 Preetinanda Panda1,2
Preetinanda Panda1,2 Bhanupratap R. Vanga2,3Ashley Lu2†Mark Fiers2†
Bhanupratap R. Vanga2,3Ashley Lu2†Mark Fiers2† Peter C. Fineran1,3Ruth Butler2Karen Armstrong1
Peter C. Fineran1,3Ruth Butler2Karen Armstrong1 Clive W. Ronson3
Clive W. Ronson3 Andrew R. Pitman1,2*
Andrew R. Pitman1,2*- 1The Bio-Protection Research Centre, Lincoln, New Zealand
- 2Plant Pathology, The New Zealand Institute for Plant and Food Research Limited, Lincoln, New Zealand
- 3Department of Microbiology and Immunology, University of Otago, Dunedin, New Zealand
Integrative and conjugative elements (ICEs) play a central role in the evolution of bacterial virulence, their transmission between bacteria often leading to the acquisition of virulence factors that alter host range or aggressiveness. Much is known about the functions of the virulence determinants that ICEs harbor, but little is understood about the cryptic effects of ICEs on their host cell. In this study, the importance of horizontally acquired island 2 (HAI2), an ICE in the genome of Pectobacterium atrosepticum SCRI1043, was studied using a strain in which the entire ICE had been removed by CRISPR-Cas-mediated genome editing. HAI2 encodes coronafacic acid, a virulence factor that enhances blackleg disease of potato stems caused by P. atrosepticum SCRI1043. As expected, deletion of HAI2 resulted in reduced blackleg symptoms in potato stems. A subsequent screen for HAI2-related ICEs in other strains of the Pectobacterium genus revealed their ubiquitous nature in P. atrosepticum. Yet, HAI2-related ICEs were only detected in the genomes of a few P. carotovorum strains. These strains were notable as blackleg causing strains belonging to two different subspecies of P. carotovorum. Sequence analysis of the ICEs in different strains of both P. atrosepticum and P. carotovorum confirmed that they were diverse and were present in different locations on the genomes of their bacterial host, suggesting that the cfa cluster was probably acquired independently on a number of occasions via chromosomal insertion of related ICEs. Excision assays also demonstrated that the ICEs in both P. atrosepticum and P. carotovorum are mobilized from the host chromosome. Thus, the future spread of these ICEs via lateral gene transfer might contribute to an increase in the prevalence of blackleg-causing strains of P. carotovorum.
Introduction
Historically, the enterobacterial phytopathogen Pectobacterium atrosepticum was considered the primary pathogen responsible for the rotting or wilting of stems on growing potato plants, referred to as blackleg (Pérombelon, 2002). Recent surveys to identify the source of blackleg symptoms in the field, however, led to the isolation of numerous species and subspecies of Pectobacterium from disease lesions. For example, P. carotovorum subsp. brasiliensis was obtained from potatoes in Brazil, where it was the major cause of blackleg (Duarte et al., 2004). P. carotovorum subsp. brasiliensis was subsequently detected in potato cropping systems in Israel (Ma et al., 2007), South Africa (van der Merwe et al., 2010), Canada (De Boer et al., 2012), New Zealand (Panda et al., 2012), Zimbabwe (Ngadze et al., 2012) and in the Netherlands (Leite et al., 2014), suggesting the pathogen has a global impact on potato. P. wasabiae was also collected from potato plants with blackleg symptoms (Pérombelon and Kelman, 1980; De Boer, 2002). Isolates of P. wasabiae were initially thought to be secondary invaders, until their vacuum infiltration into tubers was shown to induce blackleg (de Haan et al., 2008). P. wasabiae was subsequently detected in a multitude of potato growing regions including the United States, New Zealand, Iran, and South Africa (Kim et al., 2009; Pitman et al., 2010; Baghaee-Ravari et al., 2011; Moleleki et al., 2013).
The isolation of divergent Pectobacterium strains with the capacity to cause blackleg suggests that they may share common virulence determinants associated with stem infection. The genome sequence of P. atrosepticum SCRI1043 (Bell et al., 2004) identified various loci associated with virulence, including a coronafacic acid (Cfa) biosynthetic gene cluster. Cfa is a component of the toxin coronatine (Cor), an important virulence factor for some pathovars of the bacterial pathogen Pseudomonas syringae (Bender et al., 1999). Cor is formed by the conjugation of the Cfa polyketide to coronamic acid (Cma), an ethylcyclopropyl amino acid derived from isoleucine (Parry et al., 1994). Unlike P. syringae, P. atrosepticum SCRI1043 does not encode genes for the production of Cma, suggesting that it produces an alternative polyketide phytotoxin, or that Cfa acts alone during disease development. In P. syringae, Cor mimics jasmonate 12-oxo-phytodienoic acid. Jasmonate 12-oxo-phytodienoic acid is a precursor to jasmonic acid (Weiler et al., 1994). Cor acts by stimulating jasmonate response pathways (Zhao et al., 2003) and suppressing salicylic acid-mediated defenses (Uppalapati et al., 2007). The function of Cfa in P. atrosepticum is unknown, but a transposon insertion in the cfa cluster reduced the capacity of the pathogen to cause blackleg symptoms on potato (Bell et al., 2004). Thus, Cfa probably enables the pathogen to manipulate host immunity during blackleg-related infection in potato.
In P. atrosepticum SCRI1043, the cfa biosynthetic cluster is harbored on a putative horizontally acquired island, HAI2 (Bell et al., 2004). HAI2 is 97,875 bp in size, has a G+C content of 48.30% compared with 50.97% for the entire genome and has 99 predicted coding DNA sequences (CDSs; Vanga et al., 2012). It shows strong similarity to a family of integrative and conjugative elements (ICEs) that include SPI-7 from Salmonella enterica serovar Typhi TY2 (Bueno et al., 2004), PAP1 from P. aeruginosa PA14 (He et al., 2004), and PPHGI-1 from P. syringae pv. phaseolicola 1302A (Pitman et al., 2005).
Integrative and conjugative elements are typically stably integrated into the chromosome, flanked by direct repeats and inserted at the 3′ end of a tRNA gene (Hacker and Kaper, 2000). They can also excise from the chromosome, resulting in the formation of an extrachromosomal circular form that facilitates transfer between donor and recipient cells. HAI2 is no exception, integrated within the genome of P. atrosepticum SCRI1043 at a bacterial attachment site (attB0515) located within the phe-tRNA gene immediately downstream of ECA0515 (Vanga et al., 2012). HAI2 can excise at low frequency from the chromosome (Vanga et al., 2012), a process that is induced in planta (Vanga et al., 2015). Mobilization and transfer of ICEs plays a central role in the evolution of pathogens by transferring large amounts of genetic information between bacteria (Morschhäuser et al., 2000). As the cfa biosynthetic cluster has only been detected in strains of P. atrosepticum and P. carotovorum responsible for blackleg (Bell et al., 2004; Slawiak and Lojkowska, 2009; Panda et al., 2015), it is possible that independent acquisition of HAI2 has contributed to the capacity of both species to cause the disease by transferring this important virulence factor.
In this study, we examined the role of HAI2 in aggressiveness of P. atrosepticum SCRI1043 on potato stems by conducting pathogenicity tests using SCRI1043ΔHAI2, an ‘ICE-less’ strain previously generated using endogenous type I-F CRISPR-Cas chromosomal targeting (Vercoe et al., 2013; Dy et al., 2014; Richter et al., 2014). CRISPR-Cas systems include clustered regularly interspaced short palindromic repeats (CRISPRs) and their associated Cas proteins, and are an adaptive immune system in bacteria against foreign genetic elements (Dy et al., 2013). We also studied the distribution and genetic organization of the cfa biosynthetic clusters in a variety of related strains of the Pectobacterium genus from potato to understand if independent acquisition of HAI2 may have led to the capacity of different species of Pectobacterium to cause blackleg.
Materials and Methods
Bacterial Strains and DNA Manipulations
SCRI1043ΔHAI2 was used to study the function of HAI2. SCRI1043ΔHAI2 is a derivative of P. atrosepticum SCRI1043 that has the entire ICE removed from the genome using CRISPR-Cas-mediated genome targeting (Vercoe et al., 2013) followed by curing the strain of the chromosomal-targeting crRNA expression plasmid (Richter et al., 2014). An additional 87 strains of the Pectobacterium genus isolated from potato in New Zealand (Pitman et al., 2008) and a variety of type strains and genetically or biochemically characterized isolates obtained from the International Collection of Micro-organisms from Plants (ICMP), Landcare Research, New Zealand (unless otherwise stated; Supplementary Table S1) were also used to examine the distribution of HAI2 and other closely related ICEs. All bacteria were routinely grown in Lysogeny Broth medium (LB) or Minimal Medium (MM) at 28°C for 24–48 h. Genomic DNA was extracted from liquid cultures using the DNeasy tissue kit (Qiagen) following the manufacturer’s instructions.
Pathogenicity Tests on Potato
The virulence of SCRI1043ΔHAI2 was compared with wild type P. atrosepticum SCRI1043 by carrying out blackleg assays on potato stems. Potato plants of the susceptible cultivar ‘Ilam Hardy’ were grown for ∼4 weeks in a controlled growth chamber, with a 16 h photoperiod at 23°C and 80% humidity (to a height of ∼20 cm). Plants were inoculated under the second fully expanded leaf by injecting 10 μL of bacteria grown in LB into the stem with a micropipette (either 104 or 106 cells per inoculation site). A total of 22 plants were used for each treatment. Inoculation sites were covered with plastic wrap to avoid desiccation and the plants were incubated in growth chambers at 23°C and high humidity (>80%). The length of blackleg lesions was measured on each plant daily for a period of 14 days post-inoculation (dpi), with absent lesions and lesions of <0.5 mm recorded as ‘<0.5.’
Data from the blackleg assays were primarily explored graphically. Statistical analysis was carried out for the data at day 14. For the final assessment at day 14, the percentage of plants showing measurable lesions (measurable is lesions not recorded as <0.5) was analyzed with a binomial generalized linear model with a logit link (McCullagh and Nelder, 1989). Predicted percentages and associated 95% confidence limits were obtained on the logit scale and back-transformed to percentages. Also for the final assessment, the length of lesions for plants with measurable lesions was analyzed with ANOVA. Mean lesion length was obtained, along with 95% confidence limits for the means. The strains were compared within the analysis of deviance/variance, and were assessed with a X2/F-test. All analyses were carried out with GenStat (GenStat Committee, 2012).
Detection of Loci Belonging to the cfa Biosynthetic Cluster and HAI2 in Strains of Pectobacterium
The collection of Pectobacterium strains was screened for the presence of cfa6 and cfa7 by PCR. Six other loci associated with HAI2 (Vanga et al., 2012) were targeted by PCR. These included attL and attR, the direct repeats that delineate HAI2 when the ICE is integrated in the chromosome of P. atrosepticum SCRI1043 (at the 3′ end of a phe-tRNA gene between the CDS identifiers ECA0515 and ECA0615). The presence of ECA0516 (soj), ECA0525 (topB), ECA0532 (pilL) and ECA0614 (int), four genes predicted to be involved in the stable maintenance, excision, and transfer of HAI2 (Vanga et al., 2012), was also examined. The primers used for amplification of the eight loci are listed in Table 1. PCR was performed using Taq polymerase (Roche Diagnostics) in a GeneAmpR PCR System 9700 thermocycler (Applied Biosciences) with the following steps: 5 min at 95°C for initial denaturation, followed by 35 amplification cycles of 94°C for 30 s, annealing for 30 s, and 72°C for 60 s and a final extension phase at 72°C for 7 min. The annealing temperatures for each PCR are described in Table 1.
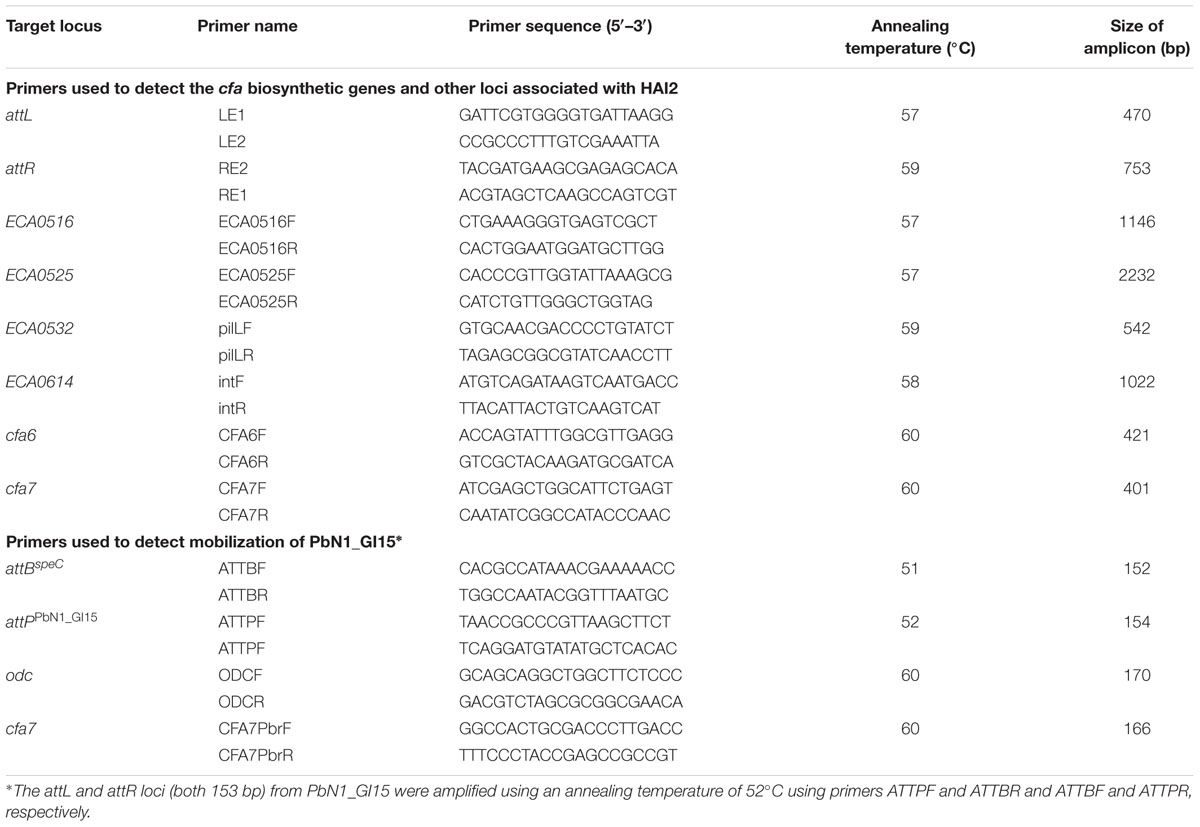
TABLE 1. Primers used in this study.
qPCR Assays to Measure Excision of HAI2-Related ICEs in Pectobacterium
Quantitative PCR (qPCR) was used to measure excision of HAI2-related ICEs in different Pectobacterium isolates by quantifying the amounts of attP, attB0515 and a reference gene located on the core genome, upstream of the ICE (Vanga et al., 2012). An attP locus is formed upon the circularization of an ICE during excision and the subsequent formation of an extrachromosomal element. Excision of the ICE results in the reconstitution of the chromosomal insertion site, attB0515, located within the phe-tRNA gene downstream of ECA0515 in P. atrosepticum SCRI1043 (and other Pectobacterium). Thus, detection of attB0515 occurs when the ICE is not inserted in this target site on the chromosome.
Excision assays were performed by growing each strain in triplicate for 24 h in MM. A single DNA sample was then extracted from each culture using the DNeasy tissue kit (Qiagen). A qPCR was performed in triplicate for each DNA sample using the iTaq SYBR Green Supermix with ROX (Biorad) and variable concentrations of each primer (Vanga et al., 2012). Amplification was performed by the 7500 Fast System (Applied Biosystems) using the following conditions: 2 min 30 s at 95°C for initial denaturation, followed by 40 cycles of 95°C for 15 s, annealing for 20 s, and 72°C for 20 s. The annealing temperatures used for attB0515, attP, and ECA0515 are described by Vanga et al. (2012). To check the integrity of the qPCR products, melting curves were carried out for each reaction using the following conditions: 15 s at 95°C, followed by annealing at reaction-specific annealing temperatures for 60 s and extension at 72°C for 1 min.
The Ct values for each qPCR were collected and the mean Ct produced by the three replicate reactions for each DNA sample was calculated using the Applied Biosystems 7500 Fast System SDS software V. 1.3. The amplification efficiency of all assays was also determined using plasmid pBP515, containing single copies of attP, attB0515, and ECA0515, linearized with SpeI. Serial dilutions of the plasmid were prepared and used as templates for qPCR to generate standard curves for each of the PCRs by plotting DNA concentration versus log (Ct) value. Amplification efficiency was determined using the slope values obtained from the standard curves and the equation E = 10(-1/slope). The ratios of attB0515 and attP (Ratt) to ECA0515 were then calculated for each qPCR, by normalizing the results for attB0515 and attP against those obtained using ECA0515, with correction for differences in amplification efficiencies (Vanga et al., 2012).
The log10 ratios for attB0515 and attP were analyzed with ANOVA (the log10 transformation was used to stabilize for variance). Differences between the mean log10 ratios for each isolate and the mean for P. atrosepticum SCRI1043 were calculated and back-transformed, to give REST (relative expression) ratios between each isolate (‘sample’) and P. atrosepticum SCRI1043 (‘control’). To identify significant differences between DNA samples from different strains of Pectobacterium, the back-transformed least significant difference (LSD) was then calculated to give a least significant ratio (LSR); that is the smallest ratio for which REST is significantly greater than 1 (1/LSR gives the largest ratio for which REST is significantly smaller than 1). All analyses were carried out with GenStat Committee (2012).
Genome Sequencing of HAI2-Related ICEs Encoding Cfa in Pectobacterium
To characterize ICEs harboring the cfa biosynthetic cluster, the genomes of P. atrosepticum ICMP 1526, the type strain for P. atrosepticum, and P. carotovorum subsp. brasiliensis ICMP 19477 were sequenced using a Roche FLX 454 sequencer by the Liverpool Advanced Genomics Facility (Liverpool University, UK). Sequences were assembled with Newbler to produce draft genome sequences to 40–50 × coverage, comprising 38 and 35 contigs for P. atrosepticum ICMP 1526 and P. carotovorum subsp. brasiliensis ICMP 19477, respectively (Panda et al., 2015). The genome of UGC32 was also sequenced using the HiSeq 2000 system from Illumina sequencing services to generate pair-end reads with a genome coverage of ∼60× (Panda et al., 2015). UGC32 was previously identified as a P. carotovorum subsp. carotovorum isolate with the capacity to cause blackleg (Slawiak and Lojkowska, 2009). The contigs for each strain were ordered and orientated with respect to the reference genome sequence of P. atrosepticum SCRI1043 (accession BX950851) using MUMmer. Annotation of the genome sequences was completed using the PGAAP pipeline1. The genome sequencing, assembly and annotation of SCRI1043ΔHAI2 were performed as described for UGC32 and gave the following statistics: number of contigs/scaffolds, 135; N50, 1,156,993.
Comparative Analysis of HAI2-Related ICEs Encoding Cfa in Pectobacterium
Geneious Pro 5.5.5 (Kearse et al., 2012) was used to collate genome sequence data for each strain, and the Blast suite version 2.2.31 (blastn using max E value 1e-1) was used to identify cfa biosynthetic clusters in each genome (using the cfa genes from P. atrosepticum SCRI1043 as a reference). The genetic structure and organization of the cfa clusters and their related ICEs were then curated manually from the annotated genomes of each strain by conducting blastx comparisons of individual CDSs with HAI2 from P. atrosepticum SCRI1043. Each ICE was delineated by the presence of direct repeats attL and attR. DNA sequences for the ICEs can be obtained from the annotated draft genome sequences of P. atrosepticum ICMP 1526 and P. carotovorum subsp. brasiliensis ICMP 19477, which have the Genbank accessions ALIV00000000 and ALIU00000000, respectively. The accession number for the draft genome of P. carotovorum subsp. carotovorum UGC32 is AODU00000000. Similarities and differences in the HAI2-related ICEs were visualized in Easyfig 2.2.2 (Sullivan et al., 2011) by performing tblastx comparisons with a cut off E value of 1e-10.
Excision of PbN1_GI15 in P. carotovorum subsp. brasiliensis ICMP 19477
Integration of PbN1_GI15 into the chromosomal target site attBspeC in P. carotovorum subsp. brasiliensis ICMP 19477 was examined by amplifying the attL (153 bp) and attR (153 bp) sites using primers ATTPF and ATTBR and ATTBF and ATTPR, respectively. Excision of PbN1_GI15 was also studied by PCR to establish whether PbN1_GI15 could mobilize from the chromosome of this bacterium. Initially, a PCR was performed using primers ATTBF and ATTBR to detect the 152-bp amplicon indicative of reconstitution of the chromosomal target site attBspeC upon mobilization of the putative ICE. A PCR strategy was then designed to amplify a 154-bp PCR product (attPPbN1_GI15) using primers ATTPF and ATTPR only when the attL and attR sites delineating PbN1_GI15 recombined during excision to form an attP site indicative of circularization. To confirm DNA extracted from P. carotovorum subsp. brasiliensis ICMP 19477 could be amplified, primers were designed to amplify the ornithine decarboxylase gene (odc) located on the core genome of this bacterium. The cfa7 gene was also targeted by PCR to re-confirm the presence of the cfa gene cluster. All primers are described in Table 1. PCR amplification conditions consisted of an initial denaturation step at 95°C for 3 min, followed by 40 cycles of 94°C for 30 s, an annealing temperature specific to each primer pair (Table 1) for 30 s, and an extension step at 72°C for 30 s. A final extension step was carried out at 72°C for 5 min. Negative PCR controls were included for PCRs with each primer set.
Results
HAI2 Is Required for Full Virulence of P. atrosepticum on Potato Stems
A role for HAI2 in virulence of P. atrosepticum SCRI1043 was examined by comparing the aggressiveness of the wild type with that of a strain in which the ICE had been removed by CRISPR-Cas-mediated genome targeting (Vercoe et al., 2013; Richter et al., 2014). The HAI2 junction of this deletion mutant had been previously sequenced. To confirm this deletion and to rule out off-target mutations of this ‘ICE-less’ strain (SCRI1043ΔHAI2), we sequenced the genome of the mutant. Assembly of the sequenced genome showed that the only large scale deletion from the genome was that of the ∼100 kb ICE HAI2 targeted using the CRISPR-Cas system (Supplementary Figure S1), demonstrating the specificity of the endogenous type I-F CRISPR-Cas system for genome editing.
Plant infection assays were subsequently performed with the ‘ICE-less’ strain or the wild type using two different starting quantities of inoculum. In the wild type, blackleg incidence increased with greater inoculum (p = 0.018): 77.3% (95% confidence limits; 55.7, 90.2) of plants showing symptoms when inoculated with 106 cells versus 50.0% (30.2, 69.8) when inoculated with 104 cells per inoculation site (Figure 1A). The concentration of the inoculum also had a significant effect on the incidence of blackleg in plants inoculated with the ‘ICE-less’ strain, with blackleg incidence reaching 45.5% (95% confidence limits; 26.5, 65.9) in plants inoculated with 106 cells of the ICE-less strain but only 22.7% (9.8, 44.3) when inoculated with 104 cells after 14 days (Figure 1A).
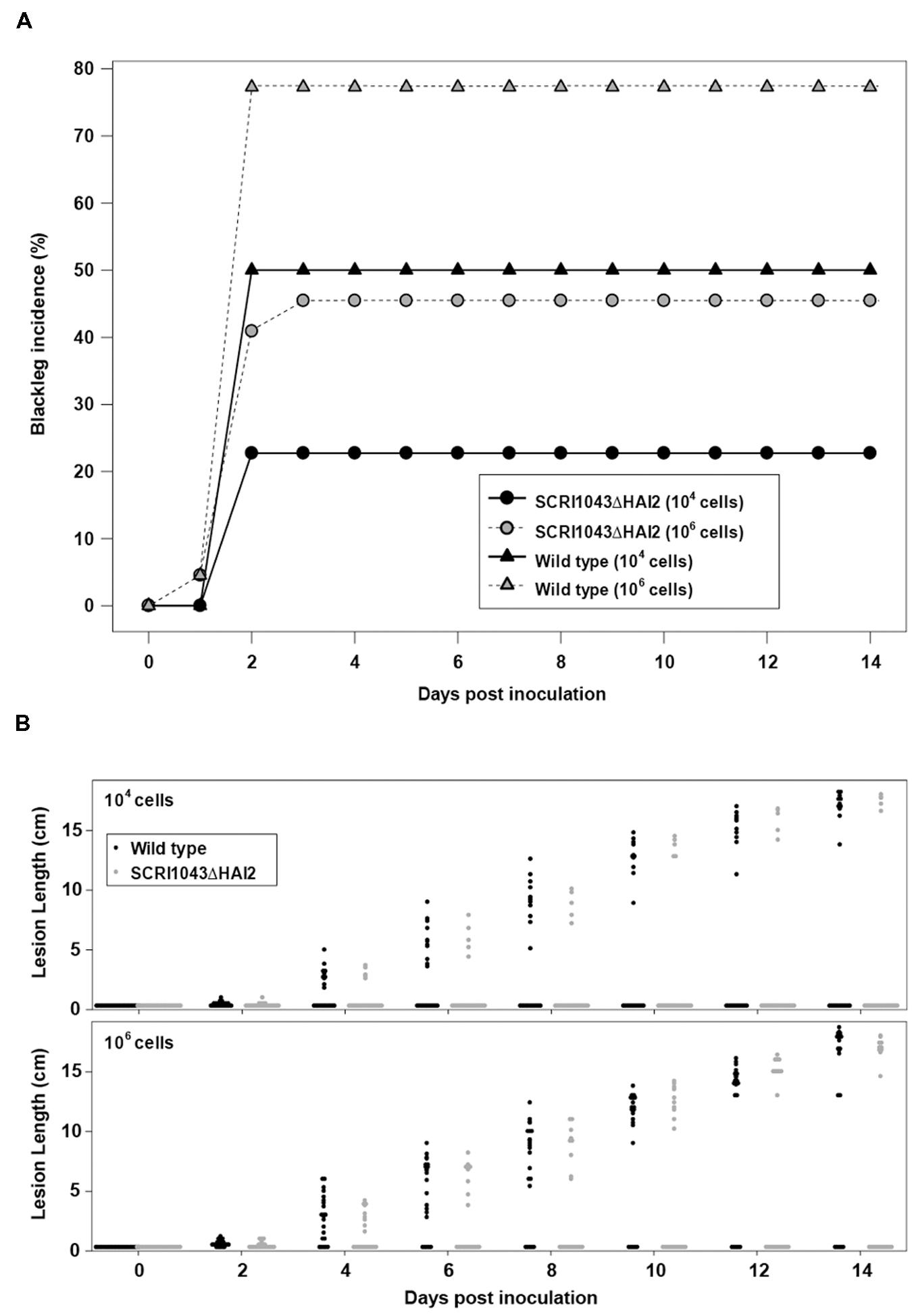
FIGURE 1. Deletion of HAI2 from Pectobacterium atrosepticum SCRI1043 reduces development of blackleg on potato stems. (A) Incidence of blackleg (% of plants with stem lesions) at each assessment for plants inoculated with either SCRI1043ΔHAI2 or the wild type. Each bacterium was inoculated into plants at either 104 or 106 cells per inoculation site. (B) Dot-histograms of lesion lengths on stems for each of 22 potato plants inoculated with either 104 or 106 cells per inoculation site of SCRI1043ΔHAI2 or the wild type. Measurements were taken every second day to 14 days post-inoculation.
Using either inoculum amounts, significantly more plants developed blackleg lesions when inoculated with the wild type compared with the ‘ICE-less’ strain (p = 0.005; Figure 1A). For plants that formed lesions, however, the lesions were similar in length whether inoculated with the wild type or the ‘ICE-less’ strain. The amount of inoculum did not alter the length of these lesions either (p > 0.4 for the strain and concentration main effects and for the interaction between them at 14 dpi; Figure 1B). Thus, the HAI2 mutant produced a lower incidence of blackleg, but once disease symptoms became established the mutation had no detectable impact on lesion development.
Detection of HAI2 and HAI2-Related ICEs in Isolates of P. atrosepticum
Given the role of HAI2 in the virulence of P. atrosepticum SCRI1043, the distribution of HAI2-like loci was examined in other potato isolates of P. atrosepticum by PCR. Products indicative of cfa6 and cfa7 were amplified using DNA from all nine isolates (Supplementary Table S1). Amplicons were also generated using primers targeted to ECA0516, ECA0525, ECA0532, and ECA0614 (indicative of the presence of HAI2) using DNA from all isolates. These data suggested that not only was the cfa cluster ubiquitous in this pathogen, but it was likely to be harbored on similar ICEs in different strains.
To examine whether the ICEs carrying the cfa cluster had integrated into the phe-tRNA gene immediately downstream of ECA0515 (attB0515) in each strain (as in SCRI1043), attempts were made to amplify attL and attR by PCR. The attL and attR loci were detected in all P. atrosepticum strains with the exception of ICMP 1526 (the type strain for P. atrosepticum; Supplementary Table S1). The inability to detect these loci in P. atrosepticum ICMP 1526 suggested that the regions close to the proximity of the ICE were sufficiently different from those in HAI2 to restrict binding of the PCR primers used for PCR amplification. Alternatively, the cfa cluster was located on a plasmid or HAI2 had integrated at a chromosomal target site other than attB0515 in P. atrosepticum ICMP 1526.
At the same time, the formation of attP and attB0515 was measured by qPCR to determine the dynamics of ICE excision in P. atrosepticum. In these experiments, both attP and attB0515 were detected in all isolates (Figure 2). The ratios for attP and attB0515 were 2.25 × 10-5 and 8.45 × 10-6 in P. atrosepticum SCRI1043, consistent with the low frequency of HAI2 excision from the phe-tRNA gene immediately downstream of ECA0515 and the formation of the circular extrachromosomal form reported previously (Vanga et al., 2012, 2015). The relative proportion of attP compared to attB0515 calculated using these ratios was 2.67. A ratio above 1 suggested that the ICE may be replicating at low copy number.
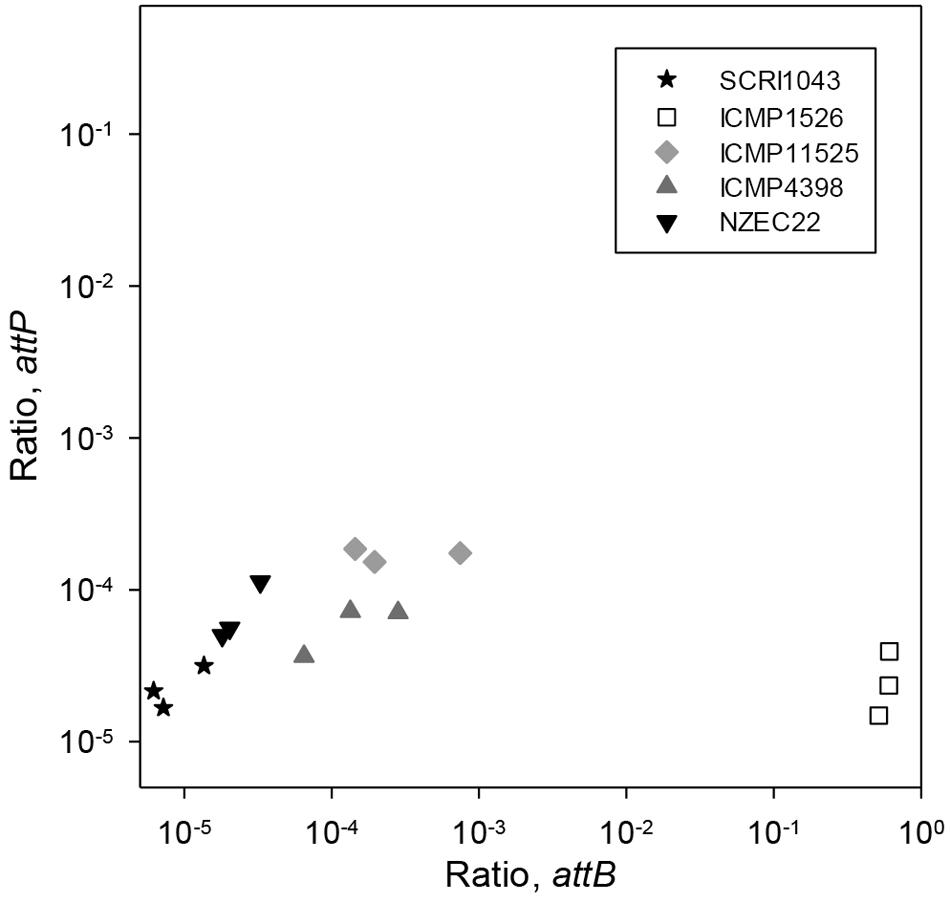
FIGURE 2. Excision of HAI2-like elements in various strains of P. atrosepticum grown in vitro. Back-transformed mean log10 ratios (normalized to ECA0515) for attP are plotted against those for attB0515 on a log10 spaced axes for P. atrosepticum strains SCRI1043, NZEC22, ICMP 11525, ICMP 4398, and ICMP 1526. Cultures were grown in MM for 24 h, in triplicate. Three qPCR reactions were performed for each DNA sample and the mean ratio was plotted. The LSD between two means on the transformed scale was 0.29 for attP and was 0.44 for attB (at the 5% significance level with 10 degrees of freedom).
Consistent with the findings in P. atrosepticum SCRI1043, in P. atrosepticum NZEC22 the relative proportion of attP (2.29 × 10-5) to attB0515 (6.81 × 10-5) was 2.97. In P. atrosepticum ICMP 4398 and ICMP 11525, however, the relative proportions of attP to attB0515 were 0.42 and 0.62, respectively. A value lower than 1 suggested that in these two strains the ICE was either highly unstable or was capable of integrating into a second chromosomal target site as well as attB0515. Furthermore, based on the attP and attB0515 ratios, ICE excision in the two strains was ∼10-fold greater than for P. atrosepticum SCRI1043 (p ranging from p < 0.001 to p < 0.05).
In P. atrosepticum ICMP 1526, attP (2.39 × 10-5) was similar (p > 0.05) to that for P. atrosepticum SCRI1043, but attB0515 was detected in almost all cells (CTattB = CtECA0515; Figure 2). Given that attL and attR were not detected in P. atrosepticum ICMP 1526 by PCR (Supplementary Table S1), and that attB0515 was detected in most cells of P. atrosepticum ICMP 1526, HAI2 was almost certainly integrated elsewhere in the genome of this strain.
Detection of HAI2-Related ICEs in Isolates of P. carotovorum Causing Blackleg
The distribution of HAI2-like ICEs and the cfa biosynthetic genes was also examined in 62 P. carotovorum isolates from potato by PCR. A cfa6 amplicon and a cfa7 product were amplified using DNA from only seven (Supplementary Table S1). These isolates were all known to cause blackleg (Slawiak and Lojkowska, 2009; Panda et al., 2012), and included the New Zealand isolates P. carotovorum subsp. brasiliensis ICMP 19477 and NZEC150, as well as the five P. carotovorum subsp. carotovorum strains from Peru that were already known to harbor the cfa biosynthetic cluster (Slawiak and Lojkowska, 2009). The attL, ECA0516, ECA0525, and ECA0532 fragments were not amplified using DNA from any of the P. carotovorum isolates, while attR and ECA0614 were only amplified from P. carotovorum subsp. carotovorum UGC32 and the other related isolates from Peru. Consistent with these results, attP was not detected using DNA from P. carotovorum in qPCR (data not shown).
The ICE Encoding Cfa in P. carotovorum subsp. brasiliensis ICMP 19477 Is Distinct from HAI2
The absence of the HAI2-related loci in isolates such as P. carotovorum subsp. brasiliensis ICMP 19477 suggested that the cfa cluster in P. carotovorum was located on a mobile element distinct from HAI2. Thus, the genetic structure and organization of the cfa biosynthetic clusters in P. atrosepticum and P. carotovorum as well as the mobile elements encoding them were examined further by sequencing the genomes of P. atrosepticum ICMP 1526, P. carotovorum subsp. brasiliensis ICMP 19477 and P. carotovorum subsp. carotovorum UGC32. P. atrosepticum ICMP 1526 was chosen for DNA sequencing, as it was the type strain for P. atrosepticum and attL and attR were not amplified using DNA from this isolate as a template in PCR. These data, along with the high amount of attB0515 detected in qPCR, suggested an alternative HAI2 insertion site in P. atrosepticum ICMP 1526. PCR also failed to amplify attL, attR, and attP from P. carotovorum subsp. brasiliensis ICMP 19477, which causes blackleg and represents one of the two lineages of P. carotovorum on potato. Similarly, the DNA sequence of UGC32 was obtained as this isolate has the capacity to cause blackleg and is purportedly characteristic of the second lineage of P. carotovorum (P. carotovorum subsp. carotovorum).
Comparison of HAI2 from P. atrosepticum SCRI1043 and the draft genome sequences from P. atrosepticum ICMP 1526, P. carotovorum subsp. brasiliensis ICMP 19477, and P. carotovorum subsp. carotovorum UGC32 revealed homologous cfa biosynthetic clusters in each of the Pectobacterium isolates (Figure 3). Their organization was highly conserved, each cluster spanning 22 kb and comprising the nine genes required for synthesis of the polyketide in P. atrosepticum SCRI1043 (cfa1 to cfa8b). The amino acid sequences of the cfa genes also had between 100 and 96% identity to those in P. atrosepticum SCRI1043 (100% for P. atrosepticum ICMP 1526 and 96% for both P. carotovorum subsp. brasiliensis ICMP 19477 and P. carotovorum subsp. carotovorum UGC32). A cfl gene, which encodes coronafacate ligase, was located adjacent to the cfa biosynthetic clusters in each strain, although the amino acid sequences they encode were highly divergent; the amino acid identity of cfl in P. carotovorum subsp. carotovorum UGC32 (when compared with the cfl gene in P. atrosepticum SCRI1043) was 93%, but only 35% in P. atrosepticum ICMP 1526 and 31% in P. carotovorum subsp. brasiliensis ICMP 19477. As in P. atrosepticum SCRI1043, however, the cma biosynthetic cluster was absent from the genomes of the three newly sequenced pathogens. As coronafacate ligase mediates conjugation of Cfa to Cma in pseudomonads, the absence of a cma biosynthetic cluster and the divergence of the cfl gene suggests that Cfa may be ligated to one or more unidentified molecules in Pectobacterium.
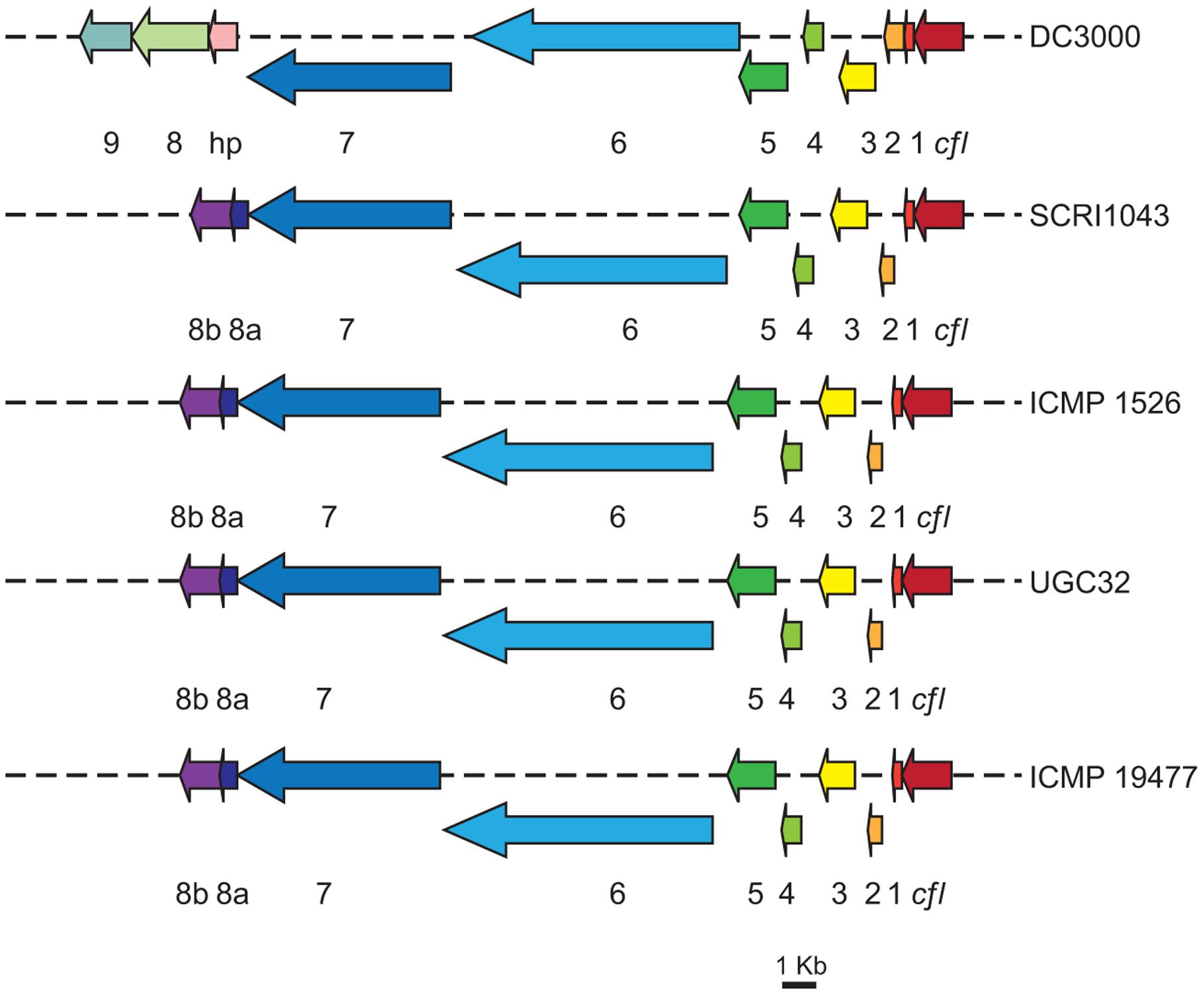
FIGURE 3. Organization of the cfa biosynthetic operons in different isolates of Pectobacterium. The cfa (1–9) and the cfl genes are illustrated (where appropriate) for Pseudomonas syringae pv. tomato DC3000, P. atrosepticum SCRI1043, P. atrosepticum ICMP 1526, P. carotovorum subsp. carotovorum UGC32 and P. carotovorum subsp. brasiliensis ICMP 19477. Colored arrows represent the predicted biosynthetic genes [scale bar: 1 Kb], hp, denotes a gene encoding a hypothetical protein predicted to have thioesterase activity, which has weak similarity to cfa8a. The cfa9 gene is found only in P. syringae pv. tomato DC3000.
The homologous cfa biosynthetic clusters in each of the Pectobacterium isolates were also highly similar to the cfa cluster from P. syringae pv. tomato DC3000 (Figure 3). Amino acid sequence comparisons showing 45–75% identity between proteins Cfl to Cfa8b from P. atrosepticum SCRI1043 and those (Cfl to Cfa8) in the pseudomonad. In contrast, cfa9 was not detected in the genomes of the Pectobacterium isolates. Cfa9 is dispensable for Cfa and Cor production in P. syringae pv. tomato DC3000, but may increase the release of enzyme-bound products from the Cor pathway (Rangaswamy et al., 1998).
As in P. atrosepticum SCRI1043, the cfa biosynthetic clusters in P. atrosepticum ICMP 1526, P. carotovorum subsp. brasiliensis ICMP 19477 and P. carotovorum subsp. carotovorum UGC32 were located on putative ICEs. As predicted from excision assays, the ICE (named Pba1526_HAI2) in P. atrosepticum ICMP 1526 had high sequence identity (99%) to HAI2 (Figure 4), but was inserted into a second phe-tRNA gene located adjacent to speC (locus tag: ECA0967). Pba1526_HAI2 was 97,216 bp, contained 97 putative CDSs (Supplementary Table S2) and was delineated by 49-bp direct repeat sequences attL and attR. Gene organization on Pba1526_HAI2 was almost identical to that on HAI2 (Figure 4; Supplementary Table S2). Synteny was lost in only one region, notably, within and immediately adjacent to pilV, which encodes a component of the type IV pilus involved in conjugation. The pilV shufflon on other mobile elements acts as a biological switch, selecting one of seven C-terminal segments of the pilV gene that confer recipient specificity during conjugation (Komano et al., 1987).
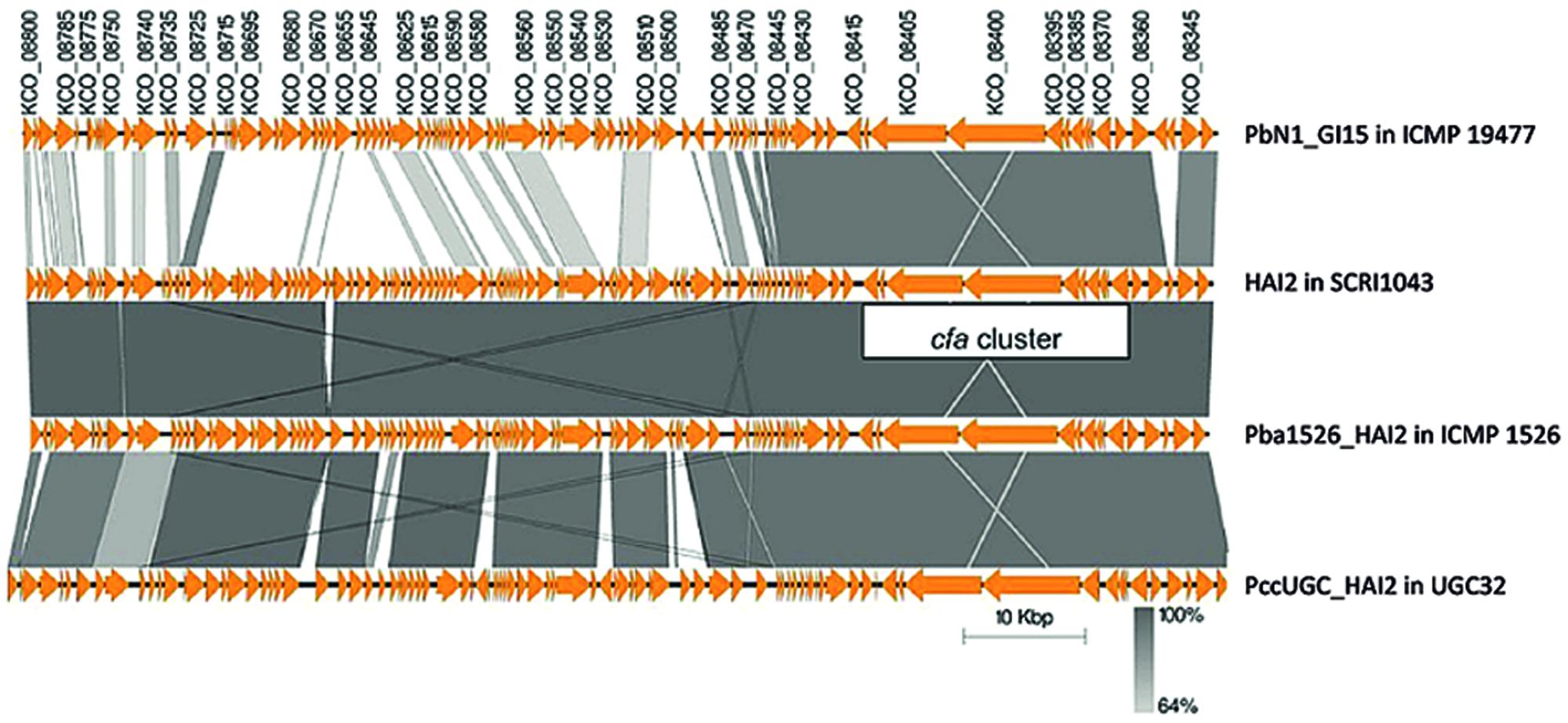
FIGURE 4. A nucleotide alignment of putative ICEs encoding coronafacic acid in strains of P. atrosepticum and P. carotovorum. The schematic representation drawn using Easyfig (Sullivan et al., 2011) shows pairwise alignments between PbN1_GI15 from P. carotovorum subsp. brasiliensis ICMP 19477, HAI2 from P. atrosepticum, SCRI1043, Pba1526_HAI2 from P. atrosepticum ICMP 1526 and PccUGC_HAI2 from P. carotovorum subsp. carotovorum UGC32. Orange arrows indicate annotated Coding Domain Sequences (CDSs). The locus tags for CDSs from the putative ICE in P. carotovorum subsp. brasiliensis ICMP 19477 are provided above the arrows, and their regions of nucleotide identity with CDSs from other putative ICEs are represented by gray shading. The genome coordinates for the ICEs in each strain are as follows: 910293 to 1008796 for PbN1_GI15, 590755 to 688630 for HAI2, 958243 to 1055429 for Pba1526_HAI2 and 591326 to 692291 for PccUGC_HAI2.
The cfa cluster was harbored on two different ICEs in P. carotovorum. PccUGC_HAI2, the ICE in P. carotovorum subsp. carotovorum UGC32, was highly similar to HAI2 (95% nucleotide identity) and located within the same target site (attB0515). PbN1_GI15, the ICE in P. carotovorum subsp. brasiliensis ICMP 19477, was distinct from HAI2 and was inserted within the phe-tRNA gene located adjacent to speC. Both PccUGC_HAI2 and PbN1_GI15 were delineated by the 49-bp direct repeats that define HAI2 and Pba1526_HAI2, which are important for insertion into either of the two target sites.
PccUGC_HAI2 was 100,966 bp in size and contained 100 putative CDSs. Of these 100 CDSs, 90 were present in the related ICE, HAI2. The functions of the 10 CDSs unique to PccUGC_HAI2 were unknown as they encode hypothetical proteins. The exception was GO33_02925, which possesses a domain with similarity to Abi_2 family proteins that include the bacteriophage resistance AbiD1 protein from Lactococcus lactis (Anba et al., 1995). PccUGC_HAI2, on the other hand, was missing eight CDSs from HAI2 including two genes (ECA0582 and ECA0583) that appear to encode a putative toxin–antitoxin system with similarity to the pemK-pemI addiction module. The protein encoded by ECA0582 had 28% amino acid identity to the factor encoded by pemK, which stabilizes plasmid R100 in bacterial populations by killing plasmid-free segregants (Tsuchimoto et al., 1992). ECA0583 produced a protein with 33% similarity to PemI, an unstable inhibitor of PemK that suppresses plasmid stabilization. No alternative toxin–antitoxin system could be identified on PccUGC_HAI2 using RASTA-Bacteria: a web-based tool for identifying toxin–antitoxin loci in prokaryotes (Sevin and Barloy-Hubler, 2007).
PbN1_GI15 was 98,504 bp and possessed a total of 93 putative CDSs (Supplementary Tables S2 and S3). Although it was distinct from HAI2, many of the CDSs in PbN1_GI15 had strong similarity to those in HAI2, especially in the region surrounding the cfa cluster between KCO_08485 (ECA0587 in HAI2) and KCO_08340 (ECA0614 in HAI2; Figure 4; Supplementary Table S2). Of the 28 putative CDSs annotated in this region of PbN1_GI15, 24 were similar to genes from HAI2, including the integrase (locus tag KCO_08340) that was located adjacent to one of the direct repeats (attR) delineating the ICE. The integrase mediates excision of HAI2 out of the chromosome (Vanga et al., 2015). ECA0612, which encoded a DinB-family protein involved in regulation of SOS-related responses, was absent from PbN1_GI15, with two putative CDSs (KCO_08350 and KCO_08355) transcribed on the complementary strand of PbN1_GI15 instead. Like ECA0612, KCO_08350 possessed a DNA-binding motif and is predicted to be involved in transcriptional regulation.
Sequence similarities between PbN1_GI15 and HAI2 were dramatically lower upstream of KCO_08485, which encodes a second putative integrase. Of most note, a cluster of seven genes was present in HAI2 but absent from PbN1_GI15. This cluster encoded the same pemIK putative toxin–antitoxin (ECA0582-83) system missing from PbN1_GI15 as well as a Type I restriction-modification system encoded by ECA0584 (methylase) and ECA0585 (restriction endonuclease). As for PccUGC_HAI2, no alternative toxin–antitoxin (or restriction modification) system could be identified on the ICE using RASTA-Bacteria (Sevin and Barloy-Hubler, 2007).
A second cluster of genes was absent in PbN1_GI15. These genes were located adjacent to the pil operon in HAI2 and were predicted to encode proteins involved in conjugal transfer (e.g., TraE). In contrast, PbN1_GI15 harbored a group of six CDSs (locus tags KCO_08550 to KCO_08525) that were absent from HAI2 that have largely no known function. The only exception was KCO_08530, which was predicted to produce an SMF protein likely involved in DNA uptake. PbN1_GI15 also encoded a RNA-dependent DNA polymerase similar to those associated with group II introns (encoded by KCO_08725) as well as a distinct type IV secretion system.
Mobilization of PbN1_GI15 in P. carotovorum subsp. brasiliensis ICMP 19477
Despite the differences between PbN1_GI15 and HAI2, PbN1_GI15 retains features of a mobile element (e.g., an integrase, direct repeats). Thus, mobilization of PbN1_GI15 was examined by PCR. Firstly, integration of PbN1_GI15 into the chromosomal target site attBspeC in P. carotovorum subsp. brasiliensis ICMP 19477 was confirmed, with amplicons indicative of both attL and attR produced (Figure 5). However, many ICEs become anchored in the genome and lose their capacity to excise. Therefore, the excision of PbN1_GI15 from the chromosome was monitored by measuring the formation of attPPbN1_GI15 and attBspeC using PCR. These reactions produced fragments indicative of both loci (Figure 5), confirming precise excision of PbN1_GI15 and reconstitution of the chromosomal target site in P. carotovorum subsp. brasiliensis ICMP 19477. The amplification of attPPbN1_GI15 was consistent with this ICE retaining the capacity to excise from the chromosome prior to transfer to donor cells. No amplicons were produced in the negative PCR controls (containing no template DNA).
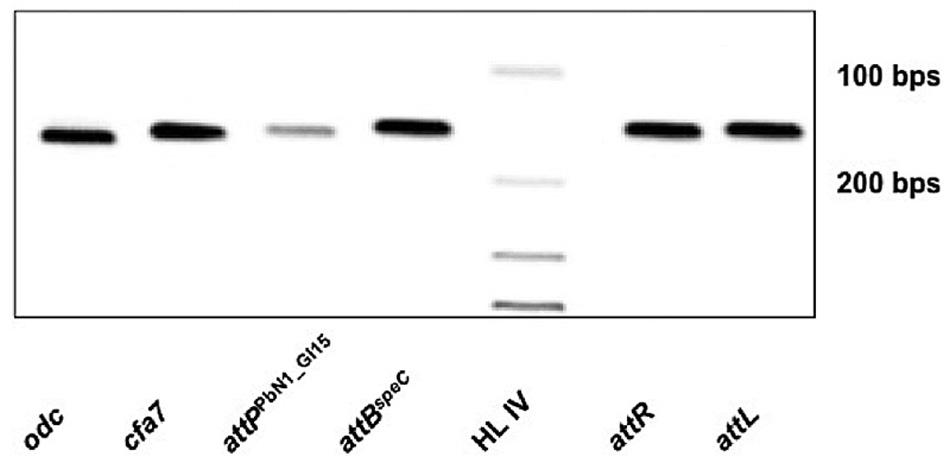
FIGURE 5. Excision of ICE PbN1_GI15 in P. carotovorum subsp. brasiliensis ICMP 19477. Electrophoresis gel image showing PCR amplicons associated with mobilization of ICE PbN1_GI15. Lane 1 (odc), ornithine decarboxylase gene; lane 2 (cfa7), coronafacic acid biosynthetic gene; lane 3 (attPPbN1_GI15), the attP site formed upon the circularization of PbN1_GI15 during excision from attBspeC; lane 4 (attBspeC), 152-bp amplicon indicative of reconstitution of the chromosomal target site attBspeC upon mobilization of PbN1_GI15; lane 5 (HL IV), Hyperladder IV (Bioline, UK); lane 6 (attR), the 153 bp sequence delimiting the right end of PbN1_GI15 when integrated within the attB site adjacent the speC; lane 7 (attL), the 153 bp sequence delimiting the left end of PbN1_GI15 when integrated within the attB site adjacent to speC.
Discussion
Deletion of HAI2 from the genome of P. atrosepticum SCRI1043 by CRISPR-Cas-mediated genome editing resulted in reduced virulence of the pathogen on potato. Previous studies of ICE biology have relied largely on the inactivation of the virulence determinants harbored on the mobile element or on screening a population for individuals in which the ICE has been lost. Targeted inactivation of virulence determinants requires complex cloning strategies and only reveals the roles of specific genes, which can overlook many unknown virulence genes. For example, two prophages in the genome of P. atrosepticum SCRI1043 harbor no known virulence determinants, yet their deletion results in a reduction in the virulence of the pathogen (Evans et al., 2010). The low frequency of both ICE excision (between 10-5 and 10-6 per cell; Buchrieser et al., 1998; Lesic et al., 2004; Sakellaris et al., 2004) and subsequent loss from the bacterial population under laboratory conditions (Lovell et al., 2009) also makes the identification of strains without an ICE challenging.
The reduction in the virulence of P. atrosepticum SCRI1043 upon deletion of HAI2 was not entirely unexpected, given that a transposon insertion within the cfa biosynthetic cluster reduces severity of blackleg in plants infected by the pathogen (Bell et al., 2004). Inactivation of PbTopo IIIβ, a topoisomerase encoded on HAI2, also reduces the incidence of the disease (Vanga et al., 2012). However, the deletion of the entire ICE did not result in the reduction in the length of blackleg symptoms described by Bell et al. (2004), which may have been because a different cultivar was used in the two plant assays or as a result of different experimental conditions. Alternatively, it may be due to hidden costs associated with retaining the ICE. ICEs can have great evolutionary benefits, but may also be disadvantageous to the cell. For example, in susceptible leaf tissues of Phaseolus vulgaris, P. syringae pv. phaseolicola exhibits reduced colony formation compared with an isogenic strain without the ICE PPHGI-1 (Godfrey et al., 2010). Reduced colony formation of the strain containing the PPHGI-1 is predicted to result from a compromise in the in planta fitness of the bacterium due to one or more plant responses triggered by the ICE.
Consistent with the findings of Slawiak and Lojkowska (2009), a screen of isolates of P. atrosepticum using PCR revealed loci associated with the cfa biosynthetic cluster in all. This, together with the ubiquitous nature of other loci associated with HAI2, suggested that the ICE and the virulence factor it harbors were acquired prior to the speciation of P. atrosepticum. Therefore, it appears that the ICE remains under strong selective pressure to be retained. Comparative genomics of P. atrosepticum SCRI1043 and P. atrosepticum ICMP 1526 confirmed the very high level of homology and synteny amongst the ICEs in P. atrosepticum. Of particular note, a candidate toxin–antitoxin system was identified in both ICEs, closely associated with a putative restriction-modification system. Toxin–antitoxin systems participate in the maintenance of ICEs. For example, retention of SXT, an ICE in Vibrio cholerae that encodes multiple antibiotic resistance genes, is promoted by mosA and mosT (Wozniak and Waldor, 2009). When integrated in the chromosome, mosAT expression is shut down by MosA, but expression of mosA and mosT is up-regulated when SXT is extrachromosomal and vulnerable to loss. Thus, SXT seems to activate a toxin–antitoxin module that minimizes formation of SXT-free cells (Wozniak et al., 2009; Wozniak and Waldor, 2010). Genome context analysis has also shown that restriction-modification genes are often located on mobile elements such as plasmids and prophages (Betlach et al., 1976; Kita et al., 2003), and some are linked to recombination-related genes such as integrases, invertases, and transposases (Anton et al., 1997). Like the toxin–antitoxin system, restriction-modification systems mediate plasmid maintenance (Kulakauskas et al., 1995) and have also been implicated in site-specific recombination (Chang and Stanley, 1977). Given that HAI2 is not essential for pathogenicity of its bacterial host, it is possible that strong selection for retention of this ICE is conferred by these addiction modules.
Comparative analyses of the ICEs in P. atrosepticum ICMP 1526 and P. atrosepticum SCRI1043 revealed they can insert into the identical 49-bp attB sites present in either of two phe-tRNA. These two phe-tRNAs are located in different regions of the chromosome, one adjacent to speC (locus tag: ECA0967) and one some distance away (adjacent to locus tag: ECA0615). Insertion of HAI2 into multiple integration sites is consistent with the findings of Lovell et al. (2009), who showed that PPHGI-1 can insert into one of two sites in the genome of P. syringae pv. phaseolicola 1448A upon transmission from the donor P. syringae pv. phaseolicola 1302A. The P. syringae pv. phaseolicola 1448A genome contains two 52-bp att sites with the same sequence as the att borders of PPHGI-1 in the 1302A genome. PPHGI-1 integrated preferentially into one of these att sites. Interestingly, HAI2 (and its relatives) was also largely detected in the insertion site adjacent to locus tag ECA0615, suggesting that this ICE preferentially integrates into one insertion site in the chromosome of P. atrosepticum. The reasons for such a preference are unknown as the two attB sites in this pathogen are identical. The impacts to the cell of insertion into different sites also remain to be studied. Nevertheless, genome context may be critical for successful chromosomal insertion given that several genes on HAI2 predicted to alter DNA topology not only influence excision but also transcription of genes both on the ICE and elsewhere in the chromosome (Vanga et al., 2012, 2015). Studying the genome-wide transcription of strains in which HAI2 has been removed or inserted at different locations would identify if the ICE has different impacts on the host bacterium depending on its position in the genome.
The cfa biosynthetic cluster was also detected in strains belonging to two subspecies of P. carotovorum (as defined using multi-locus sequence analysis by Slawiak and Lojkowska, 2009 and Panda et al., 2012). Pectobacterium carotovorum subsp. brasiliensis is a well-described blackleg pathogen (Duarte et al., 2004), so, as in P. atrosepticum, the presence of the cfa cluster in ICMP 19477 may support the invasion of the xylem by this enterobacterium. A cfa biosynthetic cluster is not harbored in the genome of P. carotovorum subsp. brasiliensis 1692 however, (Glasner et al., 2008), indicating that Cfa production is unlikely to be essential for pathogenicity of this subspecies in stems (again, similar to its role in P. atrosepticum).
The capacity of P. carotovorum subsp. carotovorum to elicit blackleg disease is controversial. Thus, the detection of the cfa biosynthetic cluster in P. carotovorum subsp. carotovorum UGC32 and its absence from so many other isolates is intriguing. Pectobacterium carotovorum subsp. carotovorum UGC32 causes blackleg (Slawiak and Lojkowska, 2009), whereas most other P. carotovorum subsp. carotovorum isolates are only able to invade potato tubers. Perhaps, as in P. atrosepticum and P. carotovorum subsp. brasiliensis, this cluster supports invasion of the xylem by blackleg causing strains of P. carotovorum subsp. carotovorum. Consistent with this hypothesis, P. carotovorum subsp. carotovorum ICMP 5702 is unable to invade the stems of potato and its genome does not encode Cfa (data not shown). A broader genome-based taxonomic revision of P. carotovorum will be required, however, to shed further light on the importance of Cfa production to this pathogen and its capacity to cause blackleg.
The detection of cfa biosynthetic clusters in only limited strains of P. carotovorum subsp. brasiliensis and P. carotovorum subsp. carotovorum indicates that the clusters were probably acquired as a result of independent lateral transfer events in these strains. Consistent with this hypothesis, DNA sequencing of the ICEs harboring the cfa biosynthetic clusters in ICMP 19477 and UGC32 demonstrated that they were distinct from HAI2. They were also distinct from one another. The low incidence of these ICEs in P. carotovorum might also result from barriers to acquisition, however. For example, CRISPR-Cas systems can act as adaptive immune systems, which provide resistance to incoming mobile elements such as plasmids and phages (Dy et al., 2014). In P. atrosepticum, HAI2 appears to be a target for its endogenous CRISPR-Cas system except that a single nucleotide mutation adjacent to the targeted sequence in HAI2 results in CRISPR-Cas avoidance (Vercoe et al., 2013). Furthermore, genome sequencing of P. carotovorum subsp. brasiliensis ICMP 19477 and P. carotovorum subsp. carotovorum UGC32 failed to identify the candidate restriction modification and toxin–antitoxin genes in PbN1_GI15 and PccUGC_HAI2, which are present in derivatives of HAI2 in P. atrosepticum. As no alternative restriction-modification and toxin–antitoxin systems could be found on these ICEs either, the ICEs detected in P. carotovorum may be less stable than their counterparts in P. atrosepticum.
Finally, excision assays confirmed mobilization of PbN1_GI15 from the chromosome of P. carotovorum subsp. brasiliensis ICMP 19477. As mobilization from the chromosome occurs in preparation for transfer of an ICE to a new host (Clark and Adelberg, 1962), future acquisition of this ICE by other strains of P. carotovorum is possible. Furthermore, given the ICE harbors the cfa biosynthetic cluster, acquisition of PbN1_GI15 would be likely to enhance the capacity of recipient strains to cause blackleg. The probability of these events occurring could be addressed using transfer assays such as those described by Lovell et al. (2009).
Author Contributions
AP, PF, KA, and CR provided substantial contributions to the conception or design of the work. AP, PF, PP, BV, RB, AL, and MF provided substantial contributions to acquisition, analysis, or interpretation of data for the work. AP, PF, KA, CR, PP, BV, RB, AL, and MF were involved in drafting the work or revising it critically for important intellectual content and provided final approval of the version to be submitted. All authors agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was funded by the Tertiary Education Commission of New Zealand through the Bio-Protection Research Centre and by a grant from the Marsden Fund administered by the Royal Society of New Zealand (RSNZ). PF was funded by a Rutherford Discovery Fellowship, RSNZ.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2016.00397
Footnotes
References
Anba, J., Bidnenko, E., Hillier, A., Ehrlich, D., and Chopin, M. C. (1995). Characterization of the lactococcal abiD1 gene coding for phage abortive infection. J. Bacteriol. 177, 3818–3823.
Anton, B. P., Heiter, D. F., Benner, J. S., Hess, E. J., Greenough, L., Moran, L. S., et al. (1997). Cloning and characterization of the Bg/II restriction–modification system reveals a possible evolutionary footprint. Gene 187, 19–27. doi: 10.1016/S0378-1119(96)00638-5
Baghaee-Ravari, S., Rahimian, H., Shams-Bakhsh, M., Lopez-Solanilla, E., Antunez-Lamaz, M., and Rodriguez-Penzuela, P. (2011). Characterization of Pectobacterium species from Iran using biochemical and molecular methods. Eur. J. Plant Pathol. 129, 413–425. doi: 10.1007/s10658-010-9704-z
Bell, K. S., Sebaihia, M., Pritchard, L., Holden, M. T. G., Hyman, L. J., Holeva, M. C., et al. (2004). Genome sequence of the enterobacterial phytopathogen Erwinia carotovora subsp atroseptica and characterization of virulence factors. Proc. Natl. Acad. Sci. U.S.A. 101, 11105–11110. doi: 10.1073/pnas.0402424101
Bender, C. L., Alarcon-Chaidez, F., and Gross, D. C. (1999). Pseudomonas syringae Phytotoxins: mode of action, regulation, and biosynthesis by peptide and polyketide synthetases. Microbiol. Mol. Biol. Rev. 63, 266–292.
Betlach, M., Hershfield, V., Chow, L., Brown, W., Goodman, H., and Boyer, H. W. (1976). A restriction endonuclease analysis of the bacterial plasmid controlling the EcoRI restriction and modification of DNA. Fed. Proc. 35, 2037–2043.
Buchrieser, C., Brosch, R., Bach, S., Guiyoule, A., and Carniel, E. (1998). The high-pathogenicity island of Yersinia pseudotuberculosis can be inserted into any of the three chromosomal asn tRNA genes. Mol. Microbiol. 30, 965–978. doi: 10.1046/j.1365-2958.1998.01124.x
Bueno, S. M., Santiviago, C. A., Murillo, A. A., Fuentes, J. A., Trombert, A. N., Rodas, P. I., et al. (2004). Precise excision of the large pathogenicity island, SPI-7, in Salmonella enterica serovar Typhi. J. Bacteriol. 186, 3202–3213. doi: 10.1128/JB.186.10.3202-3213.2004
Chang, S., and Stanley, N. C. (1977). In vivo site-specific genetic recombination promoted by EcoR1 restriction endonuclease. Proc. Natl. Acad. Sci. U.S.A. 74, 4811–4815. doi: 10.1073/pnas.74.11.4811
Clark, A. J., and Adelberg, E. A. (1962). Bacterial conjugation. Annu. Rev. Microbiol. 16, 289–319. doi: 10.1146/annurev.mi.16.100162.001445
De Boer, S. H. (2002). Relative incidence of Erwinia carotovora subsp atroseptica in stolon end and peridermal tissue of potato tubers in Canada. Plant Dis. 86, 960–964. doi: 10.1094/PDIS.2002.86.9.960
De Boer, S. H., Li, X., and Ward, L. J. (2012). Pectobacterium spp. associated with bacterial stem rot syndrome of potato in Canada. Phytopathology 102, 937–947. doi: 10.1094/PHYTO-04-12-0083-R
de Haan, E. G., Dekker-Nooren, T. C. E. M., van den Bovenkamp, G. W., Speksnijder, A. G. C. L., van der Zouwen, P. S., and van der Wolf, J. M. (2008). Pectobacterium carotovorum subsp carotovorum can cause potato blackleg in temperate climates. Eur. J. Plant Pathol. 122, 561–569. doi: 10.1007/s10658-008-9325-y
Duarte, V., De Boer, S. H., Ward, L. J., and de Oliveira, A. M. R. (2004). Characterization of atypical Erwinia carotovora strains causing blackleg of potato in Brazil. J. Appl. Microbiol. 96, 535–545. doi: 10.1111/j.1365-2672.2004.02173.x
Dy, R. L., Pitman, A. R., and Fineran, P. C. (2013). Chromosomal targeting by CRISPR-Cas systems can contribute to genome plasticity in bacteria. Mob. Genet. Elements 3, e26831.
Dy, R. L., Richter, C., Salmond, G. P. C., and Fineran, P. C. (2014). Remarkable mechanisms in microbes to resist phage infections. Ann. Rev. Virol. 1, 307–331. doi: 10.1146/annurev-virology-031413-085500
Evans, T. J., Coulthurst, S. J., Komitopoulou, E., and Salmond, G. P. (2010). Two mobile Pectobacterium atrosepticum prophages modulate virulence. FEMS Microbiol. Lett. 304, 195–202. doi: 10.1111/j.1574-6968.2010.01901.x
Glasner, J. D., Marquez-Villavicencio, M., Kim, H. S., Jahn, C. E., Ma, B., Biehl, B. S., et al. (2008). Niche-specificity and the variable fraction of the Pectobacterium pan-genome. Mol. Plant Microbe Interact. 21, 1549–1560. doi: 10.1094/MPMI-21-12-1549
Godfrey, S. A. C., Mansfield, J. W., Corry, D. S., Lovell, H. C., Jackson, R. W., and Arnold, D. L. (2010). Confocal imaging of Pseudomonas syringae pv. phaseolicola colony development in bean reveals reduced multiplication of strains containing the Genomic Island PPHGI-1. Mol. Plant Microbe Interact. 23, 1294–1302. doi: 10.1094/MPMI-05-10-0114
Hacker, J., and Kaper, J. B. (2000). Pathogenicity islands and the evolution of microbes. Annu. Rev. Microbiol. 54, 641–679. doi: 10.1146/annurev.micro.54.1.641
He, J., Baldini, R. L., Deziel, E., Saucier, M., Zhang, Q., Liberati, N. T., et al. (2004). The broad host range pathogen Pseudomonas aeruginosa strain PA14 carries two pathogenicity islands harboring plant and animal virulence genes. Proc. Natl. Acad. Sci. U.S.A. 101, 2530–2535. doi: 10.1073/pnas.0304622101
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Kim, H. S., Ma, B., Perna, N. T., and Charkowski, A. O. (2009). Phylogeny and virulence of naturally occurring type III secretion system-deficient Pectobacterium strains. Appl. Environ. Microbiol. 75, 4539–4549. doi: 10.1128/AEM.01336-08
Kita, K., Kawakami, H., and Tanaka, H. (2003). Evidence for horizontal transfer of the EcoT38I restriction-modification gene to chromosomal DNA by the P2 phage and diversity of defective P2 prophages in Escherichia coli TH38 strains. J. Bacteriol. 185, 2296–2305. doi: 10.1128/JB.185.7.2296-2305.2003
Komano, T., Kim, S. R., and Nisioka, T. (1987). Distribution of shufflon among IncI plasmids. J. Bacteriol. 169, 5317–5319.
Kulakauskas, S., Lubys, A., and Ehrlich, S. D. (1995). DNA restriction-modification systems mediate plasmid maintenance. J. Bacteriol. 177, 3451–3454.
Leite, L. N., de Haan, E. G., Krijger, M., Kastelein, P., van der Zouwen, P. S., van den Bovenkamp, et al. (2014). First report of potato blackleg caused by Pectobacterium carotovorum subsp. brasiliensis in the Netherlands. New Dis. Rep. 29, 24. doi: 10.5197/j.2044-0588.2014.029.024
Lesic, B., Bach, S., Ghigo, J. M., Dobrindt, U., Hacker, J., and Carniel, E. (2004). Excision of the high-pathogenicity island of Yersinia pseudotuberculosis requires the combined actions of its cognate integrase and Hef, a new recombination directionality factor. Mol. Microbiol. 52, 1337–1348. doi: 10.1111/j.1365-2958.2004.04073.x
Lovell, H. C., Mansfield, J. W., Godfrey, S. A. C., Jackson, R. W., Hancock, J. T., and Arnold, D. L. (2009). Bacterial evolution by genomic island transfer occurs via DNA transformation in planta. Curr. Biol. 19, 1586–1590. doi: 10.1016/j.cub.2009.08.018
Ma, B., Hibbing, M. E., Kim, H. S., Reedy, R. M., Yedidia, I., Breuer, J., et al. (2007). Host range and molecular phylogenies of the soft rot enterobacterial genera Pectobacterium and Dickeya. Phytopathology 97, 1150–1163. doi: 10.1094/PHYTO-97-9-1150
McCullagh, P., and Nelder, J. A. (1989). Generalized Linear Models, 2nd Edn. London: Chapman and Hall/CRC Press.
Moleleki, L. N., Onkendi, E. M., Mongae, A., and Kubheka, G. C. (2013). Characterisation of Pectobacterium wasabiae causing blackleg and soft rot diseases in South Africa. Eur. J. Plant Pathol. 135, 279–288. doi: 10.1007/s10658-012-0084-4
Morschhäuser, J., Köhler, G., Ziebuhr, W., Blum-Oehler, G., Dobrindt, U., and Hacker, J. (2000). Evolution of microbial pathogens. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 355, 695–704. doi: 10.1098/rstb.2000.0609
Ngadze, E., Brady, C. L., Coutinho, T. A., and Van der Waals, J. E. (2012). Pectinolytic bacteria associated with potato soft rot and blackleg in South Africa and Zimbabwe. Eur. J. Plant Pathol. 134, 533–549. doi: 10.1007/s10658-012-0036-z
Panda, P., Fiers, M. A. W. J., Armstrong, K., and Pitman, A. R. (2012). First report of blackleg and soft rot of potato caused by Pectobacterium carotovorum subsp. brasiliensis in New Zealand. New Dis. Rep. 26, 15. doi: 10.5197/j.2044-0588.2012.026.015
Panda, P., Fiers, M. A. W. J., Lu, A., Armstrong, K. F., and Pitman, A. R. (2015). Draft genome sequences of three Pectobacterium strains causing blackleg of potato: P. carotovorum subsp. brasiliensis ICMP 19477, P. atrosepticum ICMP 1526, and P. carotovorum subsp. carotovorum UGC32. Genome Announc. 3, e000874-15. doi: 10.1128/genomeA.00874-15
Parry, R. J., Mhaskar, S. V., Lin, M. T., Walker, A. E., and Mafoti, R. (1994). Investigations of the biosynthesis of the phytotoxin coronatine. Can. J. Chem. 72, 86–99. doi: 10.1139/v94-014
Pérombelon, M. C. M. (2002). Potato diseases caused by soft rot erwinias: an overview of pathogenesis. Plant Pathol. 51, 1–12. doi: 10.1046/j.0032-0862.2001.Short%20title.doc.x
Pérombelon, M. C. M., and Kelman, A. (1980). Ecology of the soft rot erwinias. Annu. Rev. Phytopathol. 18, 361–387. doi: 10.1146/annurev.py.18.090180.002045
Pitman, A. R., Harrow, S. A., and Visnovsky, S. B. (2010). Genetic characterisation of Pectobacterium wasabiae causing soft rot disease of potato in New Zealand. Eur. J. Plant Pathol. 126, 423–435. doi: 10.1007/s10658-009-9551-y
Pitman, A. R., Jackson, R. W., Mansfield, J. W., Kaitell, V., Thwaites, R., and Arnold, D. L. (2005). Exposure to host resistance mechanisms drives evolution of bacterial virulence in plants. Curr. Biol. 15, 2230–2235. doi: 10.1016/j.cub.2005.10.074
Pitman, A. R., Wright, P. J., Galbraith, M. D., and Harrow, S. A. (2008). Biochemical and genetic diversity of pectolytic enterobacteria causing soft rot disease of potatoes in New Zealand. Australas. Plant Pathol. 37, 559–568. doi: 10.1071/AP08056
Rangaswamy, V., Jiralerspong, S., Parry, R., and Bender, C. L. (1998). Biosynthesis of the Pseudomonas polyketide coronafacic acid requires monofunctional and multifunctional polyketide synthase proteins. Proc. Natl. Acad. Sci. U.S.A. 95, 15469–15474. doi: 10.1073/pnas.95.26.15469
Richter, C., Dy, R. L., McKenzie, R. E., Watson, B. N., Taylor, C., Chang, J. T., et al. (2014). Priming in the Type IF CRISPR-Cas system triggers strand-independent spacer acquisition, bi-directionally from the primed protospacer. Nucleic Acids Res. 42, 8516–8526. doi: 10.1093/nar/gku527
Sakellaris, H., Luck, S. N., Al-Hasani, K., Rajakumar, K., Turner, S. A., and Adler, B. (2004). Regulated site-specific recombination of the she pathogenicity island of Shigella flexneri. Mol. Microbiol. 52, 1329–1336. doi: 10.1111/j.1365-2958.2004.04048.x
Sevin, E. W., and Barloy-Hubler, F. (2007). RASTA-Bacteria: a web-based tool for identifying toxin-antitoxin loci in prokaryotes. Genome Biol. 8:R155. doi: 10.1186/gb-2007-8-8-r155
Slawiak, M., and Lojkowska, E. (2009). Genes responsible for coronatine synthesis in Pseudomonas syringae present in the genome of soft rot bacteria. Eur. J. Plant Pathol. 124, 353–361. doi: 10.1007/s10658-008-9418-7
Sullivan, M. J., Petty, N. K., and Beatson, S. A. (2011). Easyfig: a genome comparison visualizer. Bioinformatics 27, 1009–1010. doi: 10.1093/bioinformatics/btr039
Tsuchimoto, S., Nishimura, Y., and Ohtsubo, E. (1992). The stable maintenance system pem of plasmid R100: degradation of PemI may allow PemK protein to inhibit cell growth. J. Bacteriol. 174, 4205–4211.
Uppalapati, S. R., Ishiga, Y., Wangdi, T., Kunkel, B. N., Anand, A., Mysore, K. S., et al. (2007). The phytotoxin coronatine contributes to pathogen fitness and is required for suppression of salicylic acid accumulation in tomato inoculated with Pseudomonas syringae pv. tomato DC3000. Mol. Plant Microbe Interact. 20, 955–965. doi: 10.1094/MPMI-20-8-0955
van der Merwe, J. J., Coutinho, T. A., Korsten, L., and van der Waals, J. E. (2010). Pectobacterium carotovorum subsp. brasiliensis causing blackleg on potatoes in South Africa. Eur. J. Plant Pathol. 126, 175–185. doi: 10.1007/s10658-009-9531-2
Vanga, B. R., Butler, R. C., Toth, I. K., Ronson, C. W., and Pitman, A. R. (2012). Inactivation of PbTopo IIIβ causes hyper-excision of the Pathogenicity Island HAI2 resulting in reduced virulence of Pectobacterium atrosepticum. Mol. Microbiol. 84, 648–663. doi: 10.1111/j.1365-2958.2012.08050.x
Vanga, B. R., Ramakrishnan, P., Butler, R. C., Toth, I. K., Ronson, C. W., Jacobs, J., et al. (2015). Mobilisation of HAI2 is induced in planta in the phytopathogen Pectobacterium atrosepticum SCRI1043, and involves the putative relaxase ECA0613 and quorum sensing. Environ. Microbiol. 17, 4730–4744. doi: 10.1111/1462-2920.13024
Vercoe, R. B., Chang, J. T., Dy, R. L., Taylor, C., Gristwood, T., Clulow, J. S., et al. (2013). Cytotoxic chromosomal targeting by CRISPR-Cas systems can reshape bacterial genomes and expel or remodel pathogenicity islands. PLoS Genet. 9:e1003454. doi: 10.1371/journal.pgen.1003454
Weiler, E. W., Kutchan, T. M., Gorba, T., Brodschelm, W., Niesel, U., and Bublitz, F. (1994). The Pseudomonas phytotoxin coronatine mimics octadecanoid signaling molecules of higher-plants. FEBS Lett. 345, 9–13. doi: 10.1016/0014-5793(94)00411-0
Wozniak, R. A. F., Fouts, D. E., Spagnoletti, M., Colombo, M. M., Ceccarelli, D., Garriss, G., et al. (2009). Comparative ICE genomics: insights into the evolution of the SXT/R391 family of ICEs. PLoS Genet. 5:e1000786. doi: 10.1371/journal.pgen.1000786
Wozniak, R. A. F., and Waldor, M. K. (2009). A toxin–antitoxin system promotes the maintenance of an integrative conjugative element. PLoS Genet. 5:e1000439. doi: 10.1371/journal.pgen.1000439
Wozniak, R. A. F., and Waldor, M. K. (2010). Integrative and conjugative elements: mosaic mobile genetic elements enabling dynamic lateral gene flow. Nat. Rev. Microbiol. 8, 552–563. doi: 10.1038/nrmicro2382
Keywords: soft rot enterobacteriaceae, pathogenicity island, blackleg, CRISPR-Cas chromosomal editing, horizontal gene transfer
Citation: Panda P, Vanga BR, Lu A, Fiers M, Fineran PC, Butler R, Armstrong K, Ronson CW and Pitman AR (2016) Pectobacterium atrosepticum and Pectobacterium carotovorum Harbor Distinct, Independently Acquired Integrative and Conjugative Elements Encoding Coronafacic Acid that Enhance Virulence on Potato Stems. Front. Microbiol. 7:397. doi: 10.3389/fmicb.2016.00397
Received: 02 December 2015; Accepted: 14 March 2016;
Published: 31 March 2016.
Edited by:
Feng Gao, Tianjin University, ChinaReviewed by:
Leighton Pritchard, James Hutton Institute, UKDawn Arnold, University of the West of England, Bristol, UK
Hong-Yu Ou, Shanghai Jiaotong University, China
Copyright © 2016 Panda, Vanga, Lu, Fiers, Fineran, Butler, Armstrong, Ronson and Pitman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrew R. Pitman, YW5kcmV3LnBpdG1hbkBwbGFudGFuZGZvb2QuY28ubno=
†Present address: Ashley Lu, Laboratory for the Research of Neurodegenerative Diseases, Leuven, Belgium; Mark Fiers, VIB11 vzw Center for the Biology of Disease, Leuven, Belgium