 Mithila Ferdous1†
Mithila Ferdous1† Kai Zhou1,2,3†
Kai Zhou1,2,3† Richard F. de Boer4
Richard F. de Boer4 Alexander W. Friedrich1*
Alexander W. Friedrich1* Anna M. D. Kooistra-Smid1,4
Anna M. D. Kooistra-Smid1,4 John W. A. Rossen1
John W. A. Rossen1- 1Department of Medical Microbiology, University Medical Center Groningen, University of Groningen, Groningen, Netherlands
- 2State Key Laboratory for Diagnosis and Treatment of Infectious Diseases, The First Affiliated Hospital, College of Medicine, Zhejiang University, Hangzhou, China
- 3Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases, Hangzhou, China
- 4Department of Medical Microbiology, Certe-Laboratory for Infectious Diseases, Groningen, Netherlands
In 2011, a Shiga toxin-producing Enteroaggregative Escherichia coli (EAEC Stx2a+) O104:H4 strain caused a serious outbreak of acute gastroenteritis and hemolytic-uremic syndrome (HUS) in Germany. In 2013, E. coli O104:H4 isolates were obtained from a patient with HUS and her friend showing only gastrointestinal complaints. The antimicrobial resistance and virulence profiles of these isolates together with three EAEC Stx2a+ O104:H4 isolates from 2011 were determined and compared. Whole-genome sequencing (WGS) was performed for detailed characterization and to determine genetic relationship of the isolates. Four additional genomes of EAEC Stx2a+ O104:H4 isolates of 2009 and 2011 available on NCBI were included in the virulence and phylogenetic analysis. All E. coli O104:H4 isolates tested were positive for stx2a, aatA, and terD but were negative for escV. All, except one 2011 isolate, were positive for aggR and were therefore considered EAEC. The EAEC Stx2a+ O104:H4 isolates of 2013 belonged to sequence type (ST) ST678 as the 2011 isolates and showed slightly different resistance and virulence patterns compared to the 2011 isolates. Core-genome phylogenetic analysis showed that the isolates of 2013 formed a separate cluster from the isolates of 2011 and 2009 by 27 and 20 different alleles, respectively. In addition, only a one-allele difference was found between the isolate of the HUS-patient and that of her friend. Our study shows that EAEC Stx2a+ O104:H4 strains highly similar to the 2011 outbreak clone in their core genome are still circulating necessitating proper surveillance to prevent further outbreaks with these potentially pathogenic strains. In addition, WGS not only provided a detailed characterization of the isolates but its high discriminatory power also enabled us to discriminate the 2013 isolates from the isolates of 2009 and 2011 expediting the use of WGS in public health services to rapidly apply proper infection control strategies.
Introduction
Shiga toxin-producing Escherichia coli (STEC) are a pathogenic group of Escherichia coli (E. coli) producing the potent cytotoxin Shiga-toxin (Stx) similar to the one produced by Shigella dysenteriae serotype 1 (Gyles, 2007). STEC may cause a broad spectrum of illness, ranging from diarrhea to the potentially fatal hemolytic uremic syndrome (HUS) (Farrokh et al., 2013). A subset of STEC can cause bloody diarrhea in humans and they are known as enterohemorrhagic E. coli (EHEC) while a subset of EHEC can cause HUS and are known as HUS-associated E. coli HUSEC (Mellmann et al., 2008). In addition to Shiga toxin-converting bacteriophages, STEC may contain several other mobile genetic elements encoding virulence factors as pathogenicity islands (PAI), and a large, approximately 90 kb plasmid (pO157; Karch, 2001). Stx production is common to all HUS-associated E. coli isolates regardless of their serotype. When the toxin enters the blood stream it binds to receptors on endothelial cells abundantly present in kidneys and brain, leading to neurological sequel and/or to microvascular disease that may result in HUS (Welinder-Olsson and Kaijser, 2005).
In 2011, a large outbreak was reported in Germany caused by an Enteroaggregative E. coli (EAEC) O104:H4 strain lysogenized with the Stx2a bacteriophage and thereby becoming an EAEC/STEC hybrid strain (Bielaszewska et al., 2011; Brzuszkiewicz et al., 2011). Besides the stx2a gene, this unusual strain had virulence properties of EAEC including plasmid pAA carrying the aggregative adherence fimbriae (AAF) variant I encoded by the aggA gene whose expression is regulated by the aggR gene. In addition, it contained a protein-coat secretion system (Aat), dispersin (Aap), a putative type VI secretion system (Aai), and a rare combination of serine protease autotransporters of Enterobacteriaceae (SPATEs) genes, i.e., sepA, sigA, and pic (Rasko et al., 2011; Scheutz et al., 2011). It also contained a terD gene (tellurite resistance gene as a marker for the ter cluster) and a plasmid-borne extended spectrum beta-lactamase (ESBL) gene blaCTX-M-15 resulting in resistance to several antibiotics (Brzuszkiewicz et al., 2011; Rasko et al., 2011). However, the outbreak clone did not possess the escV gene encoding the predicted outer membrane protein and a marker for the locus of enterocyte effacement (LEE) PAI (Müller et al., 2009; Scheutz et al., 2011).
In clinical microbiology, whole genome sequencing (WGS) has already shown its value in outbreak investigations and epidemiological typing due to its high-resolution discriminatory power and detailed virulence profiling, thereby becoming more and more important in routine diagnostics (Mellmann et al., 2011; Rohde et al., 2011; Grad et al., 2012). In this study, we characterized EAEC Stx2a+ O104:H4 strains isolated from a HUS patient and her friend who traveled together to Turkey in 2013 prior to diagnosing the patient with HUS. For this, a WGS approach in parallel with routine phenotypic and genotypic laboratory methods was used. Analyses were performed to get more insight into the antibiotic resistance and virulence profiles of the isolates and to reveal their genetic relationship with the 2011 German outbreak EAEC Stx2a+ O104:H4 isolates.
Materials And Methods
E. coli Isolates used in This Study
In July 2013, four E. coli isolates were obtained from a HUS patient (isolate 338) and her friend (isolates 381-1, 381-3, and 381-4). They were compared to three EAEC Stx2a+ O104:H4 strains named 7N, 8G, and LB227103 which were a kind gift of Dr. Alexander Mellmann (Institute of Hygiene, University of Muenster, Muenster, Germany) and were isolated during the 2011 German outbreak period from stool samples of patients, submitted to the National Consulting Laboratory for HUS in Münster, Germany, between May 23 and June 2, 2011 (Bielaszewska et al., 2011; Pritchard et al., 2012). In addition, publically available genomes of five previously reported strains (TY-2482, 2011C-3494, 2009EL-2050, 2009EL-2071, and 55989) were included in virulence and phylogenetic analyses. Detailed information on the isolates used in this study is shown in Table 1.
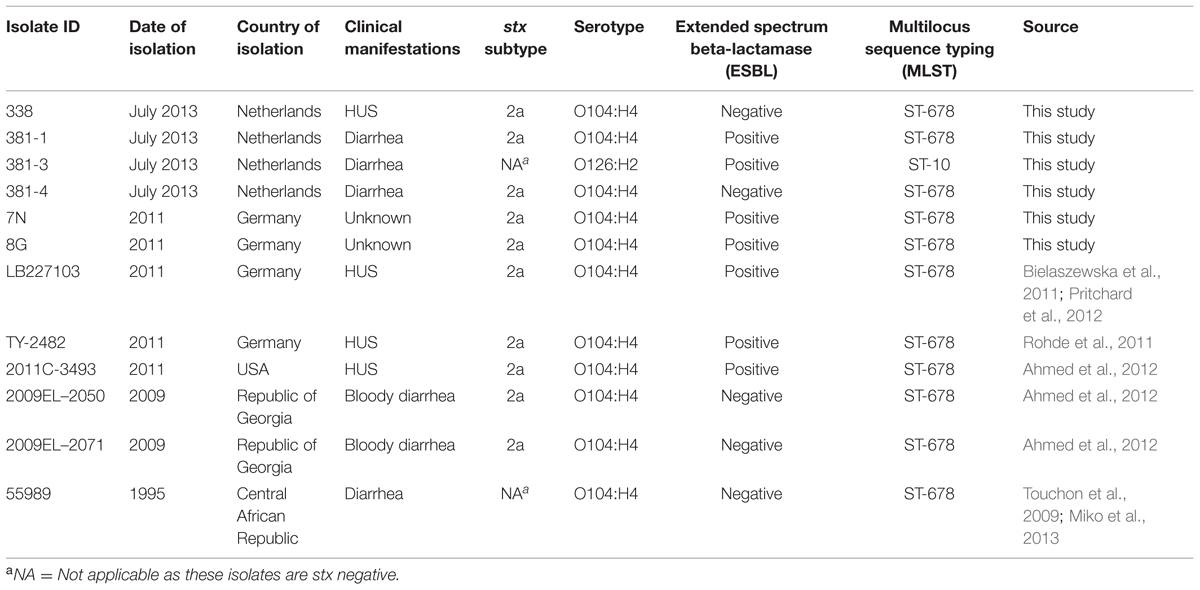
TABLE 1. Characteristics of isolates analyzed in this study.
Diagnostic Procedures
Fecal samples from the HUS patient (patient 338) and her friend (patient 381) were collected for diagnostic purposes at Certe Laboratory for Infectious Diseases as described previously (de Boer et al., 2015). Shortly, fecal samples were screened for the presence of the virulence genes stx1, stx2, and escV by real-time PCR (qPCR) and stx-positive samples were subsequently enriched in Brilliant green bile (BGB) broth. After DNA isolation from the enriched broth, qPCR was performed for detection of the EAEC pAA plasmid encoding genes aggR and aatA, and O-serogroup encoding genes (O26, O91, O103, O104, O111, O121, O145, and O157). In parallel, part of the BGB broth was plated on CHROMagar STEC and Sorbitol MacConkey agar plates (Mediaproducts BV, Groningen, the Netherlands). To obtain a pure isolate from the selective agar plates, more than 30 colonies of each patient were analyzed for the presence of stx genes by qPCR and for ESBL production by using CHROM ID ESBL agar (bioMérieux, Marcy I’Etoile, France). In addition, sorbitol fermentation and tellurite resistance of the isolates were determined by observing their ability to grow on CT-SMAC plates (Sorbitol MacConkey agar with Cefixime and Tellurite). In addition, conventional PCRs for stx-subtypes 2a, 2b, 2c, 2d, 2e, 2f, and 2g were performed to determine the subtype of the stx2 gene as described previously (Scheutz et al., 2012). Finally, a multiplex PCR targeting typical molecular features of the 2011 outbreak strain, i.e., the presence of the wzyO104 (O antigen polymerase as a marker for the E. coli O104 gene cluster), fliCH4 (encoding the H4 specific flagellin), stx2, and terD genes was performed (Bielaszewska et al., 2011; Zhang et al., 2012).
Antimicrobial Resistance
Antibiotic resistance patterns of the four isolates of 2013 (338, 381-1, 381-3, and 381-4) and three isolates of 2011 (7N, 8G, and LB227103) were analyzed using VITEK2 (bioMerieux, Marcy l’Etoile, France) and E-Test (bioMérieux, Marcy l’Etoile, France) following EUCAST guidelines. Presence of ESBL, carbapenemases and AmpCs genes was confirmed using the Check-MDR CT103 assay according to the manufacturer’s protocol (Check-Points BV Wageningen, the Netherlands).
DNA Isolation
For repetitive sequence based PCR fingerprinting and WGS, genomic DNA was extracted by the UltraClean® microbial DNA isolation kit (MO BIO Laboratories, Carlsbad, CA, USA) according to the manufacturer’s protocol. For other molecular assays DNA was extracted using a DNeasy blood and tissue kit (QIAGEN, GmbH, Hilden, Germany) following the manufacturer’s protocol.
Multilocus Sequence Typing (MLST)
Multilocus sequence typing was performed using a 3130xl Genetic Analyzer (Applied Biosystems) to sequence internal fragments of seven housekeeping genes (adk, fumC, gyrB, icd, mdh, purA, and recA) according to the protocol described in the E. coli MLST databases (http://mlst.warwick.ac.uk/mlst/dbs/Ecoli/documents/primersColi_html). The alleles and sequence types (ST) were assigned in accordance with the same database.
Virulence Properties
To assess the presence of known E. coli virulence genes, a DNA microarray analysis was performed using the E. coli genotyping combined assay kit according to the manufacturer’s protocol (Clondiag, Alere Technologies, GmbH, Jena, Germany; Geue et al., 2010).
Repetitive Sequence based PCR (rep-PCR)
Isolated DNA was amplified using the DiversiLab Escherichia kit for repetitive sequence based PCR fingerprinting following the manufacturer’s instructions (bioMérieux, Marcy-l’Etoile, France). The rep-PCR products were detected and analyzed using lab-on-a-chip microfluidics technology on an Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA). Further analysis was performed with the web-based DiversiLab software (version 3.4) using the Pearson correlation coefficient to calculate pairwise similarities among all samples. In this study, we used 95% similarity thresholds for the data analysis (Voets et al., 2013).
Whole Genome Sequencing and Analysis
A DNA library was prepared using the Nextera XT kit (Illumina, San Diego, CA, USA) according to the manufacturer’s instructions and then run on a Miseq (Illumina) for generating paired-end 250-bp reads. De novo assembly was performed using default settings of CLC Genomics Workbench v6.0.5 (CLC bio A/S, Denmark) as described previously (Ferdous et al., 2015). The MLST ST was also confirmed from the whole genome sequence by uploading the assembled genomes to the CGE MLST server (version 1.7; Larsen et al., 2012); the configuration for MLST was set to E. coli scheme 1. The resistome was generated by uploading assembled genomes to the CGE Resfinder server 2.0 (Zankari et al., 2012). The default settings for Resfinder were used except for the threshold of ID that was set to 85%. The serogenotype of the isolates was identified using CGE SerotypeFinder 1.0 (Joensen et al., 2015). The virulence genes were identified by mapping reads to a pseudomolecule generated by concatenating a set of E. coli virulence genes described before (Bielaszewska et al., 2011; Lima et al., 2013). In addition to the isolates of 2013 and 2011, the genomes of two isolates of 2009 (2009EL-2050, 2009EL-2071) were included for comparison of the virulence genes. The genome sequence of 2011C-3493 (GenBank accession no. NC_018658.1) was used as reference for extracting open reading frames (ORFs) from contigs of each strain by SeqSphere v1.0 (Ridom GmbH, Münster, Germany). Only the ORFs without premature stop codons and ambiguous nucleotides were extracted, and the sequences of those ORFs shared by all samples analyzed here were defined as the core genome for subsequent phylogenetic analysis (Leopold et al., 2014). According to the sequence identity of each ORF, a numerical allele type was assigned by SeqSphere. SeqSphere used the allelic profile formed by the combination of all alleles in each genome for constructing the minimum-spanning tree. This Whole Genome Shotgun project has been deposited at DDBJ/EMBL/GenBank under the accession number JRJF01000000 (E. coli 338), JRKD01000000 (E. coli 381-1), JRLM01000000 (E. coli 381-3), JRLD01000000 (E. coli 381-4), JRKE01000000 (E. coli 7N), JRLN00000000 (E. coli 8G), JRKF01000000 (E. coli LB227103). The version described in this paper is the first version.
Results
Demographic and Clinical Characteristics of Patients
Strain 338 was isolated from the fecal sample of a 22 year-old female patient (338) admitted to the Intensive Care Unit of the University Medical Center Groningen (UMCG) in July 2013, after returning from a holiday in Turkey. She presented with abdominal pain, bloody diarrhea, vomiting, and eventually developed HUS. Patient 381, a 24-year-old female traveling together with patient 338, showed symptoms of diarrhea, abdominal cramps, and abdominal pain but was not hospitalized.
Primary Screening Results
Direct screening of feces by qPCR revealed the presence of stx1, stx2, and escV genes (patient 338) and stx2 gene (patient 381). Additional molecular screening of DNA isolated from enriched BGB broth for other virulence genes and O-serogroups resulted in detection of aggR, aatA, and wzyO104 in both patients. Subsequently, as a result of colony screening for the presence of the stx gene and/or for ESBL production one stx2-positive non-ESBL-producing isolate (isolate 338) was obtained from patient 338, whereas two different stx2-positive isolates were recovered from patient 381: an ESBL producing isolate (381-1) and a non-ESBL producing isolate (381-4). In addition, one stx-negative but ESBL-producing isolate (381-3) was obtained from this patient. All stx-positive isolates contained the aatA and terD but not the escV gene, similar to the 2011 German outbreak isolates. No stx1-positive isolates were obtained. The aggR gene was detected in the 2013 and two of the three 2011 isolates (7N and 8G) but unexpectedly not in isolate LB227103. All stx-positive isolates were subtyped as stx2a and shared the same serotype (O104:H4) as the 2011 German outbreak isolates, whereas the stx negative ESBL producing isolate was serotype O126:H2. All isolates were sorbitol fermenting and tellurite resistant. MLST analysis showed that all 2013 EAEC Stx2a+ O104:H4 isolates belonged to ST678 as the 2011 outbreak strains, whereas the stx-negative isolate (381-3) was assigned to ST10. The screening results are summarized in Table 1.
Phenotypic and Genotypic Resistance Profiles
Isolates 338 and 381-4 (both ESBL negative) were resistant to ampicillin and trimethoprim. Isolate 381-1 (ESBL positive) was resistant to ampicillin, cefotaxime, ceftazidime, cefepime, cefuroxime, and trimethoprim and this resistance pattern was almost identical to that of the stx-negative/ESBL positive isolate 381-3 from the same patient except that the latter was susceptible to trimethoprim. The resistance pattern of isolate 381-1 differed from that of three of the 2011 outbreak isolates which were resistant to amoxicillin-clavulanic acid, tetracycline and trimethoprim/sulfamethoxazole (Table 2).
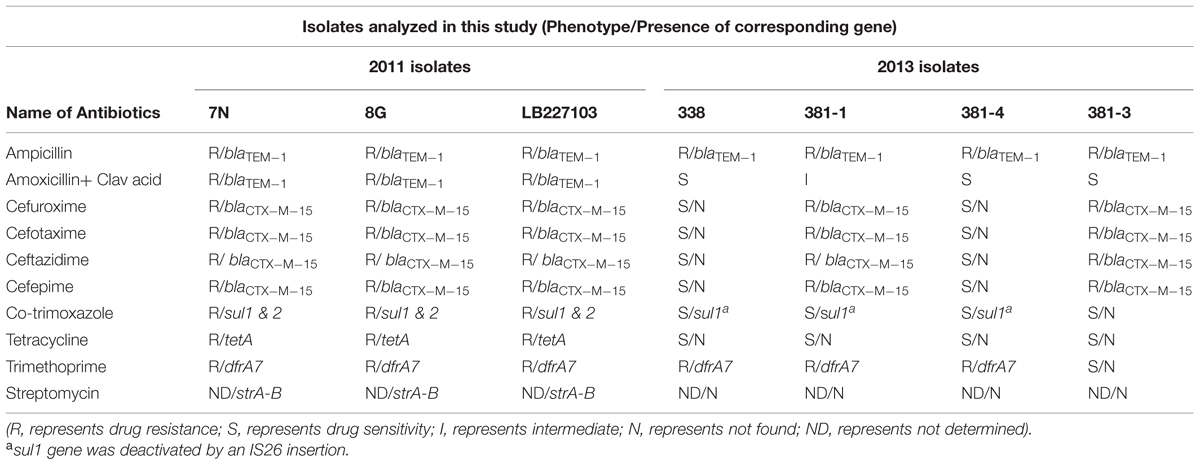
TABLE 2. Phenotypic antibiotic resistance patterns and presence of corresponding antibiotic resistance genes among the isolates.
In agreement with the results of the phenotypic resistance pattern, the resistome of the three 2013 EAEC Stx2a+ O104:H4 isolates differed from that of the 2011 isolates: the strA, strB (streptomycin resistance), sul2 (sulfonamide resistance), and tetA (tetracycline resistance) genes were only found in the 2011 outbreak isolates (Table 2). A blaCTX-M-15 gene was detected in isolate 381-1 (stx-positive) and 381-3 (stx-negative) as well as in the 2011 isolates. In addition, WGS showed the presence of the dfrA7 gene in all isolates except 381-3 resulting in the latter’s trimethoprim-sensitive phenotype. A truncated sul1 gene resulting from an insertion of IS26 was found in all EAEC Stx2a+ O104:H4 isolates.
Virulence Profile
DNA microarray results showed that the virulence profile of the 2013 isolates was similar to that of the EAEC Stx2a+ O104:H4 isolates of 2011 and 2009 used in this study. The only difference was the presence of the microcin H47 biosynthesis gene mchB and mchC, which were identified in the 2009 and 2011 strains, but were absent in the EAEC Stx2a+ O104:H4 isolates of 2013.
The results obtained by WGS were identical to that of the DNA microarray and confirmed the absence of the aggR gene in the outbreak strain LB227103. WGS revealed the presence of several additional virulence genes including aap, aaiC, aggA, lpfAO113, lpfAO26, fyuA, irp2, and set1 (not detected by PCR or microarray) in our isolates (Table 3).
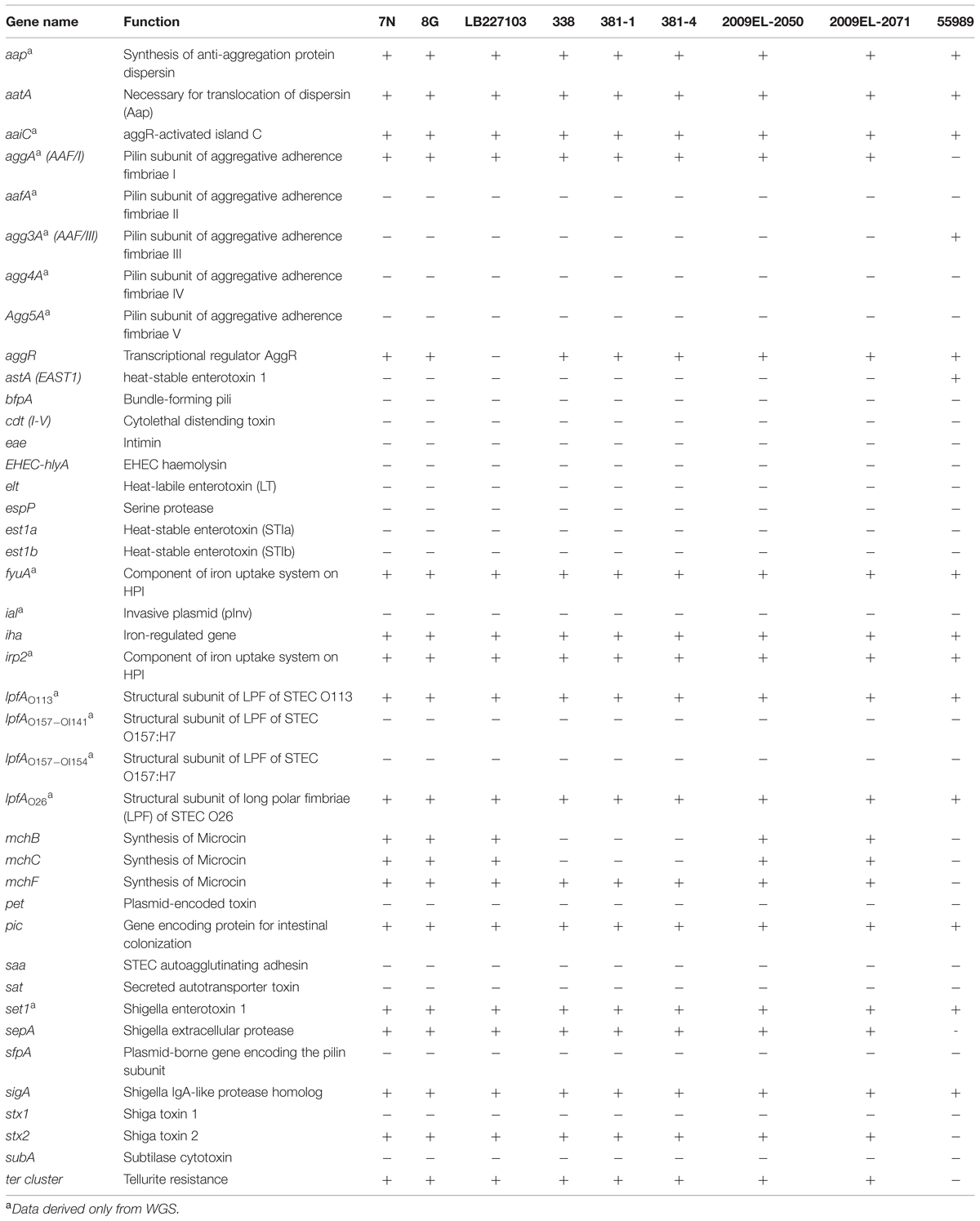
TABLE 3. Virulence genes detected by qPCR, microarray and whole-genome sequencing (WGS) analysis.
Phylogenetic Analyses
Rep-PCR analysis was used to investigate the genetic relationship between the different isolates. EAEC Stx2a+ O104:H4 isolates of 2011 and 2013 clustered into two different groups sharing a similarity of less than 95% whereas the two non O104:H4 isolates (E. coli ATCC 25922 and E. coli O126:H2) separated from the O104:H4 clusters with less than 80% similarity (Figure 1). To type the different isolates with an even higher resolution, a core-genome phylogenetic analysis based on WGS results was performed. In total 3764 ORFs were shared by all isolates analyzed and these were defined as the core genome for further phylogenetic analysis in this study. The minimum-spanning tree shows that the three 2013 EAEC Stx2a+ O104:H4 isolates clustered together with one to two-allele difference among them. One-allele difference of isolate 381-1 from isolates 338 and 381-4 was found in the gene encoding a carboxyterminal protease and was caused by a non-synonymous point mutation (1481T > A) resulting in a premature protein (Leu494Stop) in isolate 381-1. In addition, one-allele difference of isolate 381-4 from isolates 338 and 381-1 was found in a gene encoding a putative cation:proton antiport protein caused by a non-synonymous point mutation (1465A > C) resulting in an amino-acid change in the protein (Ile489Leu) in isolate 381-4. No allele differences were found between the three 2011 outbreak isolates (7N, 8G, and LB227103), and they clustered together with two other reported outbreak strains (TY-2482 and 2011C-3493) with two to three-allele difference among them (Figure 2). The 2009 isolates (2009El-2071 and 2009El-2050) were separated from each other with a five-allele difference. The cluster of 2013 O104:H4 isolates were separated from the 2009 and 2011 O104:H4 clusters with a minimum of 20 and 27 different alleles, respectively (Figure 2 and Supplementary Table S1). There were more than 193 different alleles between the typical EAEC strain 55989 (O104:H4 stx-negative) and the 2013 EAEC stx2a+ O104:H4 isolates, whereas isolate 381-3 (O126:H2, stx-negative) was far distinct from any of the O104:H4 strains analyzed, with more than 3291-allele differences (Figure 2).

FIGURE 1. Dendrogram of Escherichia coli isolates based on rep-PCR results. Stx2a+ O104:H4 isolates of 2011 and 2013 are separated into two clusters with a similarity of less than 95% whereas the non O104:H4 E. coli isolates are clearly separated with a similarity of less than 80%.
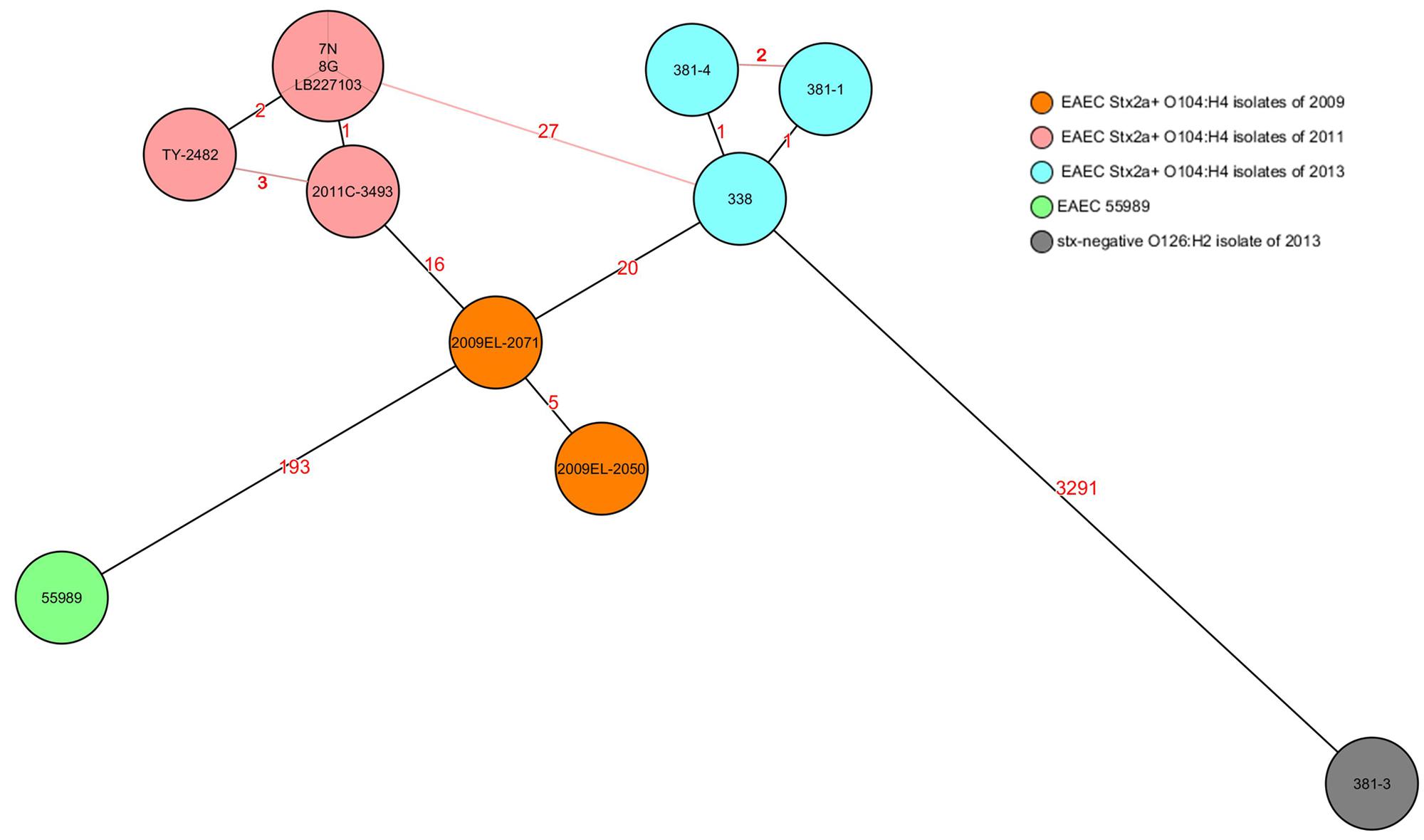
FIGURE 2. Core-genome phylogenetic analysis of E. coli strains. The minimum spanning tree was generated by SeqSphere and based on allelic profiles comparing 3764 alleles present in all analyzed strains and defined as their core genome. Numbers in the lines indicate the number of allele differences between isolates. Black lines indicate minimum distances and red lines connect isolates with more than minimum distances. Different colors represent isolates of different time periods except isolate 381-3, which was isolated in 2013 but marked in a different color as it is stx negative. EAEC 55989 was used as the hypothetical EAEC Stx2a+ O104:H4 progenitor.
Discussion
In this study, EAEC Stx2a+ O104:H4 isolates obtained from stool samples of a HUS patient and her friend, traveling together to Turkey in 2013, were characterized and compared with the 2011 German EAEC Stx2a+ O104:H4 outbreak isolates and with two isolates from cases of bloody diarrhea that occurred in the Republic of Georgia in 2009. The phylogenetic relationship of these isolates was also compared with EAEC 55989, the hypothetical EAEC Stx2a+ O104:H4 progenitor strain isolated in central Africa in 1995. Besides using established molecular typing methods routinely used in our laboratory, WGS was used. Initially, the three EAEC Stx2a+ O104:H4 isolates obtained in 2013 were suspected to be similar to the 2011 German outbreak strains based on PCR results. The 2013 isolates could not be distinguished from the 2011 outbreak strains using a multiplex screening PCR targeting O104wzy, fliCH4, stx2, and terD genes as characteristic features of the 2011 outbreak strains (Bielaszewska et al., 2011; Zhang et al., 2012). Our microarray analyses revealed more detailed information but did not allow us to definitely discriminate the 2013 EAEC Stx2a+ O104:H4 isolates from the 2011 ones. Differences found between the 2011 and 2013 EAEC Stx2a+ O104:H4 isolates included two virulence genes (mchB and mchC) and three drug-resistant genes (tetA, strA-B, and sul2). However, these genes are known to be located in multiple genomic islands and could therefore be easily lost or obtained (Grad et al., 2013; Guy et al., 2013). Although rep-PCR separated the 2011 and 2013 EAEC Stx2a+ O104:H4 isolates into two clusters, this technique is not suitable for large-scale/global outbreak typing, as the inter-laboratory reproducibility is not sufficient for this. In addition, also the intra-laboratory reproducibility may vary making it necessary for an accurate typing to include all the isolates that need to be compared within the same run (Voets et al., 2013). The high resolution of WGS enabled us to discriminate the 2013 isolates from the 2011 German outbreak in the most accurate way.
In this study, a gene-by-gene typing approach also known as whole genome MLST (wgMLST), extended MLST, MLST+ or core genome MLST (cgMLST) was used (Maiden et al., 2013). The EAEC Stx2a+ O104:H4 isolates of 2013 appear to differ from those of 2011 and 2009 by a minimum of 27 and 20 alleles, respectively. This suggests that 2013 isolates were relatively closer related to the 2009 clone of southeast Europe than to the 2011 outbreak clone. Unfortunately, there is no established threshold for the MLST+ approach to address intra- and inter-cluster differences yet. In addition, only the core genome was used for constructing the phylogenetic tree minimizing the chance of including mobile genetic elements (MGEs) in the analysis as there is no consensus on how MGEs should be taken into account in bacterial phylogeny (Daubin et al., 2002; Frost et al., 2005). As the estimated mutation rate of E. coli is reported to be 1.1 nucleotide per genome per year (Reeves et al., 2011), the 27-allele difference between the 2013 and 2011 isolates suggests that the EAEC Stx2a+ O104:H4 isolates from the different time periods belong to different clones. Moreover, only a one-allele difference was found between the isolate of the HUS patient and that of her friend, strongly supporting the hypothesis that either both patients obtained STEC from the same source or that one transmitted STEC to the other.
In addition to high resolution typing, WGS provided us with more detailed molecular characteristics of the isolates. This helped us to explain in more detail the phenotypic results of the isolates. For example, an insertion within the sul1 gene resulting in protein truncation was found in all 2013 EAEC Stx2a+ O104:H4 isolates, which could explain why the isolates were susceptible to trimethoprim/sulfamethoxazole in spite of the presence of the sul1 gene detected by the microarray. On the other hand, a sul2 gene existing in the 2011 but not the 2013 isolates may confer resistance to trimethoprim/sulfamethoxazole in spite of the fact that also the 2011 isolates carried the truncated sul1.
Notably, one of the 2013 EAEC Stx2a+ O104:H4 isolates (381-1) was blaCTX-M-15 positive as the 2011 outbreak isolates. As isolate 381-3 (stx-negative/O126:H2/blaCTX-M-15) was also obtained from the same host, it is likely that 381-1 acquired this ESBL gene from isolate 381-3 or the other way around. Our study did, however, not address a detailed analysis of the MGEs and plasmids as the aim of the study was to show the value of WGS technique in routine clinical diagnostics to identify and compare potentially pathogenic strains.
Recently, two similar isolates of EAEC Stx2a+ O104:H4 causing bloody diarrhea and HUS were reported in Belgium. Patients of both cases traveled to Tunisia and Turkey, respectively, and the infection may have been acquired in those regions (De Rauw et al., 2014). This is similar to our and other studies describing EAEC Stx2a+ O104:H4 infections associated with traveling to Turkey, Tunisia, Egypt, and North Africa (Ecdc/Efsa, 2011; Grad et al., 2013).
Our study shows that WGS has the potential to be integrated in current routine laboratories to relatively rapidly obtain reproducible and detailed information in one single step. This facilitates hospitals and public health organizations to make appropriate strategies for infection control and public health measures in real time (Reuter et al., 2013). Surely, some challenges have to be overcome before applying WSG to routine diagnostics, like the availability of user-friendly bioinformatics tools, automation of the workflow and availability of genomic databases (Sherry et al., 2013).
Conclusion
The detailed characterization of EAEC Stx2a+ O104:H4 isolates obtained in 2013 including the high resolution typing approach helped us to distinguish them from the 2011 German outbreak strains. It further supported the general idea that, although the 2011 outbreak is stopped, EAEC Stx2a+ O104:H4 strains highly similar to the 2011 outbreak strain in their core genome still circulate. They still pose a serious risk for public health being a potential source for causing a new EAEC Stx2a+ O104:H4 outbreak if not monitored carefully.
Author Contributions
Study design: MF, KZ, JR, AF
Performing Experiments: MF, KZ, RdB
Acquisition of data: MF, KZ, RdB
Analysis and interpretation of data: MF, KZ, AK-S, JR
Drafting of manuscript: MF, KZ, JR, AF
Critical revision: MF, KZ, RdB, AK-S, JR, AF
Funding
This study was partly supported by the Interreg IVa-funded projects EurSafety Heath-net (III-1-02 = 73) and SafeGuard (III-2-03 = 025) and by a University Medical Center Groningen Healthy Ageing Pilots grant.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
We thank Dr. Alexander Mellmann for providing us with E. coli O104:H4 strains of the German outbreak and Brigitte Dijkhuizen for technical assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2015.01348
References
Ahmed, S. A., Awosika, J., Baldwin, C., Bishop-Lilly, K. A., Biswas, B., Broomall, S., et al. (2012). Genomic comparison of Escherichia coli O104:H4 isolates from 2009 and 2011 reveals plasmid, and prophage heterogeneity, including Shiga toxin encoding phage stx2. PLoS ONE 7:e48228. doi: 10.1371/journal.pone.0048228
Bielaszewska, M., Mellmann, A., Zhang, W., Köck, R., Fruth, A., Bauwens, A., et al. (2011). Characterisation of the Escherichia coli strain associated with an outbreak of haemolytic uraemic syndrome in Germany, 2011: a microbiological study. Lancet Infect. Dis. 11, 671–676. doi: 10.1016/S1473-3099(11)70165-7
Brzuszkiewicz, E., Thürmer, A., Schuldes, J., Leimbach, A., Liesegang, H., Meyer, F. D., et al. (2011). Genome sequence analyses of two isolates from the recent Escherichia coli outbreak in Germany reveal the emergence of a new pathotype: entero-aggregative-haemorrhagic Escherichia coli (EAHEC). Arch. Microbiol. 193, 883–891. doi: 10.1007/s00203-011-0725-6
Daubin, V., Gouy, M., and Perrière, G. (2002). A phylogenomic approach to bacterial phylogeny: evidence of a core of genes sharing a common history. Genome Res. 12, 1080–1090. doi: 10.1101/gr.187002
de Boer, R. F., Ferdous, M., Ott, A., Scheper, H. R., Wisselink, G. J., Heck, M. E., et al. (2015). Assessing the public health risk of shiga toxin-producing Escherichia coli by use of a rapid diagnostic screening algorithm. J. Clin. Microbiol. 53, 1588–1598. doi: 10.1128/JCM.03590-14
De Rauw, K., Vincken, S., Garabedian, L., Levtchenko, E., Hubloue, I., Verhaegen, J., et al. (2014). Enteroaggregative Shiga toxin-producing Escherichia coli of serotype O104: H4 in Belgium and Luxembourg. New Microbes New Infect 2, 138–143. doi: 10.1002/nmi2.58
Ecdc/Efsa (2011). STEC/VTEC in Humans, Food and Animals in the EU/EEA. ECDC/EFSA joint Technical report NO. 23. Stockholm: ECDC. doi: 10.2900/55055
Farrokh, C., Jordan, K., Auvray, F., Glass, K., Oppegaard, H., Raynaud, S., et al. (2013). Review of Shiga-toxin-producing Escherichia coli (STEC) and their significance in dairy production. Int. J. Food Microbiol. 162, 190–212. doi: 10.1016/j.ijfoodmicro.2012.08.008
Ferdous, M., Zhou, K., Mellmann, A., Morabito, S., Croughs, P. D., de Boer, R. F., et al. (2015). Is Shiga toxin-negative Escherichia coli O157:H7 enteropathogenic or enterohaemorrhagic Escherichia coli? Comprehensive molecular aanalysis using whole– genome sequencing. J. Clin. Microbiol. 53, 3530–3538. doi: 10.1128/JCM.01899-15
Frost, L. S., Leplae, R., Summers, A. O., and Toussaint, A. (2005). Mobile genetic elements: the agents of open source evolution. Nat. Rev. Microbiol. 3, 722–732. doi: 10.1038/nrmicro1235
Geue, L., Schares, S., Mintel, B., Conraths, F. J., Müller, E., and Ehricht, R. (2010). Rapid microarray-based genotyping of enterohemorrhagic Escherichia coli serotype O156:H25/H-/Hnt isolates from cattle and clonal relationship analysis. Appl. Environ. Microbiol. 76, 5510–5519. doi: 10.1128/AEM.00743-10
Grad, Y. H., Godfrey, P., Cerquiera, G. C., Mariani-Kurkdjian, P., Gouali, M., Bingen, E., et al. (2013). Comparative genomics of recent Shiga toxin-producing Escherichia coli O104:H4: short-term evolution of an emerging pathogen. MBio 4, 1–10. doi: 10.1128/mBio.00452-12
Grad, Y. H., Lipsitch, M., Feldgarden, M., Arachchi, H. M., Cerqueira, G. C., FitzGerald, M., et al. (2012). Genomic epidemiology of the Escherichia coli O104:H4 outbreaks in Europe, 2011. Proc. Natl. Acad. Sci. 109, 3065–3070. doi: 10.1073/pnas.1121491109
Guy, L., Jernberg, C., Arvén Norling, J., Ivarsson, S., Hedenström, I., Melefors, Ö, et al. (2013). Adaptive mutations and replacements of virulence traits in the Escherichia coli O104:H4 outbreak population. PLoS ONE 8:e63027. doi: 10.1371/journal.pone.0063027
Gyles, C. L. (2007). Shiga toxin-producing Escherichia coli: an overview. J. Anim. Sci. 85, E45–E62. doi: 10.2527/jas.2006-508
Joensen, K. G., Tetzschner, A. M., Iguchi, A., Aarestrup, F. M., and Scheutz, F. (2015). Rapid and easy in silico serotyping of Escherichia coli isolates by use of whole-genome sequencing data. J. Clin. Microbiol. 53, 2410–2426. doi: 10.1128/JCM.00008-15
Karch, H. (2001). The role of virulence factors in enterohemorrhagic Escherichia coli (EHEC)–associated hemolytic-uremic syndrome. Semin. Thromb. Hemost. 27, 207–213. doi: 10.1055/s-2001-15250
Larsen, M. V., Cosentino, S., Rasmussen, S., Friis, C., Hasman, H., Marvig, R. L., et al. (2012). Multilocus sequence typing of total-genome-sequenced bacteria. J. Clin. Microbiol. 50, 1355–1361. doi: 10.1128/JCM.06094-11
Leopold, S. R., Goering, R. V., Witten, A., Harmsen, D., and Mellmann, A. (2014). Bacterial whole-genome sequencing revisited: portable, scalable, and standardized analysis for typing and detection of virulence and antibiotic resistance genes. J. Clin. Microbiol. 52, 2365–2370. doi: 10.1128/JCM.00262-14
Lima, I. F., Boisen, N., Quetz, J., da, S., Havt, A., de Carvalho, E. B., et al. (2013). Prevalence of enteroaggregative Escherichia coli and its virulence-related genes in a case-control study among children from north-eastern Brazil. J. Med. Microbiol. 62, 683–693. doi: 10.1099/jmm.0.054262-0
Maiden, M. C. J., Jansen van Rensburg, M. J., Bray, J. E., Earle, S. G., Ford, S. A., Jolley, K. A., et al. (2013). MLST revisited: the gene-by-gene approach to bacterial genomics. Nat. Rev. Microbiol. 11, 728–736. doi: 10.1038/nrmicro3093
Mellmann, A., Bielaszewska, M., Köck, R., Friedrich, A. W., Fruth, A., Middendorf, B., et al. (2008). Analysis of collection of hemolytic uremic syndrome-associated enterohemorrhagic Escherichia coli. Emerg. Infect. Dis. 14, 1287–1290. doi: 10.3201/eid1408.071082
Mellmann, A., Harmsen, D., Cummings, C. A., Zentz, E. B., Leopold, S. R., Rico, A., et al. (2011). Prospective genomic characterization of the german enterohemorrhagic Escherichia coli O104:H4 outbreak by rapid next generation sequencing technology. PLoS ONE 6:e22751. doi: 10.1371/journal.pone.0022751
Miko, A., Delannoy, S., Fach, P., Strockbine, N. A., Lindstedt, B. A., Mariani-Kurkdjian, P., et al. (2013). Genotypes and virulence characteristics of Shiga toxin-producing Escherichia coli O104 strains from different origins and sources. Int. J. Med. Microbiol. 303, 410–421. doi: 10.1016/j.ijmm.2013.05.006
Müller, D., Benz, I., Liebchen, A., Gallitz, I., Karch, H., and Schmidt, M. A. (2009). Comparative analysis of the locus of enterocyte effacement and its flanking regions. Infect. Immun. 77, 3501–3513. doi: 10.1128/IAI.00090-09
Pritchard, L., Holden, N. J., Bielaszewska, M., Karch, H., and Toth, I. K. (2012). Alignment-free design of highly discriminatory diagnostic primer sets for Escherichia coli O104:H4 outbreak strains. PLoS ONE 7:e34498. doi: 10.1371/journal.pone.0034498
Rasko, D. A., Webster, D. R., Sahl, J. W., Bashir, A., Boisen, N., Scheutz, F., et al. (2011). Origins of the E. coli strain causing an outbreak of hemolytic-uremic syndrome in Germany. N. Engl. J. Med. 365, 709–717. doi: 10.1056/NEJMoa1106920
Reeves, P. R., Liu, B., Zhou, Z., Li, D., Guo, D., Ren, Y., et al. (2011). Rates of mutation and host transmission for an Escherichia coli clone over 3 years. PLoS ONE 6:e26907. doi: 10.1371/journal.pone.0026907
Reuter, S., Ellington, M. J., Cartwright, E. J. P., Köser, C. U., Török, M. E., Gouliouris, T., et al. (2013). Rapid bacterial whole-genome sequencing to enhance diagnostic and public health microbiology. JAMA Intern. Med. 173, 1397–1404. doi: 10.1001/jamainternmed.2013.7734
Rohde, H., Qin, J., Cui, Y., Li, D., Loman, N. J., Hentschke, M., et al. (2011). E. coli O104:H4 Genome Analysis Crowd-Sourcing Consortium. Open-source genomic analysis of Shiga-toxin-producing E. coli O104:H4. N. Engl. J. Med. 365, 718–724. doi: 10.1056/NEJMoa1107643
Scheutz, F., Nielsen, E. M., Frimodt-Møller, J., Boisen, N., Morabito, S., Tozzoli, R., et al. (2011). Characteristics of the enteroaggregative Shiga toxin/verotoxin-producing Escherichia coli O104:H4 strain causing the outbreak of haemolytic uraemic syndrome in Germany, May to June 2011. Euro Surveill. 16, 19889. PubMed PMID: 21699770
Scheutz, F., Teel, L. D., Beutin, L., Piérard, D., Buvens, G., Karch, H., et al. (2012). Multicenter evaluation of a sequence-based protocol for subtyping Shiga toxins and standardizing Stx nomenclature. J. Clin. Microbiol. 50, 2951–2963. doi: 10.1128/JCM.00860-12
Sherry, N. L., Porter, J. L., Seemann, T., Watkins, A., Stinear, T. P., and Howden, B. P. (2013). Outbreak investigation using high-throughput genome sequencing within a diagnostic microbiology laboratory. J. Clin. Microbiol. 51, 1396–1401. doi: 10.1128/JCM.03332-12
Touchon, M., Hoede, C., Tenaillon, O., Barbe, V., Baeriswyl, S., Bidet, P., et al. (2009). Organised genome dynamics in the Escherichia coli species results in highly diverse adaptive paths. PLoS Genet. 5:e1000344. doi: 10.1371/journal.pgen.1000344
Voets, G. M., Leverstein-Van Hall, M. A., Kolbe-Busch, S., Van Der Zanden, A., Church, D., Kaase, M., et al. (2013). International multicenter evaluation of the diversilab bacterial typing system for Escherichia coli and Klebsiella spp. J. Clin. Microbiol. 51, 3944–3949. doi: 10.1128/JCM.01664-13
Welinder-Olsson, C., and Kaijser, B. (2005). Enterohemorrhagic Escherichia coli (EHEC). Scand. J. Infect. Dis. 37, 405–416. doi: 10.1080/00365540510038523
Zankari, E., Hasman, H., Cosentino, S., Vestergaard, M., Rasmussen, S., Lund, O., et al. (2012). Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 67, 2640–2644. doi: 10.1093/jac/dks261
Keywords: Shiga toxin-producing E. coli –STEC, enterohemorrhagic E. coli –EHEC, hemolytic uremic syndrome, outbreak, antimicrobial resistance, whole genome sequencing, phylogenetic analysis
Citation: Ferdous M, Zhou K, de Boer RF, Friedrich AW, Kooistra-Smid AMD and Rossen JWA (2015) Comprehensive Characterization of Escherichia coli O104:H4 Isolated from Patients in the Netherlands. Front. Microbiol. 6:1348. doi: 10.3389/fmicb.2015.01348
Received: 08 September 2015; Accepted: 16 November 2015;
Published: 02 December 2015.
Edited by:
Yuji Morita, Aichi Gakuin University, JapanReviewed by:
Eelco Franz, Centre for Infectious Disease Control, NetherlandsJames L. Bono, U.S. Meat Animal Research Center, USDA-ARS, USA
Copyright © 2015 Ferdous, Zhou, de Boer, Friedrich, Kooistra-Smid and Rossen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexander W. Friedrich, YWxleC5mcmllZHJpY2hAdW1jZy5ubA==
†These authors have equally contributed to this work