 Jacquelynn Benjamino
Jacquelynn Benjamino Joerg Graf
Joerg Graf- 1Department of Molecular and Cell Biology, University of Connecticut, Storrs, CT, USA
- 2Institute for Systems Genomics, University of Connecticut, Storrs, CT, USA
The hindgut of the termite Reticulitermes flavipes harbors a complex symbiotic community consisting of protists, bacteria, and archaea. These symbionts aid in the digestion of lignocellulose from the termite’s wood meal. Termite hindguts were sampled and the V4 hyper-variable region of the 16S rRNA gene was sequenced and analyzed from individual termites. The core microbiota of worker termites consisted of 69 OTUs at the 97% identity level, grouped into 16 taxa, and together accounted for 67.05% of the sequences from the bacterial community. The core was dominated by Treponema, which contained 36 different OTUs and accounted for ∼32% of the sequences, which suggests Treponema sp. have an important impact on the overall physiology in the hindgut. Bray–Curtis beta diversity metrics showed that hindgut samples from termites of the same colony were more similar to each other than to samples from other colonies despite possessing a core that accounted for the majority of the sequences. The specific tasks and dietary differences of the termite castes could have an effect on the composition of the microbial community. The hindgut microbiota of termites from the alate castes differed from the worker caste with significantly lower abundances of Treponema and Endomicrobia, which dominated the hindgut microbiota in workers and soldiers. Protist abundances were also quantified in the same samples using qPCR of the 18S rRNA gene. Parabasalia abundances dropped significantly in the winged alates and the Oxymonadida abundances dropped in both alate castes. These data suggest that the changes in diet or overall host physiology affected the protist and bacterial populations in the hindgut. The in-depth bacterial characterization and protist quantification in this study sheds light on the potential community dynamics within the R. flavipes hindgut and identified a large and complex core microbiota in termites obtained from multiple colonies and castes.
Introduction
Termites have long been studied because of their uncommon diet and complex hindgut microbiota. Researchers study the termite symbiotic system for the discovery of lignocellulases to aid in biofuel production (Tartar et al., 2009; Sethi et al., 2013), understanding of the coevolution of the host and symbionts (Hongoh et al., 2005), and the ability to manipulate and study the structure and function of a complex microbiota (Brauman et al., 2001). Termites are descendants of the wood-feeding cockroach Cryptocercus, and are separated into two groups, higher and lower termites (Dietrich et al., 2014). Lower termites contain an abundance and diversity of flagellate protozoa that aid them in the digestion of wood and higher termites have been reported not to harbor symbiotic protists (Ohkuma, 2003); however, recently, a low-abundant ciliate has been detected in the guts of several higher termites species (Rahman et al., 2015).
Reticulitermes flavipes, the Eastern subterranean termite, is indigenous to the northeastern United States and harbors a tripartite symbiosis in its hindgut consisting of protozoal, bacterial, and archaeal symbionts (Ohkuma, 2003). The digestive enzymes from R. flavipes cannot fully break down the lignocellulosic components of wood, the termites’ sole food source, while the hindgut symbionts aid the digestion of these wood particles and provide acetate as a nutrient for the host (Ohkuma, 2003). The composition of the microbiota residing in the hindgut of R. flavipes has been previously investigated using culture-independent approaches as reviewed by Scharf (Scharf, 2015). Other studies investigated the R. flavipes hindgut microbiota by pooling DNA from several termites and sequencing a variable region of the 16S rRNA gene using 454 pyrosequencing (Ohkuma, 2008). Boucias et al. (2013) reported that the community is comprised of an estimated 581 bacterial operational taxonomic units, OTUs at the 97% identity level with approximately 80% of the symbionts belonging to the phyla Spirochaetes, Elusimicrobia, Bacteroidetes, Firmicutes, and Proteobacteria. They also evaluated the maintenance and stability of the microbial community in the hindgut and discovered that after R. flavipes termites were fed either a lignocellulose or cellulose diet for seven days, 88% of the OTUs in the hindgut microbiota were preserved despite the different diets, while only 12% of the OTUs were variable (Boucias et al., 2013). Proctodeal feeding has been suggested as an important mechanism contributing to this stability of the microbial community wherein the worker caste feeds the other members in the colony via fecal transfer (Buczkowski et al., 2007).
The core microbiota is defined as the organisms shared across multiple samples obtained from the same host, which is likely to play crucial roles in the functionality of that habitat (Turnbaugh et al., 2007). The core community of any symbiotic system is important in the health and maintenance of the symbiosis. Many studies have found the presence of a core microbiota in a variety of hosts, either in the form of a taxonomic core or a functional core (encoded genes) (Huse et al., 2012; Shade and Handelsman, 2012; Turnbaugh and Gordon, 2013; Maltz et al., 2014). Knowing the composition of the core microbiota is important because it ensures the maintenance of functions within the habitat and serves as an anchor for community resistance and/or resilience (Huse et al., 2012; Shade et al., 2012). However, it should be noted that differences in the hindgut microbiota can be critical for nestmate recognition or various caste-related functions (Cleveland, 1925; Minkley et al., 2006). Determining the core in smaller animals such as insects can be more challenging as the samples can be very small and thus some tend to pool samples prior to DNA extraction. While these studies provide important insight into the complexity and stability of the community, pooling samples averages the signal and prevents detection of variation between individuals. As the resulting OTU data is averaged, determining the prevalence in the individuals comprising the sample is impossible, and thus the core microbiota cannot be accurately determined (Hamady and Knight, 2009). These studies still provide valuable information but are distinct from a “core” and we will refer to such conclusions as the common microbiota in this manuscript. The core microbiota in some insects is extremely small, for example the core consists of ten OTUs in the bed bug Cimex lectularius (Meriweather et al., 2013), two taxa in Anopheles gambiae (Wang et al., 2011), and nine taxa in the honey bee (Moran et al., 2012; Sabree et al., 2012). The common microbiota of the fungus-growing higher termite, Macrotermitinae, (Otani et al., 2014) non fungus-growing higher termites, lower termites and cockroaches is shared between eleven phyla residing in all four groups and the five most abundant phyla being Firmicutes, Bacteroidetes, Spirochaetes, Proteobacteria, and Synergistes (Dietrich et al., 2014; Otani et al., 2014).
We hypothesize that the feeding habits dictate protist abundance, which in turn affects the abundance of protist-associated bacteria such as Endomicrobia and Treponema. Caste specific micrbiotas have been shown in the honey bee where queens have a higher abundance of Parasaccharibacter apium (Kapheim et al., 2015), Alphaproteobacteria and a Firmicute (Firm-5) (Tarpy et al., 2015), while workers harbor a higher abundance of Betaproteobacteria and Gammaproteobacteria (Tarpy et al., 2015). Being a eusocial insect, R. flavipes colonies have a caste system made up of juveniles, workers, soldiers, and reproductives. The workers forage for food and return to the nest to feed other members. The soldiers’ sole purpose is to defend the colony and these individuals have an enlarged mandible, which makes it impossible for them to masticate wood (Cleveland, 1925). Select members of the worker caste morph into alates (winged termites) and harbor a dramatically reduced number of protists in their gut while preparing to swarm (Shimada et al., 2013). After which they lose their wings, pair up and become reproductive termites that establish new colonies. The microbiota in the alates is of particular importance as these termites are the reproductive caste that found new colonies and presumably are the source of the key members of the hindgut microorganisms unless they can be acquired from the environment. The king and queen reproduce for colony growth, and are also fed by the worker caste. The soldier and reproductive castes are thought to have a reduced need for hindgut protists as they do not partake in the initial breakdown of wood into its constituent parts. Therefore, these castes have fewer protists in the hindgut, while kings and queens in mature colonies have no hindgut protists (Shimada et al., 2013).
While researchers are trying to understand the functions of the termite symbionts, fundamental aspects about the microbiota are not well known including the variability in the composition of microbiota between colonies, between different castes and between individual workers obtained from the same colony. We characterized the microbiota by sequencing the V4 region of the 16S rRNA gene from the hindguts of individual R. flavipes obtained from multiple colonies. Our analysis revealed a stable microbial community within the hindgut of workers that is comprised by a large core community, while there were significant differences in the abundance of protists and in the composition of the bacterial community in different castes.
Materials and Methods
Termite Collection and Maintenance
Reticulitermes flavipes termites were either collected using cardboard traps about a month after placement, captured directly from a rotting log, or purchased. The locations of termite colonies at time of collection are as follows: Mansfield CT (CT.A, October 2011 & CT.C, August 2012), Willington CT (CT.B, October 2013), Willimantic CT (CT.D, July 2013), Groton MA (MA.B, July 2013), Woods Hole MA (MA.C, July 2013), or purchased from Connecticut Valley Biological Supply Co. in Southampton MA (MA.A, June 2013). Additional alate termites used in the qPCR assays for the caste analyses were collected from an eighth colony (April 2014, Storrs, CT, USA), along with workers from the same colony for comparison. Once in the lab, the termites were placed in plastic containers with moist, autoclaved sand and spruce. Colonies were maintained at room temperature in the dark, and the sand was moistened with water every 3–4 weeks. Each colony, with the exceptions of CT.A and CT.C, were sampled upon collection. Colonies CT.A and CT.C were sampled for 4 months following the collection date. Termites sampled were assumed to have been initially collected from the natural habitat, as no evidence of reproduction was observed during the maintenance of the colonies in the lab.
Molecular Identification of Termites
Termite DNA from each colony was used for sequencing the Cytochrome Oxidase II (COII) gene to ensure the termites were R. flavipes (Supplementary Figure 1). Primers used for COII sequencing were a modified version of A-tLEU (5′-CAGATAAGTGCATTGGATTT-3′) and B-tLYS (5′-GTTTAAGAGACCAGTACTTG-3′) from Liu and Beckenbach (1992). Sequences were aligned in Geneious 6.1.7 using a MUSCLE alignment (Kearse et al., 2012). A neighbor-joining consensus tree was created with 100 bootstraps iterations, using COII sequences from this study along with sequences from multiple Reticulitermes species from NCBI (accession numbers: KR537205-12, JF7962324.1, KM245774-5, JF796221-2, AF262607.1, AY808093.1, EU253889.1, FJ806884.1, JQ280728-36, JX142171-72, JX142149-54) (Su et al., 2006; Legendre et al., 2008; Lim and Forschler, 2012; Perdereau et al., 2013).
Sample Collection and DNA Isolation
Hindguts were removed from the termite by pulling the thorax and anus apart with forceps (Matson et al., 2007) and placed in TE buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8.0). Samples consisted of single, whole hindguts with the exception of the data for colony CT.A where each data point represents pools from five hindguts (these samples were collected before we had established an efficient methods for single hindgut sampling). Seven colonies were sampled for all analyses, excluding the caste analysis. The number of samples per colony are as follows: CT.A (11 samples), CT.B (5), CT.C (8), CT.D (4), MA.A (9), MA.B (6), MA.C (2). For the caste analysis, 19 samples were taken from seven colonies; soldiers and alates were always matched with workers from the same colony. The number of samples per caste are as follows: workers (nine samples), soldiers (5), winged alates (2), de-winged alates (3). DNA was isolated immediately after collection using a modified (the starting lysis buffer was 500 μL and the final elution volume was 30 μL AE buffer) RBB+C isolation protocol as described by Yu and Morrison (2004). This method uses repeated bead beating along with chemical and high temperature cell lysis, and DNA precipitation followed by RNA and protein removal using a QIAmp DNA Mini Kit (Qiagen®, Germantown, MD, USA) column.
PCR Amplification of 16S rRNA Gene and Library Preparation
Hindgut samples were amplified using the V4 hyper-variable region of the 16S rRNA gene using primers developed by Caporaso et al. (2012). PCR reactions included Phusion® High-Fidelity PCR Master Mix with HF Buffer (New England Biolabs Inc., Ipswich, MA, USA) (50% of total volume), 10 μM forward and reverse primers, ∼10 ng DNA, and dH2O to the final volume of 25 μL. All reactions were amplified in triplicate using the following parameters: 94°C for 3 min, followed by 30 cycles of 94°C (45 s), 50°C (60 s), and 72°C (90 s), with a final extension of 72°C for 10 min (Nelson et al., 2014). Triplicate reactions were pooled and each sample was tested for size by running a 1% agarose gel.
Amplicons were purified and size selected using Agencourt AMPure XP (Beckman Coulter Inc., Brea, CA, USA) magnetic beads (0.65 × μL of sample volume) to select for 400 bp amplicons according to manufacturer’s protocol. Samples were then quantified using a Qubit® dsDNA HS Assay (ThermoFisher Scientific Inc.). Concentrations of each sample were calculated and then diluted to 4 nM. All samples were pooled in equimolar amounts for sequencing.
Sequencing and Data Processing
Samples were sequenced using an Illumina MiSeq (Illumina, San Diego, CA, USA) with custom sequencing primers added to the reagent cartridge (Caporaso et al., 2012) and sequenced 2 × 150 bp (CT.A & CT.C) or 2 × 250 bp. Both sequencing methods sequenced the entire V4 region of the 16S rRNA gene and the same merging and quality control parameters were used on both sets of data. The reads were processed as described previously by Nelson et al. (2014). Briefly, output reads were merged to create single reads spanning the entire 254 bp of the V4 hypervariable region using SeqPrep1, and the PhiX control reads were removed by mapping to the PhiX genome. Data analysis was performed using QIIME (Caporaso et al., 2010). Low quality reads (less than Q30) were removed and operational taxonomic units (OTUs) were determined by clustering reads to the Greengenes reference 16S rRNA gene reference dataset (2013-08 release) (DeSantis et al., 2006) at a 97% identity, and then performing de novo OTU clustering on reads that failed to cluster to a reference (McDonald et al., 2011; Nelson et al., 2014). Chimeras were then removed and the dataset was filtered to remove singleton and doubleton OTUs and then OTUs present at less than 0.0005% (Bokulich et al., 2012). The data was rarified to 15,000 reads per sample in order to include all samples in this study.
Data Analysis
The core microbiota was determined using all of the samples collected from individual R. flavipes workers in this study (excluding colony CT.A). Using QIIME, with Greengenes (2013-08) and the DictDb database (Mikaelyan et al., 2015), we calculated the OTUs (at the 97% identity level) that were present in at least 95% of the samples (Huse et al., 2012). These OTUs were then paired with taxonomy to the lowest level of classification, and the sequence abundance of each core OTU was reported. The sequences in the DictDb database were shortened to only include the V4 hypervariable region and combined with the Greengenes database. The combined file was aligned to generate the aligned reference. Some sequences failed to align due to shortness in length and were removed from the unaligned reference file (5,845 sequences out of 55,394).
The samples used in the geographic analysis were all from the worker caste. After quality filtering and rarifying to 15,000 sequences per sample, alpha diversity (Shannon and Phylogenetic diversity) (Faith and Baker, 2006) and beta diversity metrics (Bray–Curtis) (Lozupone and Knight, 2005; Lozupone et al., 2006; Anderson et al., 2011) were performed using QIIME 1.8 and R 3.2.0 (R Development Core Team, 2005; Wickham, 2009; Oksanen et al., 2015). The PERMANOVA statistical analysis was performed to determine the significance of microbial community differences among the different colonies using the Bray–Curtis dissimilarity matrix in QIIME (Caporaso et al., 2010). This analysis was performed over 999 permutations and returned a Pseudo-F (f) statistic along with a p-value (p).
Reticulitermes flavipes worker, soldier, and alate hindgut samples collected from various colonies were used in the analysis of the caste microbiota. For each non-worker (soldier, alate), a worker was collected at the same time from the same colony for comparison. The winged alates were collected on February 27, 2013, and de-winged alates were collected on May 31, 2013. The bacterial taxonomic abundances were averaged for each caste, and the averages were used in the analysis. Similar to the statistical analysis done on the microbial communities from different colonies, the PERMANOVA statistical analysis was used to determine the significance of the microbial community differences between workers and soldiers, workers and winged alates, and workers and de-winged alates. The sequences (OTU assignments using Greengenes) in the caste dataset were compared to the DictDb database, which is a curated database for microbes from termites and cockroaches that provides greater taxonomic resolution, using BLASTN at the 97% identity level (Altschul et al., 1990; DeSantis et al., 2006; Mikaelyan et al., 2015). Spirochaete sequences with 100% query coverage were assigned OTUs and taxonomy using the DictDb database. Treponema sequences that did not match reference sequences in the DictDb database or Greengenes database were designated as ‘de novo’. A one-way ANOVA with a Bonferroni post-test was performed for each taxonomic grouping of Spirochaetes using GraphPad Prism version 6.0f for Mac OSX2 (GraphPad Software, San Diego, CA, USA).
Quantitative PCR (qPCR) of Protist Symbionts
Caste hindgut samples with 16S rRNA sequencing data were used for qPCR analysis. Additional alate samples (and workers from the same group) were added to the analysis, the number of samples per caste are: workers (19 samples), soldiers (3), winged alates (12), de-winged alates (6). Primer sets for the two groups of protists found in the hindgut were designed using 18S rRNA gene sequences from NCBI in Geneious (phylum Parabasalia and order Oxymonadida) (Kearse et al., 2012). The primers were then tested on hindgut contents, termite DNA, and bacterial DNA to ensure there was no amplification of termite or bacterial DNA. Primer sequences are as follows: Para361F-5′CGCGAAACTTACCCACTCG-3′, Para510R-5′TTACCGCAGCTGCTGGC-3′ and Oxy161bF-5′CGGATAGCCGTAGTAATTCTAGAGCT-3′, Oxy352bR-5′AACGTCAGGTTGATAGGTTAGAAATT-3′. All reactions were setup in a 10 μL volume including: SsoAdvanced SYBR Green Supermix (Bio-Rad Laboratories Inc., Hecrules, CA, USA) (50% of reaction volume), 10 μM forward and reverse primers (15% each of reaction volume), dH2O (10% of reaction volume) and 1 μL of DNA template. Reactions were amplified in triplicate using a CFX96 Real-Time Thermocycler (Bio-Rad Laboratories Inc., Hercules, CA, USA) with the following parameters for the Oxymonadida: 95°C (3 min), followed by 40 cycles of 95°C (30 s), 64°C (30 s), and 72°C (30 s). The parameters for the Parabasalia were the same except that the annealing temperature was 67°C. Negative controls with no template added were prepared and tested with each set of reactions. Standard curves were generated for each primer set using 102–108 copies per reaction and the real-time data was normalized to the concentration of DNA added to the PCR reaction to calculate the Ct value, representing a single hindgut and then square-root transformed for statistical analyses. A one-way ANOVA with a Bonferroni post-test was performed for each caste in both protist groups using GraphPad Prism version 6.0f for Mac OSX (GraphPad Software, San Diego, CA, USA).
Data Availability
The 16S rRNA gene sequence data was deposited in the European Nucleotide Archive (ENA) SRA under project ID PRJEB5527.
The COII gene sequence data was deposited in GenBank under accession numbers: KR537205-12.
Results
Identifying the Core Microbiota of R. flavipes
Determining a core microbiota is important for any host-associated or environmental community because one can infer the composition of the “healthy” or undisturbed community and a diseased one (Turnbaugh et al., 2007; Shade and Handelsman, 2012). In our study, we defined the core microbiota as the OTUs at the 97% identity level that were present in 95% of the samples (Huse et al., 2012). Our data showed that the core of the worker termites consisted of 69 OTUs and accounted for 67.05% of the sequences of the hindgut microbiota (Table 1). Of these OTUs, the genus Treponema, contained 41 OTUs, 5 of them being classified to the species T. primita, and accounted for almost 41.43% of the total sequences in the hindgut. The class Endomicrobia (8 OTUs) and genus Azobacteroides (3 OTUs) had an abundance of 18.50 and 3.18%, respectively. The remaining 17 OTUs fell into 13 taxa and comprised 3.95% of sequences from the hindgut. 32.95% of OTUs found in the termite hindgut varied between individuals and were not considered to be part of the core microbiota. One OTU (0.16%) was identified from the DictDb (cockroach and termite specific) database and was unassigned. Box and whisker plots showing taxon abundances (Figure 1) were created for each colony, using the taxa found in the core microbiota. These data support the average taxon abundances shown in the core (Table 1). All colonies show similar abundance patterns for each of the core taxa.
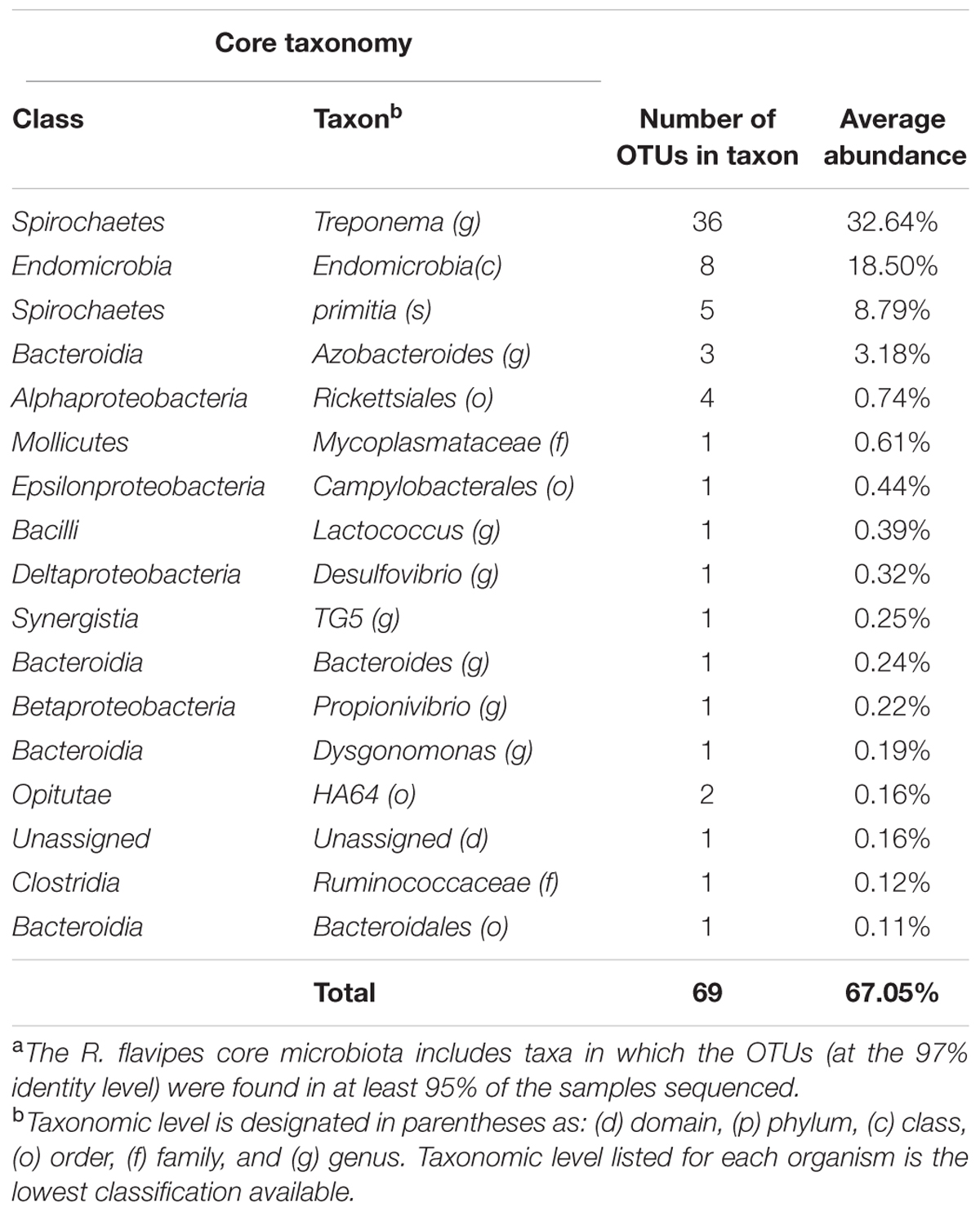
TABLE 1. The R. flavipes worker core hindgut microbiota.a
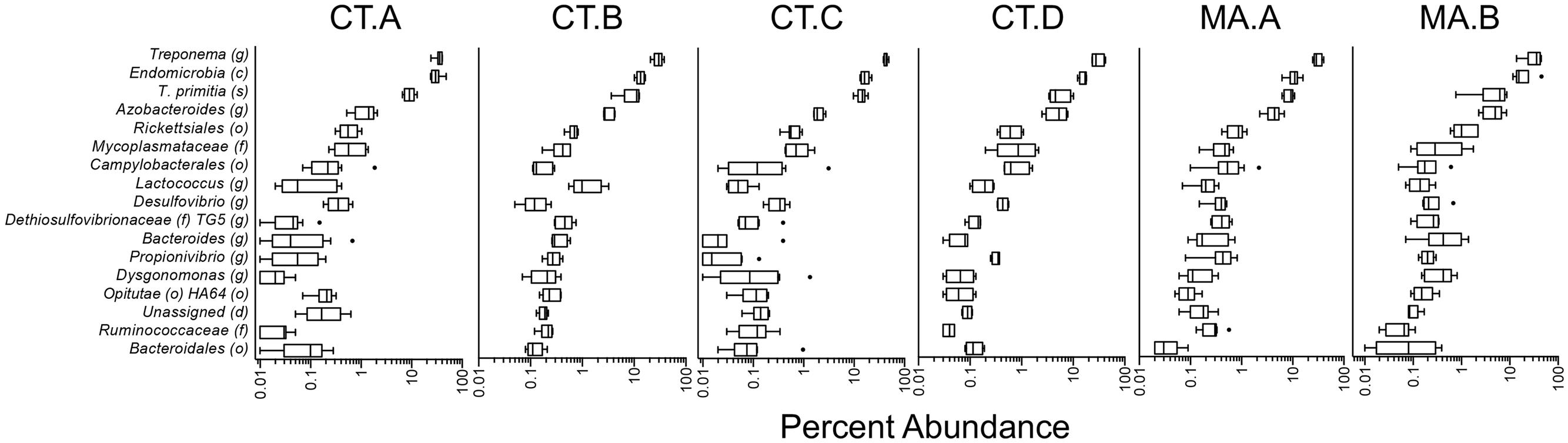
FIGURE 1. The abundance of core taxa in the worker hindgut. A box and whisker plot for each colony is shown with the abundance of the taxa found in the core microbiota. The abundance of each taxon is consistent throughout the six. The number of samples in each colony are as follows: CT.A (n = 11), CT.B (n = 5), CT.C (n = 8), CT.D (n = 4), MA.A (n = 9), MA.B (n = 6). Colony MA.C was not used in this analysis due to its small sample size (n = 2). Colony CT.A contained five pooled hindguts per sample and was not used in determining the core microbiota, however, it showed the same pattern of core taxon abundances.
Analysis of the Hindgut Microbiota Among Different Colonies
The microbiota of xylophagus insects, such as the wood-feeding cockroach, Cryptocercus kyebangensis, is shared between members of the colony through proctodeal trophalaxis (Park et al., 2002). This process is thought to create a homogenous microbial community throughout the colony, which may aid in digestion and colony health. To determine the homogeneity of the R. flavipes hindgut microbiota, individual and pooled worker hindguts were sampled from seven colonies originating in Massachusetts or Connecticut. Alpha diversity analyses were performed and the values for each sample within a colony averaged and a single value was presented for each grouping (Table 2). The Shannon index and equitability show that the microbial community is not evenly distributed. In the case of colonies CT.A and CT.C (sampled over 4 months in the lab), the microbial community becomes less complex over time (One-way ANOVA; (F(6,38) = 36.54, p < 0.0001)). The phylogenetic diversity differs among the microbial communities in different colonies, with some having a less diverse microbiota than others. At the phylum level, Spirochaetes dominate the hindgut community, with an average sequence abundance of 55% among the 45 R. flavipes workers. Elusimicrobia and Bacteroidetes are present at 24 and 10%, respectively. The remaining 11% of sequences belong to the phyla Proteobacteria, Firmicutes, Tenericutes, and Synergistes, or were unclassified Bacteria (Figure 2, Supplementary Figure 2 and Supplementary Table 1). In our study, the archaeal community was present at an abundance of less than 0.1%. This could be due to the specificity of the primers used for amplifying the V4 region of the 16S rRNA gene, but other studies using different approaches also reported that in lower termites, archaea are present at low abundances (Berchtold et al., 1999; Hongoh, 2010).
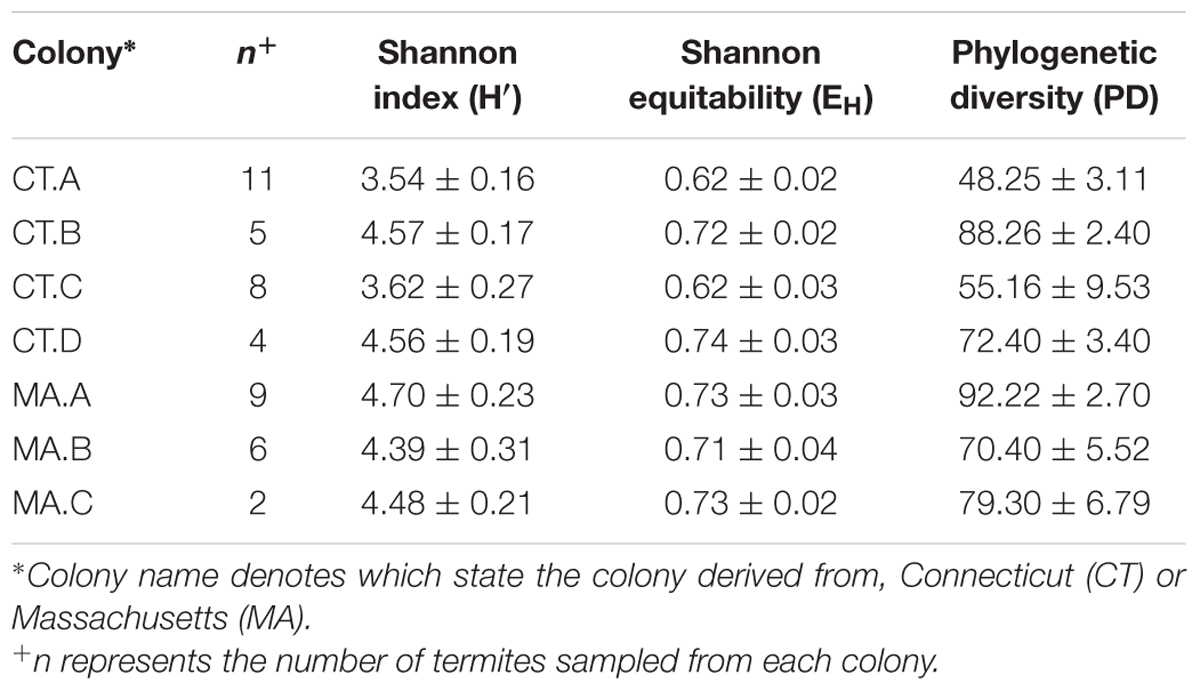
TABLE 2. Bacterial alpha diversity of the R. flavipes worker hindgut among different colonies based on the 16S rRNA amplicon.
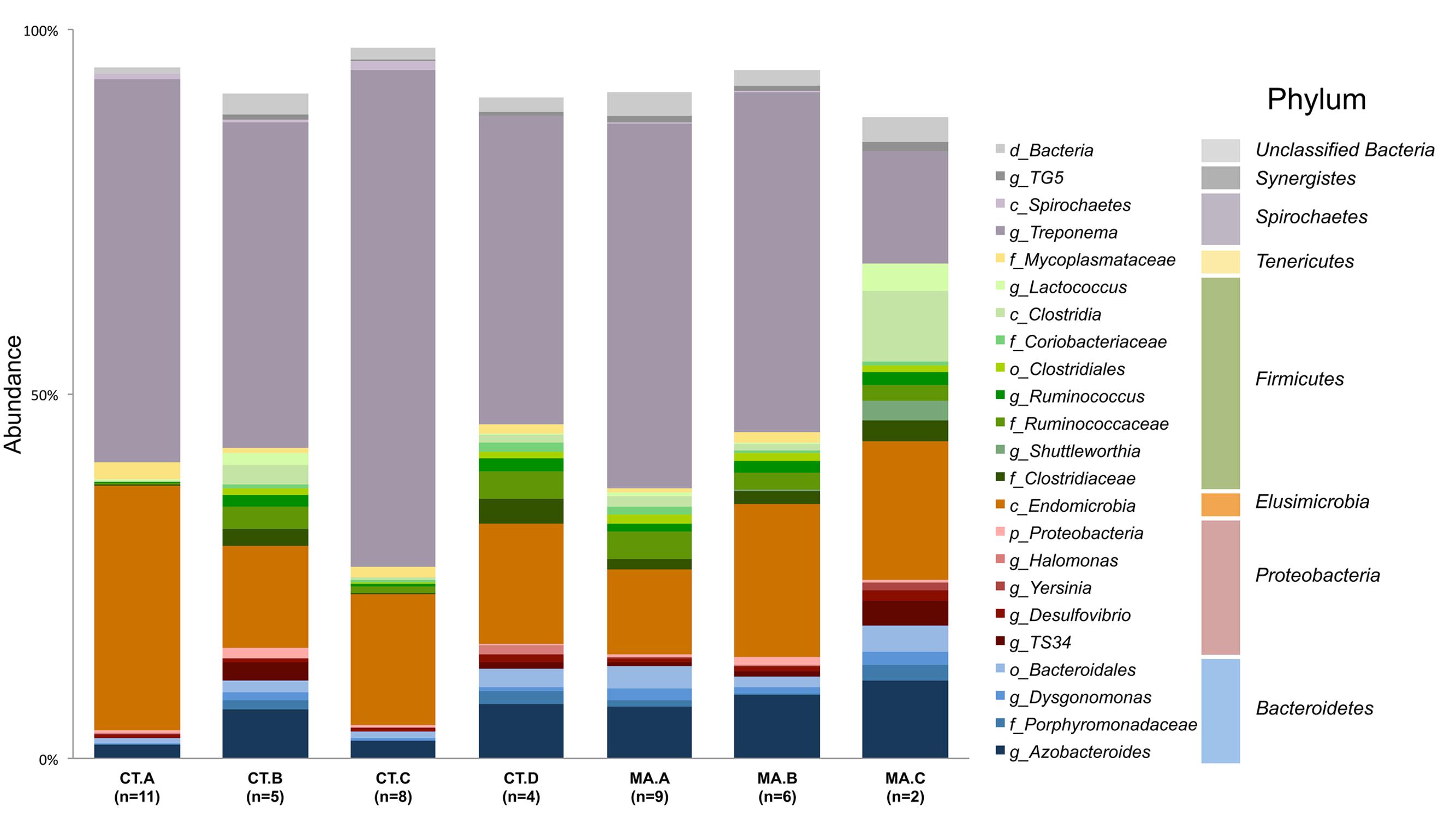
FIGURE 2. Composition of the microbiota in the R. flavipes worker hindgut. The genus Treponema and class Endomicrobia dominate the hindguts in all colonies. Taxonomic abundances from hindgut samples of the same colony were averaged together. Taxa present at a sequence abundance of 1% or higher are shown. Letters before the taxonomic name represent the taxonomic level at which that organism was identified (d-domain, c-class, f-family, o-order, g-genus).
We wanted to assess whether the microbial community residing in the hindgut of one termite was more similar to other termites in the same colony than to termites from a different colony in similar geographic locations. The Bray–Curtis beta diversity analysis was performed to determine similarities and differences in the composition of the microbiota and was used to create a NMDS (non-metric multi-dimensional scaling) plot of the 45 hindgut samples (Figure 3). The microbial communities within a colony grouped significantly closer together than to communities from other colonies (PERMANOVA, f = 8.62, p = 0.001) and there was no clustering of samples according to the state from which they originated nor according to the COII sequence of the termite.
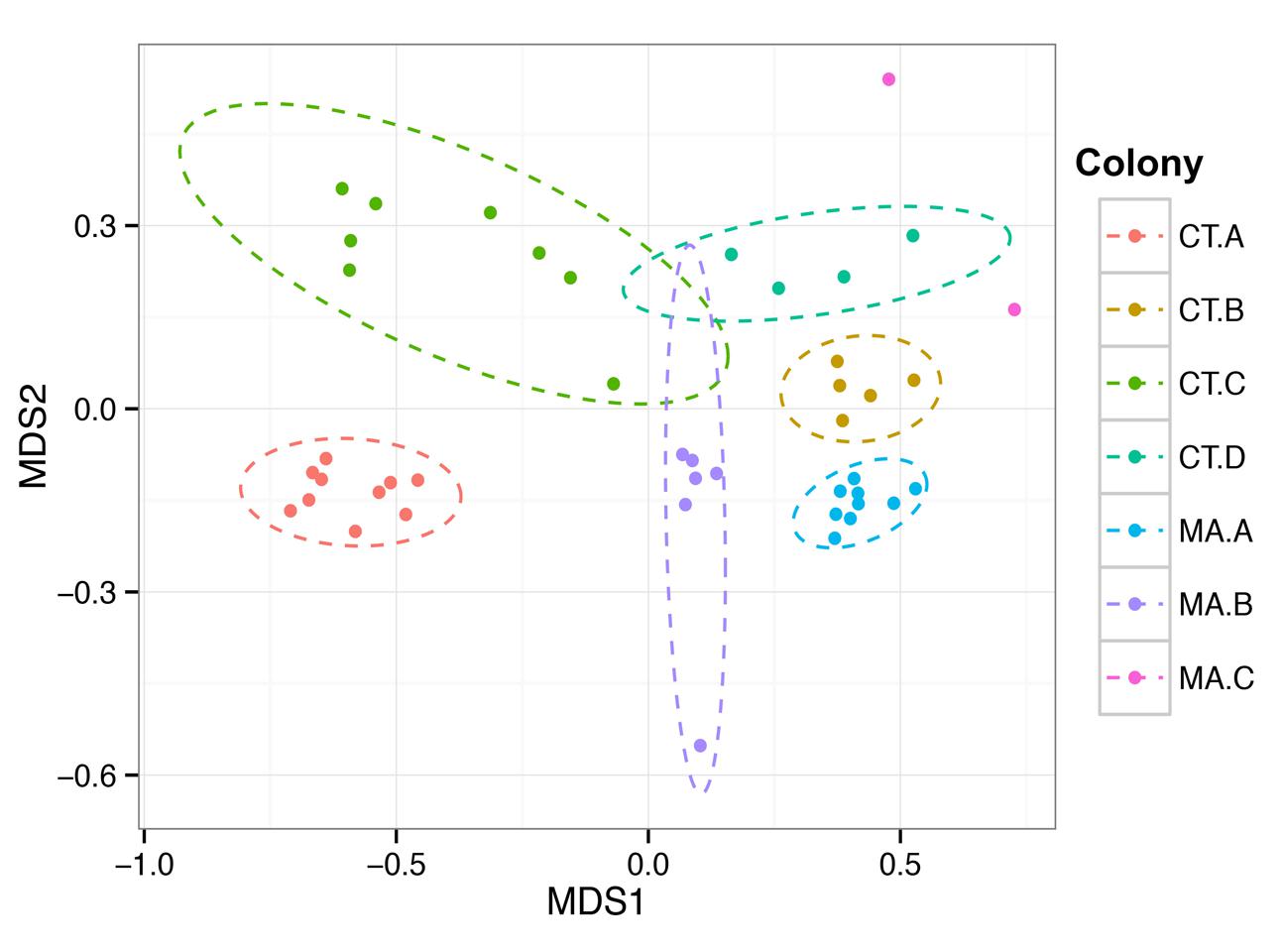
FIGURE 3. Colony specificity of the R. flavipes worker hindgut microbiota. The hindgut microbiota shows more similarity between hindguts from the same colony than between colonies (PERMANOVA, F = 6.6201, p = 0.001). Nonmetric Multi-Dimensional Scaling was performed on the hindgut microbiota from different colonies using the Bray–Curtis dissimilarity metric. The ellipses were obtained from the standard deviation within a colony, and plotted using a 95% confidence level. The differences in the MA.C colony could be due to the coastal environment compared to the inland environment of the other colonies.
Comparison of the Microbiota Among Different Castes
A termite colony is composed of various castes, each with a unique function contributing that might influence the composition of the hindgut microbiota (Lewis and Forschler, 2013). Alpha diversity analysis of the members from each caste was performed, and the evenness and richness of the microbial community in each caste were similar (Table 3). While a trend of a difference in the phylogenetic diversity of each caste was noted, it was not statistically significant, perhaps future studies with a larger sample size might support this finding.
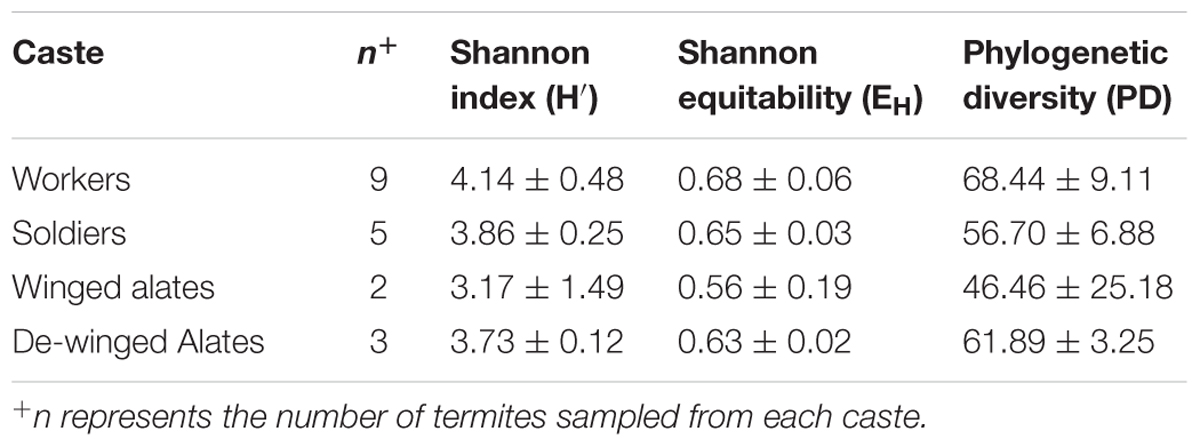
TABLE 3. Bacterial alpha diversity of the R. flavipes hindgut among different castes based on the 16S rRNA amplicon.
Each caste is known to have different diets and perform specialized functions in the colony, which suggest that the hindgut microbiota may reflect these differences (Lewis and Forschler, 2013). Averaging the sequence abundances at the taxonomic order level for each caste and comparing the values to the worker caste enabled a comparison of the microbial composition in different castes. We found differences in taxonomic abundances between alates and workers according to a PERMANOVA (winged: f = 3.59, p = 0.001; de-winged: f = 2.27, p = 0.01). Sequences representing the two dominating taxa, order Spirochaetales and class Endomicrobia, decreased in abundance in the winged alates from 48 and 22% to 11.6 and 1.1%, respectively, (Figure 4 and Supplementary Figure 3). Sequences belonging to the order Bacteroidales were found in the workers and soldiers at an abundance of less than 11% while they were present at over 20% in both the winged and de-winged alates. The orders Enterobacteriales, Flavobacteriales, and Pseudomonadales were present at average abundances of 9.7, 13.6, and 9%, respectively, in the winged alates while they were below the limit of detection in workers, soldiers, and de-winged alates (0–0.06%). The Spirochaete sequences were further classified using OTU assignments and taxonomic classifications from the DictDb database (Figure 5) (Mikaelyan et al., 2015). The sequences were classified into five groups: Treponema Ia, Ib, Ic, Ig, II, and sequences that were not similar to any in the DictDb database were labeled as de novo Treponema. The subgroup Treponema Ia were the most abundant taxon in the hindgut with relative abundances reaching up to 33%. Treponema Ib were the least abundant and most consistent subgroup with average abundances of 1% for all castes. The abundances of Treponema Ia [F(3,15) = 9.331, p = 0.001] and Treponema II [F(3,15) = 4.489, p = 0.0190] were significantly lower in the winged alates when compared to the worker caste (one-way ANOVA with Bonferonni-corrected p-values), which may indicate that Treponema Ia and II are necessary in the digestion process of workers and associated with protists.
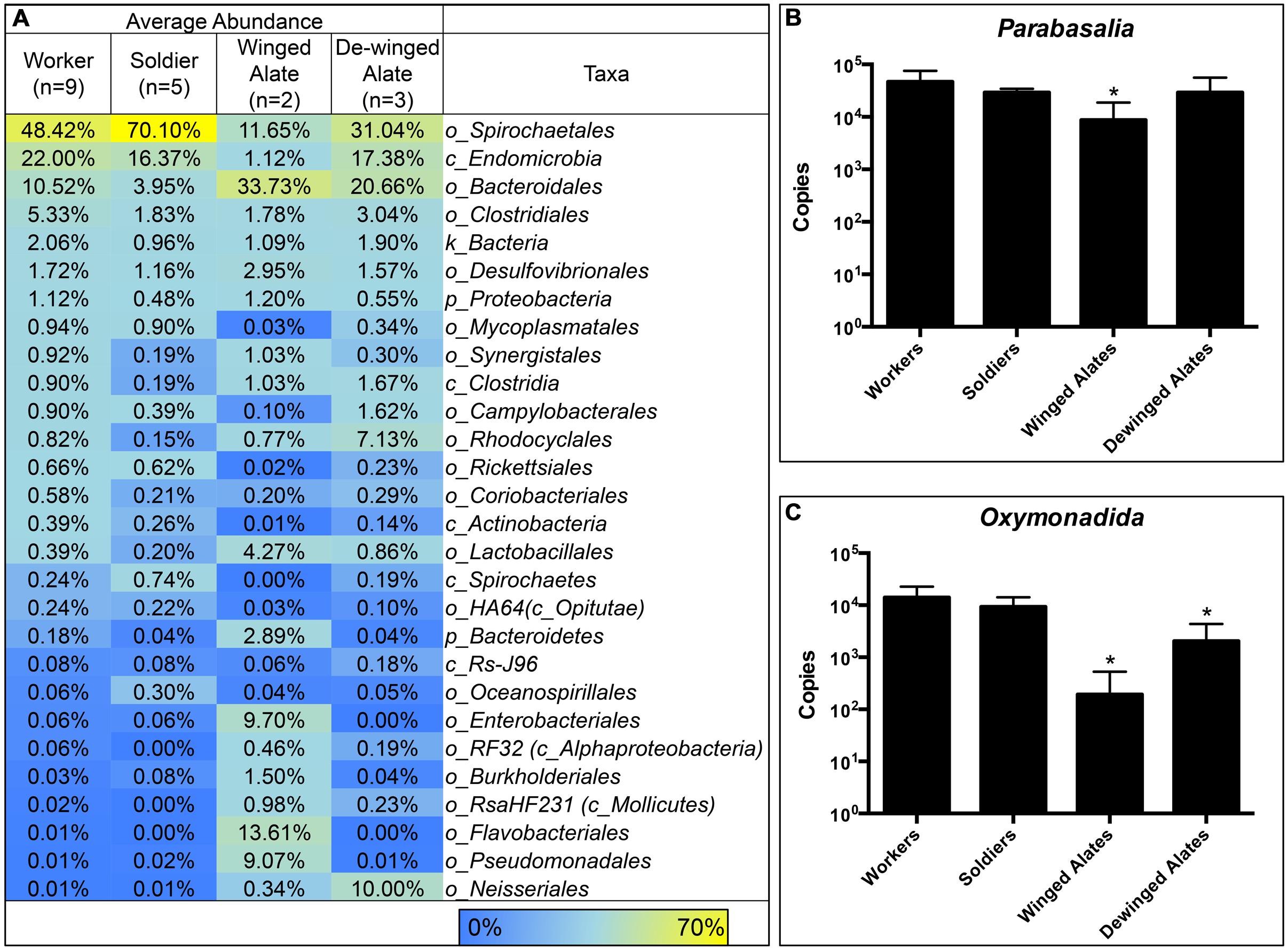
FIGURE 4. Comparison of Bacterial Taxa and Protists in R. flavipes Castes. Treponema, Endomicrobia, and protist abundances are lower in the alate castes compared to the worker caste. Hindgut samples were taken from worker and non-worker termites in various colonies. (A) Abundances for each caste were averaged and compared to the worker caste. In winged alates, the abundance of the two dominating taxa, Spirochaetales, and Endomicrobia, are lower in abundance compared to the worker caste. Enterobacteriales, Flavobacteriales, Pseudomonadales, and Neisseriales are present in very low abundances in the workers and soldiers, but were more abundant in the alate caste. Protists belonging to the phylum Parabasalia (B) and the order Oxymonadida (C) were quantified using qPCR. Each caste was compared to the worker caste using a one-way ANOVA. ∗ Indicates a significant value compared to the worker caste. (B) Parabasalia protists were less abundant in the winged alates [F(3,36) = 11.9, p < 0.0001]. (C) Oxymonadida protists were less abundant in the winged and de-winged alates compared to workers [F(3,36) = 36.94, p < 0.0001].
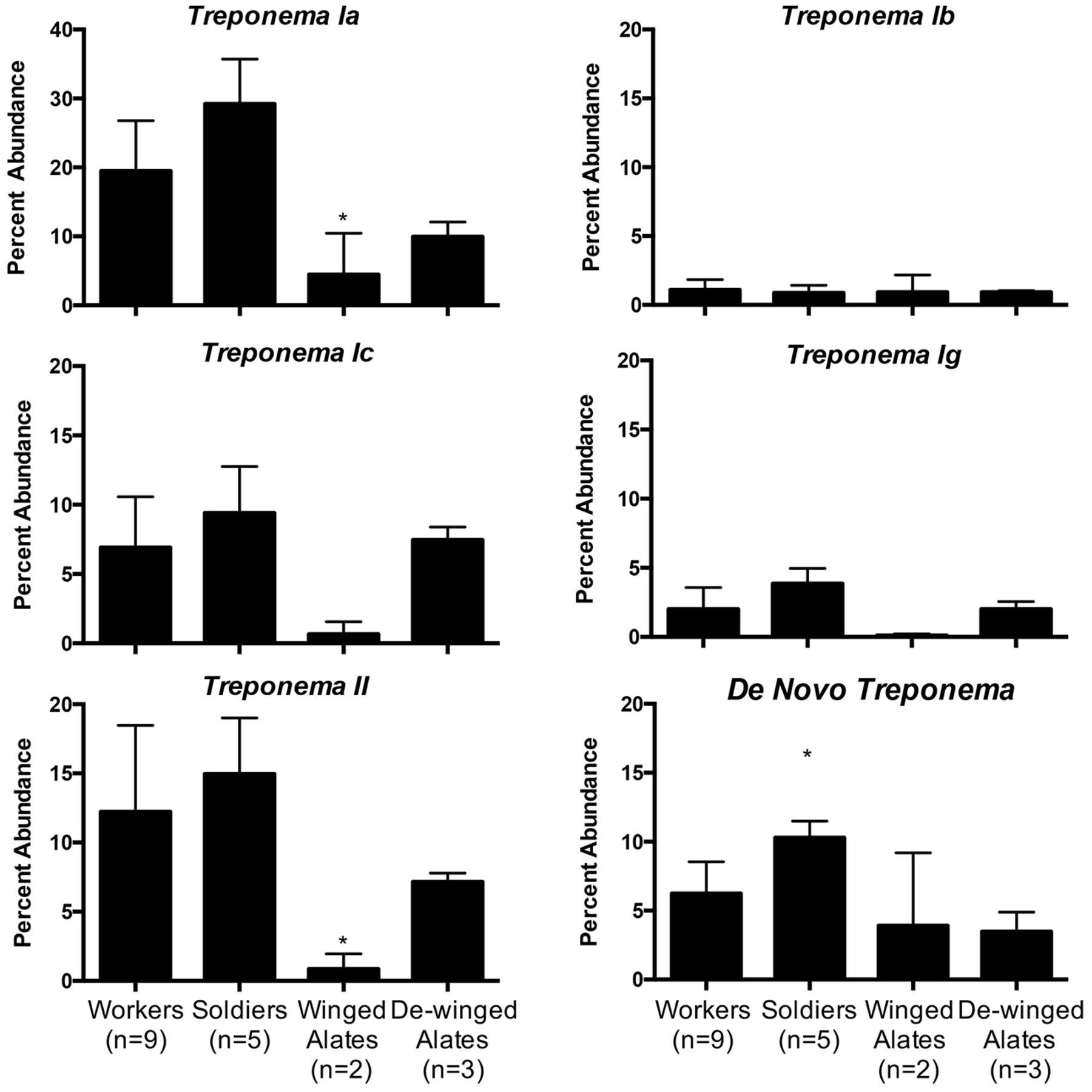
FIGURE 5. Relative abundance of Treponema groups among different castes. Five groups of Treponema differ in abundance in workers, soldiers, winged alates, and de-winged alates. OTUs classified as Treponema were categorized into groups Ia, Ib, Ic, Ig, and II according to the DictDb database. OTUs that could not be classified in DictDb were processed de novo and classified to the genus Treponema. Treponema Ia is the most abundant group while Treponema Ib is the least abundant but most consistent throughout the castes. Treponema Ia and II have significantly lower abundances in winged alates compared to workers as indicated by a one-way ANOVA [F(3,15) = 9.331, p = 0.001, and F(3,15) = 4.489, p = 0.019]. De Novo Treponema have a significantly higher abundance in soldiers compared to workers as indicated by a one-way ANOVA [F(3,15) = 7.043, p = 0.0035]. All significant data are indicated by an asterisk ∗.
Endomicrobia, along with some Spirochaetes are known protist symbionts in the termite hindgut (Iida et al., 2000; Stingl et al., 2005; Ikeda-Ohtsubo et al., 2007). The observed decrease in the abundance of these bacteria in winged alates led us to investigate the protist abundances in the same samples. Abundances of two groups of protists, phylum Parabasalia and order Oxymonadida, were determined using qPCR. The protist abundances in each caste were compared to the worker caste using a one-way ANOVA. Parabasalia abundances were 10-fold fewer in the winged alate class, compared to the worker caste [F(3,36) = 11.9, p < 0.0001]. Protists belonging to the order Oxymonadida were less abundant in the winged alates and de-winged alates [F(3,36) = 36.94, p < 0.0001]. We were interested in determining if the abundance of bacterial OTUs correlated with the abundance of protists and tested for this by calculating Pearson correlations between each protist group and the Treponema (Spirochaetes) or Endomicrobia OTUs. The two-tailed p-values were Bonferroni-corrected to account for the number of comparisons performed. Twenty-three of the 49 Treponema OTUs correlated with the Oxymonadida protists, and only ten of forty-nine correlated with the Parabasalia (p < 0.001, Supplementary Figure 4A). Of these nine Treponema OTUs were identical matches for sequences found in the DictDb database and belong to Treponema subgroups Ia, Ic, and Ig the OTU IDs are listed (Supplementary Figure 4A). Eight out of twelve Endomicrobia correlated with Parabasalia, and seven of 12 correlated with Oxymonadida protists (p < 0.004, Supplementary Figure 4B).
Discussion
The R. flavipes Core Microbiota
The presence of a core microbiota and its composition provides insight into the structure of the microbial community in the habitat of interest, and suggests the metabolic potential and conserved functions of the community (Tap et al., 2009; Huse et al., 2012; Shade and Handelsman, 2012). In termites, many groups have researched the hindgut microbial population, and the presence of a core community has been suggested (Fisher et al., 2007; Boucias et al., 2013; Huang et al., 2013; Scharf, 2015), however, the actual core microbiota remains to be defined as previous groups relied on pools of animals in their analysis which hides the variation amongst individuals. The common microbiota present in nine species of fungus-growing termites was determined, which included 42 taxa comprising eight phyla. The majority of sequences were assigned to two taxa, Bacteroidetes and Firmicutes (78.6% of sequences) (Otani et al., 2014). In a large survey, Dietrich et al. pooled gut homogenates from 3 to 10 individuals and reported the similarities and differences of the gut microbiota among cockroaches, lower termites, and higher termites. That study found that between 77 and 79% of the sequenced reads, and between 50 and 87 genus-level taxa were assigned to the shared microbiota in each of the three groups tested, cockroaches, lower termites, and higher termites (Dietrich et al., 2014).
For determining the core microbiota, we optimized the DNA extraction protocol for working with individual hindguts. This optimization allowed us to determine variation between individuals when calculating the core microbiota. Determining the composition of the hindgut microbiota from 45 termites obtained from seven different colonies (up to 250 km apart) aided in determining a taxonomic and OTU-based core community (Table 1). The more abundant taxa that we identified to comprise the core are identical to the phyla previously reported in the hindgut of R. flavipes: Spirochaetes, Elusimicrobia, Bacteroidetes, Firmicutes, and Proteobacteria (Fisher et al., 2007; Ohkuma, 2008; Boucias et al., 2013; Huang et al., 2013). Included in these phyla are the abundant taxa, Treponema, Endomicrobia, and Azobacteroides. Less abundant taxa comprising the core include Desulfovibrio, Lactococcus, Bacteroidales, which have been reported to be present in the hindgut of termites (Scharf, 2015). While future studies that expand the geographic range could reduce the number of OTUs comprising the core, the large number of animals and geographic range sampled provides an excellent baseline.
The bacterial, protist, and archaeal populations in the wood-feeding termite hindgut are known to play an integral part in the digestion of the wood meal, and without the complex bacterial community, the termite cannot survive (Raina et al., 2004; Rosengaus et al., 2011). The presence of a core microbiota suggests that each member of the core fills a niche in the termite hindgut that is consistently present despite changes in habitat, geography or food source and thus are likely to contribute to the overall health of the termite. We hypothesize that in R. flavipes, the core microbiota is made up of 69 OTUs in 16 taxa and accounts for more than 67.05% of the sequences. The dominant taxa found in the core were Treponema (41.43%) and Endomicrobia (18.50%), and were reported as part of the common microbiota by Dietrich et al (Dietrich et al., 2014). Treponema is a protist ectosymbiont as well as a free-living bacterium in the lumen of the hindgut and is the primary producer of acetate via reductive acetogenesis, which is the main nutrient for the termite host (Leadbetter et al., 1999; Graber and Breznak, 2004). Endomicrobia have been found to exist as a protist endosymbiont and a free-living bacterium in the hindgut, providing vitamins, and amino acids (Hongoh et al., 2008; Ikeda-Ohtsubo et al., 2010). The genus Azobacteroides (3.44%) was also found in the core and has been previously identified as a protist symbiont and nitrogen fixer in the gut of the termite Coptotermes formosanus (Raina et al., 2004). Each of the less abundant taxa were comprised of 1-2 OTUs and each accounted for less than 1% of the total sequences but were present in over 95% of the samples. In general, the abundances of the different taxa comprising the core followed similar patterns in the different colonies analyzed in this study (Figure 1). The core taxa were determined using colonies CT.B, CT.C, CT.D, MA.A, MA.B, and MA.C because they were represented by single hindgut samples. Although colony CT.A was not used in the calculation of the core microbiota due to pooled sampling methods, the taxa found in the core were found in similar abundances (Figure 1). The consistent detection of 13 taxa that each accounted for less than 1% of the sequences suggests an important, yet still undefined role for these low abundant organisms.
Sequencing depth will also affect the core size, as a greater sequencing depth increases the chance detection of less abundant taxa. By analyzing 15,000 sequences per sample, we had a greater likelihood of including these taxa in the core as opposed to utilizing fewer sequences, which may only detect low abundant taxa sporadically. This has important implications for previous studies of other microbial communities. The greater sampling depth provided by Illumina sequencing is likely to show a larger core microbial community by detecting less abundant sequences in different environments. Sixteen OTUs that we identified in this study were also present in the DictDb cockroach and termite symbiont reference database, which does not include data from R. flavipes but includes data from two related species, R. chinensis and R. speratus (Mikaelyan et al., 2015). A Treponema OTU was present at 3% in one sample, and another at 2% in one sample. The rest of the 14 OTUs were present at 1% or less in all samples. It is interesting that the most abundant OTUs were species-specific at least in this case. It has been reported that the hindgut protists vary depending on termite species (Ohkuma, 2008) and this may be the case for hindgut bacteria as well and has been suggested by Dietrich et al. (2014).
Analysis of the Hindgut Microbiota in Termites from Different Colonies
The microbiota has been shown to aid in host health when present in a symbiotic relationship. The maintenance of the bacterial community throughout a colony of bumble bees aids in the protection from the parasite Crithidia bombi (Koch and Schmid-Hempel, 2011). Rosengaus et al. (2011) reported the importance of the hindgut microbiota on host survival in the dampwood termite, Zootermopsis angusticollis and R. flavipes. In that study, a 64% reduction in bacterial diversity and a small, short-term reduction of gut protists occurred when the diet was supplemented with 0.005 g/mL of the antibiotic, Rifampin. Lower survival rates and a reduction of eggs, larvae, and soldiers were observed for both termite species and correlated with the reduced bacterial diversity (Rosengaus et al., 2011). In addition to proctodeal feeding, it has been shown that social grooming and deposition of fecal contents and saliva in foraging galleries spread termite hindgut bacteria throughout a colony. The bacteria (mostly Actinobacteria sp.) in these galleries have been shown to breakdown the cell walls of pathogenic fungi (Klassen, 2014) and possibly pathogenic bacteria (Carr et al., 2012). The maintenance of the R. flavipes hindgut microbiota within a colony could provide the termite with protection from microbial invaders in addition to the provision of nutrients.
Previous studies have characterized the bacterial taxonomic abundances in R. flavipes using the V1–V3 and V5–V6 region (Boucias et al., 2013; Huang et al., 2013; Nelson et al., 2014), and our study with samples from seven colonies in Connecticut and Massachusetts revealed similar taxonomic composition and abundances. The most abundant phyla represented were Spirochaetes (∼55%), Elusimicrobia (∼24%), and Bacteroidetes (∼10%), with lower abundant representatives from the phyla Firmicutes, Proteobacteria, Tenericutes, Synergistes, and unclassified Bacteria. The relative abundance of sequences in the termite hindgut varied slightly depending on which region of the 16S rRNA gene was sequenced, which is a known caveat of 16S rRNA studies (Janda and Abbott, 2007; Aird et al., 2011; Soergel et al., 2012; Nelson et al., 2014) but did not change the overall composition.
Different colonies in the same geographic area have been suggested to harbor slightly different hindgut microbiotas, which might allow termites to distinguish nest mates from invaders (Hongoh et al., 2005; Minkley et al., 2006). Beta diversity analyses in this study are consistent with this concept of nest specificity as termites within a colony show a greater similarity of the hindgut microbiota than to other colonies. Colonies CT.A and CT.C were sampled over 4 months and each grouped as a colony according to the NMDS plot. This shows the homogenous nature and stability of the hindgut microbiota within a colony that was transferred from the field and maintained in the laboratory. It was interesting to note, however, that these colonies had a lower Shannon Index (H’) which is indicative of a lower OTU richness compared to the other colonies. The Shannon Equitability (EH) was also lower in these two colonies which indicates more evenness compared to the other colonies. The lower richness and lower evenness may be due to the colonies being kept in the lab, whereas the other colonies were sampled directly from their natural habitat, which would be analogous to a “zoo” effect (Ley et al., 2008; Kohl and Dearing, 2014). A Bray–Curtis analysis of the samples in this study showed hindgut microbiotas from the same colony grouping together (Figure 3). Colony MA.C from Woods Hole, MA, shows the most differences according to relative abundances of multiple taxa among the colonies (Figure 2), however, our analysis of the COII sequences did not reveal a corresponding phylogenetic difference of the hosts. This may be due to environmental conditions such as higher salt concentration in the air, sand-rich soils, and the lack of dense forestry. Leadbetter and Breznak (1996) reported different morphotypes of methanogens found in R. flavipes hindguts in Michigan and Woods Hole, MA, USA, which coincides with our findings of differing bacterial taxa. While the core taxonomic abundances show very small differences, it can be that changes in the relative abundance of key taxa or the fluctuation of low abundance taxa between samples, which could be involved in colony recognition.
The Hindgut Microbiota Among Different Castes
While workers are the primary caste many researchers study, the soldier and alate castes play important roles in the colony. Soldiers protect the colony from invaders and cannot morph into any other caste. Alates, a form of reproductive termite, swarm to a new area to establish a new colony, wherein they will become the primary reproductives (king and queen). During the transition to winged alates, the termites lose the majority of their gut protists and rely on lipids and glycogen stored in the fat body for nutrition (Costa-Leonardo et al., 2013). Alates shed their wings after swarming to an area to start a new colony, morph into primary reproductives and give rise to juvenile termites. The primary reproductives forage on wood, feed the first generation of juveniles until they are ready to provide for the colony and transmit the symbionts to the juveniles unless some symbionts are acquired from the environment (Shimada et al., 2013). Lewis et al demonstrated that protist abundances in the hindgut differ depending on the feeding habits of the caste, with protist abundances being lower in the alate and soldier castes of three Reticulitermes species (Lewis and Forschler, 2013). This finding leads to the question of whether or not different castes with different digestive functions harbor the same hindgut bacterial community.
The dramatic drop in abundance of the protozoal symbionts Spirochaetales and Endomicrobia in the winged alates coincides with the dramatic decrease in protist numbers during this morphing stage, as shown by Shimada et al. (2013). During this time, the termite is building up fat bodies and storing more nutrients in the fat bodies as the animals are preparing to swarm and establish a new colony (Shimada et al., 2013). We performed qPCR to quantify the two protist groups found in the hindgut, Parabasalia and Oxymonadida on the same hindgut samples that we sequenced the 16S rRNA gene from. These data show a drop in abundance of both protist groups in the winged (Parabasalia and Oxymonadida) and dewinged alates (Oxymonadida), when compared to the worker caste. When evaluating whether OTUs were potential protist symbionts, seven and eight of the 12 Endomicrobia OTUs correlated with Oxymonadida and Parabasalia protists, respectively (Supplementary Figure 4B). Endomicrobia exists in the hindgut as a strict endosymbiont in both Oxymonadida and Parabasalia protists (Ohkuma et al., 2007), which likely accounts for the large percentage of OTUs correlating with either protist group. The Endomicrobia OTUs that do not correlate with the abundance of either protist group could be due to sequencing errors, PCR sensitivity, being present inside protists that do not change abundance according to caste differentiation or exist without an obligate association with protists. The Treponema OTUs correlate with the Parabasalia and Oxymonadida as well (10/49 and 23/49, respectively). Treponema are known to exist in the hindgut as protist symbionts as well as free-living, which could account for less than half of the OTUs actually correlating with either protist group (Supplementary Figure 4A) (Leadbetter et al., 1999). Using the DictDb database, OTUs belonging to the Treponema taxon were further classified into subgroups (Treponema Ia, Ib, Ic, Ig, II) and this revealed that Treponema Ia was the most abundant taxa (Figure 5). Treponema Ib was the least abundant among the five groups and was also consistent among the castes, suggesting that this group may be a free-living spirochete. The abundance of Treponema Ia and Treponema II was significantly lower in winged alates compared to the worker caste, suggesting that these may be protist symbionts. Sequences corresponding to the order Bacteroidales nearly doubled in abundance in both the winged and de-winged alates as compared to workers and soldier microbiotas, which could be a result from a greater growth rate or of the Spirochaetales and Endomicrobia sequences dropping in abundance as these are not absolute but relative values. The spike of Enterobacteriales, Flavobacteriales, and Pseudomonadales sequences in the winged alates suggests the hindgut is in an altered state in the winged alates, which may reflect the physiological needs of the alate during swarming.
Overall Characterization of the R. flavipes Hindgut Microbiota
Studying the hindgut microbiota of individual termites from multiple colonies and castes has added to the understanding of the bacterial components of this complex symbiosis. The ability to sequence many hindgut samples has allowed for a more comprehensive comparison of different colonies and various castes, as well as the determination of a core microbiota in the R. flavipes species. Defining a core microbiota in the R. flavipes hindgut has revealed the presence of relatively constant and complex bacterial populations in the hindgut of workers. The differences in the composition of the bacterial and protist communities in the winged alates and de-winged alates suggest that major changes occur in the termite digestive tract physiology in this caste, perhaps related to the animals not feeding while relying on the fat body, which would lead to “starvation” of the protists, bacteria and archaea in the hind gut. Importantly, the community cannot be depleted of the core members, as it needs to be passed on to the workers from the new colony unless they are acquired from the environment. The maintenance of such a large core community is important as it suggests that a consistent group of microorganisms participates in the complex degradation of lignocellulose in the hindgut and the provision of nutrients that this simple diet is depleted in. The termite holobiont, or the combination of host and symbionts, which together form a functional unit, is complex and likely to be even more complex as insight is gained into viruses or fungi that may be present inside the termite in addition to the archaea, bacteria, and protists (Rohwer et al., 2002; Zilber-Rosenberg and Rosenberg, 2008; Bordenstein and Theis, 2015).
Studying the bacterial and protist populations in the R. flavipes hindgut throughout different life stages, colonies, and over time provides a cohesive representation of the community dynamics. The consistent presence of sequences at low percentages suggests that these analyses need to be done with sufficient sensitivity to detect the activities of these members as well, albeit technical caveats make the analysis of less abundant or even rare taxa more challenging (Reeder and Knight, 2009). As the bacteria in the hindgut are not easily cultured outside the host, the ability to manipulate the hindgut community as a whole in vivo, for example through environmental changes or dietary changes, allows for this host to become a model for complex symbioses by revealing principles that are conserved among distantly related digestive tract symbioses (Ruby, 2008; Nelson and Graf, 2012; Maltz et al., 2014).
Author Contributions
JB performed the laboratory work and statistical analysis. JB and JG contributed the experimental design and writing of the manuscript.
Funding
This research was funded by the National Science Foundation (NSF) division of Emerging Frontiers in Research and Innovation in Multicellular and Inter-kingdom Signaling. Award number 1137249 (R. Srivastava, D. Gage, J. Graf, L. Shor, B. Mustain, and J. Leadbetter).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to thank Dr. Michael C. Nelson for creating a script to centralize the QIIME processing, and Kendra Maas of the University of Connecticut Microbial Analysis Research and Services, MARS, Facility for assisting with the use of R. The authors also wish to thank Drs., Jared Leadbetter, Michael C. Nelson, Sophie Colston, and Emily McClure for their valuable feedback on this manuscript and Charles Bridges, Dr. Daniel Gage, Dr. Jared Leadbetter, and Emily McClure for collecting termites.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2016.00171
Footnotes
References
Aird, D., Ross, M. G., Chen, W.-S., Danielsson, M., Fennell, T., Russ, C., et al. (2011). Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome Biol. 12, 1–14. doi: 10.1186/gb-2011-12-2-r18
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Anderson, M. J., Crist, T. O., Chase, J. M., Vellend, M., Inouye, B. D., Freestone, A. L., et al. (2011). Navigating the multiple meanings of β diversity: a roadmap for the practicing ecologist. Ecol. Lett. 14, 19–28. doi: 10.1111/j.1461-0248.2010.01552.x
Berchtold, M., Chatzinotas, A., Schonhuber, W., Brune, A., Amann, R., Hahn, D., et al. (1999). Differential enumeration and in situ localization of microorganisms in the hindgut of the lower termite Mastotermes darwiniensis by hybridization with rRNA-targeted probes. Arch. Microbiol. 172, 407–416. doi: 10.1007/s002030050778
Bokulich, N. A., Subramanian, S., Faith, J. J., Gevers, D., Gordon, J. I., Knight, R., et al. (2012). Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 10, 57–59. doi: 10.1038/nmeth.2276
Bordenstein, S. R., and Theis, K. R. (2015). Host biology in light of the microbiome: ten principles of holobionts and hologenomes. PLoS Biol. 13:e1002226. doi: 10.1371/journal.pbio.1002226
Boucias, D. G., Cai, Y., Sun, Y., Lietze, V.-U., Sen, R., Raychoudhury, R., et al. (2013). The hindgut lumen prokaryotic microbiota of the termite Reticulitermes flavipes and its responses to dietary lignocellulose composition. Mol. Ecol. 22, 1836–1853. doi: 10.1111/mec.12230
Brauman, A., Dore, J., Eggleton, P., Bignell, D., Breznak, J. A., and Kane, M. D. (2001). Molecular phylogenetic profiling of prokaryotic communities in guts of termites with different feeding habits. FEMS Microbiol. Ecol. 35, 27–36. doi: 10.1111/j.1574-6941.2001.tb00785.x
Buczkowski, G., Wang, C., and Bennett, G. (2007). Immunomarking reveals food flow and feeding relationships in the eastern subterranean termite, Reticulitermes flavipes (Kollar). Environ. Entomol. 36, 173–182. doi: 10.1603/0046-225X(2007)36[173:IRFFAF]2.0.CO;2
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Caporaso, J. G., Lauber, C. L., Walters, W. A., Berg-Lyons, D., Huntley, J., Fierer, N., et al. (2012). Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 6, 1621–1624. doi: 10.1038/ismej.2012.8
Carr, G., Poulsen, M., Klassen, J. L., Hou, Y., Wyche, T. P., Bugni, T. S., et al. (2012). Microtermolides A and B from termite-associated Streptomyces sp. and structural revision of vinylamycin. Org. Lett. 14, 2822–2825. doi: 10.1021/ol301043p
Cleveland, L. R. (1925). The feeding habit of termite castes and its relation to their intestinal flagellates. Biol. Bull. 48, 295–308. doi: 10.2307/1536598
Costa-Leonardo, A. M., Laranjo, L. T., Janei, V., and Haifig, I. (2013). The fat body of termites: functions and stored materials. J. Insect Physiol. 59, 577–587. doi: 10.1016/j.jinsphys.2013.03.009
DeSantis, T. Z., Hugenholtz, P., Larsen, N., Rojas, M., Brodie, E. L., Keller, K., et al. (2006). Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72, 5069–5072. doi: 10.1128/AEM.03006-05
Dietrich, C., Köhler, T., and Brune, A. (2014). The cockroach origin of the termite gut microbiota: patterns in bacterial community structure reflect major evolutionary events. Appl. Environ. Microbiol. 80, 2261–2269. doi: 10.1128/AEM.04206-13
Faith, D. P., and Baker, A. M. (2006). Phylogenetic diversity (PD) and biodiversity conservation: some bioinformatics challenges. Evol. Bioinformat. 2, 121–128.
Fisher, M., Miller, D., Brewster, C., Husseneder, C., and Dickerman, A. (2007). Diversity of gut bacteria of Reticulitermes flavipes as examined by 16S rRNA gene sequencing and amplified rDNA restriction analysis. Curr. Microbiol. 55, 254–259. doi: 10.1007/s00284-007-0136-8
Graber, J. R., and Breznak, J. A. (2004). Physiology and nutrition of Treponema primitia, an H2/CO2-acetogenic spirochete from termite hindguts. Appl. Environ. Microbiol. 70, 1307–1314. doi: 10.1128/AEM.70.3.1307-1314.2004
Hamady, M., and Knight, R. (2009). Microbial community profiling for human microbiome projects: tools, techniques, and challenges. Genome Res. 19, 1141–1152. doi: 10.1101/gr.085464.108
Hongoh, Y. (2010). Diversity and genomes of uncultured microbial symbionts in the termite gut. Biosci. Biotechnol. Biochem. 74, 1145–1151. doi: 10.1271/bbb.100094
Hongoh, Y., Deevong, P., Inoue, T., Moriya, S., Trakulnaleamsai, S., Ohkuma, M., et al. (2005). Intra- and interspecific comparisons of bacterial diversity and community structure support coevolution of gut microbiota and termite host. Appl. Environ. Microbiol. 71, 6590–6599. doi: 10.1128/AEM.71.11.6590-6599.2005
Hongoh, Y., Sharma, V. K., Prakash, T., Noda, S., Toh, H., Taylor, T. D., et al. (2008). Genome of an endosymbiont coupling N2 fixation to cellulolysis within protist cells in termite gut. Science 322, 1108–1109. doi: 10.1126/science.1165578
Huang, X.-F., Bakker, M. G., Judd, T. M., Reardon, K. F., and Vivanco, J. M. (2013). Variations in diversity and richness of gut bacterial communities of termites (Reticulitermes flavipes) fed with grassy and woody plant substrates. Microbial. Ecol. 65, 531–536. doi: 10.1007/s00248-013-0219-y
Huse, S. M., Ye, Y., Zhou, Y., and Fodor, A. A. (2012). A core human microbiome as viewed through 16S rRNA sequence clusters. PLoS ONE 7:e34242. doi: 10.1371/journal.pone.0034242
Iida, T., Ohkuma, M., Ohtoko, K., and Kudo, T. (2000). Symbiotic spirochetes in the termite hindgut: phylogenetic identification of ectosymbiotic spirochetes of oxymonad protists. FEMS Microbiol. Ecol. 34, 17–26. doi: 10.1111/j.1574-6941.2000.tb00750.x
Ikeda-Ohtsubo, W., Desai, M., Stingl, U., and Brune, A. (2007). Phylogenetic diversity of ‘Endomicrobia’ and their specific affiliation with termite gut flagellates. Microbiology 153, 3458–3465. doi: 10.1099/mic.0.2007/009217-0
Ikeda-Ohtsubo, W., Faivre, N., and Brune, A. (2010). Putatively free-living ‘Endomicrobia’ - ancestors of the intracellular symbionts of termite gut flagellates? Environ. Microbiol. Rep. 2, 554–559. doi: 10.1111/j.1758-2229.2009.00124.x
Janda, J. M., and Abbott, S. L. (2007). 16S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: pluses. Perils, and Pitfalls. J. Clin. Microbiol. 45, 2761–2764. doi: 10.1128/JCM.01228-07
Kapheim, K. M., Rao, V. D., Yeoman, C. J., Wilson, B. A., White, B. A., Goldenfeld, N., et al. (2015). Caste-specific differences in hindgut microbial communities of honey bees (Apis mellifera). PLoS ONE 10:e0123911. doi: 10.1371/journal.pone.0123911
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformation 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Klassen, J. L. (2014). Microbial secondary metabolites and their impacts on insect symbioses. Curr. Opin. Insect Sci. 4, 15–22. doi: 10.1016/j.cois.2014.08.004
Koch, H., and Schmid-Hempel, P. (2011). Socially transmitted gut microbiota protect bumble bees against an intestinal parasite. Proc. Natl. Acad. Sci. U.S.A. 108, 19288–19292. doi: 10.1073/pnas.1110474108
Kohl, K. D., and Dearing, M. D. (2014). Wild-caught rodents retain a majority of their natural gut microbiota upon entrance into captivity. Environ. Microbiol. Rep. 6, 191–195. doi: 10.1111/1758-2229.12118
Leadbetter, J. R., and Breznak, J. A. (1996). Physiological Ecology of Methanobrevibacter cuticularis sp. nov.and Methanobrevibacter curvatus sp. nov., Isolated from the Hindgut of the Termite Reticulitermes flavipes. Appl. Environ. Microbiol. 62, 3620–3631.
Leadbetter, J. R., Schmidt, T. M., Graber, J. R., and Breznak, J. A. (1999). Acetogenesis from H2 plus CO2 by spirochetes from termite guts. Science 283, 686–689. doi: 10.1126/science.283.5402.686
Legendre, F., Whiting, M. F., Bordereau, C., Cancello, E. M., Evans, T. A., and Grandcolas, P. (2008). The phylogeny of termites (Dictyoptera: Isoptera) based on mitochondrial and nuclear markers: implications for the evolution of the worker and pseudergate castes, and foraging behaviors. Mol. Phylogenet. 48, 615–627. doi: 10.1016/j.ympev.2008.04.017
Lewis, J. L., and Forschler, B. T. (2013). Protist communities from four castes and three species of Reticulitermes (Isoptera: Rhinotermitidae). Ann. Entomol. Soc. Am. 97, 1242–1251. doi: 10.1603/0013-8746(2004)097[1242:PCFFCA]2.0.CO;2
Ley, R. E., Hamady, M., Lozupone, C., Turnbaugh, P. J., Ramey, R. R., Bircher, J. S., et al. (2008). Evolution of mammals and their gut microbes. Science 320, 1647–1651. doi: 10.1126/science.1155725
Lim, S. Y., and Forschler, B. T. (2012). Reticulitermes nelsonae, a new species of subterranean termite (Rhinotermitidae) from the Southeastern United States. Insects 3, 62–90. doi: 10.3390/insects3010062
Liu, H., and Beckenbach, A. T. (1992). Evolution of the mitochondrial cytochrome oxidase II gene among 10 orders of insects. Mol. Phylogenet. Evol. 1, 41–52. doi: 10.1016/1055-7903(92)90034-E
Lozupone, C., Hamady, M., and Knight, R. (2006). UniFrac – An online tool for comparing microbial community diversity in a phylogenetic context. BMC Bioinformat. 7:371. doi: 10.1186/1471-2105-7-371
Lozupone, C., and Knight, R. (2005). UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 71, 8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005
Maltz, M. A., Bomar, L., Lapierre, P., Morrison, H. G., Mcclure, E. A., Sogin, M. L., et al. (2014). Metagenomic analysis of the medicinal leech gut microbiota. Front. Microbiol. 5:151. doi: 10.3389/fmicb.2014.00151
Matson, E., Ottesen, E., and Leadbetter, J. (2007). Extracting DNA from the gut microbes of the termite (Zootermopsis Angusticollis) and visualizing gut microbes. J. Vis. Exp. 4:e195.
McDonald, D., Price, M. N., Goodrich, J., Nawrocki, E. P., Desantis, T. Z., Probst, A., et al. (2011). An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 6, 610–618. doi: 10.1038/ismej.2011.139
Meriweather, M., Matthews, S., Rio, R., and Baucom, R. S. (2013). A 454 survey reveals the community composition and core microbiome of the common bed bug (Cimex lectularius) across an Urban Landscape. PLoS ONE 8:e61465. doi: 10.1371/journal.pone.0061465
Mikaelyan, A., Kohler, T., Lampert, M., Rohland, J., Boga, H., Meuser, K., et al. (2015). Classifying the bacterial gut microbiota of termites and cockroaches: a curated phylogenetic reference database (DictDb). Syst. Appl. Microbiol. 38, 472–482. doi: 10.1016/j.syapm.2015.07.004
Minkley, N., Fujita, A., Brune, A., and Kirchner, W. H. (2006). Nest specificity of the bacterial community in termite guts (Hodotermes mossambicus). Insect. Soc. 53, 339–344. doi: 10.1007/s00040-006-0878-5
Moran, N. A., Hansen, A. K., Powell, J. E., and Sabree, Z. L. (2012). Distinctive gut microbiota of honey bees assessed using deep sampling from individual worker bees. PLoS ONE 7:e36393. doi: 10.1371/journal.pone.0036393
Nelson, M. C., and Graf, J. (2012). Bacterial symbioses of the medicinal leech Hirudo verbana. Gut Microbes 3, 322–331. doi: 10.4161/gmic.20227
Nelson, M. C., Morrison, H. G., Benjamino, J., Grim, S. L., and Graf, J. (2014). Analysis, optimization and verification of Illumina-generated 16S rRNA gene amplicon surveys. PLoS ONE 9:e94249. doi: 10.1371/journal.pone.0094249
Ohkuma, M. (2003). Termite symbiotic systems: efficient bio-recycling of lignocellulose. Appl. Microbiol. Biotechnol. 61, 1–9. doi: 10.1007/s00253-002-1189-z
Ohkuma, M. (2008). Symbioses of flagellates and prokaryotes in the gut of lower termites. Trends Microbiol. 16, 345–352. doi: 10.1016/j.tim.2008.04.004
Ohkuma, M., Sato, T., Noda, S., Ui, S., Kudo, T., and Hongoh, Y. (2007). The candidate phylum ‘Termite Group 1’ of bacteria: phylogenetic diversity, distribution, and endosymbiont members of various gut flagellated protists. FEMS Microbiol. Ecol. 60, 467–476. doi: 10.1111/j.1574-6941.2007.00311.x
Oksanen, J., Blanchet, F. G., Kindt, R., Legendre, P., Minchin, P. R., O’hara, R. B., et al. (2015). vegan: Community Ecology Package. R Package Version 2.2–1. Available at: http://cran.r-project.org
Otani, S., Mikaelyan, A., Nobre, T., Hansen, L. H., Koné, N. G. A., Sørensen, S. J., et al. (2014). Identifying the core microbial community in the gut of fungus-growing termites. Mol. Ecol. 23, 4631–4644. doi: 10.1111/mec.12874
Park, Y. C., Grandcolas, P., and Choe, J. C. (2002). Colony composition, social behavior and some ecological characteristics of the korean wood-feeding cockroach (Cryptocercus kyebangensis). Zoo. Sci. 19, 1133–1139. doi: 10.2108/zsj.19.1133
Perdereau, E., Bagneres, A., Bankhead-Dronnet, S., Dupont, S., Zimmerman, M., Vargo, E., et al. (2013). Global genetic analysis reveals the putative native source of the invasive termite, Reticulitermes flavipes, in France. Mol. Ecol. Res. 22, 1105–1119. doi: 10.1111/mec.12140
R Development Core Team (2005). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Rahman, N. A., Parks, D. H., Wilnlner, D. L., Engelbrektson, A. L., Goffredi, S. K., Warnecke, F., et al. (2015). A molecular survey of the Australian and North American termite genera indicates that vertical inheritance is the primary force shaping termite gut microbes. Microbiome 3:5. doi: 10.1186/s40168-015-0067-8
Raina, A., Park, Y., and Lax, A. (2004). Defaunation leads to cannibalism in primary reproductives of the Formosan subterranean termite, Coptotermes formosanus (Isoptera: rhinotermitidae). Ann. Entomol. Soc. Am. 97, 753–756. doi: 10.1603/0013-8746(2004)097[0753:DLTCIP]2.0.CO;2
Reeder, J., and Knight, R. (2009). The ‘rare biosphere’: a reality check. Nat. Methods 6, 636–637. doi: 10.1038/nmeth0909-636
Rohwer, F., Seguritan, V., and Azam, F. (2002). Diversity and distribution of coral-associated bacteria. Marine Ecol. 243, 1–10. doi: 10.3354/meps243001
Rosengaus, R. B., Zecher, C. N., Schultheis, K. F., Brucker, R. M., and Bordenstein, S. R. (2011). Disruption of the termite gut microbiota and its prolonged consequences for fitness. Appl. Environ. Microbiol. 77, 4303–4312. doi: 10.1128/AEM.01886-10
Ruby, E. G. (2008). Symbiotic conversations are revealed under genetic interrogation. Nat. Rev. Microbiol. 6, 752–762. doi: 10.1038/nrmicro1958
Sabree, Z. L., Hansen, A. K., and Moran, N. A. (2012). Independent studies using deep sequencing resolve the same set of core bacterial species dominating gut communities of honey bees. PLoS ONE 7:e41250. doi: 10.1371/journal.pone.0041250
Scharf, M. E. (2015). Omic research in termites: an overview and a roadmap. Front. Genet. 6:76. doi: 10.3389/fgene.2015.00076
Sethi, A., Slack, J. M., Kovaleva, E. S., Buchman, G. W., and Scharf, M. E. (2013). Lignin-associated metagene expression in a lignocellulose-digesting termite. Insect Biochem. Mol. Biol. 43, 91–101. doi: 10.1016/j.ibmb.2012.10.001
Shade, A., and Handelsman, J. (2012). Beyond the Venn diagram: the hunt for a core microbiome. Environ. Microbiol. 14, 4–12. doi: 10.1111/j.1462-2920.2011.02585.x
Shade, A., Peter, H., Allison, S. D., Baho, D. L., Berga, M., Bürgmann, H., et al. (2012). Fundamentals of microbial community resistance and resilience. Front. Microbiol. 3:417. doi: 10.3389/fmicb.2012.00417
Shimada, K., Lo, N., Kitade, O., Wakui, A., and Maekawa, K. (2013). Cellulolytic protist numbers rise and fall dramatically in termite queens and kings during colony foundation. Eukaryot. Cell 12, 545–550. doi: 10.1128/EC.00286-12
Soergel, D. A. W., Dey, N., Knight, R., and Brenner, S. E. (2012). Selection of primers for optimal taxonomic classification of environmental 16S rRNA gene sequences. ISME J 6, 1440–1444. doi: 10.1038/ismej.2011.208
Stingl, U., Radek, R., Yang, H., and Brune, A. (2005). “Endomicrobia”: cytoplasmic symbionts of termite gut protozoa form a separate phylum of prokaryotes. Appl. Environ. Microbiol. 71, 1473–1479. doi: 10.1128/AEM.71.3.1473-1479.2005
Su, N., Ye, W., Ripa, R., Scheffrahn, R., and Giblin-Davis, R. (2006). Identification of Chilean Reticulitermes (Isoptera: rhinotermitidae) inferred from three mitochondrial gene DNA sequences and soldier morphology. Ann. Entomol. Soc. Am. 99, 352–363. doi: 10.1603/0013-8746(2006)099[0352:IOCRIR]2.0.CO;2
Tap, J., Mondot, S., Levenez, F., Pelletier, E., Caron, C., Furet, J.-P., et al. (2009). Towards the human intestinal microbiota phylogenetic core. Environ. Microbiol. 11, 2574–2584. doi: 10.1111/j.1462-2920.2009.01982.x
Tarpy, D. R., Mattila, H. R., and Newton, I. L. G. (2015). Development of the honey bee gut microbiome throughout the queen-rearing process. Appl. Environ. Microbiol. 81, 3182–3191. doi: 10.1128/AEM.00307-15
Tartar, A., Wheeler, M. M., Zhou, X., Coy, M. R., Boucias, D. G., and Scharf, M. E. (2009). Parallel metatranscriptome analyses of host and symbiont gene expression in the gut of the termite Reticulitermes flavipes. Biotechnol. Biofuels 2:25. doi: 10.1186/1754-6834-2-25
Turnbaugh, P. J., and Gordon, J. I. (2013). The core gut microbiome, energy balance and obesity. J. Physiol. 17, 4153–4158. doi: 10.1113/jphysiol.2009.174136
Turnbaugh, P. J., Ley, R. E., Hamady, M., Fraser-Ligett, C., Knight, R., and Gordon, J. I. (2007). The human microbiome project: exploring the microbial part of ourselves in a changing world. Nature 449, 804–810. doi: 10.1038/nature06244
Wang, Y., Gilbreath, T. M., Kukutla, P., Yan, G., and Xu, J. (2011). Dynamic gut microbiome across life history of the malaria mosquito Anopheles gambiae in Kenya. PLoS ONE 6:e24767. doi: 10.1371/journal.pone.0024767
Yu, Z., and Morrison, M. (2004). Improved extraction of PCR-quality community DNA from digesta and fecal samples. Biotechniques 36, 808–812.
Keywords: Reticulitermes flavipes, Core microbiota, Termite caste microbiota, 16S rRNA gene, Illumina amplicon sequencing
Citation: Benjamino J and Graf J (2016) Characterization of the Core and Caste-Specific Microbiota in the Termite, Reticulitermes flavipes. Front. Microbiol. 7:171. doi: 10.3389/fmicb.2016.00171
Received: 01 October 2015; Accepted: 01 February 2016;
Published: 17 February 2016.
Edited by:
Mike Taylor, The University of Auckland, New ZealandCopyright © 2016 Benjamino and Graf. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joerg Graf, am9lcmcuZ3JhZkB1Y29ubi5lZHU=