thermoacidophilic methanotroph Methylacidiphilum fumariolicum strain SolV. J. Bacteriol. 194, 3729–3730. doi: 10.1128/JB.00501-12
Khalifa, A., Lee, C. G., Ogiso, T., Ueno, C., Dianou, D., Demachi, T., et al. (2015). Methylomagnum ishizawai gen. nov., sp. nov., a mesophilic type I methanotroph isolated from rice rhizosphere. Int. J. Syst. Evol. Microbiol. 65, 3527–3534. doi: 10.1099/ijsem.0.000451
Khmelenina, V. N., Beck, D. A., Munk, C., Davenport, K., Daligault, H., Erkkila, T., et al. (2013). Draft genome sequence of Methylomicrobium buryatense strain 5G, a haloalkaline-tolerant methanotrophic bacterium. Genome Announc. 1, e00053–e00013. doi: 10.1128/genomeA.00053-13
Khmelenina, V. N., Kalyuzhnaya, M. G., Starostina, N. G., Suzina, N. E., and Trotsenko, Y. A. (1997). Isolation and characterization of halotolerant alkaliphilic methanotrophic bacteria from Tuva soda lakes. Curr. Microbiol. 35, 257–261. doi: 10.1007/s002849900249
Kip, N., Ouyang, W., Van Winden, J., Raghoebarsing, A., Van Niftrik, L., Pol, A., et al. (2011). Detection, isolation, and characterization of acidophilic methanotrophs from Sphagnum mosses. Appl. Environ. Microbiol. 77, 5643–5654. doi: 10.1128/AEM.05017-11
Kits, K. D., Kalyuzhnaya, M. G., Klotz, M. G., Jetten, M. S., Op Den Camp, H. J., Vuilleumier, S., et al. (2013). Genome sequence of the obligate gammaproteobacterial methanotroph Methylomicrobium album strain BG8. Genome Announc. 1, e0017013. doi: 10.1128/genomeA.00170-13
Kits, K. D., Klotz, M. G., and Stein, L. Y. (2015). Methane oxidation coupled to nitrate reduction under hypoxia by the Gammaproteobacterium Methylomonas denitrificans, sp. nov. type strain FJG1. Environ Microbiol. 17, 3219–3232. doi: 10.1111/1462-2920.12772
Kleiveland, C. R., Hult, L. T. O., Kuczkowska, K., Jacobsen, M., Lea, T., and Pope, P. B. (2012). Draft genome sequence of the methane-oxidizing bacterium Methylococcus capsulatus (Texas). J. Bacteriol. 194, 6626–6626. doi: 10.1128/JB.01656-12
Knief, C., and Dunfield, P. F. (2005). Response and adaptation of different methanotrophic bacteria to low methane mixing ratios. Environ. Microbiol. 7, 1307–1317. doi: 10.1111/j.1462-2920.2005.00814.x
Knief, C., Kolb, S., Bodelier, P. L., Lipski, A., and Dunfield, P. F. (2006). The active methanotrophic community in hydromorphic soils changes in response to changing methane concentration. Environ. Microbiol. 8, 321–333. doi: 10.1111/j.1462-2920.2005.00898.x
Knief, C., Lipski, A., and Dunfield, P. F. (2003). Diversity and activity of methanotrophic bacteria in different upland soils. Appl. Environ. Microbiol. 69, 6703–6714. doi: 10.1128/AEM.69.11.6703-6714.2003
Knief, C., Vanitchung, S., Harvey, N. W., Conrad, R., Dunfield, P. F., and Chidthaisong, A. (2005). Diversity of methanotrophic bacteria in tropical upland soils under different land uses. Appl. Environ. Microbiol. 71, 3826–3831. doi: 10.1128/AEM.71.7.3826-3831.2005
Kolb, S. (2009). The quest for atmospheric methane oxidizers in forest soils. Environ. Microbiol. Rep. 1, 336–346. doi: 10.1111/j.1758-2229.2009.00047.x
Kolb, S., Knief, C., Dunfield, P. F., and Conrad, R. (2005). Abundance and activity of uncultured methanotrophic bacteria involved in the consumption of atmospheric methane in two forest soils. Environ. Microbiol. 7, 1150–1161. doi: 10.1111/j.1462-2920.2005.00791.x
Kolb, S., Knief, C., Stubner, S., and Conrad, R. (2003). Quantitative detection of methanotrophs in soil by novel pmoA-targeted real-time PCR assays. Appl. Environ. Microbiol. 69, 2423–2429. doi: 10.1128/AEM.69.5.2423-2429.2003
Kolb, S., and Stacheter, A. (2013). Prerequisites for amplicon pyrosequencing of microbial methanol utilizers in the environment. Front. Microbiol. 4:268. doi: 10.3389/fmicb.2013.00268
Krause, S., Van Bodegom, P. M., Cornwell, W. K., and Bodelier, P. L. E. (2014). Weak phylogenetic signal in physiological traits of methane-oxidizing bacteria. J. Evol. Biol. 27, 1240–1247. doi: 10.1111/jeb.12401
Kravchenko, I. K., Kizilova, A. K., Bykova, S. A., Men'ko, E. V., and Gal'chenko, V. F. (2010). Molecular analysis of high-affinity methane-oxidizing enrichment cultures isolated from a forest biocenosis and agrocenoses. Microbiology 79, 106–114. doi: 10.1134/S0026261710010145
Kumaresan, D., Abell, G. C. J., Bodrossy, L., Stralis-Pavese, N., and Murrell, J. C. (2009). Spatial and temporal diversity of methanotrophs in a landfill cover soil are differentially related to soil abiotic factors. Environ. Microbiol. Rep. 1, 398–407. doi: 10.1111/j.1758-2229.2009.00059.x
Lau, E., Ahmad, A., Steudler, P. A., and Cavanaugh, C. M. (2007). Molecular characterization of methanotrophic communities in forest soils that consume atmospheric methane. FEMS Microbiol. Ecol. 60, 490–500. doi: 10.1111/j.1574-6941.2007.00308.x
Leng, L. Q., Chang, J. L., Geng, K., Lu, Y. H., and Ma, K. (2015). Uncultivated Methylocystis species in paddy soil Iinclude facultative methanotrophs that utilize acetate. Microb. Ecol. 70, 88–96. doi: 10.1007/s00248-014-0540-0
Letunic, I., and Bork, P. (2011). Interactive Tree Of Life v2: online annotation and display of phylogenetic trees made easy. Nucleic Acids Res. 39, W475–W478. doi: 10.1093/nar/gkr201
Li, M., Jain, S., Baker, B. J., Taylor, C., and Dick, G. J. (2014). Novel hydrocarbon monooxygenase genes in the metatranscriptome of a natural deep-sea hydrocarbon plume. Environ. Microbiol. 16, 60–71. doi: 10.1111/1462-2920.12182
Lidstrom, M. E. (1988). Isolation and characterization of marine methanotrophs. Anton Leeuw Int. J. G 54, 189–199. doi: 10.1007/BF00443577
Liebner, S., and Svenning, M. M. (2013). Environmental transcription of mmoX by methane-oxidizing Proteobacteria in a subarctic palsa peatland. Appl. Environ. Microbiol. 79, 701–706. doi: 10.1128/AEM.02292-12
Lima, A. B., Muniz, A. W., and Dumont, M. G. (2014). Activity and abundance of methane-oxidizing bacteria in secondary forest and manioc plantations of Amazonian Dark Earth and their adjacent soils. Front. Microbiol. 5:550. doi: 10.3389/fmicb.2014.00550
Lindner, A. S., Pacheco, A., Aldrich, H. C., Costello Staniec, A., Uz, I., and Hodson, D. J. (2007). Methylocystis hirsuta sp. nov., a novel methanotroph isolated from a groundwater aquifer. Int. J. Syst. Evol. Microbiol. 57, 1891–1900. doi: 10.1099/ijs.0.64541-0
Ludwig, W., Strunk, O., Westram, R., Richter, L., Meier, H., Yadhukumar, B., et al. (2004). ARB: a sofware environment for sequence data. Nucleic Acids Res. 32, 1363–1371. doi: 10.1093/nar/gkh293
Luesken, F. A., Zhu, B., Van Alen, T. A., Butler, M. K., Diaz, M. R., Song, B., et al. (2011). pmoA primers for detection of anaerobic methanotrophs. Appl. Environ. Microbiol. 77, 3877–3880. doi: 10.1128/AEM.02960-10
Lüke, C., Bodrossy, L., Lupotto, E., and Frenzel, P. (2011). Methanotrophic bacteria associated to rice roots: the cultivar effect assessed by T-RFLP and microarray analysis. Environ. Microbiol. Rep. 3, 518–525. doi: 10.1111/j.1758-2229.2011.00251.x
Lüke, C., and Frenzel, P. (2011). Potential of pmoA amplicon pyrosequencing for methanotroph diversity studies. Appl. Environ. Microbiol. 77, 6305–6309. doi: 10.1128/AEM.05355-11
Lüke, C., Krause, S., Cavigiolo, S., Greppi, D., Lupotto, E., and Frenzel, P. (2010). Biogeography of wetland rice methanotrophs. Environ. Microbiol. 12, 862–872. doi: 10.1111/j.1462-2920.2009.02131.x
Macalady, J. L., McMillan, A. M. S., Dickens, A. F., Tyler, S. C., and Scow, K. M. (2002). Population dynamics of type I and II methanotrophic bacteria in rice soils. Environ. Microbiol. 4, 148–157. doi: 10.1046/j.1462-2920.2002.00278.x
Malashenko, Y. R., Romanovskaya, V. A., Bogachenko, V. N., and Stved, A. D. (1975). Thermophilic and thermotolerant bacteria that assimilate methane. Mikrobiologiya 44, 855–862.
Martineau, C., Pan, Y., Bodrossy, L., Yergeau, E., Whyte, L. G., and Greer, C. W. (2014). Atmospheric methane oxidizers are present and active in Canadian high Arctic soils. FEMS Microbiol. Ecol. 89, 257–269. doi: 10.1111/1574-6941.12287
McDonald, I. R., Bodrossy, L., Chen, Y., and Murrell, J. C. (2008). Molecular ecology techniques for the study of aerobic methanotrophs. Appl. Environ. Microbiol. 74, 1305–1315. doi: 10.1128/AEM.02233-07
McDonald, I. R., Smith, K., and Lidstrom, M. E. (2005). Methanotrophic populations in estuarine sediment from Newport Bay, California. FEMS Microbiol. Lett. 250, 287–293. doi: 10.1016/j.femsle.2005.07.016
Morris, S. A., Radajewski, S., Willison, T. W., and Murrell, J. C. (2002). Identification of the functionally active methanotroph population in a peat soil microcosm by stable-isotope probing. Appl. Environ. Microbiol. 68, 1446–1453. doi: 10.1128/AEM.68.3.1446-1453.2002
Nazaries, L., Tate, K. R., Ross, D. J., Singh, J., Dando, J., Saggar, S., et al. (2011). Response of methanotrophic communities to afforestation and reforestation in New Zealand. ISME J. 5, 1832–1836. doi: 10.1038/ismej.2011.62
Nercessian, O., Bienvenu, N., Moreira, D., Prieur, D., and Jeanthon, C. (2005). Diversity of functional genes of methanogens, methanotrophs and sulfate reducers in deep-sea hydrothermal environments. Environ. Microbiol. 7, 118–132. doi: 10.1111/j.1462-2920.2004.00672.x
Nold, S. C., Zhou, J., Devol, A. H., and Tiedje, J. M. (2000). Pacific Northwest marine sediments contain ammonia-oxidizing bacteria in the beta subdivision of the Proteobacteria. Appl. Environ. Microbiol. 66, 4532–4525. doi: 10.1128/AEM.66.10.4532-4535.2000
Ogiso, T., Ueno, C., Dianou, D., Huy, T. V., Katayama, A., Kimura, M., et al. (2012). Methylomonas koyamae sp. nov., a type I methane-oxidizing bacterium from floodwater of a rice paddy field. Int. J. Syst. Evol. Microbiol. 62, 1832–1837. doi: 10.1099/ijs.0.035261-0
Omelchenko, M. V., Vasilyeva, L. V., Zavarzin, G. A., Savel'eva, N. D., Lysenko, A. M., Mityushina, L. L., et al. (1996). A novel psychrophilic methanotroph of the genus Methylobacter. Mikrobiologiya 65, 339–343.
Op den Camp, H. J. M., Islam, T., Stott, M. B., Harhangi, H. R., Hynes, A., Schouten, S., et al. (2009). Environmental, genomic and taxonomic perspectives on methanotrophic Verrucomicrobia. Environ. Microbiol. Rep. 1, 293–306. doi: 10.1111/j.1758-2229.2009.00022.x
Pacheco-Oliver, M., McDonald, I. R., Groleau, D., Murrell, J. C., and Miguez, C. B. (2002). Detection of methanotrophs with highly divergent pmoA genes from Arctic soils. FEMS Microbiol. Lett. 209, 313–319. doi: 10.1111/j.1574-6968.2002.tb11150.x
Parte, A. C. (2014). LPSN - list of prokaryotic names with standing in nomenclature. Nucleic Acids Res. 42, D613–D616. doi: 10.1093/nar/gkt1111
Pester, M., Friedrich, M. W., Schink, B., and Brune, A. (2004). pmoA-based analysis of methanotrophs in a littoral lake sediment reveals a diverse and stable community in a dynamic environment. Appl. Environ. Microbiol. 70, 3138–3142. doi: 10.1128/AEM.70.5.3138-3142.2004
Petersen, J. M., and Dubilier, N. (2009). Methanotrophic symbioses in marine invertebrates. Environ. Microbiol. Rep. 1, 319–335. doi: 10.1111/j.1758-2229.2009.00081.x
Poehlein, A., Deutzmann, J. S., Daniel, R., and Simeonova, D. D. (2013). Draft genome sequence of the methanotrophic Gammaproteobacterium Methyloglobulus morosus DSM 22980 strain KoM1. Genome Announc. 1, e01078–e01013. doi: 10.1128/genomeA.01078-13
Pol, A., Heijmans, K., Harhangi, H. R., Tedesco, D., Jetten, M. S. M., and Den Camp, H. J. M. O. (2007). Methanotrophy below pH1 by a new Verrucomicrobia species. Nature 450, 874–879. doi: 10.1038/nature06222
Pratscher, J., Dumont, M. G., and Conrad, R. (2011). Assimilation of acetate by the putative atmospheric methane oxidizers belonging to the USC alpha clade. Environ. Microbiol. 13, 2692–2701. doi: 10.1111/j.1462-2920.2011.02537.x
Qiu, Q., Noll, M., Abraham, W. R., Lu, Y., and Conrad, R. (2008). Applying stable isotope probing of phospholipid fatty acids and rRNA in a Chinese rice field to study activity and composition of the methanotrophic bacterial communities in situ. ISME J. 2, 602–614. doi: 10.1038/ismej.2008.34
Raggi, L., Schubotz, F., Hinrichs, K. U., Dubilier, N., and Petersen, J. M. (2013). Bacterial symbionts of Bathymodiolus mussels and Escarpia tubeworms from Chapopote, an asphalt seep in the Southern Gulf of Mexico. Environ. Microbiol. 15, 1969–1987. doi: 10.1111/1462-2920.12051
Rahalkar, M., Bussmann, I., and Schink, B. (2007). Methylosoma difficile gen. nov., sp nov., a novel methanotroph enriched by gradient cultivation from littoral sediment of Lake Constance. Int. J. Syst. Evol. Microbiol. 57, 1073–1080. doi: 10.1099/ijs.0.64574-0
Rahalkar, M., and Schink, B. (2007). Comparison of aerobic methanotrophic communities in littoral and profundal sediments of Lake Constance by a molecular approach. Appl. Environ. Microbiol. 73, 4389–4394. doi: 10.1128/AEM.02602-06
Rahman, M. T., Crombie, A., Chen, Y., Stralis-Pavese, N., Bodrossy, L., Meir, P., et al. (2011). Environmental distribution and abundance of the facultative methanotroph Methylocella. ISME J. 5, 1061–1066. doi: 10.1038/ismej.2010.190
Redmond, M. C., Valentine, D. L., and Sessions, A. L. (2010). Identification of novel methane-, ethane-, and propane-oxidizing bacteria at marine hydrocarbon seeps by stable isotope probing. Appl. Environ. Microbiol. 76, 6412–6422. doi: 10.1128/AEM.00271-10
Reeburgh, W. S. (2007). Oceanic methane biogeochemistry. Chem. Rev. 107, 486–513. doi: 10.1021/cr050362v
Ricke, P., Kolb, S., and Braker, G. (2005). Application of a newly developed ARB software-integrated tool for in silico terminal restriction fragment length polymorphism analysis reveals the dominance of a novel pmoA cluster in a forest soil. Appl. Environ. Microbiol. 71, 1671–1673. doi: 10.1128/AEM.71.3.1671-1673.2005
Rodrigues, C. F., Hiláro, A., Cunha, M. R., Weightman, A. J., and Webster, G. (2011). Microbial diversity in Frenulata (Siboglinidae, Polychaeta) species from mud volcanoes in the Gulf of Cadiz (NE Atlantic). Antonie Van Leeuwenhoek 100, 83–98. doi: 10.1007/s10482-011-9567-0
Romanovskaya, V. A., Malashenko, Y. R., and Bogachenko, V. N. (1978). Refinement of the diagnosis of the genera and species of methane-using bacteria. Mikrobiologiya 47, 120–130.
Romanovskaya, V. A., Rokitko, P. V., Shilin, S. O., and Malashenko, Y. R. (2006). Emended description of Methylomonas rubra sp. nov. Microbiology 75, 689–693. doi: 10.1134/S0026261706060117
Ruff, S. E., Arnds, J., Knittel, K., Amann, R., Wegener, G., Ramette, A., et al. (2013). Microbial communities of deep-sea methane seeps at Hikurangi continental margin (New Zealand). PLoS ONE 8:e72627. doi: 10.1371/journal.pone.0072627
Saidi-Mehrabad, A., He, Z., Tamas, I., Sharp, C. E., Brady, A. L., Rochman, F. F., et al. (2013). Methanotrophic bacteria in oilsands tailings ponds of northern Alberta. ISME J. 7, 908–921. doi: 10.1038/ismej.2012.163
Sayavedra-Soto, L. A., Hamamura, N., Liu, C. W., Kimbrel, J. A., Chang, J. H., and Arp, D. J. (2011). The membrane-associated monooxygenase in the butane-oxidizing Gram-positive bacterium Nocardioides sp. strain CF8 is a novel member of the AMO/PMO family. Environ. Microbiol. Rep. 3, 390–396. doi: 10.1111/j.1758-2229.2010.00239.x
Schloss, P. D., and Handelsman, J. (2005). Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl. Environ. Microbiol. 71, 1501–1506. doi: 10.1128/AEM.71.3.1501-1506.2005
Schloss, P. D., and Westcott, S. L. (2011). Assessing and improving methods used in operational taxonomic unit-based approaches for 16S rRNA gene sequence analysis. Appl. Environ. Microbiol. 77, 3219–3226. doi: 10.1128/AEM.02810-10
Semrau, J. D., Dispirito, A. A., and Vuilleumier, S. (2011). Facultative methanotrophy: false leads, true results, and suggestions for future research. FEMS Microbiol. Lett. 323, 1–12. doi: 10.1111/j.1574-6968.2011.02315.x
Semrau, J. D., Dispirito, A. A., and Yoon, S. (2010). Methanotrophs and copper. FEMS Microbiol. Rev. 34, 496–531. doi: 10.1111/j.1574-6976.2010.00212.x
Serrano-Silva, N., Valenzuela-Encinas, C., Marsch, R., Dendooven, L., and Alcantara-Hernandez, R. J. (2014). Changes in methane oxidation activity and methanotrophic community composition in saline alkaline soils. Extremophiles 18, 561–571. doi: 10.1007/s00792-014-0641-1
Sharp, C. E., Smirnova, A. V., Graham, J. M., Stott, M. B., Khadka, R., Moore, T. R., et al. (2014). Distribution and diversity of Verrucomicrobia methanotrophs in geothermal and acidic environments. Environ. Microbiol. 16, 1867–1878. doi: 10.1111/1462-2920.12454
Sharp, C. E., Smirnova, A. V., Kalyuzhnaya, M. G., Bringel, F., Hirayama, H., Jetten, M. S., et al. (2015). Draft genome sequence of the moderately halophilic methanotroph Methylohalobius crimeensis Strain 10Ki. Genome Announc. 3, e00644–e00615. doi: 10.1128/genomeA.00644-15
Sharp, C. E., Stott, M. B., and Dunfield, P. F. (2012). Detection of autotrophic verrucomicrobial methanotrophs in a geothermal environment using stable isotope probing. Front. Microbiol. 3:303. doi: 10.3389/fmicb.2012.00303
Sheik, C. S., Jain, S., and Dick, G. J. (2014). Metabolic flexibility of enigmatic SAR324 revealed through metagenomics and metatranscriptomics. Environ. Microbiol. 16, 304–317. doi: 10.1111/1462-2920.12165
Shigematsu, T., Hanada, S., Eguchi, M., Kamagata, Y., Kanagawa, T., and Kurane, R. (1999). Soluble methane monooxygenase gene clusters from trichloroethylene-degrading Methylomonas sp. strains and detection of methanotrophs during in situ bioremediation. Appl. Environ. Microbiol. 65, 5198–5206.
Shrestha, P. M., Kammann, C., Lenhart, K., Dam, B., and Liesack, W. (2012). Linking activity, composition and seasonal dynamics of atmospheric methane oxidizers in a meadow soil. ISME J. 6, 1115–1126. doi: 10.1038/ismej.2011.179
Sieburth, J. M., Johnson, P. W., Eberhardt, M. A., Sieracki, M. E., Lidstrom, M., and Laux, D. (1987). The first methane-oxidizing bacterium from the upper mixing layer of the deep ocean—Methylomonas pelagica sp. nov. Curr Microbiol 14, 285–293. doi: 10.1007/BF01568138
Siljanen, H. M. P., Saari, A., Krause, S., Lensu, A., Abell, G. C. J., Bodrossy, L., et al. (2011). Hydrology is reflected in the functioning and community composition of methanotrophs in the littoral wetland of a boreal lake. FEMS Microbiol. Ecol. 75, 430–445. doi: 10.1111/j.1574-6941.2010.01015.x
Singh, B. K., Tate, K. R., Kolipaka, G., Hedley, C. B., Macdonald, C. A., Millard, P., et al. (2007). Effect of afforestation and reforestation of pastures on the activity and population dynamics of methanotrophic bacteria. Appl. Environ. Microbiol. 73, 5153–5161. doi: 10.1128/AEM.00620-07
Smith, T. J., and Dalton, H. (2004). Biocatalysis by methane monooxygenase and its implications for the petroleum industry. Petrol. Biotechnol. 151, 177–192. doi: 10.1016/s0167-2991(04)80147-2
Söhngen, N. L. (1906). Über Bakterien, welche Methan als Kohlenstoffnahrung und Energiequelle gebrauchen. Centralbl. Bakt. Parasitenk. II. Abt. 15, 513–517.
Steenbergh, A. K., Meima, M. M., Kamst, M., and Bodelier, P. L. E. (2010). Biphasic kinetics of a methanotrophic community is a combination of growth and increased activity per cell. FEMS MicrobiolEcol 71, 12–22. doi: 10.1111/j.1574-6941.2009.00782.x
Stein, L. Y., Yoon, S., Semrau, J. D., Dispirito, A. A., Crombie, A., Murrell, J. C., et al. (2010). Genome sequence of the obligate methanotroph Methylosinus trichosporium strain OB3b. J. Bacteriol. 192, 6497–6498. doi: 10.1128/JB.01144-10
Stoecker, K., Bendinger, B., Schöning, B., Nielsen, P. H., Nielsen, J. L., Baranyi, C., et al. (2006). Cohn's Crenothrix is a filamentous methane oxidizer with an unusual methane monooxygenase. Proc. Natl. Acad. Sci. U.S.A. 103, 2363–2367. doi: 10.1073/pnas.0506361103
Stralis-Pavese, N., Abell, G. C., Sessitsch, A., and Bodrossy, L. (2011). Analysis of methanotroph community composition using a pmoA-based microbial diagnostic microarray. Nat. Protoc. 6, 609–624. doi: 10.1038/nprot.2010.191
Strong, P. J., Xie, S., and Clarke, W. P. (2015). Methane as a resource: can the methanotrophs add value? Environ. Sci. Technol. 49, 4001–4018. doi: 10.1021/es504242n
Suzuki, T., Nakamura, T., and Fuse, H. (2012). Isolation of two novel marine ethylene-assimilating bacteria, Haliea species ETY-M and ETY-NAG, containing particulate methane monooxygenase-like genes. Microbes Environ. 27, 54–60. doi: 10.1264/jsme2.ME11256
Svenning, M. M., Hestnes, A. G., Wartiainen, I., Stein, L. Y., Klotz, M. G., Kalyuzhnaya, M. G., et al. (2011). Genome sequence of the Arctic methanotroph Methylobacter tundripaludum SV96. J. Bacteriol. 193, 6418–6419. doi: 10.1128/JB.05380-11
Takeuchi, M., Kamagata, Y., Oshima, K., Hanada, S., Tamaki, H., Marumo, K., et al. (2014). Methylocaldum marinum sp. nov., a thermotolerant, methane-oxidizing bacterium isolated from marine sediments, and emended description of the genus Methylocaldum. Int. J. Syst. Evol. Microbiol. 64, 3240–3246. doi: 10.1099/ijs.0.063503-0
Tamas, I., Smirnova, A. V., He, Z., and Dunfield, P. F. (2014). The (d)evolution of methanotrophy in the Beijerinckiaceae - a comparative genomics analysis. ISME J. 8, 369–382. doi: 10.1038/ismej.2013.145
Tavormina, P. L., Hatzenpichler, R., McGlynn, S., Chadwick, G., Dawson, K. S., Connon, S. A., et al. (2015). Methyloprofundus sedimenti gen. nov., sp. nov., an obligate methanotroph from ocean sediment belonging to the 'deep sea-1' clade of marine methanotrophs. Int. J. Syst. Evol. Microbiol. 65, 251–259. doi: 10.1099/ijs.0.062927-0
Tavormina, P. L., Orphan, V. J., Kalyuzhnaya, M. G., Jetten, M. S., and Klotz, M. G. (2011). A novel family of functional operons encoding methane/ammonia monooxygenase-related proteins in gammaproteobacterial methanotrophs. Environ. Microbiol. Rep. 3, 91–100. doi: 10.1111/j.1758-2229.2010.00192.x
Tavormina, P. L., Ussler, W., Joye, S. B., Harrison, B. K., and Orphan, V. J. (2010). Distributions of putative aerobic methanotrophs in diverse pelagic marine environments. ISME J. 4, 700–710. doi: 10.1038/ismej.2009.155
Tavormina, P. L., Ussler, W., and Orphan, V. J. (2008). Planktonic and sediment-associated aerobic methanotrophs in two seep systems along the North American margin. Appl. Environ. Microbiol. 74, 3985–3995. doi: 10.1128/AEM.00069-08
Tavormina, P. L., Ussler, W., Steele, J. A., Connon, S. A., Klotz, M. G., and Orphan, V. J. (2013). Abundance and distribution of diverse membrane-bound monooxygenase (Cu-MMO) genes within the Costa Rica oxygen minimum zone. Environ. Microbiol. Rep. 5, 414–423. doi: 10.1111/1758-2229.12025
Tchawa Yimga, M., Dunfield, P. F., Ricke, P., Heyer, J., and Liesack, W. (2003). Wide distribution of a novel pmoA-like gene copy among type II methanotrophs, and its expression in Methylocystis strain SC2. Appl. Environ. Microbiol. 69, 5593–5602. doi: 10.1128/AEM.69.9.5593-5602.2003
Theisen, A. R., Ali, M. H., Radajewski, S., Dumont, M. G., Dunfield, P. F., McDonald, I. R., et al. (2005). Regulation of methane oxidation in the facultative methanotroph Methylocella silvestris BL2. Mol. Microbiol. 58, 682–692. doi: 10.1111/j.1365-2958.2005.04861.x
Theisen, A. R., and Murrell, J. C. (2005). Facultative methanotrophs revisited. J. Bacteriol. 187, 4303–4305. doi: 10.1128/JB.187.13.4303-4305.2005
Trotsenko, Y. A., and Khmelenina, V. N. (2002). Biology of extremophilic and extremotolerant methanotrophs. Arch. Microbiol. 177, 123–131. doi: 10.1007/s00203-001-0368-0
Trotsenko, Y. A., and Murrell, J. C. (2008). Metabolic aspects of aerobic obligate methanotrophy. Adv. Appl. Microbiol. 63, 183–229. doi: 10.1016/S0065-2164(07)00005-6
Tsubota, J., Eshinimaev, B. T., Khmelenina, V. N., and Trotsenko, Y. A. (2005). Methylothermus thermalis gen. nov., sp nov., a novel moderately thermophilic obligate methanotroph from a hot spring in Japan. Int. J. Syst. Evol. Microbiol. 55, 1877–1884. doi: 10.1099/ijs.0.63691-0
Urmann, K., Schroth, M. H., Noll, M., Gonzalez-Gil, G., and Zeyer, J. (2008). Assessment of microbial methane oxidation above a petroleum-contaminated aquifer using a combination of in situ techniques. J. Geophys. Res. 113, G2006. doi: 10.1029/2006JG000363
van Teeseling, M. C. F., Pol, A., Harhangi, H. R., Van Der Zwart, S., Jetten, M. S. M., and Den Camp, H. J. M. O. (2014). Expanding the verrucomicrobial methanotrophic world: description of three novel species of Methylacidimicrobium gen. nov. Appl Environ Microbiol 80, 6782–6791. doi: 10.1128/AEM.01838-14
Vigliotta, G., Nutricati, E., Carata, E., Tredici, S. M., De Stefano, M., Pontieri, P., et al. (2007). Clonothrix fusca Roze 1896, a filamentous, sheathed, methanotrophic gamma-proteobacterium. Appl. Environ. Microbiol. 73, 3556–3565. doi: 10.1128/AEM.02678-06
Vorobev, A. V., Baani, M., Doronina, N. V., Brady, A. L., Liesack, W., Dunfield, P. F., et al. (2011). Methyloferula stellata gen. nov., sp. nov., an acidophilic, obligately methanotrophic bacterium possessing only a soluble methane monooxygenase. Int. J. Syst. Evol. Microbiol. 61, 2456–2463. doi: 10.1099/ijs.0.028118-0
Vuilleumier, S., Khmelenina, V. N., Bringel, F., Reshetnikov, A. S., Lajus, A., Mangenot, S., et al. (2012). Genome sequence of the haloalkaliphilic methanotrophic bacterium Methylomicrobium alcaliphilum 20Z. J. Bacteriol. 194, 551–552. doi: 10.1128/JB.06392-11
Wartiainen, I., Hestnes, A. G., McDonald, I. R., and Svenning, M. M. (2006a). Methylobacter tundripaludum sp. nov., a methane-oxidizing bacterium from Arctic wetland soil on the Svalbard islands, Norway (78 degrees N). Int. J. Syst. Evol. Microbiol. 56, 109–113. doi: 10.1099/ijs.0.63728-0
Wartiainen, I., Hestnes, A. G., McDonald, I. R., and Svenning, M. M. (2006b). Methylocystis rosea sp. nov., a novel methanotrophic bacterium from Arctic wetland soil, Svalbard, Norway (78 degrees N). Int. J. Syst. Evol. Microbiol. 56, 541–547. doi: 10.1099/ijs.0.63912-0
Watsuji, T. O., Yamamoto, A., Takaki, Y., Ueda, K., Kawagucci, S., and Takai, K. (2014). Diversity and methane oxidation of active epibiotic methanotrophs on live Shinkaia crosnieri. ISME J. 8, 1020–1031. doi: 10.1038/ismej.2013.226
Wendeberg, A., Zielinski, F. U., Borowski, C., and Dubilier, N. (2012). Expression patterns of mRNAs for methanotrophy and thiotrophy in symbionts of the hydrothermal vent mussel Bathymodiolus puteoserpentis. ISME J. 6, 104–112. doi: 10.1038/ismej.2011.81
Whittenbury, R., and Dalton, H. (1981). “The methylotrophic bacteria,” in The Prokaryotes, eds M. P. Starr, H. Stolph, H. G. Trüper, A. Balows, and H. G. Schlegel (Berlin: Springer-Verlag KG), 894–902.
Whittenbury, R., Davies, S. L., and Davey, J. F. (1970a). Exospores and cysts formed by methane-utilizing bacteria. J. Gen. Microbiol. 61, 219–226.
Whittenbury, R., and Krieg, N. R. (1984). “Family Methylococcaceae,” in Bergey's Manual of Systematic Bacteriology, eds N. R. Krieg and J. G. Holt (Baltimore: The Williams & Wilkins), 256–261.
Whittenbury, R., Phillips, K. C., and Wilkinson, J. F. (1970b). Enrichment, isolation and some properties of methane-utilizing bacteria. J. Gen. Microbiol. 61, 205–218.
Wieczorek, A. S., Drake, H. L., and Kolb, S. (2011). Organic acids and ethanol inhibit the oxidation of methane by mire methanotrophs. FEMS Microbiol. Ecol. 77, 28–39. doi: 10.1111/j.1574-6941.2011.01080.x
Wise, M. G., McArthur, J. V., and Shimkets, L. J. (2001). Methyolosarcina fibrata gen. nov., sp. nov. and Methylosarcina quisquiliarum sp. nov., novel type I methanotrophs. Int. J. Syst. Evol. Microbiol. 51, 611–621. doi: 10.1099/00207713-51-2-611
Yan, T. F., Zhou, J. Z., and Zhang, C. L. L. (2006). Diversity of functional genes for methanotrophs in sediments associated with gas hydrates and hydrocarbon seeps in the Gulf of Mexico. FEMS Microbiol. Ecol. 57, 251–259. doi: 10.1111/j.1574-6941.2006.00122.x
Yoon, S., Im, J., Bandow, N., Dispirito, A. A., and Semrau, J. D. (2011). Constitutive expression of pMMO by Methylocystis strain SB2 when grown on multi-carbon substrates: implications for biodegradation of chlorinated ethenes. Environ. Microbiol. Rep. 3, 182–188. doi: 10.1111/j.1758-2229.2010.00205.x
Yun, J. L., Zhang, H. X., Deng, Y. C., and Wang, Y. F. (2015). Aerobic methanotroph diversity in Sanjiang Wetland, Northeast China. Microb. Ecol. 69, 567–576. doi: 10.1007/s00248-014-0506-2
Zbinden, M., Shillito, B., Le Bris, N., De Montlaur, C. D., Roussel, E., Guyot, F., et al. (2008). New insigths on the metabolic diversity among the epibiotic microbial communitiy of the hydrothermal shrimp Rimicaris exoculata. J. Exp. Mar. Biol. Ecol. 359, 131–140. doi: 10.1016/j.jembe.2008.03.009
Keywords: methanotrophic bacteria, pmoA, diversity, phylogeny, habitat specificity, ecological niche
Citation: Knief C (2015) Diversity and Habitat Preferences of Cultivated and Uncultivated Aerobic Methanotrophic Bacteria Evaluated Based on pmoA as Molecular Marker. Front. Microbiol. 6:1346. doi: 10.3389/fmicb.2015.01346
Received: 30 September 2015; Accepted: 16 November 2015;
Published: 15 December 2015.
Edited by:
Svetlana N. Dedysh, Winogradsky Institute of Microbiology, Russia Academy of Science, RussiaReviewed by:
Marc Gregory Dumont, University of Southampton, UKLevente Bodrossy, CSIRO Ocean and Atmosphere, Australia
Paul Bodelier, Netherlands Institute of Ecology, Netherlands
Copyright © 2015 Knief. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Claudia Knief, a25pZWZAdW5pLWJvbm4uZGU=