 Belén Carbonetto1
Belén Carbonetto1 Mónica C. Fabbro1
Mónica C. Fabbro1 Mariela Sciara2
Mariela Sciara2 Analía Seravalle2Guadalupe Méjico2
Analía Seravalle2Guadalupe Méjico2 Santiago Revale1María S. Romero1
Santiago Revale1María S. Romero1 Bianca Brun1Marcelo Fay2Fabián Fay2
Bianca Brun1Marcelo Fay2Fabián Fay2 Martin P. Vazquez1*
Martin P. Vazquez1*- 1Genomics and Bioinformatics, Instituto de Agrobiotecnología de Rosario – CONICET, Rosario, Argentina
- 2Centro de Diagnostico Medico de Alta Complejidad (CIBIC), Rosario, Argentina
The human microbiota is the collection of microorganisms living in or on the human body. An imbalance or dysbiosis in these microbial communities can be associated with a wide variety of human diseases (Petersen and Round, 2014; Pham and Lawley, 2014; Zaura et al., 2014). Moreover, when the microbiota of the same body sites is compared between different healthy individuals, specific microbial community features are apparent (Li et al., 2012; Yatsunenko et al., 2012; Oh et al., 2014; Relman, 2015). In addition, specific selective pressures are found at distinct body sites leading to different patterns in microbial community structure and composition (Costello et al., 2009; Consortium, 2012b; Zhou et al., 2013). Because of these natural variations, a comprehensive characterization of the healthy microbiota is critical for predicting alterations related to diseases. This characterization should be based on a broad healthy population over time, geography, and culture (Yatsunenko et al., 2012; Shetty et al., 2013; Leung et al., 2015; Ross et al., 2015). The study of healthy individuals representing different ages, cultural traditions, and ethnic origins will enable to understand how the associated microbiota varies between populations and respond to different lifestyles. It is important to address these natural variations in order to later detect variations related to disease.
During the last decade researchers from around the world have characterized and defined geographical differences in the microbiota’s composition of healthy adults. The most important projects so far have been the Human Microbiome Project (HMP; Peterson et al., 2009), MetaHIT (Qin et al., 2010; Arumugam et al., 2011) and the American Gut Project1. Despite the big effort conducted, these major projects are based on US and European populations and exclude other ethnic groups, socio-economic, geographic, and cultural settings. Although some projects tried to fill this gap (Yatsunenko et al., 2012; Schnorr et al., 2014; Clemente et al., 2015; Leung et al., 2015; Obregon-Tito et al., 2015; Ross et al., 2015), there are still no records on human microbiota of urban middle-income populations in South America. Here, we characterized the microbiota of six different body habitats: palatine tonsils, saliva, buccal mucosa, throat, anterior nares, and gut from samples of 20 healthy middle-income men and women between 20 and 50 years living in Rosario City, Argentina. Genomic DNA was purified from each sample and the 16S rRNA gene V1–V3 region was amplified. Amplicons were sequenced on a 454 GS-FLX+ Titanium platform and produced a total of 360,177 reads. We compared microbial community composition and structure data with the HMP dataset. Results showed that the microbiota of buccal mucosa, palatine tonsils, and gut differed between the Argentine and US populations. We make available the first 16S rRNA profile dataset of human body microbiota of an Argentine urban cohort. Our results support the idea that the generation of more and larger local datasets of healthy individuals is needed in order to analyze dysbiosis related to disease in middle-income urban population.
Methods
Sampling
Samples were swabbed in a non-invasive manner from palatine tonsils, saliva, buccal mucosa, throat, and anterior nares; gastrointestinal tract samples were collected from stool samples. Twenty healthy subjects, men and women between 20 and 50 years-old living in Rosario city in the central region of Argentina, were recruited. Subjects donated blood to examine the presence of viral markers and metadata was collected by medical examination in order to select healthy individuals (Supplementary Table S1, also see Supplementary material for inclusion/exclusion criteria of healthy subjects). Samples were collected between May and August 2013 in a single visit and sent immediately to the lab where they were processed.
Ethics, Consent, and Permissions
The study was approved by the Institutional Ethics Committee from the Hospital Italiano Garibaldi in Rosario, Argentina. Moreover, a consent form with information about the study, including the rights, risks, and benefits involved in participating in the study was signed by each individual.
DNA Extraction and Sequencing
Total genomic DNA was extracted from 200 mg of each fresh stool sample using QIAamp (Qiagen, Valencia, CA, USA) DNA Stool Mini Kit following manufacturer’s instructions. Palatine tonsils, saliva, buccal mucosa, throat, and anterior nares swabs were resuspended in 200 μl sterile saline solution. Genome DNA was extracted from this solution using QIAamp (Qiagen, Valencia, CA, USA) DNA Mini Kit following manufacturer’s instructions.
For the construction of pyrotag libraries the V1–V3 hyper variable regions of the 16S rRNA gene was amplified using the 27F (5′-AGAGTTTGATCCTGGCTCAG-3′) and 534R (5′-ATTACCGCGGCTGCTGG-3′) tagged primers (Consortium, 2012a). Samples were amplified using two rounds of PCR: a first round to amplify the 16S rRNA gene (30 cycles) and a second round to add barcodes for sample identification (10 cycles). PCR reactions were performed following the procedures detailed in Rascovan et al. (2013). Duplicated reactions were performed in both rounds of PCR to reduce amplification biases and then pooled. Amplicons were cleaned using Ampure DNA capture beads (Agencourt- Beckman Coulter, Inc.), quantified using Quant-iT PicoGreen dsDNA Kit (Invitrogen Molecular Probes, Inc., Eugene, OR, USA) and pooled in equimolar concentrations before sequencing on a Genome Sequencer FLX (454-Roche Applied Sciences) using 454 GS FLX+ chemistry according to the manufacturer’s instructions.
Amplicon Sequence Processing, OTU Classification, and Taxonomic Assignment
We chose the HMP dataset to compare with our data since it is the most complete reference collection of 16S ribosomal RNA gene sequences collected from sites across the human body (Consortium, 2012a). We are aware that the DNA extraction method used by the HMP is different from the used in this project (MoBio PowerSoil DNA Isolation Kit vs. QIAamp DNA kits). Nevertheless, recent results showed that both DNA extraction methods are reproducible enough for the analysis of bacterial community diversity of human samples (Wagner Mackenzie et al., 2015). Taking this into account we decided to use the HMP data for our comparison demonstration. The HMP dataset based on the amplification of the V1–V3 hyper variable regions of the 16S rRNA gene was downloaded from the HMP website2. Our dataset and the HMP dataset were processed using the QIIME v1.8 analysis pipeline (Caporaso et al., 2010b). For comparative purposes the same number of individuals (N = 20) was selected randomly from the HMP dataset. A random selection of 1000 reads per sample was done using a custom-made script. The command split libraries.py was used for demultiplexing and quality filtering. Reads with more than 10 mismatches in the forward primer sequence and more than eight mismatches in the reverse primer sequence were removed. Up to two mismatches were allowed in barcode sequences. Homopolymers longer than 6 bp were excluded. The mean qual score used was 25Q. The size of quality score window was set up to 50 bp. If the average score of a continuous set of 50 nucleotides fell below 25Q, the sequence was discarded. Minimum sequence length allowed was 200 bp. Filtered sequences were then clustered into operational taxonomic units (OTUs) using the pick_otus.py script with the Uclust method at 97% sequence similarity (Edgar, 2010). OTU representative sequences were aligned using PyNast algorithm with QIIME default parameters (Caporaso et al., 2010a). Phylogenetic trees containing the aligned sequences were then produced using FastTree (Price et al., 2009). Richness alpha diversity metrics and rarefaction curves were calculated by sub-sampling the OTU tables at different depths and counting the resulting number of phylotypes using 10 iterations per sample. Phylogeny-based beta diversity distances between OTUs were calculated using weighted Unifrac (Lozupone and Knight, 2005; Lozupone et al., 2007). Taxonomic classification of sequences was done with Ribosomal Database Project (RDP) Classifier using the Greengenes V13.5 database and a 50% confidence threshold (Wang et al., 2007).
Numerical Analyses
Unifrac phylogenetic pairwise distances among samples were visualized with principal coordinates analysis (PCoA). Analysis of similarity statistics (ANOSIM) was calculated to test a priori sampling groups. Mann–Whitney non-parametric tests were performed to elucidate differences in taxa abundances. All calculations were carried out with R packages ‘BiodiversityR’ and ‘Vegan.’
Results
Argentine Human Microbiota Overview
Results showed that OTU richness differed between body habitats. Saliva presented the higher richness and the anterior nares was the less diverse site (Figure 1A). Beta diversity also revealed differences in community structure between body habitats (Figure 1B). The PCoA visualization revealed a separation of the data in three main groups: oropharyngeal region, nose and gut (ANOSIM, p < 0.05).
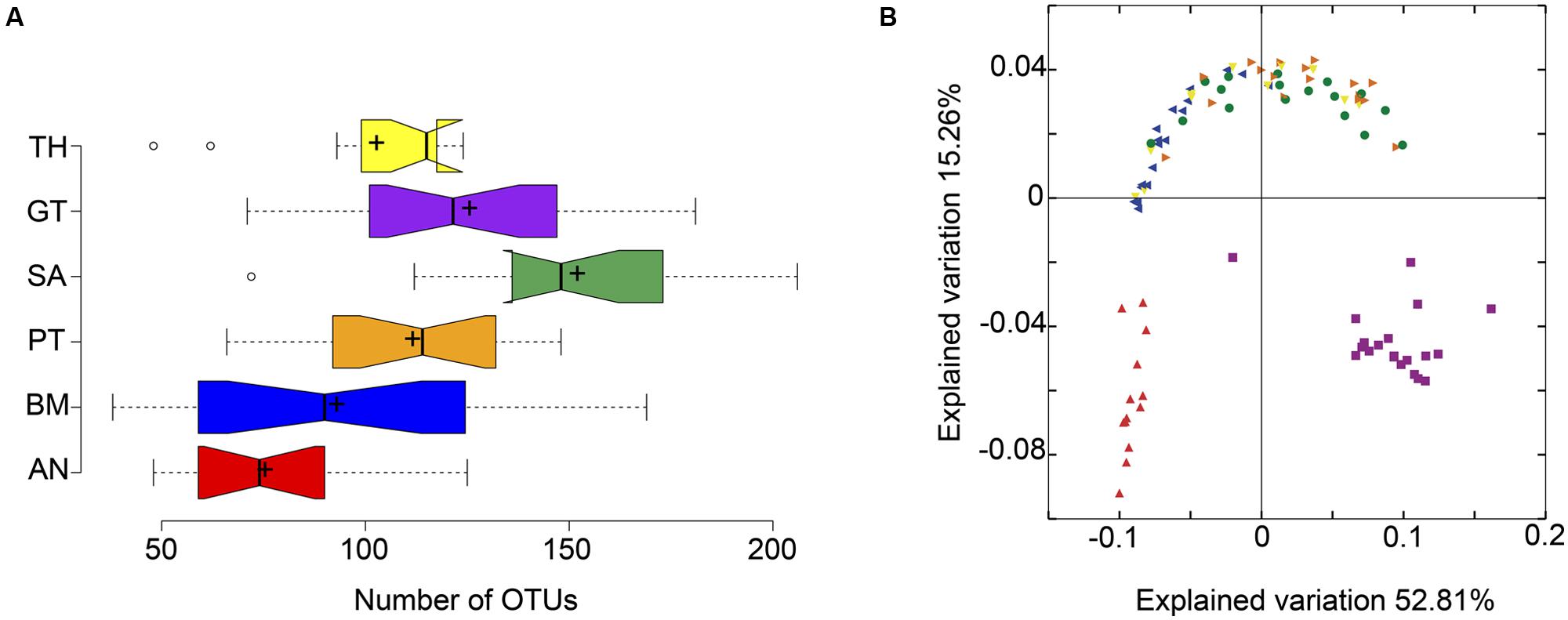
FIGURE 1. Diversity analysis of the Argentine human microbiota. (A) Alpha diversity analysis based on OTU richness, BM, buccal mucosa; TH, throat; PT, palatine tonsils; AN, anterior nares; SA, saliva; GT, gut. (B) Beta diversity analysis based on weighted Unifrac pairwise distances, each color represents the same body part as in (A).
Comparison with the HMP Dataset
We present here a simple analysis showing differences with the HMP dataset in order to encourage the use of the Argentine dataset for comparison purposes. We observed that the microbiota of the Argentine and US populations differed in composition and community structure. Beta diversity results based on weighted Unifrac distances showed that the microbiota of the buccal mucosa, the palatine tonsils, and the gastrointestinal tract differed between Argentine and US individuals (Supplementary Figure S1, ANOSIM, p < 0.05). Moreover, the taxonomic composition of these body habitats showed differences between populations. We observed differences in the abundance of the most predominant taxa (Figure 2). For example, the abundance of Bacteroidaceae family was higher in the US gut microbiota, while Ruminococcaceae, Lachnospiraceae, Rikenellaceae, and Prevotellaceae were more abundant in the Argentine gut microbiota. It is known that the variation in the levels of the three main taxa in the gut microbiota can define enterotypes (Arumugam et al., 2011). The higher relative abundance of three genera defines the enterotypes: bacteroides defines enterotype 1, Prevotella defines enterotype 2 and of Ruminococcus defines enterotype 3. Although we found differences in Bacteroidaceae and Ruminococcaceae abundances (Figure 2, Mann–Withney p < 0.05) and Bacteroides, Ruminococcus, and Prevotella abundances (Mann–Withney p < 0.05, data not shown) between populations, both populations can be assigned to enterotype 1 (Supplementary Figure S2). This is, to our knowledge the first time gut microbiota of middle-income populations of North and South America are compared. Differences in Prevotella and Bacteroides abundances between southamerican and US populations were previously reported. However, in these studies the microbiota of Ameridian individuals living in small villages near the Amazonas with the microbiota of western citizens living in metropolitan areas were compared (Yatsunenko et al., 2012). Regarding the buccal mucosa microbiota, we observed that Streptococcaceae family was more abundant in the Argentine population while Gemellaceae family was higher in the US individuals (Figure 2, Mann–Withney p < 0.05). The abundance of Streptococcaceae family was also higher in the palatine tonsils of argentine individuals. Our results encourage the idea that the human microbiota ecosystem has multiple states of equilibrium and that these variations are present between healthy populations. Moreover, it is probable that these multiple states are related to different lifestyles, location, ethnics, cultural tradition, age, and gender.
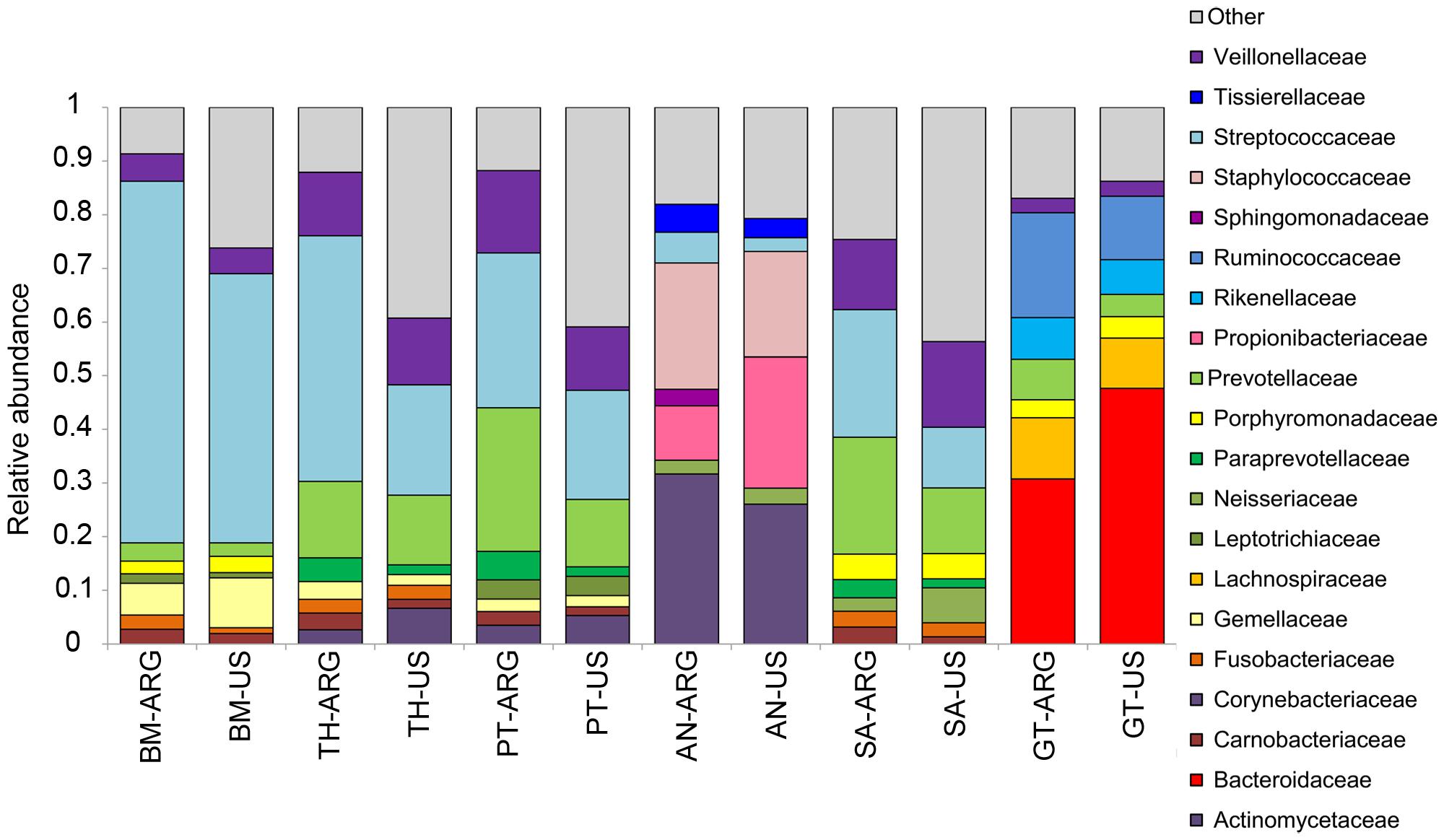
FIGURE 2. Taxonomic profiles. Twenty top abundant bacterial families in the Argentine and US (HMP) human microbiota. ARG, Argentine population; BM, buccal mucosa; TH, throat; PT, palatine tonsils; AN, anterior nares; SA, saliva; GT, gut.
Conclusion
Here, we present the first dataset based on human microbiota samples of an urban middle-income population in South America. We characterized the microbiota of six different body habitats: palatine tonsils, saliva, buccal mucosa, throat, anterior nares and gut from samples of healthy individuals living in a metropolitan area in Argentina. Our initial findings revealed differences in the structure and composition of the microbial communities compared to the US urban population.
By sharing our data, we want to actively encourage its reuse for comparison purposes. This will ultimately result in novel biological insights on the variability of the microbiota of healthy individuals across populations worldwide. Moreover, the understanding of the human microbiota ecosystem in a health-associated state will help to answer questions related to the role of the microbiota in disease.
Data Access
Raw datasets are available in the NCBI SRA database under the accession number: SRP062999. Datasets can be downloaded as fasta or fastq files. The datasets are part of the NCBI Bioproject PRJNA2935213.
Author Contributions
BC and MF analyzed the data and performed the numerical and statistical analyses. BC wrote the manuscript. SR processed the sequence raw data. MF and MS designed the protocol and criteria for healthy individual selection. MS wrote the protocol for recruiting the individuals, supervised the recruiting process, and collected the metadata for the individuals. GM performed DNA preparation from human samples. MR and BB performed amplicon library preparation and sequencing. FF directed the project and supervised the study. MV directed the project, conceived the study, supervised the bioinformatics analysis and supervised the manuscript.
Funding
Funding for this work was provided by Agencia Nacional de Promoción Científica y Tecnológica in Argentina.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2016.00051
Footnotes
- ^http://americangut.org/
- ^ftp://public-ftp.hmpdacc.org/HMQCP/seqs_v13.fna.gz
- ^http://www.ncbi.nlm.nih.gov/bioproject/PRJNA293521/
References
Arumugam, M., Raes, J., Pelletier, E., Le Paslier, D., Yamada, T., Mende, D. R., et al. (2011). Enterotypes of the human gut microbiome. Nature 473, 174–180. doi: 10.1038/nature09944
Caporaso, J. G., Bittinger, K., Bushman, F. D., Desantis, T. Z., Andersen, G. L., and Knight, R. (2010a). PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26, 266–267. doi: 10.1093/bioinformatics/btp636
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010b). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Clemente, J. C., Pehrsson, E. C., Blaser, M. J., Sandhu, K., Gao, Z., Wang, B., et al. (2015). The microbiome of uncontacted Amerindians. Sci. Adv. 1, e1500183. doi: 10.1126/sciadv.1500183
Consortium, T. H. M. P. (2012a). A framework for human microbiome research. Nature 486, 215–221. doi: 10.1038/nature11209
Consortium, T. H. M. P. (2012b). Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214. doi: 10.1038/nature11234
Costello, E. K., Lauber, C. L., Hamady, M., Fierer, N., Gordon, J. I., and Knight, R. (2009). Bacterial community variation in human body habitats across space and time. Science 326, 1694–1697. doi: 10.1126/science.1177486
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Leung, M. H. Y., Wilkins, D., and Lee, P. K. H. (2015). Insights into the pan-microbiome: skin microbial communities of Chinese individuals differ from other racial groups. Sci. Rep. 5, 11845. doi: 10.1038/srep11845
Li, K., Bihan, M., Yooseph, S., and Methé, B. A. (2012). Analyses of the microbial diversity across the human microbiome. PLoS ONE 7:e32118. doi: 10.1371/journal.pone.0032118
Lozupone, C., and Knight, R. (2005). UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 71, 8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005
Lozupone, C. A., Hamady, M., Kelley, S. T., and Knight, R. (2007). Quantitative and qualitative β diversity measures lead to different insights into factors that structure microbial communities. Appl. Environ. Microbiol. 73, 1576–1585. doi: 10.1128/AEM.01996-06
Obregon-Tito, A. J., Tito, R. Y., Metcalf, J., Sankaranarayanan, K., Clemente, J. C., Ursell, L. K., et al. (2015). Subsistence strategies in traditional societies distinguish gut microbiomes. Nat. Commun. 6, 6505. doi: 10.1038/ncomms7505
Oh, J., Byrd, A. L., Deming, C., Conlan, S., Program, N. C. S., Kong, H. H., et al. (2014). Biogeography and individuality shape function in the human skin metagenome. Nature 514, 59–64. doi: 10.1038/nature13786
Petersen, C., and Round, J. L. (2014). Defining dysbiosis and its influence on host immunity and disease. Cell. Microbiol. 16, 1024–1033. doi: 10.1111/cmi.12308
Peterson, J., Garges, S., Giovanni, M., Mcinnes, P., Wang, L., Schloss, J. A., et al. (2009). The NIH human microbiome project. Genome Res. 19, 2317–2323. doi: 10.1101/gr.096651.109
Pham, T. A. N., and Lawley, T. D. (2014). Emerging insights on intestinal dysbiosis during bacterial infections. Curr. Opin. Microbiol. 17, 67–74. doi: 10.1016/j.mib.2013.12.002
Price, M. N., Dehal, P. S., and Arkin, A. P. (2009). FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 26, 1641–1650. doi: 10.1093/molbev/msp077
Qin, J., Li, R., Raes, J., Arumugam, M., Burgdorf, K. S., Manichanh, C., et al. (2010). A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464, 59–65. doi: 10.1038/nature08821
Rascovan, N., Carbonetto, B., Revale, S., Reinert, M., Alvarez, R., Godeas, A., et al. (2013). The PAMPA datasets: a metagenomic survey of microbial communities in Argentinean pampean soils. Microbiome 1, 21. doi: 10.1186/2049-2618-1-21
Relman, D. A. (2015). The human microbiome and the future practice of medicine. JAMA 314, 1127–1128. doi: 10.1001/jama.2015.10700
Ross, M., Muzny, D., Mccormick, J., Gibbs, R., Fisher-Hoch, S., and Petrosino, J. (2015). 16S gut community of the Cameron County Hispanic Cohort. Microbiome 3, 1–11. doi: 10.1186/s40168-015-0072-y
Schnorr, S. L., Candela, M., Rampelli, S., Centanni, M., Consolandi, C., Basaglia, G., et al. (2014). Gut microbiome of the Hadza hunter-gatherers. Nat. Commun. 5, 3654. doi: 10.1038/ncomms4654
Shetty, S., Marathe, N., and Shouche, Y. (2013). Opportunities and challenges for gut microbiome studies in the Indian population. Microbiome 1, 24. doi: 10.1186/2049-2618-1-24
Wagner Mackenzie, B., Waite, D. W., and Taylor, M. W. (2015). Evaluating variation in human gut microbiota profiles due to DNA extraction method and inter-subject differences. Front. Microbiol. 6:130. doi: 10.3389/fmicb.2015.00130
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/AEM.00062-07
Yatsunenko, T., Rey, F. E., Manary, M. J., Trehan, I., Dominguez-Bello, M. G., Contreras, M., et al. (2012). Human gut microbiome viewed across age and geography. Nature 486, 222–227. doi: 10.1038/nature11053
Zaura, E., Nicu, E. A., Krom, B. P., and Keijser, B. J. F. (2014). Acquiring and maintaining a normal oral microbiome: current perspective. Front. Cell. Infect. Microbiol. 4:85. doi: 10.3389/fcimb.2014.00085
Keywords: human microbiome, dysbiosis, South America, healthy microbiota, amplicon sequencing
Citation: Carbonetto B, Fabbro MC, Sciara M, Seravalle A, Méjico G, Revale S, Romero MS, Brun B, Fay M, Fay F and Vazquez MP (2016) Human Microbiota of the Argentine Population- A Pilot Study. Front. Microbiol. 7:51. doi: 10.3389/fmicb.2016.00051
Received: 30 October 2015; Accepted: 12 January 2016;
Published: 01 February 2016.
Edited by:
M. Pilar Francino, FISABIO – Public Health, Valencian Health Department, SpainReviewed by:
Elisabeth Margaretha Bik, Stanford University School of Medicine, USAAymé Spor, INRA, UMR1347 Agroécologie, France
Copyright © 2016 Carbonetto, Fabbro, Sciara, Seravalle, Méjico, Revale, Romero, Brun, Fay, Fay and Vazquez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Martín P. Vazquez, bWFydGluLnZhenF1ZXpAaW5kZWFyLmNvbQ==