Abstract
The viral enzyme integrase (IN) is essential for the replication of human immunodeficiency virus type 1 (HIV-1) and represents an important target for the development of new antiretroviral drugs. In this study, we focused on the N-terminal domain (NTD), which is mainly involved into protein oligomerization process, for the development and synthesis of a library of overlapping peptide sequences, with specific length and specific offset covering the entire native protein sequence NTD IN 1–50. The most potent fragment, VVAKEIVAH (peptide 18), which includes a His residue instead of the natural Ser at position 39, inhibits the HIV-1 IN activity with an IC50 value of 4.5 μM. Amino acid substitution analysis on this peptide revealed essential residues for activity and allowed us to identify two nonapeptides (peptides 24 and 25), that show a potency of inhibition similar to the one of peptide 18. Interestingly, peptide 18 does not interfere with the dynamic interplay between IN subunits, while peptides 24 and 25 modulated these interactions in different manners. In fact, peptide 24 inhibited the IN-IN dimerization, while peptide 25 promoted IN multimerization, with IC50 values of 32 and 4.8 μM, respectively. In addition, peptide 25 has shown to have selective anti-infective cell activity for HIV-1. These results confirmed peptide 25 as a hit for further development of new chemotherapeutic agents against HIV-1.
Introduction
Integrase (IN) is a key enzyme for the integration of the HIV-1 genome into the host cell chromosome and, therefore, a very promising target for anti-AIDS drug design. IN performs two essential catalytic reactions. The first one takes place in the cytoplasm within particles termed preintegration complex (PIC). IN cleaves the conserved dinucleotides GT from the 3′ ends (3′-processing) of the double-stranded viral DNA, generating reactive 3′-hydroxyl DNA ends. Following translocation to the nucleus, IN uses the hydroxyl ends as a substrate reaction for a nucleophilic attack integrating the viral genome in the host chromosomal DNA (strand-transfer reaction, STIN) (Semenova et al., 2006; Delelis et al., 2008).
IN is a protein of 32 kDa organized in three domains (Wang et al., 2001; Chiu and Davies, 2004). The N-terminal domain (NTD, residues 1–50) contains a zinc-binding H12H16C40C43 motif and contributes to protein oligomerization process (Engelman and Craigie, 1992; Cai et al., 1997). The central domain (CCD, residues 51–212) encloses the enzyme active site characterized by the catalytic triad D64D116E152, which is highly conserved in all INs, with the two residues D64 and D116 forming a coordination complex with divalent ions like Mn2+ and Mg2+ (Goldgur et al., 1998; Chen et al., 2000). The C-terminal domain (CTD, residues 213–288), which is the least conserved domain between retroviral INs, binds non-specifically to DNA and is also involved in IN multimerization (Chen et al., 2000).
Despite the CCD contains the catalytic site, in the absence of the NTD and CTD domains, IN can only catalyze the disintegration reaction in vitro, while it needs both the NTD and CTD in a dimeric complex to catalyze 3′-processing and strand-transfer reactions (Lia et al., 2011). Until now, only inhibitors targeting the catalytic site of IN with a specific effect on strand-transfer process (INSTIs) have been identified and developed (Pommier et al., 2005; Savarino, 2006; Dayam et al., 2008; Katlama and Murphy, 2012; Esposito and Tramontano, 2013). In 2007, the INSTI Raltegravir became the first IN inhibitor approved for use in the treatment of HIV infected patients (Evering and Markowitz, 2007; Summa et al., 2008). More recently, the FDA also approved Stribild, a single-tablet regimen HIV medication containing four drug combination of Elvitegravir, another HIV INSTI, Cobicistat, a CYP3A inhibitor, and Emtricitabine and Tenofovir DF, both HIV nucleoside analog reverse transcriptase inhibitors (Shimura et al., 2008; Brinson, 2013).
The emergence of the resistance to these drugs gives raise to the pressing need of novel IN inhibitors (Mesplède et al., 2012). However, absence of information on the structures of a full-sized enzyme, on its complex with DNA and also on the PIC composition and operation complicates and slows the search for novel IN inhibitors.
To overcome these drawbacks, targeting allosteric sites of the protein including interaction site of IN with cellular co-factor essential for integration, for example the LEDGF/p75-IN interaction, or oligomerization/multi oligomerization sites might represent alternative approaches to IN inhibition (Zhao et al., 2003; Li et al., 2006; Al-Mawsawi and Neamati, 2011; Maes et al., 2012; Christ and Debyser, 2013; Long et al., 2013; Di Santo, 2014).
In this context, compounds that target the IN N-terminal domain would be effective at disrupting IN function through a range of mechanisms, including allosteric or oligomerization inhibition. We considered this domain an important starting point for the identification of peptide inhibitors in order to clarify some points on IN function in viral replication, such as the elucidation of HIV-1 IN polymerization state, or its potentiality in the structure-based design.
Here we present the design and synthesis of peptides targeting the integration events by directly inhibiting IN or its multimerization process. The synthesized peptides were assayed in vitro for their ability to inhibit IN strand transfer activity and for the capability to inhibit IN dimerization or to promote IN multimerization. Finally, the most potent compounds, conveniently conjugated with cell-penetrating fragment Tat, were assayed in MT-4 cells for determining anti-HIV infective activity.
Materials and methods
Nα-Fmoc-protected amino acids, Rink amide-resin, HOAt, HOBt, HBTU, DIEA, piperidine, and trifluoroacetic acid were purchased from Iris Biotech (Germany). Rink Amide-ChemMatrix resin was purchased from Biotage AB (Sweden). Peptide synthesis solvents, reagents, as well as CH3CN for HPLC were reagent grade and were acquired from commercial sources and used without further purification unless otherwise noted.
Peptide synthesis
The synthesis of IN analogs was performed according to the solid phase approach using standard Fmoc methodology in a manual reaction vessel and automated microwave synthesizer (Wang et al., 1989; Malik et al., 2010). The first amino acid, Nα-Fmoc-Xaa-OH (Nα-Fmoc-Asp(OtBu)-OH, Nα-Fmoc-Glu(OtBu)-OH, Nα-Fmoc-His(N(im)trityl(Trt))-OH, Nα-Fmoc-Trp(Boc)-OH, Nα-Fmoc-Ala-OH, Nα-Fmoc-Leu-OH, Nα-Fmoc-Val-OH, Nα-Fmoc-Lys(Boc)-OH, Nα-Fmoc-Cys(Trt)-OH, Nα-Fmoc-Met-OH, Nα-Fmoc-Ser(tBu)-OH, was linked on to the Rink resin (100–200 mesh, 1% DVB, 0.59 mmol/g) previously deprotected by a 25% piperidine solution in DMF for 30 min. The following protected amino acids were then added stepwise. Each coupling reaction was accomplished using a three-fold excess of amino acid with HBTU (3 eq.) and HOBt (3 eq.) in the presence of DIEA (6 eq.). The Nα-Fmoc protecting groups were removed by treating the protected peptide resin with a 25% solution of piperidine in DMF (1 × 5 min and 1 × 25 min). The peptide resin was washed three times with DMF and the next coupling step was initiated in a stepwise manner. The peptide resin was washed with DCM (3 ×), DMF (3 ×), and DCM (3 ×), and the deprotection protocol was repeated after each coupling step. In addition, after each step of deprotection and after each coupling step, Kaiser test was performed to confirm the complete removal of the Fmoc protecting group, respectively, and to verify that complete coupling has occurred on all the free amines on the resin. The N-terminal Fmoc group was removed as described above, and the peptides were acetylated adding a solution of Ac2O/DCM (1:3) shaking for 30 min. Finally the peptides were released from the resin with TFA/iPr3SiH/H2O (90:5:5) for 3 h. The resin was removed by filtration, and the crude peptide was recovered by precipitation with cold anhydrous ethyl ether to give a white powder and then lyophilized.
Microwave peptides synthesis
The peptides Tat-18, Tat-24, and Tat-25 were synthesized using a Biotage Syro Wave fully automated microwave and parallel peptide synthesizer or assembled on the Automated Microwave Peptide Synthesizer from Biotage AB (Initiator + AlstraTM).
Peptides were synthesized on a Rink Amide-ChemMatrix resin (150 mg, loading 0.4–0.6 mmol/g), previously deprotected with 25% piperidine/DMF (1 × 3 min, 1 × 10 min) at room temperature. The resin was then washed with DMF (4 × 4.5 ml). The following protected amino acids were then added on to the resin stepwise. Coupling reactions were performed using Nα-Fmoc amino acids (3.0 eq., 0.5 M), using as coupling reagent HBTU (3eq, 0.6 M), HOAt (3eq, 0.5 M), and DIEA (6eq, 2 M) in N-methyl-2-pyrrolidone (NMP). All couplings were achieved for 10 min at 75°C (2 ×) and 2 × 45 min at RT for histidine and cysteine couplings to avoid the epimerization. After each coupling step, the Fmoc protecting group was removed as described above. The resin was washed with DMF (4 × 4.5 ml) after each coupling and deprotection step. Finally peptides were released as described above.
Purification and characterization
All crude peptides were purified by RP-HPLC on a preparative C18-bonded silica column (Phenomenex Jupiter 100 proteo 90 Å, 100 × 21.20 mm, 10 μm) using a Shimadzu SPD 10A UV−Vis detector, with detection at 210 and 254 nm. The column was perfused at a flow rate of 15 mL/min with solvent A (10%, v/v, water in 0.1% aqueous TFA), and a linear gradient from 10 to 90% of solvent B (80%, v/v, acetonitrile in 0.1% aqueous TFA) over 25 min was adopted for peptide elution. Analytical purity, retention time (tR) and peptides molecular weights were determined by ESI mass spectrometry in Shimadzu a LC-MS 2010 instrument fitted with Phenomenex, Aeris XB-C18 column (150 mm × 4.60, 3.6 μm).
IN activity inhibition assay
Express Biotech International Company performed this assay using its HIV-1 IN Assay kit (Xpressbio Life Science Products, USA. www.xpressbio.com). Briefly, Streptavidin coated 96-well plates were coated with a double-stranded HIV-1 LTR U5 donor substrate (DS) oligonucleotide containing an end-labeled biotin. Full-length recombinant HIV-1 integrase protein was then loaded onto this oligo substrate. Compounds were added to the reaction and then a different double-stranded target substrate (TS) oligo containing 3′-end modifications was added to the plate. The sequence of DS DNA and TS DNA substrates used in this assay were
5′-P ACC CTT TTA GTC AGT GTG GAA AAT CTC TAG
CAG T
GAA AAT CAG TCA CAC CTT TTA GAG ATC GTC A
and
5′ TGA CCA AGG GCT AAT TCA CT
ACT GGT TCC CGA TTA GAS GA
respectively (Hazuda et al., 1994 and Engelman et al., 1993). The HIV-1 IN cleaves the terminal two bases from the exposed 3′-end of the HIV-1 LTR DS and then catalyzes a strand-transfer reaction to integrate the DS into the TS. The products of the reaction were detected colorimetrically using an HRP-labeled antibody directed against the TS 3′-end modification. Percent inhibition in the IN assay was calculated and IC50 values were determined.
HTRF-based IN subunit exchange assay
Full-length IN and LEDGF proteins were expressed in E. coli BL21(DE3) and were purified as described (Kessl et al., 2012; Esposito et al., 2015; Tintori et al., 2015). His- and FLAG-tagged INs, were mixed in 25 mM Tris (pH 7.4) buffer containing 150 mM NaCl, 2 mM MgCl2, 0.1% Nonidet P-40, 1 mg/ml BSA. Test compounds were then added to the mixture. A mixture of anti-His6-XL665 and anti-FLAG-EuCryptate antibodies were then added to the reaction and, after an incubation, the plate are read and the HTRF signal is calculated from the 665/620 nm ratio.
Circular dichroism (CD)
All CD spectra were recorded using a JASCO J710 spectropolarimeter at 20°C between λ = 260–190 nm (1 mm path, 1 nm bandwidth, 4 accumulations, and 100 nm min−1 scanning speed). Measurements were performed with peptides in H2O (0.100 mM, pH 7.4) or in in 50% TFE/water solution.
Cell-based assays
The HIV-1 strain NL4.3 was obtained from the AIDS Research and Reference Reagent Program (Division of AIDS, NIAID, NIH, USA). The HIV-2 strain ROD was obtained from the Medical Research Council (MRC, London, UK). The anti-HIV assay in MT-4 cells has been described previously (Vermeire et al., 2004). MT-4 cells (1 × 106 cells/ml; 50 μl of volume) were pre-incubated for 30 min at 37°C with the test compounds in 96-well plates (Falcon, BD Biosciences). The laboratory HIV strains (NL4.3 and ROD) were added according to the 50% tissue culture infectious dose (TCID50) of the viral stock. Cellular cytopathicity was scored microscopically in MT-4 cells 5 days post-infection and IC50s were calculated spectrophotometrically using MTS/PES. For the latter viability assay, the Cell-Titer96 Aqueous One Solution Proliferation Assay (Promega, Leiden, The Netherlands) kit was used.
Results
Design
Starting from HIV-1 IN NTD we synthesized an overlapping peptide library to generate a library of peptide sequences of specific length and specific offset, to cover the entire native protein sequence NTD IN 1–50 (peptides 1–15, Figure 1). The solution structure of NTD has been solved by 3D and 2D NMR spectroscopy, classifying NTD as an “all alpha helix” structure. In fact, it consists of three α helices, which range from residues 6–15, 19–25, and 31–39, stabilized by a Zn2+ ion coordinated with the HHCC motif (evidenced in Figure 1). Ile5, Ala8, Ile28, Ala33, and Ile36 form part of the hydrophobic core, which explains their importance for structural integrity (Eijkelenboom et al., 1997). As showed in Figure 1, peptide 2 and peptides 6 and 7 include the 1rd α-helix and 2rd α-helix respectively, while peptide 11 forms the 3rd α-helix of the NTD, and incorporates the Cys40 of the HHCC motif.
Figure 1
The relevance of both a α-helix structure and a Zn2+ chelating group on the inhibitory activity was assessed by the synthesis of three peptides derived from peptide 11, in which the Cys residue was deleted (peptide 16) or substituted by His amino acid a residue also involved in the Zn2+ coordination (peptide 17) (Wolfe et al., 2001; Liao et al., 2013). The same His was also used to replace the C-terminal Ser residue of peptide 16 leading to peptide 18.
Biological studies: HIV-1 IN catalytic activity inhibition
The synthetized peptides 1–18 were tested in vitro for their ability to inhibit the HIV-1 IN. Results showed that peptide 18 was the most potent of the series and inhibit the HIV-1 IN activity with an IC50 values of 4.5 μM (Table 1). Peptides 9 and 12 containing the VVAK and VA fragments of peptide 18, respectively, and peptide 2, which includes the 6–13 fragment of the first α-helix, maintained a certain inhibitory activity. In this assay, we use Raltegravir as a controls.
Table 1
| Peptide | Sequence | HPLC | ESI MS | IN inhibition |
|---|---|---|---|---|
| k' | Found | IC50(μM) ±SD | ||
| 1 | 1FLDGIDKAQD | 4.70 | 1163.1 | NA |
| 2 | 4GIDKAQDEHE | 5.03 | 1182.5 | 162±9 |
| 3 | 7KAQDEHEKYH | 4.63 | 1326.7 | NE |
| 4 | 10DEHEKYHSNW | 4.75 | 1385.5 | NA |
| 5 | 13EKYHSNWRA | 5.02 | 1363.7 | NE |
| 6 | 16HSNWRAMASD | 2.94 | 1215.5 | NE |
| 7 | 19WRAMASDFNL | 2.30 | 1251.7 | NE |
| 8 | 22MASDFNLPPV | 1.30 | 1131.6 | NA |
| 9 | 25DFNLPPVVAK | 3.70 | 1140.6 | 213 ± 12 |
| 10 | 28LPPVVAKEIV | 4.40 | 1105.8 | NA |
| 11 | 31VVAKEIVASC | 2.12 | 1059.7 | NA |
| 12 | 34KEIVASCDKC | 3.06 | 1136.5 | 477 ± 23 |
| 13 | 37VASCDKCQLK | 2.80 | 1135.5 | NA |
| 14 | 40CDKCQLKGEA | 2.60 | 1135.4 | NA |
| 15 | 43CQLKGEAM | 3.70 | 920.3 | NA |
| 16 | 31VVAKEIVAS | 2.50 | 956.5 | NA |
| 17 | VVAKEIVASH VVAKEIVAH | 5.22 | 1094.0 | NA |
| 18 | VVAKEIVAH | 3.50 | 1007.0 | 4.5 ± 0.9 |
| Raltegravir | 0.18 ± 0.01 |
Sequence, analytical data and effect of peptides 1–18 on HIV-1 IN activities.
All peptide are acetylated and amidate at N terminal and C terminal respectively.
k' [(peptide retention time – solvent retention time)/solvent retention time]. Peptide concentration μM.
NE, No effect. The peptide demonstrated no significant effect in the IN assay, where the percent activity for the compounds was found to be 100 ± 7% (As example see the dose-response curve of peptide 5 in Supplementary Material).
NA, Not Applicable, 50% inhibition by the compound was not achieved (As example see the dose-response curve of peptide 8 in Supplementary Material).
Base on the ability of inhibiting HIV-1 IN showed by peptide 18, we synthesized three peptides, in which Val at position 1, 2, and 7 was substituted with Phe (peptides 19, 20, and 21, respectively). In fact, Phe and Val differ in participating helical assembly of the peptide molecules, and have different hydrophobicity degree (Javadpour and Barkley, 1997; Azmi et al., 2013). In addition, Phe could be involved in additional stacking interactions with the viral target.
Subsequently, we established the contribution of the various amino acid residues to the antiviral activity of peptide 18 (peptides 22–28) through an L-Ala scanning analysis. The results of the biological evaluation of these peptides are summarized in Table 2.
Table 2
| Peptide | Sequence | HPLC | ESI-MS | IN inhibition |
|---|---|---|---|---|
| k' | Found | IC50(μM) | ||
| 18 | VVAKEIVAH | 3.50 | 1007 | 4.5 ± 0.9 |
| 19 | FVAKEIVAH | 4.40 | 1054.4 | NA |
| 20 | VFAKEIVAH | 4.50 | 1054.5 | 375 ± 19 |
| 21 | VVAKEIFAH | 4.20 | 1054.2 | 504 ± 22 |
| 22 | AVAKEIVAH | 3.90 | 978.6 | NA |
| 23 | VAAKEIVAH | 4.20 | 978.4 | NA |
| 24 | VVAAEIVAH | 4.84 | 949.5 | 20.8 ± 1.3 |
| 25 | VVAKAIVAH | 4.05 | 948.6 | 13.0 ± 1.0 |
| 26 | VVAKEAVAH | 1.39 | 964.5 | NA |
| 27 | VVAKEIAAH | 3.90 | 978.5 | NA |
| 28 | VVAKEIVAA | 4.89 | 939.5 | NA |
| Raltegravir | 0.24 ± 0.02 |
Sequence, analytical data, and effect of peptides 19–28 on HIV-1 IN activities.
All peptide are acetylated and amidate at C terminal and N terminal respectively.
k' [(peptide retention time – solvent retention time)/solvent retention time]. Peptide concentration μM.
NA = Not Applicable, 50% inhibition by the compound was not achieved (as example see the dose-response curve of peptide 19 in Supplementary Material).
Replacement of the Val residue for the bulky-aromatic Phe led to a strong reduction of the activity of the corresponding peptides 19–21 compared with 18, indicating the importance of this ramified amino acid on the HIV-1 IN inhibition capability. This result was also confirmed with the data from the Alanine scanning study. In fact, the substitution of Val1 or Val2 or Val7 residues by Ala produced a dramatic loss of the HIV-1 IN inhibitory activity of the corresponding analogs (22, 23, and 27). The most interesting result was obtained with peptides 24 and 25. In these cases, the substitution by Ala of Lys4 or Glu5, respectively, produced only a weak decrease of HIV-1 IN inhibitor ability if compared to the hit peptide, suggesting that a lack of polar side chains in these positions or the introduction of low hindrance, lipophilic features is well accepted.
Conformational studies
Circular Dichroism was used to determine the conformational-activity relationships of peptide 18 and its analogs (22–28). Analysis of spectra acquired in water solution indicated that all peptides are random coil in water (data not shown). In contrast, spectra acquired in 50% TFE/water solution (TFE, trifluoroethanol) indicate that peptides have high tendency to fold as α helix in fluoroalcohol solution (Figure 2). In particular, all peptides have similar high helical content (about 40%) except peptide 25 whose helical content is lower (about 10%).
Figure 2
IN-IN dimerization and IN multimerization studies
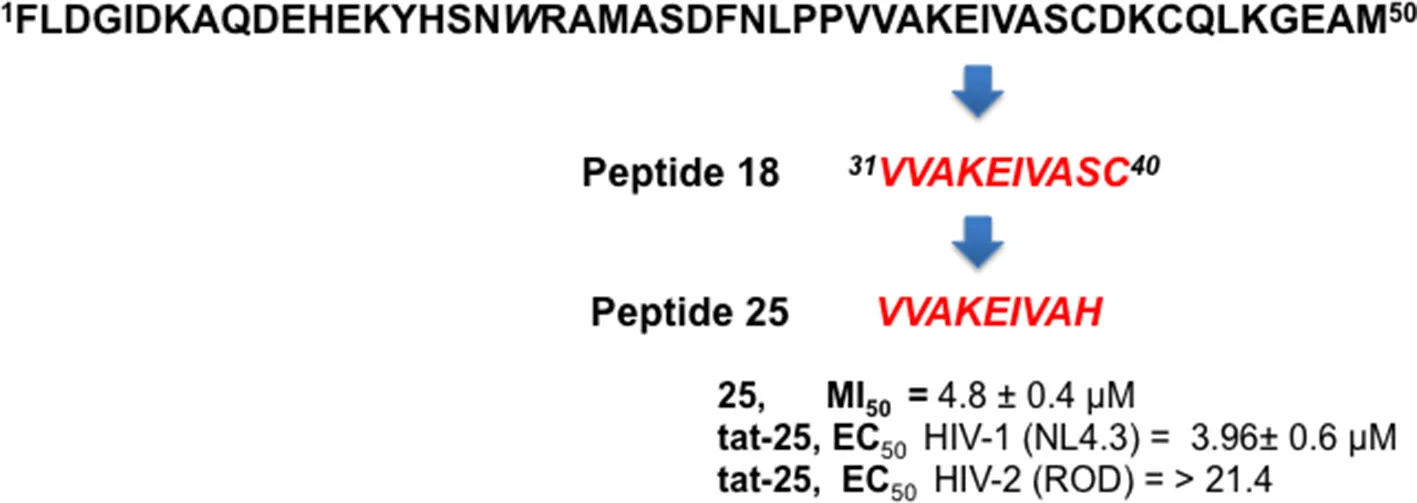
Taking in consideration that these peptides represent fragments of NTD domain, which is involved in different and essential process of the IN activity (Engelman and Craigie, 1992; Cai et al., 1997), we wondered whether they were able to inhibit the IN-IN dimerization or stabilizing the IN in the multimeric form. In fact, it has been reported that allosteric compounds inhibit IN activity binding a site that is different from CCD binding site and working as disruptors of the IN dimer formation (Christ and Debyser, 2013). To verify whether active peptides 18, 24, and 25 can modulate the dynamic interplay between IN subunits, we tested them using an HTRF-based IN subunit exchange assay. In this assay two IN preparation are used: a first His-tagged IN preparation, and a second Flag-tagged IN preparation. In both preparations the purified IN is at equilibrium as a dimer; such dimers go through monomers exchange. Hence, in the assay the monomer exchange of the two different preparations leads to the formation of a dimer with one His-tagged subunit and one Flag-tagged subunit. The formation of this “hybrid” dimer can be monitored in the HTRF-based assay as described (Esposito et al., 2015; Tintori et al., 2015). When the assay is performed in the presence of a compound inhibiting dimerization, the HTRF signal decreases as the concentration of compound increases, whereas in the presence of a compound promoting IN multimerization (association of dimers) the HTRF signal increases as the concentration of the compound increases. We found that peptides 24 and 25 modulate the interactions between the IN subunits in different manners using the allosteric inhibitor LEDGIN-6 as a control (Table 3). In fact, peptide 24 was able to inhibit the IN-IN dimer formation with an IC50 value of 32 μM, but did not affect IN multimerization, while peptide 25 promoted IN multimerization, with a concentration of compound stimulating the Multimerization Increase by 50% (MI50 value) of 4.8 μM, but did not inhibited IN multimerization. While the reason for this different behavior was not clear, it demonstrates that IN multimerization can be differently modulated by interfering with the NTD sequence. Differently, peptide 18 was not able to modulate the dimer/multimer formation. Hence, the three peptides showed three different modes of IN inhibition in biochemical assays.
Table 3
| Compounds | Sequence | IN-IN subunit exchange | IN-multim |
|---|---|---|---|
| aIC50(μM) | cMI50(μM) | ||
| 18 | 31VVAKEIVAH | >100 (100%)b | >100 (100%) |
| 24 | VVAAEIVAH | 32 ± 3 | >100 (100%) |
| 25 | VVAKAIVAH | >100 (100%) | 4.8 ± 0.4 |
| LEDGIN-6 | 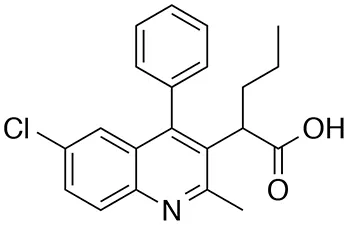 | >100 (100%) | 10 ± 1 |
Effects of peptides 18, 24, and 25 on the HIV-1 IN dimerization and multimerization process.
Compound concentration required to inhibit the HIV-1 IN-IN subunit exchange by 50%.
Percentage of control measured in the presence of 100 μM concentration.
Compound concentration required to inhibit the multimerization increase by 50%.
Cell-based HIV replication assay
The most potent peptides 18, 24, and 25, conveniently conjugated with the cell-penetrating fragment Tat, were evaluated for their capability to inhibit the HIV-1 replication in CD4+ MT-4 cells (Table 4). The conjugated peptides 18 and 24 were devoid of antiviral activity when evaluated against HIV replication. In contrast, 25 showed marked inhibitory potency with good selectivity against HIV-1 (IC50 value in the low micromolar range, accordingly with the results on IN catalysis and dimerization, and selective index of 5.4), but not against HIV-2.
Table 4
| Peptides | ECa50 (μM) | CCb50 (μM) | |
|---|---|---|---|
| HIV-1 (NL4.3) | HIV-2 (ROD) | MT-4 | |
| Tat-18 | >105 | >105 | 105 |
| Tat-24 | >50 | >50 | >50 |
| Tat-25 | 4.0 ± 0.6 | >21.4 | 21.4 ± 2.0 |
Evaluation of the synthesized peptides against HIV-1 and HIV-2 replication in MT-4 cell cultures.
50% Effective concentration, or compound concentration required to inhibit HIV-induced cytopathogenic effect in MT-4 cell culture.
Compound concentration required to reduce by 50% MT-4 cell viability.
Discussion
A library of peptides was developed from the NTD sequence (1–50) by using a mimotopic strategy. Preliminary results of the in vitro integrase assay indicated that the fragment VVAKEIVAH (peptide 18), which includes a His residue instead of the natural Ser at position 39, reduced significantly the HIV-1 IN activity showing an IC50 value of 4.5 μM. Peptide 18 is included into the sequence of the 3rd α-helix of the NTD, which is located on the dimerization interface within the context of the crystal structure of this domain (Cai et al., 1997). Accordingly, we observed significant changes in potency of this peptide when lipophilic and ramified residues involved in the interaction between subunits, such as Val1, Val2, Ile6, and Val7 are substituted. Ala scanning showed also that loss of a lysine positively charge or a glutamate negatively charge does not modify substantially the inhibitory activity of the resulting peptides (24 and 25, respectively). According to the circular dichroism results, the inhibitory activity of these compounds does not seem to be related to their ability to form helical structures. In fact, inactive peptides 22, 23, 26, 27, and 28 all show similar helical content as 18 and 24. On the other hand, compound 25 shows a significant reduction of helical tendency but is approximately as active as 18. Recently, an approach to develop new HIV-1 IN inhibitors that bind in a pocket different from the active binding site, lead to the discovery of new drugs that are able to interfere with the IN dynamic interplay, in two possible ways (Kessl et al., 2012). In the first one, the molecules interfere with the IN dimer-dimer formation, while the second, the molecules promote the IN multimerization. In this case, IN is trapped in a multimeric form in which the movements of the individual subunits are restricted, and, as a consequence, also the catalytic process. We tested peptides 18, 24, and 25 on HTRF assays to determine their potential mechanism of actions and to verify whether they were capable to inhibit the HIV-1 IN. Peptide 24 was able to inhibit the IN-IN dimer formation, while peptide 25 stabilizes the multimeric form of IN. Differently, peptide 18 was not able to modulate the dimer/multimer formation. These data suggest that the modulation of IN dimerization and/or multimerization process could be determined by the net charge of the synthetized peptides, or more specifically by their ability to form intermolecular salt bridges between Glu5 (peptide 24) or Lys4 (peptide 25) residues and Lys or Asp/Glu residues of different subunits, respectively (Michel et al., 2009; Lia et al., 2011). This hypothesis, however, is less probable for peptide 18, in which the formation of an intramolecular salt bridge between adjacent residues Lys4 and Glu5 prevents their interactions with acid and basic residues of other subunits. In this case, the in vitro inhibitory activity of peptide 18 could be related to its ability of inhibiting IN-DNA interactions.
Interestingly, peptide 25, conjugated with the cell-penetrating Tat fragment, is the only compound into this series able to inhibit the HIV-1-induced cytopathogenic effect in MT-4 cell. Our results indicate that the presence of a negative charge (Glu5 side chain) is detrimental for the inhibition of HIV replication in target CD4 T cells (analogs 18 and 24), while the positively charged Lys4 residue (peptide 25) plays an important role in the potential antiviral activity. In addition, this peptide has shown to have potent and selective antiviral activity for HIV-1. Although the antiviral activity is about five-fold lower than the concentration of its cellular toxicity, peptide 25 represents a suitable lead compound for the development of novel derivatives with improved toxicity profile.
Overall, our data confirm the hypothesis that peptides derived from the protein of interest may be used to identify domains which are important for catalytic activity, protein−protein interactions, and/or multimerization process (Li et al., 2006; Lia et al., 2011). Data in Tables 1–3 reflect also the difficulties in identifying inhibitor compounds whose biological target is the viral integration process (Liao et al., 2010). In fact, the modification or replacement of a single amino acid in the sequence of the most potent compound in this series (peptide 25), determines the diminution or loss of the target activity. The identification of the amino acid residues implicated in the inhibition of IN activity highlights the importance of studies that identify such residues and investigates possible roles for them (Li et al., 2006).
In conclusion, this study shows that the modified 31–39 sequence of the N-terminal domain of HIV integrase (VVAKAIVAH, peptide 25) is able to interfere with functional integration of the HIV-1 enzyme. This peptide conveniently conjugated (peptide tat-25) inhibits the HIV activity in cell culture probably due to its ability to stabilize the IN multimeric form. Our peptide, one of the few in cell active peptides reported to date, could be considered as a suitable hit compound in the development of novel anti-HIV1 peptidomimetic agents with a mechanism of action different to that the approved anti-IN drugs.
Funding
This work was supported by grants from the Italian Ministry of Education (MIUR) (PRIN n°2010-11E61J12000210001), the FWO (G-485-08 and G.0528.12 N), the KU Leuven (PF/10/018 and GOA/10/014) and the EU FP7 project CHAARM (no. 242135).
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Statements
Author contributions
IGM, and PC were responsible for the idea and design of peptides; IGM, PC, MS, ET, and DS designed research; MS, AS, FE, MCS, EV, MC performed research; IGM, PC, ET, DS, MS, AS, FE, MCS, EV, AB, and MM supervised experiments and analyzed data; IGM, MS, AS, MCS, ET, and DS wrote the paper; IGM, PC, PG, AC, EN, ET, FE corrected the manuscript.
Acknowledgments
We are grateful to S. Claes, D. Ceusters, E. Van Kerckhove and E. Fonteyn for excellent technical assistance.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2016.00845
- IN
integrase
- HIV-1
human immunodeficiency virus type 1
- PIC
preintegration complex
- NTD
N-terminal domain
- STIN
strand transfer integrase reaction
- CCD
catalytic core domain
- CTD
C-terminal domain
- INSTIs
integrase strand transfer inhibitors (INSTIs)
- HOAt
1-Hydroxy-7-azabenzotriazole
- HOBt
hydroxybenzotriazole
- HBTU, N, N, N'
N'-tetramethyl-O-(1H-benzotriazol-1-yl)uraniumhexafluorophosphate
- DIEA, N
N-diisopropylethylamine
- DMF
dimethylformamide.
Abbreviations
Graphical abstract
References
1
Al-MawsawiL. Q.NeamatiN. (2011). Allosteric inhibitor development targeting HIV-1 integrase. Chem. Med. Chem.6, 228–241. 10.1002/cmdc.201000443
2
AzmiS.SrivastavaS.MishraN. N.TripathiJ. K.ShuklaP. K.GhoshJ. K. (2013). Characterization of antimicrobial, cytotoxic, and antiendotoxin properties of short peptides with different hydrophobic amino acids at “a” and “d” positions of a heptad repeat sequence. J. Med. Chem.56, 924–939. 10.1021/jm301407k
3
BrinsonC. (2013). Stribild, a single tablet regimen for the treatment of HIV disease. Comb. Prod. Ther.3, 1–8. 10.1007/s13556-013-0001-y
4
CaiM.ZhengR.CaffreyM.CraigieR.CloreG. M.GronenbornA. M. (1997). Solution structure of the N-terminal zinc binding domain of HIV-1 integrase. Nat. Struct. Biol.4, 567–577. 10.1038/nsb0797-567
5
ChenJ. C.KrucinskiJ.MierckeL. J.Finer-MooreJ. S.TangA. H.LeavittA. D.et al. (2000). Crystal structure of the HIV-1 integrase catalytic core and C-terminal domains: a model for viral DNA binding. Proc. Natl. Acad. Sci. U.S.A.97, 8233–8238. 10.1073/pnas.150220297
6
ChiuT. K.DaviesD. R. (2004). Structure and function of HIV-1 integrase. Curr. Top. Med. Chem.4, 965–977. 10.2174/1568026043388547
7
ChristF.DebyserZ. (2013). The LEDGF/p75 integrase interaction, a novel target for anti-HIV therapy. Virology435, 102–110. 10.1016/j.virol.2012.09.033
8
DayamR.GundlaR.Al-MawsawiL. Q.NeamatiN. (2008). HIV-1 integrase inhibitors: 2005−2006 update. Med. Res. Rev.28, 118–154. 10.1002/med.20116
9
DelelisO.CarayonK.SaïbA.DeprezE.MouscadetJ. F. (2008). Integrase and integration: biochemical activities of HIV-1 integrase. Retrovirology5:114. 10.1186/1742-4690-5-114
10
Di SantoR. (2014). Inhibiting the HIV Integration Process: Past, Present, and the Future. J. Med. Chem.57, 539–566. 10.1021/jm400674a
11
EijkelenboomA. P. A. M.VandenentF. M. I.VosA.DoreleijersJ. F.HardK.TulliusT. D. (1997). The solution structure of the amino-terminal HHCC domain of HIV-2 integrase: a three-helix bundle stabilized by zinc. Curr. Biol.7, 739–746. 10.1016/S0960-9822(06)00332-0
12
EngelmanA.BushmanF. D.CraigieR. (1993). Identification of discrete functional domains of HIV-1 integrase and their organization within an active multimeric complex. EMBO J.12, 3269–3275.
13
EngelmanA.CraigieR. (1992). Identification of conserved amino acid residues critical for human immunodeficiency virus type 1 integrase function in vitro. J. Virol.66, 6361–6369.
14
EspositoF.TramontanoE. (2013). Past and future. Current drugs targeting HIV-1 integrase and reverse transcriptase-associated ribonuclease H activity: single and dual active site inhibitors. Antiv. Chem. Chemother. 23, 129–144. 10.3851/IMP2690
15
EspositoF.TintoriC.FerrareseR.CabidduG.CoronaA.CeresolaE. R.et al. (2015). Kuwanon-L as a new allosteric HIV-1 integrase inhibitor: molecular modeling and biological evaluation. Chembiochem16, 2507–2512. 10.1002/cbic.201500385
16
EveringT. H.MarkowitzM. (2007). Raltegravir (MK-0518): an integrase inhibitor for the treatment of HIV-1. Drugs Today43, 865–877. 10.1358/dot.2007.43.12.1146063
17
GoldgurY.DydaF.HickmanA. B.JenkinsT. M.CraigieR.DaviesD. R. (1998). Three new structures of the core domain of HIV-1 integrase: an active site that binds magnesium. Proc. Natl. Acad. Sci. U.S.A.95, 9150–9154. 10.1073/pnas.95.16.9150
18
HazudaD. J.HastingsJ. C.WolfeA. L.EminiE. A. (1994). A novel assay for the DNA strand-transfer reaction of HIV-1 integrase. Nucleic Acid Res.2, 1121–1122. 10.1093/nar/22.6.1121
19
JavadpourM. M.BarkleyM. D. (1997). Self-assembly of designed antimicrobial peptides in solution and micelles. Biochemistry36, 9540–9549. 10.1021/bi961644f
20
KatlamaC.MurphyR. (2012). Dolutegravir for the treatment of HIV. Expert Opin. Invest. Drugs21, 523–530. 10.1517/13543784.2012.661713
21
KesslJ. J.JenaN.KohY.Taskent-SezginH.SlaughterA.FengL.et al. (2012). Multimode, cooperative mechanism of action of allosteric HIV-1 integrase inhibitors. J. Biol. Chem.287, 16801–16811. 10.1074/jbc.M112.354373
22
LiH. Y.ZawahirZ.SongL. D.LongY. Q.NeamatiN. (2006). Sequence-based design and discovery of peptide inhibitors of HIV-1 integrase: insight into the binding mode of the enzyme. J. Med. Chem.49, 4477–4486. 10.1021/jm060307u
23
LiaX.KrishnanaL.CherepanovbP.EngelmanaA. (2011). Structural biology of retroviral DNA integration. Virology411, 194–205. 10.1016/j.virol.2010.12.008
24
LiaoC.MarchandC.BurkeT. R.Jr.PommierY.NicklausM. C. (2010). Authentic HIV-1 integrase inhibitors. Future Med. Chem.2, 1107–1122. 10.4155/fmc.10.199
25
LiaoS. M.DuQ. S.MengJ. Z.Zong-Wen PangZ. W.HuangR. B. (2013). The multiple roles of histidine in protein interactions. Chem. Cent. J.7:44. 10.1186/1752-153X-7-44
26
LongY. Q.HuangS. X.ZawahirZ.XuZ. L.LiH.SanchezT. W.et al. (2013). Design of cell-permeable stapled peptides as HIV-1 integrase inhibitors. J. Med. Chem.56, 5601–5612. 10.1021/jm4006516
27
MaesM.LoyterA.FriedlerA. (2012). Peptides that inhibit HIV-1 integrase by blocking its protein–protein interactions. FEBS J.279, 2795–2809. 10.1111/j.1742-4658.2012.08680.x
28
MalikL.ToftengA. P.PedersenS. L.SørensenK. K.JensenK. J. (2010). Automated ‘X-Y’ robot for peptide synthesis with microwave heating: application to difficult peptide sequences and protein domains. J. Pept. Sci.16, 506–512. 10.1002/psc.1269
29
MesplèdeT.QuashieP. K.WainbergM. A. (2012). Resistance to HIV integrase inhibitors. Curr. Opin. HIV AIDS7, 401–408. 10.1097/COH.0b013e328356db89
30
MichelF.CrucifixC.GrangerF.EilerS.MouscadetJ. F.KorolevS.et al. (2009). Structural basis for HIV-1 DNA integration in the human genome, role of the LEDGF/P75 cofactor. Embo J.28, 980–991. 10.1038/emboj.2009.41
31
PommierY.JohnsonA. A.MarchandC. (2005). Integrase inhibitors to treat HIV/AIDS. Nat. Rev. Drug Discov.4, 236–248. 10.1038/nrd1660
32
SavarinoA. (2006). A historical sketch of the discovery and development of HIV–1 integrase inhibitors. Review of the history, of the discovery, and development of HIV-1 IN inhibitors. Expert Opin. Investig. Drugs.15, 1507–1522. 10.1517/13543784.15.12.1507
33
SemenovaE. A.JohnsonA. A.MarchandC.PommierY. (2006). Integration of human immunodeficiency virus as a target for antiretroviral therapy. Curr. Opin. HIV AIDS1, 380–387. 10.1097/01.COH.0000239850.14991.f9
34
ShimuraK.KodamaE.SakagamiY.MatsuzakiY.WatanabeW.YamatakaK.et al. (2008). Broad antiretroviral activity and resistance profile of the novel human immunodeficiency virus integrase inhibitor elvitegravir (JTK-303/GS-9137). J. Virol.82, 764–774. 10.1128/JVI.01534-07
35
SummaV.PetrocchiA.BonelliF.CrescenziB.DonghiM.FerraraM.et al. (2008). Discovery of raltegravir, a potent, selective orally bioavailable HIV integrase inhibitor for the treatment of HIV-AIDS infection. J. Med. Chem.51, 5843–5855. 10.1021/jm800245z
36
TintoriC.EspositoF.MorrealeF.MartiniR.TramontanoE.BottaM. (2015). Investigation on the sucrose binding pocket of HIV-1 Integrase by molecular dynamics and synergy experiments. Bioorg. Med. Chem. Lett.25, 3013–3016. 10.1016/j.bmcl.2015.05.011
37
VermeireK.PrincenK.HatseS.De ClercqE.DeyK.BellT. W.et al. (2004). CADA, a novel CD4-targeted HIV inhibitor, is synergistic with various anti-HIV drugs in vitro. AIDS18, 2115. 10.1097/00002030-200411050-00003
38
WangJ. Y.LingH.YangW.CraigieR. (2001). Structure of a two-domain fragment of HIV-1 integrase: implications for domain organization in the intact protein. EMBO J.20, 7333–7343. 10.1093/emboj/20.24.7333
39
WangS. S.TamJ. P.WangB. S.MerrifieldR. B. (1989). Enhancement of peptide coupling reactions by 4-Dimethylaminopyridine. Int. J. Pept. Protein Res.18, 459–467. 10.1111/j.1399-3011.1981.tb03007.x
40
WolfeS. A.GrantR. A.Elrod-EricksonM.PaboC. O. (2001). Beyond the ‘recognition code’: structures of two Cys2His2 zinc finger/TATA box complexes. Structure (Camb.)9, 717–723. 10.1016/S0969-2126(01)00632-3
41
ZhaoL.O'ReillyM. K.ShultzM. D.ChmielewskiJ. (2003). Interfacial peptide inhibitors of HIV-1 integrase activity and dimerization. Bioorg. Med. Chem. Lett.13, 1175–1177. 10.1016/S0960-894X(03)00040-4
Summary
Keywords
HIV-1, integrase, N-terminal domain, peptides, inhibitors
Citation
Sala M, Spensiero A, Esposito F, Scala MC, Vernieri E, Bertamino A, Manfra M, Carotenuto A, Grieco P, Novellino E, Cadeddu M, Tramontano E, Schols D, Campiglia P and Gomez-Monterrey IM (2016) Development and Identification of a Novel Anti-HIV-1 Peptide Derived by Modification of the N-Terminal Domain of HIV-1 Integrase. Front. Microbiol. 7:845. doi: 10.3389/fmicb.2016.00845
Received
26 February 2016
Accepted
22 May 2016
Published
10 June 2016
Volume
7 - 2016
Edited by
Slobodan Paessler, University of Texas Medical Branch, USA
Reviewed by
Takao Masuda, Tokyo Medical and Dental University, Japan; Veljko Veljkovic, Vinča Institute of Nuclear Sciences, Serbia;
Updates
Copyright
© 2016 Sala, Spensiero, Esposito, Scala, Vernieri, Bertamino, Manfra, Carotenuto, Grieco, Novellino, Cadeddu, Tramontano, Schols, Campiglia and Gomez-Monterrey.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pietro Campiglia pcampigl@unisa.it;
This article was submitted to Virology, a section of the journal Frontiers in Microbiology
†These authors have contributed equally to this work.
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.