Abstract
The proteomes of the metabolically versatile and poorly characterized Gram-positive bacterium Desulfotomaculum reducens MI-1 were compared across four cultivation conditions including sulfate reduction, soluble Fe(III) reduction, insoluble Fe(III) reduction, and pyruvate fermentation. Collectively across conditions, we observed at high confidence ~38% of genome-encoded proteins. Here, we focus on proteins that display significant differential abundance on conditions tested. To the best of our knowledge, this is the first full-proteome study focused on a Gram-positive organism cultivated either on sulfate or metal-reducing conditions. Several proteins with uncharacterized function encoded within heterodisulfide reductase (hdr)-containing loci were upregulated on either sulfate (Dred_0633-4, Dred_0689-90, and Dred_1325-30) or Fe(III)-citrate-reducing conditions (Dred_0432-3 and Dred_1778-84). Two of these hdr-containing loci display homology to recently described flavin-based electron bifurcation (FBEB) pathways (Dred_1325-30 and Dred_1778-84). Additionally, we propose that a cluster of proteins, which is homologous to a described FBEB lactate dehydrogenase (LDH) complex, is performing lactate oxidation in D. reducens (Dred_0367-9). Analysis of the putative sulfate reduction machinery in D. reducens revealed that most of these proteins are constitutively expressed across cultivation conditions tested. In addition, peptides from the single multiheme c-type cytochrome (MHC) in the genome were exclusively observed on the insoluble Fe(III) condition, suggesting that this MHC may play a role in reduction of insoluble metals.
Introduction
Microbial sulfate and metal reduction are thought to be ancient processes that today are major drivers of nutrient cycles in anaerobic environments (Bird et al., 2011; Pereira et al., 2011; Hori et al., 2015). Decades of research have focused on understanding the proteins involved in these electron transfer pathways, and Gram-negative bacteria have mainly served as model organisms in studies. However, it is becoming increasingly understood that sulfate-reducing organisms (SROs) and dissimilatory metal-reducing organisms (DMROs) are phylogenetically diverse, and Gram-positive bacteria are suspected to be major contributors to these processes in natural settings (Petrie et al., 2003; Suzuki et al., 2003; Cardenas et al., 2010; Williamson et al., 2013; Newsome et al., 2014).
Studies on the Gram-negative genus Desulfovibrio have been foundational in elucidating mechanisms involved in microbial sulfate reduction. Core proteins in the sulfate reduction pathway (sulfate adenyltransferase, APS reductase, sulfite reductase) are conserved amongst SROs. However, several other proteins described in the pathway of sulfate reduction in Desulfovibrio do not contain homologs in the genomes of Gram-positive SROs, implying that divergent mechanisms are involved (Pereira et al., 2011; Grein et al., 2013). Even less is understood regarding metal reduction in Gram-positive bacteria. Studies on Gram-negative metal-reducing representatives, namely Geobacter and Shewanella species, have defined the current understanding of microbial metal reduction. A critical trait for metal reduction shared amongst model metal reducers is an abundance of genes encoding multiheme c-type cytochromes (MHCs; Wall and Krumholz, 2006; Shi et al., 2009; Sharma et al., 2010). Gram-positive bacteria, however, rarely contain multiple MHCs (Sharma et al., 2010).
The Desulfotomaculum genus is composed of anaerobic, Gram-positive, endospore-forming SROs. These Peptococcaceae are widely distributed in the environment, and the number of identified species as well as sequenced genomes is continually expanding (Aüllo et al., 2013; Kuever et al., 2014; Visser et al., 2014). Characterized species display considerable metabolic versatility, and some have been shown to reduce metals (Tebo and Obraztsova, 1998; Haouari et al., 2008; Barton et al., 2015). Certain Desulfotomaculum species completely oxidize organic substrates to CO2, whereas others are incomplete oxidizers. The sulfite reductases from these two groups are phylogenetically distinct, and it has been suggested that complete oxidizers acquired their sulfite reductase laterally from a deltaproteobacterial donor (Klein et al., 2001; Zverlov et al., 2005).
Desulfotomaculum reducens MI-1 was isolated from a heavy-metal contaminated site and is a species of marked interest as a Gram-positive SRO that also has the capability to reduce metals including Fe(III) and Mn(IV; Tebo and Obraztsova, 1998). As one of only a few organisms reported to conserve energy from utilization of U(VI) and Cr(VI) as electron acceptors, D. reducens is also of interest for bioremediation applications (Tebo and Obraztsova, 1998; Wall and Krumholz, 2006; Barton et al., 2015). Previous work in D. reducens has helped to elucidate the mechanism of Fe(III) reduction. It was proposed that Fe(III)-oxide reduction during pyruvate fermentation occurs through a soluble electron shuttle, as direct contact was found to not be required for Fe(III) reduction on this condition (Dalla Vecchia et al., 2014b). Conversely, direct contact was found to be required for Fe(III)-oxide reduction with lactate as electron donor and is therefore not mediated by a soluble shuttle (Dalla Vecchia et al., 2014a). Understanding of the proteins involved in Fe(III) reduction in D. reducens is less developed. The genome contains one annotated MHC (Dred_0700-1, nitrite reductase), a likely candidate for Fe(III) reduction (Junier et al., 2010). However, studies have analyzed its expression levels and found it to not be induced in the presence of soluble Fe(III) or U(VI) relative to pyruvate fermentation conditions (Junier et al., 2011; Dalla Vecchia et al., 2014a,b). One of these studies also analyzed the surface proteins (or surfaceome) of D. reducens while fermenting pyruvate or reducing soluble Fe(III) with lactate as electron donor and identified no peptides for the MHC (Dalla Vecchia et al., 2014a). However, no mRNA/protein-based analyses of the MHC during insoluble Fe(III) reduction have been performed.
In this study, we compared the proteomes of D. reducens during sulfate, soluble Fe(III), and insoluble Fe(III) reduction (all with lactate as electron donor), and pyruvate fermentation. The genome of D. reducens was sequenced in 2010, allowing for genomic predictions to be tested with proteomic measurements (Junier et al., 2010). The D. reducens genome contains an abundance of proteins that contain oxidoreductase-related annotations but lack specific functional annotations. There is increasing understanding of the types of domains and proteins that drive anaerobic respiration (Grein et al., 2013). In D. reducens, however, experimental information is not available in order to determine the function of many predicted redox proteins. Furthermore, in recent years, pathways of flavin-based electron bifurcation (FBEB) have been described in anaerobic microorganisms. FBEB is now regarded as a third mode of energy conservation, along with respiration and fermentation (Herrmann et al., 2008; Buckel and Thauer, 2013). The D. reducens genome contains homology to described FBEB pathways, and comparative proteomic analysis provides a tool for predicting whether these pathways are contributing to the metabolism of D. reducens. We report here the first global proteomic comparison of a Desulfotomaculum species. In fact, to the best of our knowledge this is the first full-proteome comparative analysis of any Gram-positive organism focused on either sulfate or metal-reducing conditions.
Materials and methods
Biomass preparation
D. reducens MI-1 was purchased from the American Type Culture Collection (ATCC® BAA-1160™) and grown anaerobically at 30°C in batch culture with an 80/20 N2/CO2 headspace in Widdel Low Phosphate (WLP) media (pH 7) as described (Otwell et al., 2015). Cultures were grown with either 28 mM sodium sulfate, 25 mM Fe(III)-citrate, or 35 mM Fe(III)-oxide with 20 mM sodium lactate as electron donor. Fermentation cultures were grown with 20 mM pyruvic acid. Lactate only and Fe(III)-citrate only controls did not exhibit growth. For each cultivation condition analyzed, D. reducens was transferred to fresh growth medium at least three times prior to harvesting in order to ensure accurate representation of the proteome for the given cultivation condition. Cell growth was tracked with fluorescence microscopy by staining with acridine orange and the ferrozine assay for quantification of Fe(II) or the Cline assay for quantification of sulfide when appropriate (Lovley and Phillips, 1987; Strocchi et al., 1992). Representative growth curves for each culture condition are displayed in Supplementary Figure 1. Cells were harvested anaerobically using an anaerobic glove bag during mid-late exponential phase and cell pellets were stored at −80°C until protein extraction.
Protein extraction and digestion
Harvested cell pellets were suspended in 50 mM NH4HCO3 (pH 8) and lysed in the presence of 100–200 μL of 0.1 mm zirconia/silica beads (BioSpec Products, Bartlesville, OK) as described (Callister et al., 2006) with the following modifications. Bead beating was performed using a Bullet Blender (Next Advance, Averill Park, NY) operated for 3 min at speed 8. The lysate was collected at 4500 g for 5 min at 4°C and the beads were rinsed with 100 μL of 50 mM NH4HCO3 (pH 8). The combined lysates were transferred to a 1.5-mL microcentrifuge tube and cellular debris was separated from soluble lysate material by centrifuging at 10,000 g for 15 min at 4°C. Protein concentrations within the soluble lysate were measured using the Bicinchoninic Acid (BCA) Protein Assay and then normalized to equal concentration (Smith et al., 1985). Proteins were denatured and digested by following the FASP digest method (Wiśniewski et al., 2009) without alkylation, then washed using C18 SPE chromatography according to previously established protocols (Callister et al., 2008). Equal portions of peptides (100 μg) from biological replicates acquired for each cultivation condition were pooled, then separated using C18 reverse phase high pH fractionation according to previously published protocols (Wang et al., 2011). Ninety-six fractions were collected from each pooled sample and dried overnight in a SpeedVac. Peptides within each well were suspended in 100 ml of 50% methanol and combined into 12 fractions, concentrated to dryness then suspended in nanopure water to achieve a final concentration of 0.1 mg/ml. Additionally, peptides from the unfractionated samples were transferred to MicroSolv ALS vials and diluted with nanopure water to a final concentration of 0.1 μg/μL.
Proteomic data generation
Mass spectra were generated for both fractionated (60 fractions in total) and unfractionated peptide samples (30 samples originating from 2 to 3 biological replicates per cultivation condition and three technical replicates per biological replicate). The unfractionated samples were randomized and blocked prior to data generation. For both sample types 7 mg of peptides was injected to an Agilent LC system (Agilent Technologies, Santa Clara, CA) coupled to a hybrid ion trap Orbitrap Velos mass spectrometer (Thermo Scientific, San Jose, CA) equipped with an ion funnel and electrospray ionization (ESI) interface. Conditions for peptide separation and HPLC operating conditions have been previously published (Sowell et al., 2008; Robidart et al., 2013). Orbitrap spectra were collected from 400 to 2000 m/z at a resolution of 100 k followed by data-dependent ion trap tandem mass (MS/MS) spectra generation of the six most abundant ions using 35% collision energy (CID). Additional mass spectrometer operating conditions have been previously described (Sowell et al., 2008; Robidart et al., 2013).
Data analysis
Peptide sequences were assigned to MS/MS spectra using the MSGF search algorithm (Kim et al., 2008) and the translated D. reducens MI-1 annotated genome sequence downloaded (September 18, 2012) from the U.S. Department of Energy's Joint Genome Institute's (JGI) IMG database (Markowitz et al., 2014). From measured peptides, an empirical peptide database was generated for use as a library to match high-resolution parent ion spectra (i.e., AMT tag approach Lipton et al., 2002) generated by the Orbitrap Velos instrument from the unfractionated samples. These unfractionated peptide samples were used to obtain label-free abundance measurements for use in relative quantification comparisons. The area under each peptide peak, constructed from ion intensities (ion current) measured across instrument scans, was used to represent the arbitrary abundances of peptides (Lipton et al., 2002; Sowell et al., 2008). The dataset of peptides and their associated abundances, resulting from matching to the library, was filtered to achieve a false discovery rate of =5% using established protocols (Stanley et al., 2011). Peptides matching to multiple proteins were filtered out unless the matched proteins were identical, duplicated genes. Peptide abundances were log transformed (base 2) and protein abundance was estimated as the mean of its measured peptides' log-transformed abundances across replicates. A protein was considered positively observed if identified by at least two unique peptide sequences in replicates from a given cultivation condition. If detected, the protein's mean abundance was compared between different cultivation conditions and p-values assigned using ANOVA; part of the InfernoRDN (previously known as DAnTE, Polpitiya et al., 2008) proteomics analysis software tools (http://omics.pnl.gov/software/InfernoRDN). Abundance measurements for a protein were required to be present in at least 50% of replicates in order for its means to be compared. Proteins were considered differentially abundant if their mean peptide intensity had a >2-fold change calculated as the difference between means across the conditions being compared, with a p ≤ 0.05. Along with the cultivation conditions described, the AMT tag approach was used to analyze Fe(III)-oxide cultures harvested at 8 days. This data was excluded from the analysis as the 6-day samples matched closer with the growth phase of the other conditions. All raw data is deposited and publicly available in MassIVE (Mass spectrometry Interactive Virtual Environment) with accession number MSV000079501 and ProteomeXchange with accession number PXD003605.
Results: global proteome of D. reducens
The genome of D. reducens encodes 3324 predicted proteins, and across the four conditions analyzed in our study, 1268 proteins were confidently identified, representing ~38% of predicted proteins. This includes 1064 proteins with functional annotations and 204 hypothetical proteins. Table 1 displays a global overview of the proteomic data presented.
Table 1
| Cultivation condition | Total proteins identified | Average log2 peptide ion intensity | Proteins unique to condition | Proteins significantly increased compared to pyruvate |
|---|---|---|---|---|
| Pyruvate fermentation | 1104 | 19.87 (SD 1.49) | 113 (Supplementary Table 1a) | NA |
| Sulfate reduction (lactate as ED) | 1046 | 19.85 (SD 1.63) | 81 (Supplementary Table 1b) | 109 (Supplementary Table 2a) |
| Fe(III)-citrate reduction (lactate as ED) | 582 | 19.11 (SD 1.69) | 23 (Supplementary Table 1c) | 29 (Supplementary Table 2b) |
| Fe(III)-oxide reduction (lactate as ED) | 724 | 19.58 (SD 1.66) | 35 (Supplementary Table 1d) | 54 (Supplementary Table 2c) |
Global overview of D. reducens comparative proteomic analysis.
Biological and technical replicates of each condition cluster tightly together when visualizing full-proteome relatedness in a hierarchical clustering-based heat map, demonstrating consistency in the data (Figure 1). A Venn diagram (created in Venny 2.0, Oliveros, 2007) shows overlap of observed proteins across conditions tested, and displays a “core” proteome of 465 proteins identified on every condition (Supplementary Figure 2). In evaluating the number of proteins observed for each cultivation condition, more total proteins were identified on the pyruvate fermentation and sulfate reduction conditions than on the Fe(III) reduction conditions, as there are 1104, 1046, 582, and 724 total proteins observed on pyruvate, sulfate, Fe(III)-citrate, and Fe(III)-oxide respectively (Table 1). 113 proteins were observed solely on the pyruvate condition, 81 on sulfate, 23 on Fe(III)-citrate, and 35 on Fe(III)-oxide (Table 1). These proteins observed exclusively on a single condition are displayed in Supplementary Table 1. Research on model metal-reducing organisms has identified oxidoreductases as well as porin-like proteins that together function as a conduit for extracellular electron transfer, and therefore proteins with either of these predicted functions are highlighted for the Fe(III) reduction conditions (Richardson et al., 2012; Shi et al., 2012; Liu et al., 2014). Proteins that display significant fold increases (>2-fold change, p < 0.05) relative to pyruvate during sulfate, Fe(III)-citrate, and Fe(III)-oxide reduction conditions are provided in Supplementary Table 2. This includes 109 proteins significantly increased on sulfate relative to pyruvate, 29 on Fe(III)-citrate, and 54 on Fe(III)-oxide. Again, potential oxidoreductases and porin-like proteins are highlighted for the Fe(III) reduction conditions. A summary of all proteomic data presented in Supplementary Tables is displayed in Table 2.
Figure 1
Table 2
| Supplementary Table Number | Content |
|---|---|
| 1a-d | List of proteins unique to each cultivation condition |
| 2a-c | Abundance comparisons for proteins significantly increased compared to pyruvate |
| 3 | Abundance comparisons for proteins potentially involved in lactate and/or pyruvate utilization. |
| 4 | Abundance comparisons for annotated hydrogenases |
| 5 | Abundance comparisons for putative respiration-related proteins |
| 6 | Abundance comparisons for additional clusters of proteins significantly increased during sulfate reduction |
Summary of proteomic data presented in Supplementary Tables.
Along with lower total protein numbers on the Fe(III) reduction conditions, average ion intensities are lower compared to the sulfate and pyruvate conditions. Comparing average log2 fold change of average ion intensities across all proteins observed on both pyruvate and sulfate conditions yields a value of ~0.03 (pyruvate to sulfate), demonstrating that neither condition displayed significantly higher ion intensities. However, the average log2fold change of all proteins identified on pyruvate and Fe(III)-citrate is 1.34 (pyruvate to Fe(III)-citrate), showing that overall, abundances are >2-fold lower on Fe(III)-citrate. The average log2 fold change between the pyruvate and Fe(III)-oxide conditions is 0.78 (pyruvate to Fe(III)-oxide). These differences are reflected in the average ion intensity (log2) for all proteins detected, which is 19.87 (SD 1.49) for the pyruvate condition, 19.85 (SD 1.63) for sulfate, 19.11 (SD 1.69) for Fe(III)-citrate, and 19.58 (SD 1.66) for Fe(III)-oxide (Table 1). Also reflecting this variance between conditions are abundance patterns of certain housekeeping genes, including the RNA polymerase. For instance, RpoB (Dred_0207) is increased on pyruvate 1.8-fold (p < 0.01) relative to sulfate, 5.8-fold (p < 0.01) relative to Fe(III)-citrate, and 3.0-fold (p < 0.01) relative to Fe(III)-oxide. The β' (Dred_0208) and α (Dred_0243) subunits display similar abundance patterns. The 36 ribosomal proteins encoded in this region are nearly equal in abundance on pyruvate and sulfate (1.1-fold increased on pyruvate relative to sulfate on average), but significantly decreased in abundance on Fe(III) conditions. These ribosomal proteins are increased on average 5.8-fold on pyruvate conditions relative to Fe(III)-citrate and 1.6-fold relative to Fe(III)-oxide. This is likely a reflection of the slower growth rates observed on Fe(III)-reduction conditions compared with sulfate reduction and pyruvate fermentation (Supplementary Figure 1).
Our comparative proteomic analysis has revealed many potentially redox-related proteins from D. reducens. For instance, seven heterodisulfide reductase (hdr)-containing loci were identified in the genome, which are genomic regions rich with other predicted redox-related proteins including ferredoxins, proteins with iron-sulfur binding domains, proteins with flavin-binding domains, and annotated oxidoreductases (Junier et al., 2010). High numbers of unique peptides and differential protein abundance patterns across conditions were observed for proteins encoded within the hdr-containing loci. Identification patterns of proteins within these loci are displayed in Figure 2, and abundance comparisons for proteins of interest are displayed in Table 4. Proteomic data from various proteins, including proteins within these hdr-containing loci, are discussed below.
Figure 2
Discussion
Energy production and catabolism of organic carbon in D. reducens
A novel method of lactate metabolism involving a FBEB complex in Acetobacterium woodii (Awo_c08730, 20, and 10) was recently described (Weghoff et al., 2015). The researchers suggested that the complex has the same role in many anaerobes, and Dred_0367-9 is the homologous region in D. reducens. These proteins are currently annotated as the beta and alpha subunits of electron transfer flavoproteins (Dred_0367-8) and an FAD-linked oxidase domain-containing protein (Dred_0369). A high number of unique peptides for each of these proteins was detected on all conditions, with the lowest number observed on pyruvate. The average ion intensity of each protein in the operon Dred_0367-9 is higher on lactate-fed sulfate reduction cultures than pyruvate fermentation cultures, with p < 0.002, 0.08, and 0.03 respectively. Abundance comparisons for all conditions are displayed in Supplementary Table 3. The annotated pathway for lactate oxidation in the D. reducens genome contains lactate dehydrogenase (LDH) Dred_2797, but no peptides for this protein were observed in our study. Based on the lack of detection of the annotated LDH, the high sequence similarity between Dred_0367-9 and the FBEB LDH complex, and the confident identification of these proteins in lactate-grown cultures, we propose that Dred_0367-9 is how D. reducens oxidizes lactate to pyruvate, producing NADH in the process (Figure 3A). It is important to note that in A. woodii, it was proposed that reduced ferredoxin is regenerated from NADH by reverse electron transfer, mediated by the Rnf complex (Weghoff et al., 2015). As the genome of D. reducens does not encode this complex, however, reduced ferredoxin would need to be regenerated through other means (see below).
Figure 3
Three potential pathways for pyruvate utilization are predicted in the annotated genome of D. reducens, encoded by genes Dred_0047-50, Dred_2750-3, and Dred_1893 (Junier et al., 2010). Proteomic data revealed that the pyruvate-ferredoxin oxidoreductase pathway (Dred_0047-50) is expressed during both lactate oxidation and pyruvate fermentation and that the pyruvate-formate lyase pathway (Dred_2750-3) is expressed only during pyruvate fermentation. The third predicted pathway, through the annotated pyruvate dehydrogenase Dred_1893 (a pseudogene), was not observed (Figure 3B, Supplementary Table 3). The pyruvate-ferredoxin oxidoreductase pathway (Dred_0047-50) yields acetate, CO2, and H2 during fermentative growth. H2 is not produced during lactate oxidation, and instead this pathway is proposed to produce reduced ferredoxin, which could feed into the LDH complex described above (Junier et al., 2010). High numbers of unique peptides were observed for Dred_0047-9 on both lactate and pyruvate-fed cultures and the proteins do not display strong differential abundance patterns across conditions (Supplementary Table 3). Peptides from Dred_0050, an annotated 4Fe-4S ferredoxin, are detected in every condition (and are the highest in abundance on Fe(III)-citrate conditions), but these peptides are also present in another annotated 4Fe-4S ferredoxin (Dred_2822) and therefore cannot be distinguished. The monomeric hydrogenase Dred_1794 was identified as a likely candidate for production of hydrogen during pyruvate fermentation based on transcriptome patterns, and our proteomic data support this hypothesis (Junier et al., 2010). Dred_1794 is significantly increased on pyruvate relative to all other conditions (5 to 8-fold increase, p < 0.01). Abundance ratios for this hydrogenase, as well as all six hydrogenases annotated in the genome of D. reducens, are displayed in Supplementary Table 4. Our data suggest that the second predicted pyruvate utilization pathway, the acetate and formate yielding pyruvate-formate lyase pathway (Dred_2750-3) is active during pyruvate fermentation. Previously reported experimental evidence supports this finding, as small amounts of formate are shown to accumulate during pyruvate fermentation (Dalla Vecchia et al., 2014b). Dred_2751 is detected only on the pyruvate condition and Dred_2750 and Dred_2752 are most abundant during fermentative growth relative to all other conditions (~3-and 2-fold increased relative to sulfate conditions, p < 0.01 and 0.1 respectively). Peptides from Dred_2753 were not detected on any condition (Supplementary Table 3). A model illustrating lactate and/or pyruvate oxidation in D. reducens based on proteomic findings is presented in Figure 3B.
The genome of D. reducens contains an eleven-subunit, proton-translocating NADH: quinone oxidoreductase (Nuo, Dred_2036-46), predicted to be involved in NADH oxidation for cellular respiration (Junier et al., 2010). On sulfate reduction conditions as well as pyruvate fermentation, four of the subunits were identified (B, C, D, and I), while only the D subunit was observed on Fe(III)-oxide. During Fe(III)-citrate reduction, not a single subunit of Nuo was identified with any confidence, providing evidence that the Nuo complex is not being utilized during this type of growth in D. reducens. Cellular respiration generates ATP by ATP synthase, and all but two subunits of the F-type H+-transporting ATPase were identified in our study. The six identified proteins of the ATP synthase (Dred_3149-56) were most abundant during the sulfate reduction condition and decreased in abundance on Fe(III)-citrate relative to all other conditions (Supplementary Table 5). While Fe(III) was initially reported to serve as an electron acceptor for D. reducens, more recent reports have suggested that Fe(III) acts as an electron dump rather than a true respiratory electron acceptor (Dalla Vecchia et al., 2014a,b). Our proteomic findings on the Nuo complex as well as the ATP synthase support the idea that D. reducens may not be truly respiring Fe(III). We propose that the downregulation of energy production-related proteins during Fe(III) reduction in D. reducens indicates that Fe(III) serves as a less suitable cultivation condition for D. reducens than sulfate reduction or pyruvate fermentation. This is supported by less vigorous growth of D. reducens on Fe(III), as evidenced by substantially lower growth rates and biomass yields (Supplementary Figure 1).
Proteome of D. reducens during sulfate reduction
Our proteomic analysis revealed that core proteins involved in sulfate reduction in D. reducens (and conserved between Gram-negative and Gram-positive SROs) are consistent in abundance across conditions. Furthermore, we have identified key clusters of proteins that are highly abundant on sulfate relative to other conditions, and we hypothesize that these are involved in the respiratory process. Some of these clusters include the hdr-containing loci introduced previously (Figure 2). Figure 4 summarizes the predicted pathway of electron transfer during sulfate reduction in D. reducens based on proteomic evidence.
Figure 4
The core sulfate reduction pathway involves activation of sulfate to adenosine 5′-phosphosulfate (APS) by sulfate adenyltransferase (Dred_0635), processing of the pyrophosphate formed by a pyrophosphatase (Dred_2985), reduction of APS to sulfite by APS reductase (Dred_0636-7), and reduction of sulfite to sulfide by dissimilatory-type sulfite reductase DsrAB (Dred_3186-7, DsrB and DsrA; Pereira et al., 2011; Grein et al., 2013). Our data reveals that in most cases, the core sulfate-reducing machinery is not differentially expressed in D. reducens across the cultivation conditions tested (Table 3, Figure 4).
Table 3
| Locus tag | Protein name | Log2 Sulf/Pyr | Log2 Sulf/Fe(III)–citrate | Log2 Sulf/Fe(III)–oxide | PSORTb localization |
|---|---|---|---|---|---|
| CORE SULFATE REDUCTION MACHINERY | |||||
| Dred_0635 | Sulfate adenyltransferase (Sat) | −0.28 | 0.16 | −1.03 | C |
| Dred_0636 | Adenylylsulfate reductase subunit beta (AprB) | −0.25 | 0.59 | 0.27 | U |
| Dred_0637 | Adenylylsulfate reductase subunit alpha (AprA) | 0.16 | 0.39 | −0.23 | C |
| Dred_2985 | Membrane-bound proton-translocating pyrophosphatase | 0.40 | 3.97 | 1.60 | CM |
| Dred_3186 | Sulfite reductase, dissimilatory-type beta subunit (DsrB) | 0.42 | 0.99 | 0.36 | C |
| Dred_3187 | Sulfite reductase, dissimilatory-type alpha subunit (DsrA) | 0.26 | 0.33 | 0.03 | C |
| ADDITIONAL PUTATIVE SULFATE REDUCTION PROTEINS | |||||
| Dred_0638 | 4Fe-4S ferredoxin iron-sulfur binding domain-containing protein (QmoA) | 1.07 | 0.77 | 0.21 | C |
| Dred_0639 | Methyl-viologen-reducing hydrogenase, delta subunit (QmoB) | 0.49 | 0.08 | −0.04 | C |
| Dred_3185 | Putative dissimilatory sulfite reductase subunit D (DsrD) | −0.02 | 4.03 | 3.35 | C |
| Dred_3197 | DsrC family protein | 0.32 | 1.43 | 1.15 | C |
| Dred_3198 | Hypothetical protein (DsrK) | −0.90 | 3.37 | 0.87 | C |
| Dred_3199 | Nitrate reductase, gamma subunit (DsrM) | 0.47 | NI | −0.14 | CM |
| Table key: | |||||
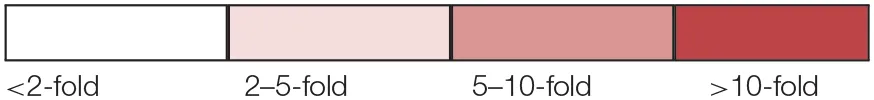 | |||||
Log2 abundance comparisons of proteins putatively involved in dissimilatory sulfate reduction in D. reducens.
Protein identification is based on detection of at least two unique peptides in a biological replicate and peptide detection in at least 50% of replicates. P-values for all ratios highlighted for abundance changes >2-fold are < 0.01. NI, protein not identified. Localization is from PSORTb (Yu et al., 2010). C, cytoplasmic; CM, cytoplasmic membrane; E, extracellular; CW, cell wall; U, unknown.
Outside of the core enzymes, distinct differences are observed between the genomes of Gram-positive and Gram-negative SROs, as outlined in-depth in recent reviews (Pereira et al., 2011; Grein et al., 2013). In particular, transmembrane complexes that have been ascribed to electron transfer during sulfate reduction in model Desulfovibrio species, many of which contain MHC components, are not conserved in Gram-positives. One of these transmembrane complexes is QmoABC, shown to transfer electrons to the APS reductase. The genome of D. reducens contains predicted orthologs to QmoA (Dred_0638) and QmoB (Dred_0639), but is missing the transmembrane subunit QmoC, suggesting that other mechanisms are involved in electron transfer to APS reductase. As was observed for the core sulfate-reducing machinery, Dred_0638-9 is similarly abundant across all cultivation conditions (Table 3, Figure 4). Another transmembrane complex of interest is the five-subunit DsrMKJOP, described in Desulfovibrio to transfer electrons to the sulfite reductase. The genome of D. reducens encodes predicted orthologs only to two subunits and these are rather distant. Dred_3198 shares 37% sequence identity across 81% query coverage to DsrK from Desulfovibrio vulgaris and Dred_3199 shares 26% identity across 74% to DsrM (http://blast.ncbi.nlm.nih.gov). Both proteins do not exhibit any discernable differential abundance on sulfate, pyruvate, and Fe(III)-oxide, but Dred_3198 is significantly lower in abundance on Fe(III)-citrate relative to sulfate (10.3-fold higher on sulfate, p < 0.01) and Dred_3199 was not identified on Fe(III)-citrate. DsrC (Dred_3197) is a protein conserved in all SROs sequenced to date, positioned next to the Dsr complex, and found to interact with the sulfite reductase (Venceslau et al., 2014). DsrC was recently shown to form a trisulfide intermediate and to serve as a co-substrate for DsrAB (Santos et al., 2015). In D. reducens, DsrC is increased >2-fold on sulfate vs. both Fe(III) conditions (Table 3).
Proteins in heterodisulfide reductase-containing loci are abundant in sulfate reduction proteome
Our proteomic analysis revealed specific clusters of proteins that are abundant during sulfate reduction relative to the other cultivation conditions. Interestingly, three of these clusters fall into the hdr-containing loci displayed in Figure 2, specifically Dred_0633-4, Dred_0689-90, and Dred_1325-30 (Table 4). Dred_0633-4, a predicted operon in locus IV, is significantly increased relative to Fe(III) reduction conditions but not pyruvate fermentation conditions. However, the positioning of Dred_0633 (an HdrC-type protein) and Dred_0634 (an HdrB-type protein) next to key sulfate reduction proteins including the sulfate adenyltransferase (Dred_0635), the APS reductase subunits (Dred_0636-7, AprBA), and QmoAB (Dred_0638-9) suggests an involvement in sulfate reduction. Due to their genomic localization near the AprBA, these Hdr-like proteins may be involved in electron transfer to this reductase (Figure 4). In fact, in all sequenced Desulfotomaculum genomes, orthologs of these two Hdr-like proteins exist next to the sulfate adenyltransferase and APS reductase, as determined by the IMG neighborhood viewer (https://img.jgi.doe.gov).
Table 4
| Locus tag | Protein name | Log2 Sulf/Pyr | Log2 Sulf/Fecit | Log2 Sulf/Feox | Localization |
|---|---|---|---|---|---|
| LOCUS IV: Dred_0633-4, SULFATE-INDUCED | |||||
| Dred_0633 | Putative heterodisulfide reductase, C subunit (HdrC) | −0.33 | 2.57 (<0.01) | 2.62 | C |
| Dred_0634 | CoB–CoM heterodisulfide reductase (HdrB) | 0.23 | 2.27 (<0.01) | 1.02 | C |
| LOCUS V: Dred_0689-90, SULFATE-INDUCED | |||||
| Dred_0689 | Hypothetical protein | 0.23 | 3.76 (<0.01) | 2.74 | C |
| Dred_0690 | hypothetical protein (HdrD) | 0.01 | 2.35 (<0.01) | 1.05 | CM |
| LOCUS VI: Dred_1325-30, SULFATE-INDUCED | |||||
| Dred_1325 | Heterodisulfide reductase, C subunit (HdrC) | 1.88 (<0.01) | 6.02 (<0.01) | 0.06 | C |
| Dred_1326 | Hypothetical protein (hdrB) | 1.25 (<0.01) | 4.1 (<0.01) | 3.81 (<0.01) | C |
| Dred_1327 | 4Fe-4S ferredoxin iron-sulfur binding domain-containing protein (HdrA1) | 1.03 (<0.01) | 2.98 (<0.01) | 2.54 (<0.01) | C |
| Dred_1328 | 4Fe-4S ferredoxin iron-sulfur binding domain-containing protein (HdrA) | 0.67 (<0.01) | 4.36 (<0.01) | 2.9 (<0.01) | C |
| Dred_1329 | Methyl-viologen-reducing hydrogenase, delta subunit (MvhD) | 0.75 (<0.01) | 4.6 (<0.01) | 4.1 (<0.01) | U |
| Dred_1330 | Formate dehydrogenase | 0.85 (<0.01) | 3.09 (<0.01) | 2.11 (<0.01) | C |
| Locus tag | Protein name | Log2 Fecit/pyr | Log2 Fecit/Sulf | Log2 Fecit/Feox | Localization |
| LOCUS I: NO SIGNIFICANT DIFFERENTIAL ABUNDANCE PATTERN | |||||
| Dred_0137/0143 | 4Fe-4S ferredoxin iron-sulfur binding domain-containing protein (HdrA) | −0.29 | −0.77 (<0.01) | −0.24 | U |
| Dred_0138 | pseudogene | NI | NI | NI | |
| Dred_0139/0146 | 4Fe-4S ferredoxin iron-sulfur binding domain-containing protein (FeS) | −0.85 (0.01) | −0.62 | 0.71 (0.04) | CM |
| Dred_0140/0147 | 4Fe-4S ferredoxin iron-sulfur binding domain-containing protein (FeS) | −0.26 | −0.60 (0.01) | −0.47 (0.02) | C |
| Dred_0141/0148 | Oxidoreductase FAD/NAD(P)-binding subunit | −1.07 (<0.01) | −1.61 (<0.01) | 0.09 | C |
| Dred_0142 | Hypothetical protein | −0.96 (<0.01) | −1.28 (<0.01) | 1.48 (<0.01) | C |
| Dred_0144 | Methyl-viologen-reducing hydrogenase, delta subunit (MvhD) | 1.37 | 0.62 | 0.47 | U |
| Dred_0145 | Heterodisulfide reductase subunit (HdrD) | −0.7 (0.03) | −1.61 (<0.01) | −0.43 | U |
| LOCUS III: Dred_0432-3, Fe(III)-CITRATE-INDUCED | |||||
| Dred_0432 | Hypothetical protein (FeS) | Fecit only (1 unique pep) | C | ||
| Dred_0433 | CoB–CoM heterodisulfide reductase (HdrD) | 1.80 (<0.01) | NI | NI | CM |
| LOCUS VI: Dred_1778-84, Fe(III)-CITRATE-INDUCED | |||||
| Dred_1778 | Electron transfer flavoprotein subunit beta (EtfB) | 0.01 | −0.02 | 2.73 (<0.01) | C |
| Dred_1779 | Electron transfer flavoprotein subunit alpha (eEfA) | 0.23 | 0.24 | 3.58 (<0.01) | U |
| Dred_1780 | 3-hydroxybutyryl-CoA dehydrogenase | 1.22 (<0.01) | 0.39 (0.01) | 3.29 (<0.01) | C |
| Dred_1781 | Enoyl-CoA hydratase/isomerase | 2.12 (<0.01) | 2.12 (<0.01) | 4.07 (<0.01) | C |
| Dred_1782 | butyryl-CoA dehydrogenase | 0.62 (0.02) | −0.17 | 1.84 (<0.01) | C |
| Dred_1783 | Hypothetical protein (HdrD) | 0.98 (<0.01) | 0.87 (<0.01) | 1.87 (<0.01) | CM |
| Dred_1784 | Acetyl-CoA acetyltransferase | 0.71 (<0.01) | 0.41 | 3.49 (<0.01) | C |
Log2 proteomic abundance comparisons for proteins discussed from heterodisulfide reductase (hdr)-containing loci.
Protein identification is based on detection of at least two unique peptides in a biological replicate (unless noted otherwise) and peptide detection in at least 50% of replicates. All significant p-values (<0.05) are shown. NI, protein not identified. Localization is from PSORTb (Yu et al., 2010). C, cytoplasmic; CM, cytoplasmic membrane; E, extracellular; CW, cell wall; U, unknown.
A significant increase in abundance is also observed for the predicted operon Dred_0689-90 on the sulfate reduction condition relative to Fe(III) reduction conditions but not relative to pyruvate conditions. Dred_0689 is annotated as a hypothetical protein and Dred_0690 is an HdrD-like protein (Figure 2 and Table 4). Both of these proteins have annotations that suggest involvement in lactate oxidation, described by IMG as L-lactate utilization proteins LutC and LutB respectively, which suggests an involvement in lactate-fed sulfate reduction rather than pyruvate fermentation. Furthermore, the hdr-containing locus is conserved across Desulfotomaculum species, and several species contain an adjacent L-lactate transport protein. The locus is also conserved in species of Desulfosporosinus and Desulfitobacterium (the latter of which reduces sulfite but not sulfate), suggesting that these proteins may be involved with lactate utilization and/or electron transfer to the sulfite reductase (https://img.jgi.doe.gov).
A particularly striking hdr-containing locus is Dred_1325-30 (locus VI in Figure 2), where a significant increase in abundance is observed across the six proteins on sulfate relative to all other cultivation conditions (with p < 0.05; Table 4). For instance, compared with the Fe(III)-citrate condition, these proteins are increased >20-fold on average. The six proteins of this locus are completely conserved only in other species of Desulfotomaculum that are incomplete lactate oxidizers (https://img.jgi.doe.gov). Intriguingly, there is similarity between Dred_1325-30 and the FlxABCD-HdrABC cluster, recently shown in Desulfovibrio vulgaris Hildenborough to be essential for NADH oxidation during sulfate reduction with ethanol as electron donor and suggested to reduce DsrC through FBEB (Ramos et al., 2015). The Hdr components are completely conserved, as are parts of the Flx components. While Dred_1325-30 is missing an ortholog to FlxA (the predicted NADH dehydrogenase), Dred_1327 is a much larger protein than the HdrA from Desulfovibrio and contains multiple partial HdrA domains as well as putative NADH-binding domains (MicrobesOnline, Dehal et al., 2010). This large protein may be performing the NADH-oxidizing function in the cluster, while Dred_1328 (also an HdrA-type protein) reduces ferredoxin (Figure 5A). Therefore, it is possible that this cluster may be performing a similar function in D. reducens, utilizing FBEB in order to improve the energetic favorability of sulfate reduction. Another hdr-containing locus in D. reducens (Dred_0137-48) shows similarity to the Dred_1325-30 cluster and in fact encodes components of Flx missing from Dred_1325-30 (Figure 5B). Dred_0137-48 is missing the HdrB and HdrC components of the cluster, but it is possible that this locus coordinates with Dred_1325-30, oxidizing NADH and working together to reduce ferredoxin and DsrC. The Dred_0137-48 locus is not differentially abundant across conditions in most cases (see further description below), unlike the Dred_1325-30 cluster (Table 4). Additional clusters of interest on the sulfate reduction condition (not hdr- containing) are displayed in Supplementary Table 6.
Figure 5
Proteome of D. reducens during Fe(III) reduction
All previous RNA and/or protein-based analyses of Fe(III) reduction in D. reducens have used soluble Fe(III) due to technical challenges associated with culturing and sample preparation on insoluble Fe(III) (Dalla Vecchia et al., 2014a,b). In this study, we have analyzed the proteomes of D. reducens cultivated on both soluble Fe(III)-citrate and insoluble Fe(III)-oxide, allowing for a valuable comparison of external electron acceptors.
Proteins in heterodisulfide reductase-containing loci are abundant in soluble Fe(III) reduction proteome
Three hdr-containing loci are highlighted in the proteome of D. reducens during soluble Fe(III) reduction. This includes proteins within locus VII (Dred_1777-86) locus III (Dred_0427-33), and locus I (Dred_0137-48) (Figure 2). While the electron transfer proteins (Dred_1778-9) are similarly abundant on Fe(III)-citrate, sulfate, and pyruvate, Dred_1780-4 represents the most increased cluster of three or more proteins on Fe(III)-citrate relative to pyruvate (Table 4). It is increased in abundance ~2.2-fold across the five proteins (all p < 0.02). In addition, while the Fe(III)-citrate condition overall has nearly half of the total unique peptides observed on the sulfate and pyruvate condition, and accordingly tends to have less peptides detected for each protein, the number of unique peptides detected across Dred_1778-84 is significantly higher on Fe(III)-citrate than all other conditions. Averaged across technical triplicates and biological duplicates, the mean number of peptides/proteins detected across the seven proteins is 42.9 on the Fe(III)-citrate condition, 17.2 on pyruvate, 20.8 on sulfate, and 8.0 on Fe(III)-oxide. Five proteins in this cluster (Dred_1782, Dred_1784, and Dred_1778-80) fall into the top 20 proteins across the Fe(III)-citrate proteome with respect to highest unique peptide counts. This result is surprising because these genes are predicted to be involved in butyrate oxidation, which is not the electron donor for any cultures analyzed in this study. It is possible that these proteins are using a different substrate than their annotation suggests, or that these enzymes are acting in the opposite direction, converting acetyl-CoA to butyrl-CoA (Figure 6). In fact, proteins within this cluster are similar to one of the most well-described FBEB systems, the clostridial butyryl-CoA dehydrogenase/electron transferring flavoprotein (BcdA–EtfBC) complex (Li et al., 2008; Buckel and Thauer, 2013). This complex is known to catalyze the electron bifurcation from NADH to ferredoxin and crotonyl-CoA. The proteins Dred_1778-9 (electron transfer flavoproteins) and Dred_1782 (butyryl-CoA dehydrogenase) match closely to this complex, while orthologs to Dred_1780-2 and Dred_1784 catalyze the reactions leading to this FBEB. In this scheme, a protein such as the formate acetyltransferase Dred_0039 (observed on all conditions and most abundant on pyruvate) could be converting pyruvate to acetyl-CoA. Then, following FBEB, reduced ferredoxin would be available for other reductive processes in the cell. A full visual comparing the BcdA-EtfBC complex in Clostridium kluyveri DSM 555 with the D. reducens locus is displayed in Figure 6 (Buckel and Thauer, 2013). D. reducens also contains proteins that are redundant to components of this FBEB system and these are unique to the Fe(III)-citrate condition. Also important to note, the described clostridial FBEB system lacks Hdr-like proteins. The placement of the HdrD-like protein Dred_1783 in this annotated butyrate-oxidizing cluster is unique to D. reducens (https://img.jgi.doe.gov).
Figure 6
In the hdr-containing locus III (Dred_0427-33), peptides for proteins encoded between Dred_0427-31 were not identified in our study. Dred_0432-3, however, is significant on the Fe(III)-citrate condition (Table 4). Dred_0432 was only identified on pyruvate and Fe(III)-citrate and is increased ~3.5-fold on Fe(III)-citrate (p < 0.01). Dred_0433 is completely unique to the Fe(III)-citrate condition. The identification of Dred_0432 on Fe(III)-citrate is based on a single unique peptide in 5/6 replicates. Dred_0433 is localized to the cytoplasmic membrane as predicted by PSORTb (Yu et al., 2010). The predicted operon is from Dred_0431-3, and Dred_0431 is a predicted permease. Dred_0432-3 encompasses the Hdr component of the locus, and interestingly, both proteins have similarity to the L-lactate utilization protein LutB, as described by IMG. As additional evidence toward a role of the proteins in Fe(III) reduction, the closest orthologs to Dred_0432-3 are all from other Gram-positive metal-reducers, specifically Desulfosporosinus and Desulfitobacterium species. In these other genera, the genes are encoded next to five permease/transport proteins (https://img.jgi.doe.gov). Dred_0432-3 is not conserved in other Desulfotomaculum species.
The hdr-containing locus Dred_0137-48 is also of interest. While many proteins within the locus are similar to Dred_1325-30 (Figure 5) our proteomic abundance data reveals that Dred_0137-48 is not strongly differentially abundant across conditions and that a high number of unique peptides are observed on all conditions (Table 4). Interestingly, the Dred_0137-148 locus contains one of three redox proteins predicted as putative Fe(III) reductases in the analysis of the surfaceome of D. reducens (Dalla Vecchia et al., 2014a). This is the ferredoxin Dred_0143, which has an unknown localization based on PSORTb, but contains one transmembrane helix as predicted by the transmembrane helix (TMH) prediction algorithm TMHMM Server v. 2.0 (www.cbs.dtu.dk/services/TMHMM). A duplication is encoded within this locus which is only observed in two other Desulfotomaculum species, and Dred_0137 is identical to Dred_0143. The authors did not observe significant differential abundance of this protein between conditions tested (pyruvate fermentation and Fe(III)-citrate with lactate as electron donor), which is consistent with our findings (Table 4). The high expression of this locus on all conditions (in contrast to the downregulation of Dred_1325-30 on Fe(III) conditions) supports the possible involvement of Dred_0143 in Fe(III) reduction proposed by the surfaceome study (Dalla Vecchia et al., 2014a). The other two proteins identified in the surfaceome study as potential Fe(III) reductases are not supported by our proteomic data. This includes Dred_0462, a subunit of the membrane-bound, trimeric hydrogenase Dred_0461-3 (Supplementary Table 3). Dred_0462 was suggested as a putative Fe(III) reductase, although the authors observed a decrease in its abundance during Fe(III)-reduction relative to pyruvate fermentation. Similarly in our study, both Dred_0462 and Dred_0463 (the catalytic domain) are increased during pyruvate fermentation relative to Fe(III)-reduction and most abundant during sulfate-reduction (with p-values of 0.02 and 0.01 relative to pyruvate; Supplementary Table 3). From these findings, it is most likely that this hydrogenase is utilized during sulfate reduction and/or pyruvate fermentation by D. reducens. Finally, the only redox-related protein that increased in abundance during Fe(III)-citrate reduction vs. pyruvate fermentation in the surfaceome study was the alkyl hydroperoxide reductase Dred_1533. We observed no peptides for this protein on any condition.
Additional proteins of interest on the Fe(III) reduction condition
Dred_0701, annotated as the sole multiheme c-type cytochrome (Dred_0700-1) was identified only on the Fe(III)-oxide condition. A single peptide was identified in four out of six replicates, whereas no peptides were detected from this protein on any other condition. This is the first evidence of metal reduction-related MHC expression in D. reducens. This finding emphasizes the importance of testing environmentally relevant conditions like insoluble Fe(III), as previous analyses of the MHC were performed during soluble Fe(III) and U(VI) reduction. Potentially related to this finding, the protein most increased in abundance on Fe(III)-oxide relative to pyruvate is porphobilinogen deaminase (Dred_2163, HemC), a protein in the heme biosynthesis pathway (~136.1-fold increase, p < 0.01). This protein was identified by a single peptide in each replicate of the Fe(III)-oxide condition. Two other proteins in this pathway are also highest in abundance on the Fe(III)-oxide condition relative to all other conditions (Dred_2160 and Dred_2162, HemB and HemD respectively).
Finally, our comparative proteomic analysis provides support for the involvement of the NADH:flavin oxidoreductase (Dred_2421) in Fe(III) reduction, a protein that our group previously identified as a metal reductase based on functional screens of the D. reducens proteome (Otwell et al., 2015). Multiple unique peptides were observed for this protein only on Fe(III)-oxide, so therefore with stringent filtering criteria Dred_2421 is unique to the Fe(III)-oxide condition (Supplementary Table 1d). Including data for proteins identified by a single unique peptide in at least 50% of the replicates, Dred_2421 is actually most abundant on the Fe(III)-citrate condition, increased ~1.6-fold (p < 0.01) relative to pyruvate and ~2.3-fold (p < 0.01) relative to the sulfate condition. Comparing the single peptide identified on all conditions, Dred_2421 is ~3 fold more abundant on Fe(III)-citrate than Fe(III)-oxide. The closest orthologs to this protein are not from Desulfotomaculum species, but instead from other genera of Gram-positive metal reducers, specifically Desulfitobacterium and Deuslfosporosinus species. Interestingly, in Desulfitobacterium species, this NADH:flavin oxidoreductase is inserted within the hdr-containing locus orthologous to Dred_1778-84 (https://img.jgi.doe.gov).
Conclusions
Comparative proteomic analysis of D. reducens cultivated on varied conditions has revealed multiple insights into the metabolism of this Gram-positive organism. Our proteomic dataset allows us to analyze predictions made by the annotated genome and form stronger hypotheses about protein function. A greater number of proteins were observed while D. reducens was either reducing sulfate (with lactate as electron donor) or fermenting pyruvate compared with Fe(III)-reducing conditions. The soluble and insoluble Fe(III)-reducing proteomes, analyzed for the first time in a Gram-positive organism, were distinctive from one another, a result consistent with findings in the Gram-negative metal-reducing organisms Geobacter sulfurreducens and Geobacter bemidjiensis (Ding et al., 2008; Merkley et al., 2015). Peptides for the sole MHC annotated in the genome (Dred_0700-1) were detected only on the insoluble Fe(III) condition, and an enzyme involved in heme biosynthesis was upregulated >100-fold on Fe(III)-oxide relative to pyruvate. Certain clusters of proteins were significantly differentially abundant across cultivation conditions studied. Several of these clusters include Hdrs, and our study has suggested potential involvement of these hdr-containing loci in metabolic processes in D. reducens including sulfate and Fe(III) reduction.
While comparative expression analyses (mRNA and protein-based) are accepted methods for highlighting genes/proteins of interest, these approaches have limitations. Namely, differential expression is not necessarily directly linked with function (Price et al., 2013). For instance, our analysis of putative sulfate reduction-related proteins in D. reducens revealed that most of these proteins are not differentially abundant on cultivation conditions tested, which is not the result we expected. Nonetheless, comparative proteomic analysis offers a method for hypothesis generation regarding protein function and is especially useful in poorly characterized organisms such as D. reducens.
Funding
This project was funded by the Department of Energy's Office of Biological and Environmental Research within the Office of Science, project number DE-SC0006644. A portion of this research was funded by the Department of Energy's (DOE) Office of Biological and Environmental Research (OBER) Pan-omics program and performed in the Environmental Molecular Sciences Laboratory at Pacific Northwest National Laboratory (PNNL). The Environmental Molecular Sciences Laboratory is a U.S. Department of Energy (DOE) Office of Biological and Environmental Research national scientific user facility on the PNNL campus. PNNL is a multi-program national laboratory operated by Battelle for the DOE under contract DE-AC05-76RL01830.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Statements
Author contributions
AO participated in conception of the study, experimental work, analysis and interpretation of proteomic data, and wrote the first draft of the manuscript. SC participated in conception of the study, global proteomic analysis, and reviewed and edited the manuscript. EZ participated in global proteomic analysis and reviewed and edited the manuscript. RS participated in global proteomic analysis and reviewed and edited the manuscript. RR participated in conception of the study, global proteomic analysis, and reviewed and edited the manuscript.
Acknowledgments
The authors would like to acknowledge Erika Mudrak of the Cornell Statistical Consulting Unit for helpful assistance with statistical analyses.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2016.00191
References
1
AülloT.Ranchou-PeyruseA.OllivierB.MagotM. (2013). Desulfotomaculum spp. and related gram-positive sulfate-reducing bacteria in deep subsurface environments. Front. Microbiol.4:362. 10.3389/fmicb.2013.00362
2
BartonL.Tomei-TorresF.XuH.ZoccoT. (2015). Metabolism of metals and metalloids by the sulfate-reducing bacteria, in Bacteria-Metal Interactions, ed Saffarini ChamD. (Cham: Springer International Publishing), 57–83.
3
BirdL. J.BonnefoyV.NewmanD. K. (2011). Bioenergetic challenges of microbial iron metabolisms. Trends Microbiol.19, 330–340. 10.1016/j.tim.2011.05.001
4
BuckelW.ThauerR. K. (2013). Energy conservation via electron bifurcating ferredoxin reduction and proton/Na(+) translocating ferredoxin oxidation. Biochim. Biophys. Acta1827, 94–113. 10.1016/j.bbabio.2012.07.002
5
CallisterS. J.McCueL. A.TurseJ. E.MonroeM. E.AuberryK. J.SmithR. D.et al. (2008). Comparative bacterial proteomics: analysis of the core genome concept. PLoS ONE3:e1542. 10.1371/journal.pone.0001542
6
CallisterS. J.NicoraC. D.ZengX.RohJ. H.DominguezM. A.TavanoC. L.et al. (2006). Comparison of aerobic and photosynthetic Rhodobacter sphaeroides 2.4.1 proteomes. J. Microbiol. Methods67, 424–436. 10.1016/j.mimet.2006.04.021
7
CardenasE.WuW.-M.LeighM. B.CarleyJ.CarrollS.GentryT.et al. (2010). Significant association between sulfate-reducing bacteria and uranium-reducing microbial communities as revealed by a combined massively parallel sequencing-indicator species approach. Appl. Environ. Microbiol.76, 6778–6786. 10.1128/AEM.01097-10
8
Dalla VecchiaE.ShaoP. P.SuvorovaE.ChiappeD.HamelinR.Bernier-LatmaniR. (2014a). Characterization of the surfaceome of the metal-reducing bacterium Desulfotomaculum reducens. Front. Microbiol.5:432. 10.3389/fmicb.2014.00432
9
Dalla VecchiaE.SuvorovaE. I.MaillardJ.Bernier-LatmaniR. (2014b). Fe(III) reduction during pyruvate fermentation by Desulfotomaculum reducens strain MI-1. Geobiology12, 48–61. 10.1111/gbi.12067
10
DehalP. S.JoachimiakM. P.PriceM. N.BatesJ. T.BaumohlJ. K.ChivianD.et al. (2010). Microbesonline: an integrated portal for comparative and functional genomics. Nucleic Acids Res.38, D396–D400. 10.1093/nar/gkp919
11
DingY.-H. R.HixsonK. K.AklujkarM. A.LiptonM. S.SmithR. D.LovleyD. R.et al. (2008). Proteome of Geobacter sulfurreducens grown with Fe(III) oxide or Fe(III) citrate as the electron acceptor. Biochim. Biophys. Acta1784, 1935–1941. 10.1016/j.bbapap.2008.06.011
12
GreinF.RamosA. R.VenceslauS. S.PereiraI. A. C. (2013). Unifying concepts in anaerobic respiration: insights from dissimilatory sulfur metabolism. Biochim. Biophys. Acta1827, 145–160. 10.1016/j.bbabio.2012.09.001
13
HaouariO.FardeauM.-L.CayolJ.-L.CasiotC.Elbaz-PoulichetF.HamdiM.et al. (2008). Desulfotomaculum hydrothermale sp. nov., a thermophilic sulfate-reducing bacterium isolated from a terrestrial Tunisian hot spring. Int. J. Syst. Evol. Microbiol. 58, 2529–2535. 10.1099/ijs.0.65339-0
14
HerrmannG.JayamaniE.MaiG.BuckelW. (2008). Energy conservation via electron-transferring flavoprotein in anaerobic bacteria. J. Bacteriol.190, 784–791. 10.1128/JB.01422-07
15
HoriT.AoyagiT.ItohH.NarihiroT.OikawaA.SuzukiK.et al. (2015). Isolation of microorganisms involved in reduction of crystalline iron(III) oxides in natural environments. Front. Microbiol.6:386. 10.3389/fmicb.2015.00386
16
JunierP.JunierT.PodellS.SimsD. R.DetterJ. C.LykidisA.et al. (2010). The genome of the Gram-positive metal- and sulfate-reducing bacterium Desulfotomaculum reducens strain MI-1. Environ. Microbiol.12, 2738–2754. 10.1111/j.1462-2920.2010.02242.x
17
JunierP.VecchiaE. D.Bernier-LatmaniR. (2011). The response of Desulfotomaculum reducens MI-1 to U(VI) exposure: a transcriptomic study. Geomicrobiol. J.28, 483–496. 10.1080/01490451.2010.512031
18
KimS.GuptaN.PevznerP. A. (2008). Spectral probabilities and generating functions of tandem mass spectra: a strike against decoy databases. J. Proteome Res.7, 3354–3363. 10.1021/pr8001244
19
KleinM.FriedrichM.RogerA. J.HugenholtzP.FishbainS.AbichtH.et al. (2001). Multiple lateral transfers of dissimilatory sulfite reductase genes between major lineages of sulfate-reducing prokaryotes. J. Bacteriol.183, 6028–6035. 10.1128/JB.183.20.6028-6035.2001
20
KueverJ.VisserM.LoefflerC.BollM.WormP.SousaD. Z.et al. (2014). Genome analysis of Desulfotomaculum gibsoniae strain Groll(T) a highly versatile Gram-positive sulfate-reducing bacterium. Stand. Genomic Sci.9, 821–839. 10.4056/sigs.5209235
21
LiF.HinderbergerJ.SeedorfH.ZhangJ.BuckelW.ThauerR. K. (2008). Coupled ferredoxin and crotonyl coenzyme A (CoA) reduction with NADH catalyzed by the butyryl-CoA dehydrogenase/Etf complex from Clostridium kluyveri. J. Bacteriol.190, 843–850. 10.1128/JB.01417-07
22
LiptonM. S.Paša-TolićL.AndersonG. A.AndersonD. J.AuberryD. L.BattistaJ. R.et al. (2002). Global analysis of the Deinococcus radiodurans proteome by using accurate mass tags. Proc. Natl. Acad. Sci. U.S.A.99, 11049–11054. 10.1073/pnas.172170199
23
LiuY.WangZ.LiuJ.LevarC.EdwardsM. J.BabautaJ. T.et al. (2014). A trans-outer membrane porin-cytochrome protein complex for extracellular electron transfer by Geobacter sulfurreducens PCA. Environ. Microbiol. Rep.6, 776–785. 10.1111/1758-2229.12204
24
LovleyD. R.PhillipsE. J. P. (1987). Rapid assay for microbially reducible ferric iron in aquatic sediments. Appl. Environ. Microbiol.53, 1536–1540.
25
MarkowitzV. M.ChenI.-M. A.PalaniappanK.ChuK.SzetoE.PillayM.et al. (2014). IMG 4 version of the integrated microbial genomes comparative analysis system. Nucleic Acids Res.42, D560–D567. 10.1093/nar/gkt963
26
MerkleyE. D.WrightonK. C.CastelleC. J.AndersonB. J.WilkinsM. J.ShahV.et al. (2015). Changes in protein expression across laboratory and field experiments in Geobacter bemidjiensis. J. Proteome Res.14, 1361–1375. 10.1021/pr500983v
27
NewsomeL.MorrisK.LloydJ. R. (2014). The biogeochemistry and bioremediation of uranium and other priority radionuclides. Chem. Geol.363, 164–184. 10.1016/j.chemgeo.2013.10.034
28
OliverosJ. C. (2007). VENNY. An Interactive Tool for Comparing Lists with Venn Diagrams. Available online at: http://bioinfogp.cnb.csic.es/tools/venny/index.html
29
OtwellA. E.SherwoodR. W.ZhangS.NelsonO. D.LiZ.LinH.et al. (2015). Identification of proteins capable of metal reduction from the proteome of the Gram-positive bacterium Desulfotomaculum reducens MI-1 using an NADH-based activity assay. Environ. Microbiol.17, 1977–1990. 10.1111/1462-2920.12673
30
PereiraI. A. C.RamosA. R.GreinF.MarquesM. C.da SilvaS. M.VenceslauS. S. (2011). A comparative genomic analysis of energy metabolism in sulfate reducing bacteria and archaea. Front. Microbiol.2:69. 10.3389/fmicb.2011.00069
31
PetrieL.NorthN. N.DollhopfS. L.BalkwillD. L.KostkaJ. E. (2003). Enumeration and characterization of iron(III)-reducing microbial communities from acidic subsurface sediments contaminated with uranium(VI). Appl. Environ. Microbiol.69, 7467–7479. 10.1128/AEM.69.12.7467-7479.2003
32
PolpitiyaA. D.QianW.-J.JaitlyN.PetyukV. A.AdkinsJ. N.CampD. G.et al. (2008). DAnTE: a statistical tool for quantitative analysis of -omics data. Bioinformatics24, 1556–1558. 10.1093/bioinformatics/btn217
33
PriceM. N.DeutschbauerA. M.SkerkerJ. M.WetmoreK. M.RuthsT.MarJ. S.et al. (2013). Indirect and suboptimal control of gene expression is widespread in bacteria. Mol. Syst. Biol.9, 660. 10.1038/msb.2013.16
34
R Development Core Team (2012). R: A Language and Environment for Statistical Computing. Vienna, Austria: the R Foundation for Statistical Computing. Available online at: http://www.R-project.org/
35
RamosA. R.GreinF.OliveiraG. P.VenceslauS. S.KellerK. L.WallJ. D.et al. (2015). The FlxABCD-HdrABC proteins correspond to a novel NADH dehydrogenase/heterodisulfide reductase widespread in anaerobic bacteria and involved in ethanol metabolism in Desulfovibrio vulgaris Hildenborough. Environ. Microbiol.17, 2288–2305. 10.1111/1462-2920.12689
36
RichardsonD. J.ButtJ. N.FredricksonJ. K.ZacharaJ. M.ShiL.EdwardsM. J.et al. (2012). The “porin-cytochrome” model for microbe-to-mineral electron transfer. Mol. Microbiol.85, 201–212. 10.1111/j.1365-2958.2012.08088.x
37
RobidartJ.CallisterS. J.SongP.NicoraC. D.WheatC. G.GirguisP. R. (2013). Characterizing microbial community and geochemical dynamics at hydrothermal vents using osmotically driven continuous fluid samplers. Environ. Sci. Technol.47, 4399–4407. 10.1021/es3037302
38
SantosA. A.VenceslauS. S.GreinF.LeavittW. D.DahlC.JohnstonD. T.et al. (2015). A protein trisulfide couples dissimilatory sulfate reduction to energy conservation. Science350, 1541–1545. 10.1126/science.aad3558
39
SharmaS.CavallaroG.RosatoA. (2010). A systematic investigation of multiheme c-type cytochromes in prokaryotes. J. Biol. Inorg. Chem.15, 559–571. 10.1007/s00775-010-0623-4
40
ShiL.RichardsonD. J.WangZ.KerisitS. N.RossoK. M.ZacharaJ. M.et al. (2009). The roles of outer membrane cytochromes of Shewanella and Geobacter in extracellular electron transfer. Environ. Microbiol. Rep.1, 220–227. 10.1111/j.1758-2229.2009.00035.x
41
ShiL.RossoK. M.ClarkeT. A.RichardsonD. J.ZacharaJ. M.FredricksonJ. K. (2012). Molecular underpinnings of Fe(III) oxide reduction by Shewanella Oneidensis MR-1. Front. Microbiol.3:50. 10.3389/fmicb.2012.00050
42
SmithP. K.KrohnR. I.HermansonG. T.MalliaA. K.GartnerF. H.ProvenzanoM. D.et al. (1985). Measurement of protein using bicinchoninic acid. Anal. Biochem.150, 76–85. 10.1016/0003-2697(85)90442-7
43
SowellS. M.NorbeckA. D.LiptonM. S.NicoraC. D.CallisterS. J.SmithR. D.et al. (2008). Proteomic analysis of stationary phase in the marine bacterium “Candidatus Pelagibacter ubique”. Appl. Environ. Microbiol.74, 4091–4100. 10.1128/AEM.00599-08
44
StanleyJ. R.AdkinsJ. N.SlyszG. W.MonroeM. E.PurvineS. O.KarpievitchY. V.et al. (2011). A statistical method for assessing peptide identification confidence in accurate mass and time tag proteomics. Anal. Chem.83, 6135–6140. 10.1021/ac2009806
45
StrocchiA.FurneJ. K.LevittM. D. (1992). A modification of the methylene blue method to measure bacterial sulfide production in feces. J. Microbiol. Methods15, 75–82. 10.1016/0167-7012(92)90071-B
46
SuzukiY.KellyS. D.KemnerK. M.BanfieldJ. F. (2003). Microbial populations stimulated for hexavalent uranium reduction in uranium mine sediment. Appl. Environ. Microbiol.69, 1337–1346. 10.1128/AEM.69.3.1337-1346.2003
47
TeboB. M.ObraztsovaA. Y. (1998). Sulfate-reducing bacterium grows with Cr(VI), U(VI), Mn(IV), and Fe(III) as electron acceptors. FEMS Microbiol. Lett.162, 193–198. 10.1111/j.1574-6968.1998.tb12998.x
48
VenceslauS. S.StockdreherY.DahlC.PereiraI. A. C. (2014). The “bacterial heterodisulfide” DsrC is a key protein in dissimilatory sulfur metabolism. Biochim. Biophys. Acta1837, 1148–1164. 10.1016/j.bbabio.2014.03.007
49
VisserM.ParshinaS. N.AlvesJ. I.SousaD. Z.PereiraI. A. C.MuyzerG.et al. (2014). Genome analyses of the carboxydotrophic sulfate-reducers Desulfotomaculum nigrificans and Desulfotomaculum carboxydivorans and reclassification of Desulfotomaculum carboxydivorans as a later synonym of D. nigrificans. Stand. Genomic Sci.9, 655–675. 10.4056/sigs.4718645
50
WallJ. D.KrumholzL. R. (2006). Uranium reduction. Annu. Rev. Microbiol.60, 149–166. 10.1146/annurev.micro.59.030804.121357
51
WangY.YangF.GritsenkoM. A.WangY.ClaussT.LiuT.et al. (2011). Reversed-phase chromatography with multiple fraction concatenation strategy for proteome profiling of human MCF10A cells. Proteomics11, 2019–2026. 10.1002/pmic.201000722
52
WeghoffM. C.BertschJ.MüllerV. (2015). A novel mode of lactate metabolism in strictly anaerobic bacteria. Environ. Microbiol.17, 670–677. 10.1111/1462-2920.12493
53
WilliamsonA. J.MorrisK.ShawS.ByrneJ. M.BoothmanC.LloydJ. R. (2013). Microbial reduction of Fe(III) under alkaline conditions relevant to geological disposal. Appl. Environ. Microbiol.79, 3320–3326. 10.1128/AEM.03063-12
54
WiśniewskiJ. R.ZougmanA.NagarajN.MannM. (2009). Universal sample preparation method for proteome analysis. Nat. Methods6, 359–362. 10.1038/nmeth.1322
55
YuN. Y.WagnerJ. R.LairdM. R.MelliG.ReyS.LoR.et al. (2010). PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics26, 1608–1615. 10.1093/bioinformatics/btq249
56
ZverlovV.KleinM.LückerS.FriedrichM. W.KellermannJ.StahlD. A.et al. (2005). Lateral gene transfer of dissimilatory (bi)sulfite reductase revisited. J. Bacteriol.187, 2203–2208. 10.1128/JB.187.6.2203-2208.2005
Summary
Keywords
Fe(III) reduction, sulfate reduction, Desulfotomaculum reducens, gram-positive bacteria, comparative proteomic analysis, flavin-based electron bifurcation, heterodisulfide reductase
Citation
Otwell AE, Callister SJ, Zink EM, Smith RD and Richardson RE (2016) Comparative Proteomic Analysis of Desulfotomaculum reducens MI-1: Insights into the Metabolic Versatility of a Gram-Positive Sulfate- and Metal-Reducing Bacterium. Front. Microbiol. 7:191. doi: 10.3389/fmicb.2016.00191
Received
26 December 2015
Accepted
03 February 2016
Published
19 February 2016
Volume
7 - 2016
Edited by
Inês A. Cardoso Pereira, Instituto de Tecnologia Química e Biológica and Universidade Nova de Lisboa, Portugal
Reviewed by
Julien Maillard, École Polytechnique Fédérale de Lausanne, Switzerland; Morgan N. Price, Lawrence Berkeley National Laboratory, USA
Updates
Copyright
© 2016 Otwell, Callister, Zink, Smith and Richardson.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anne E. Otwell annie.otwell@gmail.com
This article was submitted to Microbial Physiology and Metabolism, a section of the journal Frontiers in Microbiology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.