 Sou Miyake
Sou Miyake David K. Ngugi
David K. Ngugi Ulrich Stingl
Ulrich Stingl- Marine Microbial Ecology, Red Sea Research Center, King Abdullah University of Science and Technology, Thuwal, Saudi Arabia
Epulopiscium is a group of giant bacteria found in high abundance in intestinal tracts of herbivorous surgeonfish. Despite their peculiarly large cell size (can be up to 600 μm), extreme polyploidy (some with over 100,000 genome copies per cell) and viviparity (whereby mother cells produce live offspring), details about their diversity, distribution or their role in the host gut are lacking. Previous studies have highlighted the existence of morphologically distinct Epulopiscium cell types (defined as morphotypes A to J) in some surgeonfish genera, but the corresponding genetic diversity and distribution among other surgeonfishes remain mostly unknown. Therefore, we investigated the phylogenetic diversity of Epulopiscium, distribution and co-occurrence in multiple hosts. Here, we identified eleven new phylogenetic clades, six of which were also morphologically characterized. Three of these novel clades were phylogenetically and morphologically similar to cigar-shaped type A1 cells, found in a wide range of surgeonfishes including Acanthurus nigrofuscus, while three were similar to smaller, rod-shaped type E that has not been phylogenetically classified thus far. Our results also confirmed that biogeography appears to have relatively little influence on Epulopiscium diversity, as clades found in the Great Barrier Reef and Hawaii were also recovered from the Red Sea. Although multiple symbiont clades inhabited a given species of host surgeonfish and multiple host species possessed a given symbiont clade, statistical analysis of host and symbiont phylogenies indicated significant cophylogeny, which in turn suggests co-evolutionary relationships. A cluster analysis of Epulopiscium sequences from previously published amplicon sequencing dataset revealed a similar pattern, where specific clades were consistently found in high abundance amongst closely related surgeonfishes. Differences in abundance may indicate specialization of clades to certain gut environments reflected by inferred differences in the host diets. Overall, our analysis identified a large phylogenetic diversity of Epulopiscium (up to 10% sequence divergence of 16S rRNA genes), which lets us hypothesize that there are multiple species that are spread across guts of different host species.
Introduction
Epulopiscium is an unusual group of enteric symbionts found in high abundance amongst herbivorous surgeonfishes (family: Acanthuridae; Fishelson et al., 1985; Montgomery and Pollak, 1988a; Clements et al., 1989). They were initially identified as protists due to their extremely large cell size (up to ∼600 μm; Fishelson et al., 1985), but close examination of their ultrastructural features suggested them to be prokaryotic (Clements and Bullivant, 1991). Their status as bacteria was then confirmed by 16S rRNA gene analysis, which indicated Clostridium species as their closest relatives (Angert et al., 1993), and more specifically the lineage encompassing the cluster XIVb Clostridia (Collins et al., 1994). In the absence of isolates, various culture-independent studies shed light on their highly unusual characteristics, such as large cell size, extreme polyploidy (up to 50,000–120,000 copies of single-copy marker genes in mature cells; Mendell et al., 2008), and for some of them, a viviparous reproduction cycle, where live offspring emerge from mother cells (Fishelson et al., 1985; Angert and Clements, 2004; Ward et al., 2009).
Earlier study from the Great Barrier Reef (GBR) noted a high morphological diversity of Epulopiscium-like giant bacteria in certain surgeonfishes, and categorized these into 10 ‘morphotypes’ according to their shape, size and reproductive strategy (Clements et al., 1989; summarized in Table 1). Briefly, type A were described as largest cigar-shaped cells that can reach up to ∼600 μm and form internal daughter cells; type B cells were more rounded in shape (described as oval-shape) and smaller (<230 μm); types C and D cells were similar in shape to type A, but smaller (40–80 and 8–40 μm, respectively); type E cells were rod-shaped; type F cells were small cigar-shaped cells that seem to reproduce exclusively through multiple phase-bright internal structures (up to 7); type G cells seem to reproduce exclusively by binary fission; type H were heterogeneous cells that contain two distinct internal structures; type I cells were defined by mode of reproduction (binary fission) and the presence of elongate internal structures; and type J were elongate/string-like cells. Note that the phase-bright internal structures described by Clements et al. (1989) were later demonstrated to be endospores by Flint et al. (2005).
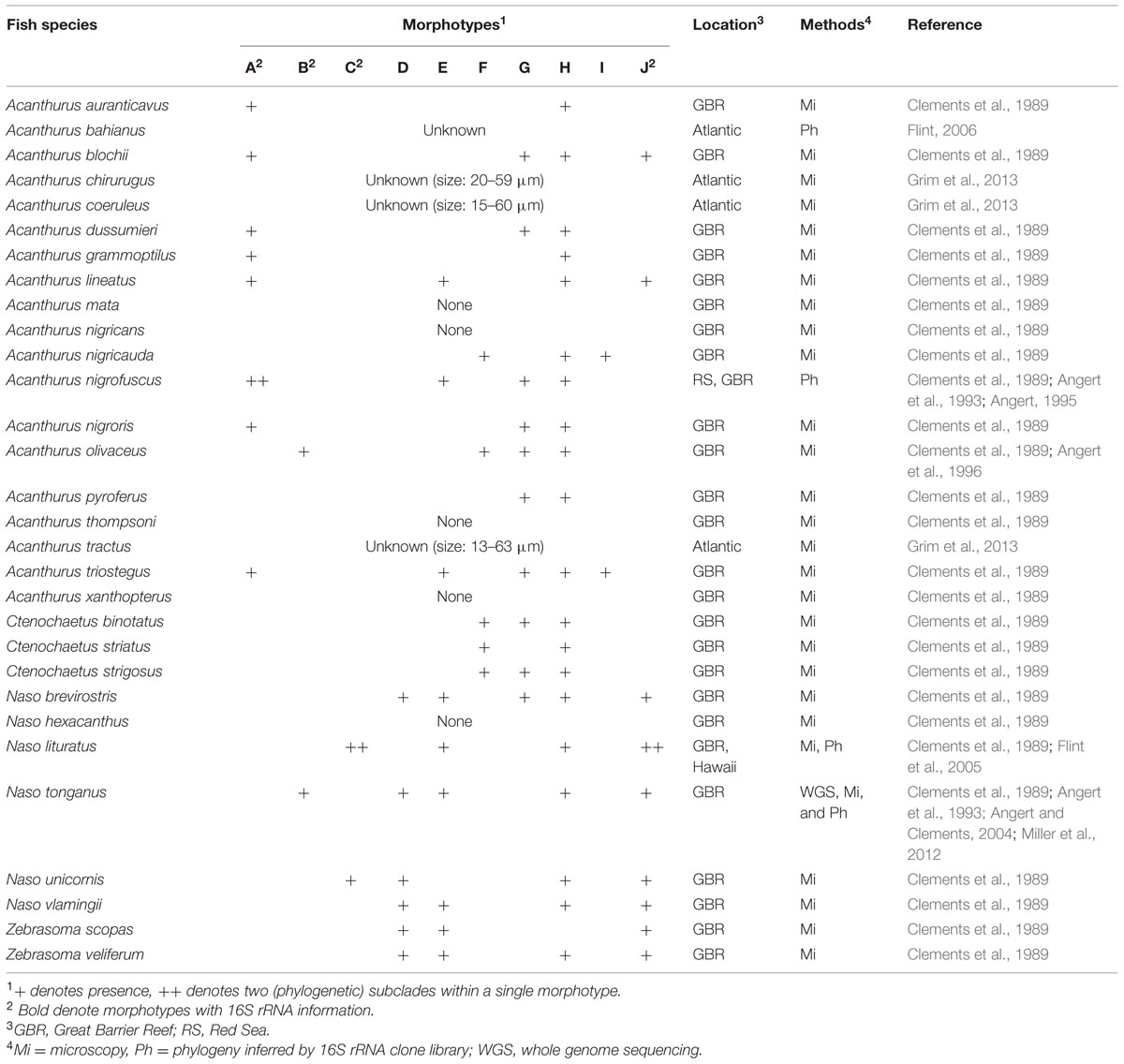
TABLE 1. Previously described diversity of Epulopiscium.
However, only four of these morphotypes (A, B, C, and J) have been phylogenetically classified at the 16S rRNA gene level (Angert et al., 1993; Flint et al., 2005), of which three were further split into subclades (A1, A2, C1, C2, J1, and J2) due to large sequence divergences. The large cigar-shaped type A (70–417 μm) had the greatest sequence divergence (>9%), and was separated into two completely different phylogenetic groups (A1 and A2; Angert et al., 1993). Yet, both type A1 and A2 cells found in Acanthurus nigrofuscus from geographically disparate locations (the Red Sea and the Pacific) showed a very high 16S rRNA gene sequence similarity (>98%; Angert et al., 1993). Whilst much less divergent, type C — smaller, oval/cigar-shaped and phylogenetically closer to A2 — and type J, the elongate and string-like cells (10–240 μm), were further split into C1, C2, J1, and J2, respectively.
In terms of the distribution, Epulopiscium have only been directly observed in herbivorous and detritivorous surgeonfish species from the GBR (Table 1; Clements et al., 1989), and this was partially confirmed by the high relative proportions of Epulopiscium-like reads (up to 99% in Naso elegans) in these fishes in a recent 16S rRNA gene-based pyrosequencing survey conducted in the Red Sea (Miyake et al., 2015). However, some sequences were also recovered from non-herbivorous surgeonfish as well as non-surgeonfishes, albeit at extremely low abundances (<3%); these rare occurrences have been postulated to represent allochthonous populations. Importantly, Clements et al. (1989) also reported association of specific Epulopiscium morphotypes with certain host species, in particular related to the host feeding categories. For instance, the type A cells were found exclusively in herbivorous surgeonfishes grazing on hard substrata and mixed substrata, while type F cells were only found in sand-grazing and detritivorous species.
Additionally, host–symbiont cophylogenetic relationships that pertain to coevolution between surgeonfishes and Epulopiscium remain unexplored. The life cycle of Epulopiscium has been reported to be diurnal and strongly linked to the daily activity of the host fish (Angert and Clements, 2004; Flint et al., 2005), suggesting that the relationship is highly intimate that may support a coevolutionary relationship. To date, direct studies on host and enteric symbiont phylogenies are primarily from plant or invertebrate hosts (Dale and Moran, 2006; Moran et al., 2008). For instance, Noda et al. (2007) found strict phylogenetic associations between termites (Rhinotermitidae), cellulolytic gut symbiont Pseudotrichonympha (protists) and intracellular bacterial symbionts of the protists. Similarly, a strong connection between host nematodes and the developmental stages of their gut symbionts, Xenorhabdus sp. and Photorhabdus sp. (Forst and Nealson, 1966), was reflected by complete congruence in their phylogenies (Maneesakorn et al., 2011). Other examples that are suggestive of coevolutionary relationship between the host and specific gut symbionts have also been reported (e.g., Russell et al., 2009 that explored the evolution of gut symbionts in herbivorous ants). The surgeonfish-Epulopiscium symbiotic relationship is likely to be more complex, as multiple symbiont morphotypes were found among multiple host species (Clements et al., 1989).
Thus far, little is understood about the diversity and abundance of Epulopiscium in surgeonfishes at the molecular level. In this study we therefore conducted 16S rRNA gene surveys of Epulopiscium in several unrelated surgeonfishes from the Red Sea to assess their phylogenetic diversity and distribution among the different hosts. These datasets allowed us to address the following three questions: (1) how extensive is the phylogenetic diversity and the abundance of Epulopiscium in different surgeonfishes from the same locale? (2) Do the different Epulopiscium morphotypes represent coherent phylogenetic clusters? Lastly, (3) do specific phylogenetic clusters preferentially associate with certain host fishes (cophylogeny), suggesting their coevolution?
Materials and Methods
Sample Collection and DNA Extraction
Fish specimens were collected from Al Fahal reef (E38.57935, N22.18344) on the Saudi Arabian coast of the central Red Sea. This research was carried out under the general auspices of King Abdullah University of Science and Technology’s (KAUST) arrangements for marine research with the Saudi Arabian Coast Guard and the Saudi Arabian Presidency of Meteorology and Environment. These are the relevant Saudi Arabian authorities governing all sea-going research actions in the Saudi marine environment. KAUST has negotiated a general and broad permission for marine research in Saudi Arabian Red Sea waters with these two agencies and thus there is no permit number to provide. The study was approved by the Internal Bioethic Committee (IBEC) at KAUST under the permit 15IBEC31_Stingl. All fishes were collected within an hour (8–9 am) to minimize time-dependent effects, and were immediately placed on ice for dissection in the laboratory later on the same day. Three replicates per species of surgeonfishes: A. nigrofuscus, Acanthurus sohal, Acanthurus gahhm, Ctenochaetus striatus, N. elegans, Naso unicornis, Naso hexacanthus, Zebrasoma desjardinii and Zebrasoma xanthurum, as well as two parrotfish species, Chlorurus sordidus and Scarus niger, and a rabbitfish species, Siganus stellatus, were captured for the analysis. From the same coordinate, coral pieces were collected in a sterile bag, which was later subject to air blowing in the laboratory to collect mucus layer and biofilms, to test for presence of Epulopiscium-like bacteria outside the host using Epulopiscium-specific PCR primer designed in this study as described below. Once the fishes were transported back to the laboratory, the processing of the fin and gut samples (including DNA extractions) were performed as previously described in Miyake et al. (2015), with the exception that aliquots of all gut samples were also fixed in 4% formaldehyde for microscopy.
The dietary information of the host fishes is provided in Supplementary Table S1. The fishes were categorized into: (i) macroscopic brown algae feeders with high SCFA profiles in the hindgut region; (ii) turfing and filamentous red and green algae feeders with moderate SCFA; (iii) zooplankton feeders with moderate SCFA; (iv) detritus and sedimentary material feeders (detritivores) with low levels of SCFA; and (v) omnivores that feed on both detritus and planktons that seem to show individual differences. Most of the in-depth dietary analysis was conducted in the Indo-Pacific or western Indian Ocean, but was confirmed for the fishes in this study by a brief analysis of stomach contents (Supplementary Table S2). A. nigrofuscus was classified as turfing and filamentous red and green algae feeders, although epiphytic diatoms are also consumed seasonally in the Red Sea (Fishelson et al., 1987; Montgomery et al., 1989). A. sohal was also classified as turfing and filamentous red and green algae feeders as reported by Vine (1974) and Alwany et al. (2005). Z. xanthurum has also been reported to use similar dietary sources to A. nigrofuscus and thus classified as turfing and filamentous red and green algae feeder (Montgomery et al., 1989). In contrast, much less work has been conducted on Z. desjardinii, but the few papers available confirmed them to share the same diet as Z. xanthurum (Alwany, 2008; Cernohorsky et al., 2015), which was corroborated by stomach content analysis (Supplementary Table S1). Ct. striatus was classified as detritivore (e.g., Purcell and Bellwood, 1993; Choat et al., 2002, 2004; Crossman et al., 2005; Marshell and Mumby, 2012) although as with A. nigrofuscus, seasonal increase in epiphytic diatoms have been reported (Montgomery and Galzin, 1993). N. elegans and N. unicornis have been reported to be macroscopic brown algae feeders (Choat et al., 2002, 2004; Crossman et al., 2005; Pinault et al., 2013; Cernohorsky et al., 2015), while N. hexacanthus has been reported to be zooplankton feeder (e.g., Choat et al., 2002). Lastly, A. gahhm has often been referred to as detrital grazer (much like Ct. striatus; Afeworki et al., 2013), but was classified as omnivore here as a few have been observed to feed presumably on plankton in the water column (Miyake, personal observations; Choat, personal communications) — much like its sister species A. xanthopterus. This has also been confirmed by brief gut content analysis in this study (Supplementary Tables S1 and S2) and in Miyake et al. (2015).
Host Phylogeny: PCR, DNA Sequencing, and Phylogenetic Analysis
Fish fin DNA was amplified by polymerase-chain reaction (PCR) using four genetic markers: cytochrome c oxidase (CO1), mitochondrial cytochrome b (cytb), 16S (mt16S), and nuclear intron (ETS2) as described by Klanten et al. (2004). Supplementary Table S3 provides details on primers and PCR conditions. Sequencing, downstream analysis and phylogenetic reconstructions were performed according to Miyake et al. (2015), with the exception that the four genetic markers were separately aligned and concatenated to four-gene-per-sequence alignments as recommended by Gadagkar et al. (2005).
Epulopiscium: PCR, Cloning, DNA Sequencing, and Phylogenetic Analysis
Initially, full-length 16S rRNA gene sequences of gut bacterial DNA were PCR-amplified, cloned, and sequenced as described by Ngugi and Stingl (2012) using the universal bacterial primer (EUB, 27F/1492R) on gut bacterial DNA from the sampled fishes (Supplementary Table S3). The cleaned-up PCR products were bi-directionally sequenced (using the original PCR primers) on an Applied Biosystems 3730× l DNA Analyzer at the Bioscience Core Lab of King Abdullah University of Science and Technology (KAUST). Raw sequences were trimmed using SEQUENCHER 4.9 (Gene Codes Corporation) and initially aligned based on SILVA SSUREF database (ver. 119) in MOTHUR, and chimeric (using the UCHIME algorithm; Edgar et al., 2011) as well as identical sequences were removed. The processed sequences were re-aligned and classified online by SINA (ver. 1.2.9) using the SILVA SEED database (Pruesse et al., 2012). From these, sequences classified as Epulopiscium were then imported into ARB (ver. 5.1-org-6213; Ludwig et al., 2004) for phylogenetic analysis.
These full-length Epulopiscium sequences were then used to design Epulopiscium-specific PCR primers in ARB. The designed primer (579uF/1232R) targeted all known Epulopiscium sequences (both from this study and from the SILVA database) as well as Metabacterium polyspora, spanning the hypervariable regions V4-V7. In silico PCR of the primer on SILVA database online (Klindworth et al., 2013) showed 14 hits with no mismatch, and 6 hits with 1 mismatch. Most of these were Epulopiscium, but also included a few from closely related Cellulosilyticum sequences. The primer did not amplify with Escherichia coli DNA, but consistently amplified with the gut DNA that contained Epulopiscium in the earlier 16S rRNA clone libraries. The primer was used for PCR, cloning and sequencing of the Epulopiscium fraction of the gut microbiota, as described above (Supplementary Table S3). The downstream analysis was conducted as described for the full-length sequences above, except that redundant sequences were removed by UCLUST (Edgar, 2010) with 99% criterion and imported to ARB for phylogenetic analysis.
Epulopiscium phylogenetic analysis was conducted as recommended by Peplies et al. (2008), whereby full-length 16S rRNA gene sequences were used to form the backbone of the phylogenetic tree, and populated by shorter 16S rRNA gene sequences derived from Epulopiscium group-specific PCR primer designed in this study. Specifically, the maximum likelihood (ML) tree of full-length 16S full-length, together with reference sequences of Epulopiscium and closely related M. polyspora from the SILVA database (SSU Ref NR 119), were constructed with 1,000 bootstrap replicates. The topology of the tree was confirmed by neighbor joining (NJ, with 1,000 replicates) as well as Bayesian inference methods [BI, in Geneious v8.0.4 (http://www.geneious.com/)]. The Epulopiscium partial sequences (derived using the 579uF/1232R primer set) were then added to the full-length 16S phylogenetic tree using the parsimony function in ARB, without allowing change in the overall tree topology, for more robust phylogeny.
The distinct nodes within the tree that showed sequence diversity of ∼3% were clustered together as ‘clades.’ Sequences that did not affiliate with previously described phylotypes (A1, A2, B, C, J1, and J2) were designated as clades RS01-RS11, where RS stands for the Red Sea. Furthermore, the species delimitation plugin (ver.1.03) in Geneious was used to reaffirm different phylogenetic groups (Masters et al., 2011). The tool utilizes bootstrapping/Bayesian posterior probability and Rosenberg’s probability of reciprocal monophyly (PAB; Rosenberg, 2007) to assess if different phylogenetic groups are statistical supported. PAB is the probability that species A, represented by a sequences in a clade of a + b sequences, will be reciprocally monophyletic with the remaining b sequences under the null model of random coalescence. Different clades were then tested to observe similarity and difference in their morphology as described below.
Fluorescence Microscopy Using DAPI and FISH
A combination of 4′,6-diamidino-2-phenylindole (DAPI) stain and an Epulopiscium-specific fluorescent in situ hybridization (FISH) probe 1432R, designed by Angert et al. (1993) was used to assess the morphological diversity of Epulopiscium and to identify the predominant morphotypes in different surgeonfishes. All samples were prepared as described in Angert et al. (1993). Bright-field and fluorescence microscopy were performed using a Zeiss LSM 710 upright confocal microscope. The lengths of DAPI-stained Epulopiscium cells in each sample were measured at 10 randomly selected spots on the slide, and differences in cell size between hosts were assessed by Analysis of Variance (ANOVA) and Tukey’s HSD post hoc tests. The cell counts recorded ranged from 24 cells in Ct. striatus to 275 in A. sohal.
Furthermore, clade-specific FISH probes were designed and used to investigate the morphologies of the newly discovered clades in A. nigrofuscus, A. sohal, and Z. desjardinii. The FISH probes were designed for RS01-RS08, while A2 and B probes were also re-designed to capture all the sequences from each clade. Probes could not be designed for clades RS09 and RS10 due to the relatively unstable nature of their phylogenetic branches and the lack of suitable target matches in their 16S rRNA hypervariable regions. The design, preparation, optimization, and evaluation of the probes, as well as FISH were performed as described by Daims et al. (2005). Briefly, we constructed probes complementary to 16S rRNA sequences that are 15–25 nucleotides in length using the ‘design probe’ function in ARB. All probes captured 100% of the sequences in their targeted group without any mismatches, but at least 1 or 2 mismatches against non-target sequences, depending on the position of the mismatch. For probes that were used together in the same FISH analysis, we ensured that the target location did not overlap (as indicated by E. coli position; Supplementary Table S3). Because of the large size of Epulopiscium cells, it was easy to distinguish potentially unspecific hybridization of the probe to non-Epulopiscium organisms under the microscope. For each probe, different concentrations of formamide were used to obtain maximum hybridization stringency. Additionally, for probes with non-target Epulopiscium sequences containing 1 or 2 mismatches, unlabeled competitor probes that matched 100% to these non-targets were used to prevent unspecific hybridization of non-targets (as indicated in Supplementary Table S3). Pure E. coli or Bacillus culture did not hybridize to any of these probes, nor did they hybridize to any giant Epulopiscium-like cells of different morphology. All probes were labeled with fluorescein (Fluo) or cyanine 3 (Cy3) fluorescent dyes at the 5′ end, with both Fluo and Cy3 used for each probe. The optimum FISH protocol as well as further information on hybridisation protocols is described in Supplementary Table S3. Probes for RS04 and RS07 were disregarded from the study as no hybridization was observed, although there remains a possibility that these clades were not present in the prepared samples. For FISH positive clades, cell size was recorded and statistically assessed for significant differences between clades using ANOVA and Tukey’s honest significant difference (HSD) tests.
Abundance and Distribution Analysis
To robustly assess the relative abundance and distribution of different Epulopiscium clades in surgeonfishes, we re-analyzed pyrosequencing data recently published by Miyake et al. (2015), specifically focusing on the Epulopiscium-related reads in these datasets. Briefly, the reads from surgeonfishes were aligned and classified based on the SILVA SSURef database (ver. 119), and those identified as Epulopiscium were selected (at 80% confidence threshold). These Epulopiscium sequences were then reclassified to the different Epulopiscium clades identified by full-length 16S rRNA gene phylogeny in this study (see above), using the naïve Bayesian classification method at 80% confidence cut-off (Wang et al., 2007), as implemented in MOTHUR. Sequences that could not be confidently assigned to the clades were labeled as unclassified. The sequence assignment to clades was further validated using Pplacer, which locates reads onto a reference phylogeny by maximizing the phylogenetic likelihood or the posterior probability (Matsen et al., 2010). Clade-specific abundances (for each sample) relative to the total bacterial pyrosequence reads per sample and the clustering of related samples were determined using R (ver. 3.1.3) based on a Bray–Curtis dissimilarity matrix. The resulting matrix was then subjected to hierarchical clustering by complete linkage method and visualized by UPGMA dendogram. The statistical R package, Adonis (McArdle and Anderson, 2001), was then used to test for significant differences in Epulopiscium clade abundance between host genera, species and diet (as categorized above) at 1,000 permutations.
Cophylogeny
Furthermore, we investigated the cophylogenetic relationship between surgeonfishes and Epulopiscium. While various methods for studying host–symbiont cophylogeny are well-documented (Stevens, 2004), conventional cost-based methods were impractical for this study because of the complexity of the surgeonfish-Epulopiscium relationship, where multiple symbionts were found amongst multiple hosts, making the cost calculation computationally intensive. Importantly, cost-based methods require some understanding on the host–symbiont relationship for deriving assumptions of relative costs of different evolutionary events (e.g., co-speciation, host switching, duplication, and lineage sorting), which are still lacking for this system.
For our purpose, we elected to use two independent global fit models, specifically the Procrustean Approach to Cophylogeny (PACo; Balbuena et al., 2013) and AxParaFit (Legendre et al., 2002; Stamatakis et al., 2007), to statistically assess the host–symbiont cophylogeny. PACo places the host and symbiont phylogenies into separate distance matrices, which are then transformed by principle coordinates. These are then superimposed by Procrustes method using the information on the host–symbiont link. The analysis yields a global goodness-of-fit statistic, whose significance can be established by a randomization procedure. Similarly, AxParaFit is essentially a principle component analysis that uses permutations to calculate the probability of obtaining a null hypothesis. The null hypothesis can be rejected for any interaction with a random tip mapping of 500 iterations. The output of both models indicates the significance of the overall host–symbiont relationship and of interactions between individual links that contribute to the overall pattern. PACo was run in R-project (ver. 3.1.1) using the script provided by Balbuena et al. (2013), and AxParaFit was run through CopyCat GUI (Meier-Kolthoff et al., 2007), both with 105 permutations.
Data Accessibility
Sequences of fish fin genetic markers (CO1, cytb, mt16S, and ETS2) have been submitted to GenBank under accession numbers KT953164 – KT953199 (cytb), KT952598 – KT952633 (mt16S), KT953200 – KT953235 (ETS2). Sequences for CO1 were not submitted due to being identical to previously submitted sequences (KJ658899 – KJ658957). Non-redundant Epulopiscium 16S rRNA sequences have been submitted under accession numbers KT952527 – KT952597 for ‘full-length’ and KT952879 – KT953163 for ‘partial’ sequences. Although not used in this study, the representative 16S rRNA sequences of the non-Epulopiscium enteric bacteria have also been submitted to GenBank under accession numbers KT952634 – KT952878.
Results
Surgeonfish Phylogeny and Diet
Nine surgeonfish (A. gahhm, A. nigrofuscus, A. sohal, Ct. striatus, N. elegans, N. hexacanthus, N. unicornis, Z. desjardinii, and Z. xanthurum) and three non-surgeonfish species (Ch. sordidus, Sc. niger, and Si. stellatus) totaling 36 individuals were collected from the central Red Sea (Supplementary Table S2). We used four marker genes, CO1, CytB, ETS2, and mt16S (aligned to 658, 800, 547, and 599 bases, respectively) to accurately resolve the phylogeny of these fishes. The phylogeny constructed from concatenating these genetic markers was consistent across NJ, ML and BI methods with high bootstrap support (>80%; Figure 1). The overall topology was consistent with previous morphological (Winterbottom and Mclennan, 1993) and phylogenetic studies (Klanten et al., 2004; Sorenson et al., 2013). In contrast to our previous work that only used CO1 gene, A. gahhm and Ct. striatus clustered together within the Acanthurus clade because CO1 is relatively conserved amongst surgeonfishes (Miyake et al., 2015).
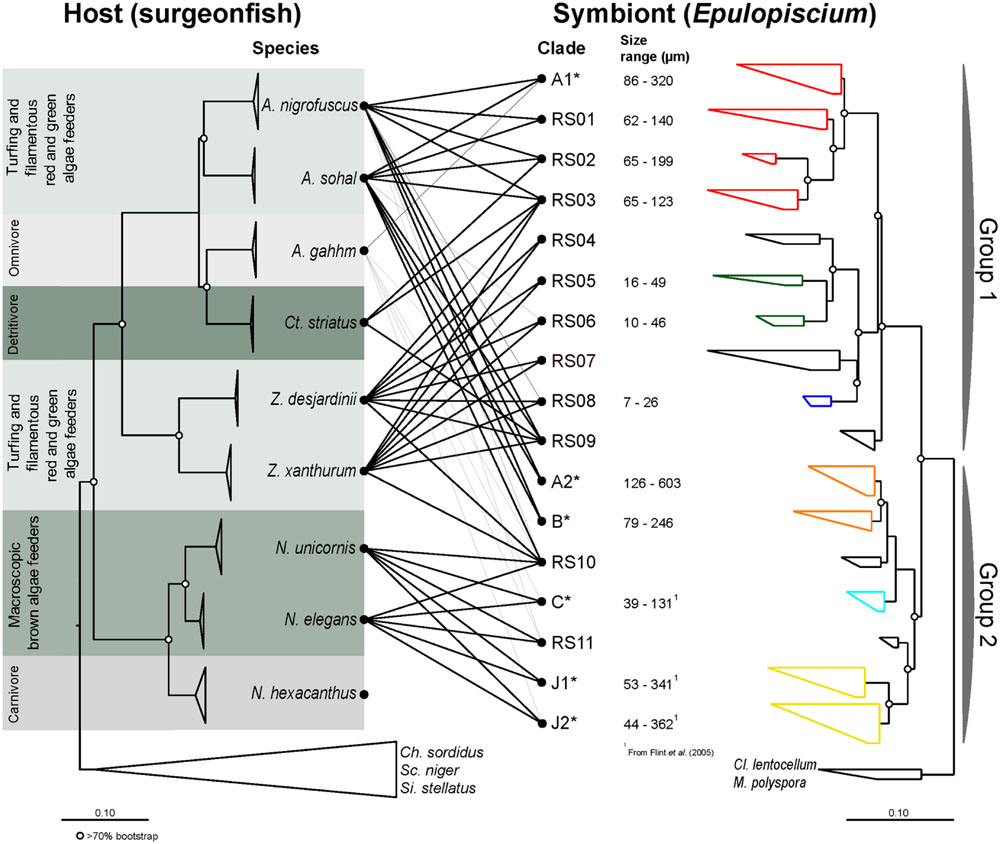
FIGURE 1. Maximum-likelihood (ML) phylogenies of surgeonfishes (left) from the central Red Sea and their putative Epulopiscium-like symbionts (right) based on trees constructed using multiple marker genes (CO1, CytB, ETS2, and mt16S) and the 16S rRNA gene respectively. The diet category is as according to Choat et al. (2002, 2004). Lines in the middle represent host–symbiont associations observed, with black lines based on sequences derived from clone libraries (this study) or gray lines based on pyrosequencing data (Miyake et al., 2015). Epulopiscium clades that have previously been morphologically characterized elsewhere (in Hawaii or the Great Barrier Reef) are starred (∗). Open circles denote branches with bootstrap support values of >70%. The color on the symbiont tree indicates their known morphology: (1) red denotes cigar-shaped clades; (2) green and blue denote short-elongate clades; (3) orange denotes oval/cigar-shaped clades; (4) light blue denotes small cigar-shaped clades; and (5) yellow denotes long-elongate clades.
The stomach content analysis (Supplementary Table S1) was consistent with previous studies, and confirmed that: N. elegans and N. unicornis are macroscopic brown algae feeders, A. nigrofuscus, A. sohal, Z. desjardinii, and Z. xanthurum are turfing and filamentous red and green algae feeders, N. hexacanthus is a zooplankton feeder, Ct. striatus as well as three non-surgeonfishes are detritus and sedimentary material feeders, and A. gahmm is an omnivore (Supplementary Table S1). For turfing and filamentous red and green algae feeders, Acanthurus species possessed higher proportion of red algae, while Zebrasoma species possessed more green algae. Our own brief analysis of the stomach for this study was broadly consistent with their results, except that A. nigrofuscus gut contained small amount of sediments (Supplementary Table S2). Unfortunately, one individual each from A. gahhm, A. nigrofuscus, N. hexacanthus, and Sc. niger had empty stomachs.
Morphological Diversity Identified by DAPI and FISH Staining
We used DAPI and FISH [probe 1432R, designed by Angert et al. (1993)] to evaluate the cell size distribution and identify dominant morphotypes of Epulopiscium in different species of surgeonfish. We observed no giant Epulopiscium-like cells (typically identified as cigar-shaped) or FISH signals in any of the three replicates of A. gahhm or N. hexacanthus, and from only one of Ct. striatus replicates. For other surgeonfishes, the recorded cell sizes and shapes indicated that phylogenetically closely related surgeonfish species were predominated by morphologically similar Epulopiscium cells as previously addressed by Clements et al. (1989; Supplementary Figure S1). These cell sizes and shapes were consistent among replicates, although the relative abundances differed. In general, Epulopiscium cells in A. nigrofuscus and A. sohal were significantly larger than those in other surgeonfishes (Tukey’s HSD test on ANOVA, P < 0.001; Supplementary Figure S1B). Most of them were cigar-shaped and resembled morphotype A, averaging 112 μm in A. nigrofuscus and 189 μm in A. sohal, respectively. In both fishes, giant cells showed a bimodal distribution, where two distinct size clusters of cigar-shaped cells (approximately >150 μm and <150 μm) were distinguishable.
In contrast, the guts of Z. desjardinii and Z. xanthurum had predominantly rod-shaped Epulopiscium cells (average size 28 and 31 μm, respectively), similar to type E cells found in Zebrasoma scopas (Clements et al., 1989), while the two herbivorous Naso species (N. elegans and N. unicornis) were dominated by small cigar-shaped (51 μm, resembling type C) and elongate/string-like cells (57 μm, resembling type J; Supplementary Figure S1A). In both Zebrasoma and Naso species described here, as well as in Ct. striatus, few small cigar-shaped cells (>25 μm) were also observed.
Epulopiscium Phylogenetic Diversity in the Red Sea
Consistent with the microscopy study above, no Epulopiscium-like sequences were recovered from clone libraries of A. gahhm, N. hexacanthus, or non-surgeonfishes (Ch. sordidus, Sc. niger, and Si. stellatus), while they were only recovered from Epulopiscium-specific clone libraries in Ct. striatus. For more comprehensive taxonomic composition of gut microbiota from different fishes refer to Miyake et al. (2015). The extensive phylogenetic tree of full-length representative sequences (at 99% sequence identity cut-off) is shown in Supplementary Figure S2.
Eighty two full-length (1,434 aligned positions) and 285 partial (680 aligned positions) non-redundant Epulopiscium-like 16S rRNA gene sequences were used to construct the phylogenetic trees (Figures 1 and 2; also Supplementary Table S4). Overall, the phylogenetic analysis indicated that Epulopiscium in surgeonfishes from the central Red Sea were phylogenetically diverse and divergent, with pairwise sequence difference of >10%. Two phylogenetically discrete clusters of Epulopiscium sequences – Group 1 and Group 2, were observed in our phylogenetic analyses, which was statistically supported by Rosenberg’s PAB test for reciprocal monophyly (PAB = 2.5 × 10-25) and by high bootstrap values (>70%) for the branch nodes (Figure 2). The two groups divided further into 17 distinct monophyletic clades, which had nodes with >70% bootstrap support and a pairwise sequence divergence of ∼3% (Supplementary Table S5). The exceptions were clades RS05/06, RS10, and C, which had bootstrap support <70%, suggesting that the underlying sequences are either highly heterogeneous or are under sampled. Clades previously only identified from other geographically distinct locations (the GBR and Hawaii) were also recovered here (clades A1, A2, B, C, J1, and J2; Supplementary Figure S2). However, in clades where previously published Red Sea sequences were present (A1 and A2; Angert et al., 1993, 1996), the sequences recovered in this study clustered closer to the Red Sea sequences relative to those from the other regions, suggesting an effect of biogeography at sub-clade level. This was not as prominent in A1 where pairwise sequence divergence ranged between 0.8 and 1.6% amongst the Red Sea sequences, but differed by 1.1–1.6% compared to the GBR sequences. However, for A2, the sequences differed by as much as 2.1% between the Red Sea and the GRB, but only 0.4% amongst Red Sea sequences (Supplementary Table S5).
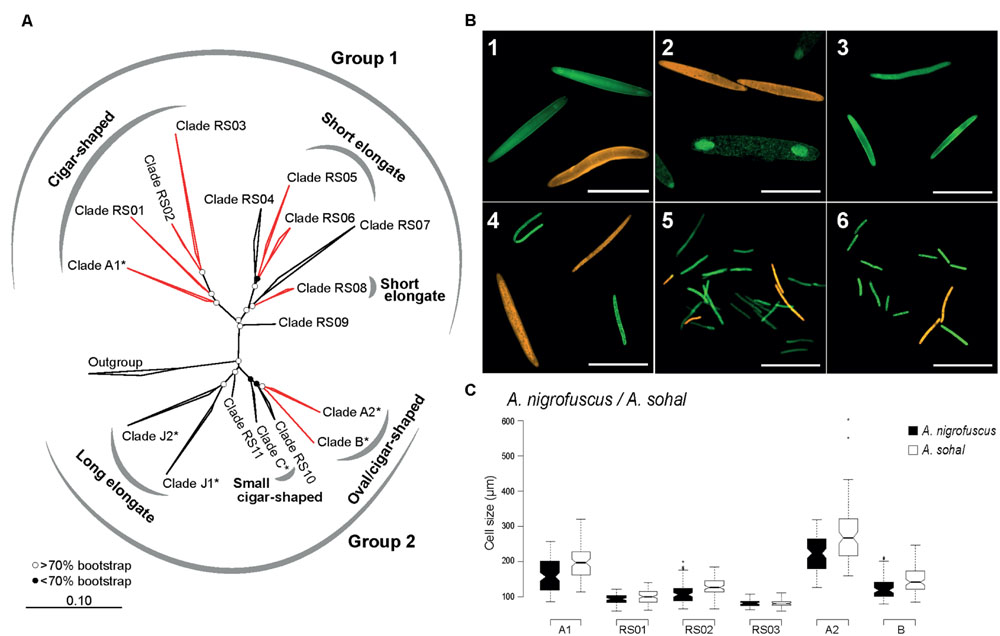
FIGURE 2. Phylogenetic and morphological diversity of Epulopiscium-like 16S rRNA gene sequences from the central Red Sea. (A) ML 16S rRNA phylogeny. Open and filled circles denote branches with bootstrap support values of >70 or <70%, respectively, based on maximum-likelihood and Bayesian trees. Epulopiscium clades that have previously been morphologically characterized elsewhere (in Hawaii or the Great Barrier Reef) are starred (∗), and clades investigated by FISH are labeled in red. (B) FISH of several clades in A. sohal and Z. desjardinii: 1) A1 (Cy3-labeled, orange) and A2 (fluorescein-labeled, green) in A. sohal, 2) A2 (orange) and B (green) in A. sohal – note that A2 cells are internal offspring that still resides within a mother cell, 3) RS01 in A. sohal (green), 4) RS02 (orange) and RS03 (green) in A. sohal – RS03 cell on the top left corner seems to be undergoing binary fission, 5) RS05 (green) and RS06 (orange) in Z. desjardinii, 6) RS06 (orange) and RS08 (green) in Z. desjardinii. The scale bars represent 100 μm in images 1–4 and 50 μm in images 5 and 6. (C) Cell size distribution of different cigar-shaped clades found in A. sohal and A. nigrofuscus.
Our study identified eleven new phylogenetic clades (RS01 to RS11) in addition to six that have been previously characterized (i.e., A1, A2, B, C, J1, and J2). Except for sequences within A1, which encompasses previously published sequences, all other clades in Group 2 (RS01 to RS09) essentially represent monophyletic groups of sequences that have not been reported before. Although clade C was previously classified into two clades (C1 and C2; Flint et al., 2005), we did not see a clear separation between C1 and C2 upon inclusion of additional sequences.
Is Epulopiscium Phylogeny Consistent with Morphology?
Given the enormous phylogenetic diversity of Epulopiscium, we wanted to analyze the shapes and sizes of the novel clades using newly designed clade-specific FISH probes (Supplementary Table S3) along with those reported in Angert et al. (1993) and Flint et al. (2005). For this purpose, we used gut preparations from A. nigrofuscus, A. sohal and Z. desjardinii, for the simple fact that these fishes harbored novel Epulopiscium clades for which probes were designed. Consistent with the earlier phylogenetic data, FISH analyses also showed that clades A1, A2, B, RS01, and RS02 were abundant in A. nigrofuscus and A. sohal but not in Z. desjardinii, while RS05, RS06, and RS08 were found in Z. desjardinii but not in A. nigrofuscus and A. sohal (Figure 2B). Contrastingly, RS03 was only detected from the Acanthurus hosts (albeit in low abundance), but not in Zebrasoma, despite sequences retrieved from all three species. No hybridization was observed for RS07-specific probes from all these fishes.
In terms of morphology, A1 and A2 were large cigar-shaped cells, while most B cells were considerably smaller and were more rounded in shape as previously reported by Angert et al. (1993; Figure 2B). However, although A1 and A2 were both cigar-shaped, the former was often not perfectly symmetrical, while the latter was consistently cigar-shaped and laterally symmetrical. Type B cells, which were reported to be ‘oval’ shape — at least in Naso tonganus (Ward et al., 2009), were also laterally symmetrical, similar to closely related A2. Additionally, these three clades (A1, A2, and B) were significantly different in size (ANOVA with Tukey’s HSD, P < 0.01), with A2 being the largest (126–603 μm) and B the smallest (79–246 μm; Figure 2C). Interestingly, there were also host-specific cell size differences, where average sizes were larger in A. sohal than in A. nigrofuscus for all three clades (average of 161 and 196 μm for A1; 224 and 280 μm for A2 and 124 and 147 μm for B in A. nigrofuscus and A. sohal, respectively; ANOVA, P < 0.01).
Clades RS01, RS02, and RS03 were also cigar-shaped similar to closely related A1, but were significantly smaller in size and less numerous (ANOVA with Tukey’s HSD, P < 0.01). Often the cells from these clades were less ‘oval’ in shape than A1/A2/B, but nevertheless were rounded at the polar ends, and thus qualified as ‘cigar-shaped’ for which probes were designed. There was a significant size difference between RS02 (120 μm) and RS03 (84 μm) cells (ANOVA, P < 0.01), but this may be due to the lack of adequate sample size, particularly as RS03 was rarely observed. Interestingly, RS03 seems to undergo binary fission (Figure 2), which was not observed for RS01 or RS02. Clades A1, RS01, RS02, and RS03 can be grouped together due to having the same morphology and phylogenetic position. This assumption was supported by the high bootstrap value (100%) and significant Rosenberg’s PAB value (PAB = 9.0 × 10-12).
In contrast, cells from the RS05, RS06, and RS08 clades in Z. desjardinii were rod-shaped (Figure 2B). There was no significant size (or shape) difference between these three clades, although RS05 was the most abundant clade observed. The species delimitation analysis demonstrated that these three clades probably belong to the same group (Rosenberg’s PAB = 4.2 × 10-4, bootstrapping = 81%), which is also reflected by their morphological similarity and phylogenetic placement, but the large sequence divergence (>8%) indicates that they are not part of the same ‘species.’ Unfortunately, RS04 and RS07 probe did not hybridize to any cells. Corroboration analyses between morphologies and phylogenies could not be conducted for other clades, due to unstable phylogeny (as was the case for RS09, 10, and 11), or lack of hybridisation (RS04 and RS07).
Relative Abundance and Distribution of Epulopiscium
Considering the observed phylogenetic and morphological diversity of Epulopiscium, we were then prompted to examine in detail the relative abundance and distribution of different clades using a previously published high-throughput 16S rRNA gene dataset in surgeonfishes from different reefs within the central Red Sea (Miyake et al., 2015). A total of 84,550 Epulopiscium-like reads (from 178,187 bacterial reads) were retrieved and assigned to the 17 phylogenetic clades (Figure 3; Supplementary Table S4). Only about 4.3% of Epulopiscium-like reads were not assignable to the phylogenetically defined clades. However, given the high classification threshold that we used (80%), it is likely that most of the Epulopiscium diversity within the sampled surgeonfishes was covered in the reconstructed phylogenetic tree. All 17 clades identified from the clone libraries were also found in the amplicon dataset. The pattern of distribution was consistent between the Bayesian and Pplacer classifications, adding further support.
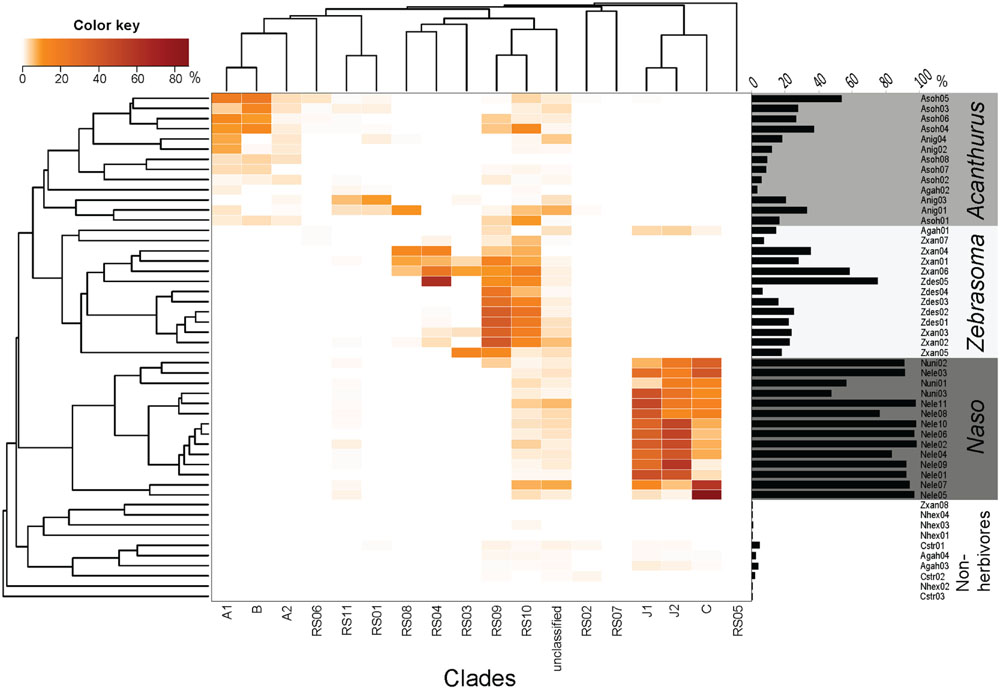
FIGURE 3. Relative sequence abundance of different Epulopiscium clades from pyrosequencing data of Miyake et al. (2015). The Bray–Curtis hierarchical clustering (left) and the Epulopiscium relative abundance (right) are also shown. Epulopiscium clade abundance formed four main clusters that consisted of herbivorous (Acanthurus, Zebrasoma, and Naso species) and non-herbivorous surgeonfishes.
A differential clustering of clade-specific Epulopiscium communities was observed in herbivorous hosts, driven by formation of phylogenetically related groupings (Adonis, P < 0.001). Acanthurus species (A. nigrofuscus and A. sohal) clustered together due to the dominance of clades A1, A2 and B, whereas Naso species (N. elegans and N. unicornis) groped together principally due to the predominance of C, J1 and J2 clades. Though Zebrasoma species (Z. desjardinii and Z. xanthurum) also grouped together – due to the dominance of clades RS09 and RS10 – many of these clades also occurred in the Acanthurus and Naso species (e.g., RS05, RS06, RS09, and RS10), albeit at lower abundances. One exception was Z. xanthurum sample number 8 (Zxan08) that clustered together with non-herbivorous surgeonfish (Figure 3). The non-herbivorous surgeonfishes (Ct. striatus, N. hexacanthus and two replicates of A. gahhm) clustered together probably due to the fact that they possessed low numbers of Epulopiscium reads. Some A. gahhm, whose Epulopiscium abundance varied considerably amongst replicates, clustered with herbivorous Acanthurus or Zebrasoma. Additionally, the observed clustering was also significantly correlated to the host diet (Adonis, P < 0.001).
Host–Symbiont Cophylogeny
Visualization of Epulopiscium clade distribution by Procrustes superimposition plot illustrated that the three main phylogenetic branches of herbivorous (and detritivorous) surgeonfishes — Acanthurus/Ctenochaetus, Naso and Zebrasoma — possessed distinctive Epulopiscium clades (Supplementary Figure S3). Furthermore, the host and symbiont phylogenies were overall statistically significantly correlated (m2 = 0.31, P < 0.001 based on 105 iterations using PACo; Balbuena et al., 2013). The jackknifed-squared residuals were strongest for the species in the Naso and Zebrasoma genera, which implies stronger association with their resident Epulopiscium clades (Supplementary Figure S4). Of 45 interactions, 22 were above the median squared residual value. In particular Z. xanthurum-RS03, A. nigrofuscus-RS01, N. elegans-J2, and Z. desjardinii-RS09 showed the largest jackknifed squared residuals. Similarly, AxParaFit (Stamatakis et al., 2007) also confirmed significant global association between the host and symbiont phylogenies (P = 0.001, ParaFit Global = 0.004 at 999 permutations). Most of the significant individual links in PACo analysis were also significant with ParaFit Link1 test for individual links, which identifies the individual associations responsible for the global cophylogenetic congruence (those with P < 0.05 are shaded in Supplementary Figure S4). However, some interactions below the median squared residual value in PACo analysis were significant with ParaFit Link1 test (e.g., A. nigrofuscus-RS03). The strength of congruence increased further when only three most abundant symbiont clades per host were considered (P < 0.001).
Discussion
Epulopiscium Phylogenetic Diversity
Previous studies based on morphological data have indicated that Epulopiscium are morphologically diverse in a number of herbivorous surgeonfishes (e.g., Clements et al., 1989), but only a few morphotypes have been phylogenetically classified (Angert, 1995; Flint, 2006). The uncovered phylogenetic diversity of Epulopiscium-like bacteria in surgeonfishes from the Red Sea at 16S rRNA gene level varied by as much as 10%, which is well over the threshold used for the genus level taxonomic ranking (Stackerbrandt and Goebel, 1994; Schloss and Handelsman, 2005; Kim et al., 2014). Thus, the two discrete phylogenetic branches encompassing Epulopiscium fishelsoni (Angert et al., 1993) and related bacteria, which we confirm here, potentially consist of two genera, each containing multiple species. It remains to be seen whether the novel clades identified in this study are indeed specific to the Red Sea.
No effect of biogeography was evident at clade-level, as similar sequences as those recovered from the GBR and Hawaii (A1, A2, B, C, J1, and J2) were also found in this study. There seems to be some clustering of Red Sea sequences at a subclade level (for instance, A1 sequences from the Red Sea were closer to each other than those from GBR), but this may be caused by the lack of sequences from other regions, and thus would require similar surveys of Epulopiscium from reefs around the world to validate. A topographically similar Epulopiscium phylogenetic tree has been constructed from the gut of A. bahianus in the Atlantic, but the sequences remain unpublished (Flint, 2006). Recently, smaller Epulopiscium-like cells, closely resembling the morphotypes F and G, have been described in A. chirurgus, A. coeruleus, and A. tractus from the US Virgin Islands (Grim et al., 2013); however, they have not yet been phylogenetically characterized and it remains to be seen whether they fall within the uncharacterized branches of the phylogeny described here.
Although, not all Epulopiscium clades were morphologically characterized, our data suggest that specific morphotypes are also phylogenetically distinct. This was exemplified by cigar-shaped (A1, RS01, RS02, and RS03) and rod-shaped morphotypes (RS05, RS06, and RS08) in this study, as well as string-shaped (type J) and oval-shaped clades from Flint et al. (2005), which were all phylogenetically unique. Rod-shaped clades (RS05, RS06, and RS08) are likely to represent morphotype E, based on descriptions by Clements et al. (1989), while clades RS01-03 seem to represent type D or I [smaller cigar-shaped morphotypes as described by Clements et al. (1989)], but differed by their size range, presence/absence of internal structures or apparent evidence for binary fission.
Epulopiscium Clade Abundances in Different Host Species
Epulopiscium clades showed a host-specific abundance pattern amongst herbivorous surgeonfishes which was significantly correlated to the host’s phylogeny and inferred diet. This has also been reported for the community composition of the entire gut microbiota (Miyake et al., 2015), but here we extend this finding to a particular group of highly unusual and abundant symbionts. Relationships amongst host feeding categories and Epulopiscium morphotypes have been reported previously (Clements et al., 1989). In light of the high abundance of Epulopiscium in most of these herbivorous surgeonfishes (Miyake et al., 2015), the previous clustering patterns observed in the gut microbiota are likely to be influenced by them, with this study further providing the source for that covariance — that is, the differences in Epulopiscium clades among the hosts. One exception to the clustering was a single individual of Z. xanthurum (Zxan08) that clustered with non-herbivorous surgeonfishes. This is most likely caused by the extremely low abundance of Epulopiscium in this sample (∼0.3%). The actual clades found in Zxan08 were similar to those in other Z. xanthurum samples (RS03, RS04, RS10, and unclassified), and they in fact clustered together if only clade composition (disregarding the abundance) of clades were considered (data not shown). Either way, this particular individual seems to be an exception to the rule, which was also indicated by a significantly different gut microbiota compared to other Z. xanthurum samples (Miyake et al., 2015).
The Epulopiscium community within A. nigrofuscus clustered to those of phylogenetically related turfing and filamentous algae feeder (A. sohal), while the total gut microbiota resembled more closely to that of non-herbivorous fishes (A. gahhm, Ct. striatus, N. hexacanthus, Ch. sordidus, Sc. niger, and Si. stellatus). These non-herbivorous fishes possessed a high proportion of allochthonous, transient bacteria according to BLAST search of the closest known relatives (Miyake et al., 2015). Similarly, A. gahhm showed inconsistencies in the clustering of Epulopiscium clades, where one individual clustered with Acanthurus, one with Zebrasoma and two with non-herbivorous surgeonfishes. This is likely due to the fact that they are omnivorous that feed on a range of diet, which was confirmed by a stomach content analysis (Supplementary Table S1). The role of environmental and host genetic factors in shaping the individuality in the gut microbiota composition has been reported in mice previously (Benson et al., 2010).
Additionally, because closely related hosts have similar nutritional ecology, we cannot conclusively distinguish the relative contribution of the phylogeny (i.e., coevolution) to that of co-occurrence due to the host’s diet in shaping the clustering pattern. However, different clades dominated in A nigrofuscus/A. sohal compared to Z. desjardinii/Z. xanthurum, despite all being turfing and filamentous red and green algae feeders indicating that host-dependence in shaping the Epulopiscium population. The role of host phylogeny in shaping the gut microbiota as a whole has been explored at subspecies level in zebrafish (Roeselers et al., 2011) and Trinidadian guppies (Sullam et al., 2015), and to species level in surgeonfish (Miyake et al., 2015), while much broader connections have been made for vertebrates (Ley et al., 2008), although host diet is also implicated. Likewise, we argue that in the surgeonfish-Epulopiscium symbiosis, both host phylogeny and diet are important factors in shaping the distribution and abundance of Epulopiscium. This uncertainty can be resolved by looking at a wider number of surgeonfishes with varying diet.
The Host–Symbiont Cophylogeny
The phylogenetic data presented here indicated that Epulopiscium form complex associations with surgeonfishes, where multiple symbiont clades were found within a given host species and multiple host species harbored a given symbiont clade. The pattern was markedly different to the sensu stricto one-to-one host–symbiont associations exemplified by some intracellular symbionts that stems from mother-to-offspring vertical transmission of the symbiont over an evolutionary time scale (e.g., Baumann et al., 1995; Degnan et al., 2004; Hughes et al., 2007; Ikeda-Ohtsubo and Brune, 2009). The fact that Epulopiscium has been found in genus Naso, the most extant genus of surgeonfish, suggest that the host–symbiont association could be as old as ∼50 million years ago when surgeonfish as a family started to diversify (Sorenson et al., 2013). The direct transfer of symbiont between host mother-offspring has not been observed in Epulopiscium, nor any parental care of the larvae has been reported, leaving little room for vertical transmission. Instead, they are likely to be acquired post partum from the environment, yet certain clades consistently dominate in the specific host species. They have however, neither been detected in the surrounding seawater (Ngugi et al., 2012; Moitinho-Silva et al., 2013; Thompson et al., 2013), nor in coral mucus and biofilm (Roder et al., 2014, 2015; this study). One possible hypothesis is that transmission might occur by coprophagy, in which organisms consume the feces (Frankenberg and Smith, 1967). Interestingly, coprophagy is reported as the mode of transmission the guinea pig symbiont, M. polyspora — the deepest phylogenetic sister group of Epulopiscium (Angert et al., 1996). Indeed, coprophagy has been reported in certain species of adult and juvenile surgeonfishes (e.g., A. nigrofuscus and Z. scorpas; Bailey and Robertson, 1982; Clements, 1997). As noted previously, there remains a possibility that the displayed cophylogeny may be due to an effect of host diet. Elsewhere in vertebrates, the Gram-positive enteric bacterium Lactobacillus reuteri has been studied extensively due to its beneficial attributes in human guts (Walter et al., 2011). It is autochthonous in wide variety of hosts, and studies have shown the genomic basis for host specialization (Frese et al., 2011). The phylogenetic analysis of L. reuteri from six different host species (human, mouse, rat, pig, chicken, and turkey) clearly reflected the host origin, illustrating a host-driven coevolution of the symbiont (Oh et al., 2010).
The Importance of Different Epulopiscium Clades
The significant cophylogenetic relationship and clade-specific distribution pattern of Epulopiscium clades in the different hosts highlight the importance of Epulopiscium in herbivorous surgeonfishes, although previous report has indicated that they are able to survive without the symbiont, at least for a few days (Montgomery and Pollak, 1988b). Nevertheless, the drop in enteric pH has been associated with the presence of Epulopiscium in A. nigrofuscus, which implies that they do indeed play a role in the host gastrointestinal regulation (Pollak and Montgomery, 1994).
One hypothesis for the huge diversity yet host-specific distribution of Epulopiscium in surgeonfishes is that the different clusters are specialized for the specific gut condition. For instance, A1, A2, and B may be especially adapted for uptake/breakdown and degradation of different polysaccharides or nutrients resulting from the host specializing in turfing and filamentous algae, while C, J1, and J2 may be better adapted for macroalgae feeders. The diversification of the symbionts due to host specialization has been reported in other systems, particularly in the enteric symbiont L. reuteri of mammalian hosts (Oh et al., 2010; Frese et al., 2011). However, the studied hosts were phylogenetically distant (rodents and humans), and there was no clear morphological differentiation in these symbionts. In addition, various enzymes (beta-galactosidase, beta-glucosidase, mannanase, pullulanase/neopullulanase, and xylobiase) responsible for the breakdown of carbohydrates and complex hemicellulose have been found in the published draft genome of morphotype B (Miller et al., 2011), while the closely related Cellulosilyticum lentocellum is also known to be cellulose degrading (Miller et al., 2011). However, this does not necessarily say that Epulopiscium themselves are directly responsible for the breakdown of algal matter, but rather, they are likely to be one of a series of players that together form a consortia to breakdown the food consumed by the host (Flint et al., 2008). Indeed, such complex microbial system has been studied extensively in termites (e.g., Warnecke et al., 2007; Brune, 2014), while host-specific gut microbiota has been reported for the Red Sea surgeonfishes (Miyake et al., 2015). By adapting their abilities to digest certain types of macronutrients available in specific host gut, Epulopiscium might have evolved into diverse clades to exploit different niche environments. To better understand the metabolic diversity of the clades, a better understanding of the constituents in the food resources of different surgeonfishes (e.g., carbohydrate composition) and in situ metabolite profiles in their guts, together with a detailed analysis of the genetic repertoires of different Epulopiscium clades would be pivotal.
Conclusion
This study revealed the phylogenetic diversity and distribution of Epulopiscium in surgeonfishes from the Red Sea. Eleven previously unexplored clades of Epulopiscium were identified and characterized, with multiple ‘species’ apparently existing within multiple ‘genera’ of Epulopiscium. Surgeonfish and Epulopiscium are intricately associated, as evident by clear dominance of specific clades in a given host species, as well as significant cophylogeny. The clades are likely adapted to specific conditions in the host gut, presumably driven by the diet. Given that various enzymes responsible for the breakdown of carbohydrates and complex hemicellulose have been found in the draft genome (Miller et al., 2012), we speculate further that specific clades found in high abundance may be adapted to the specific gut environment of the host. The study highlighted the importance of Epulopiscium to herbivorous surgeonfishes and their host-specific clade specialization, but further investigation into the functional repertoires of different Epulopiscium will be essential to unravel their role in the host gut.
Author Contributions
SM helped to design the study, conducted the experiments and wrote the draft. DN was involved in data analyses and edited the manuscript. US designed the project, and edited the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This study was supported by King Abdullah University of Science and Technology (KAUST) through baseline funding to US.
Acknowledgments
The research reported here was supported by King Abdullah University of Science and Technology (KAUST). We would like to thank Dr. John Howard Choat and Dr. William D. Robbins (James Cook University, Australia) for providing us with the host fish diet information and discussions related to the host diet. We also thank Dr. Matthew Cahill on his invaluable input and expertise, and Professor Michael L. Berumen, Dr. Joseph DiBattista, and Tane H. Sinclair-Taylor for assistance with fish sampling. We also extend our thanks to the KAUST Coastal and Marine Resources Core Lab and the Bioscience Core Lab for their technical assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2016.00285
References
Afeworki, Y., Videler, J. J., and Bruggemann, J. H. (2013). Seasonally changing habitat use patterns among roving herbivorous fishes in the southern Red Sea: the role of temperature and algal community structure. Coral Reefs 32, 475–485. doi: 10.1007/s00338-012-1000-2
Alwany, M., Thaler, E., and Stachowitsch, M. (2005). Territorial behaviour of Acanthurus sohal and Plectroglyphidodon leucozona on the fringing Egyptian Red Sea reefs. Environ. Biol. Fishes 72, 321–334. doi: 10.1007/s10641-004-2587-0
Alwany, M. A. (2008). Behavioural responses towards different light intensities of two Red Sea surgeonfishes. Egyptian J. Exp. Biol. (Zool.) 4, 199–204.
Angert E. R. (1995). Molecular Phylogenetic Analyses of Epulopiscium-like Symbionts, Ph.D. thesis, Department of Biology, Indiana University, Bloomington, IN.
Angert, E. R., Brooks, A. E., and Pace, N. R. (1996). Phylogenetic analysis of Metabacterium polyspora: clues to the evolutionary origin of daughter cell production in Epulopiscium species, the largest bacteria. J. Bacteriol. 178, 1451–1456.
Angert, E. R., and Clements, K. D. (2004). Initiation of intracellular offspring in Epulopiscium. Mol. Microbiol. 51, 827–835. doi: 10.1046/j.1365-2958.2003.03869.x
Angert, E. R., Clements, K. D., and Pace, N. R. (1993). The largest bacterium. Nature 362, 239–241. doi: 10.1038/362239a0
Bailey, T. G., and Robertson, D. R. (1982). Organic and caloric levels of fish feces relative to its consumption by coprophagous reef fishes. Mar. Biol. 69, 45–50. doi: 10.1007/BF00396959
Balbuena, J. A., Miguez-Lozano, R., and Blasco-Costa, I. (2013). PACo: a novel procrustes application to cophylogenetic analysis. PLoS ONE 8:e61048. doi: 10.1371/journal.pone.0061048
Baumann, P., Baumann, L., Lai, C. Y., Rouhbakhsh, D., Moran, N. A., and Clark, M. A. (1995). Genetics, physiology, and evolutionary relationships of the genus Buchnera: intracellular symbionts of aphids. Annu. Rev. Microbiol. 49, 55–94. doi: 10.1146/annurev.mi.49.100195.000415
Benson, A. K., Kelly, S. A., Legge, R., Ma, F., Low, S. J., Kim, J., et al. (2010). Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc. Natl. Acad. Sci. U.S.A. 107, 18933–18938. doi: 10.1073/pnas.1007028107
Brune, A. (2014). Symbiotic digestion of lignocellulose in termite guts. Nat. Rev. Microbiol. 12, 168–180. doi: 10.1038/nrmicro3182
Cernohorsky, N. H., McClanahan, T. R., Babu, I., and Horsák, M. (2015). Small herbivores suppress algal accumulation on Agatti atoll, Indian Ocean. Coral Reefs 34, 1023–1035. doi: 10.1007/s00338-015-1331-x
Choat, J. H., Clements, K. D., and Robbins, W. D. (2002). The trophic status of herbivorous fishes on coral reefs - I: dietary analyses. Mar. Biol. 140, 613–623. doi: 10.1007/s00227-001-0715-3
Choat, J. H., Robbins, W. D., and Clements, K. D. (2004). The trophic status of herbivorous fishes on coral reefs - II. Food processing modes and trophodynamics. Mar. Biol. 145, 445–454.
Clements, K. D. (1997). “Fermentation and gastrointestinal microorganisms in fishes,” in Gastrointestinal Microbiology, eds R. I. Mackie and B. A. White (Berlin: Springer), 156–198.
Clements, K. D., and Bullivant, S. (1991). An unusual symbiont from the gut of surgeonfishes may be the largest known prokaryote. J. Bacteriol. 173, 5359–5362.
Clements, K. D., Sutton, D. C., and Choat, J. H. (1989). Occurrence and characteristics of unusual protistan symbionts from surgeonfishes (acanthuridae) of the great barrier-reef, Australia. Mar. Biol. 102, 403–412. doi: 10.1007/BF00428493
Collins, M. D., Lawson, P. A., Willems, A., Cordoba, J. J., Fernandez-Garayzabal, J., Garcia, P., et al. (1994). The phylogeny of the genus Clostridium – proposal of 5 new genera and 11 new species combinations. Int. J. Syst. Bacteriol. 44, 812–826. doi: 10.1099/00207713-44-4-812
Crossman, D. J., Choat, J. H., and Clements, K. D. (2005). Nutritional ecology of nominally herbivorous fishes on coral reefs. Mar. Ecol. Prog. Ser. 296, 129–142. doi: 10.3354/meps296129
Daims, H., Stoecker, K., and Wagner, M. (2005). “Fluorescence in situ hybridization for the detection of prokaryotes,” in Advanced Methods in Molecular Microbial Ecology, eds A. M. Osborn and C. J. Smith (Abingdon: Bios-Garland)213–239.
Dale, C., and Moran, N. A. (2006). Molecular interactions between bacterial symbionts and their hosts. Cell 126, 453–465. doi: 10.1016/j.cell.2006.07.014
Degnan, P. H., Lazarus, A. B., Brock, C. D., and Wernegreen, J. J. (2004). Host-symbiont stability and fast evolutionary rates in an ant-bacterium association: cospeciation of camponotus species and their endosymbionts, candidatus blochmannia. Syst. Biol. 53, 95–110. doi: 10.1080/10635150490264842
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Fishelson, L., Montgomery, W. L., and Myrberg, A. H. (1987). Biology of surgeonfish Acanthurus nigrofuscus with emphasis on changeover in diet and annual gonadal cycles. Mar. Ecol. Prog. Ser. 39, 37–47. doi: 10.3354/meps039037
Fishelson, L., Montgomery, W. L., and Myrberg, A. A. (1985). A unique symbiosis in the gut of tropical herbivorous surgeonfish (acanthuridae, teleostei) from the red-sea. Science 229, 49–51.
Flint, H. J., Bayer, E. A., Rincon, M. T., Lamed, R., and White, B. A. (2008). Polysaccharide utilization by gut bacteria: potential for new insights from genomic analysis. Nat. Rev. Microbiol. 6, 121–131. doi: 10.1038/nrmicro1817
Flint, J. F. (2006). Molecular Methods to Investigate Symbiotic Bacteria of Gastrointestinal Systems. New York, NY: Cornell University.
Flint, J. F., Drzymalski, D., Montgomery, W. L., Southam, G., and Angert, E. R. (2005). Nocturnal production of endospores in natural populations of epulopiscium-like surgeonfish symbionts. J. Bacteriol. 187, 7460–7470. doi: 10.1128/JB.187.21.7460-7470.2005
Forst, S., and Nealson, K. (1996). Molecular biology of the symbiotic-pathogenic bacteria Xenorhabdus spp. and Photorhabdus spp. Microbiol. Rev. 60, 21–43.
Frankenberg, D., and Smith, K. L. (1967). Coprophagy in marine animals1. Limnol. Oceanogr. 12, 443–450. doi: 10.1007/s00114-010-0659-x
Frese, S. A., Benson, A. K., Tannock, G. W., Loach, D. M., Kim, J., Zhang, M., et al. (2011). The evolution of host specialization in the vertebrate gut symbiont Lactobacillus reuteri. PLoS Genet. 7:e1001314. doi: 10.1371/journal.pgen.1001314
Gadagkar, S. R., Rosenberg, M. S., and Kumar, S. (2005). Inferring species phylogenies from multiple genes: concatenated sequence tree versus consensus gene tree. J. Exp. Zool. Part B Mol. Dev. Evol. 304B, 64–74. doi: 10.1002/jez.b.21026
Grim, J. N., Nemeth, D., and Montgomery, W. L. (2013). The occurrence of Epulopiscium-like eubacteria in the intestines of surgeonfish from the US virgin islands, western Atlantic Ocean. Mar. Biodiversity Rec. 6:e79. doi: 10.1017/S1755267213000559
Hughes, J., Kennedy, M., Johnson, K. P., Palma, R. L., and Page, R. D. M. (2007). Multiple cophylogenetic analyses reveal frequent cospeciation between pelecaniform birds and Pectinopygus lice. Syst. Biol. 56, 232–251. doi: 10.1080/10635150701311370
Ikeda-Ohtsubo, W., and Brune, A. (2009). Cospeciation of termite gut flagellates and their bacterial endosymbionts: trichonympha species and “Candidatus Endomicrobium trichonymphae.” Mol. Ecol. 18, 332–342. doi: 10.1111/j.1365-294X.2008.04029.x
Kim, M., Oh, H. S., Park, S. C., and Chun, J. (2014). Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int. J. Syst. Evol. Microbiol. 64, 346–351. doi: 10.1099/ijs.0.059774-0
Klanten, S. O., van Herwerden, L., Choat, J. H., and Blair, D. (2004). Patterns of lineage diversification in the genus Naso (Acanthuridae). Mol. Phylogenet. Evol. 32, 221–235. doi: 10.1016/j.ympev.2003.11.008
Klindworth, A., Pruesse, E., Schweer, T., Peplies, J., Quast, C., Horn, M., et al. (2013). Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41:e1. doi: 10.1093/nar/gks808
Legendre, P., Desdevises, Y., and Bazin, E. (2002). A statistical test for host-parasite coevolution. Syst. Biol. 51, 217–234. doi: 10.1080/10635150252899734
Ley, R. E., Hamady, M., Lozupone, C., Turnbaugh, P. J., Ramey, R. R., Bircher, J. S., et al. (2008). Evolution of mammals and their gut microbes. Science 320, 1647–1651. doi: 10.1126/science.1155725
Ludwig, W., Strunk, O., Westram, R., Richter, L., Meier, H., and Yadhukumar. (2004). ARB: a software environment for sequence data. Nucleic Acids Res. 32, 1363–1371. doi: 10.1093/nar/gkh293
Maneesakorn, P., An, R. S., Daneshvar, H., Taylor, K., Bai, X. D., Adams, B. J., et al. (2011). Phylogenetic and cophylogenetic relationships of entomopathogenic nematodes (Heterorhabditis: Rhabditida) and their symbiotic bacteria (Photorhabdus: Enterobacteriaceae). Mol. Phylogenet. Evol. 59, 271–280. doi: 10.1016/j.ympev.2011.02.012
Marshell, A., and Mumby, P. J. (2012). Revisiting the functional roles of the surgeonfish Acanthurus nigrofuscus and Ctenochaetus striatus. Coral Reefs 31, 1093–1101. doi: 10.1007/s00338-012-0931-y
Masters, B. C., Fan, V., and Ross, H. A. (2011). Species delimitation - a geneious plugin for the exploration of species boundaries. Mol. Ecol. Resour. 11, 154–157. doi: 10.1111/j.1755-0998.2010.02896.x
Matsen, F. A., Kodner, R. B., and Armbrust, E. V. (2010). pplacer: linear time maximum-likelihood and bayesian phylogenetic placement of sequences onto a fixed reference tree. BMC Bioinformatics 11:538. doi: 10.1186/1471-2105-11-538
McArdle, B. H., and Anderson, M. J. (2001). Fitting multivariate models to community data: a comment on distance-based redundancy analysis. Ecology 82, 290–297. doi: 10.1890/0012-9658(2001)082[0290:FMMTCD]2.0.CO;2
Meier-Kolthoff, J. P., Auch, A. F., Huson, D. H., and Goker, M. (2007). CopyCat: cophylogenetic analysis tool. Bioinformatics 23, 898–900. doi: 10.1093/bioinformatics/btm027
Mendell, J. E., Clements, K. D., Choat, J. H., and Angert, E. R. (2008). Extreme polyploidy in a large bacterium. Proc. Natl. Acad. Sci. U.S.A. 105, 6730–6734. doi: 10.1073/pnas.0707522105
Miller, D. A., Suen, G., Bruce, D., Copeland, A., Cheng, J. F., Detter, C., et al. (2011). Complete genome sequence of the cellulose-degrading bacterium Cellulosilyticum lentocellum. J. Bacteriol. 193, 2357–2358. doi: 10.1128/JB.00239-11
Miller, D. A., Suen, G., Clements, K. D., and Angert, E. R. (2012). The genomic basis for the evolution of a novel form of cellular reproduction in the bacterium Epulopiscium. BMC Genomics 13:265. doi: 10.1186/1471-2164-13-265
Miyake, S., Ngugi, D. K., and Stingl, U. (2015). Diet strongly influences the gut microbiota of surgeonfishes. Mol. Ecol. 24, 656–672. doi: 10.1111/mec.13050
Moitinho-Silva, L., Bayer, K., Cannistraci, C. V., Giles, E. C., Ryu, T., Seridi, L., et al. (2013). Specificity and transcriptional activity of microbiota associated with low and high microbial abundance sponges from the Red Sea. Mol. Ecol. 23, 1348–1363. doi: 10.1111/mec.12365
Montgomery, W. L., and Galzin, R. (1993). Seasonality in gonads, fat deposits and condition of tropical surgeonfishes (Teleostei, Acanthuridae). Mar. Biol. 115, 529–536. doi: 10.1007/BF00349359
Montgomery, W. L., Myrberg, A. A., and Fishelson, L. (1989). Feeding ecology of surgeonfishes (Acanthuridae) in the northern red-sea, with particular reference to Acanthurus nigrofuscus (Forsskal). J. Exp. Mar. Biol. Ecol. 132, 179–207. doi: 10.1016/0022-0981(89)90127-5
Montgomery, W. L., and Pollak, P. E. (1988a). Epulopiscium-fishelsoni Ng, N-Sp, a protist of uncertain taxonomic affinities from the gut of an herbivorous reef fish. J. Protozool. 35, 565–569. doi: 10.1111/j.1550-7408.1988.tb04153.x
Montgomery, W. L., and Pollak, P. E. (1988b). Gut anatomy and Ph in a red-sea Surgeonfish, Acanthurus nigrofuscus. Mar. Ecol. Prog. Ser. 44, 7–13. doi: 10.3354/meps044007
Moran, N. A., McCutcheon, J. P., and Nakabachi, A. (2008). Genomics and evolution of heritable bacterial symbionts. Annu. Rev. Genet. 42, 165–190. doi: 10.1146/annurev.genet.41.110306.130119
Ngugi, D. K., Antunes, A., Brune, A., and Stingl, U. (2012). Biogeography of pelagic bacterioplankton across an antagonistic temperature-salinity gradient in the Red Sea. Mol. Ecol. 21, 388–405. doi: 10.1111/j.1365-294X.2011.05378.x
Ngugi, D. K., and Stingl, U. (2012). Combined analyses of the ITS loci and the corresponding 16S rRNA genes reveal high micro- and macrodiversity of SAR11 populations in the Red Sea. PLoS ONE 7:e50274. doi: 10.1371/journal.pone.0050274
Noda, S., Kitade, O., Inoue, T., Kawai, M., Kanuka, M., Hiroshima, K., et al. (2007). Cospeciation in the triplex symbiosis of termite gut protists (Pseudotrichonympha spp.), their hosts, and their bacterial endosymbionts. Mol. Ecol. 16, 1257–1266. doi: 10.1111/j.1365-294X.2006.03219.x
Oh, P. L., Benson, A. K., Peterson, D. A., Patil, P. B., Moriyama, E. N., Roos, S., et al. (2010). Diversification of the gut symbiont Lactobacillus reuteri as a result of host-driven evolution. ISME J. 4, 377–387. doi: 10.1038/ismej.2009.123
Peplies, J., Kottmann, R., Ludwig, W., and Glockner, F. O. (2008). A standard operating procedure for phylogenetic inference (SOPPI) using (rRNA) marker genes. Syst. Appl. Microbiol. 31, 251–257. doi: 10.1016/j.syapm.2008.08.003
Pinault, M., Loiseau, N., Chabanet, P., Durville, P., Magalon, H., Quod, J. P., et al. (2013). Marine fish communities in shallow volcanic habitats. J. Fish Biol. 82, 1821–1847. doi: 10.1111/jfb.12110
Pollak, P. E., and Montgomery, W. L. (1994). Giant bacterium (Epulopiscium-fishelsoni) influences digestive enzyme-activity of an herbivorous surgeonfish (Acanthurus nigrofuscus). Compar. Biochem. Physiol. Physiol. 108, 657–662. doi: 10.1016/0300-9629(94)90352-2
Pruesse, E., Peplies, J., and Glockner, F. O. (2012). SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28, 1823–1829. doi: 10.1093/bioinformatics/bts252
Purcell, S. W., and Bellwood, D. R. (1993). A functional-analysis of food procurement in 2 surgeonfish species, Acanthurus nigrofuscus and Ctenochaetus striatus (Acanthuridae). Environ. Biol. Fishes 37, 139–159. doi: 10.1007/BF00000589
Roder, C., Arif, C., Daniels, C., Weil, E., and Voolstra, C. R. (2014). Bacterial profiling of white plague disease across corals and oceans indicates a conserved and distinct disease microbiome. Mol. Ecol. 23, 965–974. doi: 10.1111/mec.12638
Roder, C., Bayer, T., Aranda, M., Kruse, M., and Voolstra, C. R. (2015). Microbiome structure of the fungid coral Ctenactis echinata aligns with environmental differences. Mol. Ecol. 24, 3501–3511. doi: 10.1111/mec.13251
Roeselers, G., Mittge, E. K., Stephens, W. Z., Parichy, D. M., Cavanaugh, C. M., Guillemin, K., et al. (2011). Evidence for a core gut microbiota in the zebrafish. ISME J. 5, 1595–1608. doi: 10.1038/ismej.2011.38
Rosenberg, N. A. (2007). Statistical tests for taxonomic distinctiveness from observations of monophyly. Evolution 61, 317–323. doi: 10.1111/j.1558-5646.2007.00023.x
Russell, J. A., Moreau, C. S., Goldman-Huertas, B., Fujiwara, M., Lohman, D. J., and Pierce, N. E. (2009). Bacterial gut symbionts are tightly linked with the evolution of herbivory in ants. Proc. Natl. Acad. Sci. U.S.A. 106, 21236–21241. doi: 10.1073/pnas.0907926106
Schloss, P. D., and Handelsman, J. (2005). Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl. Environ. Microbiol. 71, 1501–1506. doi: 10.1128/AEM.71.3.1501-1506.2005
Sorenson, L., Santini, F., Carnevale, G., and Alfaro, M. E. (2013). A multi-locus timetree of surgeonfishes (Acanthuridae, Percomorpha), with revised family taxonomy. Mol. Phylogenet. Evol. 68, 150–160. doi: 10.1016/j.ympev.2013.03.014
Stackerbrandt, E., and Goebel, B. M. (1994). Taxonomic note: a place for DNA-DNA reassociation and 16S rRNA sequence analysis in the present species definition in bacteriology. Int. J. Syst. Bacteriol. 44, 846–849. doi: 10.1099/00207713-44-4-846
Stamatakis, A., Auch, A. F., Meier-Kolthoff, J., and Goker, M. (2007). AxPcoords & parallel AxParafit: statistical co-phylogenetic analyses on thousands of taxa. BMC Bioinformatics 8:405. doi: 10.1186/1471-2105-8-405
Stevens, J. (2004). Computational aspects of host-parasite phylogenies. Brief. Bioinform. 5, 339–349. doi: 10.1093/bib/5.4.339
Sullam, K. E., Rubin, B. E., Dalton, C. M., Kilham, S. S., Flecker, A. S., and Russell, J. A. (2015). Divergence across diet, time and populations rules out parallel evolution in the gut microbiomes of Trinidadian guppies. ISME J. 9, 1508–1522. doi: 10.1038/ismej.2014.231
Thompson, L. R., Field, C., Romanuk, T., Ngugi, D., Siam, R., E., Dorry H., et al. (2013). Patterns of ecological specialization among microbial populations in the Red Sea and diverse oligotrophic marine environments. Ecol. Evol. 3, 1780–1797. doi: 10.1002/ece3.593
Vine, P. J. (1974). Effects of Algal Grazing and Aggressive-Behavior of Fishes Pomacentrus-lividus and Acanthurus sohal on coral-reef ecology. Mar. Biol. 24, 131–136. doi: 10.1007/BF00389347
Walter, J., Britton, R. A., and Roos, S. (2011). Host-microbial symbiosis in the vertebrate gastrointestinal tract and the Lactobacillus reuteri paradigm. Proc. Natl. Acad. Sci. U.S.A. 108, 4645–4652. doi: 10.1073/pnas.1000099107
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/AEM.00062-07
Ward, R. J., Clements, K. D., Choat, J. H., and Angert, E. R. (2009). Cytology of terminally differentiated Epulopiscium mother cells. DNA Cell Biol. 28, 57–64. doi: 10.1089/dna.2008.0801
Warnecke, F., Luginbühl, P., Ivanova, N., Ghassemian, M., Richardson, T. H., Stege, J. T., et al. (2007). Metagenomic and functional analysis of hindgut microbiota of a wood-feeding higher termite. Nature 450, 560–565. doi: 10.1038/nature06269
Keywords: Epulopiscium, coevolution, surgeonfish, Red Sea, 16S rRNA
Citation: Miyake S, Ngugi DK and Stingl U (2016) Phylogenetic Diversity, Distribution, and Cophylogeny of Giant Bacteria (Epulopiscium) with their Surgeonfish Hosts in the Red Sea. Front. Microbiol. 7:285. doi: 10.3389/fmicb.2016.00285
Received: 07 October 2015; Accepted: 23 February 2016;
Published: 14 March 2016.
Edited by:
Shana Goffredi, Occidental College, USAReviewed by:
Kendall D. Clements, University of Auckland, New ZealandEsther Angert, Cornell University, USA
Copyright © 2016 Miyake, Ngugi and Stingl. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ulrich Stingl, dWxpLnN0aW5nbEBrYXVzdC5lZHUuc2E=; dWxpc3RpbmdsQGdtYWlsLmNvbQ==