 Xiao-Yan Yang1,2
Xiao-Yan Yang1,2 Ke He2Gaofei Du2
Ke He2Gaofei Du2 Xiaohui Wu2
Xiaohui Wu2 Guangchuang Yu2Yunlong Pan1Gong Zhang2*
Guangchuang Yu2Yunlong Pan1Gong Zhang2* Xuesong Sun2*Qing-Yu He2*
Xuesong Sun2*Qing-Yu He2*- 1The First Affiliated Hospital of Jinan University, Guangzhou, China
- 2Key Laboratory of Functional Protein Research of Guangdong Higher Education Institutes, Institute of Life and Health Engineering, College of Life Science and Technology, Jinan University, Guangzhou, China
Streptococcus pneumoniae (S.pneumoniae) is a major human pathogen causing morbidity and mortality worldwide. Efficiently acquiring iron from the environment is critical for S. pneumoniae to sustain growth and cause infection. There are only three known iron-uptake systems in Streptococcal species responsible for iron acquisition from the host, including ABC transporters PiaABC, PiuABC, and PitABC. Besides, no other iron-transporting system has been suggested. In this work, we employed our newly established translating mRNA analysis integrated with proteomics to evaluate the possible existence of novel iron transporters in the bacterium. We simultaneously deleted the iron-binding protein genes of the three iron-uptake systems to construct a piaA/piuA/pitA triple mutant (Tri-Mut) of S. pneumoniae D39, in which genes and proteins related to iron transport should be regulated in response to the deletion. With ribosome associated mRNA sequencing-based translatomics focusing on translating mRNA and iTRAQ quantitative proteomics based on the covalent labeling of peptides with tags of varying mass, we indeed observed a large number of genes and proteins representing various coordinated biological pathways with significantly altered expression levels in the Tri-Mut mutant. Highlighted in this observation is the identification of several new potential iron-uptake ABC transporters participating in iron metabolism of Streptococcus. In particular, putative protein SPD_1609 in operon 804 was verified to be a novel iron-binding protein with similar function to PitA in S. pneumoniae. These data derived from the integrative translatomics and proteomics analyses provided rich information and insightful clues for further investigations on iron-transporting mechanism in bacteria and the interplay between Streptococcal iron availability and the biological metabolic pathways.
Introduction
Streptococcus pneumoniae (S. pneumoniae) is a Gram-positive bacterium and a major human pathogen. The bacterium is carried asymptomatically in the nasopharynx by up to 70% human population, establishing septicemia, and respiratory tract infections if given the chance to access deeper tissues (Mitchell, 2000; Ong et al., 2013). The capacity of bacterial pathogens such as S. pneumoniae to capture iron from the host environment is essential for the establishment of infection (Schaible and Kaufmann, 2004), as the host strictly limits iron sources as a part of its innate defense against invading pathogens. In order to ensure efficient uptake of iron sources, bacterial pathogens have evolved high-affinity iron-uptake ATP-binding cassette (ABC) transporters, with regulated expressions of a range of genes, in response to the limited iron concentration.
ABC transporters share two transmembrane domains (TMDs) that usually form the ligand binding sites with specificity, and two nucleotide-binding domains (NBDs) that bind and hydrolyze ATP to drive the translocation of the bound ligands, transporting a large number of substrates across cellular membranes (Davidson et al., 2008). These ABC transporters are frequently redundant in a strain for efficiently acquiring iron; for example, at least four iron-uptake ABC transporters have been found in Staphylococcus aureus (Hammer and Skaar, 2011). In Gram-positive bacteria, substrate-binding proteins (SBPs) of ABC transporters are typical lipoproteins located on cell surface (Hutchings et al., 2009). In S. pneumoniae, three ABC transporters, including PiaABC, PiuABC, and PitABC with lipoproteins PiaA, PiuA, and PitA as SBPs, have been identified to be involved in the acquisition of iron, respectively responsible for transporting ferrichrome (Fch), heme, and ferric irons (Brown et al., 2001a, 2002). Recently, two membrane proteins (gi|15901633 and gi|15900732) (Romero-Espejel et al., 2013) and one secreted protein (GAPDH) (Vázquez-Zamorano et al., 2014) have been identified as hemoglobin and haem-binding proteins. Intriguingly, in Brown et al. (2002) study, the piuB/piaA/pitA triple mutant poorly grew in iron-restricted medium, but its growth can be restored by adding FeCl3 (Brown et al., 2002). We therefore speculated that there are one or more novel iron-uptake systems to sustain the growth of piuB/piaA/pitA triple mutant strain. Apart from these, no other novel iron-uptake systems have been suggested to date and the detailed molecular mechanism of iron transportation in S. pneumoniae is poorly understood.
To obtain comprehensive information about iron regulation and search for potential iron transporters in S. pneumoniae, we used translatomics together with proteomics to assess the global changes in gene and protein expressions in response to the deletions of the three SBP lipoproteins. Translatomics here refers to our newly established translating mRNA analysis, in which mRNAs in the ribosome are purified and sequenced to obtain the comprehensive information on the genes being translated into proteins (Wang et al., 2013b; Zhang et al., 2014). Under steady-states, the abundances of translating mRNAs are highly correlated to the expression levels of proteins on genome-wide scale, and the preferentially translated genes dictate the cellular functions and phenotypes (Wang et al., 2013b; Zhang et al., 2014). With high sequencing coverage and sensitivity, translatome sequencing is capable to detect full-length translating mRNAs corresponding to low abundance proteins, poorly soluble proteins and easily degradable proteins that usually missed by proteomics, and thus becomes an important method complementary to proteomics in identifying protein alterations (Zhang et al., 2014; Zhong et al., 2014).
In this work, we constructed the piaA/piuA/pitA triple mutant (Tri-Mut) and investigated the effects of the simultaneous deletion of the three SBP genes on pneumococcal growth in various media. We then compared both the translatomes and proteomes in parallel between Tri-Mut and the corresponding wild-type (WT) strain of S. pneumoniae D39 using ribosome associated mRNA analysis integrated with iTRAQ-based proteomics. Quantitative results revealed that a large number of genes and proteins are affected by the gene deletions, among which a novel iron-binding protein SPD_1609 belonging to a potential iron-uptake ABC transporter, operon 804, in S. pneumoniae was identified and verified.
Materials and Methods
Ethics Approvals
The sheep blood was purchased from Ruite (Guangzhou, China), all procedures were performed in accordance with the China Animal Experimentation and Welfare Ethics Committee.
Bacterial Strains, Growth Media, and Culture Conditions
S. pneumoniae D39 strain was routinely cultured in Todd-Hewitt broth (Oxiod, UK) containing 0.5% yeast extract (THY) or grew on Columbia agar (Difco, USA) containing 5% sheep blood (Ruite, China) at 37°C with 5% CO2. When necessary, appropriate antibiotics were added to media: erythromycin (Erm) at 0.2 μg/mL, chloramphenicol (Cm) at 4 μg/mL, spectinomycin (Spec) at 100 μg/mL, tetracycline (Tet) at 3.5 μg/mL. The iron-restricted medium was produced by adding 5% Chelex-100 (Bio-Rad) to THY for 8 h with continuous agitation, followed by filter sterilization to remove the Chelex-100 and supplementation with 100 μM CaCl2 and 2 mM MgCl2. The iron content in the medium after Chelex-100 treatment was determined by inductively coupled plasma mass spectrometry (ICP-MS, Thermo Scientific, USA). When required, 20 μM FeCl3, hemin, or Fch was added to the iron-restricted medium.
Construction of the piaA/piuA/pitA Triple Mutant Strain (Tri-Mut)
The primers used for this work were listed in Table S1. Tri-Mut was constructed using the previously described method (Wach, 1996; Bayle et al., 2011). Competent cells of WT-D39 strain was transformed with long flanking homology-polymerase chain reaction (LFH-PCR) products, consisting of an antibiotic resistance cassette (Erm, Cm, or Spec) flanked by 500 bp long fragments homologous to the ends of each target gene, piaA(SPD_0915), piuA(SPD_1652), or pitA (SPD_0226). Tri-Mut was made by replacing piaA, piuA, and pitA genes of WT with gene encoding resistance to erythromycin (Erm), chloramphenicol (Cm), and spectinomycin (Spec), respectively. Transformant was selected with adequate antibiotic-containing Columbia sheep blood agar plates, and confirmed by PCR and Western blotting. The piaA/piuA double mutant and piaA/piuA/1609 triple mutant (Tri-Mut2) were also constructed using a similar method. These mutations were stable after six sequential passages in THY medium without antibiotic selection.
Ribosome Associated mRNAs Purification
S. pneumoniae WT and Tri-Mut strains were cultured in normal THY medium, and collected at exponential growth phase. WT and Tri-Mut cells were pre-treated with 100 μg/mL chloramphenicol for 20 min and then pelleted by centrifugation (6000 g, 20 min, 4°C), the supernatants were removed, and the pellets were resuspended in 5 mL pre-chilled Buffer B [50 mM Hepes, 500 mM KOAc, 24 mM Mg (OAc)2, 100 μg/mL chloramphenicol, pH 7.4] supplemented with 10 mg/mL lysozyme. After 20 min ice-bath, samples were frozen by liquid nitrogen and disrupted using mechanical grinding. Cell lysates were treated with RNase-free DNase I (Thermo Scientific, USA) for 15 min on ice, and the debris was removed by centrifugation at 18,000 rpm for 15 min at 4°C. Supernatants (3 mL) were layered on 12 mL of 35% sucrose buffer, Ribosome associated mRNAs were pelleted after ultracentrifugation (Beckman Coulter SW 70 Ti rotor) at 42,000 rpm for 5 h at 4°C. Ribosome associated mRNAs were isolated using the TRIzol RNA extraction reagent (Ambion, USA) according to the manufacturer's instructions. Genomic DNA was removed by treatment with RNase-free DNase I. The 23S, 16S, and 5S rRNAs were removed using the Ribo-Zero magnetic kit (Gram-Positive Bacteria, Epicentre, USA). The rRNA-depleted mRNA was verified by agarose gel electrophoresis.
Sequencing and Data Analysis
The ribosome associated mRNA libraries were generated using NEBNext® mRNA Library Prep Master Mix Set for Illumina (BioLabs, USA) as directed by the manufacturer. The purified libraries were sequenced on an Illumina HiSeq 2000 sequencer for 50 cycles. The reads that passed the Illumina filter were mapped to S. pneumoniae D39 reference genome (NC_008533.1) using FANSe2 program (http://bioinformatics.jnu.edu.cn/software/fanse2/) (Xiao et al., 2014) with the following criteria: max read length = 60; max error = 3; indel detection = on; best position = on; min. seed length = 8; memory reduction = on. Differential expression analyses between groups were conducted using edgeR (Robinson et al., 2010). A combined criterion of |log2(fold change)|≥ 1 and a p < 0.05 was adopted to judge the significance of differentially translated gene (DTG) between WT and Tri-Mut. Two biological replicates were performed.
Protein Preparation, iTRAQ Labeling, and Proteomics Analysis
Proteins were extracted from the WT and Tri-Mut strains cultivated in normal THY medium at exponential growth phase in accordance with our previously reported method (Yang et al., 2015). Two hundred microgram proteins from each sample were dissolved in an equal volume of sample buffer, followed by disulfide reduction with 10 mM of dithiothreitol (56°C, 1 h) and alkylation with 55 mM of iodoacetamide (25°C, 40 min in dark). For each sample, 20 μg proteins were quantified by 10% SDS-PAGE, then 150 μg proteins were precipitated with 4 volume of ice-cold acetone at −20°C for 2 h and collected by centrifugation (2000 g, 5 min, 4°C). The pellet was resuspended in 40 μL dissolution buffer (0.5% TAB, 1 M urea), digested with trypsin (1:25 w/w at 0 h, 1:50 w/w at 3 h) (Promega, USA) at 37°C for 18 h and then lyophilized.
The iTRAQ labeling of the peptide samples were performed using an iTRAQ Reagent 4-plex kit (AB SCIEX, USA) according to the manufacturer's protocol. Two biological replicates for WT were labeled with 114-, 115-, and two biological replicates for Tri-Mut were labeled with 116-, 117-. After incubation for 2 h, the labeled peptides with respective isobaric tags were dried to ~20 μL with a vacuum centrifuge. The labeled WT and Tri-Mut replicate samples were 1:1 pooled (114 vs. 116, 115 vs. 117), and cleaned up using Strata-X 33u polymeric reversed phase column (10 mg/mL, Phenomenex, USA). Desalted peptides were resuspended with buffer A (5% acetonitrile, 0.1% formic acid) and detected using an ABSCIEX Triple-TOF 5600 mass spectrometer (AB SCIEX, USA) coupled with a Nanospray III source and a pulled quartz tip. The parameters were used in the mass spectrometer as described previously (Yin et al., 2013).
The data (.mgf) were acquired from raw data (.wiff) by AB SCIEX MS Data Converter V1.1 software, then identified and quantified via ProteinPilot™ Software 4.5. The quantitative analysis parameters were set as follow: Sample Type, iTRAQ 4 plex (Peptide Labeled); Cys. Alkylation, Iodoacetic acid; Digestion, Trypsin; Instrument, Triple-TOF 5600; ID Focus, Biological modifications; Database, S. pneumoniae D39_.fasta.fasta; Search Effort, Thorough; Detected Protein Threshold [Unused ProtScore (Conf)] > 1.30 (95.0%). The criteria of fold chang >1.20 or < 0.83 combined with p < 0.05 was used to define the differentially expressed protein (DEP) between WT and Tri-Mut in two biological replicates.
Functional Category and Network Analysis
The biological processes of the DTGs and DEPs were analyzed according to published data or closely related homologs, combined with (Yu et al., 2012) on the Gene Ontology (GO) database searches using clusterProfiler (v1.12.0) (http://www.bioconductor.org/) of S. pneumoniae D39 (Sun et al., 2010). And the interaction networks for DTGs and DEPs were constructed by the STRING as described previously (von Mering et al., 2005, 2007) (http://string-db.org/). The following parameter settings were used: organism S. pneumoniae D39; confidence threshold 0.70; no more than 10 interactors. The networks were represented with Cytoscape.
Immunization Experiments and Western Blotting Analysis
Purified His6-PiaA, His6-PiuA, His6-PitA, His6-PsaA, and SPD_1609 proteins were used as antigens for the immunization experiments to generate multiclonal antibodies in mice according to the previous report (Brown et al., 2001b). For Western blotting analysis, equal amounts of proteins were separated with 10–20% SDS-PAGE electrophoresis prior to transfer onto polyvinylidenedifluoride (PVDF) membranes (Millipore, USA), then probed with the mouse specific multiclonal antibodies and horseradish peroxidase (HRP)-conjugated goat anti-mouse secondary antibodies. The results were visualized by Clarity™ Western ECL Substrate (Bio-Rad, USA) and quantified using ImageMaster 2D Platinum 6.0. PsaA protein was used as a loading control.
Analysis of Intracellular Metal Concentration
Bacteria at exponential growth phase (OD600 = 0.4~0.5) were pelleted and washed three times with 1 × PBS which pretreated with chelex-100 resin. Subsequently, the cell pellets were dried using a Scanvac Freeze Dryer (Labgene Scientific, Switzerland) and the dry weights were measured. The dry cell mass was resuspended in 65% nitric acid, then heated to 95°C for 20 min. Samples were then diluted to 2% nitric acid and centrifuged at 13,200 g for 30 min, the supernatants were collected and submitted for ICP-MS analysis. Metal contents of samples were normalized to dry weight of cells (ng of Fe per mg cells). All data were evaluated with at least three independent biological experiments.
Statistical Analysis
Statistical analysis was carried out using two tailed, unpaired Student's t-test and assigned p ≤ 0.05 (denoted by *), p ≤ 0.01 (denoted by **) and p ≤ 0.001 (denoted by ***) by GraphPad Prism version 5.01. Data presented are the mean ± SEM of at least three biological replicates.
Results and Discussion
The Growth of the Tri-Mut was Disturbed in Iron-Depleted Media
In S. pneumoniae, three operons (pia, piu, pit) encode ABC transporters known as iron-uptake systems, each contains one lipoprotein iron receptor (SBP), one ATPase and two transmembrane permease proteins (Brown et al., 2001a, 2002). In order to search for new iron-uptake related proteins, we deleted the three critical SBP genes piaA/piuA/pitA in WT-D39 strain using homologous recombination (Figure 1A) to construct the Tri-Mut, with a presumption that genes/proteins related to the iron uptake must be potentially regulated in response to the deletion. PCR (Figure S1) and Western blotting were used to confirm the complete absence of the three SBPs in Tri-Mut (Figure 1B).
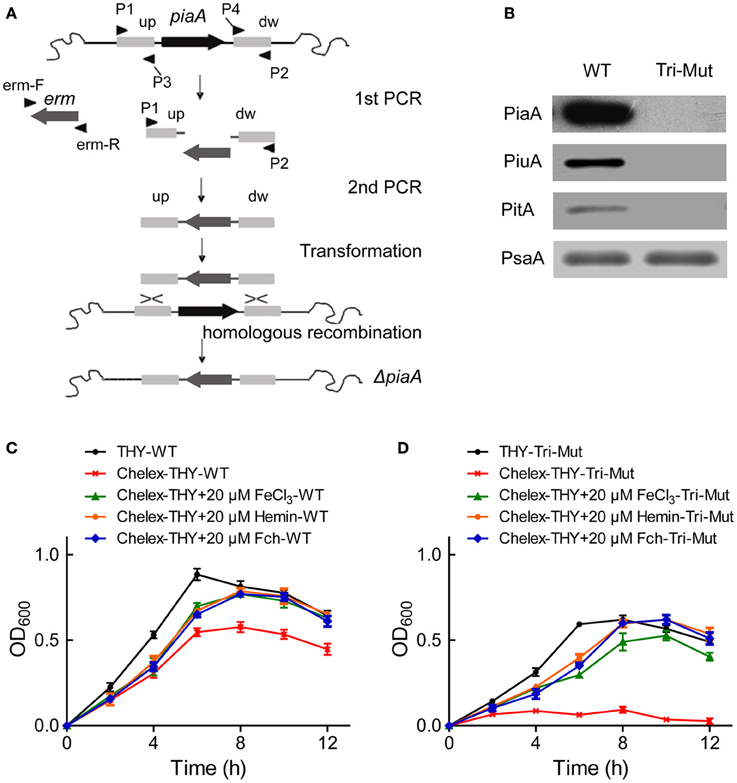
Figure 1. Construction, verification, and characterization of piaA/piuA/pitA triple mutant (Tri-Mut) of S. pneumoniae. (A) Strategy map of gene knock-out. (B) Verification of the simultaneous deletion of the three genes piaA/piuA/pitA in Tri-Mut strain by Western blotting. PsaA protein was used as loading control. (C) The growth curves of WT strain in various media with or without iron source. (D) The growth curves of Tri-Mut in various media in the presence or absence of iron source.
We then evaluated the bacterial growth of the Tri-Mut as compared to WT in various media. As shown in Figure 1C, WT strain reasonably had a delayed growth in Chelex-treated iron-restricted medium, and adding any of FeCl3, hemin, and Fch can recover the growth. As comparison, Tri-Mut had a decreased growth even in normal THY medium and almost did not grow in iron-restricted medium; interestingly, adding iron sources restored the bacterial growth (Figure 1D). This observation suggests the existence of other iron-acquisition channels that assimilated iron for the bacterial growth in the absence of the three primary SBPs in Tri-Mut. Indeed, potential iron-related genes, and proteins can be stimulated to compensate the defect of the normal iron-uptake systems in Tri-Mut. These genes and proteins were probably up-regulated in Tri-Mut and should be identified by both translatomics and proteomics through the comparison between WT and Tri-Mut.
Screening for DTGs and DEPs by Translatomics and Proteomics
We firstly used mRNA sequencing to screen for the affected genes in Tri-Mut. A total of 1454 genes in WT and 1666 genes in Tri-Mut were mapped with ≥10 reads, representing ~70.3 and 80.5% of the 2069 predicted genes in the S. pneumoniae D39 genome, respectively. The differences in gene expression between WT and Tri-Mut with two biological replicates were calculated by edgeR, resulting in totally 1601 genes (Figure 2A). Using a cutoff of 2.0-fold change and p < 0.05, 793 genes were considered as differentially translated genes (DTGs) (Figure 2B), including 430 up-regulated DTGs and 363 down-regulated DTGs in Tri-Mut. More than half of genes in Tri-Mut were regulated as that the simultaneous deletion of three major iron transporter genes in the bacterium seriously destroyed the iron-uptake ability of Tri-Mut and thus affected many important biological pathways.
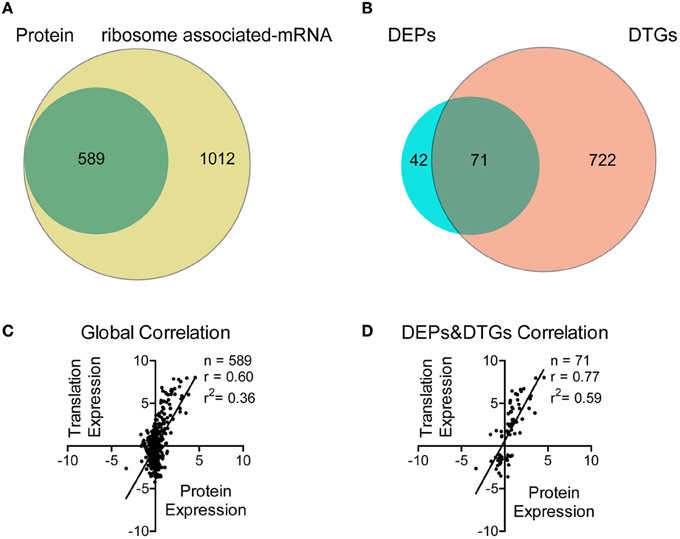
Figure 2. Correlation analysis between proteomics and translatomics. (A) Venn diagram of the number of proteins and mRNAs quantified using quantitative proteomics and mRNA-Seq, respectively. (B) Venn diagram of the number of DEPs and DTGs, respectively. (C) Scatterplot of the relationship between the fold changes of proteins and mRNAs (Tri-Mut vs. WT strain, log2-transformed) quantified in both data sets. (D) Scatterplot of the relationship between the fold changes of DEPs and DTGs (Tri-Mut vs. WT strain, log2-transformed) quantified in both data sets.
iTRAQ-based proteomics was then used to identify the proteins with altered expressions in Tri-Mut. Using ProteinPilot™ Software, we detected a total of 589 proteins with isotopic labels for comparison in two biological replicates (Figure 2A). Relative quantitative analysis revealed 113 DEPs with p < 0.05, including 52 DEPs with increased abundance (≥1.20 fold) and 61 DEPs with decreased abundance (≤0.83) in Tri-Mut (Figure 2B). The complete list of the DTGs and DEPs is provided in Tables S2, S3 in the Supplemental Materials. The global correlation between translatomics and proteomics data was visualized as scatter-plots (Figure 2C), showing a moderate but significant correlation (r = 0.60; n = 589, p < 0.001). Moreover, the degree of correlation between DEPs and DEGs also exhibited a good correlation (r = 0.77; n = 71, p < 0.001, Figure 2D).
As shown in Figure 2A, the number of genes observed by translatomics were more than those identified by proteomics because ribosome associated mRNA sequencing is independent of physical and chemical properties of proteins. For example, many transmembrane proteins such as SPD_0088, SPD_0089, SPD_1607 were solely detected in mRNA-seq (Table S2), probably due to their low solubility and thus failed to be identified by mass spectrometry. Moreover, we noted that almost all genes of an operon could be found in the translatomics data, but only one or two proteins encoded by the operon genes were identified by proteomics (Tables S2, S3). These results exhibited that full-length translating mRNA analysis possesses higher identification efficiency than proteomics in prokaryotic systems. Indeed, proteins are the end products of genes that carry out biological functions, the genome-wide and bias-free measurements on translational regulation provide a global view for us to assess the networks involved in the iron metabolism, favorable to the identification of new potential iron-binding proteins.
Integrative Analysis of DTGs and DEPs
Obviously, the DTGs and DEPs that were simultaneously identified by both translatomics and proteomics are most confidential and promising molecules to be followed up, meaning that they were regulated in both the translating mRNA and protein levels at the same time. By overlapping the two parts of data, we found that 71 DEPs were also identified as DTGs, among which 59 proteins shared the same change trend at both levels; 40 proteins were significantly up-regulated in Tri-Mut while 19 proteins were highly expressed in WT (Figure 2B). Literature searches and related homologs analyses combined with GO analysis were carried out for the biological process of these 59 DEPs and DTGs (Figure 3). These DEPs and DTGs were annotated to various cellular processes including translation, protein folding or secretion, DNA replication or transcription, amino acid metabolism, carbohydrate metabolism, fatty acid metabolism, oxidation–reduction process, and sugar/ion transport (Figure 3 and Table 1).
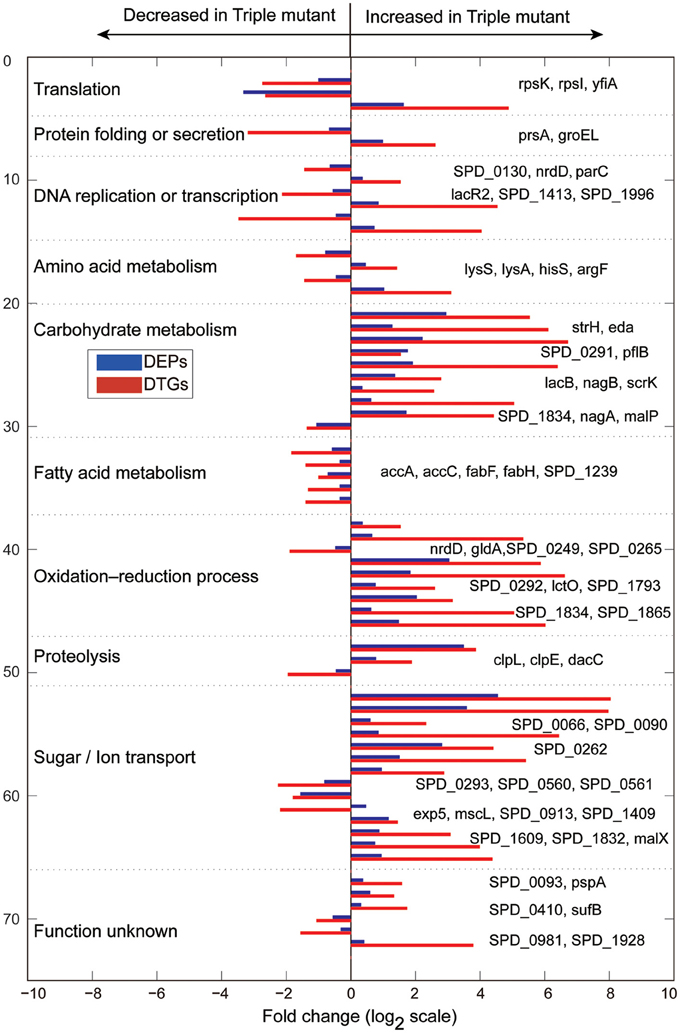
Figure 3. Biological processes of the DTGs and DEPs between WT and Tri-Mut of S. pneumoniae, which were analyzed according to published data or closely related homologs, combined with the Gene Ontology (GO) database searches using clusterProfiler (v1.12.0) of S. pneumoniae D39.
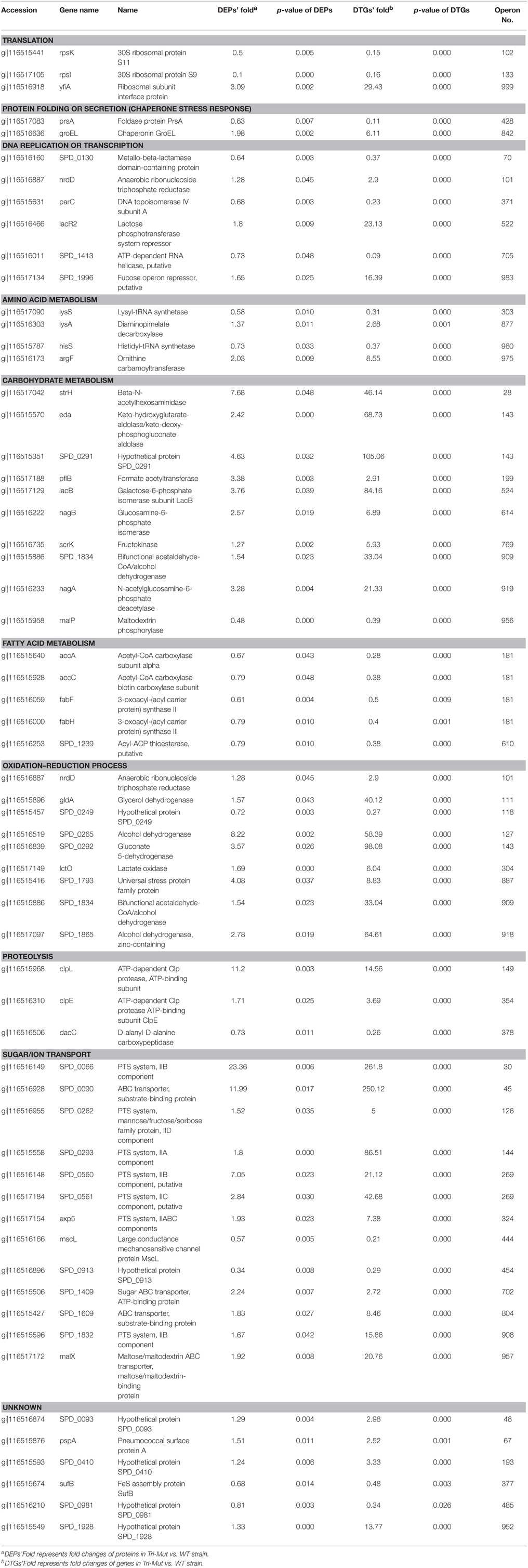
Table 1. Summary of DEPs and DTGs.
We next constructed the interaction networks of the 59 DEPs and DTGs by using STRING database (http://string-db.org/). As shown in Figure 4, most of the DEPs in the map have direct or indirect linkages to each other, forming a large interaction network, suggesting that they are most likely to cooperate in implementing specific biological functions. It is worth noting that five DEPs, including acetyl-CoA carboxylase subunit alpha (AccA), acetyl-CoA carboxylase biotin carboxylase subunit (AccC), 3-oxoacyl-(acyl carrier protein) synthase II (FabF), 3-oxoacyl-(acyl carrier protein) synthase III (FabH), and acyl-ACP thioesterase (putative SPD_1239), involved in fatty acid metabolism as a sub-network, were suppressed in Tri-Mut (Figure 4). Consistently, most genes of this operon within fatty acid metabolism pathway were down-regulated at translational level (Table S2), collectively suggesting that iron is the important cofactor in this process.
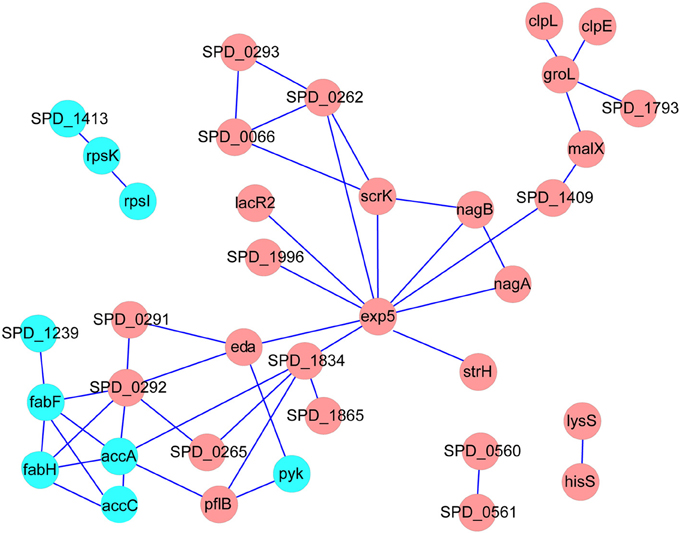
Figure 4. The interaction networks of DEPs and DTGs were constructed by the STRING database, using confidence level of 0.70, no more than 10 interactors, and the networks were represented with Cytoscape. Red represents the proteins up-regulated in Tri-Mut, blue represents the proteins down-regulated in Tri-Mut.
In addition, the down-regulation of ribosomal proteins was also detected in Tri-Mut, implicating that iron deficiency inhibited protein biosynthesis, as observed in Arabidopsis exposed to iron-deficient conditions (Wang et al., 2013a).
Moreover, seven proteins belong to PTS systems were also induced in Tri-Mut both at mRNA and protein levels (Table 1). The PTS system is traditionally considered as a sugar uptake and phosphorylation system, also mediating catabolite repression in bacteria (Siebold et al., 2001; Lengeler and Jahreis, 2009). Usually, a PTS system is comprised of two cytoplasmic energy-coupling proteins, the Enzyme I (EI) and the histidine-containing protein (HPr), and carbohydrate specific Enzymes II (EII), which is consisted of one or two hydrophobic integral membrane domains (IIC and IID) and two hydrophilic domains (IIA and IIB), responsible for transport- and catalysis-concomitant phosphorylation of the carbohydrate (Deutscher et al., 2006). Previous studies have suggested that PTS system can be considered as the “nerve system” of the bacteria, which employs elaborate signal transduction pathway, responding to external stimuli as well as the internal metabolic status (Aboulwafa and Saier, 2013). The PTS system broadly highly expressed in Tri-Mut, possibly providing carbohydrate to maintain bacterial growth in iron-starved status. This phenomenon had also been found in Arabidopsis in responding to iron-deficient stress (Zargar et al., 2015). Nevertheless, whether the PTS system has a side-function involved in iron uptake awaits further investigations.
Identification of Unknown Iron-Uptake Proteins in S. pneumoniae
We supposed that potential iron-uptake proteins would be substrate transporters that were highly expressed in the Tri-Mut to acquire iron and maintain bacterial growth, thus we focused on the specifically up-regulated DEPs and DTGs in Tri-Mut. We paid particular attention to the up-regulated DEPs and DTGs assigned to the category of “sugar/ion transport” and “hypothetical protein.” The “sugar/ion transport” category highly expressed in Tri-Mut contains mainly six PTS systems and five ABC transporters.
In particular, up-regulated ABC transporters here included two sugar ABC transporter proteins, SPD_1409, malX, and two putative conserved ABC transporter SBPs, SPD_0090, and SPD_1609. SPD_0090 protein is located immediately adjacent to two predicted permease proteins SPD_0088 and SPD_0089 of the same transport system (operon 45), while SPD_1609 gene couples with four neighboring genes: MgtC/SapB family protein (SPD_1606), permease protein (SPD_1607), ATP-binding protein (SPD_1608), and conserved hypothetical protein (SPD_1610), forming another ABC transport system (operon 804) (Figure 5A). Supportively, translatomics data demonstrated that all the genes of operons 45 and 804 were up-regulated in Tri-Mut (Table S2).
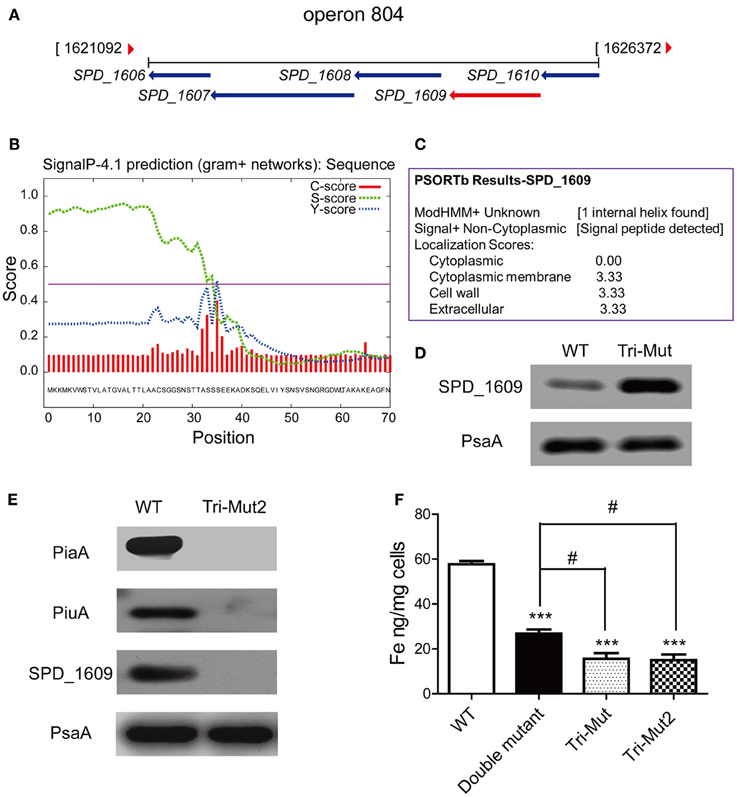
Figure 5. The operon 804 is probably a novel iron-uptake system required for Streptococcal growth. (A) Organization of operon 804 in the S. pneumoniae D39 genome. (B) The SignalP 4.1 analysis on SPD_1609 protein. (C) The Psortb analysis on SPD_1609 protein. (D) Verification of SPD_1609 protein up-regulation in piaA/piuA/pitA Tri-Mut by Western blotting. (E) Verification of the complete absence of the three genes piaA/piuA/1609 in Tri-Mut2 by Western blotting. (F) ICP-MS analysis of iron contents in WT strain, piaA/piuA double mutant, Tri-Mut, and Tri-Mut2, respectively. Results are the mean of 3 experiments; error bars are SEM, ***p < 0.001 vs. WT strain; #p < 0.05 vs. piaA/piuA double mutant.
Further protein sequence alignment revealed that SPD_0090 is highly similar to the SBPs of sugar ABC transporters in many Gram-positive bacteria (Table S4), while SPD_1609 shows a high similarity with the SBPs of iron ABC transporters in Streptococcus and Bacillus species (Table S5). Moreover, SPD_1609 is a lipoprotein anchored into the cell membrane that contains a typical lipoprotein signal peptide cleavage site (between position 34 and 35 of SPD_1609 protein sequence), which was predicted by SignalP-4.1 (Figure 5B) and Psortb (Figure 5C). These analytical results render SPD_0090 and SPD_1609 appropriate candidates of the SBPs in unknown iron-transport systems, they are potential iron-binding proteins that were up-regulated for iron acquisition compensating to the defect of the primary iron SBPs in Tri-Mut.
Validation of Novel Iron-Uptake Protein SPD_1609 in S. pneumoniae
We selected one of the candidates, SPD_1609, for functional validation. We firstly prepared a multiclonal antibody against SPD_1609, and confirmed the up-regulated expression of SPD_1609 in Tri-Mut by Western blotting analysis (Figure 5D). To investigate the biological function of SPD_1609 and exclude the interference of two major iron transporters PiaA and PiuA, we knocked out SPD_1609 gene in piaA/piuA double mutant to generate piaA/piuA/1609 triple mutant (Tri-Mut2) for the assessment of its biological function. Western blotting experiments shown in Figure 5E confirmed the successful deletion of the three genes piaA/piuA/1609 at the same time. We also tried several times to construct pitA/piaA/piuA/1609 tetra mutant, but failed to obtain the mutant strain as it almost did not grow in THY medium. One possible reason is that the simultaneous deletion of the four genes in the bacterium resulted in a complete loss of iron uptake ability. The knock-out of SPD_1609 gene ensured the complete inactivation of operon 804, the function of the potential iron-transport system can then be evaluated by the comparison among WT, the piaA/piuA double mutant, and Tri-Mut2.
We next used ICP-MS to determine the intracellular iron levels in WT, piaA/piuA double mutant, Tri-Mut and Tri-Mut2 under identical normal growth conditions. The results are shown in Figure 5F. The deletion of any of the SBP genes in the bacterium would reasonably result in impaired iron acquisition of the mutant strains. As expected, the piaA/piuA double mutant presented a substantially reduced intracellular iron concentration as compared to WT. Under this background, deleting one more gene, either pitA or SPD_1609, further depressed the iron-acquisition ability of the bacterium, leading to the similar decreased levels of intracellular iron in both the triple mutants (Figure 5F). This observation implicates that SPD_1609 may play an iron-binding function similar to PitA; the absence of either the proteins caused an equivalent effect of impairment on iron acquisition. Taken together, these results provided evidences that operon 804 containing SPD_1609 may function as a novel Streptococcal iron-transport system, which works as an important backup for iron metabolism especially when the three known iron-transport systems were inactivated in S. pneumoniae.
Conclusions
In this work, we performed translatomics integrated with proteomics analysis to screen for potential novel iron-transporting proteins in Gram-positive bacteria using S. pneumoniae as a model strain. By deleting the genes of three known iron-binding SBPs, a great number of both conventional and previously unreported genes were affected in the Tri-Mut and their corresponding protein alterations were detected by proteomics. With a speculation that potential iron-binding proteins should be up-regulated to compensate the deletion of the three primary SBP genes, we focused on those highly expressed and overlapped DTGs and DEPs in Tri-Mut. Among a number of these up-regulated DEPs, functionally putative proteins SPD_0090 and SPD_1609 were identified to be the potential candidates of novel iron-transporting proteins. Further validations confirmed that SPD_1609 in operon 804 is probably an iron-binding protein similar to PitA. We thus demonstrated a powerful strategy for the search of potential new functional molecules, providing a number of promising candidate genes and proteins related to bacterial iron acquisition. Further validations and investigations on these molecules with putative functions may help to comprehensively understand the iron-transport mechanism, shedding light on the interplay between iron availability and the biological metabolic pathways in bacteria.
The raw data of ribosome associated mRNA sequencing have been uploaded on SRA website (SRP067291). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (Vizcaíno et al., 2014) via the PRIDE partner repository with the dataset identifier PXD003313.
Author Contributions
XY perfomed experiments and wrote paper. KH and GD performed experiments. XW, GY, and YP analyzed data. GZ, XS, and QH instructed experiments and revised paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Yibing Yin and Dr. Xuemei Zhang from Chongqing Medical University for their help in constructing mutant strains. This work was supported by the National Natural Science Foundation of China (21271086, to QH; 21571082, to XS), Guangdong Natural Science Research Grant (32213027/32215077, to QH; 2015A030313334 to XS), the Pearl River Rising Star of Science and Technology of Guangzhou City (2011J2200003, to XS), and The Open Fund of The First Affiliated Hospital, Jinan University, Guangzhou.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2016.00078
Table S1. The primer sequences of genes.
Table S2. The genes identified by ribosome associated mRNA sequencing-based translatomics.
Table S3. The proteins identified by iTRAQ-based proteomics.
Table S4. Proteins with high identity and similarity with SPD_0090 based on protein sequence alignment using Position-Specific Iterated BLAST (PSI-BLAST) searches.
Table S5. Proteins with high identity and similarity with SPD_1609 based on protein sequence alignment using Position-Specific Iterated BLAST (PSI-BLAST) searches.
Figure S1. Verification of piaA, piuA, pitA, erm, spec, and cm gene by PCR in piaA/piuA/pitA triple mutant strain. Lane 1 and 8, DL 2000 DNA Marker; lane 2, piaA of WT; lane 3, piaA of Tri-Mut; lane 4, piuA of WT; lane 5, piuA of Tri-Mut; lane 6, pitA of WT; lane 7, pitA of Tri-Mut; lane 9, erm of WT; lane 10, erm of Tri-Mut; lane 11, spec of WT; lane 12, spec of Tri-Mut; lane 13, cm of WT; lane 14, cm of Tri-Mut.
References
Aboulwafa, M., and Saier, M. H. Jr. (2013). Lipid dependencies, biogenesis and cytoplasmic micellar forms of integral membrane sugar transport proteins of the bacterial phosphotransferase system. Microbiology 159, 2213–2224. doi: 10.1099/mic.0.070953-0
Bayle, L., Chimalapati, S., Schoehn, G., Brown, J., Vernet, T., and Durmort, C. (2011). Zinc uptake by Streptococcus pneumoniae depends on both AdcA and AdcAII and is essential for normal bacterial morphology and virulence. Mol. Microbiol. 82, 904–916. doi: 10.1111/j.1365-2958.2011.07862.x
Brown, J. S., Gilliland, S. M., and Holden, D. W. (2001a). A Streptococcus pneumoniae pathogenicity island encoding an ABC transporter involved in iron uptake and virulence. Mol. Microbiol. 40, 572–585. doi: 10.1046/j.1365-2958.2001.02414.x
Brown, J. S., Gilliland, S. M., Ruiz-Albert, J., and Holden, D. W. (2002). Characterization of pit, a Streptococcus pneumoniae iron uptake ABC transporter. Infect. Immun. 70, 4389–4398. doi: 10.1128/IAI.70.8.4389-4398.2002
Brown, J. S., Ogunniyi, A. D., Woodrow, M. C., Holden, D. W., and Paton, J. C. (2001b). Immunization with components of two iron uptake ABC transporters protects mice against systemic Streptococcus pneumoniae infection. Infect. Immun. 69, 6702–6706. doi: 10.1128/IAI.69.11.6702-6706.2001
Davidson, A. L., Dassa, E., Orelle, C., and Chen, J. (2008). Structure, function, and evolution of bacterial ATP-binding cassette systems. Microbiol. Mol. Biol. Rev. 72, 317–364, table of contents. doi: 10.1128/MMBR.00031-07
Deutscher, J., Francke, C., and Postma, P. W. (2006). How phosphotransferase system-related protein phosphorylation regulates carbohydrate metabolism in bacteria. Microbiol. Mol. Biol. Rev. 70, 939–1031. doi: 10.1128/MMBR.00024-06
Hammer, N. D., and Skaar, E. P. (2011). Molecular mechanisms of Staphylococcus aureus iron acquisition. Annu. Rev. Microbiol. 65, 129–147. doi: 10.1146/annurev-micro-090110-102851
Hutchings, M. I., Palmer, T., Harrington, D. J., and Sutcliffe, I. C. (2009). Lipoprotein biogenesis in Gram-positive bacteria: knowing when to hold ′em, knowing when to fold ′em. Trends Microbiol. 17, 13–21. doi: 10.1016/j.tim.2008.10.001
Lengeler, J. W., and Jahreis, K. (2009). Bacterial PEP-dependent carbohydrate: phosphotransferase systems couple sensing and global control mechanisms. Contrib. Microbiol. 16, 65–87. doi: 10.1159/000219373
Mitchell, T. J. (2000). Virulence factors and the pathogenesis of disease caused by Streptococcus pneumoniae. Res. Microbiol. 151, 413–419. doi: 10.1016/S0923-2508(00)00175-3
Ong, C. L., Potter, A. J., Trappetti, C., Walker, M. J., Jennings, M. P., Paton, J. C., et al. (2013). Interplay between manganese and iron in pneumococcal pathogenesis: role of the orphan response regulator RitR. Infect. Immun. 81, 421–429. doi: 10.1128/IAI.00805-12
Robinson, M. D., McCarthy, D. J., and Smyth, G. K. (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. doi: 10.1093/bioinformatics/btp616
Romero-Espejel, M. E., González-López, M. A., and Olivares-Trejo Jde, J. (2013). Streptococcus pneumoniae requires iron for its viability and expresses two membrane proteins that bind haemoglobin and haem. Metallomics 5, 384–389. doi: 10.1039/c3mt20244e
Schaible, U. E., and Kaufmann, S. H. (2004). Iron and microbial infection. Nat. Rev. Microbiol. 2, 946–953. doi: 10.1038/nrmicro1046
Siebold, C., Flükiger, K., Beutler, R., and Erni, B. (2001). Carbohydrate transporters of the bacterial phosphoenolpyruvate: sugar phosphotransferase system (PTS). FEBS Lett. 504, 104–111. doi: 10.1016/S0014-5793(01)02705-3
Sun, X., Ge, F., Xiao, C. L., Yin, X. F., Ge, R., Zhang, L. H., et al. (2010). Phosphoproteomic analysis reveals the multiple roles of phosphorylation in pathogenic bacterium Streptococcus pneumoniae. J. Proteome Res. 9, 275–282. doi: 10.1021/pr900612v
Vázquez-Zamorano, Z. E., González-López, M. A., Romero-Espejel, M. E., Azuara-Liceaga, E. I., Lopez-Casamichana, M., and Olivares-Trejo Jde, J. (2014). Streptococcus pneumoniae secretes a glyceraldehyde-3-phosphate dehydrogenase, which binds haemoglobin and haem. Biometals 27, 683–693. doi: 10.1007/s10534-014-9757-0
Vizcaíno, J. A., Deutsch, E. W., Wang, R., Csordas, A., Reisinger, F., Rios, D., et al. (2014). ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 32, 223–226. doi: 10.1038/nbt.2839
von Mering, C., Jensen, L. J., Kuhn, M., Chaffron, S., Doerks, T., Krüger, B., et al. (2007). STRING 7–recent developments in the integration and prediction of protein interactions. Nucleic Acids Res. 35, D358–D362. doi: 10.1093/nar/gkl825
von Mering, C., Jensen, L. J., Snel, B., Hooper, S. D., Krupp, M., Foglierini, M., et al. (2005). STRING: known and predicted protein-protein associations, integrated and transferred across organisms. Nucleic Acids Res. 33, D433–D437. doi: 10.1093/nar/gki005
Wach, A. (1996). PCR-synthesis of marker cassettes with long flanking homology regions for gene disruptions in S. cerevisiae. Yeast 12, 259–265.
Wang, J., Lan, P., Gao, H., Zheng, L., Li, W., and Schmidt, W. (2013a). Expression changes of ribosomal proteins in phosphate- and iron-deficient Arabidopsis roots predict stress-specific alterations in ribosome composition. BMC Genomics 14:783. doi: 10.1186/1471-2164-14-783
Wang, T., Cui, Y., Jin, J., Guo, J., Wang, G., Yin, X., et al. (2013b). Translating mRNAs strongly correlate to proteins in a multivariate manner and their translation ratios are phenotype specific. Nucleic Acids Res. 41, 4743–4754. doi: 10.1093/nar/gkt178
Xiao, C. L., Mai, Z. B., Lian, X. L., Zhong, J. Y., Jin, J. J., He, Q. Y., et al. (2014). FANSe2: a robust and cost-efficient alignment tool for quantitative next-generation sequencing applications. PLoS ONE 9:e94250. doi: 10.1371/journal.pone.0094250
Yang, X. Y., Zhang, L., Liu, J., Li, N., Yu, G., Cao, K., et al. (2015). Proteomic analysis on the antibacterial activity of a Ru(II) complex against Streptococcus pneumoniae. J. Proteomics 115, 107–116. doi: 10.1016/j.jprot.2014.11.018
Yin, S., Xue, J., Sun, H., Wen, B., Wang, Q., Perkins, G., et al. (2013). Quantitative evaluation of the mitochondrial proteomes of Drosophila melanogaster adapted to extreme oxygen conditions. PLoS ONE 8:e74011. doi: 10.1371/journal.pone.0074011
Yu, G., Wang, L. G., Han, Y., and He, Q. Y. (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287. doi: 10.1089/omi.2011.0118
Zargar, S. M., Kurata, R., Inaba, S., Oikawa, A., Fukui, R., Ogata, Y., et al. (2015). Quantitative proteomics of Arabidopsis shoot microsomal proteins reveals a cross-talk between excess zinc and iron deficiency. Proteomics 15, 1196–1201. doi: 10.1002/pmic.201400467
Zhang, G., Wang, T., and He, Q. (2014). How to discover new proteins-translatome profiling. Sci. China Life Sci. 57, 358–360. doi: 10.1007/s11427-014-4618-1
Keywords: translatomics, proteomics, S. pneumoniae, iron-acquisition system, iron-transporting protein
Citation: Yang X-Y, He K, Du G, Wu X, Yu G, Pan Y, Zhang G, Sun X and He Q-Y (2016) Integrated Translatomics with Proteomics to Identify Novel Iron–Transporting Proteins in Streptococcus pneumoniae. Front. Microbiol. 7:78. doi: 10.3389/fmicb.2016.00078
Received: 06 September 2015; Accepted: 15 January 2016;
Published: 03 February 2016.
Edited by:
Weiwen Zhang, Tianjin University, ChinaCopyright © 2016 Yang, He, Du, Wu, Yu, Pan, Zhang, Sun and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gong Zhang, emhhbmdnb25nQGpudS5lZHUuY24=;
Xuesong Sun, dHN1bnhzQGpudS5lZHUuY24=;
Qing-Yu He, dHF5aGVAam51LmVkdS5jbg==