 William D. Leavitt
William D. Leavitt Alexander S. Bradley
Alexander S. Bradley André A. Santos
André A. Santos Inês A. C. Pereira
Inês A. C. Pereira David T. Johnston
David T. Johnston- 1Department of Earth and Planetary Sciences, Harvard University, Cambridge, MA, USA
- 2Department of Earth and Planetary Sciences, Washington University in St. Louis, St. Louis, MO, USA
- 3Bacterial Energy Metabolism Laboratory, Instituto de Tecnologia Química e Biológica António Xavier, Universidade Nova de Lisboa, Oeiras, Portugal
The precise interpretation of environmental sulfur isotope records requires a quantitative understanding of the biochemical controls on sulfur isotope fractionation by the principle isotope-fractionating process within the S cycle, microbial sulfate reduction (MSR). Here we provide the only direct observation of the major (34S/32S) and minor (33S/32S, 36S/32S) sulfur isotope fractionations imparted by a central enzyme in the energy metabolism of sulfate reducers, dissimilatory sulfite reductase (DsrAB). Results from in vitro sulfite reduction experiments allow us to calculate the in vitro DsrAB isotope effect in 34S/32S (hereafter, ) to be 15.3 ± 2‰, 2σ. The accompanying minor isotope effect in 33S, described as , is calculated to be 0.5150 ± 0.0012, 2σ. These observations facilitate a rigorous evaluation of the isotopic fractionation associated with the dissimilatory MSR pathway, as well as of the environmental variables that govern the overall magnitude of fractionation by natural communities of sulfate reducers. The isotope effect induced by DsrAB upon sulfite reduction is a factor of 0.3–0.6 times prior indirect estimates, which have ranged from 25 to 53‰ in 34εDsrAB. The minor isotope fractionation observed from DsrAB is consistent with a kinetic or equilibrium effect. Our in vitro constraints on the magnitude of is similar to the median value of experimental observations compiled from all known published work, where 34εr−p = 16.1‰ (r–p indicates reactant vs. product, n = 648). This value closely matches those of MSR operating at high sulfate reduction rates in both laboratory chemostat experiments ( 17.3 ± 1.5‰, 2σ) and in modern marine sediments ( 17.3 ± 3.8‰). Targeting the direct isotopic consequences of a specific enzymatic processes is a fundamental step toward a biochemical foundation for reinterpreting the biogeochemical and geobiological sulfur isotope records in modern and ancient environments.
Introduction
Microbial sulfate reduction provides a critical link between Earth's surface sulfur, carbon, iron, and oxygen cycles (Thode et al., 1961; Holland, 1973; Garrels and Lerman, 1981; Canfield, 2001a). This metabolism is comprised of a set of enzymes working in concert to reduce sulfate () to sulfide (H2S) (Peck, 1961; Pereira et al., 2011; Figure 1). During this transformation, MSR generates 34S/32S, 33S/32S, 36S/32S, 18O/16O, and 17O/16O stable isotope fractionations (Harrison and Thode, 1958; Kaplan and Rittenberg, 1964; Kemp and Thode, 1968; Chambers et al., 1975; Goldhaber and Kaplan, 1975; Fritz et al., 1989; Canfield, 2001b; Sim et al., 2011b; Leavitt et al., 2013), the biochemical source of which is unclear (Chambers and Trudinger, 1979). To construct a biochemically constrained perspective of sulfur isotope fractionations during MSR requires we quantify how material moves through the metabolic network, and gain an understanding of the isotope effect(s) associated with each constituent enzymatic step (Hayes, 2001).
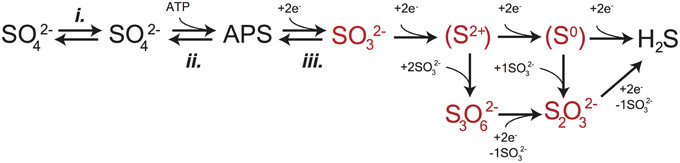
Figure 1. A schematic capturing the central role of DsrAB in MSR. The in vivo dissimilatory sulfate reduction pathway, where red highlighted steps represent sulfite reduction by DsrAB in the absence of DsrC, as targeted here in vitro. The constituent steps of MSR relevant to S isotope fractionation are likely APS reduction to sulfite by APSr, sulfite reduction (the subject of this study) by DsrAB, and the terminal production of sulfide by DsrC/DsrMKJOP. The pathway is described in detail in the text.
The enzyme catalyzed reaction network of MSR is represented in Figure 1. Sulfate is first imported into the cytoplasm by a variety of transporters (Cypionka, 1994; Piłsyk and Paszewski, 2009; Figure 1), and subsequently activated to a high-energy intermediate, adenosine 5′-phosphosulfate (APS). The latter reaction generates pyrophosphate (PPi) at the expense of ATP by the enzyme sulfate-adenylyl transferase (Sat) (Peck, 1962). APS is reduced to sulfite () through a two-electron transfer by the soluble cytoplasmic enzyme APS oxidoreductase (ApsR) (Peck, 1959), which is linked to energy conservation by the membrane-bound complex QmoABC (Pires et al., 2003). APS reduction is highly reversible, depending on the in vivo or in vitro conditions (Peck, 1960). Sulfite has several potential fates. Sulfite can either be re-oxidized to sulfate (directly or via APS) or further reduced to sulfide by DsrAB with the involvement of DsrC (Oliveira et al., 2008b). A critical step is during the reduction of sulfite when it binds the iron of the siroheme in the DsrAB active site. The subsequent reduction occurs via electron transfer from an adjacent Fe-S cluster (Oliveira et al., 2008a,b; Parey et al., 2010). In vivo, DsrAB has been proposed to generate intermediate valence sulfur, which is then bound to DsrC and converted to sulfide via DsrC/MK (Oliveira et al., 2008b; Venceslau et al., 2013, 2014; Santos et al., 2015). Sulfide then leaves the cell by diffusion (as H2S) (Mathai et al., 2009) or through anion transport (as HS− or S2−) (Czyzewski and Wang, 2012), though the mechanism of sulfide exit in MSR is still not well known. In instances when DsrC is unavailable (e.g., when DsrAB is pure in vitro) or limiting (e.g., intracellular sulfite is in excess of reduced DsrC), intermediates such as thiosulfate () may become important, likely due to the reaction of sulfite with sulfide (in vivo) or the partially reduced sulfur from DsrAB (in vitro) (Chambers and Trudinger, 1975; Drake and Akagi, 1976, 1977, 1978; Kim and Akagi, 1985). A few examples exist where thiosulfate is a key component in closing S mass balance during in vivo MSR (Sass et al., 1992; Leavitt et al., 2014; Price et al., 2014), and in one instance trithionate is observed () (Sass et al., 1992), though it is not clear in this case it is a physiological product. Under these conditions, accumulation and excretion of such compounds as thiosulfate may be important (Bradley et al., 2011). It is within this broader biochemical and physiological context that we examine the isotopic consequences of sulfite reduction by DsrAB, which as outlined above, is central to the biochemistry of dissimilatory sulfate reduction.
Reduction of sulfite by DsrAB breaks three of the four S-O bonds in the original sulfate (Venceslau et al., 2014). As such, the isotope effect of DsrAB likely plays a significant role in setting the overall fractionations observed from MSR (Harrison and Thode, 1958; Rees, 1973; Farquhar et al., 2003; Brunner and Bernasconi, 2005). Measured enzyme-specific isotope effects are lacking for MSR and the S cycle in general. Such information has been transformative for the study of other biogeochemical elements like carbon. For example, experimental work quantifying the 13C/12C effect of RuBisCO (Park and Epstein, 1960; Farquhar et al., 1982; Tcherkez et al., 2006), the core enzyme in carbon fixation, has greatly advanced the applicability of carbon isotope biogeochemistry. More specifically, understanding the fractionation associated with RuBisCO allowed greater insight into modern (Hayes, 1993) and ancient (Hayes et al., 1999) carbon cycling, and facilitated a better understanding of primary productivity in the both modern (Laws et al., 1995) and ancient (Pagani et al., 2009) oceans. Similar enzyme-specific approaches have also proven greatly informative in studies of methanogenesis (Scheller et al., 2013), nitrate assimilation (Karsh et al., 2012) and nitrogen fixation (Sra et al., 2004). With these studies as a guide, we look to further unlock the sulfur cycle through targeting a key microbial sulfate reduction enzyme, DsrAB.
To close the knowledge gap between whole-cell observations and enzyme-catalyzed reactions, as well as to turn natural isotope records into catalogs of environmental information, we conduct the first enzyme-specific sulfur isotope experiments. Here we report the sulfur isotope fractionation factors associated with in vitro sulfite reduction by the dissimilatory sulfite reductase enzyme (DsrAB). Using these results and a new multi-component isotope distillation model, we are able to place improved constraints on the root of sulfur isotope fractionation during MSR. This refines our understanding of the predominant biological process responsible for generating environmental S isotope records throughout geological history.
Experimental Methods Summary
We conducted a series of closed system in vitro sulfite reduction experiments with purified DsrAB from Desulfovibrio vulgaris str. Hildenborough (DSM 644) and Archaeoglobus fulgidus. These enzymes are structurally similar and evolutionarily related (Parey et al., 2013), and we chose them to attempt to determine conservation of isotope fractionation in D. vulgaris and A. fulgidus DsrAB. The complete isolation and purification details are available in the Supplementary Material.
DsrAB experiments were conducted in vitro under strictly anoxic conditions with H2, [NiFe] hydrogenase, and methyl viologen as the electron donation system. Key considerations in experimental design are: (i) to provide enough sulfur at each time point for isotopic characterization of residual reactant and products; (ii) to provide the proper reaction conditions to allow for optimal DsrAB activity (pH = 7.1, T = 20° or 31°C for D. vulgaris and 65°C for A. fulgidus); (iii) to ensure hydrogenase activity is not inhibited by the experimental pH (optimum at pH 7.5, activity significantly depleted below pH 6.5, so we chose pH 7.1, to account for optima of both DsrAB and [NiFe]-hydrogenase); and finally (iv) to ensure the sulfite to hydrogen ratio strongly favors sulfite reduction. Experiments setup is detailed in the Supplementary Material.
Each experiment was performed in duplicate and sampled as sulfite was consumed (reaction progress tracked as fSO3, equivalent to the fraction of remaining sulfite). The reaction consumed sulfite to form products thiosulfate and trithionate, with no detectable sulfide. Thiosulfate and trithionate concentrations were quantified following published cyanolysis protocols (Kelly and Wood, 1994), where we used a modified “Fuschin” method (Grant, 1947) to quantify sulfite and a modified Cline method (Cline, 1969) to measure sulfide. All quantification and experimental methods are fully detailed in the Supplementary Material. In addition to concentrations, we measured the major and minor sulfur isotopic compositions of three operationally defined and precipitated pools: sulfite (both initial and residual reactant), product sulfonate (from trithionate or thiosulfate) and the “reduced sulfur” products (central and terminal sulfurs in trithionate and thiosulfate, respectively). Complete IUPAC definitions of each S reservoir, along with all isotopic measurement methods and error propagation calculations are fully articulated in the Supplementary Material.
Isotope Notation
The variability in 34S of a measured pool is reported in standard delta notation (for instance δ34S, in ‰ units), where 34S/32S of the sample is the relative difference from a standard (Hayes, 1983), and is reported as the isotopic offset between two measured pools of sulfur, 34ε (=103x(34α-1)), still in ‰ units. Fractionation factors (α's and associated ε's) are annotated with a subscript to denote the process of interest or pools being related, such as , , 34εr−p, or 34εSO4−H2S. The same nomenclature convention is followed when a minor isotope, 33S, is included. The only exception is the addition of one new term, 33λ, which is approximately the slope of a line on a plot of δ33S vs. δ34S (Miller, 2002; Farquhar et al., 2003), but can be simply interpreted as a measure of mass-dependent minor isotope fractionation. Mathematical definitions are provided below.
Fractionation Modeling
Calculation of the isotopic fractionation imposed by the reduction of sulfite through DsrAB requires tracking the concentration of the reactant, accumulation of the products, and determining the isotopic composition of all as the reaction progressed. This necessitates the application of a closed-system model in order to calculate fractionation factors. Determining the intrinsic isotope effect associated with a closed system reaction can be approached in a number of ways. Normally, in a system where one reactant is consumed in order to generate a single product, a Rayleigh model is employed (Nakai and Jensen, 1964; Mariotti et al., 1981). This approach assumes that the reaction of interest is unidirectional, generates only one product, and that the fractionation factor is invariant throughout the reaction. In this case, the isotope effect is calculated as a function of the isotope ratio, R, of the starting composition (Rao) and evolving product pool (Rp, defined below), equal to the mass balance on sulfite:
In this solution, f tracks the fractional amount of reactant remaining (). For our experiments, we define fSO3:
In the specific case of our experiments and the reduction of sulfite by DsrAB, however, the standard closed-system isotope distillation models (Equation 1) requires expansion. Recall that the in vitro reaction involves the accumulation of two products (trithionate and thiosulfate). Each of these products further contains sulfur moieties in more than one oxidation state (Suh and Akagi, 1969; Kobayashi et al., 1974; Drake and Akagi, 1976, 1977, 1978). This means that, rather than Rp being the isotope composition of a single product pool, we define it as the mass-weighted sum of the oxidized (Rox) and reduced (Rred) products in trithionate and thiosulfate:
Here the reduced and oxidized pools are the operationally defined reservoirs discussed above. In the mass balance accounting equation we introduce a term to quantify the ratio of products, j (Figure 2). The concentrations of sulfite, trithionate, and thiosulfate were measured at each time-point, ensuring the closure of mass balance and validating the use of a relative mass term. The j term is thus the fraction of products residing in thiosulfate:
For isotopic measurements we quantitatively separated the oxidized moieties from trithionate and thiosulfate from the partially reduced moieties of both products. There were no available methods to separate trithionate and thiosulfate and isolate each S site within those products (a target for future work). We then measured the isotopic compositions of the pooled oxidized and pooled reduced products. As the goal is to identify the fractionation between the residual sulfite and either the oxidized (3xαox) or reduced (3xαred) moieties in trithionate and thiosulfate, we present the general equation, (3xαz):
where z is either ox or red. This solution is then translated into standard 3xε notation. Fractionation factors are then related in triple isotope space with:
a term which finds common application in mass-dependent studies (Young et al., 2002; Farquhar et al., 2003). Finally, we note the models assumptions: (1) sulfite and its isomers carry the same isotopic composition as each other, (2) the isotopic composition of the sulfonate groups (in trithionate and thiosulfate) are isotopically identical, and (3) similar to 2, the isotopic composition of reduced sulfur in trithionate and thiosulfate are isotopically identical.
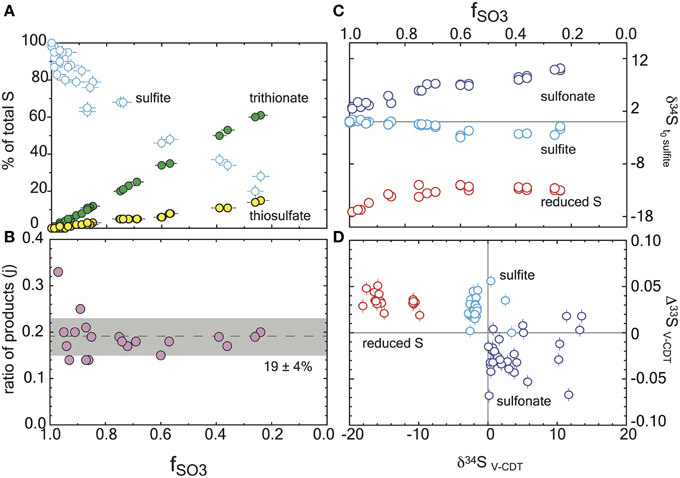
Figure 2. Reaction progress during sulfite reduction with D. vulgaris DsrAB in vitro. Errors are included in all measurements (2σ) and are smaller than the symbol if not seen. (A) The mol fraction of sulfur in each sulfur product pool as a function of reaction progress, fSO3. Mass balance conservation is discussed in the text. (B) The ratio of products at each time point, demonstrating the constancy of the reaction scheme (denoted as j in the model). This is the ratio of the slopes of the products from (A). (C) Major isotope data for each operationally defined sulfur pool as a function of reaction progress, and normalized to the initial sulfite composition. (D) A triple isotope cross plot of the data presented in frame C, normalized to V-CDT.
The complexity added above in equation 6 allows for numerous products for a given reaction, but still assumes that the fractionation factors involved are static over the time series of the experiment (fSO3) and that there is only one reaction present. If this is true, then the model prediction will match the observation over all values of fSO3. Although, we observed a statistically invariant ratio of thiosulfate to trithionate production throughout the reaction (j in Figure 2), suggesting a static set of reactions through the entire experiment, it appears that the net fractionation factor was indeed time-dependent. In the event of an evolving α, the fractionation factor early in the experiment, where the concentration of products remains low, most closely approximates the isotope effect of DsrAB solely reducing sulfite. We explore this time-dependence further in the Supplementary Material for all sampled points on the reaction progress coordinate (fSO3). Thus, for extracting the intrinsic isotope effect associated with enzymatic reduction of sulfite, we focused on data where fSO3 > 0.85. To do so, we have used our modified Rayleigh-type isotope distillation model in which we account for the production of reduced and oxidized sulfur within aqueous products trithionate and thiosulfate. Procedures for error propagation associated with these calculations are described in the Supplementary Material.
Results
We tracked all S pools at each time point. Mass balance was satisfied within ±10% of the initially provided sulfite in every experiment, and within ±5% in 27 of 33 experiments (Figure 2A). The majority of this variance is due to analytical error in the sulfite quantifications. In experiments with the D. vulgaris DsrAB, the products were generated with a mean of 19% of the product sulfur forming thiosulfate and the remainder accumulating as trithionate (Figure 2B). This is consistent with previous reports (Drake and Akagi, 1976, 1978), and expected given the absence of active DsrC in these experiments. Some inactive DsrC does accompany the D. vulgaris DsrAB during isolation and purification (Oliveira et al., 2008a,b, 2011), however there is no means to recycle this component, and as such, it is not a functional part of the experiment. Therefore, the in vitro sulfite reduction reactions produce thionates rather than sulfide. In our experiments, sulfite was always in excess of H2, and therefore was not limiting at any point.
To extend our studies to a different taxonomic form of the enzyme, we also experimented on the DsrAB from the thermophilic archaeon A. fulgidus. This enzyme operates at higher temperature and lacks DsrC in the complex (Schiffer et al., 2008). A. fulgidus experiments showed consistent loss of sulfite and accumulation of products between replicates at each time point. Unlike D. vulgaris DsrAB experiments, however, only small quantities of product were generated. From these experiments we were able to resolve a complete sample set (i.e., sulfite, sulfonate, and reduced S) from one time-point and partial sets from another (i.e., sulfite and sulfonate). Special efforts were made to correct data available on A. fulgidus experiments (see the Supplementary Material). The results using DsrAB from A. fulgidus are consistent with those of D. vulgaris, but with a large calculated uncertainty. We therefore focus our interpretations on the results from the D. vulgaris experiments.
We use the concentrations of sulfite, trithionate, and thiosulfate, as well as the isotopic compositions of each operationally defined product to solve for the fractionations associated with DsrAB. The calculated 34εDsrAB for sulfite reduction by the D. vulgaris DsrAB is 15.3 ± 2.0‰ (2σ, Figure 3), where the concurrent fractionation associated with the generation of the sulfonate is −3.2 ± 0.8‰ (2σ). The 34εDsrAB from A. fulgidus is generally consistent with the D. vulgaris experiments, yielding a reductive fractionation of 16‰ (2σ from 22 to 12‰) at 65°C. Large and asymmetric errors on the A. fulgidus data are the result of exceptionally small sample sizes, which also precluded the collection of 33S data (see the Supplementary Material). Together, these experiments demonstrate a broad consistency in fractionation by DsrAB over a wide range of temperatures (20 and 30°C for D. vulgaris, and 65°C for A. fulgidis) and across two Domains of life.
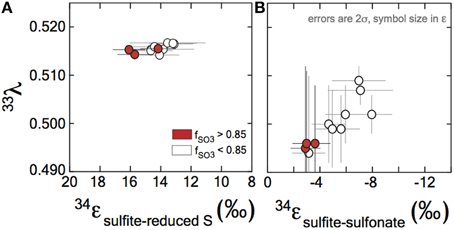
Figure 3. Sulfur isotope fractionation during sulfite reduction by DsrAB. (A) The fractionation between sulfite and reduced sulfur (within trithionate and thiosulfate) facilitated by in vivo DsrAB. Initial values of 34εDsrAB when fSO3 is >0.85 are near 15‰. A small amount of variability accompanies changes in fSO3 to < 0.85. Data from A. fulgidus overlap D. vulgaris DsrAB data (see Figure A2 in the Supplementary Material). The minor sulfur isotope fractionation, 33λDsrAB, is stable near 0.515. (B) Fractionation factors for sulfonate generation are much smaller in 34ε and carry the opposite sign. (A,B) Both the 34ε and 33λ evolve over the time course of the experiment, only after f SO3 < 0.85. Data and application to calculations are further discussed in the text.
Fractionation of 33S between sulfite and reduced sulfur by D. vulgaris DsrAB is reported as 33λDsrAB, with a calculated result of 0.5150 ± 0.0012 (2σ) over the initial range of fSO3. The conversion of sulfite to sulfonate yielded a calculated 33λDsrAB that changes as the reaction progressed, from 0.495 ± 0.017 (2σ) at fSO3 > 0.85, toward 0.510 at fSO3 < 0.85. The experimental error on 33λDsrAB is inversely related to the magnitude of 33εDsrAB (Johnston et al., 2007), thus is larger for sulfonate generation. We interpret the observed fractionation factors between sulfite and reduced S as representing the binding and reduction of sulfite by DsrAB. The fractionation associated with sulfonate production is more difficult to uniquely diagnose given the wide array of potential biotic and abiotic reactions.
Discussion
Microbial sulfate reduction is a major process in the sulfur cycle and generates characteristic isotopic fractionations. These fractionations are critical in tracing the movement of sulfur within natural settings (marine and lacustrine). Determining the isotope effects associated with key enzymes in this pathway is critical to disentangling biological and physical controls on the distribution of sulfur isotopes among environmental pools of sulfur. In this study we provide the first constraints (34ε and 33λ) on the isotope effects associated with one such enzyme: DsrAB, the central redox enzyme in dissimilatory sulfate reduction. This experimental constraints generated herein provides insight and critical boundary conditions for understanding sulfur isotope fractionation by sulfate reducers. Fortunately, half a century of research on sulfur isotope fractionation by MSR in vivo puts in place a series of useful observations that help to guide our interpretation as to the role of DsrAB. This in turn allows significantly greater access to the information locked in sulfur isotope records.
Fractionation at the Cellular Scale
The rich literature of whole cell isotope fractionation data associated with MSR lacks information about kinetic isotope effects associated with specific enzymes within the metabolism. Cellular-level observations include secular and spatial trends in sulfur isotope records attributed to changes in the environmental conditions at the site of MSR, and degree to which biogenic sulfide is preserved in marine sediments (Holland, 1973; Canfield and Farquhar, 2009; Leavitt et al., 2013). The environmental variables most commonly invoked to explain isotopic variability are aqueous sulfate and organic carbon concentrations (Goldhaber and Kaplan, 1975; Habicht et al., 2002; Bradley et al., 2015). Both of these variables ultimately contribute to the net reduction rate and carry independent biological thresholds, one of which ultimately becoming rate-limiting (Bradley et al., 2011, 2015). More specifically, variability in these substrates is manifested as changes in the cell-specific rates of MSR in both the laboratory and natural environment (Chambers et al., 1975; Goldhaber and Kaplan, 1975; Leavitt et al., 2013). In laboratory experiments and natural marine and lacustrine systems, volumetric sulfate reduction rates scale primarily as a function of the availability of sulfate relative to common electron donors like organic carbon (Chambers et al., 1975; Goldhaber and Kaplan, 1975; Sim et al., 2011b; Leavitt et al., 2013). Indeed, sulfate can be non-limiting even in environments with as little as μM sulfate (Nakagawa et al., 2012; Gomes and Hurtgen, 2013; Crowe et al., 2014; Bradley et al., 2015), assuming organic matter is more limiting to allow a fractionation to occur (Wing and Halevy, 2014; Bradley et al., 2015). Constrained whole cell (in vivo) laboratory experiments demonstrate that when electron donors are limiting, the magnitude of fractionation between sulfate and sulfide (34ε) carries a nonlinear inverse relationship with cell-specific sulfate reduction rates (Harrison and Thode, 1958; Kaplan and Rittenberg, 1964; Chambers et al., 1975; Sim et al., 2011b; Leavitt et al., 2013). Thus, the range of isotopic compositions produced and preserved in natural environments are interpreted as an output of intracellular rates, which scales with enzyme activity associated with microbial sulfate reduction (Goldhaber and Kaplan, 1975; Leavitt et al., 2013).
In addition to following a rate relationship, fractionation in MSR isotope studies often approaches characteristic upper and lower fractionation limits. Recent experimental work at low sulfate reduction rates captures a 34εMSR (the net isotope effect of microbial sulfate reduction) of nearly 70‰ (Canfield et al., 2010; Sim et al., 2011a). This magnitude of fractionation approaches the theoretical low temperature equilibrium prediction of 71.3 to 67.7‰ between 20° and 30°C (Tudge and Thode, 1950; Farquhar et al., 2003), inspiring research more directly comparing the biologically catalyzed reversibility of MSR enzymes and that of equilibrium (Wing and Halevy, 2014; see also Rees, 1973; Farquhar et al., 2003, 2008; Brunner and Bernasconi, 2005; Johnston et al., 2007; Mangalo et al., 2008; Bradley et al., 2011, 2015; Holler et al., 2011). These studies are fueled by the knowledge that direct (abiotic) equilibration between sulfate and sulfide at Earth surface temperatures is exceedingly slow, with a half-life of exchange estimated at 1.1 × 1010 (at 30°C) to 1.6 × 1012 years (at 20°C; these values are extrapolated from Ames and Willard, 1951). Thus, large fractionations between sulfate and sulfide at Earth surface conditions strongly suggests a role for biology, and likely multiple enzyme catalyzed steps with associated fractionations.
Most experiments with sulfate reducing microorganisms result in isotope fractionations much smaller than would be predicted from abiotic equilibrium estimates. Indeed, more than half a century of research and 648 observations from in vivo MSR experiments capture a median isotope fractionation of 34εMSR = 16.1‰ (in both sulfate and sulfite reduction experiments: Figure 4). In fact, half of experimental data fall between 10 and 22.5‰. This is consistent with the phenomenology of laboratory experiments being conducted at significantly higher sulfate reduction rates than occur in most natural settings. However, given that all these experiments occurred with the same biochemical network, any enzyme-level explanation for the range of fractionations observed at both high and low sulfate reduction rates must be internally consistent.
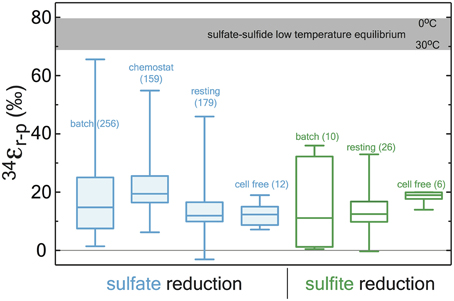
Figure 4. A box and whisker plot of previously published sulfate (blue) and sulfite (green) reduction experiments. All data is binned by experimental approach. The whiskers reflect the entire range of the data, with the boxes reflecting the middle 50% of the data. The median of the data is represented by the bar dividing the box. The bar running across the top is a temperature dependent prediction based on low temperature thermodynamic equilibrium (Farquhar et al., 2003). The statistical method and output are detailed in the Supplementary Material along with the compiled data (http://dx.doi.org/10.6084/m9.figshare.1436115), where all compiled values are from the following sources: (Thode et al., 1951; Ford, 1957; Harrison and Thode, 1957, 1958; Jones and Starkey, 1957; Kaplan and Rittenberg, 1964; Kemp and Thode, 1968; Krouse et al., 1968; Chambers et al., 1975; McCready, 1975; McCready et al., 1975; Smock et al., 1998; Bottcher et al., 1999; Bolliger et al., 2001; Detmers et al., 2001; Farquhar et al., 2003; Kleikemper et al., 2004; Habicht et al., 2005; Johnston, 2005; Canfield, 2006; Hoek et al., 2006; Knöller et al., 2006; Johnston et al., 2007; Mangalo et al., 2007, 2008; Pallud et al., 2007; Davidson et al., 2009; Sim et al., 2011a,b, 2012, 2013; Leavitt et al., 2013, 2014).
Fractionation at the Intracellular Scale
As described above, a suite of enzymes and cofactors drives dissimilatory sulfate reduction. During the reduction of sulfate to sulfide, sulfur isotope effects are likely to result primarily from transformations that involve the making or breaking S related bonds. Initial steps in sulfate reduction, such as transport into the cell and activation via a reaction with ATP to generate APS (Figure 1; Fritz, 2002), do not involve the formation of new S linkages, and are not predicted to be associated with primary isotope effects. Influence on the expressed isotopic fractionation due to transport limitation is, however, conceivable. That is, the concentration of sulfate in the cell may influence the expression of downstream isotope effects, altering the net observed 34εMSR. Sulfate transporters may also induce an isotope effect associated with varying membrane fluidity or other strain-specific optima, in response to changing temperature (Kaplan and Rittenberg, 1964; Canfield, 2006), pH (Furusaka, 1961), or as environmental sulfate concentrations become metabolically limiting (Habicht et al., 2005; see discussion in Bradley et al., 2015).
Primary isotope effects are predicted where bonds are made or broken. APS reductase catalyzes a two-electron exchange that breaks a S-O bond during reduction of APS to generate free sulfite. From the crystal structure of ApsR (Fritz, 2002), it is apparent that the enzyme binds with the APS bound sulfur directly on a nitrogen in the FAD (flavin adenine dinucloetide) cofactor. The product sulfite is then available to interact with DsrAB. This heterodimeric enzyme binds sulfite in an active site containing siroheme. The formation of the Fe-S bond between siroheme and sulfite may be the critical reaction controlling isotope fractionation. Following this, sulfite is reduced by the transfer of two electrons to form a S2+ intermediate (Santos et al., 2015). Under in vivo conditions, the sulfur intermediate was suggested to be withdrawn from the DsrAB complex by the small transfer protein DsrC (Oliveira et al., 2008b; Venceslau et al., 2014), and this has been recently demonstrated (Santos et al., 2015). Under in vitro conditions, DsrC is generally absent, and the reduced sulfur in the active site may react with excess sulfite, forming thiosulfate and trithionate (Figure 1; Drake and Akagi, 1976). DsrC is independently regulated in vivo (Karkhoff-Schweizer et al., 1993), and generates the terminal sulfide from DsrAB bound sulfur derived from sulfite (Venceslau et al., 2014). The relative importance of this protein has only been realized in the last few years (Oliveira et al., 2008b; Venceslau et al., 2014), and has an unconstrained isotope effect.
In general, the magnitude of the thermodynamically predicted sulfur isotope effect scales positively with the number of bonds are made or broken (Tudge and Thode, 1950; Bigeleisen and Wolfsberg, 1958). As described above, sulfite reduction by DsrAB is a central enzyme in MSR, breaking three S-O bonds (Oliveira et al., 2008b; Venceslau et al., 2014), and therefore knowing the fractionation associated with this step is critical to any predictive MSR isotope model (c.f. Rees, 1973; Brunner and Bernasconi, 2005; Farquhar et al., 2007, 2008; Johnston et al., 2007; Bradley et al., 2011, 2015; Wing and Halevy, 2014). Our direct constraint on the fractionation imposed by sulfite reduction indicates that the published assignments of 25‰ (Harrison and Thode, 1958; Rees, 1973) and 53‰ (Brunner and Bernasconi, 2005) for DsrAB are significant over-estimates. It is perhaps not surprising, given that previous appraisals were generated through various indirect approaches (Harrison and Thode, 1958; Rees, 1973; Farquhar et al., 2003; Brunner and Bernasconi, 2005; Johnston et al., 2007). As previously stated (Chambers and Trudinger, 1979), the lack of direct experimental constraints on enzyme-specific fractionation factors leave significant uncertainty in metabolic, geochemical, and geological models that simply assign values to each enzymatic step.
Enzymatic Constrains (This Study)
Our measured 34εDsrAB value for sulfite reduction (15.3 ± 2.0‰) is large enough to account for a large portion of the fractionations observed in published whole-cell MSR experiments over the last 65 years (median of 16.1‰, n = 648; Figure 5). As noted previously, laboratory experiments carry a strong bias toward higher rates of sulfate reduction, and as such, the data compilation should be viewed in this light. As most recently articulated through a series of chemostat experiments (Sim et al., 2011a; Leavitt et al., 2013), the consequence of elevated metabolic rate is a smaller relative 34ε. In isotope biogeochemistry, relationships like this often depend on the single slowest overall rate-limiting step within a metabolism (Mariotti et al., 1981; Hayes, 1993). The fractionation limit at high metabolic rates in cultures (34ε = 17.3 ± 1.3‰), marine sediments (34ε = 17.3 ± 3.8‰) and DsrAB are statistically indistinguishable (Figure 5). This similarity is consistent with DsrAB as a rate-limiting step explaining the majority of observed fractionation (Figure 5). However, this interpretation omits complexity associated with the metabolic network.
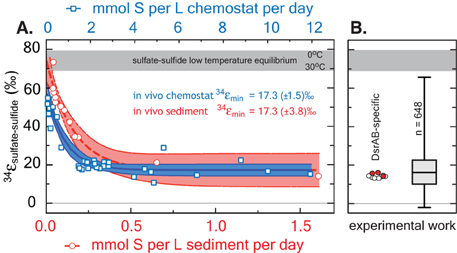
Figure 5. A comparison between modern marine (environmental) and laboratory (experimental) S isotope fractionations, as a function of sulfate reduction rate. These data are further referenced to a statistical distribution of published experimental fractionation data. (A) Fractionation as a function of volumetric sulfate reduction rate from axenic continuous culture experiments (blue squares and regression; Leavitt et al., 2013) and modern marine sediments (red circles and regression; Goldhaber and Kaplan, 1975). Solid lines are mean values with shaded regions representing the 95% confidence interval around a non-linear regression. While the upper fractionation limits are offset, perhaps due to differences in biomass per volume of sediment vs. volume of chemostat, the limits approached at high reduction rates are statistically indistinguishable at 17.3‰. (B) The fractionation associated with DsrAB experiments, color-coded as in Figure 3 and on the same isotope scale. Also included is a box-whisker treatment of all measured 34ε (n = 648) sulfate and sulfite reduction experiments compiled in Figure 4. Here, the median value is 16.1‰, also statistically indistinguishable from chemostat and modern marine sediments limits at elevated rates of sulfate reduction. Included for reference is the theoretical sulfate-sulfide equilibrium fractionation (gray bar) for 0–30°C (Farquhar et al., 2003).
This raises an essential question: how does the DsrAB constraint change our understanding of the possible range of fractionations imposed by MSR? For instance, if we assume that DsrAB is the slowest reaction in MSR, then the metabolic steps preceding sulfite () will necessarily approach equilibrium (Wing and Halevy, 2014). The thermodynamic predictions would then require an accompanying fractionation approaching 25‰ between sulfate and sulfite at biologically relevant temperatures (the equilibrium fractionation estimate; Farquhar et al., 2003). At its simplest, this effect would be additive with that of DsrAB (25 + 15.3‰, or 40.3‰), which encompasses a majority of experimental MSR isotopic fractionations. However, the lower plateau in fractionation approached at high rates, stemming from the calculations using both modern marine sediment and chemostat data (see the Supplementary Material)—is less than half of this magnitude. That is, the lower plateaus for in vivo fractionation at high rates of 17.3‰ allows only a ~2‰ fractionation partitioned among these upstream steps, if in fact DsrAB is fully expressed. This smaller “upstream” kinetic fractionation is, however, consistent with the few loose estimates from crude cell extracts and resting cell studies (i.e., not purified enzymes), which putatively suggests a 34ε of 4–15‰ for the cumulative activation of sulfate to APS and reduction to sulfite (Ford, 1957; Kemp and Thode, 1968). This may suggest that the sulfate-sulfite conversion (in vivo and in vitro) reflects a predominately kinetic (rather than equilibrium) control. This requires, however, we loosen the degree to which DsrAB is called upon to fully control the net MSR isotopic fractionation, invoking some delicate balance between upstream reactions and that of DsrAB, but maintaining the model where enzyme kinetics (especially DsrAB) will win out over equilibrium effects as sulfate reduction rates move from low to high.
An alternate explanation is that APS reductase (ApsR) is the rate-limiting step under sulfate replete conditions (Rees, 1973). If this is the case, fractionation imposed by DsrAB is unexpressed, as it is downstream of ApsR (Rees, 1973; Hayes, 2001). If fractionation associated with APS reductase is near 17‰, it could alone account for most of the observed fractionation observed at high sulfate reduction rates. The fractionation imposed by ApsR has not been directly measured, though can be preliminarily estimated from the discussion above. However, evidence against ApsR as the rate-limiting step is shown by studies indicating reversibility of the ApsR (Peck, 1960) and the sulfate reduction pathway (Chambers and Trudinger, 1975; Holler et al., 2011). Recent studies using oxygen isotopes as tracers have demonstrated that some intracellular sulfite is oxidized in vivo back to sulfate (Mangalo et al., 2007, 2008; Einsiedl, 2008; Farquhar et al., 2008; Turchyn et al., 2010). These studies demonstrate that sulfite re-oxidation is commonplace in MSR and often quantitatively significant (Antler et al., 2013). This reoxidation is inconsistent with ApsR being rate-limiting under the range conditions tested.
Any explanation for the net MSR isotopic fractionation must also account for the large fractionations observed at low sulfate reduction rates. These large fractionations are common in nature, and require another type of mechanism. These isotopic fractionations approach but do not reach the theoretical equilibrium values for sulfur isotope exchange between sulfate and sulfide (Figures 4, 5; Tudge and Thode, 1950; Farquhar et al., 2003; Johnston et al., 2007). In this context, further analysis to understand intracellular thermodynamics is critical (the redox pairs responsible for various reactions, see Wing and Halevy, 2014), along with measurements of the intrinsic isotope effects of other key enzymes in the metabolic network, including ApsR and DsrC. In that sense, this study represents a key first step.
In parallel to examining the 34ε effects, measuring minor S isotope (33S/32S) fractionation provides additional information about the class of reaction mechanism associated with in vitro DsrAB activity. In our experiments, the conversion of sulfite to sulfonate carries a 33λ of from ~0.496 (±0.012, 2σ), evolving toward 0.510 as the reaction proceeds. Sulfite reduction via DsrAB has an invariant 33λ of 0.5150 (±0.0012, 2σ). In cases where 33λ is 0.515, a purely equilibrium fractionation is often inferred but not required, while values less than this require kinetic effects (Young et al., 2002; Farquhar et al., 2003). In this framework, in vitro sulfonate formation falls under kinetic control, while formation of reduced S could be interpreted as a kinetic or equilibrium reaction. Thus, specific predictions for the DsrAB enzyme require more detailed modeling of the structure and function of the DsrAB enzymatic active site. This level of analysis—where inroads joining empirical work with theory are constructed—is present in analogous systems (Karsh et al., 2012), but absent within the S cycle. Nonetheless, the work here provides the only triple-isotope constraints on enzyme-specific fractionation factors in both MSR and the global biogeochemical S cycle. Further, this approach may prove useful in other enzymatic systems where elements with ≥3 stable isotopes are involved (e.g., O, Fe, Ca, Mg, Se, Zn, and Mo).
Conclusions
Direct constraints on enzymatic isotope effects, when placed in context of laboratory and field observations, represent a key step toward improving our understanding of how environmental factors come to control biochemical sulfur isotope fractionations in nature. Experimental results indicate that the kinetic isotope effect generated by dissimilatory sulfite reductase, the enzymatic core of MSR, generates less than a quarter of the maximum fractionation observed in sulfate reduction experiments and modern marine sediments. However, the 34εDsrAB aligns nicely with the vast majority of experimental data generated over the last 65 years, as well as chemostat and marine sediment studies sampling high rates of sulfate reduction. The consistency between these published fractionations and the DsrAB isotope effect suggests a fundamental role of this enzyme in setting sulfur isotope compositions. This work highlights the need for further consideration of the allied enzymes in MSR and the likelihood of abiological (and/or equilibrium) effects as microbial sulfate reduction rates slow. Though questions remain, placing quantitative constraints on a core component of sulfate reduction—DsrAB—represent a fundamentally new direction in exploring experimental and environmental sulfur isotope records today and throughout Earth history.
Detailed Methods
Operational Definitions of S Moieties
In this study we measured the concentrations of three pools: sulfite, trithionate, and thiosulfate; hydrogen sulfide was not detected. We measured the major and minor sulfur isotopic compositions of three operationally defined pools: “reactant” sulfite (initial and residual), product “sulfonate,” and “reduced product” S. What we refer to as the pooled product “sulfonate” sulfurs are known in inorganic chemistry as sulfuryl groups (O2S-X2), where one of the X's represents an O−/OH and the other a S in oxidation state 0 (trithionate) or −1 (thiosulfate), meaning the outer sulfuryl-S's are in approximately oxidation state +5 (thiosulfate) or +4 (trithionate), with initial and residual reactant sulfite sulfur in the standard +4. The sulfonate S differs from sulfite S in that it is bound to either an approximately −1 valent sulfur in thiosulfate (S-S(O)32−) (Vairavamurthy et al., 1993) or as two sulfonates each bound to one sulfur of valence approximately 2+, in trithionate ((O3S-S-SO3)2−). In this study we refer to the 0 and −1 oxidation state sulfurs from trithionate and thiosulfate, respectively, as the “reduced product” S pool. They are grouped by our operational extraction (see below). For explicit definitions and nomenclature refer to the “IUPAC Goldbook” (McNaught and Wilkinson, 1997).
Enzyme Purification and In vitro Experiments
DsrAB Isolation and Purification
DsrAB was purified from Desulfovibrio vulgaris Hildenborough (DSM 644) and Archaeoglobus fulgidus cells grown in a 30 or 300 L batch culture in a modified lactate/sulfate medium (Oliveira et al., 2008a) at iBET (Instituto de Biologia Experimental Tecnológica; www.ibet.pt), grown at 37 or 80°C, respectively. The soluble cell fraction was obtained as previously described (Le Gall et al., 1994; Oliveira et al., 2008a). All purification procedures were performed under atmosphere at 4°C using an AKTA FPLC (Amersham Biotech Pharmacia) with two buffers, (A) 20 mM TrisHCl and (B) 50 mM TrisHCl with 1 M of NaCl (both pH 7.6 and containing 10% glycerol). Buffer (A) was used to equilibrate the columns and buffer (B) to generate the ionic strength gradient. The soluble cell fraction was loaded into a Q-Sepharose fast-flow (XK50/30) column, and a stepwise salt gradient applied, with the DsrAB-containing fraction eluting at 300 mM NaCl. The characteristic DsrAB (“desulfoviridin”) absorption peak at 630 nm was used to track the protein, as previously described (Wolfe et al., 1994; Marritt and Hagen, 1996). DsrAB-containing fractions were then loaded into a Q-Sepharose fast-flow (26/10) column and eluted in 250 mM NaCl. To verify enzyme purity, the final DsrAB-containing sample was analyzed by 12% SDS-PAGE gel electrophoresis. DsrC is present in the DsrAB preparation from D. vulgaris, but remains functionally inactive during in vitro assays as previously described (Oliveira et al., 2008b), and also due to the lack of DsrMKJOP (Venceslau et al., 2014). Thus, we refer only to the “DsrAB” fraction in the D. vulgaris experiments. In the A. fulgidus experiments, DsrAB is free of any DsrC. Protein was quantified by the method of Bradford (Bradford, 1976). The Desulfovirio gigas [NiFe] hydrogenase used in all assays was purified as described previously (Romão et al., 1997).
To ensure the activity of purified DsrAB was not strongly influenced by the high initial concentration of sulfite used in the fractionation experiments (10 or 15 mM), we performed small-volume kinetic assays under the same conditions as for isotope measurements. Sulfite alone was measured by HPLC on monobromobimane (MBBr) derivatized samples (Newton et al., 1981). Once the sulfite concentrations for each initial and final (0 and 2 h) time points were sampled, derivatized, measured and calculated, we applied a non-linear regression formulated from the standard Michaelis-Menten equation, solving for the Vmax and Km.
D. vulgaris DsrAB In vitro Fractionation Experiments in Detail
To determine the DsrAB-specific S isotope fractionation factors we designed and executed a series of batch (closed-system) sulfite reduction experiments. The key considerations in experimental design are: (i) to provide enough sulfite at t0 to ensure we generate significant enough quantities of all the product pools so we can measure, at high precision and accuracy, and at multiple [time] points on f, the multiple S isotopic composition of each pool (i.e., 2 μmols of S per pool per SF6 measurement on the DI-IRMS, which means 2 μmols per fluorination reaction); (ii) to provide the proper reaction conditions to allow DsrAB optimal activity for goal i (pH = 7.1, T = 20 or 31°C); (iii) to ensure hydrogenase activity is not inhibited by the pH chosen (optimum above pH 7.5, activity significantly depleted below pH 6.5, so we chose pH 7.1, to account for optima of both DsrAB and [NiFe]-Hydrogenase); and finally (iv) to ensure the sulfite to hydrogen ratio favors sulfite over reductant capacity (i.e., pH2 in the headspace relative to [sulfite]t0), such that no more than 75% of t0 sulfite is consumed to all products, and less than 50% to the reduced S (dictated by the amount of H2 in the headspace. Finally, (v) determining the sampling interval to ensure proper distribution of points along f, such that applying a closed system distillation model is possible, and statistically robust. Data plotted in Figure 3 represents experimental results that met all of these conditions. The full experimental results (33 experiments) are contained as a supplemental file (http://dx.doi.org/10.6084/m9.figshare.1436115).
All experiments were prepared in an anaerobic chamber. In vitro reactions were carried out in 100 mL acid-washed, autoclave-sterilized, borosilicate glass bottles sealed with butyl-rubber septa and aluminum crimps. Each bottle contained 50% reaction buffer and 50% gaseous headspace. This was done to sufficient H2 was present in the headspace to reduce at most 50% of the sulfite, based on an estimate using Henry's law and available solubility constants for H2 at the given preparation temperatures and headspace pressures. During manipulations in the anaerobic chamber the chamber gas was initially 95:5 N2:H2. Upon removing the reaction mixture-filled bottles from the chamber, these were capped and crimped, and headspace completely exchanged with deoxygenated 100% Ar, then finally exchanged for 100% H2 to initiate the experiments. Experimental buffer is 50 mM phosphate buffer (KPi) prepared at pH 6.9 ± 0.05, with final pH is 7.1 ± 0.05 following the addition of the stock Na2SO3 solution (the reaction is therefore initiated at 7.1). All reaction solutions contained the following: 50 mM KPi buffer (final pH 7.1 ± 0.05), 10 or 15 mM sodium sulfite, 0.832 mM methyl viologen, 242 nm or 315 nm of D. vulgaris DsrAB (calculated to give the same activity depending on the DsrAB aliquot selected), and 8.25 nm [NiFe] hydrogenase (297 U/mg). All experimental mixtures and reagents were prepared in previously boiled 18.2 MΩ water, cooled under O2-free N2.
A. fulgidus DsrAB In vitro Fractionation Experiments in Detail
To extend our studies to a different taxonomic form of the enzyme, we used DsrAB from the thermophilic archaeon A. fulgidus. This enzyme operates at higher temperature and does not have DsrC present in the complex (Schiffer et al., 2008). The results from these experiments are significantly limited compared to those with D. vulgaris, due to too few time points to apply the closed-system model (specifically due to significantly low sample sizes of reduced S for isotope measurements, note the effort made to correct the two data points on A. fulgidus reduced-S). Nevertheless, the results obtained are comparable, when considering the measured δ34S. The values for these experiments are presented with D. vulgaris values in Figure A2.
A. fulgidus DsrAB experiments were conducted 15 mM initial sulfite and 65°C, and showed consistent loss of sulfite and accumulation of products between replicates. We selectively precipitated, separated, and directly measured the 32S-33S-34S-36S compositions from the residual reactant (“sulfite S”) and the “sulfonate S” ((SO3)x). Only the 34S/32S compositions of the “reduced product S” [(S)y] reservoirs were measured (again, due to significantly small reduced-S samples recovered). From these experiments we were able to get a complete set of samples (i.e., sulfite, sulfonate, and reduced S) from one time-point and partial sets from another (i.e., sulfite and sulfonate). We are unable to calculate the for A. fulgidus directly using our model of sulfite reduction experiments due to the dearth of time points (points on f). However, the A. fulgidus isotope values agree with those measured sulfite, sulfonate, and reduced S moieties for D. vulgaris (Figure A2). This general agreement between D. vulgaris and A. fulgidus DsrAB, independent of temperature or phylogenetic origin is perhaps unsurprising, given that previous theoretical predictions deemphasize the role of temperature in determining the magnitude of kinetic isotope effects (Bigeleisen and Wolfsberg, 1958). Furthermore, these enzymes tightly share active site structures (Oliveira et al., 2008b; Parey et al., 2013).
Analytical Methods and Data Handling
Quantification of Dissolved Species
To quantify sulfite and bisulfite concentration in solution we adapted a protocol to quantify SO2 dissolved in water (Grant, 1947), referred to as the “Fuschin” assay from here foreword. Our protocol is specific to the in vitro DsrAB assay conditions. It was determined that matrix matching between samples and standards and the exclusion of oxygen is critical to a successful and reliable assay. Furthermore, we determined trithionate, thiosulfate, sulfate, and zinc sulfide solids do not interact with this color-reagent in the assay. The Fuschin assay is useful over a range of 0–40 nanomoles of sulfite in the final assay volume of 1 mL. Standards of sodium sulfite (Na2SO3 anhydrous, analytical grade) were prepared immediately before the assay is performed in deoxygenated water (boiled and degassed with N2) or KPi buffer. The reaction mixture is composed of 0.04% w/v Pararosaniline HCl (analytical grade) in 10% H2SO4 (analytical grade) v/v, prepared stored in an aluminum-foil wrapped tube or amber-glass bottle at 4°C; and 3.7% formaldehyde (HCHO) prepared fresh each day by diluting 37% (stock) formaldehyde 1:10 water. The reaction is performed on the bench working under N2 flow, or in an anaerobic chamber. A detailed step-by-step protocol is available in Leavitt (2014).
Trithonate and thiosulfate were measured by a modified cyanolysis protocol (Sörbo, 1957; Kelly et al., 1969; Kelly and Wood, 1994). We primarily employed the method of Kelly and Wood (1994) modified in the following manner: the reaction volumes were reduced to 10 rather than 25 mL's (still in volumetric flasks) and we used nitric rather than perchloric acid. Nitric acid was used in the original version of this method (Sörbo, 1957), allowing us to avoid the significant hazards of working with significant volumes of perchloric acid. Samples were added to the reaction buffer to fit within the range of ferric thiocyanate standards (prepared from potassium thiocyanate as a simple standard and thiosulfate as a reaction standard) from 5 to 25 μM (final concentration in the 10 mL reaction), as well as “blanks” prepared from the in vitro assay reaction buffer (50 mM potassium phosphate buffer at pH 7). This is typically 400 μL of in vitro solution added to the 10 mL cyanolysis reaction, in duplicate per method (2X thiosulfate determinations and 2X trithionate determinations). A detailed step-by-step protocol is available in Leavitt (2014).
For sulfide quantifications, we preserved samples in zinc acetate (2% w/v) from each closed system reaction, using a modified Cline method (Cline, 1969). Analytical grade sodium sulfide (>98.9% Na2S*9H2O) was used as the standard, and prepared in deoxygenated (boiled and N2-sparged) in vitro reaction buffer, by precipitating the sulfide with excess zinc acetate (anhydrous), mimicking our sampling protocol. A detailed step-by-step protocol is available in Leavitt (2014). In all samples no sulfide was detected above the determined detection limit of 6.25 μM. From the literature reports where membrane fractions were omitted (Drake and Akagi, 1978), signifying a lack of DsrC re-cycling mechanism (Oliveira et al., 2008b; Bradley et al., 2011), we expected little to no sulfide. This is further supported by the closure of S mass balance at each time-point from each experiment, within analytical error (see main text). Blanks were prepared identically to those in the cyanolysis protocol.
Sample Preparation for S Isotope Analysis
All S-bearing samples for S-isotope analyses (ultimately as SF6 and/or SO2) were removed from the in vitro reaction solution (50 mM potassium phosphate) following a sequential precipitation protocol, inspired by that of Smock et al. (1998). Our protocol reflects our specific experimental setup and the pools we aimed to isolate and purify: sulfite (and any trace sulfate), sulfonate, and reduced product S. Samples of the residual reactant (sulfite) and pooled products (reduced product S and sulfonate S) were removed from the in vitro reaction mixture by sequential precipitation and filtration or centrifugation to isolate solid-phases Ag2S(s) (reduced product), BaSO3(s) (sulfite), BaSO4(s) (sulfonate) by the extraction scheme modified from our recent work (Leavitt et al., 2014) and detailed elsewhere (Leavitt, 2014). Samples were captured as BaSO3(s), BaSO4(s), or Ag2S(s), respectively. Sub-samples of the reduced product Ag2S were directly fluorinated (after the below washing steps were carried out to ensure clean Ag2S), or in the case of the sulfonate S-pool, collected from the AVS residue and converted from BaSO4 to Ag2S by the method of Thode (Thode et al., 1961), according to the protocol we recently published (Leavitt et al., 2013). Novel to this study: all sulfite samples (reacted as BaSO3(s)) were oxidized with peroxide prior to “Thode”-reduction (detailed protocols for these methods are available in Leavitt, 2014). All samples entering the elemental analyzer isotope ratio mass spectrometer (EA-IRMS), and combusted to and analyzed as SO2, are prepared as dry BaSO3(s), BaSO4(s), or Ag2S(s). All samples for quadruple S isotope analysis enter the fluorination line as pure dry Ag2S(s).
Major S-isotope (34S/32S) Ratios Measurements
Continuous flow isotope ratio mass spectrometric (CF-IRMS) measurements of the three S-bearing pools of interest, sulfite, sulfonate and reduced product S, were performed as follows: 0.4 mg (±0.05 mg) BaSO3, BaSO4, or Ag2S were converted to SO2 by combustion at 1040°C in the presence of excess V2O5 (Elemental Analyzer, Costech ECS 4010) and analyzed by continuous flow isotope ratio mass spectrometry (SD = ±0.3‰; Thermo-Finnegan DELTA V Plus). All samples yielded clean chromatography and most m/z 66 amplitudes (corresponding primarily to the (16O34S16O)+ ionization product) within the range of in-run standards (IAEA: S1, S2, and S3 for Ag2S or SO5, SO6, and NBS-127 for BaSO4 and BaSO3). Some sulfite precipitates (BaSO3_sulfite), though not any of the sulfate or sulfide precipitates (BaSO4_sulfonate or Ag2S_reduced product) produced atypical weight to m/z 66 response to what we regularly note with lab standards of BaSO3 or BaSO4 – specifically the signal was less than predicted, likely due to occlusion of phosphates (from the in vitro reaction buffer) in the barium sulfite matrix. As a result, we use the m/z 66 to BaSO3 weight ratio (mg of BaSO3 per unit area of the m/z 66 peak) to calculate the desired sample weight to achieve standard signal size, re-weighed and re-combusted/measured the requisite samples, and in all cases achieved m/z 66 peak areas in the range of our IAEA BaSO4 and in-house BaSO3 standards. Each standard is measured at least 4x in-run and each sample 2-3x (when sufficient sample is available). This simplifies the scale-conversion calculation for taking samples referenced in-run to in-house standard tank gas (HAR1SO2) and ultimately to the international reference frame (V-CDT).
Multiple S-isotope (33S/32S, 34S/32S, 36S/32S) Ratio Measurements
Duel-inlet (DI-IRMS) measurements of all four stable S isotopes (32S, 33S, 34S, 36S) from the three S-bearing pools of interest, sulfite, sulfonate and reduced product S, were performed as previously described (Leavitt et al., 2013, 2014). Briefly, all samples for quadruple S-isotope analysis, prepared dry and clean Ag2S (described above), were fluorinated under 10X excess F2 to produce SF6, which is then purified cryogenically (distilled at −107°C) and chromatographically (on a 6′ molecular sieve 5 Å inline with a 6′ HayeSepQ 1/8″-stainless steel column, detected by TCD). Purified SF6 was measured as (m/z of 127, 128, 129, and 131) on a Thermo-Finnegan Scientific MAT 253 (SD: δ34S ±0.2, Δ33S ±0.006‰, Δ36S ±0.15‰). All isotope ratios are reported in parts per thousand (‰) as experimentally paired sulfates and sulfides measured. Long-term running averages and standard deviations are calculated from measures of IAEA standards: S1, S2, S3 for sulfides or NBS-127, SO5, SO6 for sulfates. Isotope calculations and notation are detailed in the text. Standard deviations for each value is estimated as reported previously (Johnston et al., 2007) with previous inaccuracies in the transcription corrected here.
Scale-Compression Correction Calculations for Small S Samples
Experiments with A. fulgidus yielded small amounts of product (0.35 to 0.01 mg), which required additional data handling during isotope analysis. Given the small size of these samples, we ran each sample only once and bracketed the samples (n = 2) with a series of standards: IAEA S1, S2 and S3 (n = 16, 14, and 14, respectively) run over a size series that captured the sample sizes, all by CF-IRMS only. As expected, we observed that the measured isotopic composition of the standards varied non-linearly as a function of signal intensity (monitored as peak integrated areas and peak intensities on m/z 64 and 66 for and 48 and 50 for SO+). The size dependence on the isotopic composition (handled as 50R and 66R, which are the 50/48 and 66/64, respectively) scale compression is calculated as a proportional change. For SO (correction factor in Figure A3) it scales as: . We focus on SO here as these samples yielded sharper chromatography on slightly different sized signals (due to resistor differences between SO and SO2 cups − 3 × 1010 Ω and 1 × 1010 Ω respectively). Thus, using the three IAEA standards, we developed a correction whereby we solve (in the standards) for the non-linear features of the data as it relates to signal intensity (here monitored as the peak integrated area on mass 48 − 32SO). This is shown in Figure A3. After this correction is applied to 50Rmeasured, SO data is converted to an SO2 scale (see Figure A3), which is a linear transfer function again derived from IAEA standard data. The final correction places all the data (now on a SO2 scale against in-house reference SO2 tank gas) to the V-CDT scale. To review, we perform the following steps (1) correcting the 50R on SO for sample size, (2) convert 50R to 64R (against tank gas), and finally (3) convert all data to a VCDT scale.
The largest source of error in this treatment is associated with the sample size correction. As such, we propagate the error associated with the fit in Figure A3 to determine the uncertainty in the final isotope value. As expected, for small samples this error is quite large (Figure A2), with the value decreasing in absolute magnitude as signal intensity (peak integrated area) increases. We also compare these error estimates to the calculated shot noise for this measurement (pink line in Figure A3d). As is presented below, our regressed error is in excess of the shot noise limit. Similarly, the error on the population of standards that were used in deriving this fit is 1‰ (n = 44).
The Closed-System Distillation Model for a More Complex Network
There exists the possible mixing of multiple fractionation factors later in the experiment (f < 0.85). The approach outlined in the main text yielded results in which the observed fractionation factor between sulfite and reduced pools appeared to change as a function of f, when f < 0.85—that is, later in the reaction, when back-reactions are more likely (Figure A4). One explanation for this apparent behavior is that the reduced pool is not the product of a single set of reactions but of multiple reactions. The most plausible explanation for this is that some fraction of the reduced pool is derived from the sulfonate pool rather than being derived solely from sulfite, particularly later in the in vitro experiment (i.e., at values of f < 0.85). Previous work (Drake and Akagi, 1977, 1978; Parey et al., 2010) has demonstrated that DsrAB is capable of reducing trithionate to thiosulfate, and thiosulfate to sulfite and sulfide, which was confirmed with the D. vulgaris enzyme.
We can constrain the magnitudes of the fractionation factor related to the conversion of the sulfonate to reduced S through the following steps. First, utilizing the framework given above to solve for αred for the time points where f is nearest to 1. As these measurements are obtained at the lowest concentrations of product, we assume that this result gives an estimate for αred that reflects the production of reduced S from sulfite only, with minimal input of reduced S derived from sulfonate. Second, we write an equation for Rred as a function of αred, RSO3, and Rox:
where XSO3 is the fraction of Rred generated directly from sulfite and αunk is the unknown fractionation factor between Rox and Rred. This equation is then rewritten and solved for αunk as a function of the other parameters over a range of values of XSO3 (0.01–0.99). This does not yield a unique solution for the unknown fractionation, but constrains its value given the relative importance of the contribution to the reduced sulfur pool of both sulfonate and sulfite. We assume that the relative contribution of the secondary reaction is invariant over the course of the reaction, thereby manifesting as no change in j.
Error Propagation Calculation for the Closed System Model Estimates
The error associated with calculations of 33λ (approximately the slope of a δ33S vs. δ34S line) is highly sensitive to the length of the line (total 34S range, 34ε) and modestly related to the residual around a mass-dependent theoretical prediction (the standard deviation on Δ33S is often used here (Farquhar et al., 2003; Johnston et al., 2007). To approximate the standard deviation (σ) associated with our 33λ calculation, we propagate our measurement errors (δ34S, concentration, etc.). We keep with the presumption that mass-dependence will dictate the δ33S, once the new δ34S is calculated. This stems from the fact that the error in δ34S and δ33S are highly correlated, meaning that the error in Δ33S is significantly smaller (0.008‰) than that for δ33S (0.1‰). As our fractionation factor model is based on a closed-system distillation equation (see above), we perform an error propagation on an equation of the form: Rf = (R0) (f(α−1)), where we are most interested in accounting for the analytical errors on the isotope measurement (σR, 0.2‰/1000) and the uncertainty on f. The second term is critical here as we are independently determining f from concentration measurements in the experiment, with a standard deviation on sulfite concentration measurements of 3%. We use this value moving forward as a metric of σf. To simplify the presentation, we let X = (α − 1) and Z = f. Following typical error propagation for power law and multiplicative relations (Bevington and Robinson, 2003 p. 43–46), we find:
which then can substitute into the final form of:
The ηεσRf is then converted into ‰ units (through multiplying by 1000) so that it can be inserted into the updated (Johnston et al., 2007) error equation for 33λ, presented here:
which can be broken down into:
and
As noted above, the error on lambda σλ is dependent on the δ34S and Δ33S. The subscript RFL represents the reference fractionation line, which for 33λ is 0.515, and for 36λ is 1.90. Data for δ36S are not discussed in the text, as they yield the same conclusions as δ33S, but are included here in Dataframe S1 (https://github.com/bradleylab/DsrAB_enzyme_models).
In total, this leaves our error estimate a function of the following five variables: 34ε, σR, f, σf, and α. The final error is not an evenly weighted sum of these variables, and in the case presented here, most heavily influenced by the error in concentration data (σf). A sensitivity analysis (Figure A6) on this exercise demonstrates that the errors in f far outweigh the analytical uncertainty in a measurement of R, and dominate the magnitude of the final σRf.
Data Compilations and Statistical Analysis
Compilation and Statistical Analysis of Pure-Culture MSR Fractions
To place our DsrAB enzyme-specific fractionation factor in context with the previous 65 years of pure-culture experimental work, we compile all available observations from studies using axenic cultures of MSR (Figure 4), in the following experimental systems: batch (closed-system, in vivo, whole-cell), chemostat (open-system, in vivo, whole-cell), resting (closed-system, in vivo, whole-cell, not growing), cell-free (closed-system, ex vivo crude cell extracts, not growing). From these four types of experiments we further subdivide experiments into where sulfate was reduced to sulfide or sulfite was reduced to sulfide. We count each experimental determination () and compile them all in the supplemental data files (http://dx.doi.org/10.6084/m9.figshare.1436115), from experiments where less than 10% of the reactant S-species was consumed. Herein we calculate and present column statistics (box-whisker plots in Figure 4) using Prism5c (GraphPad, San Diego, CA). The key finding here is that the majority of the means from each set of experiments is significantly less than the previous estimates for the fractionation factor associated with DsrAB (25 to 53‰, Harrison and Thode, 1957; Rees, 1973; Farquhar et al., 2003; Brunner and Bernasconi, 2005; Johnston et al., 2007), and that the mean values from all 650+ experimental determinations, regardless of experiment type or whether it was a sulfate-sulfide or sulfite-sulfide experiments, the grand mean for 34εr−p falls at 17.9‰ (median at 16.1‰), with the 25th and 75th percentile's falling at 10‰ and 22.5‰, respectively (Figure 5)—these are all well within the maximum fractionation accounted for by the sum of our DsrAB value (15.3‰) and our literature derived range for sulfate reduction to sulfite (4 to 15‰), for a total of 19.3 to 30.3‰ (see main text).
Literature Estimates of Fractionation during Sulfate Activation to Sulfite
The upstream kinetic isotope fractionation, the result of enzyme mediated sulfate/sulfite exchange in cell-free extract experiments, is between 4 and 15 ‰ (compilation files: http://dx.doi.org/10.6084/m9.figshare.1436115). The mean of these experiments is = 9.5‰, CI95% = 7.2 to 11.9‰, with and n = 12 (column statistics are also permanently available at: http://dx.doi.org/10.6084/m9.figshare.1436115) (Ford, 1957; Harrison and Thode, 1958; Kaplan and Rittenberg, 1964; Kemp and Thode, 1968). Deconvolving this aggregated fractionation factor () in vitro is a target for future pure enzyme experiments focusing on the constituent steps (enzyme specific 34ε), as well as the minor isotope fractionations associate with each (i.e., 33λ's).
These values represent the fractionation across the sum of the steps incorporating sulfate activation to APS and its concomitant reduction to sulfite (Figure 1). It is important to note that these values were determined using crude-cell extracts, rather than purified enzymes, and not measured over a range of reaction progress (as in Figure 2). Further, available data do not allow for the evaluation of mass balance closure, as we have done here for DsrAB. Given our present understanding of the enzymes involved in this process (Bradley et al., 2011; Pereira et al., 2011), sulfate transport into the cytoplasm followed by activation to APS (Sat) are not likely to directly impact S-isotope compositions, whereas the reduction of APS (APSr) most likely does, due to the breaking of a S-O bond. The sum of transport, Sat and APSr fractionations sit immediately upstream of the DsrAB. Both of these constraints ( and ) are interpreted in the context of the MSR data compiled from the literature, which includes lab experiments, natural waters and sediments, as discussed in the main text (Figure 5).
Statistical Analysis of Laboratory Chemostat and Marine Sediment Fractionations
To apply the compiled sedimentary sulfate reduction rates from Goldhaber and Kaplan (1975), we re-plot their log-scale values to a linear scaling (Figure 5) and apply the same non-linear regression one-phase decay model () from our recent work on fractionation—rate relationships in MSR (Leavitt et al., 2013), minimizing variance to arrive at the following parameters: Y0 = 73‰, plateau = 17.3‰, and a decay-constant (K) of 6.4 (Figure 5). For the chemostat (open-system) MSR data in the study where we derived this regression model (Leavitt et al., 2013), we re-scale the cell-specific MSR rates to basic volumetric fluxes by multiplying out the number of cells at each sampling point, using the chemostat values from our recent study (Leavitt et al., 2013). Applying the same one-phase decay model and minimize variance, we calculate the following parameters: Y0 = 56.5‰, plateau = 17.3‰, and a decay-constant (K) of 0.054. All regressions were calculated using Prism5c (GraphPad, San Diego, CA).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Thanks to Sofia Venceslau and Fabian Grein for discussions on MSR biochemistry and DsrC cycling; Isabel Pacheco for the purified hydrogenase; to Erin Beirne and Andy Masterson for expert assistance in measurements and separation chemistry. Thanks to Ann Pearson, David Fike and Itay Halevy for comments that greatly improved our interpretations and presentation. We especially thank John Hayes for exceptionally in-depth feedback. This work was funded by an NSF-GRFP (WL), NSF-EAR-1225980 (DJ, IP), the Sloan Foundation (DJ), the Agouron Institute (AB), PTDC/QUI-BIQ/100591/2008 and PTDC/BBB-BQB/0684/2012 (IP), UID/Multi/04551/2013 (to ITQB) funded by Fundacao para a Ciencia e Tecnologia (FCT, Portugal), Washington University in St. Louis (AB) and the Steve Fossett Postdoctoral Fellowship at Washington University in St. Louis (WL).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2015.01392
References
Ames, D. P., and Willard, J. E. (1951). The Kinetics of the Exchange of Sulfur between Thiosulfate and Sulfite1. J. Am. Chem. Soc. 73, 164–172.
Antler, G., Turchyn, A. V., Rennie, V., Herut, B., and Sivan, O. (2013). Coupled sulfur and oxygen isotope insight into bacterial sulfate reduction in the natural environment. Geochim. Cosmochim. Acta 118, 98–117. doi: 10.1016/j.gca.2013.05.005
Bevington, P. R., and Robinson, D. K. (2003). Data Reduction and Error Analysis. New York, NY: McGraw–Hill.
Bigeleisen, J., and Wolfsberg, M. (1958). Theoretical and experimental aspects of isotope effects in chemical kinetics. Adv. Chem. Physics 1, 15–76.
Bolliger, C., Schroth, M. H., Bernasconi, S. M., Kleikemper, J., and Zeyer, J. (2001). Sulfur isotope fractionation during microbial sulfate reduction by toluene-degrading bacteria. Geochim. Cosmochim. Acta 65, 3289–3298. doi: 10.1016/S0016-7037(01)00671-8
Bottcher, M. E., Sievert, S. M., and Kuever, J. (1999). Fractionation of sulfur isotopes during dissimilatory reduction of sulfate by a thermophilic gram-negative bacterium at 60 C. Arch. Microbiol. 172, 125–128.
Bradford, M. M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254. doi: 10.1016/0003-2697(76)90527-3
Bradley, A. S., Leavitt, W. D., and Johnston, D. T. (2011). Revisiting the dissimilatory sulfate reduction pathway. Geobiology 9, 446–457. doi: 10.1111/j.1472-4669.2011.00292.x
Bradley, A. S., Leavitt, W. D., Schmidt, M., and Knoll, A. H. (2015). Patterns of sulfur isotope fractionation during Microbial Sulfate Reduction. Geobiology. doi: 10.1111/gbi.12149. [Epub ahead of print].
Brunner, B., and Bernasconi, S. M. (2005). A revised isotope fractionation model for dissimilatory sulfate reduction in sulfate reducing bacteria. Geochim. Cosmochim. Acta 69, 4759–4771. doi: 10.1016/j.gca.2005.04.015
Canfield, D. E. (2001a). Biogeochemistry of sulfur isotopes. Rev. Mineral. Geochem. 43, 607–636. doi: 10.2138/gsrmg.43.1.607
Canfield, D. E. (2001b). Isotope fractionation by natural populations of sulfate-reducing bacteria. Geochim. Cosmochim. Acta 65, 1117–1124. doi: 10.1016/S0016-7037(00)00584-6
Canfield, D. E. (2006). Models of oxic respiration, denitrification and sulfate reduction in zones of coastal upwelling. Geochim. Cosmochim. Acta 70, 5753–5765. doi: 10.1016/j.gca.2006.07.023
Canfield, D. E., and Farquhar, J. (2009). Animal evolution, bioturbation, and the sulfate concentration of the oceans. Proc. Natl. Acad. Sci. U.S.A. 106, 8123–8127. doi: 10.1073/pnas.0902037106
Canfield, D. E., Farquhar, J., and Zerkle, A. L. (2010). High isotope fractionations during sulfate reduction in a low-sulfate euxinic ocean analog. Geology 38, 415–418. doi: 10.1130/G30723.1
Chambers, L. A., and Trudinger, P. A. (1975). Are thiosulfate and trithionate intermediates in dissimilatory sulfate reduction? J. Bacteriol. 123, 36–40.
Chambers, L. A., and Trudinger, P. A. (1979). Microbiological fractionation of stable sulfur isotopes: a review and critique. Geomicrobiol. J. 1, 249–293.
Chambers, L. A., Trudinger, P. A., Smith, J. W., and Burns, M. S. (1975). Fractionation of sulfur isotopes by continuous cultures of Desulfovibrio desulfuricans. Can. J. Microbiol. 21, 1602–1607.
Cline, J. D. (1969). Spectrophotometric determination of hydrogen sulfide in natural waters. Limnol. Oceanogr. 14, 454–458.
Crowe, S., Paris, G., Katsev, S., Jones, C., Kim, S. T., Zerkle, A. L., et al. (2014). Sulfate was a trace constituent of Archean seawater. Science 346, 735–739. doi: 10.1126/science.1258966
Czyzewski, B. K., and Wang, D.-N. (2012). Identification and characterization of a bacterial hydrosulphide ion channel. Nature 483, 494–497. doi: 10.1038/nature10881
Davidson, M. M., Bisher, M. E., Pratt, L. M., Fong, J., Southam, G., Pfiffner, S. M., et al. (2009). Sulfur isotope enrichment during maintenance metabolism in the thermophilic sulfate-reducing bacterium desulfotomaculum putei. Appl. Environ. Microbiol. 75, 5621–5630. doi: 10.1128/AEM.02948-08
Detmers, J., Brüchert, V., Habicht, K., and Kuever, J. (2001). Diversity of sulfur isotope fractionations by sulfate-reducing prokaryotes. Appl. Environ. Microbiol. 67, 888–894. doi: 10.1128/AEM.67.2.888-894.2001
Drake, H. L., and Akagi, J. M. (1976). Product analysis of bisulfite reductase activity isolated from Desulfovibrio vulgaris. J. Bacteriol. 126, 733–738.
Drake, H. L., and Akagi, J. M. (1977). Bisulfite reductase of Desulfovibrio vulgaris: explanation for product formation. J. Bacteriol. 132, 139–143.
Drake, H. L., and Akagi, J. M. (1978). Dissimilatory reduction of bisulfite by Desulfovibrio vulgaris. J. Bacteriol. 136, 916–923.
Einsiedl, F. (2008). Effect of NO2- on stable isotope fractionation during bacterial sulfate reduction. Environ. Sci. Technol. 43, 82–87. doi: 10.1021/es801592t
Farquhar, G. D., O'leary, M. H., and Berry, J. A. (1982). On the relationship between carbon isotope discrimination and the intercellular carbon dioxide concentration in leaves. Funct. Plant Biol. 9, 121–137.
Farquhar, J., Canfield, D. E., Masterson, A., Bao, H., and Johnston, D. T. (2008). Sulfur and oxygen isotope study of sulfate reduction in experiments with natural populations from Faellestrand, Denmark. Geochim. Cosmochim. Acta 72, 2805–2821. doi: 10.1016/j.gca.2008.03.013
Farquhar, J., Johnston, D. T., and Wing, B. A. (2007). Implications of conservation of mass effects on mass-dependent isotope fractionations: influence of network structure on sulfur isotope phase space of dissimilatory sulfate reduction. Geochim. Cosmochim. Acta 71, 5862–5875. doi: 10.1016/j.gca.2007.08.028
Farquhar, J., Johnston, D. T., Wing, B. A., Habicht, K. S., Canfield, D. E., Airieau, S., et al. (2003). Multiple sulphur isotopic interpretations of biosynthetic pathways: implications for biological signatures in the sulphur isotope record. Geobiology 1, 27–36. doi: 10.1046/j.1472-4669.2003.00007.x
Ford, R. W. (1957). Sulphur Isotope Effects in Chemical and Biological Processes. PhD Thesis. McMaster University.
Fritz, G. (2002). Structure of adenylylsulfate reductase from the hyperthermophilic Archaeoglobus fulgidus at 1.6-A resolution. Proc. Natl. Acad. Sci. U.S.A. 99, 1836–1841. doi: 10.1073/pnas.042664399
Fritz, P., Basharmal, G. M., Drimmie, R. J., and Ibsen, J. (1989). Oxygen isotope exchange between sulphate and water during bacterial reduction of sulphate. Chem. Geol. 79, 99–105.
Furusaka, C. (1961). Sulphate transport and metabolism desulphovibrio desulphuricans. Nature 192, 427–429.
Garrels, R. M., and Lerman, A. (1981). Phanerozoic cycles of sedimentary carbon and sulfur. Proc. Natl. Acad. Sci. U.S.A. 78, 4652–4656.
Goldhaber, M. B., and Kaplan, I. R. (1975). Controls and consequences of sulfate reduction rates in recent marine sediments. Soil Sci. 119, 42–55.
Gomes, M. L., and Hurtgen, M. T. (2013). Sulfur isotope systematics of a euxinic, low-sulfate lake: evaluating the importance of the reservoir effect in modern and ancient oceans. Geology 41, 663–666. doi: 10.1130/G34187.1
Habicht, K., Gade, M., Thamdrup, B., Berg, P., and Canfield, D. E. (2002). Calibration of sulfate levels in the Archean Ocean. Science 298, 2372–2374. doi: 10.1126/science.1078265
Habicht, K. S., Salling, L., Thamdrup, B., and Canfield, D. E. (2005). Effect of low sulfate concentrations on lactate oxidation and isotope fractionation during sulfate reduction by archaeoglobus fulgidus strain Z. Appl. Environ. Microbiol. 71, 3770–3777. doi: 10.1128/AEM.71.7.3770-3777.2005
Harrison, A. G., and Thode, H. G. (1957). The kinetic isotope effect in the chemical reduction of sulphate. Transac. Faraday Soc. 53, 1648–1651.
Harrison, A. G., and Thode, H. G. (1958). Mechanism of the bacterial reduction of sulphate from isotope fractionation studies. Transac. Faraday Soci. 54, 84–92.
Hayes, J. M. (1983). “Practices and principles of isotopic measurements in organic geochemistry,” in Organic Geochemistry of Contemporaneous and Ancient Sediments, Great Lakes Section – Society of Economic Paleontologists and Mineralogists, ed W. G. Meinschein (Bloomington. IN).
Hayes, J. M. (1993). Factors controlling 13 C contents of sedimentary organic compounds: principles and evidence. Marine Geol. 113, 111–125.
Hayes, J. M. (2001). Fractionation of carbon and hydrogen isotopes in biosynthetic processes. Rev. Mineral. Geochem. 43, 225–277. doi: 10.2138/gsrmg.43.1.225
Hayes, J. M., Strauss, H., and Kaufman, A. J. (1999). The abundance of 13C in marine organic matter and isotopic fractionation in the global biogeochemical cycle of carbon during the past 800 Ma. Chem. Geol. 161, 103–125.
Hoek, J., Reysenbach, A.-L., Habicht, K., and Canfield, D. (2006). Effect of hydrogen limitation and temperature on the fractionation of sulfur isotopes by a deep-sea hydrothermal vent sulfate-reducing bacterium. Geochim. Cosmochim. Acta 70, 5831–5841. doi: 10.1016/j.gca.2006.07.031
Holland, H. D. (1973). Systematics of the isotopic composition of sulfur in the oceans during the Phanerozoic and its implications for atmospheric oxygen. Geochim. Cosmochim. Acta 37, 2605–2616.
Holler, T., Wegener, G., Niemann, H., Deusner, C., Ferdelman, T. G., Boetius, A., et al. (2011). Carbon and sulfur back flux during anaerobic microbial oxidation of methane and coupled sulfate reduction. Proc. Natl. Acad. Sci. U.S.A. 108, E1484–E1490. doi: 10.1073/pnas.1106032108
Johnston, D. T. (2005). Multiple sulfur isotope fractionations in biological systems: a case study with sulfate reducers and sulfur disproportionators. Am. J. Sci. 305, 645–660. doi: 10.2475/ajs.305.6-8.645
Johnston, D. T., Farquhar, J., and Canfield, D. E. (2007). Sulfur isotope insights into microbial sulfate reduction: when microbes meet models. Geochim. Cosmochim. Acta 71, 3929–3947. doi: 10.1016/j.gca.2007.05.008
Jones, G. E., and Starkey, R. L. (1957). Fractionation of stable isotopes of sulfur by microorganisms and their role in deposition of native sulfur. Appl. Microbiol. 5, 111.
Kaplan, I. R., and Rittenberg, S. C. (1964). Microbiological fractionation of sulphur isotopes. J. Gen. Microbiol. 34, 195–212.
Karkhoff-Schweizer, R. R., Bruschi, M., and Voordouw, G. (1993). Expression of the gamma-subunit gene of desulfoviridin-type dissimilatory sulfite reductase and of the alpha- and beta-subunit genes is not coordinately regulated. Eur. J. Biochem. 211, 501–507. doi: 10.1111/j.1432-1033.1993.tb17576.x
Karsh, K. L., Granger, J., Kritee, K., and Sigman, D. M. (2012). Eukaryotic assimilatory nitrate reductase fractionates N and O isotopes with a ratio near unity. Environ. Sci. Technol. 46, 5727–5735. doi: 10.1021/es204593q
Kelly, D. P., Chambers, L. A., and Trudinger, P. A. (1969). Cyanolysis and spectrophotometric estimation of trithionate in mixture with thiosulfate and tetrathionate. Anal. Chem. 41, 898–901.
Kelly, D. P., and Wood, A. P. (1994). Synthesis and determination of thiosulfate and polythionates. Methods Enzymol. 243, 475–501.
Kemp, A., and Thode, H. G. (1968). The mechanism of the bacterial reduction of sulphate and of sulphite from isotope fractionation studies. Geochim. Cosmochim. Acta 32, 71–91.
Kim, J. H., and Akagi, J. M. (1985). Characterization of a trithionate reductase system from Desulfovibrio vulgaris. J. Bacteriol. 163, 472–475.
Kleikemper, J., Schroth, M., Bernasconi, S. M., Brunner, B., and Zeyer, J. (2004). Sulfur isotope fractionation during growth of sulfate-reducing bacteria on various carbon sources. Geochim. Cosmochim. Acta 68, 4891–4904. doi: 10.1016/j.gca.2004.05.034
Knöller, K., Vogt, C., Richnow, H.-H., and Weise, S. M. (2006). Sulfur and oxygen isotope fractionation during benzene, toluene, ethyl benzene, and xylene degradation by sulfate-reducing bacteria. Environ. Sci. Technol. 40, 3879–3885. doi: 10.1021/es052325r
Kobayashi, K., Yasuhide, S., and Ishimoto, M. (1974). Biochemical studies on sulfate-reducing Bacteria XIII. Sulfite reductase fromdesulfovibrio vulgaris-mechanism of trithionafe, thiosulfate, and sulfide formation and enzymatic properties. J. Biochem. 75, 519–529.
Krouse, H. R., McCready, R. G. L., Husain, S. A., and Campbell, J. N. (1968). Sulfur isotope fractionation and kinetic studies of sulfite reduction in growing cells of salmonella heidelberg. Biophys. J. 8, 109–124. doi: 10.1016/S0006-3495(68)86478-1
Laws, E. A., Popp, B. N., Bidigare, R. R., Kennicutt, M. C., and Macko, S. A. (1995). Dependence of phytoplankton carbon isotopic composition on growth rate and [CO 2) aq: theoretical considerations and experimental results. Geochim. Cosmochim. Acta 59, 1131–1138.
Leavitt, W. D. (2014). On the Mechanisms of Sulfur Isotope Fractionation During Microbial Sulfate Reduction. PhD Thesis. Harvard University.
Leavitt, W. D., Cummins, R., Schmidt, M. L., Sim, M. S., Ono, S., Bradley, A. S., et al. (2014). Multiple sulfur isotope signatures of sulfite and thiosulfate reduction by the model dissimilatory sulfate-reducer, Desulfovibrio alaskensis str. G20. Front. Microbiol. 5:591. doi: 10.3389/fmicb.2014.00591
Leavitt, W. D., Halevy, I., Bradley, A. S., and Johnston, D. T. (2013). Influence of sulfate reduction rates on the Phanerozoic sulfur isotope record. Proc. Natl. Acad. Sci. U.S.A. 110, 11244–11249. doi: 10.1073/pnas.1218874110
Le Gall, J., Payne, W. J., Chen, L., Liu, M. Y., and Xavier, A. V. (1994). Localization and specificity of cytochromes and other electron transfer proteins from sulfate-reducing bacteria. Biochimie 76, 655–665.
Mangalo, M., Einsiedl, F., Meckenstock, R. U., and Stichler, W. (2008). Influence of the enzyme dissimilatory sulfite reductase on stable isotope fractionation during sulfate reduction. Geochim. Cosmochim. Acta 72, 1513–1520. doi: 10.1016/j.gca.2008.01.006
Mangalo, M., Meckenstock, R. U., Stichler, W., and Einsiedl, F. (2007). Stable isotope fractionation during bacterial sulfate reduction is controlled by reoxidation of intermediates. Geochim. Cosmochim. Acta 71, 4161–4171. doi: 10.1016/j.gca.2007.06.058
Mariotti, A., Germon, J. C., Hubert, P., Kaiser, P., Letolle, R., Tardieux, A., et al. (1981). Experimental determination of nitrogen kinetic isotope fractionation: some principles; illustration for the denitrification and nitrification processes. Plant Soil 62, 413–430. doi: 10.1007/BF02374138
Marritt, S. J., and Hagen, W. R. (1996). Dissimilatory sulfite reductase revisited. The desulfoviridin molecule does contain 20 iron ions, extensively demetallated sirohaem, and an S= 9/2 iron-sulfur cluster. Eur. J. Biochem. 238, 724–727. doi: 10.1111/j.1432-1033.1996.0724w.x
Mathai, J. C., Missner, A., Kügler, P., Saparov, S. M., Zeidel, M. L., Lee, J. K., et al. (2009). No facilitator required for membrane transport of hydrogen sulfide. Proc. Natl. Acad. Sci. U.S.A. 106, 16633–16638. doi: 10.1073/pnas.0902952106
McCready, R. G. L. (1975). Sulfur Isotope Fractionation by Desulfovibrio and Desulfotomaculum species. Geochim. Cosmochim. Acta 39, 1395–1401.
McCready, R. G. L., Laishley, E. J., and Krouse, H. R. (1975). Stable isotope fractionation by Clostridium pasteurianum. 1.34 S/32S: inverse isotope effects during SO42-and SO32-reduction. Can. J. Microbiol. 21, 235–244.
McNaught, A. D., and Wilkinson, A. (1997). Compendium of Chemical Terminology, International Union of Pure and Applied Chemistry. Oxford: Blackwell Science.
Miller, M. F. (2002). Isotopic fractionation and the quantification of 17 O anomalies in the oxygen three-isotope system: an appraisal and geochemical significance. Geochim. Cosmochim. Acta 66, 1881–1889. doi: 10.1016/S0016-7037(02)00832-3
Nakagawa, M., Ueno, Y., Hattori, S., Umemura, M., Yagi, A., Takai, K., et al. (2012). Seasonal change in microbial sulfur cycling in monomictic Lake Fukami-ike, Japan. Limnol. Oceanogr. 57, 974–988. doi: 10.4319/lo.2012.57.4.0974
Nakai, N., and Jensen, M. L. (1964). The kinetic isotope effect in the bacterial reduction and oxidation of sulfur. Geochim. Cosmochim. Acta 28, 1893–1912. doi: 10.1016/0016-7037(64)90136-X
Newton, G. L., Dorian, R., and Fahey, R. C. (1981). Analysis of biological thiols: derivatization with monobromobimane and separation by reverse-phase high-performance liquid chromatography. Anal. Biochem. 114, 383–387.
Oliveira, T. F., Franklin, E., Afonso, J. P., Khan, A. R., Oldham, N. J., Pereira, I. A. C., et al. (2011). Structural insights into dissimilatory sulfite reductases: structure of desulforubidin from desulfomicrobium norvegicum. Front. Microbiol. 2:71. doi: 10.3389/fmicb.2011.00071
Oliveira, T. F., Vonrhein, C., Matias, P. M., Venceslau, S. S., Pereira, I. A. C., and Archer, M. (2008a). Purification, crystallization and preliminary crystallographic analysis of a dissimilatory DsrAB sulfite reductase in complex with DsrC. J. Struct. Biol. 164, 236–239. doi: 10.1016/j.jsb.2008.07.007
Oliveira, T. F., Vonrhein, C., Matias, P. M., Venceslau, S. S., Pereira, P. M., and Archer, M. (2008b). The crystal structure of Desulfovibrio vulgaris dissimilatory sulfite reductase bound to DsrC provides novel insights into the mechanism of sulfate respiration. J. Biol. Chem. 283, 34141–34149. doi: 10.1074/jbc.M805643200
Pagani, M., Liu, Z., LaRiviere, J., and Ravelo, A. C. (2009). High Earth-system climate sensitivity determined from Pliocene carbon dioxide concentrations. Nat. Geosci. 3, 27–30. doi: 10.1038/ngeo724
Pallud, C., Meile, C., Laverman, A. M., Abell, J., and Van Cappellen, P. (2007). The use of flow-through sediment reactors in biogeochemical kinetics: methodology and examples of applications. Marine Chem. 106, 256–271. doi: 10.1016/j.marchem.2006.12.011
Parey, K., Fritz, G., Ermler, U., and Kroneck, P. M. H. (2013). Conserving energy with sulfate around 100 °C – structure and mechanism of key metal enzymes in hyperthermophilic Archaeoglobus fulgidus. Metallomics 5, 302. doi: 10.1039/c2mt20225e
Parey, K., Warkentin, E., Kroneck, P. M., and Ermler, U. (2010). Reaction cycle of the dissimilatory sulfite reductase from Archaeoglobus fulgidus. Biochemistry 49, 8912–8921. doi: 10.1021/bi100781f
Park, R., and Epstein, S. (1960). Carbon isotope fractionation during photosynthesis. Geochim. Cosmochim. Acta 21, 110–126.
Peck, H. D. Jr. (1959). The ATP-dependent reduction of sulfate with hydrogen in extracts of Desulfovibrio desulfuricans. Proc. Natl. Acad. Sci. U.S.A. 45, 701.
Peck, H. D. Jr. (1960). Adenosine 5′-Phosphosulphate as an intermediate in the oxidation of thiosulfate by Thiobacillus thioparus. Proc. Natl. Acad. Sci. U.S.A. 46, 1053.
Peck, H. D. (1961). Enzymatic basis for assimilatory and dissimilatory sulfate reduction. J. Bacteriol. 82, 933–939.
Peck, H. D. (1962). The role of adenosine-5′-phosphosulfate in the reduction of sulfate to sulfite by Desulfovibrio desulfuricans. J. Biol. Chem. 237, 198–203.
Pereira, I. A. C., Ramos, A. R., Grein, F., Marques, M. C., Da Silva, S. M., and Venceslau, S. S. (2011). A comparative genomic analysis of energy metabolism in sulfate reducing bacteria and archaea. Front. Microbiol 2:69. doi: 10.3389/fmicb.2011.00069
Piłsyk, S., and Paszewski, A. (2009). Sulfate permeases—phylogenetic diversity of sulfate transport. Acta Biocim. Pol. 56, 375–384.
Pires, R. H., Lourenço, A. I., Morais, F., Teixeira, M., Xavier, A. V., Saraiva, L. M., et al. (2003). A novel membrane-bound respiratory complex from Desulfovibrio desulfuricans ATCC 27774. Biochim. Biophys. Acta 1605, 67–82. doi: 10.1016/S0005-2728(03)00065-3
Price, M. N., Ray, J., Wetmore, K. M., Kuehl, J. V., Bauer, S., Deutschbauer, A. M., et al. (2014). The genetic basis of energy conservation in the sulfate-reducing bacterium Desulfovibrio alaskensis G20. Front. Microbiol. 5:557. doi: 10.3389/fmicb.2014.00577
Rees, C. (1973). A steady-state model for sulphur isotope fractionation in bacterial reduction processes. Geochim. Cosmochim. Acta 37, 1141–1162.
Romão, C. V., Pereira, I. A. C., Xavier, A. V., LeGall, J., and Teixeira, M. (1997). Characterization of the [NiFe] Hydrogenase from the Sulfate ReducerDesulfovibrio vulgarisHildenborough. Biochem. Biophys. Res. Commun. 240, 75–79. doi: 10.1006/bbrc.1997.7598
Santos, A. A., Venceslau, S. S., Grein, F., Leavitt, W. D., Dahl, C., Johnston, D. T., et al. (2015). A protein trisulfide couples dissimilatory sulfate reduction to energy conservation. Science.
Sass, H., Steuber, J., Kroder, M., Kroneck, P., and Cypionka, H. (1992). Formation of thionates by freshwater and marine strains of sulfate-reducing bacteria. Arch. Microbiol. 158, 418–421.
Scheller, S., Goenrich, M., Thauer, R. K., and Jaun, B. (2013). Methyl-Coenzyme M reductase from methanogenic archaea: isotope effects on the formation and anaerobic oxidation of methane. J. Am. Chem. Soc. 135, 14975–14984. doi: 10.1021/ja406485z
Schiffer, A., Parey, K., Warkentin, E., Diederichs, K., Huber, H., Stetter, K. O., et al. (2008). Structure of the dissimilatory sulfite reductase from the hyperthermophilic archaeon archaeoglobus fulgidus. J. Mol. Biol. 379, 1063–1074. doi: 10.1016/j.jmb.2008.04.027
Sim, M. S., Bosak, T., and Ono, S. (2011a). Large sulfur isotope fractionation does not require disproportionation. Science 333, 74–77. doi: 10.1126/science.1205103
Sim, M. S., Ono, S., and Bosak, T. (2012). Effects of iron and nitrogen limitation on sulfur isotope fractionation during microbial sulfate reduction. Appl. Environ. Microbiol. 78, 8368–8376. doi: 10.1128/AEM.01842-12
Sim, M. S., Ono, S., Donovan, K., Templer, S. P., and Bosak, T. (2011b). Effect of electron donors on the fractionation of sulfur isotopes by a marine Desulfovibrio sp. Geochim. Cosmochim. Acta. 75, 4244–4259.
Sim, M. S., Wang, D. T., Zane, G. M., Wall, J. D., Bosak, T., and Ono, S. (2013). Fractionation of sulfur isotopes by Desulfovibrio vulgaris mutants lacking hydrogenases or type I tetraheme cytochrome c3. Front. Microbiol. 4:171. doi: 10.3389/fmicb.2013.00171
Smock, A., Bottcher, M., and Cypionka, H. (1998). Fractionation of sulfur isotopes during thiosulfate reduction by Desulfovibrio desulfuricans. Arch. Microbiol. 169, 460–463.
Sörbo, B. O. (1957). A colorimetric method for the determination of thiosulfate. Biochim. Biophys. Acta 23, 412–416.
Soriano, A., and Cowan, J. A. (1995). Sulfite reductase: active site residues are “noncatalytic.” comparison of reaction energetics for enzyme- and siroheme-catalyzed reduction of inorganic substrates. J. Am. Chem. Soc. 117, 4724–4725. doi: 10.1021/ja00121a038
Sra, A. K., Hu, Y., Martin, G. E., Snow, D. D., Ribbe, M. W., and Kohen, A. (2004). Competitive 15N kinetic isotope effects of nitrogenase-catalyzed dinitrogen reduction. J. Am. Chem. Soc. 126, 12768–12769. doi: 10.1021/ja0458470
Suh, B., and Akagi, J. M. (1969). Formation of thiosulfate from sulfite by desulfovibrio vulgaris. J. Bacteriol. 99, 210–215.
Tcherkez, G. G. B., Farquhar, G. D., and Andrews, T. J. (2006). Despite slow catalysis and confused substrate specificity, all ribulose bisphosphate carboxylases may be nearly perfectly optimized. Proc. Natl. Acad. Sci. U.S.A. 103, 7246–7251. doi: 10.1073/pnas.0600605103
Thode, H. G., Kleerekoper, H., and McElcheran, D. (1951). Isotope fractionation in the bacteria reduction of sulfate. Res. (Lond.) 4, 581–582.
Thode, H. G., Monster, J., and Dunford, H. B. (1961). Sulphur isotope geochemistry. Geochim. Cosmochim. Acta 25, 159–174.
Tudge, A. P., and Thode, H. G. (1950). Thermodynamic properties of isotopic compounds of sulphur. Can. J. Res. 28, 567–578.
Turchyn, A. V., Brüchert, V., Lyons, T. W., Engel, G. S., Balci, N., Schrag, D. P., et al. (2010). Kinetic oxygen isotope effects during dissimilatory sulfate reduction: a combined theoretical and experimental approach. Geochim. Cosmochim. Acta 74, 2011–2024. doi: 10.1016/j.gca.2010.01.004
Vairavamurthy, A., Manowitz, B., Luther Iii, G. W., and Jeon, Y. (1993). Oxidation state of sulfur in thiosulfate and implications for anaerobic energy metabolism. Geochim. Cosmochim. Acta 57, 1619–1623.
Venceslau, S. S., Cort, J. R., Baker, E. S., Chu, R. K., Robinson, E. W., Dahl, C., et al. (2013). Redox states of Desulfovibrio vulgaris DsrC, a key protein in dissimilatory sulfite reduction. Biochem. Biophys. Res. Commun. 441, 732–736. doi: 10.1016/j.bbrc.2013.10.116
Venceslau, S. S., Stockdreher, Y., Dahl, C., and Pereira, I. A. C. (2014). The “bacterial heterodisulfide” DsrC is a key protein in dissimilatory sulfur metabolism. Biochim. Biophys. Acta 1837, 1148–1164. doi: 10.1016/j.bbabio.2014.03.007
Wing, B. A., and Halevy, I. (2014). Intracellular metabolite levels shape sulfur isotope fractionation during microbial sulfate respiration. Proc. Natl. Acad. Sci. U.S.A. 115, 18116–18125.
Wolfe, B. M., Lui, S. M., and Cowan, D. (1994). Desulfoviridin, a multimeric-dissimilatory sulfite reductase from Desulfovibrio vulgaris (Hildenborough) Purification, characterization, kinetics and EPR studies. Eur. J. Biochem. 223, 79–89. doi: 10.1111/j.1432-1033.1994.tb18968.x
Young, E. D., Galy, A., and Nagahara, H. (2002). Kinetic and equilibrium mass-dependent isotope fractionation laws in nature and their geochemical and cosmochemical significance. Geochim. Cosmochim. Acta 66, 1095–1104. doi: 10.1016/S0016-7037(01)00832-8
Appendix
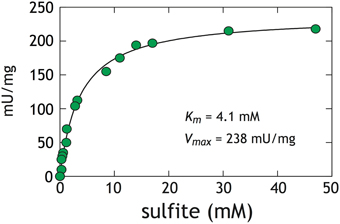
Figure A1. Activity assays with D. vulgaris DsrAB. The difference between initial and final concentration of sulfite after 2 h was used to calculate the rate. The Michaelis-Menten equation () was solved for experimental Km and Vmax under our conditions. The analytical error is less than the size of the symbols (2σ = 1μM). One unit (U) is defined as the quantity of enzyme that catalyzes the conversion of one micromol of substrate per minute. At both 10 and 15 mM initial sulfite we are assured to be well above the apparent DsrAB Km for sulfite. Reaction inhibition was not observed at sulfite concentrations as high as 50 mM (Wolfe et al., 1994; Soriano and Cowan, 1995).
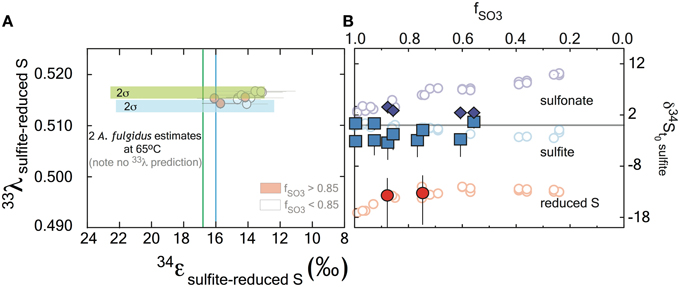
Figure A2. The major isotope ratios (δ34S) reported relative to the composition of sulfite at t0, for the A. fulgidus (closed symbols) and D. vulgaris (open symbols) experiments both as a function of reaction progress. The samples sets show a general consistency, particularly at the reduced S sites, despite the significant offet in temperature (20–30°C for D. vugaris relative to 65°C for A. fulgidus), consistent with kinetic theory (Bigeleisen and Wolfsberg, 1958), where temperature should impart a minimal effect over this range. The asymmetric error bars on reduced S moieties are a function of the non-linear correction for small sample sizes available for isotope.
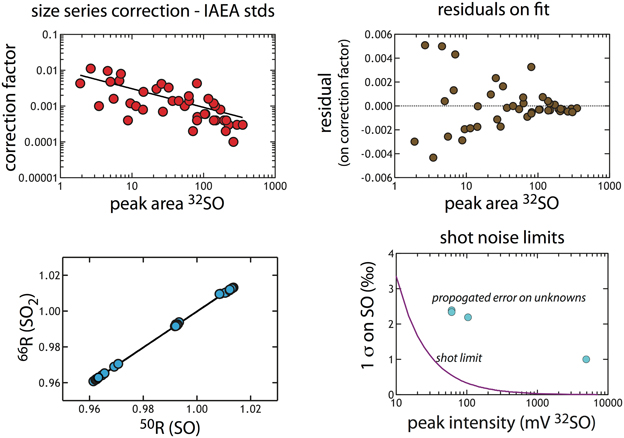
Figure A3. The size series correction calculated from IAEA standards (n = 44). (A) Plots the correction (value from against peak-integrated area). (A) Shows the calculated residual around that fit, demonstrating a symmetric distribution that scales with peak area. This is an illustration of the goodness of the fit. (C) The regression used to convert SO data to an SO2 scale. (D) The calculated shot noise for SO as a function of signal intensity (peak height in mV). This precision limit is below that which we propagate through the correction, and is provided for reference here.
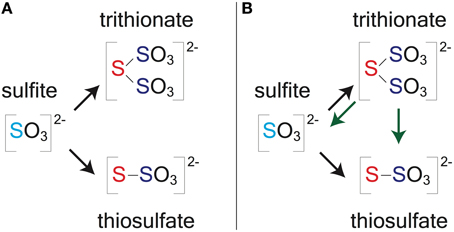
Figure A4. Reaction topology for closed-system models. (A) The simple model for early in the experiments (f > 0.85). (B) Shows a more complex reaction model that may be applicable later in the closed-system experiments (f < 0.85). Sulfite is present in light blue, reduced S is in red and the sulfonate moieties are in purple. Black (A,B) arrows represent the simple reduction of sulfite by DsrAB and then formation of thiosulfate and trithionate by combination of that partially reduced S with other sulfite molecules. The green arrows (B) represent the more complex scheme in which trithionate is then further reduced, releasing a sulfite and a thiosulfate.
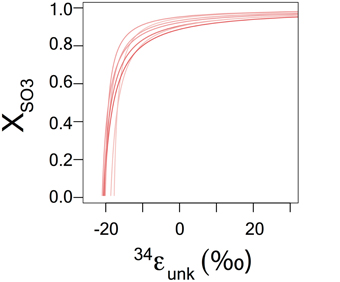
Figure A5. The relationship between the magnitude of a secondary fractionation () and the proportion of reduced sulfur deriving directly from sulfite reduction (XSO3). The balance (1- XSO3) is from the parallel reduction of bound sulfonate. Errors estimates are from the propagation calculation, incorporating both isotope and concentration analysis analytical errors (see below).
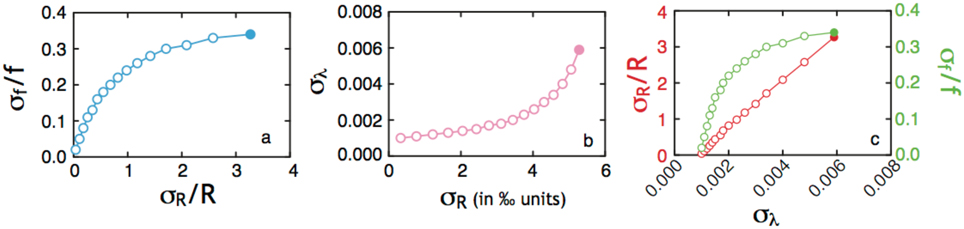
Figure A6. A series of frames (A–C) describing the origin and sensitivity of the controls on the resultant error in δ34S and 33λ plotted in Figures A2, A3. For this analysis, 34ε is set to 15.3‰ to be consistent with the reductive branch of the DsrAB experiments, the analytical precision of an isotope measurement is 0.2‰ in δ34S and 0.008‰ in Δ33S, and f is allowed to vary from 0.6 to 0.9 in 0.02 increments (noted by circles). This range in f is chosen to reflect the experimental range, and the circle is filled where f = 0.9. In (A), the covariance of the relative error in f (σf/f) is show to correlate with the error in the relative isotope ratio, σR/R. As the calculated error in the R derived from the Rayleigh equation (σR) increases, the consequence is an increase in the error in 33λ (B). Finally, the relationship between the errors in both f and R, as they contribute to the error on 33λ, are presented in frame C. Frames B,C do not approach the origin as a result of f not approaching the limits of 0 and 1, and also due to the multivariate nature of the propagation.
Keywords: microbial sulfate reduction, enzyme-specific isotope fractionation, minor sulfur isotopes, global sulfur cycle, dissimilatory sulfite reductase
Citation: Leavitt WD, Bradley AS, Santos AA, Pereira IAC and Johnston DT (2015) Sulfur Isotope Effects of Dissimilatory Sulfite Reductase. Front. Microbiol. 6:1392. doi: 10.3389/fmicb.2015.01392
Received: 06 July 2015; Accepted: 23 November 2015;
Published: 24 December 2015.
Edited by:
Orit Sivan, Ben Gurion University of the Negev, IsraelReviewed by:
Mustafa Yucel, Middle East Technical University, TurkeyTimothy Ferdelman, Max Planck Institute for Marine Microbiology, Germany
Florian Einsiedl, Technische Universität München, Germany
Copyright © 2015 Leavitt, Bradley, Santos, Pereira and Johnston. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: William D. Leavitt, d2xlYXZpdHRAZXBzLnd1c3RsLmVkdQ==
†These authors have contributed equally to this work.