 Bo Shu
Bo Shu Weicai Li
Weicai Li Yongzan Wei
Yongzan Wei- Key Laboratory of Tropical Fruit Biology, Ministry of Agriculture, South Subtropical Crops Research Institute, Chinese Academy of Tropical Agricultural Science, Zhanjiang, China
Trunk girdling can increase carbohydrate content above the girdling site and is an important strategy for inhibiting new shoot growth to promote flowering in cultivated litchi (Litchi chinensis Sonn.). However, girdling inhibits carbohydrate transport to the root in nearly all of the fruit development periods and consequently decreases root absorption. The mechanism through which carbohydrates regulate root development in arbuscular mycorrhiza (AM) remains largely unknown. Carbohydrate content, AM colonization, and transcriptome in the roots were analyzed to elucidate the interaction between host litchi and AM fungi when carbohydrate content decreases. Girdling decreased glucose, fructose, sucrose, quebrachitol, and starch contents in the litchi mycorrhizal roots, thereby reducing AM colonization. RNA-seq achieved approximately 60 million reads of each sample, with an average length of reads reaching 100 bp. Assembly of all the reads of the 30 samples produced 671,316 transcripts and 381,429 unigenes, with average lengths of 780 and 643 bp, respectively. Litchi (54,100 unigenes) and AM fungi unigenes (33,120 unigenes) were achieved through sequence annotation during decreased carbohydrate content. Analysis of differentially expressed genes (DEG) showed that flavonoids, alpha-linolenic acid, and linoleic acid are the main factors that regulate AM colonization in litchi. However, flavonoids may play a role in detecting the stage at which carbohydrate content decreases; alpha-linolenic acid or linoleic acid may affect AM formation under the adaptation process. Litchi trees stimulated the expression of defense-related genes and downregulated symbiosis signal-transduction genes to inhibit new AM colonization. Moreover, transcription factors of the AP2, ERF, Myb, WRKY, bHLH families, and lectin genes altered maintenance of litchi mycorrhizal roots in the post-symbiotic stage for carbohydrate starvation. Similar to those of the litchi host, the E3 ubiquitin ligase complex SCF subunit scon-3 and polyubiquitin of AM fungi were upregulated at the perceived stages. This occurrence suggested that ubiquitination plays an important role in perceiving carbohydrate decrease in AM fungi. The transcription of cytochrome b-245 and leucine-rich repeat was detected in the DEG database, implying that the transcripts were involved in AM fungal adaptation under carbohydrate starvation. The transcriptome data might suggest novel functions of unigenes in carbohydrate shortage of mycorrhizal roots.
Introduction
Arbuscular mycorrhiza represents widespread mutualistic association between soil-borne fungi of the Phylum Glomeromycota and most land plants. AM fungi play a key role in the life of host plants; the fungi supply mineral nutrients to the roots (biofertilizers), influence plant development (bioregulators), and enable mycorrhizal plants to overcome biotic and abiotic stresses (bioprotectors; Parniske, 2008; Smith and Read, 2008). The host plant provides carbohydrates to obligate biotrophic fungi to enable the fungi to complete their life cycle. Plants could allocate up to 30% of their photosynthetic carbohydrates to AM fungi, which in return, provides up to 80% of plant phosphate and nitrogen. Moreover, the two partners guarantee a “fair trade” of mineral nutrients against carbohydrates (Kiers et al., 2011; Fellbaum et al., 2012). Although carbohydrates provided by host plants play important roles in the life cycle of AM fungi, knowledge on processes involved in the host carbohydrate regulation for AM fungi colonization remains limited.
Litchi (Litchi chinensis Sonn.), which belongs to the Sapindaceae family, is an important woody mycorrhizal fruit tree in southern China. The area for cultivation of litchi reached 553,000 ha in 2015. The main cultivars of litchi (L. chinensis Sonn. Feizixiao and L. chinensis Sonn. Guiwei) normally flower in March and mature in June in the Guangdong province of China. Trunk girdling increases carbohydrate content above the girdling site and decreases the uptake of water and mineral nutrition; this strategy is important to inhibit new shoot growth and promote flowering in cultivated litchi (Yuan and Huang, 1993; Huang et al., 2003). Trunk girdling is generally performed in the second half of November before the period of flower-bud differentiation and wound healing in half a year. However, girdling adversely affects carbohydrate transport to the root of litchi, thereby restricting litchi mycorrhizal root development. This technique also reduces mycorrhizal root absorption, consequently decreasing the quality of fruit or inducing fruit drop during fruit development. Hence, the coping strategy of litchi mycorrhizal root under carbohydrate starvation must be elucidated to improve culture techniques.
Carbohydrates in plant tissues are diverse throughout evolution; the different kinds of carbohydrates include glucose, fructose, sucrose, sorbitol, and starch in the roots of the apple tree (Tromp, 1983) as well as glucose, fructose, sucrose, quebrachitol, and starch in the root of the litchi tree (Wang et al., 2013). All types of carbohydrates starvation may affect the pre- and post-symbiotic interactions of the AM fungi and the host plant. To date, several molecular signals are identified and used to recognize AM fungi and host plant in the pre-symbiotic period. Flavonols, strigolactones, and hydroxy fatty acids from the host plant and lipochito-oligosaccharides, short-chain chitin oligomers, and steroids from the AM fungi are dialog components (Bécard et al., 1992; Besserer et al., 2008; Maillet et al., 2011; Nagahashi and Douds, 2011; Genre et al., 2013; Bucher et al., 2014; Sun et al., 2015). Molecules and related genes in the post-symbiotic stage are more complex than those in the pre-symbiotic period. Genes, which are related to phytohormone biosynthesis and signal transduction, transport systems of nutrient and carbohydrate, transcription factor, and other processes, all participate in the post-symbiotic stages (De Hoff et al., 2009; Hogekamp et al., 2011; Handa et al., 2015). To analyze the influence of carbohydrate starvation on the litchi mycorrhizal root, this study focused on mycorrhizal root carbohydrates (glucose, fructose, sucrose, quebrachitol, and starch) to determine the degree of carbohydrate decrease and measure AM colonization rate to clarify whether carbohydrates reduce AM colonization. Based on the above analysis, this study also tested the transcriptome data to screen for interesting transcripts involved in the interaction between AM fungi and host litchi under carbohydrate starvation.
Materials and Methods
Plant Material and Cultivation Conditions
Six litchi trees with middle growth vigor were selected from 200 trees (cultivar Feizixiao, grafted on 12-year-old Huaizhi rootstock) and cultivated in the experimental orchard of the South Subtropical Crops Research Institute, Zhanjiang, China (mineral nutrition is shown in Supplementary Table 1). The trees received standard horticultural practices (such as weeding and irrigating) as well as disease and insect control. Three of the six trees were girdled in November (The girdling picture was shown in Supplementary Figure S1), and the remaining trees were used as control. Root (diameter < 1.5 mm) was sampled to 20–30 cm soil thickness, where fibrous roots were overdispersed, from the four directions of each tree; duplicate samples were obtained from each tree. The root samples were immediately obtained after girdling treatment (0 day, 0 D) as well as 1 week, 2 weeks, 1 month, and 2 months (1 W, 2 W, 1 M, and 2 M, respectively) thereafter. Thirty samples were acquired in the experiment. The samples were washed with sterile water, wiped dry with gauze at harvest, frozen in liquid nitrogen, and then immediately stored at -80°C.
Root Mycorrhizal Colonization
A fraction of the fresh roots was fixed in formalin/acetic acid/ethanol (FAA, 13:5:200 [v/v/v]) for 24 h to determine the degree of AM colonization. The roots were cleared in 10% (w/v) KOH at 99°C for 1.5 h and stained with 0.05% (w/v) trypan blue in lactophenol by using methods previously described by Phillips and Hayman (1970). The structures of AM fungi were examined under a compound light microscope (Olympus-BH-2). Fungal colonization was estimated using magnified intersection method (McGonigle et al., 1990). Total AM and arbuscule colonization rate were quantified by examining 200 intersections for each sample.
Measurements of Soluble Sugars and Starch
Soluble sugars were measured using HPLC analyses. Determinations were performed as described by Wang et al. (2013) and Wang T.D. et al. (2014). About 1 g of the roots were homogenized, extracted three times with 85% (v/v) ethanol (3 mL each), and then centrifuged at 6000 × g for 10 min. The pooled supernatant was rotary evaporated to dryness and resuspended in 4 mL of distilled water. The precipitate was then analyzed for starch content. Sugars were detected by Agilent 1200 HPLC system (Agilent Technologies, Waldbronn, Germany) equipped with a refractive index detector and a transgenomic CARB Sep Coregel 87C column (CHO-99-5860). The ultra-pure water was the mobile phase at a flow rate of 0.8 mL/min. The sugars were identified by comparing their retention times with those of authentic standards. The concentrations of individual sugars were quantified using peak areas and calibration curves derived from the standards.
The precipitates were combined with 2 mL of water, and the mixture was gelatinized for 15 min in boiled water. The mixture was cooled to 20°C and then added with 2 mL of 9.2 M perchloric acid. The mixture was stirred for 15 min, combined with 4 mL of water, and centrifuged at 3500 × g for 10 min. The supernatant was transferred to a 50 mL volumetric flask. The precipitate was combined with 2 mL of 4.6 M perchloric acid, and the resulting mixture was stirred for 15 min. The mixture was added with 5 mL of water and centrifuged at 3500 × g for 10 min. The supernatant was then transferred to the same 50 mL volumetric flask. The precipitate was washed twice with 5 mL of water, and the water was also transferred to a 50 mL volumetric flask. The solution in the 50 mL volumetric flask was analyzed for starch content according to Wang T.D. et al. (2014).
Total RNA Extraction and RNA Sequencing
Total RNA was isolated from the mycorrhizal root of litchi by using TRIzol reagent (Invitrogen, USA) and treated with DNase I to eliminate genomic DNA contamination. Three biological replicates were prepared for each treatment, and RNA of 30 samples was obtained. RNA quality was determined using Agilent 2100 Bioanalyzer. After the total RNA extraction and DNase I treatment, magnetic beads with Oligo (dT) were used to isolate mRNA. The isolated mRNA was mixed with the fragmentation buffer and then fragmented into short fragments. cDNA was synthesized using the mRNA fragments as templates. Short fragments were purified and resolved with elution buffer for end reparation and addition of single nucleotide A (adenine). The short fragments were connected with adapters. Suitable fragments were selected for PCR amplification as templates. During the quality control, Agilent 2100 Bioanalyzer and ABI StepOnePlus Real-Time PCR System were used for quantification and qualification of the sample library. Finally, 30 libraries were sequenced using the Illumina HiSeq 2000 system.
Transcript Assembly and Functional Annotation
Prior to bioinformatic analysis, the raw sequences were filtered to remove reads that contained only the adaptor sequences, those with more than 5% unknown nucleotides, and the low-quality reads with more than 20% bases having a quality value ≤ 10. Because litchi genome has not been published and multiplex types of AM fungi which might show sequence difference from Rhizophagus irregularis are existed in litchi mycorrhizal root, de novo assembly was performed by the Beijing Genomics Institute using the short-read assembly program for both litchi and AM fungi by using the Trinity software (version 2.0.6; Grabherr et al., 2011). The Blast software (version 2.2.30+) with evalue 1e-3 -num_alignments 1 was used for separating the transcript of litchi and AM fungi by against the genome (R. irregularis DAOM 197198, Gloin11) and nr database2.
TransDecoder software (version r20140704) was employed to predict the transcript open reading frame with -m50 setting. Functional annotation of the unigenes was performed using the nr database, the Swiss-Prot protein database3, the KEGG database4, uniref 905, and Gloin1 by using BLASTx with an E-value < 10-5 by Trinotate software (version 20140708). When a unigene did not align to any of the above databases, Hmmscan (HMMER) software (version 3.1) was used for annotation through functional domain prediction (Grabherr et al., 2011).
Read Mapping and Quantification of Gene Expression
Reads containing adaptors, reads with more than 10% unknown nucleotides, and low-quality reads with more than 50% bases with a quality value ≤ 5 were removed to obtain uncontaminated sequences. The uncontaminated sequences from each sample were mapped to the assembly transcripts by using Bowtie software (version: 1.0.1; parameters: mismatch = 2). The files of the bam format were achieved and used to calculate for the number of reads mapped on the transcript by RSEM software (version: v1.2.17). The number of mapped and filtered reads for each unigene was calculated to obtain the corresponding FPKM values (Li and Dewey, 2011). DEGs between two samples were determined using the FDR threshold of <0.001, an absolute log twofold change value of >1.0, and a P-value of <0.01 by Edger (version: 3.10.2). The GO and KEGG pathway analyses of DEGs were accomplished using GOseq software (version: 3.0) and KOBAS software (version: 2.0; Young et al., 2010; Xie et al., 2011). The heat maps of the selected DEGs from litchi and AM fungi were constructed using mev software (v4.9.0). The flow chart for RNA-seq analysis was shown in Supplementary Figure S2.
Statistical Analysis
Experimental data were statistically analyzed using ANOVA through SAS 8.1 software (SAS Institute, Cary, NC, USA). The probabilities of significance were used to determine significance among the treatments, and the least-significant difference (p < 0.05) was used to compare the data.
Results
Carbohydrates
The HPLC results showed that sucrose concentration (1.59–46.96 mg/g⋅FW) was higher than the other carbohydrate contents in the litchi mycorrhizal roots. Variation in glucose, fructose, sucrose, and quebrachitol contents in the control and girdled mycorrhizal roots exhibited the same pattern. This finding revealed that continuous decrease began at 0 D to 2 W after girdling and then remained stable from 2 W to 2 M. The concentrations of glucose, fructose, sucrose, and quebrachitol in the control were significantly higher than those in the girdled group from 1 W to 2 M. In contrast to other carbohydrates, starch concentration in the girdling treatment continuously decreased and significantly differed from 1 to 2 M compared with that of the control (Figure 1).
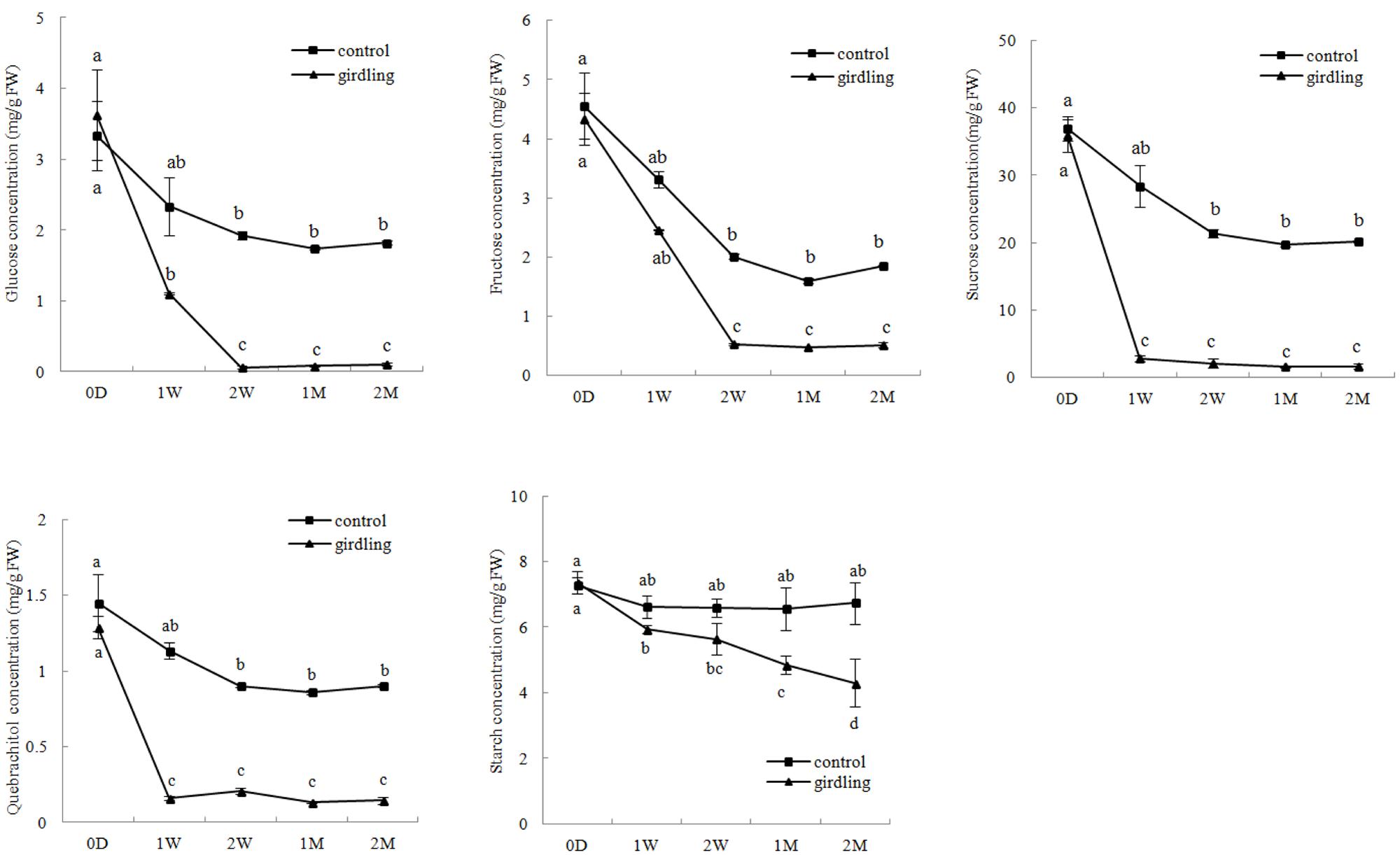
FIGURE 1. Carbohydrate variation in the litchi mycorrhizal roots after girdling treatment. The C represents control, and the G represents girdling; 0 D, 1 W, 2 W, 1 M, 2 M represent the time after treatment (0 day, 1 week, 2 weeks, 1 month, and 2 months).
Root Mycorrhizal Colonization
The rates of total, arbuscular, and vesicular colonization rates of litchi were 15.33–36.00%, 3.97–10.43%, and 3.10–3.90%, respectively, under field conditions. Variation in the total and arbuscular colonization rates continuously decreased from 0 D to 2 M. The total and arbuscular colonization rates were significantly higher in the control than those in the girdling treatment at 1 and 2 M. Vesicular colonization was not significantly different from total and arbuscular colonizations after girdling (Figure 2).
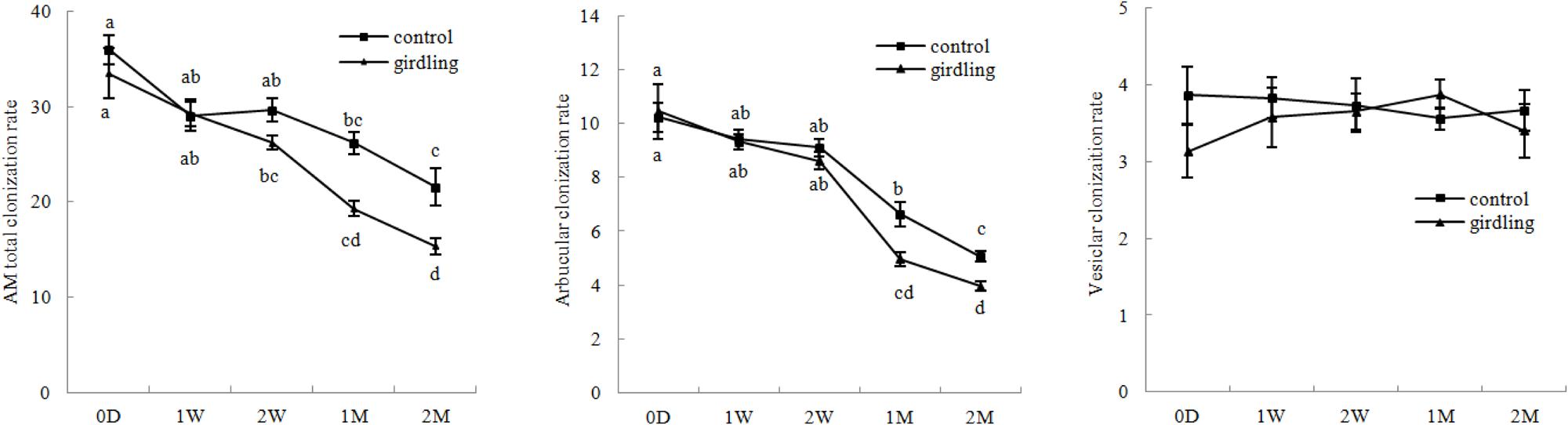
FIGURE 2. AM colonization variation in the litchi mycorrhizal roots after girdling treatment. The C represents control, and the G represents girdling; 0 D, 1 W, 2 W, 1 M, 2 M represent the time after treatment (0 day, 1 week, 2 weeks, 1 month, and 2 months).
Sequence Assembly and Annotation
Thirty samples were sequenced using the Illumina genome analyzer Hiseq 2000. After quality checking and data cleaning, approximately 60 million reads of each sample were obtained, with average lengths reaching 100 bp and GC content of 44–46% (Supplementary Table 2). Assembly of all the reads of the 30 samples generated 671,316 transcripts and 381,429 unigenes, with average lengths of 780 and 643 bp, respectively. A total of 308824 unigenes were annotated among all unigenes (Table 1). These transcriptome data were submitted to sequence read archive of NCBI with the NO. SRX1518711.
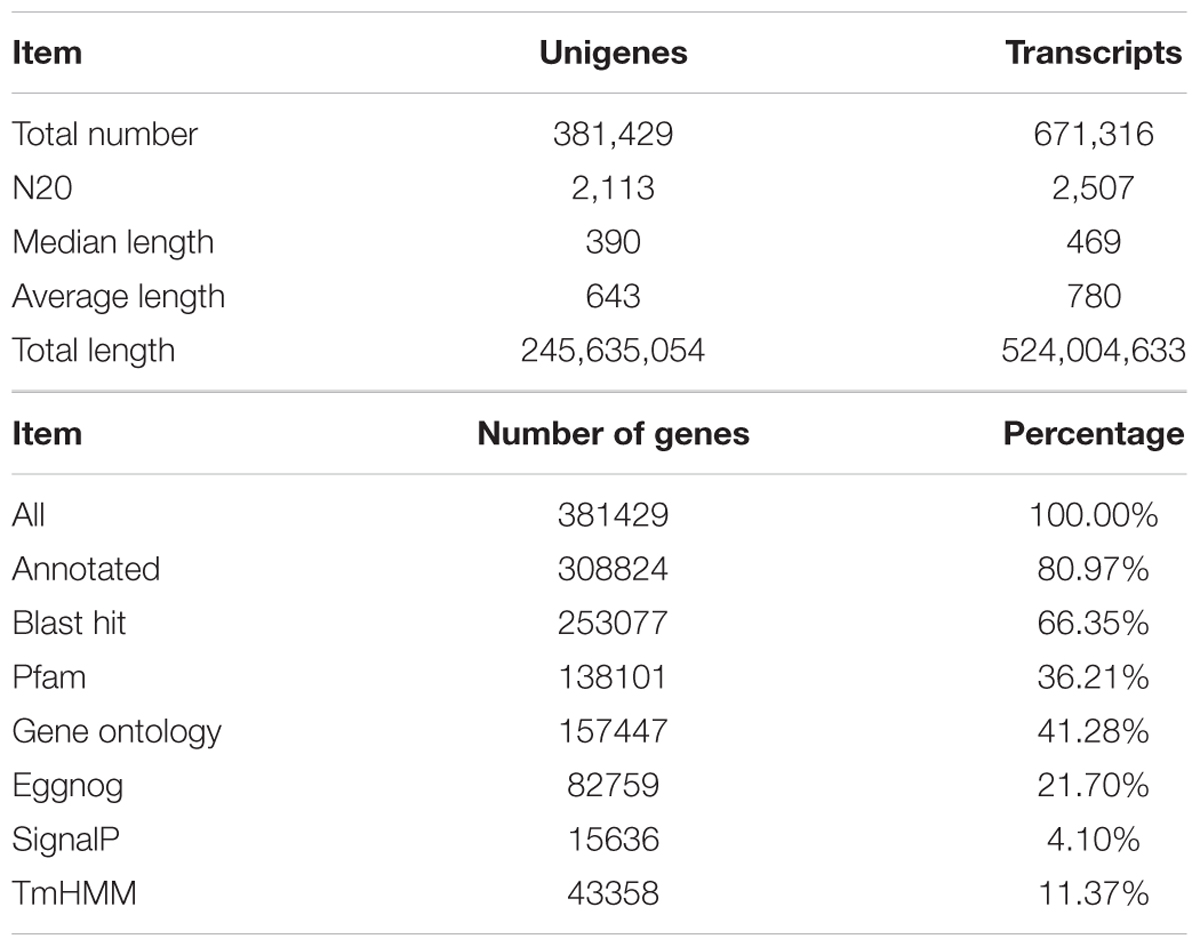
TABLE 1. Summary of read numbers based on the RNA-Seq data from the mycorrhizal roots of litchi after girdling treatment.
Approximately 262,619 unique sequences were annotated through BLASTx (cut-off E-value10-5) search of four public databases, namely, nr, Swiss-Prot protein, KEGG, and uniref 90. Of the sequences, 260,019 unique sequences were annotated with reference to the nr database, whereas 2,600 unigenes were annotated using the other databases (Figure 3A). The results of the annotation indicated that 9.66% (25,137) of the annotated sequences exhibited “very strong homology” (E-value < 10-100), 15.08% (39,623) exhibited “strong homology” (10-100 < E-value < 10-50), and 70.26% (174,042) displayed “homology” (10-50< E-value < 10-5) to the available sequences (Figure 3B). The unique sequences of AM fungi achieved top matches to sequences from R. irregularis DAOM181602 and R. irregularis DAOM197198w. The unique sequences of litchi exhibited top matches to sequences from Citrus sinensis and Citrus clementina (Figure 3C).
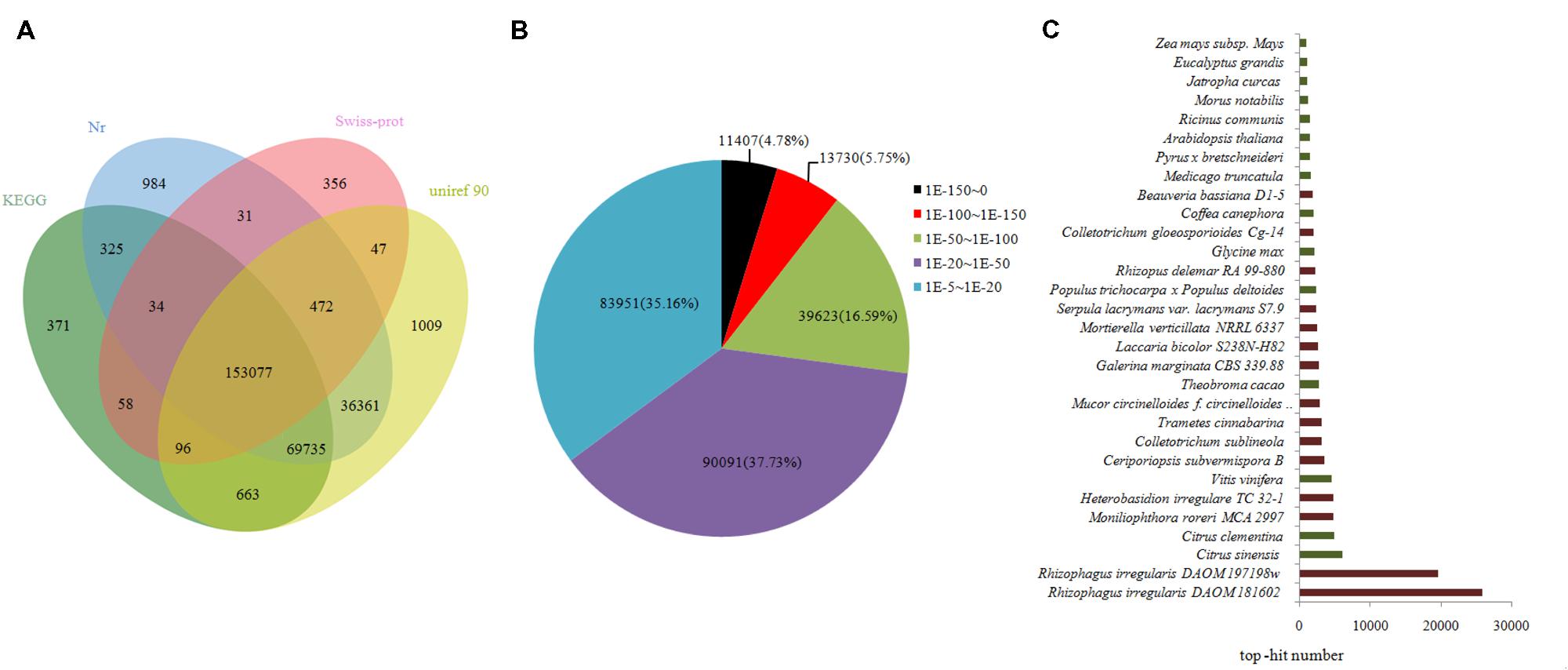
FIGURE 3. Outcome of homology search of unigenes against the nr, KEGG, Swiss-prot, and uniref 90 databases. (A) Venn diagram of unigene numbers annotated by BLASTx with an E-value threshold of 10-5 against protein databases. The numbers in the circles indicate unigenes numbers annotated by single or multiple databases. (B) E-value distribution of the top BLAST hits for each unique sequence. (C) Species distribution of the top BLAST hits for all homologous sequences. Those indicated in brown denote AM fungi, and those in green correspond to litchi in (C).
The nr, GO, KEGG, and uniref 90 databases were used to classify the functions of the predicted unigenes. The unigenes of litchi (54,100 unigenes) and AM fungi (33,120 unigenes) were classified into three main categories as follows: “cellular component,” “molecular function,” and “biological process” (Figure 4). Meanwhile, the host litchi and AM fungi obtained numerous unigenes annotated as “cell and organelle” in the “cellular component” category; “binding” and “catalytic activities” in the “molecular function” category; and “metabolic process,” “cellular process,” “response to stimulus,” and “biological regulation” as four subcategories in the “biological process” category. However, the rates of the same subcategories differed between litchi and AM fungi. Within the “cellular component,” the subcategory “extracellular region part” attained a rate of less than 1% in litchi but more than 1% in fungi. The subcategory “symplost” showed more than 1% rate in litchi but less than 1% in AM fungi. Within the “molecular function” category, the subcategory of “chemoattractant” resulted in 0.01% rate in fungi but was not found in the litchi component. The subcategory of “nutrition reviser” was more than 0.01% in litchi and less than 0.01% in AM fungi. Within “biological process,” the subcategory of “immune system process” in litchi unigenes was more than 1% but less than 1% in fungi (Figure 4).
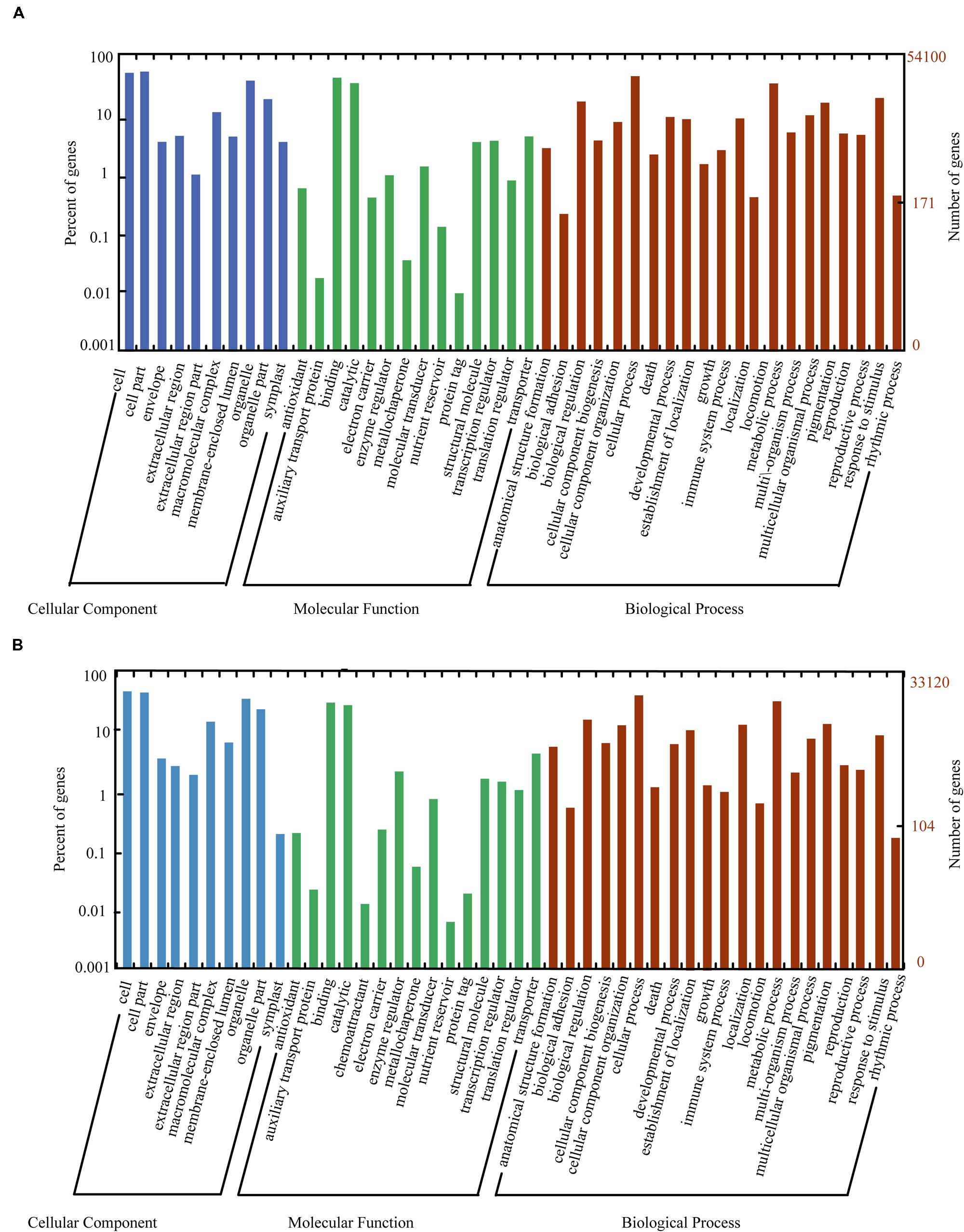
FIGURE 4. Histogram of GO classifications for litchi (A) and AM fungi (B) transcripts in mycorrhizal roots. The unigenes corresponded to three main categories: “biological process,” “cellular component,” and “molecular function.” The left- and right-hand y-axes indicate the percentage and number of annotated unigenes, respectively.
Differential Gene Expression Analysis
The DEGs after girdling were assessed by pair-wise comparisons of all time points with the expression fold (log 2 Ratio ≥ 1) and FDR ≤ 10-3 as the thresholds in litchi and AM fungi (Table 2). The number of DEGs increased with prolonged girdling. Thirty-four DEGs of litchi (34 upregulated and 0 upregulated) and only one DEG of AM fungi (1 upregulated and 0 downregulated) were detected between 0 D and 1 W. Moreover, 442 DEGs of litchi (286 upregulated and 156 downregulated) and 742 DEGs of AM fungi (742 upregulated and 0 downregulated) were identified at 0 D and 2 M after pair-wise comparison (Table 2).
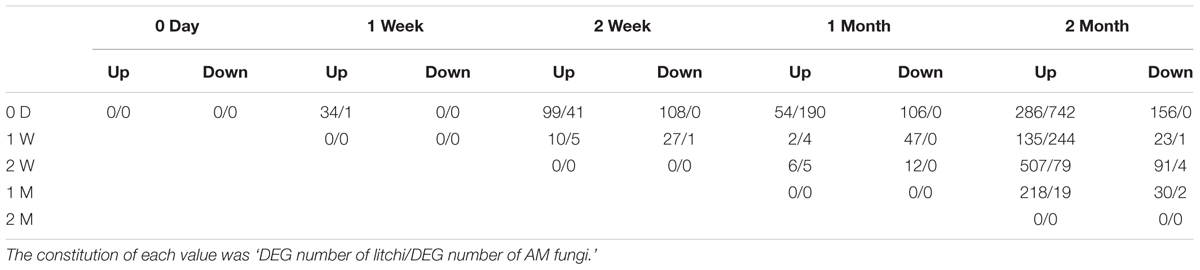
TABLE 2. The quantity of DEGs in litchi mycorrhizal roots after girdling treatment.
KEGG Pathway Enrichment Analysis of Differentially Expressed Genes
The main biological process and related unigenes were screened from host litchi and AM fungi during carbohydrate starvation. Most upregulated DEGs at 0 D versus those at 1 W as well as at 0 D versus those at 2 W were mapped to carbohydrate metabolism, organismal systems (plant–pathogen interaction), and amino-acid metabolism for litchi. In particular, the upregulated DEGs were mapped to carbohydrate metabolism pathways, such as pyruvate metabolism (ko00620), amino sugar and nucleotide sugar metabolism (ko00520), and starch and sucrose metabolism (ko00500). For organismal systems, the upregulated DEGs were mapped to the plant–pathogen interactions (ko04626). The amino-acid metabolism, such as phenylalanine metabolism (ko00360) as well as valine, leucine, and isoleucine biosynthesis (ko00290) were upregulated (Supplementary Table 3). The downregulated DEGs at 0 D versus those at 1 W and at 0 D versus those at 2 W were clustered into biosynthesis of other secondary metabolites, carbohydrate metabolism, and endocrine system. Most downregulated transcripts were then mapped to the biosynthesis of other secondary metabolites in flavonoid biosynthesis (ko00941), flavone and flavonol biosynthesis (ko00944), and phenylpropanoid biosynthesis (ko00940); carbohydrate metabolism, including starch and sucrose metabolism (ko00500); and amino sugar and nucleotide sugar metabolism (ko00520; Supplementary Table 3). In the adaptation process, most upregulated DEGs at 0 D versus those at 1 M and at 0 D versus those at 2 M in litchi were clustered into lipid metabolism and biosynthesis of other secondary metabolites. The upregulated transcripts mapped to lipid metabolism were grouped under alpha-linolenic acid metabolism (ko00592), linoleic acid metabolism (ko00591), and sphingolipid metabolism (ko00600). The biosynthesis of other secondary metabolites, including phenylpropanoid (ko00940); stilbenoid, diarylheptanoid, gingerol (ko00945); and flavonoids, (ko00941) were also upregulated. The downregulated DEGs were mapped to several clusters under carbohydrate metabolism and immunity. The transcripts mapped to carbohydrate metabolism were grouped under starch and sucrose metabolism (ko00500), amino sugar and nucleotide sugar metabolism (ko00520), and pentose phosphate pathway (ko00030). Meanwhile, the transcripts mapped to the immune system were grouped under antigen processing and presentation (ko04612) and the NOD-like receptor signaling pathway (ko04621; Supplementary Table 3).
The upregulated DEGs at 0 D versus 1 W and at 0 D versus 2 W in AM fungi were mapped to several pathways under genetic information processing and carbohydrate metabolism. Genetic information processing included protein processing in the endoplasmic reticulum (ko04141) and ribosome (ko03010), as well as ubiquitin-mediated proteolysis (ko00290). Under carbohydrate metabolism, glyoxylate and dicarboxylate metabolism (ko00630), citrate cycle (tricarboxylic acid cycle; ko00020), and pyruvate metabolism (ko00620) were noted. The upregulated DEGs at 0 D versus those at 1 M and at 0 D versus those at 2 M in the AM fungi were mapped to genetic information processing, amino acid metabolism, lipid metabolism, and signal transduction. The genetic information processing included RNA transport (ko03013), RNA degradation (ko03018), mismatch repair (ko03430), protein processing in the endoplasmic reticulum (ko04141), and ubiquitin-mediated proteolysis (ko04120). Meanwhile, amino acid metabolism included alanine, aspartate, and glutamate metabolism (ko00250) as well as arginine and proline metabolism (ko00330). For lipid metabolism, fatty-acid biosynthesis (ko00061), alpha-linolenic acid metabolism (ko00592), and biosynthesis of unsaturated fatty acids (ko01040) were found. For signal transduction, MAPK (ko04011), Ras (ko04014), and sphingolipid signaling pathways (ko04071) were noted. The downregulated DEGs of the AM fungi were few; these DEGs were noted at 1 W versus those at 2 W (TR92985| c2_g2, putative cruciform DNA binding protein), at 1 W versus those at 2 M (TR92985| c2_g2, putative cruciform DNA binding protein), at 2 W versus those at 2 M (TR5997| c0_g2, putative protein far1-related sequence 10; TR155121| c0_g1, putative pyruvate decarboxylase; TR146357| c0_g2 and TR95299| c0_g2 uncharacterized protein), and at 1 M versus those at 2 M (TR92985| c2_g2, putative cruciform DNA binding protein; TR95299| c0_g2, uncharacterized protein). Only TR155121| c0_g1 was mapped to the glycolysis/gluconeogenesis pathway (ko00010; Supplementary Table 3).
Genes Including Ubiquitination, Transcription Factor, and with Repeated Domain
Ubiquitination-related unigenes, transcription factor, and unigenes with repeated domains in the host litchi were selected to construct heat map A. Ubiquitination-related unigenes, transcription factor, unigenes with repeated domains, and unigenes related to chitin synthesis in the AM fungi were used to construct heat map B. The heat map of the host litchi revealed that all selected unigenes were divested into three main subclusters. Unigenes in subcluster I were upregulated by girdling on 2 W and 1 M; unigenes in subcluster II were induced by girdling on 2 M; and unigenes in subcluster III were downregulated by girdling from 0 D to 2 M (Figure 5A). The heat map of AM fungi showed that all of the selected unigenes were clustered into three main subclusters. Unigenes in subcluster I were downregulated by girdling from 0 D to 1 W but were upregulated from 2 W to 2 M. Unigenes in subcluster II were downregulated by girdling on 1 W but upregulated from 2 W to 2 M. Furthermore, unigenes in subcluster III were upregulated by girdling on 2 M (Figure 5B).
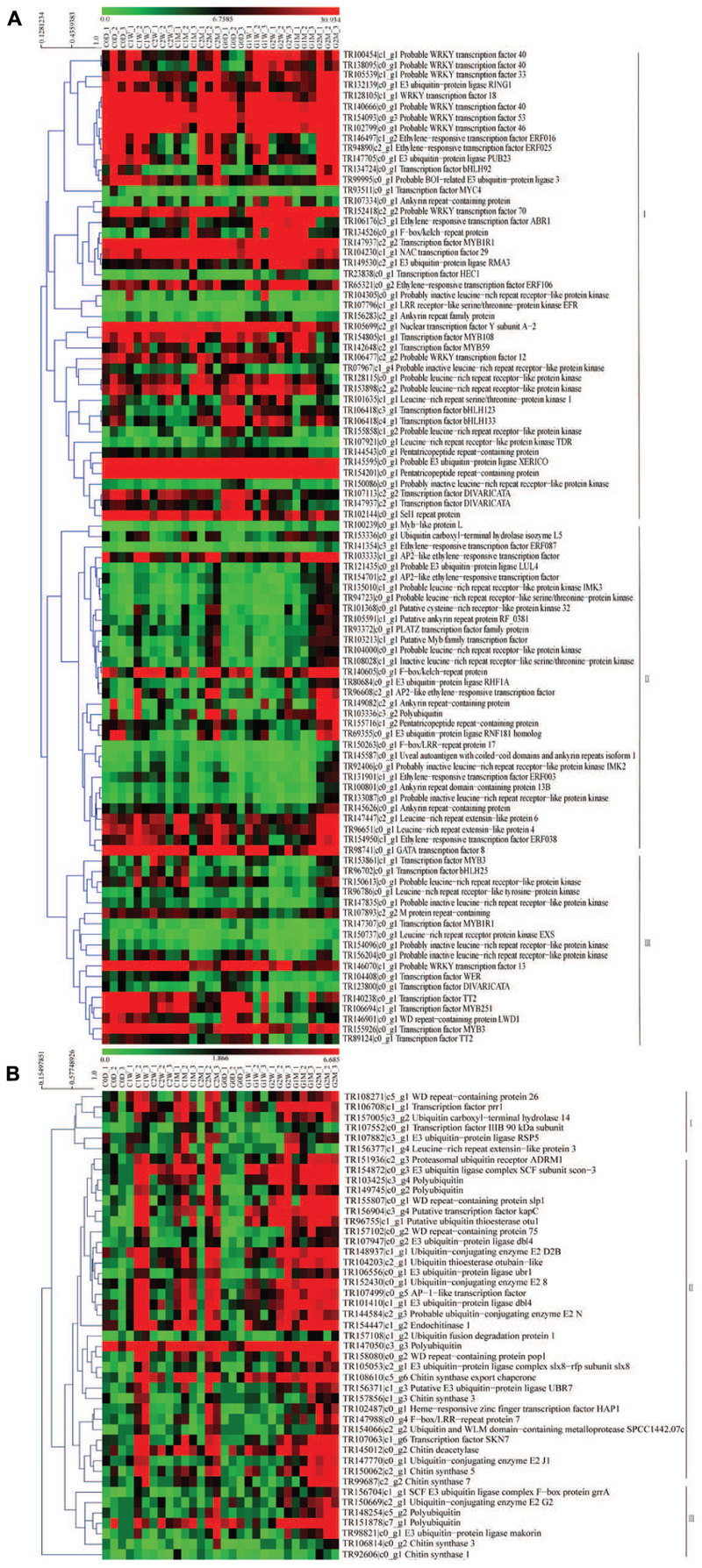
FIGURE 5. Heat map of the genes including ubiquitination, transcription factor and repeated domain expression for litchi (A); heat map of the genes including ubiquitination, transcription factor, repeated domain expression, and chitin synthesis for AM fungi (B). The C represents control and the G represents girdling; 0 D, 1 W, 2 W, 1 M, 2 M represent the time after treatment (0 day, 1 week, 2 weeks, 1 month, and 2 months). The heat map was constructed based on the FPKM values which showed on the top of the figures by the software mev (v4.9.0).
Clustering Results of Time-Course Data from RNA-Seq by STEM Analysis
DEGs with similar expression patterns were clustered into six distinct subclusters for AM fungi and host litchi, and DEGs grouped in the same subcluster may be functionally correlated. Most DEGs in the host litchi were clustered into subclusters I and II, which were upregulated in the girdling treatment (Figure 6A). The expression levels of DEGs in subcluster III were stable in the control but downregulated in the girdling treatment. The expression levels of DEGs in subcluster IV were stable in the control but induced in girdling at 2 W. The expression of DEGs in subcluster V were similar to those in the control and girdling treatment from 0 D to 1 M, but the expression levels in the latter were strong upregulated at 2 M. The expression levels of DEGs in subcluster VI of the control were downregulated from 0 D to 2 M but were induced by girdling at 2 M (Figure 6A). Similar to those in the host litchi, the numbers of genes assigned among the six clusters in the AM fungi were statistically significant (Figure 6B). The DEGs of subcluster I were upregulated from 1 W in the girdling treatment, and those of subcluster II were upregulated from 2 W. The expression patterns of DEGs in subcluster III were upregulated after 2 W of girdling and then downregulated from 2 W to 2 M. The expression patterns of DEGs in subcluster IV were also upregulated after 2 W of girdling and maintained at high levels from 2 W to 2 M. The expression levels of DEGs in subcluster V were lower than those in the control from 0 D to 2 W but were induced by girdling from 2 W to 2 M. The expression patterns of DEGs in subcluster VI showed the same patterns in the control and girdling groups, but the expression levels in the latter were higher than those in the former (Figure 6B; Unigenes of each subcluster were displayed in Supplementary Table 4).
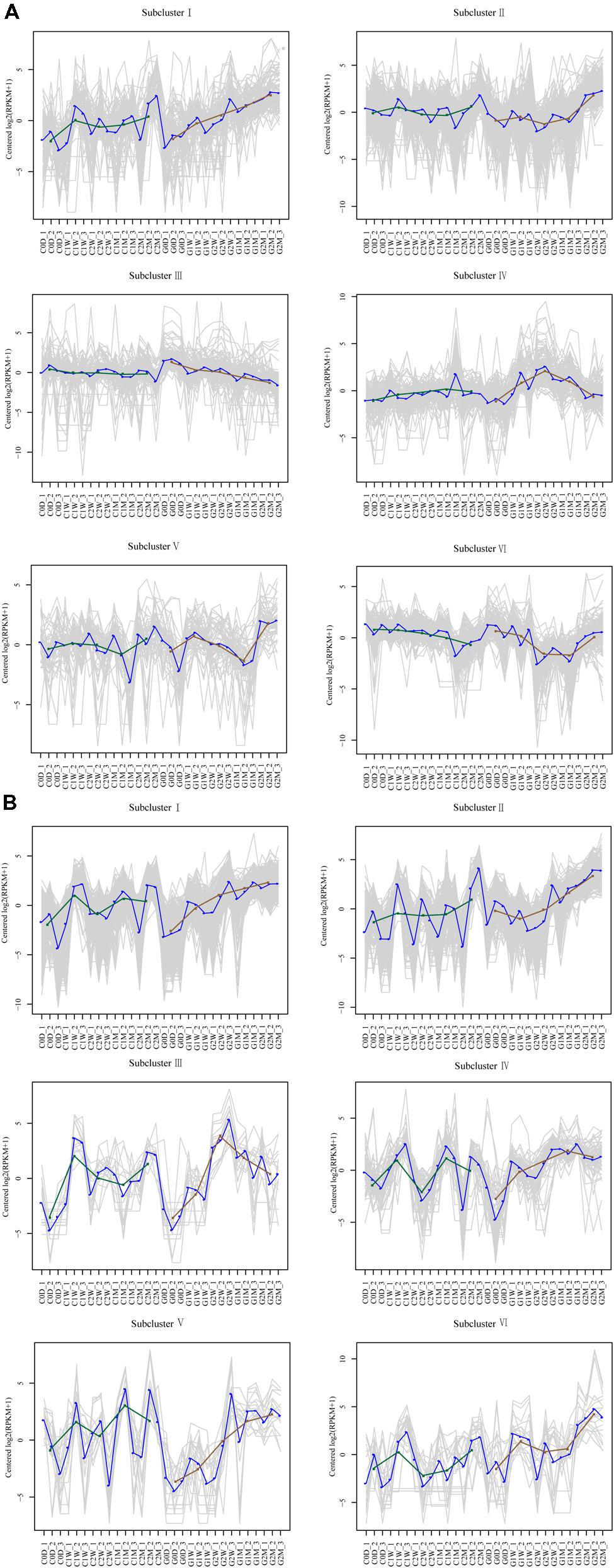
FIGURE 6. Clustering results of time-course data from RNA-seq by short time-series expression miner analysis for litchi (A) and AM fungi (B). Each box corresponds to one of the model temporal expression profiles, and the unigene belonging to each corresponding subcluster is shown in Supplementary Table 4. The C represents control, and the G represents girdling; 0 D, 1 W, 2 W, 1 M, 2 M represent the time after treatment (0 day, 1 week, 2 weeks, 1 month, and 2 months).
Discussion
RNA-seq is a technique used to detect low-expressing reads (Garber et al., 2011; Wang C. et al., 2014) and identify novel transcripts in AM with DEG screening (Fiorilli et al., 2015; Handa et al., 2015). In the present experiment, transcripts obtained from each sample included litchi- and fungi-derived transcript as well as transcripts that originated from other microorganisms. The R. irregularis genome, genomes of model plants, and those stored in other databases were used to distinguish the transcripts, whether they were derived from litchi, AM fungi, or other organisms, through BlastP prediction (Tisserant et al., 2013). RNA-seq analysis showed approximately 60 million reads for each sample, with average read length of 100 bp. The assembly of all reads of the 30 samples produced 671,316 transcripts and 381,429 unigenes, with average lengths of 780 and 643 bp, respectively. BlastX analysis revealed that 10.85% (25,920) of the unigenes were top matched to sequences from AM fungus R. irregularis DAOM 181602, and 8.22% (19,627) were matched to sequences from AM fungus R. irregularis DAOM 197198. The unigenes of litchi (54,100 unigenes) and AM fungi (33,120 unigenes) were identified by sequence annotation during the last period of carbohydrate decrease (Figure 4). More than 1,300 DEGs from host litchi and 1,000 DEGs from AM fungi were noted for the corresponding carbohydrate decrease (Supplementary Figure S2; Supplementary Table 4). The researches on transcriptome-related AM symbiosis were obtained by different methods, such as SuperSAGE analysis, Genechip and RNA-seq. SuperSAGE analysis showed 32,808 SuperSAGE Tag sequences matched to Nicotiana attenuata transcriptome database and 3,698 Tag sequences matched to R. irregularis. Genechip was used for DEGs screening in transcriptome of Medicago truncatula and Solanum lycopersicum mycorrhizal root (Gaude et al., 2012; Ruzicka et al., 2012; Bonneau et al., 2013; Hogekamp and Küster, 2013). It identified 512 DEGs between AM colonized and non-colonized cells in M. truncatula root (Gaude et al., 2012); 174 DEGs between mycorrhizal wild-type and non-mycorrhizal rmc roots irrespective of the N treatment of Solanum lycopersicum under field condition (Ruzicka et al., 2012). RNA-seq analysis identified 2,210 fungal and tomato sequence assemblies from mycorrhizal roots through comparing the wild-type and rmc root samples under field condition (Ruzicka et al., 2013); 3,641 genes differentially expressed during AM development in Lotus japonicus and approximately 80% of which were upregulated (Handa et al., 2015); 3,949 DEGs between mycorrhizal roots of large lateral roots and fine lateral roots in Oryza sativa (Fiorilli et al., 2015). The numbers of annotated sequences of host litchi and AM fungi in present study compared with those annotated sequences and DEGs in the biological process of AM fungi and other host plants as well as DEGs involved in carbohydrate starvation suggested that RNA-seq is an effective method for identifying transcript variation in litchi mycorrhizal roots under decreased carbohydrate conditions (Table 2).
Previous research suggested that plants can detect, discriminate, and reward the optimal fungal partners with high carbohydrate contents. Moreover, the fungal partners encourage cooperation by increasing nutrient transfer to the hosts to provide high amounts of carbohydrates (Kiers et al., 2011; Fellbaum et al., 2012). Mineral nutrients against carbohydrates between partners were characteristic of both sucrose starvation and hexose-enhanced conditions. Sucrose is the main sugar for transport in M. truncatula; the lines displayed up to 10-fold reduction in the expression levels of the M. truncatula sucrose synthase gene (MtSucS1) in the roots (Baier et al., 2010). The lines exhibited decreased numbers of internal hyphae, vesicles, and arbuscules, which caused an overall stunted aboveground growth under inorganic phosphorus limitations (Baier et al., 2010). Hexoses, especially glucose, are the main carbohydrates for mineral nutrition from AM fungi (Helber et al., 2011). Invertase derived from overexpressing yeast (Saccharomyces cerevisiae) in tobacco (N. tabacum) alc::cwINV increased the hexose concentration in the root. However, the colonization of Glomus intraradices and the level of fungus-specific palmitvaccenic acid, which indicate fungal carbohydrate supply, or plant phosphate content, did not increase (Schaarschmidt et al., 2007). These results implied that sufficient carbohydrates are available in mycorrhizal roots under normal conditions, but carbohydrate starvation inhibited AM development. As predicted, girdling decreased starch concentration as well as monosaccharide, disaccharide, and sugar alcohol contents after the treatment. AM colonization rate matched carbohydrate variation, which continuously decreased after the girdling treatment from 2 W to 2 M (Figures 1 and 2). Besides decreased carbohydrate content and AM colonization, DEGs generated by girdling also suggested that both the litchi root and AM fungi underwent carbohydrate starvation. Biological processes related to carbohydrate metabolism varied after the girdling treatment; these processes include glyoxylate and dicarboxylate metabolism (ko00630), citrate cycle (ko00020), and pentose phosphate pathway (ko00030) in AM fungi; as well as starch and sucrose metabolism (ko00500) and amino sugar and nucleotide sugar metabolism (ko00520) in litchi (Supplementary Table 3). The results, including carbohydrate content, AM colonization, and pathways in both host plant and AM fungi, suggested that girdling regulated litchi mycorrhizal root development by decreasing carbohydrate loss.
Carbohydrate shortage in symbiotic roots lowers AM fungi colonization and decreases the proportion of functional arbuscules (Hayman, 1974; Vierheilig et al., 2002). Girdling inhibits mycorrhizal development in both symbiotic partners. We speculate that upregulated DEGs are required for the detection and adaptation of carbohydrate stress. By contrast, downregulated DEGs in symbiotic partners may be related to carbohydrate exchange under normal conditions. Flavonoids are not strictly required for all combinations of host plant and AM fungi recognition (Bécard et al., 1995). However, the downregulated DEGs at 0 D versus 1 W and at 0 D versus 2 W in host litchi were clustered into flavonoid, flavone, and flavonol biosynthesis as systemic processes. The evidence may suggest that flavonoid was the main factor that regulated AM formation in litchi. Compared with the downregulated DEGs, most of the upregulated DEGs were clustered into stress-inducible and resistance-related pathways, which included the NADP-dependent malic enzyme gene, endochitinase PR4, the basic form of the pathogenesis-related protein 1 LRR receptor-like serine/threonine-protein kinase EFR, cyclic nucleotide-gated ion channel 1, lignin-forming anionic peroxidase, and peroxidase 72 (Gallou et al., 2012; Tromas et al., 2012; Miyata et al., 2014). These data implied that the host litchi stimulated defense-related genes in response to fungal disease when its seceded carbohydrates decreased. In the adaptation process, the downregulated DEGs were mapped to several clusters, including antigen processing and presentation (heat shock protein 82; Heat shock 70 kDa protein; putative heat shock protein HSP 90-beta-3; nuclear transcription factor Y subunit A-2), plant hormone signal transduction (transcription factor MYC4; probable protein phosphatase 2C 24; regulatory protein NPR5), NOD-like receptor signaling pathway (heat shock protein 82; putative heat shock protein HSP 90-beta-3), and MAPK signaling pathway (heat shock 70 kDa protein). The data showed that when carbohydrate shortage, the litchi might downregulate the symbiosis signal-transduction genes. The most upregulated DEGs at 0 D versus those at 1 M and at 0 D versus those at 2 M in the litchi were system clustered into lipid metabolism processes, such as alpha-linolenic acid and linoleic acid metabolism (linoleate 13S-lipoxygenase 2-1; allene oxide cyclase 2; linoleate 13S-lipoxygenase 2-1; 4-coumarate–CoA ligase-like 5; linoleate 13S-lipoxygenase 2-1; TR107818| c4_g1, cytochrome P450 83A1; Supplementary Table 3 and Figure 6). Certain 2-hydroxy fatty acids comprise the putative categories of root exudate signals perceived by Gigaspora species; several genes related to fatty-acid and lipid metabolism that are highly upregulated in AM roots were identified (Gaude et al., 2012). Whether alpha-linolenic acid and linoleic acid could regulate AM colonization under decreased carbohydrate conditions in litchi mycorrhizal tree requires additional study. In addition to the signal molecules and defense-related genes in the symbiotic process, the DEGs involved in AM maintenance, such as transcription factors (Figure 5A), lectin and chitinase genes were also screened (Figure 6A; Supplementary Table 4). The functions of the transcription factors in the AP2, ERF, Myb, WRKY, and bHLH families as well as lectin genes were involved in cellular reprogramming during AM development (De Hoff et al., 2009; Hogekamp et al., 2011; Handa et al., 2015). This finding suggested that carbohydrate shortage potentially influence AM formation, maintenance, and systematic functioning from pre-symbiotic to post-symbiotic periods in litchi roots.
For AM fungal component, the E3 ubiquitin ligase complex SCF subunit scon-3 and putative ubiquitin thioesterase otu1 were identified in the upregulated DEGs at 0 D versus those at 1 W and at 0 D versus those at 2 W. The E3 ubiquitin ligase was found to ubiquitinate Arabidopsis receptor kinase flagellin sensing 2, which was also identified as agents interacting with symbiotic RLKs in L. japonicus (Robatzek et al., 2006; Mbengue et al., 2010; Den Herder et al., 2012). Both the genes encoding the E3 ubiquitin ligase complex SCF subunit scon-3 and putative ubiquitin thioesterase otu1 were upregulated in AM fungi upon recognition of carbohydrate decrease, which illustrated the ubiquitination was important in the process of AM fungi perceived carbohydrate decreased. The KEGG pathway of upregulated DEGs at 0 D versus those at 1 M and at 0 D versus those at 2 M in the AM fungi suggested that AM fungi not only relied on the variation of carbohydrate metabolism for the adaptation process but also upregulated the transcription in arginine and proline metabolism, alanine, aspartate, and glutamate metabolism and lipid metabolism for adopting carbohydrate starvation (Supplementary Table 3). The unigenes mapped to lipid metabolism and genes encoding chitin syntheses varied during fungal adaptation under carbohydrate starvation (Figure 6). The two categories of unigenes possibly related to lipochito-oligosaccharides, short-chain chitin oligomers, and thyroid, which may be correlated to the recognition of host plant and AM fungi (Bates et al., 2012; Kobae et al., 2014; Wewer et al., 2014). Interestingly, the transcripts of cytochrome b-245 and LRR were noted in the DEGs of AM fungi (Supplementary Table 3; Figure 6). R. irregularis possesses over 200 CYPs according to domain prediction using the InterPro database (Park et al., 2008; Moktali et al., 2012; Tisserant et al., 2013). This number is relatively large for CYPs of a fungal species. Previous studies showed that CYPs include heme–thiolate proteins, which are located in the endoplasmic reticulum and catalyze the oxidation of various organic compounds, such as lipids and sterols (Črešnar and Petrič, 2011). The members of the CYP51 family are well-conserved housekeeping genes that participate in the 14-demethylation of sterol precursors (van den Brink et al., 1998; Črešnar and Petrič, 2011). The CYP diversification may be related to various metabolic processes and possible fungal adaptation to the soil environment and host plant roots. In addition to CYPs, LRR transcripts were noted in the DEG database entries on AM fungi. Various surfaces of the leucine-rich repeat LRR ectodomain superstructure are utilized for interaction with the cognate ligand in both plant and animal receptors. Arabidopsis LRR receptor-like kinase FLS2 and rice receptor kinase-like protein Xa21 possess large ectodomains that comprise 28 LRRs and 23 LRRs, respectively (Song et al., 1995; Gómez-Gómez and Boller, 2000), and are directly involved in elicitor binding (Chinchilla et al., 2006; Lee et al., 2009). Because the LRRs form versatile binding domains for plant proteins were involved in the process of plant–microbe interaction, the LRRs in the AM fungi binding the secreted proteins from host plant needed further study.
Conclusion
Girdling decreased the glucose, fructose, sucrose, quebrachitol, and even starch concentrations in the litchi mycorrhizal roots, which induced a decrease in AM colonization. In this study, we revealed the gene expression profiles of both host litchi and AM fungi during the decreased carbohydrate conditions by RNA-seq analysis. Both transcripts of litchi (54,100 unigenes) and AM fungi (33,120 unigenes) were identified in the period of decreased carbohydrates. The DEG analysis of transcriptomes identified potential novel unigenes of both host litchi and AM fungi. DEG analysis showed that flavonoids, alpha-linolenic acid, and linoleic acid were the main factors that regulated AM colonization in litchi. However, the flavonoids might play a role in the recognition of the stages of decreasing carbohydrate content, and alpha-linolenic acid or linoleic acid may affect AM formation under the process of adaptation. Litchi trees stimulated the expression of the defense-related genes (NADP-dependent malic enzyme gene, endochitinase PR4, basic form of pathogenesis-related protein 1, LRR receptor-like serine/threonine-protein kinase EFR, cyclic nucleotide-gated ion channel 1, lignin-forming anionic peroxidase, and peroxidase 72) and downregulated the symbiosis signal-transduction genes (heat-shock protein 82, heat-shock 70 kDa protein, putative heat-shock protein HSP 90-beta-3, transcription factor MYC4, probable protein phosphatase 2C 24, and regulatory protein NPR5) to inhibit new AM colonization. In addition to new AM colonization, carbohydrate shortage changed the transcription factor in the AP2, ERF, Myb, WRKY, bHLH families and lectin genes of litchi, which influenced AM maintenance in the post-symbiotic stage. Similar to those of the litchi host, the E3 ubiquitin ligase complex SCF subunit scon-3 and polyubiquitin of AM fungi were all upregulated at the perceived stages. This occurrence suggested that the ubiquitination process plays an important role in the AM fungal recognition of carbohydrate decrease. The transcription of cytochrome b-245 and LRRs were noted in the DEGs database, implying that these transcripts play important roles in the process of AM fungal adaptation under carbohydrate starvation despite the gene function still being largely unknown.
Author Contributions
The data was interpreted and the article was drafted by BS. The manuscript was revised by SS. Meanwhile, the experiments were conceived and designed by BS and SS. BS, LL, and YW performed the experiments. BS and WL analyzed the data.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the National Natural Science Fund of China (Project No. 31401818) and founded on the Basic Scientific Research Project of Non-profit Central Research Institutions (No. SSCRI-1630062014006) and the China Litchi and Longan Industry Technology Research System (Project No.CARS-33-11).
Abbreviations
AM, arbuscular mycorrhiza; COG, cluster of orthologous groups of proteins; CYP, cytochrome P450; DEG, differentially expressed gene; FDR, false discovery rate; FPKM, fragments per kilobase of exon per million fragments mapped; Gloin1, Rhizophagus irregularis genome assembly; GO, gene ontology; HPLC, high-performance liquid chromatography; KEGG, Kyoto Encyclopedia of Genes and Genomes; LRR, leucine-rich repeats; nr, NCBI non-redundant (nr) database.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2016.00408
FIGURE S1 | The girdling litchi trees in the experimental orchard. The girdling wound was deep to xylem with 0.5 cm width.
FIGURE S2 | Flow chart of RNA-seq analysis.
Footnotes
- ^http://genome.jgi.doe.gov/Gloin1/Gloin1.home.html
- ^http://www.ncbi.nlm.nih.gov
- ^http://www.expasy.ch/sprot
- ^http://www.genome.jp/kegg
- ^http://www.uniprot.org/uniref/?query=&fil=identity:0.9
References
Baier, M. C., Keck, M., Gödde, V., Niehaus, K., Küster, H., and Hohnjec, N. (2010). Knockdown of the symbiotic sucrose synthase MtSucS1 affects arbuscule maturation and maintenance in mycorrhizal roots of Medicago truncatula. Plant Physiol. 152, 1000–1014. doi: 10.1104/pp.109.149898
Bates, P. D., Fatihi, A., Snapp, A. R., Carlsson, A. S., Browse, J., and Lu, C. (2012). Acyl editing and headgroup exchange are the major mechanisms that direct polyunsaturated fatty acid flux into triacylglycerols. Plant Physiol. 160, 1530–1539. doi: 10.1104/pp.112.204438
Bécard, G., Douds, D. D., and Pfeffer, P. E. (1992). Extensive in vitro hyphal growth of vesicular-arbuscular mycorrhizal fungi in the presence of CO2 and flavonols. Appl. Environ. Microbiol. 58, 821–825.
Bécard, G., Taylor, L. P., Douds, D. D., Pfeffer, P. E., and Doner, L. W. (1995). Flavonoids are not necessary plant signal compounds in arbuscular mycorrhizal symbioses. Mol. Plant Microbe Ineract. 8, 252–258. doi: 10.1094/mpmi-8-0252
Besserer, A., Bécard, G., Jauneau, A., Roux, C., and Séjalon-Delmas, N. (2008). GR24, a synthetic analog of strigolactones, stimulates the mitosis and growth of the arbuscular mycorrhizal fungus Gigaspora rosea by boosting its energy metabolism. Plant Physiol. 148, 402–413. doi: 10.1104/pp.108.121400
Bonneau, L., Huguet, S., Wipf, D., Pauly, N., and Truong, H. N. (2013). Combined phosphate and nitrogen limitation generates a nutrient stress transcriptome favorable for arbuscular mycorrhizal symbiosis in Medicago truncatula. New Phytol. 199, 188–202. doi: 10.1111/nph.12234
Bucher, M., Hause, B., Krajinski, F., and Küster, H. (2014). Through the doors of perception to function in arbuscular mycorrhizal symbioses. New Phytol. 204, 833–840. doi: 10.1111/nph.12862
Chinchilla, D., Bauer, Z., Regenass, M., Boller, T., and Felix, G. (2006). The Arabidopsis receptor kinase FLS2 binds flg22 and determines the specificity of flagellin perception. Plant Cell 18, 465–476. doi: 10.1105/tpc.105.036574
Črešnar, B., and Petrič, S. (2011). Cytochrome P450 enzymes in the fungal kingdom. BBA-Proteins Proteom. 1814, 29–35. doi: 10.1016/j.bbapap.2010.06.020
De Hoff, P. L., Brill, L. M., and Hirsch, A. M. (2009). Plant lectins: the ties that bind in root symbiosis and plant defense. Mol. Genet. Genom. 282, 1–15. doi: 10.1007/s00438-009-0460-8
Den Herder, G., Yoshida, S., Antolín-Llovera, M., Ried, M. K., and Parniske, M. (2012). Lotus japonicus E3 ligase SEVEN IN ABSENTIA4 destabilizes the symbiosis receptor-like kinase SYMRK and negatively regulates rhizobial infection. Plant Cell 24, 1691–1707. doi: 10.1105/tpc.110.082248
Fellbaum, C. R., Gachomo, E. W., Beesetty, Y., Choudharib, S., Strahanc, G. D., Pfefferc, P. E., et al. (2012). Carbon availability triggers fungal nitrogen uptake and transport in arbuscular mycorrhizal symbiosis. Proc. Natl. Acad. Sci. U.S.A. 109, 2666–2671. doi: 10.1073/pnas.1118650109
Fiorilli, V., Vallino, M., Biselli, C., Faccio, A., Bagnaresi, P., and Bonfante, P. (2015). Host and non-host roots in rice: cellular and molecular approaches reveal differential responses to arbuscular mycorrhizal fungi. Front. Plant Sci. 6:636. doi: 10.3389/fpls.2015.00636
Gallou, A., Declerck, S., and Cranenbrouck, S. (2012). Transcriptional regulation of defence genes and involvement of the WRKY transcription factor in arbuscular mycorrhizal potato root colonization. Funct. Integr. Genomic 12, 183–198. doi: 10.1007/s10142-011-0241-4
Garber, M., Grabherr, M. G., Guttman, M., and Trapnell, C. (2011). Computational methods for transcriptome annotation and quantification using RNA-seq. Nat. Methods 8, 469–477. doi: 10.1038/nmeth.1613
Gaude, N., Bortfeld, S., Duensing, N., Lohse, M., and Krajinski, F. (2012). Arbuscule-containing and non-colonized cortical cells of mycorrhizal roots undergo a massive and specific reprogramming during arbuscular mycorrhizal development. Plant J. 69, 510–528. doi: 10.1111/j.1365-313x.2011.04810.x
Genre, A., Chabaud, M., Balzergue, C., Puech-Pagès, V., Novero, M., Rey, T., et al. (2013). Short-chain chitin oligomers from arbuscular mycorrhizal fungi trigger nuclear Ca2+ spiking in Medicago truncatula roots and their production is enhanced by strigolactone. New Phytol. 198, 190–202. doi: 10.1111/nph.12146
Gómez-Gómez, L., and Boller, T. (2000). FLS2: an LRR receptor–like kinase involved in the perception of the bacterial elicitor flagellin in Arabidopsis. Mol. Cell. 5, 1003–1011. doi: 10.1016/S1097-2765(00)80265-8
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Handa, Y., Nishide, H., Takeda, N., Suzuki, Y., Kawaguchi, M., and Saito, K. (2015). RNA-seq transcriptional profiling of an arbuscular mycorrhiza provides insights into regulated and coordinated gene expression in Lotus japonicus and Rhizophagus irregularis. Plant Cell Physiol. 56, 1490–1511. doi: 10.1093/pcp/pcv071
Hayman, D. S. (1974). Plant growth responses to vesicular-arbuscular mycorrhiza.VI. Effect of light and temperature. New Phytol. 73, 71–80.
Helber, N., Wippel, K., Sauer, N., Schaarschmidt, S., Hause, B., and Requena, N. (2011). A versatile monosaccharide transporter that operates in the arbuscular mycorrhizal fungus Glomus sp is crucial for the symbiotic relationship with plants. Plant Cell 23, 3812–3823. doi: 10.1105/tpc.111.089813
Hogekamp, C., Arndt, D., Pereira, P., Becker, J. D., Hohnjec, N., and Küster, H. (2011). Laser microdissection unravels cell-type-specific transcription in arbuscular mycorrhizal roots, including CAAT-Box transcription factor gene expression correlating with fungal contact and spread. Plant Physiol. 157, 2023–2043. doi: 10.1104/pp.111.186635
Hogekamp, C., and Küster, H. (2013). A roadmap of cell-type specific gene expression during sequential stages of the arbuscular mycorrhiza symbiosis. BMC Genomics 14:306. doi: 10.1186/1471-2164-14-306
Huang, X. M., Wang, H. C., and Yuan, W. Q. (2003). Effects of twig girdling at different stages on new shoot growth and carbon nutrient reservation. Acta Hortic. Sin. 30, 192–194.
Kiers, E. T., Duhamel, M., Beesetty, Y., Mensah, J. A., Franken, O., Verbruggen, E., et al. (2011). Reciprocal rewards stabilize cooperation in the mycorrhizal symbiosis. Science 333, 880–882. doi: 10.1126/science.1208473
Kobae, Y., Gutjahr, C., Paszkowski, U., Kojima, T., Fujiwara, T., and Hata, S. (2014). Lipid droplets of arbuscular mycorrhizal fungi emerge in concert with arbuscule collapse. Plant Cell Physiol. 55, 1945–1953. doi: 10.1093/pcp/pcu123
Lee, S. W., Han, S. W., Sririyanum, M., Park, C. J., Seo, Y. S., and Ronald, P. C. (2009). A type I–secreted, sulfated peptide triggers XA21-mediated innate immunity. Science 326, 850–853. doi: 10.1126/science.1173438
Li, B., and Dewey, C. N. (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12:323. doi: 10.1186/1471-2105-12-323
Maillet, F., Poinsot, V., André, O., Puech-Pagès, V., Haouy, A., Gueunier, M., et al. (2011). Fungal lipochitooligosaccharide symbiotic signals in arbuscular mycorrhiza. Nature 469, 58–63. doi: 10.1038/nature09622
Mbengue, M., Camut, S., de Carvalho-Niebel, F., Deslandes, L., Froidure, S., Klaus-Heisen, D., et al. (2010). The Medicago truncatula E3 ubiquitin ligase PUB1 interacts with the LYK3 symbiotic receptor and negatively regulates infection and nodulation. Plant Cell 22, 3474–3488. doi: 10.1105/tpc.110.075861
McGonigle, T. P., Miller, M. H., Evans, D. G., Fairchild, G. L., and Swan, J. A. (1990). A new method which gives an objective measure of colonization of roots by vesicular-arbuscular mycorrhizal fungi. New Phytol. 115, 495–501. doi: 10.1111/j.1469-8137.1990.tb00476.x
Miyata, K., Kozaki, T., Kouzai, Y., Ozawa, K., Ishii, K., Asamizu, E., et al. (2014). The bifunctional plant receptor, OsCERK1, regulates both chitin-triggered immunity and arbuscular mycorrhizal symbiosis in rice. Plant Cell Physiol. 55, 1864–1872. doi: 10.1093/pcp/pcu129
Moktali, V., Park, J., Fedorova-Abrams, N., Park, B., Choi, J., Lee, Y. H., et al. (2012). Systematic and searchable classification of cytochrome P450 proteins encoded by fungal and oomycete genomes. BMC Genomics 13:525. doi: 10.1186/1471-2164-13-525
Nagahashi, G., and Douds, D. D. (2011). The effects of hydroxy fatty acids on the hyphal branching of germinated spores of AM fungi. Fungal Boil. 115, 351–358. doi: 10.1016/j.funbio.2011.01.006
Park, J., Lee, S., Choi, J., Ahn, K., Park, B., Park, J., et al. (2008). Fungal cytochrome P450 database. BMC Genomics 9:402. doi: 10.1186/1471-2164-9-402
Parniske, M. (2008). Arbuscular mycorrhiza: the mother of plant root endosymbioses. Nat. Rev. Microbiol. 6, 763–775. doi: 10.1038/nrmicro1987
Phillips, J. M., and Hayman, D. S. (1970). Improved procedures for clearing roots and staining parasitic and vesicular-arbuscular mycorrhizal fungi for rapid assessment of infection. Trans. Br. Mycol. Soc. 55, 158–161. doi: 10.1016/s0007-1536(70)80110-3
Robatzek, S., Chinchilla, D., and Boller, T. (2006). Ligand-induced endocytosis of the pattern recognition receptor FLS2 in Arabidopsis. Gene Dev. 20, 537–542. doi: 10.1101/gad.366506
Ruzicka, D. R., Chamala, S., Barriosmasias, F. H., Martin, F., Smith, S., Jackson, L. E., et al. (2013). Inside arbuscular mycorrhizal roots – molecular probes to understand the symbiosis. Plant Genome 6, 494–494. doi: 10.3835/plantgenome2012.06.0007
Ruzicka, D. R., Hausmann, N. T., Barrios-Masias, F. H., Jackson, L. E., and Schachtman, D. P. (2012). Transcriptomic and metabolic responses of mycorrhizal roots to nitrogen patches under field conditions. Plant Soil 350, 145–162. doi: 10.1007/S11104-011-0890-Z
Schaarschmidt, S., González, M. C., Roitsch, T., Strack, D., Sonnewald, U., and Hause, B. (2007). Regulation of arbuscular mycorrhization by carbon. The symbiotic interaction cannot be improved by increased carbon availability accomplished by root-specifically enhanced invertase activity. Plant Physiol. 143, 1827–1840. doi: 10.1104/pp.107.096446
Smith, S. E., and Read, D. J. (2008). Mycorrhizal Symbiosis, 3nd Edn. New York, NY: Academic Press. doi: 10.1016/B978-012370526-6.50003-9
Song, W. Y., Wang, G. L., Chen, L. L., Kim, H. S., Pi, L. Y., Holsten, T., et al. (1995). A receptor kinase-like protein encoded by the rice disease resistance gene, XA21. Science 270, 1804–1806. doi: 10.1126/science.270.5243.1804
Sun, J., Miller, J. B., Granqvist, E., Wiley-Kalil, A., Gobbato, E., Maillet, F., et al. (2015). Activation of symbiosis signaling by arbuscular mycorrhizal fungi in legumes and rice. Plant Cell 27, 828–838. doi: 10.1105/tpc.114.131326
Tisserant, E., Malbreil, M., Kuo, A., Kohler, A., Symeonidi, A., Balestrini, R., et al. (2013). Genome of an arbuscular mycorrhizal fungus provides insight into the oldest plant symbiosis. Proc. Natl. Acad. Sci. U.S.A. 110, 20117–20122. doi: 10.1073/pnas.1313452110
Tromas, A., Parizot, B., Diagne, N., Champion, A., Hocher, V., Cissoko, M., et al. (2012). Heart of endosymbioses: transcriptomics reveals a conservedgenetic program among arbuscular mycorrhizal, actinorhizal and legume-rhizobial symbioses. PLoS ONE 7:e44742. doi: 10.1371/journal.pone.0044742
Tromp, J. (1983). Nutrient reserves in roots of fruit trees, in particular carbohydrates and nitrogen. Plant Soil. 71, 401–413 doi: 10.1007/BF02182682
van den Brink, H. M., van Gorcom, R. F. M., van den Hondel, C. A. M. J. J., and Punt, P. J. (1998). Cytochrome P450 enzyme systems in fungi. Fungal Genet. Biol. 23, 1–17. doi: 10.1006/fgbi.1997.1021
Vierheilig, H., Bago, B., Lerat, S., and Piché, Y. (2002). Shoot-produced, light dependent factors are partially involved in the expression of the arbuscular mycorrhizal (AM) status of AM host and non-host plants. J. Plant Nutr. Soil Sci. 165, 21–25. doi: 10.1002/1522-2624(200202)165:1<21::AID-JPLN21>3.0.CO;2-9
Wang, H. C., Wu, Z. H., Huang, X. M., Hu, G. B., and Chen, H. B. (2013). Determination of quebrachitol Litchi chinensis and Dimocarpus longan in sapindacea family. J. S. China Agric. Univ. 34, 315–319.
Wang, C., Gong, B., Bushel, P. R., Thierry-Mieg, J., Thierry-Mieg, D., Xu, J., et al. (2014). The concordance between RNA-seq and microarray data depends on chemical treatment and transcript abundance. Nat. Biotechnol. 32, 926–932. doi: 10.1038/nbt.3001
Wang, T. D., Zhang, H. F., Wu, Z. C., Li, J. G., Huang, X. M., and Wang, H. C. (2014). Sugar uptake in the aril of litchi fruit depends on the apoplasmic post-phloem transport and the activity of proton pumps and the putative transporter LcSUT4. Plant Cell Physiol. 56, 377–387. doi: 10.1093/pcp/pcu173
Wewer, V., Brands, M., and Dörmann, P. (2014). Fatty acid synthesis and lipid metabolism in the obligate biotrophic fungus Rhizophagus irregularis during mycorrhization of Lotus japonicus. Plant J. 79, 398–412. doi: 10.1038/nbt.3001
Xie, C., Mao, X., Huang, J., Ding, Y., Wu, J., Dong, S., et al. (2011). KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 39, W316–W322. doi: 10.1093/nar/gkr483
Young, M. D., Wakefield, M. J., Smyth, G. K., and Oshlack, A. (2010). Method Gene Ontology Analysis for RNA-Seq: accounting for selection bias. Genome Biol. 11:R14. doi: 10.1186/gb-2010-11-2-r14
Keywords: litchi, carbohydrates, arbuscular mycorrhizal colonization, RNA-seq, transcripts
Citation: Shu B, Li W, Liu L, Wei Y and Shi S (2016) Transcriptomes of Arbuscular Mycorrhizal Fungi and Litchi Host Interaction after Tree Girdling. Front. Microbiol. 7:408. doi: 10.3389/fmicb.2016.00408
Received: 13 January 2016; Accepted: 14 March 2016;
Published: 30 March 2016.
Edited by:
Pietro Daniele Spanu, Imperial College London, UKReviewed by:
Raffaella Balestrini, Consiglio Nazionale delle Ricerche, ItalyBiswapriya Biswavas Misra, University of Florida, USA
Copyright © 2016 Shu, Li, Liu, Wei and Shi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shengyou Shi, c3N5NzI5OUAxNjMuY29t