 Qi Ye
Qi Ye Jianan Liu
Jianan Liu Jinzhou Du
Jinzhou Du Jing Zhang
Jing Zhang- State Key Laboratory of Estuarine and Coastal Research, East China Normal University, Shanghai, China
Submarine groundwater (SGD) is one of the most significant pathways for the exchange of groundwater and/or source of nutrients, metals and carbon to the ocean, subsequently cause deleterious impacts on the coastal ecosystems. Microorganisms have been recognized as the important participators in the biogeochemical processes in the SGD. In this study, by utilizing 16S rRNA-based Illumina Miseq sequencing technology, we investigated bacterial diversity and distribution in both fresh well water and brackish recirculated porewater along the coasts in the Yellow Sea. The results showed that Actinobacteria and Betaproteobacteria, especially Comamonas spp. and Limnohabitans spp. were dominated in fresh well samples. Distinct patterns of bacterial communities were found among the porewater samples due to different locations, for examples, Cyanbacteria was the most abundant in the porewater samples far from the algal bloomed areas. The analysis of correlation between representative bacterial taxonomic groups and the contexture environmental parameters showed that fresh well water and brackish porewater might provide different nutrients to the coastal waters. Potential key bacterial groups such as Comamonas spp. may be excellent candidates for the bioremediation of the natural pollutants in the SGD. Our comprehensive understanding of bacterial diversity in the SGD along the coasts of the Yellow Sea will create a basis for designing the effective clean-up approach in-situ, and provide valuable information for the coastal management.
Introduction
Submarine groundwater discharge (SGD) includes both fresh meteoric groundwater to the ocean by terrestrially-driven directly and recirculated seawater by permeable sediment (Garcia-Solsona et al., 2010). More and more works showed that SGD is one of the most important pathways for the exchange of groundwater and/or source of dissolved compounds (e.g., nutrients, metals, carbon) to the ocean, which may cause negative impacts on the coastal ecosystems (Burnett et al., 2006; Moore, 2010). Nitrogen input through the SGD may contribute to the eutrophication of coastal regions (Moore, 2010). Several case studies showed that increased nutrient supply via SGD might be a key factor for initiating and fueling the persistent harmful algal blooms (HABs) (LaRoche et al., 1997; Hu et al., 2006; Lee and Kim, 2007; Smith and Swarzenski, 2012).
The Yellow Sea, which is as a shallow semi-enclosed water body of the Northwest Pacific Ocean, shows such a strong seasonality in the nutrients concentration and their components (Liu et al., 2013). SGD has been determined as a main nutrient source to the Yellow Sea, which is one of the largest continental shelves in the world (Kim et al., 2005; Waska and Kim, 2011). Microorganisms have been shown as the important components mediating chemical reactions in the SGD in some concerned cases (Boehm et al., 2004; Paytan et al., 2004; Halliday and Gast, 2010). Recent studies focused on the bacterial communities in the surrounding sea water during macroalgal blooms of Ulva prolifera in the Yellow Sea (Burke et al., 2011; Liu et al., 2011; Zhang et al., 2014). For example, Analysis of nifH gene clone library revealed that heterotrophic nitrogen fixers, mostly represented by Vibrio-related Gammaproteobacteria, were dominated in both of surface waters that were covered and non-covered with massive macroalgal canopies of U. prolifera in the Yellow Sea in the summer of 2011 (Zhang et al., 2014). However, for our knowledge, up to now, there are few reports on the bacterial compositions in SGD along the coast of the Yellow Sea.
Microorganisms play pivotal roles in biogeochemical cycles in marine ecosystems (DeLong and Karl, 2005; Zehr and Kudela, 2011). It was reported that several studies using 16S rRNA (Fields et al., 2005), functional genes (Yan et al., 2003; Santoro et al., 2006, 2008), microarray (Waldron et al., 2009), and metagenomics approaches (Hemme et al., 2010) to reveal bacterial communities in groundwater systems. The decreased diversity was observed in groundwater contaminated with higher levels of nitric acid and uranium waste in the Field Research Center (FRC) of the U.S. Department of Energy Environmental Remediation Science Program (Oak Ridge, TN, USA) (Fields et al., 2005). Ammonia-oxidizing bacterial and archaeal abundance as well as denitrifier communities were characterized along a nitrate and salinity gradient in groundwater samples at Huntington Beach, CA, USA (Santoro et al., 2006, 2008). Metagenomical analysis showed that a groundwater microbial community composed of clonal denitrifying γ- and β-proteobacterial populations in an extreme low-pH environment contaminated with high levels of uranium, nitric acid, technetium and organic solvents (Hemme et al., 2010). Biodegradation or biotransformation of pollutants with microorganisms is the most potential cost-effective bioremediation technology in the natural environments (Fields et al., 2005; Hwang et al., 2009). Pilot-scale uranium in situ bioremediation experiment suggested that the indigenous microbial communities can be successfully stimulated after 2 years of biostimulation with ethanol in FRC groundwater ecosystem, U(VI) levels were reduced from the initial concentration of 50 mg/L to below drinking water standard (< 30 μg/L) (Xu et al., 2010). Zhang et al. (2015) reported that amendment of the slow-release polylactate hydrogen-release compound (HRC) stimulated FRC groundwater microbial communities associated with HRC degradation and reduction of , Cr(VI), Fe(III), and . In the present work, by using 16S rRNA gene-based Miseq Illumina sequencing approach, we focused on the bacterial compositions in both fresh well water and recirculated brackish porewater along the coast of the Yellow Sea. The goal is to explore the key bacterial groups, which can play their potential ecological roles in nitrogen and carbon flux by SGD to the coastal areas. The relationship between representative taxonomic groups and contexture environmental parameters were also investigated. Our understanding on the potential bacterial candidates for bioremediation will help us design the effective clean-up technology for coastal environmental management.
Materials and Methods
Sample Collection and Measurements of Physic-Chemical Parameters
Groundwater samples (well and pore water) in our field observation were collected from wells and beach in May, 2014. Well samples along the shore were pumped from the wells utilizing the water pump; pore water samples from offshore zone were collected from a push-point piezometer using a peristaltic pump (Charette and Allen, 2006). The temperature, salinity and pH of groundwater were measured directly in the field using a portable salinometer with multiple parameters (Germany, multi 350i). Sampling depth of each sample site ranged from 0.4 m to > 20 m (Table 1).
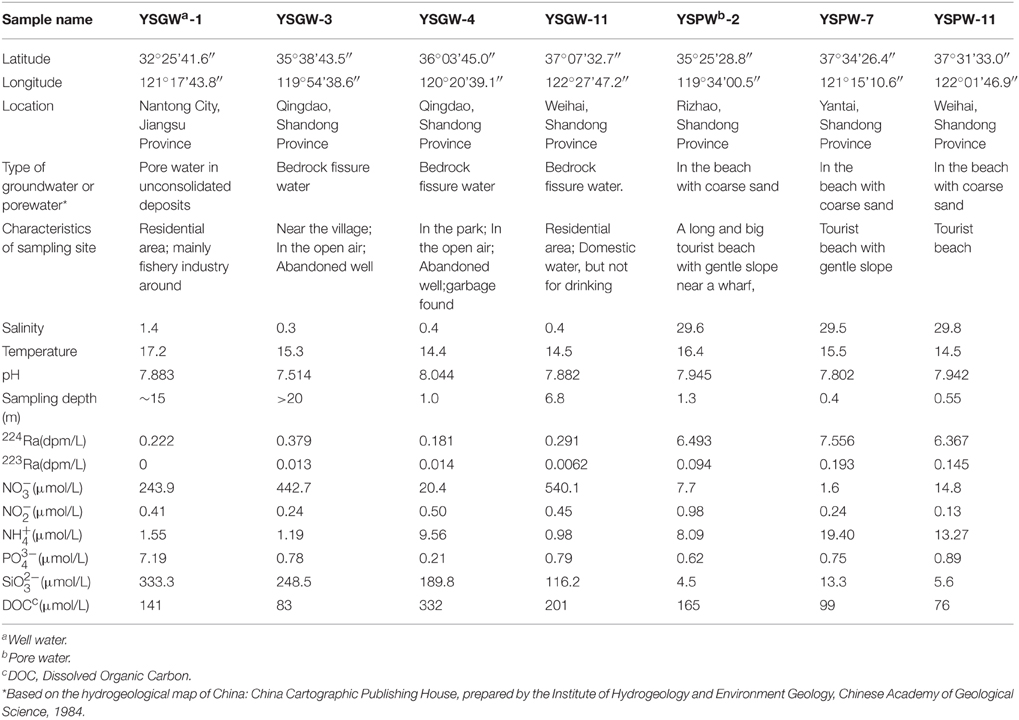
Table 1. Site descriptions and chemical measurement.
Four well water samples (~40 L) and three pore water samples (~20 L) were collected for Ra isotope measurement. After filtrating (pore size: 0.5 μm), Ra isotopes in the water were extracted using a MnO2-impregnated acrylic fiber column (20 g), then the column with Mn-fiber was immediately placed in the Radium Delayed Coincidence Counter (RaDeCC) to measure the short-lived isotope 223Ra and 224Ra in the field (Moore and Arnold, 1996).
Corresponding water samples (~60 mL) for nutrient analysis were collected with polyethylene bottles filtered through 0.45 μm cellulose acetate filters. Then the filtrates were poisoned with saturated HgCl2 and stored in the dark. The nutrient concentrations (, , , , and ) were then analyzed using an auto-analyzer (Model: Skalar SANplus146) (Liu et al., 2005). DOC samples were filtered via clean Nylon filter (pore size: 0.45 μM) immediately after collection and kept at −20°C until analysis. In the lab, DOC samples were measured with a TOC analyzer (Shimadzu® TOC-LCPH).
Replicate, 300-mL of each submarine groundwater sample were collected on a 0.22 μm pore size polycarbonate filter (Nuclepore Track-Etched Membrane, Whatman). The filter was placed in a sterile 1.5-mL microcentrifuge tube and immediately store at −20°C.
DNA Extraction, PCR, and Sequencing
Total DNA was extracted from the filter using a MoBio PowerWater® DNA Isolation Kit (MOBIO Laboratories, Carlsbad, CA, USA) according to the manufacturer's instruction. DNA from two independent extractions were combined, and their concentration and purity were measured spectrophotometrically with NanoDrop ND2000.
Minimum numbers of PCR cycles were performed and three independent PCR mixtures were pooled for each sample to decrease PCR bias. Briefly, bacterial V4-V5 hypervariable regions of 16S rRNA genes were then amplified using the specific barcoded universal primer pairs 515F (5′-GTGCCAGCMGCCGCGG-3′) and 907R (5′-CCGTCAATTCMTTTRAGTTT-3′) (Xiong et al., 2012). Cycling conditions were 2 min at 95°C, followed by 25 cycles with 30 s at 95°C, 30 s at 55°C, and 30 s at 72°C, and a final extension period of 5 min at 72°C.
PCR products were purified using the AxyPreDNA gel extraction kit (Axygen Biosciences, USA) following the manufacturer's protocol and then quantified by QuantiFluorTM-ST (Promega, USA). Reaction mixtures were pooled in equimolar ratios and paired-end reads were generated on an Ilumina Miseq PE250 (Majorbio Bio-Pharm Technology Co., Ltd., Shanghai, China).
Sequence Data Process, OTU Cluster, and Taxonomic Assignment
Raw fastq files were demultiplexed, quality-filtered using QIIME (version 1.17) (Caporaso et al., 2010) with the criteria as described previously (Li et al., 2014). Read data of each sample was imported in Fastq format according to index barcode sequence. Operational Taxonomic Units (OTUs) were clustered at 97% similarity using UPARSE (version 7.1 http://drive5.com/uparse/). Chimeric sequences were identified and removed using UCHIME. By using the usearch_global command, the number of reads from each sample assigned to each OTU was generated in an “OTU table.” Representative 16S rRNA gene sequence of each OTU was analyzed by RDP Classifier (http://rdp.cme.msu.edu/) against the Silva (SSU115) 16S rRNA database with 70% confidence threshold.
Phylogenetic Analyses
The sequences of the representative OTUs obtained in this study were compared to those in the National Center for Biotechnology Information nucleotide database by using BLAST searching. The closest sequences and selected reference sequences were downloaded and aligned using Clustal W. Phylogenetic tree was created in MEGA6 using the neighbor-joining algorithm with a bootstrap test of 1000 replicates and maximum composite likelihood model (Tamura et al., 2013).
Statistical Analyses
Alpha diversity metrics and coverage were calculated using the Mothur Program (Schloss et al., 2009). R program was used to plot Heatmap figure (R Development Core Team., 2013). Bray-Curtis dissimilarities were calculated based on genus/sample abundance matrix using function vegdist of the vegan package in R (Oksanen et al., 2007). Hierarchical clustering of the samples was performed with the complete method using the function hclust of stats package in R (R Development Core Team., 2013).
Pearson's correlation coefficient was calculated using SPSS software, corresponding heatmap was plotted using gplots package in R (Oksanen et al., 2007; Warnes et al., 2012).
Nucleotide Sequence Accession Numbers
Paired end Illumina sequence data from this study were submitted to the NCBI Sequence Read Archive (SRA) under accession number SRP064231.
Results
Site Description and Physic-chemical Characteristics
Fresh well water and brackish porewater of submarine groundwater samples were selected in this study. Wells along the shore were dug by native habitants; Porewater within Yellow Sea offshore area were collected from ~50 to 130 cm below the sediment-water interface (Table 1).
Based on the types of submarine groundwater and physicochemical parameters, seven samples were separated into two groups: Group W included four samples (YSGW1, YSGW3, YSGW4, and YSGW11) collected from the wells, the salinity of well waters ranged from 0.3 to 1.4‰, indicating their freshwater environment; Group P included three porewater samples (YSPW2, YSPW7, and YSPW11) with relative higher salinity (~30‰) (Table 1, Figure 1).
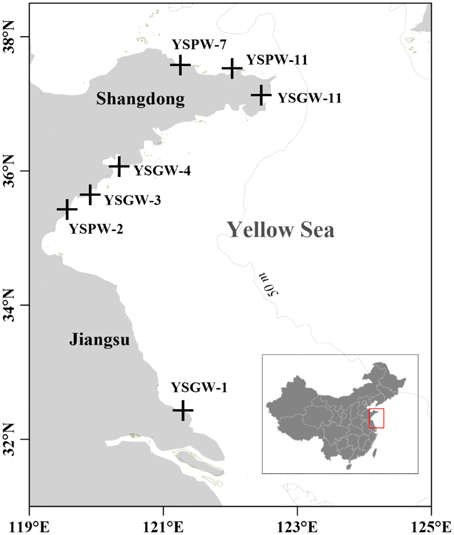
Figure 1. Map showing the sampling stations of submarine groundwater along the coasts of the Yellow Sea.
The other hydrological properties and chemical parameters of submarine groundwater are also summarized in Table 1. Fresh well waters had an average value of 0.269 and 0.0083 dpm/L for 224Ra and 223Ra. However, pore water had a higher value for 224Ra and 223Ra, was 6.805 and 0.144 dpm/L, respectively. Several nutrients in the specific well water samples were much higher than those in the pore water, for examples, nitrate concentrations ranged from 243.9 to 540.1 μmol/L within Group W except sample YSGW 4 (20.4 μmol/L), but still higher than those in porewater samples (1.6–14.8 μmol/L). Ammonium concentration ranged 8.09–19.40 μmol/L in porewater samples, and 0.98–1.55 μmol/L within Group W except sample YSGW 4 (9.56 μmol/L). Porewater sample YSGW 2 had the highest nitrite concentration and well water sample YSGW 1 had the highest phosphate concentration. The silicate concentrations in porewater samples were below 15 μmol/L, while the concentrations in well samples were much higher (116.2–333.3 μmol/L). No distinct pattern of DOC concentrations between well water and porewater samples, but YSGW 4 had the highest DOC concentration. The other parameters such as pH and temperature were relatively constant at all sites.
Bacterial Diversity and Cluster Analysis
We obtained a total of 87,475 high quality bacterial V4-V5 Illumina sequences, with an average read length of 396 bp from seven submarine groundwater samples.
According to 97% identity cutoff, there were 1078 OTUs in the complete data set. Among them, only 23 OTUs were found in all well water samples and 169 OTUs in porewater samples. Good's coverage was 99~99.8% for all samples, representing almost whole range of bacterial diversity. The Chao, ACE value, and Shannon evenness all indicated that the α-diversity of bacterial community is lower in the well water samples than in porewater samples (Table 2).
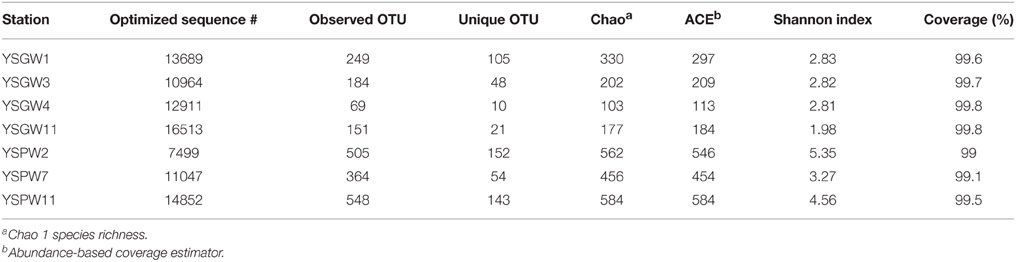
Table 2. Diversity and richness estimators for Illumina libraries.
The hierarchical heatmap at the bacterial genus level showed that seven samples could be organized into two main groups: The first group was composed of four well water samples, YSGW1 and YSGW4 were clustered together, and YSGW3 and YSGW11 formed another cluster; the second group was composed three porewater samples, turbid porewater YSPW2 was clustered separately from clear porewater YSPW7 and YSPW11 (Figure 2).
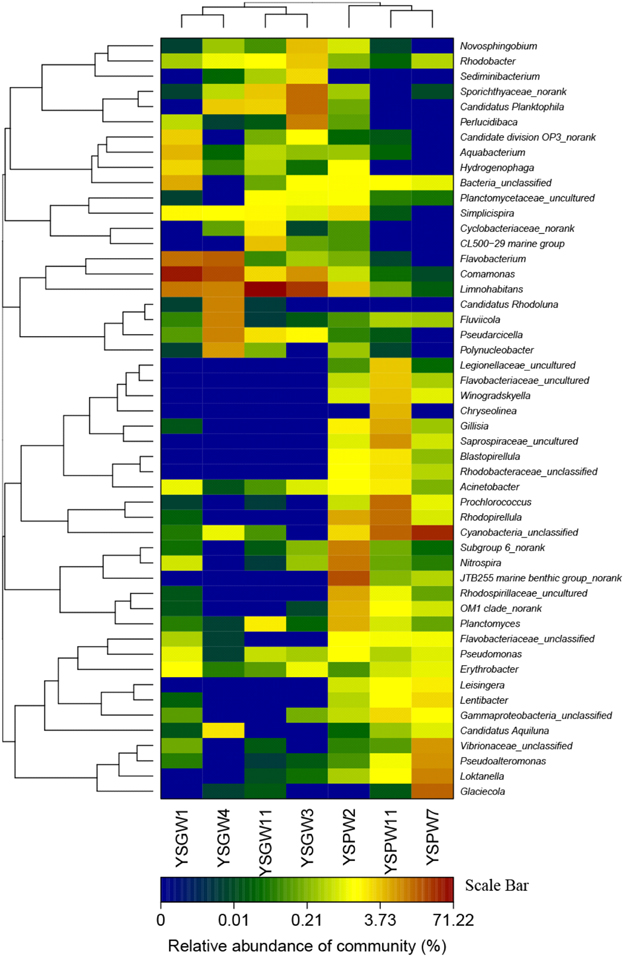
Figure 2. Heatmap showing the relative abundance and distribution of genus-based OTU illumina reads. The color code indicates relative abundance, ranging from blue (low abundance) to black to brown (high abundance).
Bacterial Distribution
Figure 3 showed that there were some variations in the percentage composition of illumina sequences among the samples. Proteobacteria was the most abundant phylum in all samples (43.7~76.7%) except YSPW11 (25.9%). Bacteriodetes, which was the most abundant phylum in YSPW11 (27.5%), was also distributed in the other samples. Actinobacteria was the other prevalent phylum in wellwater samples YSGW3 (27.2%), YSGW4 (16.7%), and YSGW11 (11.6%). Planctomycetes and Acidobacteria formed the second (17.2%) and third (12%) dominant groups after Proteobacteria within YSPW2. Cyanobacteria represented 32.8 and 22.7% of sequences within porewater samples YSGW7 and YSGW11. Interestingly, Cyanobacteria was almost absent from the other five samples.
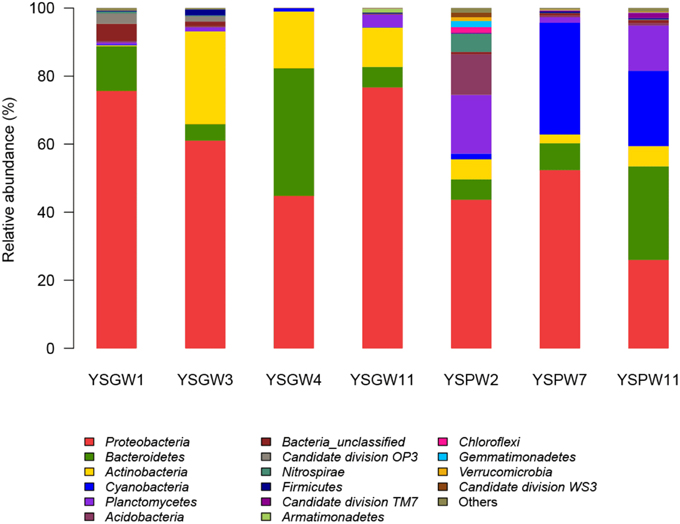
Figure 3. Phylum-level taxonomic distribution. Bars represented the percentage of Illumina tag composition represented by each phylum. Bacterial taxa represented by less than 1% reads are pooled as “other.”
Phylogenetic Analyses
Main Groups of Actinobacteria and Betaproteobacteria within Group W Samples
OTUs related to Actinobacteria and Bataproteobacteria were frequently detected within fresh well water samples (Figure 4). The actinobacterial sequences of OTU 1070 and 603 was the most frequently retrieved from YSGW3, which were identical to the environmental clones from Taihu (China) (HQ653856) (Zeng et al., 2012a) and Bourget (French) (FJ447766) (Debroas et al., 2009). Sequences of three OTUs 17, 123, and 185 were obtained from YSGW4, had 99.5, 99.7, 99.0% similarity with three representative planktonic freshwater Actinobacteria Rhodoluna lacicola strain MWH-EgelM2-3.2 (FJ545223) (Sharma et al., 2009), Candidatus Rhodoluna limnophila (NR_125490) (Hahn, 2009), Candidatus Aquiluna rubra (NR_125489) (Hahn et al., 2004) respectively (Figure 4).

Figure 4. Neighbor-joining tree showing phylogenetic relationships among the representative OTUs obtained in this study and reference 16S rRNA sequences retrieved from the NCBI GenBank for Actinobacteria and Betaproteobacteria. These OTUs were represented by more than 1% reads from a single station and/or from multiple stations within well water samples. The OTUs obtained in this study are shown in bold type. The numbers in parentheses indicate the percentage composition of reads in each station in the following order: (YSGW1, YSGW3, YSGW4, YSGW11) (YSPW2, YWPW7, YSPW11). The scale bar represents the estimated number of nucleotide changes per sequence position. The numbers at the nodes show the bootstrap values obtained after 1000 resamplings, only values of > 50 are shown. Accession numbers for the reference sequences are shown in parentheses.
Class Betaproteobacteria accounted for 38.7–70.5% within Group W, and there is a niche separation among different genus within Betaproteobaceria (Figure 4). Both YSGW1 and YSGW4 were characterized by codominance of genera Comamonas (43.6 and 22.8%), and Limnohabitans (11.1 and 9.8%), Limnohabitans was the most abundant genus in YSGW3 (28.6%) and YSGW11 (64.1%). Representative OTU286 had 99% similarity with Comamonas testosteroni strain CNB-2 (NR_102841), which is capable of degrading aromatic compounds (Ma et al., 2009), and OTU 883 had 99.5% similarity with nitrogen cycling Betaproteobacteria Limnohabitans sp. Rim28 (HE600684) (Kasalickı et al., 2013) and 99.7% similarity with Limnohabitans parvus strain II-B4 (NR_125542). OTU 896 accounted for 6.55% of total bacteria in YSGW4, but seldom detected in other samples, had 99% similarity with Polynucleobacter acidiphobus strain MWH-PoolGreenA3 (NR_125545), which may utilize autochthonous substrate sources primarily supplied by phytoplankton (Hahn et al., 2011).
Main Groups of Gammaproteobacteria, Cynobacteria, and Planctomycetes within Group P Samples
Gammaproteobacteria, Cyanobacteria or Planctomycetes was dominant in one or two Group P samples (Figure 5). The sequences of OTU 874, representing 30.4 and 7.6% in YSPW7 and YSPW11 respectively, were remotely related to the cyanobacterium Calothrix desertica strain PCC 7102 (NR_114995) (86% similarity) (Sihvonen et al., 2007). OTU 874 was also associated phylogenetically (100% similarity) with a cyanobacterial clone PROA52S_10 (GQ916428) recovered from the Florida west surface coastal water with high cell abundance of brevetoxin-producing dinoflagellate Karenia brevis (Jones et al., 2010). In addition, OTU 67 containing 12.5% sequences in YSGW11, had 97% similarity with Prochlorococcus marinu SS120 (NR_074172) with a nearly minimal oxyphototrophic genome (Dufresne et al., 2003).
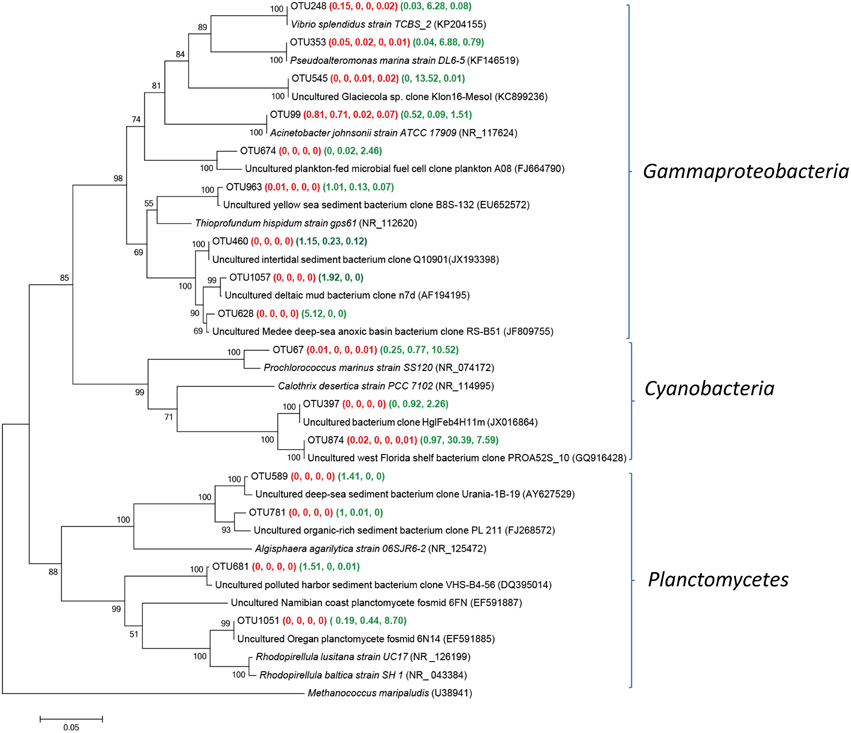
Figure 5. Neighbor-joining tree showing phylogenetic relationships among the representative OTUs obtained in this study and reference 16S rRNA sequences retrieved from the NCBI GenBank for Gammaproteobacteria, Cyanobacteria, and Planctomycetes. These OTUs were represented by more than 1% reads from a single station and/or from multiple stations within porewater samples. The OTUs obtained in this study are shown in bold type. The numbers in parentheses indicate the percentage composition of reads in each station in the following order: (YSGW1, YSGW3, YSGW4, YSGW11) (YSPW2, YWPW7, YSPW11). The scale bar represents the estimated number of nucleotide changes per sequence position. The numbers at the nodes show the bootstrap values obtained after 1000 resamplings, only values of > 50 are shown. Accession numbers for the reference sequences are shown in parentheses.
OTU 248, 353, and 545 gave the 100% match to Gammaproteobacteria Vibrio splendidus strain TCBS_2 (KP204155), Pseudoalteromonas marina strain DL6-5 (KF146519) (Wang et al., 2014), and Uncultured Glaciecola sp. clone Klon16-MesoI (KC899236) (Scheibner et al., 2014). Sequences of these three OTUs accounted for 26.68% of total bacteria in YSGW7. Within class Gammaproteobacteria, we found OTU628 matched sedimentary clone from the Medee deep-sea hypersaline anoxic basin (JF809755) (Akoumianaki et al., 2012) at 99.2% similarity and has its highest relative abundance in YSPW2 (Figure 5).
Approximately 8.7% sequences of YSPW11 were grouped into OTU1051, showed 100% identity with fosmid 6N14 (EF591885), which was retrieved from 200 m depth off the coast of Oregon (Woebken et al., 2007), they were also related to Rhodopirellula baltica strain SH 1T (NR_043384) encoding high numbers of sulfatases (~95% similarity) (Glöckner et al., 2003).
Main Groups of Bacteriodetes Detected in Both Group W and P Samples
Bacteriodetes was frequently observed in all studied submarine groundwater samples, however, the main groups of Bacteroidetes were different between Group W and P samples (Figure 6). Five Flavabacterium related OTUs were found in YSGW1 and YSGW4, indicating microbial degradation of high-molecular-weight organic matter in these samples (Kirchman, 2002). Two OTUs 19 and 275 in YSGW4 matched uncultured environmental clones from Lake Zurich (HE574353 and HE574358), which may participate in N-acetyl-glucosamine uptake (Eckert et al., 2012). OTU 219 had a significantly higher abundance in the YSGW4 and 100% identity to uncultured Flectobacillus clone ZS-2-379 (FN668111) (Van den Wyngaert et al., 2011), members of the grazing-resistant Flectobacillus genus had the ability to defense filament-forming predators (Šimek et al., 2010a). OTUs 159 and 700 in YSGW11 were closely affiliated with clones recovered from macroalgal surface (GU451453 and GU451530) (Lachnit et al., 2011).
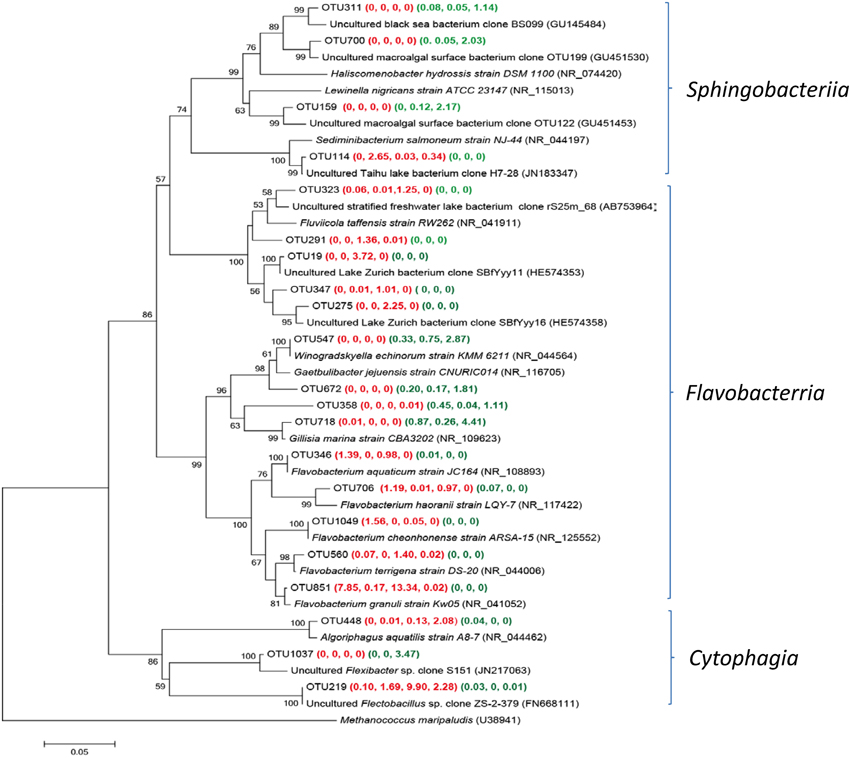
Figure 6. Neighbor-joining tree showing phylogenetic relationships among the representative OTUs obtained in this study and reference 16S rRNA sequences retrieved from the NCBI GenBank for Bacteriotedes. These OTUs were represented by more than 1% reads from a single station and/or from multiple stations within well water or porewater samples. The OTUs obtained in this study are shown in bold type. The numbers in parentheses indicate the percentage composition of reads in each station in the following order: (YSGW1, YSGW3, YSGW4, YSGW11) (YSPW2, YWPW7, YSPW11). The scale bar represents the estimated number of nucleotide changes per sequence position. The numbers at the nodes show the bootstrap values obtained after 1000 resamplings, only values of >50 are shown. Accession numbers for the reference sequences are shown in parentheses.
Correlation Analysis between Bacterial Taxonomic Groups and Environmental Parameters
In order to investigate whether some bacterial taxonomic groups are characteristics of specific environmental conditions, we calculated the Pearson's correlation coefficient between bacterial taxonomic groups and contextual environmental parameters (Gobet et al., 2012). The selected phyla/classes were divided into two groups: rare taxonomic group is defined as the phylum/class represented by more than 1% reads only from a single station, whereas abundant taxonomic groups is defined as the phylum/class represented by more than 1% reads from at least two stations (Table S1). Within abundant taxonomic groups, we found significant Pearson's correlation coefficients between isotope 224Ra tracers and Phyla including Cyanobacteria, Betaproteobacteria, and Gammaproteobacteria. Salinity is an important environmental parameter which significantly influenced the Betaproteobacteria (−0.849). Pearson's correlation coefficient between Betaproteobacteria and nitrate (0.76)/ammonium (−0.774) suggested increased in sequences as nitrate concentration got higher and ammonium concentration got lower, indicating ammonium oxidation by Betaproteobacteria. Cyanobacteria was positively correlated with the ammonium concentration (0.863) suggesting nitrogen fixation by Cyanobacteria. Sequences within class Cytophagia had highly negative correlation with temperature but positive with DOC concentration, suggested organic matter degradation of this low temperature favorable bacterial group. Interestingly, six out of nine rare taxonomic groups were detected in porewater YSPW2, and these six groups had highly positive correlation with nitrite concentration (Figure 7).
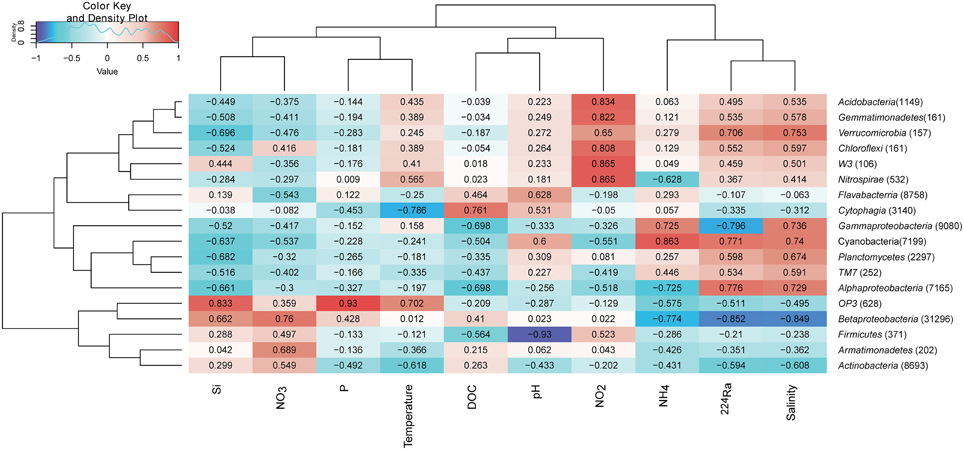
Figure 7. Environmental factors associated with variations of the bacterial community structure at the phylum level. In order to obtain a higher resolution, the Proteobacteria phylum level was separated into Alpha, Beta, and Gamma-Proteobacteria classes. Phylum Bacteriodetes was separated into classes Flavabacterria and Cytophagia. The total number of sequences in each phylum is indicated in parentheses. Pearson's correlation coefficients between -1 and 1 are shown in the rectangle, which indicates correlations between phylum/class sequence abundance and selected environmental parameters. For example, a firebrick colored rectangle (0.76) between nitrate and Betaproteobacteria indicates a higher number of sequences with increasing nitrate concentration, a blue rectangle (−0.774) between Betaproteobacteria and ammonium indicates a higher number of sequences with decreasing ammonium concentration. The color code indicates Pearson's correlation coefficients, ranging from blue (−1) to white (0) to firebrick (1). The density showed the distribution of Pearson's correlation coefficients between −1 and 1. Si, silicate; P, phosphate; NO2, nitrite; NO3, nitrate; NH4, ammonium; 224Ra, Radium isotope tracer; DOC, dissolved organic carbon.
Discussion
Distinct Bacterial Communities within Porewater Samples
In this study, different locations may result in distinct bacterial communities among the porewater samples, for example, YSPW 2 was close to the regions where the green tides frequently break out, and there were specific bacterial taxonomic groups only detected in this site, but lacked some phyla which were abundant in other two porewater samples (Figure 5). One of the negative effects of the green tides on the marine ecosystem was the production of toxic hydrogen sulfide into surrounding environments (Lyons et al., 2014). The second most sequences in turbid YSPW 2 porewater sample were affiliated with JTB255 cluster, which was considered as the putative sulfide-oxidizing bacteria (Bowman and McCuaig, 2003). It is proposed that the toxic hydrogen sulfide could be detoxified in the porewater.
The Cyanobacteria was the most abundant group with OTU 874 comprising 30.4% in YSPW 7 and 7.6% in YSPW11, but only 0.97% in YSGW 2. Sequences of OTU 874 were remotely related to the heterocystous diazotrophic cyanobacterium Calothrix genus (Zehr, 2011), and 100% identical to the environmental clone PROA52S 10 from Karenia brevis dominated surface water (Jones et al., 2010), showing this uncultured cyanobacterial group might pose the putative ecological functions to different phytoplankton blooms. In addition, the most abundant OTU 67 in YSGW 11, had 97% similarity with the non-diazotrophic picocyanobacterium Prochlorococcus marinus strain SS12, suggesting the cohabitation of two groups of cyanobacteria in this sample. Unicellular phototroph Prochlorococcus marinus strain SS12 is well known for its characteristics of the tiny cell size, autotrophic metabolism and adaption to low light niche (Dufresne et al., 2003), this low-light-adapted ecotype may make it possible to survive in the porewater at dimmer light.
It has been shown that microalgal growth was linked to nutrients released by the nitrogen fixing cyanobacteria. For instance, following aerolian iron input, nitrogen fixer Trichodesmium produced dissolved organic nitrogen (DON) for stimulating the growth of Karenia brevis, subsequently caused the dinoflagellate bloom in the Gulf of Mexico waters (Lenes et al., 2001). Jones et al. (2010) found the higher abundance of cyanobacteria in the zero/low K. brevis libraries than those of medium/high K. brevis libraries. The Cyanobacteria was also detected in the surrounding water during an early Ulva bloom (Liu et al., 2011). Notably, cyanobacteria usually grow better at the early stage of macroalgae blooms, this could be explained that allelopathic chemicals produced by periphyton biofilms suppress cyanobacterial growth in macroalgae dominated waters (Wu et al., 2011). This may explained lower abundant cyanobacteria detected in YSPW 2. We speculated that the newly fixed N released by the Cyanobacteria in surface water and porewater might be significant in initiating the algal blooms. Considering high abundance of cyanobacteria detected in porewater samples, SGD became the potential nutrient source for adjacent coastal areas.
Bacterial community structures associated with the surface of Ulva prolifera and in surrounding seawater in Jiaozhou Bay during the Ulva bloom phases in 2008 were reported (Liu et al., 2011). They found Orders Alteromonadales, Flavobacteriales, and Rhodobacterales were the common bacterial groups on the surface of U. prolifera, and Alteromonaldales was also dominant in the seawater. The genus Glaciecola within Alteromonadales was dominated not only in the algal surface but also in the seawater. In porewater sample YSGW7, sequences affiliated with three orders accounted for 42.7% of total bacteria. Ra isotope analysis showed recirculated groundwater discharge in Group P samples, it was likely that these bacteria received from the seawater. An indoor mesocosm experiment with natural plankton communities from the western Baltic Sea revealed that Glaciecola sp. was the single most abundant taxon during the phytoplankton peak in the cold water (Scheibner et al., 2014). High abundance of Glaciecola cells has also been observed in temperate North Sea during a spring bloom (Teeling et al., 2012). The considerable presence of Genus Glaciecola during the blooming phase of the diatoms in different studied regions suggested its significant roles in consuming dissolved organic carbon released by phytoplankton. YSGW7 contained 13.5% Glaciecola related sequences, indicated that SGD was significant source of the carbon flux to the coastal water. Meanwhile, Gammaproteobacteria was observed as dominated bacterial group during the phytoplankton peak (Teeling et al., 2012; Scheibner et al., 2014), the coexistence of Gammaproteobacteria and Cyanobacteria in our studied porewater sample was probably intensified contributions of the nutrient and DOC by SGD to the occurring of macroalgae bloom in the coastal areas.
Several studies of bacterial communities on the surface of marine macroalgae have shown the distinctive compositions of planctomycetes could be observed in association with different marine macroalgae (Lage and Bondoso, 2011; Bondoso et al., 2014). The second most abundant OTU 1051 in YSPW 11 was closely related to Planctomycetes Rhodopirellula Baltic strain SH 1T, this strain was also frequently found in epiphytic communities of many macroalgae in a widespread geographic locations (Lage and Bondoso, 2011). The genome of Rhodopirellula baltic strain SH 1T contains a large number of sulfatases, which are known to be involved in carbon recycling of complex sulfated heteropolysaccharides (Glöckner et al., 2003). The ability of Planctomycetes to degrade sulfatated heteropolysaccharides provides us the efficient pathway to obtain polysaccharides with lower molecular weights in porewaters, which were proved to possess important pharmacological activities such as antioxidant activities (Qi et al., 2005; Costa et al., 2010; Li et al., 2013).
The Putative Ecological Role of Genus Limnohabitans in the Fresh Well Water
Betaproteobacteria and Actinobacteria constituted the two major groups in four fresh well water samples, in agreement with the finding that a few phylogenetic clusters are dominant in a variety of freshwater ecosystems (Allgaier and Grossart, 2006; Newton et al., 2011). Our correlation analysis confirmed Betaproteobacteria favored lower salinity condition (Figure 7).
Members of Genus Limnohabitans within Betaproteobacteria were frequently predominant in freshwater bacterioplankton communities (Šimek et al., 2010b). We reported that Bacteria of the genus Limnohabitans were the major group in the fresh well samples (Figure 3). Limnohabitans spp. are the well-studied opportunistic bacterial groups (Šimek et al., 2005), with fast living generation time, specialization on low molecular weight dissolved organic matter, and highly vulnerability to size selective predation (Salcher, 2013). Autochthonous algal-derived organic matters and products of the photolysis of dissolved materials are two main groups of substrates for the growth of members of Genus Limnohabitans (Šimek et al., 2011). Genus Limnohabitants was identified as an important player in in carbon flow to higher trophic levels since they can be consumed by food for heterotrophic nanoflagellates (Šimek et al., 2013). More recent study showed Limnohabitans Species Strains Rim28 and Rim47 had a great metabolic versatility, including photosynthesis, autotrophic carbon fixation, ammonium oxidation and sulfur oxidization (Zeng et al., 2012b). The higher nitrate concentrations in YSGW3 and YSGW 11 was in good agreement with the most abundant sequences affiliated with Limnohabitans sp. Rim 28, which may serve as ammonium oxidizer in these two samples. Fresh well waters provide another potential sources for both nitrogen and carbon flux to the coastal waters.
Bacterial Candidate for Bioremediation in Well Waters
Fresh well sample YSGW 4 is located in a public park in Shandong province, garbage such as mosquito and discarded beverage bottles in the well were observed during the sampling time. Compared to the other three fresh water samples, YSGW 4 exhibited relatively lower nitrate and higher DOC concentrations. The co-occurrence of sequences closely related to Actinobacteria Rhodoluna lacicola strain MWH-EgelM2-3.2 (FJ545223), Flavobacterium granuli strain kw05 (NR_041052), and uncultured Flectobacillus clone ZS-2-379 (FN668111) (Figure 4). The presence of actinorhodopsins in Rhodoluna lacicola strain MWH-EgelM2-3.2 suggested this strain to be photoheterotrophic bacteria, surviving under low nutrient and energy conditions (Sharma et al., 2008). Flavobacterium granuli was isolated from granules used in a wastewater treatment plant (Aslam et al., 2005); Members of genus Flectobacillus usually act as the direct competitors in situ (Šimek et al., 2010a). The characteristics of these three groups indicated the hostile environment of YSGW 4 sampling site. The most abundant sequences in YSGW 4 were affiliated with C. testoteroni strain CNB-2. The complete genome of strain CNB-2 revealed its genetic versatility adaption to diverse habitats, based on its ability to metabolize a variety of compounds, such as carboxylic acids and aromatic compounds, utilize nitrate and ammonia as nitrogen source, resistant to drug and heavy metals (Ma et al., 2009). C. testosteroni strain CNB-1 has been used successfully rhizoremediation of 4-chloronitrobenzene (CNB) polluted soil (Liu et al., 2007). Other Comamonas spp. were also used for environmental applications (Tobajas et al., 2012; Wu et al., 2015). The high abundance of C. testosteroni related sequences in our samples indicated that this species could be selected as potential microbes for bioremediation in the highly polluted fresh well waters before flowing into coastal water.
Linking Bacterial Community with Environmental Variables
Based on our analysis of relationship between bacterial taxonomic groups and contexture environmental parameters, we found Betaproteobacteria in well water had highly negative correlation with ammonium, but had positive correlation with nitrate; in contrast, Cyanobacteria in porewater had highly positively correlation with ammonium. These results supported the possibilities that fresh well water and brackish porewater may provide nitrate and ammonium respectively. Only YSPW 2 contained more than 1% sequences within six rare bacterial taxonomic groups (Figure 7), which had highly positively correlation with nitrite, showing these groups in YSPW2 had the abilities to cycle nitrogen.
Four well water samples were collected from three different eco-environments impacted by heavy human activities (Table 1). We detected less diverse bacterial groups in the well water samples than those in the porewater samples (Table 2), suggesting the stressful conditions in the well water due to anthropogenic activities. Comamonas spp. and Limnohabitans spp. were dominated in fresh well samples, but were almost absent from the brackish porewater samples, which indicates these groups favored lower salinity environment. Distinctive bacterial patterns were detected among the three porewater samples collected from the different tourist beaches, for example, six rare bacterial taxonomic groups were mainly found in YSGW 2, which may have been due to niche selections.
Conclusion
Our study documented bacterial community structures in the both fresh well water and brackish porewater along the coasts of the Yellow Sea. We provided perspective into the correlations between representative bacterial taxonomic groups and environmental parameters, fresh well water and brackish porewater provided the different nitrogen sources to the coastal water. Potential key bacterial groups such as Comamonas testosteroni may be excellent candidates for bioremediation of the natural pollutants in different niches. Further studies will explore the shifts of indigenous microbial community in in-situ bioremediation experiments in the submarine groundwater.
Author Contributions
JD designed the experiments; JL collected the samples; QY and JL performed the experiments and analyzed the data; QY, JL, JD, and JZ wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We greatly appreciate our colleagues of the biogeochemical group in SKLEC/ECNU for sampling, in-situ and laboratory measurements of physic-chemical parameters. We thank two reviewers for their constructive comments on this manuscript. This study is funded by the Key Project of Chinese National Programs for Fundamental Research and Development (973 Program) (2011CB409801) and the National Natural Science Foundation of China (41376089).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2015.01519
References
Akoumianaki, I., Nomaki, H., Pachiadaki, M., Kormas, K. A., Kitazato, H., and Tokuyama, H. (2012). Low bacterial diversity and high labile organic matter concentrations in the sediments of the Medee deep-sea hypersaline anoxic basin. Microbes. Environ. 27, 504–508. doi: 10.1264/jsme2.ME12045
Allgaier, M., and Grossart, H. P. (2006). Diversity and seasonal dynamics of Actinobacteria populations in four lakes in northeastern Germany. Appl. Environ. Microb. 72, 3489–3497. doi: 10.1128/aem.72.5.3489-3497.2006
Aslam, Z., Im, W. T., Kim, M. K., and Lee, S. T. (2005). Flavobacterium granuli sp. nov., isolated from granules used in a wastewater treatment plant. Int. J. Syst. Evol. Microb. 55, 747–751. doi: 10.1099/ijs.0.63459-0
Boehm, A. B., Shellenbarger, G. G., and Paytan, A. (2004). Groundwater discharge: potential association with fecal indicator bacteria in the surf zone. Environ. Sci. Technol 38, 3558–3566. doi: 10.1021/es035385a
Bondoso, J., Balagué, V., Gasol, J. M., and Lage, O. M. (2014). Community composition of the Planctomycetes associated with different macroalgae. FEMS Microb. Ecol. 88, 445–456. doi: 10.1111/1574-6941.12258
Bowman, J. P., and McCuaig, R. D. (2003). Biodiversity, community structural shifts, and biogeography of prokaryotes within Antarctic continental shelf sediment. Appl. Environ. Microb. 69, 2463–2483. doi: 10.1128/aem.69.5.2463-2483.2003
Burke, C., Thomas, T., Lewis, M., Steinberg, P., and Kjelleberg, S. (2011). Composition, uniqueness and variability of the epiphytic bacterial community of the green alga Ulva australis. ISME J. 5, 590–600. doi: 10.1038/ismej.2010.164
Burnett, W. C., Aggarwal, P. K., Aureli, A., Bokuniewicz, H., Cable, J. E., Charette, M. A., et al. (2006). Quantifying submarine groundwater discharge in the coastal zone via multiple methods. Sci. Total. Environ. 367, 498–543. doi: 10.1016/j.scitotenv.2006.05.009
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Charette, M. A., and Allen, M. C. (2006). Precision ground water sampling in coastal aquifers using a direct-push, shielded-screen well-point system. Groundw. Monit. Rem. 26, 87–93. doi: 10.1111/j.1745-6592.2006.00076.x
Costa, L. S., Fidelis, G. P., Cordeiro, S. L., Oliveira, R. M., Sabry, D. A., Câmara, R. B. G., et al. (2010). Biological activities of sulfated polysaccharides from tropical seaweeds. Biomed. Pharmacother. 64, 21–28. doi: 10.1016/j.biopha.2009.03.005
Debroas, D., Humbert, J. F., Enault, F., Bronner, G., Faubladier, M., and Cornillot, E. (2009). Metagenomic approach studying the taxonomic and functional diversity of the bacterial community in a mesotrophic lake (Lac du Bourget–France). Environ. Microbiol. 11, 2412–2424. doi: 10.1111/j.1462-2920.2009.01969.x
DeLong, E. F., and Karl, D. M. (2005). Genomic perspectives in microbial oceanography. Nature 437, 336–342. doi: 10.1038/nature04157
Dufresne, A., Salanoubat, M., Partensky, F., Artiguenave, F., Axmann, I. M., Barbe, V., et al. (2003). Genome sequence of the cyanobacterium Prochlorococcus marinus SS120, a nearly minimal oxyphototrophic genome. Proc. Natl. Acad. Sci. U.S.A. 100, 10020–10025. doi: 10.1073/pnas.1733211100
Eckert, E. M., Salcher, M. M., Posch, T., Eugster, B., and Pernthaler, J. (2012). Rapid successions affect microbial N-acetyl-glucosamine uptake patterns during a lacustrine spring phytoplankton bloom. Environ. Microbiol. 14, 794–806. doi: 10.1111/j.1462-2920.2011.02639.x
Fields, M. W., Yan, T., Rhee, S. K., Carroll, S. L., Jardine, P. M., Watson, D. B., et al. (2005). Impacts on microbial communities and cultivable isolates from groundwater contaminated with high levels of nitric acid–uranium waste. FEMS Microbiol. Ecol. 53, 417–428. doi: 10.1016/j.femsec.2005.01.010
Garcia-Solsona, E., García-Orellana, J., Masqué, P., Garcés, E., Radakovitch, O., Mayer, A., et al. (2010). An assessment of karstic submarine groundwater and associated nutrient discharge to a Mediterranean coastal area (Balearic Islands, Spain) using radium isotopes. Biogeochemistry 97, 211–229. doi: 10.1007/s10533-009-9368-y
Glöckner, F. O., Kube, M., Bauer, M., Teeling, H., Lombardot, T., Ludwig, W., et al. (2003). Complete genome sequence of the marine planctomycete Pirellula sp. strain 1. Proc. Natl. Acad. Sci. U.S.A. 100, 8298–8303. doi: 10.1073/pnas.1431443100
Gobet, A., Böer, S. I., Huse, S. M., Van Beusekom, J. E., Quince, C., Sogin, M. L., et al. (2012). Diversity and dynamics of rare and of resident bacterial populations in coastal sands. ISME J. 6, 542–553. doi: 10.1038/ismej.2011.132
Hahn, M. W. (2009). Description of seven candidate species affiliated with the phylum Actinobacteria, representing planktonic freshwater bacteria. Int. J. Syst. Evol. Microb. 59, 112–117. doi: 10.1099/ijs.0.001743-0
Hahn, M. W., Lang, E., Brand, U., and Spröerb, C. (2011). Polynucleobacter acidiphobus sp. nov., a representative of an abundant group of planktonic freshwater bacteria. Int. J. Syst. Evol. Microb. 61, 788–794. doi: 10.1099/ijs.0.023929-0
Hahn, M. W., Stadler, P., Wu, Q. L., and Pöckl, M. (2004). The filtration–acclimatization method for isolation of an important fraction of the not readily cultivable bacteria. J. Microbiol. Meth. 57, 379–390. doi: 10.1016/j.mimet.2004.02.004
Halliday, E., and Gast, R. J. (2010). Bacteria in beach sands: an emerging challenge in protecting coastal water quality and bather health. Environ. Sci. Technol. 45, 370–379. doi: 10.1021/es102747s
Hemme, C. L., Deng, Y., Gentry, T. J., Fields, M. W., Wu, L., and Barua, S. (2010). Metagenomic insights into evolution of a heavy metal-contaminated groundwater microbial community. ISME J. 4, 660–672. doi: 10.1038/ismej.2009.154
Hu, C., Muller-Karger, F. E., and Swarzenski, P. W. (2006). Hurricanes, submarine groundwater discharge, and Florida's red tides. Geophys. Res. Let. 33, L11601. doi: 10.1029/2005GL025449
Hwang, C., Wu, W., Gentry, T. J., Carley, J., Corbin, G. A., Carroll, S. L., et al. (2009). Bacterial community succession during in situ uranium bioremediation: spatial similarities along controlled flow paths. ISME J. 3, 47–64. doi: 10.1038/ismej.2008.77
Jones, K. L., Mikulski, C. M., Barnhorst, A., and Doucette, G. J. (2010). Comparative analysis of bacterioplankton assemblages from Karenia brevis bloom and nonbloom water on the west Florida shelf (Gulf of Mexico, USA) using 16S rRNA gene clone libraries. FEMS Microbiol. Ecol. 73, 468–485. doi: 10.1111/j.1574-6941.2010.00914.x
Kasalickı, V., Jezbera, J., Hahn, M. W., and Šimek, K. (2013). The diversity of the Limnohabitans genus, an important group of freshwater bacterioplankton, by characterization of 35 isolated strains. PLoS ONE 8:e58205. doi: 10.1371/journal.pone.0058209
Kim, G., Ryu, J. W., Yang, H. S., and Yun, S. T. (2005). Submarine groundwater discharge (SGD) into the Yellow Sea revealed by 228 Ra and 226 Ra isotopes: implications for global silicate fluxes. Earth Planet. Sci. Lett. 237, 156–166. doi: 10.1016/j.epsl.2005.06.011
Kirchman, D. L. (2002). The ecology of Cytophaga–Flavobacteria in aquatic environments. FEMS Microbiol. Ecol. 39, 91–100. doi: 10.1016/s0168-6496(01)00206-9
Lachnit, T., Meske, D., Wahl, M., Harder, T., and Schmitz, R. (2011). Epibacterial community patterns on marine macroalgae are host-specific but temporally variable. Environ. Microbiol. 13, 655–665. doi: 10.1111/j.1462-2920.2010.02371.x
Lage, O. M., and Bondoso, J. (2011). Planctomycetes diversity associated with macroalgae. FEMS Microbiol. Ecol. 78, 366–375. doi: 10.1111/j.1574-6941.2011.01168.x
LaRoche, J., Nuzzi, R., Waters, R., Wyman, K., Falkowski, P., and Wallace, D. (1997). Brown tide blooms in Long Island's coastal waters linked to interannual variability in groundwater flow. Global Change Biol. 3, 97–410. doi: 10.1046/j.1365-2486.1997.00117.x
Lee, Y. W., and Kim, G. (2007). Linking groundwater-borne nutrients and dinoflagellate red-tide outbreaks in the southern sea of Korea using a Ra tracer. Estuar. Coast Shelf Sci. 71, 309–317. doi: 10.1016/j.ecss.2006.08.004
Lenes, J. M., Darrow, B. P., Cattrall, C., Heil, C. A., Callahan, M., and Vargo, G. A. (2001). Iron fertilization and the Trichodesmiumresponse on the West Florida shelf. Limnol. Oceanogr. 46, 1261–1277. doi: 10.4319/lo.2001.46.6.1261
Li, B., Liu, S., Xing, R., Li, K., Li, R., Qin, Y., et al. (2013). Degradation of sulfated polysaccharides from Enteromorpha prolifera and their antioxidant activities. Carbohydr. Polym. 92, 1991–1996. doi: 10.1016/j.carbpol.2012.11.088
Li, Y. F., Chen, Y. R., Yang, J. L., Bao, W. Y., Guo, X., Liang, X., et al. (2014). Effects of substratum type on bacterial community structure in biofilms in relation to settlement of plantigrades of the mussel Mytilus coruscus. Int. Biodeter. Biodegr. 96, 41–49. doi: 10.1016/j.ibiod.2014.08.012
Liu, L., Jiang, C. Y., Liu, X. Y., Wu, J. F., Han, J. G., and Liu, S. J. (2007). Plant–microbe association for rhizoremediation of chloronitroaromatic pollutants with Comamonas sp. strain CNB-1. Environ. Microbiol. 9, 465–473. doi: 10.1111/j.1462-2920.2006.01163.x
Liu, M., Dong, Y., Zhao, Y., Zhang, G., Zhang, W., and Xiao, T. (2011). Structures of bacterial communities on the surface of Ulva prolifera and in seawaters in an Ulva blooming region in Jiaozhou Bay, China. World J. Microb. Biot. 27, 1703–1712. doi: 10.1007/s11274-010-0627-9
Liu, M., Xiao, T., Sun, J., Wei, H., Wu, Y., Zhao, Y., et al. (2013). Bacterial community structures associated with a natural spring phytoplankton bloom in the Yellow Sea. China. Deep Sea Res. II 97, 85–92. doi: 10.1016/j.dsr2.2013.05.016
Liu, S. M., Zhang, J., Chen, H. T., and Zhang, G. S. (2005). Factors influencing nutrient dynamics in the eutrophic Jiaozhou Bay, North China. Prog. Oceanogr. 66, 66–85. doi: 10.1016/j.pocean.2005.03.009
Lyons, D. A., Arvanitidis, C., Blight, A. J., Chatzinikolaou, E., Guy-Haim, T., Kotta, J., et al. (2014). Macroalgal blooms alter community structure and primary productivity in marine ecosystems. Global Change Biol. 20, 2712–2724. doi: 10.1111/gcb.12644
Ma, Y. F., Zhang, Y., Zhang, J. Y., Chen, D. W., Zhu, Y., Zheng, H., et al. (2009). The complete genome of Comamonas testosteroni reveals its genetic adaptations to changing environments. Appl. Environ. Microbiol. 75, 6812–6819. doi: 10.1128/AEM.00933-09
Moore, W. S. (2010). The effect of submarine groundwater discharge on the ocean. Annu. Rev. Mar. Sci. 2, 59–88. doi: 10.1146/annurev-marine-120308-081019
Moore, W. S., and Arnold, R. (1996). Measurement of 223Ra and 224Ra in coastal waters using a delayed coincidence counter. J. Geophys. Res. 101, 1321–1329.
Newton, R. J., Jones, S. E., Eiler, A., McMahon, K. D., and Bertilsson, S. (2011). A guide to the natural history of freshwater lake bacteria. Microbiol. Mol. Biol. R. 75, 14–49. doi: 10.1128/mmbr.00028-10
Oksanen, J., Kindt, R., Legendre, P., O'Hara, B., Stevens, M. H. H., Oksanen, M. J., et al. (2007). The Vegan Package. Community Ecology Package. 631–637. R package version 2.3-2. Available online at: http://cran.r-project.org/
Paytan, A., Boehm, A. B., and Shellenbarger, G. G. (2004). Bacterial contamination and submarine groundwater discharge—a possible link. Environ. Chem. 1, 29–30. doi: 10.1071/en04002
Qi, H., Zhao, T., Zhang, Q., Li, Z., Zhao, Z., and Xing, R. (2005). Antioxidant activity of different molecular weight sulfated polysaccharides from Ulva pertusa Kjellm (Chlorophyta). J. Appl. Phycol. 17, 527–534. doi: 10.1007/s10811-005-9003-9
R Development Core Team. (2013). R: A Language and Environment for Statistical Computing. Available online at: http://www.r-project.org/
Salcher, M. M. (2013). Same but different: ecological niche partitioning of planktonic freshwater prokaryotes. J. Limnol. 73, 74–87. doi: 10.4081/jlimnol.2014.813
Santoro, A. E., Boehm, A. B., and Francis, C. A. (2006). Denitrifier community composition along a nitrate and salinity gradient in a coastal aquifer. Appl. Environ. Microbiol. 72, 2102–2109. doi: 10.1128/aem.72.3.2102-2109.2006
Santoro, A. E., Francis, C. A., De Sieyes, N. R., and Boehm, A. B. (2008). Shifts in the relative abundance of ammonia-oxidizing bacteria and archaea across physicochemical gradients in a subterranean estuary. Environ. Microbiol. 10, 1068–1079. doi: 10.1111/j.1462-2920.2007.01547.x
Scheibner, M., Dörge, P., Biermann, A., Sommer, U., Hoppe, H. G., and Jürgens, K. (2014). Impact of warming on phyto-bacterioplankton coupling and bacterial community composition in experimental mesocosms. Environ. Microbiol. 16, 718–733. doi: 10.1111/1462-2920.12195
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/aem.01541-09
Sharma, A. K., Sommerfeld, K., Bullerjahn, G. S., Matteson, A. R., Wilhelm, S. W., Jezbera, J., et al. (2009). Actinorhodopsin genes discovered in diverse freshwater habitats and among cultivated freshwater Actinobacteria. ISME J. 3, 726–737. doi: 10.1038/ismej.2009.13
Sharma, A. K., Zhaxybayeva, O., Papke, R. T., and Doolittle, W. F. (2008). Actinorhodopsins: proteorhodopsin-like gene sequences found predominantly in non-marine environments. Environ. Microbiol. 10, 1039–1056. doi: 10.1111/j.1462-2920.2007.01525.x
Sihvonen, L. M., Lyra, C., Fewer, D. P., Rajaniemi-Wacklin, P., Lehtimäki, J. M., Wahlsten, M., et al. (2007). Strains of the cyanobacterial genera Calothrix and Rivularia isolated from the Baltic Sea display cryptic diversity and are distantly related to Gloeotrichia and Tolypothrix. FEMS Microbiol. Ecol. 61, 74–84. doi: 10.1111/j.1574-6941.2007.00321.x
Šimek, K., Horòák, K., Jezbera, J., Mašín, M., Nedom, J., Gasol, J. M., et al. (2005). Influence of top-down and bottom-up manipulations on the R-BT065 subcluster of β-Proteobacteria, an abundant group in bacterioplankton of a freshwater reservoir. Appl. Environ. Microbiol. 71, 2381–2390. doi: 10.1128/AEM.71.5.2381-2390.2005
Šimek, K., Kasalickı, V., Horòák, K., Hahnn, M. W., and Weinbauerm, M. G. (2010a). Assessing niche separation among coexisting Limnohabitans strains through interactions with a competitor, viruses, and a bacterivore. Appl. Environ. Microbiol. 76, 1406–1416. doi: 10.1128/AEM.02517-09
Šimek, K., Kasalickı, V., Jezbera, J., Jezberová, J., Hejzlar, J., and Hahn, M. W. (2010b). Broad habitat range of the phylogenetically narrow R-BT065 cluster, representing a core group of the betaproteobacterial genus Limnohabitans. Appl. Environ. Microbiol. 76, 631–639. doi: 10.1128/AEM.02203-09
Šimek, K., Kasalickı, V., Jezbera, J., Horòák, K., Nedoma, J., Hahn, M. W., et al. (2013). Differential freshwater flagellate community response to bacterial food quality with a focus on Limnohabitans bacteria. ISME J. 7, 1519–1530. doi: 10.1038/ismej.2013.57
Šimek, K., Kasalickı, V., Zapomìlová, E., and Horòák, K. (2011). Alga-derived substrates select for distinct betaproteobacterial lineages and contribute to niche separation in Limnohabitans strains. Appl. Environ. Microbiol. 77, 7307–7315. doi: 10.1128/AEM.05107-11
Smith, C. G., and Swarzenski, P. W. (2012). An investigation of submarine groundwater–borne nutrient fluxes to the west Florida shelf and recurrent harmful algal blooms. Limnol. Oceanogr. 57, 471–485. doi: 10.4319/lo.2012.57.2.047
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Teeling, H., Fuchs, B. M., Becher, D., Klockow, C., Gardebrecht, A., Bennke, C. M., et al. (2012). Substrate-controlled succession of marine bacterioplankton populations induced by a phytoplankton bloom. Science 336, 608–611. doi: 10.1126/science.1218344
Tobajas, M., Monsalvo, V. M., Mohedano, A. F., and Rodriguez, J. J. (2012). Enhancement of cometabolic biodegradation of 4-chlorophenol induced with phenol and glucose as carbon sources by Comamonas testosteroni. J. Environ. Manage. 95, S116–S121. doi: 10.1016/j.jenvman.2010.09.030
Van den Wyngaert, S., Salcher, M. M., Pernthaler, J., Zeder, M., and Posch, T. (2011). Quantitative dominance of seasonally persistent filamentous cyanobacteria (Planktothrix rubescens) in the microbial assemblages of a temperate lake. Limnol. Oceanog. 56, 97–109. doi: 10.4319/lo.2011.56.1.0097
Waldron, P. J., Wu, L., Nostrand, J. D. V., Schadt, C. W., He, Z., Watson, D. B., et al. (2009). Functional gene array-based analysis of microbial community structure in groundwaters with a gradient of contaminant levels. Environ. Sci. Technol. 43, 3529–3534. doi: 10.1021/es803423p
Wang, W., Zhong, R., Shang, D., and Shao, Z. (2014). Indigenous oil-degrading bacteria in crude oil-contaminated seawater of the Yellow sea, China. Appl. Microbio. Biotech. 98, 7253–7269. doi: 10.1007/s00253-014-5817-1
Warnes, G. R., Bolker, B., Bonebakker, L., Gentleman, R., Liaw, W. H. A., Lumley, T., et al. (2012). gplots: Various R Programming Tools for Plotting Data (Comprehensive R Archive Network). R package version 2.8.0. Available online at: http://cran.r-project.org/web/packages/gplots/index.html
Waska, H., and Kim, G. (2011). Submarine groundwater discharge (SGD) as a main nutrient source for benthic and water-column primary production in a large intertidal environment of the Yellow Sea. J. Sea Res. 65, 103–113. doi: 10.1016/j.seares.2010.08.001
Woebken, D., Teeling, H., Wecker, P., Dumitriu, A., Kostadinov, I., and DeLong, E. F. (2007). Fosmids of novel marine Planctomycetes from the Namibian and Oregon coast upwelling systems and their cross-comparison with planctomycete genomes. ISME J. 1, 419–435. doi: 10.1038/ismej.2007.63
Wu, Y., Liu, J., Yang, L., Chen, H., Zhang, S., Zhao, H., et al. (2011). Allelopathic control of cyanobacterial blooms by periphyton biofilms. Environ. Microbiol. 13, 604–615. doi: 10.1111/j.1462-2920.2010.02363.x
Wu, Y., Shukal, S., Mukherjee, M., and Cao, B. (2015). Involvement in denitrification is beneficial to the biofilm lifestyle of comamonas testosteroni: a mechanistic study and its environmental implications. Environ. Sci. Technol. 49, 11551–11559. doi: 10.1021/acs.est.5b03381
Xiong, J., Liu, Y., Lin, X., Zhang, H., Zeng, J., Ho, J., et al. (2012). Geographic distance and pH drive bacterial distribution in alkaline lake sediments across Tibetan Plateau. Environ. Microbiol. 14, 2457–2466. doi: 10.1111/j.1462-2920.2012.02799.x
Xu, M., Wu, W. M., Wu, L., He, Z., Van Nostrand, J. D., Deng, Y., et al. (2010). Responses of microbial community functional structures to pilot-scale uranium in situ bioremediation. ISME J. 4, 1060–1070. doi: 10.1038/ismej.2010.31
Yan, T. F., Fields, M. W., Wu, L., Zu, Y., Tiedje, J. M., and Zhou, J. (2003). Molecular diversity and characterization of nitrite reductase gene fragments (nirK and nirS) from nitrate-and uranium-contaminated groundwater. Environ. Microbiol. 5, 13–24. doi: 10.1046/j.1462-2920.2003.00393.x
Zehr, J. P. (2011). Nitrogen fixation by marine cyanobacteria. Trends Microbiol. 19, 162–173. doi: 10.1016/j.tim.2010.12.004
Zehr, J. P., and Kudela, R. M. (2011). Nitrogen cycle of the open ocean: from genes to ecosystems. Annu. Rev. Mar. Sci. 3, 197–225. doi: 10.1146/annurev-marine-120709-142819
Zeng, J., Bian, Y., Xing, P., and Wu, Q. L. (2012a). Macrophyte species drive the variation of bacterioplankton community composition in a shallow freshwater lake. Appl. Environ. Microbiol. 78, 177–184. doi: 10.1128/aem.05117-11
Zeng, Y., Kasalickı, V., Šimek, K., and KoblíŽek, M. (2012b). Genome sequences of two freshwater betaproteobacterial isolates, Limnohabitans species strains Rim28 and Rim47, indicate their capabilities as both photoautotrophs and ammonia oxidizers. J. Bacteriol. 194, 6302–6303. doi: 10.1128/jb.01481-12
Zhang, P., Van Nostrand, J. D., He, Z., Chakraborty, R., Deng, Y., Curtis, D., et al. (2015). A slow-release substrate stimulates groundwater microbial communities for long-term in-situ Cr (VI) reduction. Environ. Sci. Technol. 49, 12922–12931. doi: 10.1021/acs.est.5b00024
Keywords: Yellow Sea, submarine groundwater, bacteria, nutrient, bioremediation
Citation: Ye Q, Liu J, Du J and Zhang J (2016) Bacterial Diversity in Submarine Groundwater along the Coasts of the Yellow Sea. Front. Microbiol. 6:1519. doi: 10.3389/fmicb.2015.01519
Received: 29 September 2015; Accepted: 17 December 2015;
Published: 08 January 2016.
Edited by:
Maria Dittrich, University of Toronto, CanadaReviewed by:
Jun-Jie Zhang, Chinese Academy of Sciences, ChinaDayananda Chandrappa, University of Exeter, UK
Copyright © 2016 Ye, Liu, Du and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jinzhou Du, anpkdUBza2xlYy5lY251LmVkdS5jbg==