 Vera Thiel1*†
Vera Thiel1*† Jason M. Wood2
Jason M. Wood2 Millie T. Olsen2
Millie T. Olsen2 Marcus Tank1†
Marcus Tank1† Christian G. Klatt2,3
Christian G. Klatt2,3 David M. Ward2
David M. Ward2 Donald A. Bryant1,4*
Donald A. Bryant1,4*- 1Department of Biochemistry and Molecular Biology, The Pennsylvania State University, University Park, PA, USA
- 2Department of Land Resources and Environmental Sciences, Montana State University, Bozeman, MT, USA
- 3Agricultural Research Service, United States Department of Agriculture, University of Minnesota, Saint Paul, MN, USA
- 4Department of Chemistry and Biochemistry, Montana State University, Bozeman, MT, USA
Microbial-mat communities in the effluent channels of Octopus and Mushroom Springs within the Lower Geyser Basin at Yellowstone National Park have been studied for nearly 50 years. The emphasis has mostly focused on the chlorophototrophic bacterial organisms of the phyla Cyanobacteria and Chloroflexi. In contrast, the diversity and metabolic functions of the heterotrophic community in the microoxic/anoxic region of the mat are not well understood. In this study we analyzed the orange-colored undermat of the microbial community of Mushroom Spring using metagenomic and rRNA-amplicon (iTag) analyses. Our analyses disclosed a highly diverse community exhibiting a high degree of unevenness, strongly dominated by a single taxon, the filamentous anoxygenic phototroph, Roseiflexus spp. The second most abundant organisms belonged to the Thermotogae, which have been hypothesized to be a major source of H2 from fermentation that could enable photomixotrophic metabolism by Chloroflexus and Roseiflexus spp. Other abundant organisms include two members of the Armatimonadetes (OP10); Thermocrinis sp.; and phototrophic and heterotrophic members of the Chloroflexi. Further, an Atribacteria (OP9/JS1) member; a sulfate-reducing Thermodesulfovibrio sp.; a Planctomycetes member; a member of the EM3 group tentatively affiliated with the Thermotogae, as well as a putative member of the Arminicenantes (OP8) represented ≥1% of the reads. Archaea were not abundant in the iTag analysis, and no metagenomic bin representing an archaeon was identified. A high microdiversity of 16S rRNA gene sequences was identified for the dominant taxon, Roseiflexus spp. Previous studies demonstrated that highly similar Synechococcus variants in the upper layer of the mats represent ecological species populations with specific ecological adaptations. This study suggests that similar putative ecotypes specifically adapted to different niches occur within the undermat community, particularly for Roseiflexus spp.
Introduction
Microbial mat communities inhabiting the effluent channels of Octopus and Mushroom Springs within the Lower Geyser Basin at Yellowstone National Park (YNP) have been studied for nearly 50 years (Brock, 1967; Ward et al., 2012). In these studies, the chlorophototrophic bacterial populations, i.e., chlorophyll-based phototrophs including members of the Cyanobacteria, Chloroflexi and the newly discovered Chloracidobacterium (Cab.) thermophilum and “Candidatus Thermochlorobacter (Tcb.) aerophilum,” have generally been the main focus (Bauld and Brock, 1973; Nold and Ward, 1996; Bryant et al., 2007; van der Meer et al., 2007; Steunou et al., 2008; Becraft et al., 2011; Klatt et al., 2011, 2013b; Liu et al., 2011, 2012; Tank and Bryant, 2015a,b). In contrast, the diversity and metabolic functions of the heterotrophic community in the microoxic/anoxic region of the mat are not well understood.
Using cultivation-based methods, early studies focused on the dominant Cyanobacteria and phototrophic Chloroflexi (Bauld and Brock, 1973; Bateson and Ward, 1988). Over time, these studies were extended by a variety of molecular methods with increasing molecular resolution. A pioneering molecular study targeting 16S rRNA gene sequences directly indicated a greater diversity of uncultivated bacteria in the mat than previously realized (Ward et al., 1990). However, only recently have metagenomic (Klatt et al., 2011), metatranscriptomic (Liu et al., 2011, 2012; Klatt et al., 2013b) and metametabolomic (Kim et al., 2015) analyses led to a holistic overview, in terms of the organisms present and their functional potentials, of the major taxa inhabiting the upper 2 mm of the 60–65°C regions of the Mushroom Spring microbial mats (Figure 1). The microbial community of the upper green mat layer contains eight dominant bacterial populations, of which six are chlorophototrophs (Klatt et al., 2011). Oxygenic cyanobacteria from the genus Synechococcus have been shown to be the predominant primary producers in these communities by in situ studies of bicarbonate fixation and nitrogen fixation (Steunou et al., 2008) using stable and radioactive isotopes (Bateson and Ward, 1988; Nübel et al., 2002; van der Meer et al., 2007). In addition, anoxygenic photoheterotrophic members of the Roseiflexus spp. have been suggested to perform inorganic carbon fixation (van der Meer et al., 2003, 2005, 2007, 2010; Klatt et al., 2007, 2013b). Synechococcus spp. fix CO2 and synthesize and excrete metabolites that are then consumed by (photo)heterotrophic members of the community, including several members of the Chloroflexi, and presumably Roseiflexus spp. (Anderson et al., 1987; Bateson and Ward, 1988; Kim et al., 2015). Collectively, cyanobacteria and Roseiflexus spp. account for the majority of the biomass of the upper 0–2 mm portion of the mat community. Two additional members of the phylum Chloroflexi, Chloroflexus sp. and an apparently phototrophic, “Anaerolineae-like” organism (“Ca. Roseilinea gracile”; Tank et al., in press), as well as two recently discovered aerobic/microaerophilic, anoxygenic photoheterotrophs, Cab. thermophilum (Bryant et al., 2007; Garcia Costas et al., 2012a,b; Tank and Bryant, 2015a,b) and “Ca. Tcb. aerophilum” (Liu et al., 2012), also occur in the upper photic layer of the mat.

Figure 1. Sampling site at Mushroom Spring, Yellowstone National Park, and microbial mat core (adapted from Kim et al., 2015).
Early studies on the processes and organisms involved in aerobic and anaerobic decomposition of the mat have been discussed in a review by Ward et al. (1992; and earlier papers cited therein). Since the discovery of the aerobic heterotroph Thermus aquaticus (Brock and Freeze, 1969) many aerobic (e.g., Thermomicrobium roseum; Jackson et al., 1973) and anaerobic fermentative and sulfate-reducing bacteria were cultivated from these mats (e.g., Bacillus stearothermophilis, Thermoanaerobium brockii, Thermoanaerobacter ethanolicus, Thermodesulfotobacterium commune; see Ward et al., 1992 for primary references). Many of the latter were sought with the hope that thermophiles would be useful for biofuel production. However, critical review indicated that most of these isolates had not been cultivated from highly diluted mat samples, and thus their importance to the community remained unknown (see Ward et al., 1998). Indeed, with one exception, Thermomicrobium roseum (Wu et al., 2009), the genomes of these organisms did not recruit reads with high identity values from metagenomic analysis of the upper mat layer (Klatt et al., 2011). Only two low-abundance, unidentified heterotrophic bacteria lacking the genes needed to synthesize chlorophyll (Chl) were detected in the upper mat community represented by metagenomic bins (Klatt et al., 2011). Nevertheless, heterotrophs, together with the photoheterotrophic and photomixotrophic community members, can be considered potential consumers of metabolites produced by cyanobacteria and possibly other mat inhabitants. In more recent years, the activity and diversity of sulfate-reducing bacteria of the microbial mats have been more intensively studied. Dillon et al. (2007) showed that an active sulfur cycle occurs in the mat community despite very low sulfate concentrations. The highest rates of sulfate respiration were reportedly associated with Thermodesulfovibrio-like organisms and were measured close to the surface of the mat late in the day when photosynthetic oxygen production had ceased. Additionally, methane production has been detected in numerous alkaline siliceous hot spring microbial mats in YNP (Ward, 1978; Sandbeck and Ward, 1981, 1982). Methanogenic archaea (~107 to 108 ml−1) have been enumerated in small cores of Octopus Spring mats, which in combination with the detection of low levels of archaeal lipids, suggests that methanogenesis occurred in situ in those mats (Ward, 1978; Sandbeck and Ward, 1981; Ward et al., 1985). The relative rarity of these organisms compared to Synechococcus (on the order of 1% or less) suggests that these terminal anaerobes receive little of the energy recycled during decomposition of the mat (Ward et al., 1989).
The first revolution of molecular microbial ecology enabled the study of uncultured bacterial diversity through amplification, sequencing and phylogenetic analysis of ribosomal RNA genes (Olsen et al., 1986; Ward et al., 1990; Amann et al., 1995; Hugenholtz and Pace, 1996; Hugenholtz et al., 1998a; Pace, 2009). Through such studies, our perspective on microbial diversity has increased enormously over the past three decades, and the impact of culture-independent studies on the emerging view of bacterial diversity cannot be overstated (Hugenholtz et al., 1998a). Ward and coworkers reported the presence of a number of uncultured bacterial lineages in their first molecular microbial diversity study of the mat community of Octopus Spring (Ward et al., 1990). Over the course of the past 25 years, several of those initially unidentified ribosomal RNA sequences have been associated with chlorophototrophic mat members (OS-A and B with Synechococcus spp., OS-C with Roseiflexus sp., OS-D with Cab. thermophilum, and OS-E with “Ca. Tcb. aerophilum”), whereas many others (OS-F, OS-G, OS-H, OS-K, OS-L, OS-M, OS-N, OS-R) still have not been identified and were not detected in the metagenome of the upper green layer (Klatt et al., 2011).
“Red-layer” communities, which may often be “orange” in color as is the case for the mats of Mushroom Spring, have been shown to contain novel chlorophototrophs (Boomer et al., 2000, 2002), whose pigments exhibit unusual in vivo absorption spectra (Boomer et al., 2000), but these communities have not yet been studied in detail. As part of a comparative study of YNP hot spring microbial mat communities, a 45-Mbp metagenome based on Sanger sequencing revealed some initial insights into the composition of the undermat microbial community of Mushroom Spring (Klatt et al., 2013a). Compared to the upper green layer, fewer Synechococcus spp., a greater number of Roseiflexus spp., and several presumed anaerobic or fermentative organisms within the Bacteroidetes and Thermodesulfobacteria were identified. The undermat community contained a Thermotoga-like population as well as several low G+C organisms that could not be characterized (Klatt et al., 2013a). Low coverage and a small number of long scaffolds above the threshold used in most clustering analyses (>10 kb) limited the application of metagenomic binning approaches (Klatt et al., 2013a) and indicated that additional studies with much deeper sequencing would be needed to define the undermat community.
The overall goal of this research is to investigate the complete microbial mat community at Mushroom Spring and to develop a comprehensive understanding of the microbial ecology of the microbial mats of this hot spring. The specific objectives of this study were to analyze the orange-colored undermat community, to identify those organisms that are present, and to facilitate an active integration of these mostly heterotrophic members into models of the mat community. This paper describes the composition and diversity of the Mushroom Spring undermat community based on rRNA-amplicon (iTag) and deep metagenomic sequencing analyses, with an initial focus on the identity and taxonomic diversity of the community members. A description of the metabolic potential and putative interactions, including a metabolic description of the entire microbial mat community, will be published separately.
Materials and Methods
The samples were collected on August 10th, 2011 from a chlorophototrophic microbial mat in an effluent channel of the siliceous and slightly alkaline Mushroom Spring in YNP, WY (USA). The samples were collected using a #4 cork borer at a site where the water above the mat was 60°C (Figure 1). The microbial mat is made up of an upper green layer (1–2 mm thick), which mainly consists of different chlorophototrophic bacteria, and an orange-colored undermat layer (Figure 1). Genomic DNA was extracted from the orange-colored undermat layer (~3–5 mm depth; DNA from below this level was too degraded to analyze). The metagenome as well as 16S rRNA gene PCR amplicons were sequenced at the DOE Joint Genome Institute (JGI) using HiSeq and MiSeq Illumina technologies. The iTtag sequences were analyzed at two different identity levels. All reads were clustered into operational taxonomic units (OTUs) with 97% sequence identity cutoff by using USEARCH, but they were also analyzed after dereplication (i.e., clustered by 100% nt identity, see Supplementary Materials). RDP Classifier (Wang et al., 2007; Cole et al., 2009), BLAST searches (Altschul et al., 1990) and phylogenetic analyses (Ludwig et al., 2004) were used to identify sequences. Microdiversity was assessed using the number of highly abundant dereplicated sequences, and the “oligotyping pipeline” (http://merenlab.org/projects/oligotyping/). HiSeq metagenomic reads were assembled and then clustered into bins by oligonucleotide frequency pattern analyses using ESOM (Dick et al., 2009). Metagenomic bins were treated as partial genomes of single taxa and were taxonomically affiliated using Amphoranet (http://pitgroup.org/amphoranet/, Kerepesi et al., 2014) to assess the phylogenetic marker genes present in each bin. Detailed descriptions of the methods for DNA extraction, library construction, sequencing, and data analyses are found in the Supplementary Materials.
Results
We used deep sequencing of rRNA gene amplicons (iTags) and total environmental DNA to study the subsurface community of the chlorophototrophic microbial mat at Mushroom Spring. We describe the diversity and community composition on both levels, based on “OTUs” (Figures 2A, 3A, Table 1 and Table S1) and based on “dereplicated iTag” sequences (Figures 2B, 3B, Table 2) in Section “16S rRNA Gene Amplicons (iTags),” as well as on metagenomic bins obtained based on oligonucleotide frequency patterns in Section “Metagenome Sequencing” (Figure 4, Table 3). An overview of the most important taxa detected in each phylum will be presented in Section “Overview of Phyla and Taxa Detected in the Mushroom Spring Undermat.” Each iTag OTU was found to represent a variable number of dereplicated iTtag sequences, which is interpreted as representing different degrees of microdiversity within a taxon (Figure 2B, Table 1). Members of 20 different phyla were identified (Figure 5 and Figure S1, Table S1). Organisms of the phylum Chloroflexi dominated the microbial undermat community in both read abundance and diversity (Tables 1, 2, and Table S1, Figures 2A,B). Thirteen out of seventeen members of the microbial mat detected in previous 16S rRNA gene sequence cloning and DGGE studies (OS types, Table 4; Ward et al., 1990, 1992; Weller et al., 1992; Ferris et al., 1996b, 1997; Ferris and Ward, 1997), as well as relatives of ribosomal sequence types derived from a previous undermat study (Klatt et al., 2013a, Figure 5 and Figure S1) were detected in this study and thus confirmed as members of a compositionally and temporally stable microbial community.
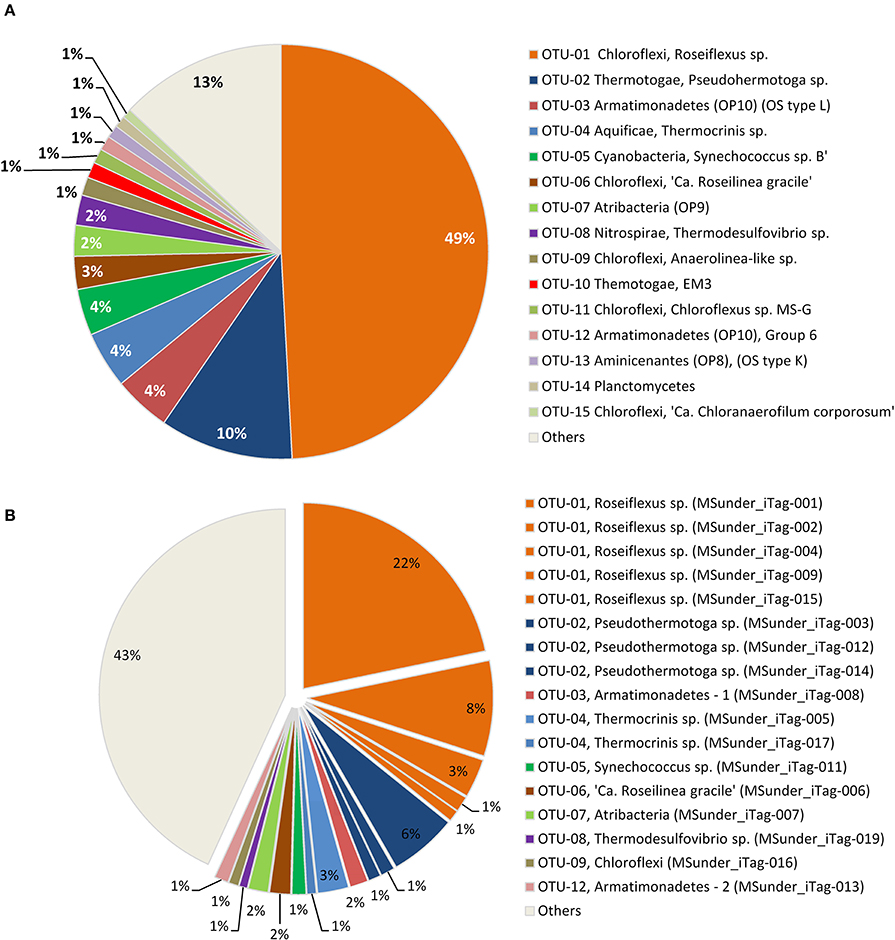
Figure 2. Relative abundance of (A) the 15 most abundant 97% OTUs, and (B) the 17 most abundant dereplicated iTag sequences in the Mushroom Spring undermat 16S rRNA gene amplicon (iTag) analysis. All less abundant OTUs (<1,000 reads each) are shown combined as “Others.”
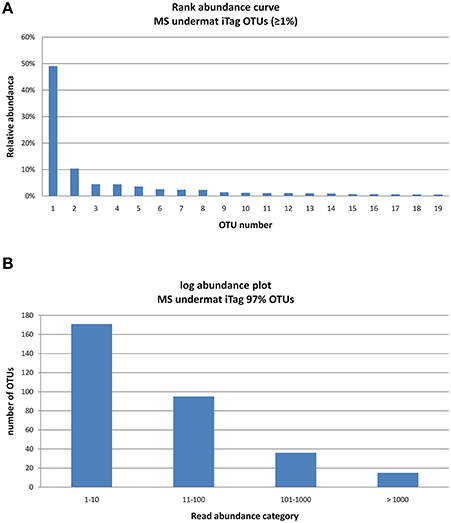
Figure 3. Rank abundance curve of the 15 very abundant OTUs (>1,000 reads) obtained from the undermat 16S rRNA gene amplicon study (relative read counts) (A), and Log abundance plot of the all 317 OTUs obtained from the undermat 16S rRNA gene amplicon study (B). 15 OTUs (= 5%) were detected more than 1,000 times.
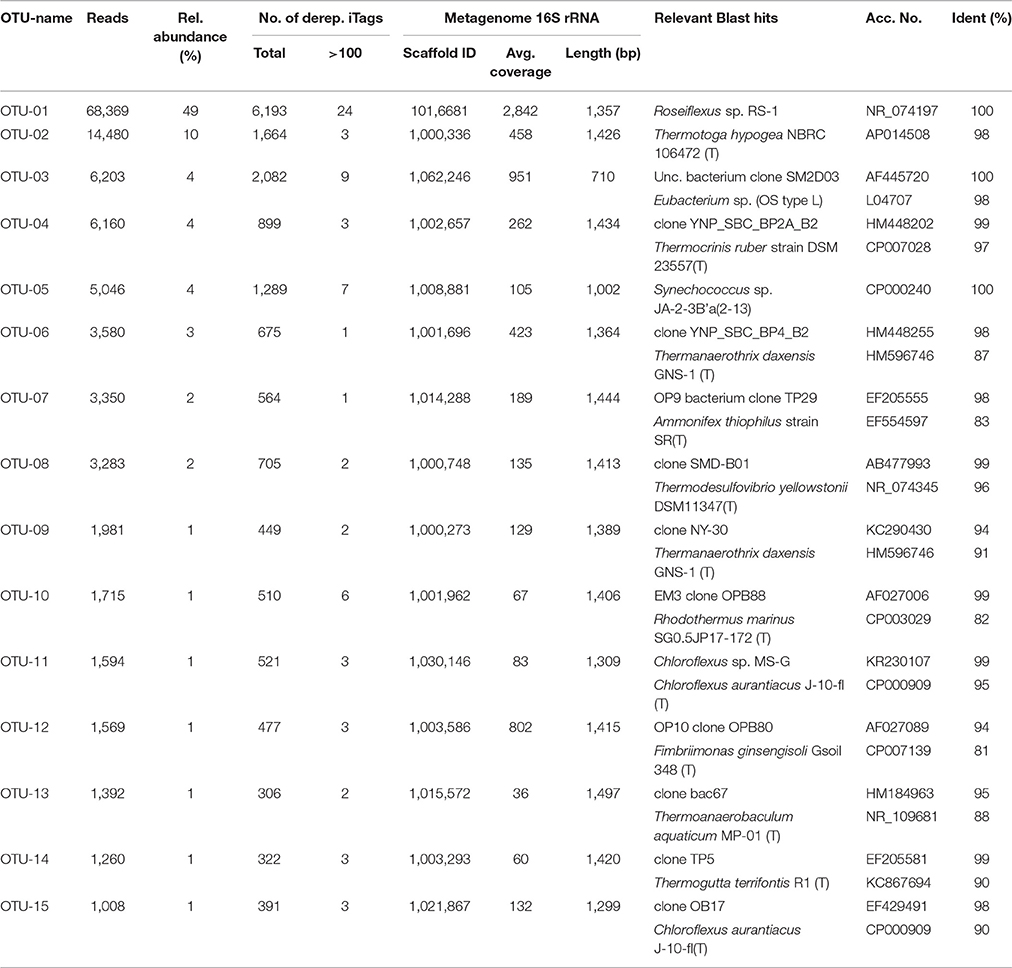
Table 1. Most abundant OTUs (97% nt identity), number of reads and relative abundance, microdiversity in terms of represented dereplicated iTag sequences, corresponding metagenome sequences and next relatives determined by BLAST search.
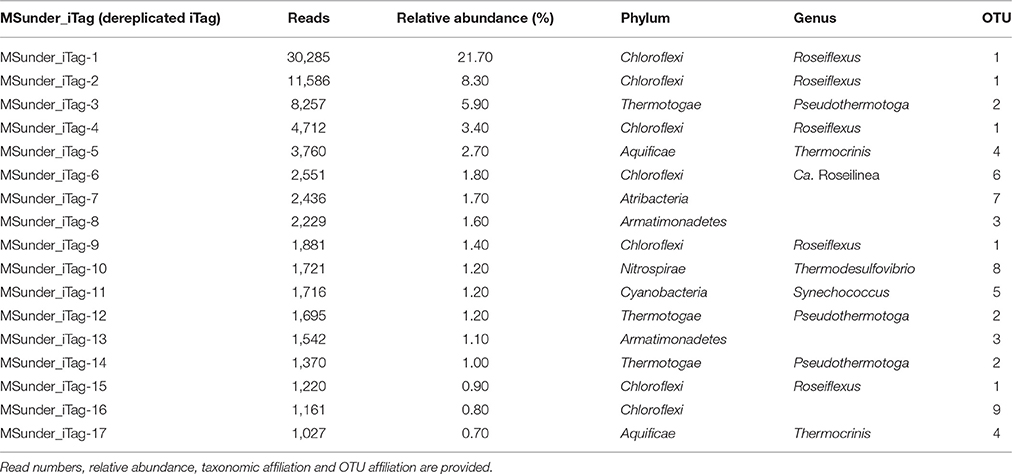
Table 2. Most abundant dereplicated iTag sequences (100% nucleotide identity) detected in the Mushroom Spring undermat.
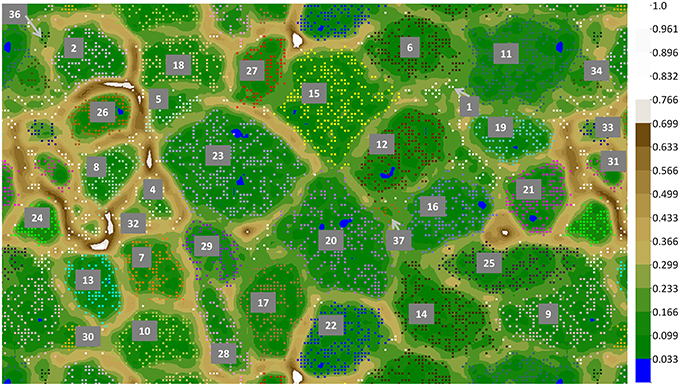
Figure 4. ESOM binning of Mushroom Spring assembled undermat metagenome sequences >5 kb. In order to show complete bins, the map is shown in tiled mode, displaying four connected partial copies of the grid with some redundancy of data points. Repeating colors imply repeating bins. Complete bins are labeled with numbers (number code and details of the bins is found in Table 3). Additional partial copies of the bins due to the tiled display are not labeled. The color gradient code on the right visualizes relative height values; the largest height normalized to 1.
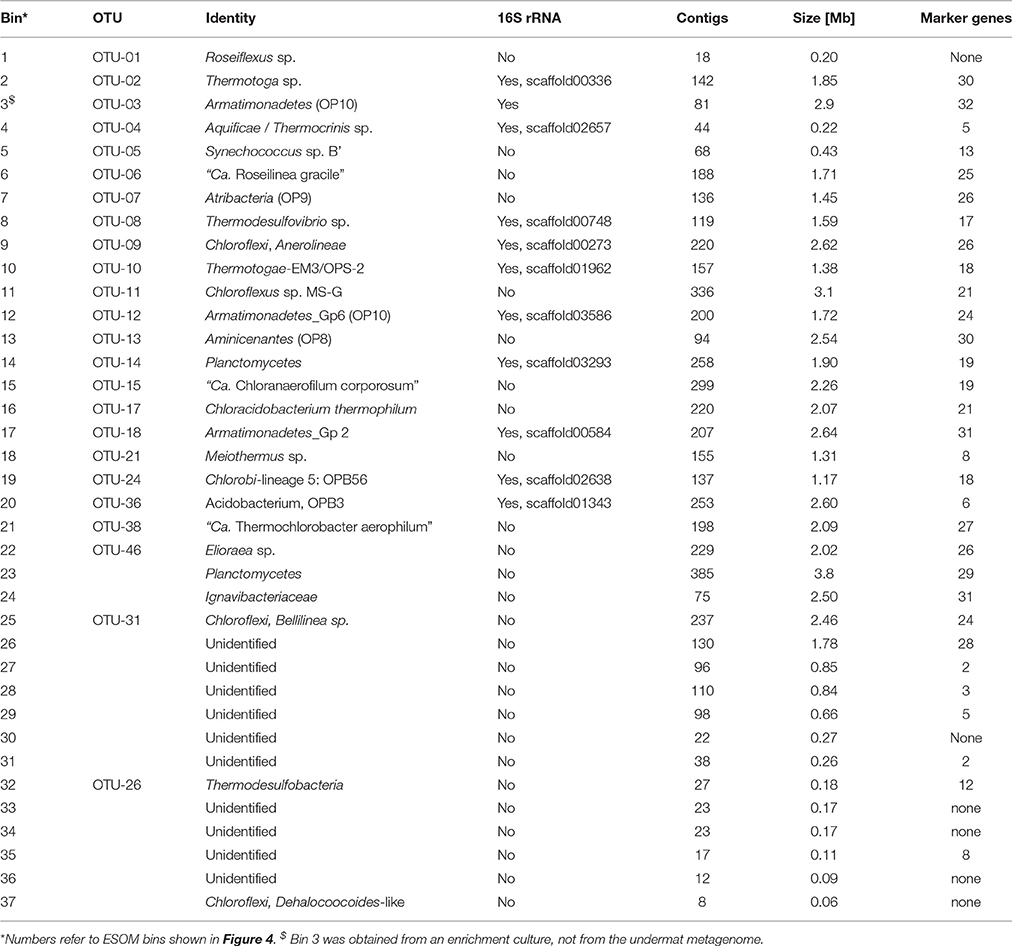
Table 3. Metagenome bins recovered based on tetranucleotide frequencies.

Figure 5. Phylogenetic tree based on 16S rRNA gene sequences showing the phylogenetic relationship between members of the phylum Chloroflexi (A) and Bacteroidetes-Chlorobi (B) phyla and sequences obtained from the Mushroom Spring microbial undermat community. The tree was generated based on the Maximum Likelihood method using the phyML software included in the ARB package. Percentage numbers on nodes refer to 100 bootstrap pseudoreplicates conducted. Only values >50% are shown. Bold sequences were obtained from Mushroom or Octopus Spring in this or previous studies. Red bold labels indicate sequences obtained in this study. Blue bold labels indicate “OS type” sequences from previous studies. OTU numbers shown refer to the most abundant OTU represented by the sequence. Only sequences with length >1,000 bp were used for phylogenetic calculations. Sequence length <1,000 bp are given in (gray) in the labels and corresponding sequences were added using the Parsimony method without changing tree topology.
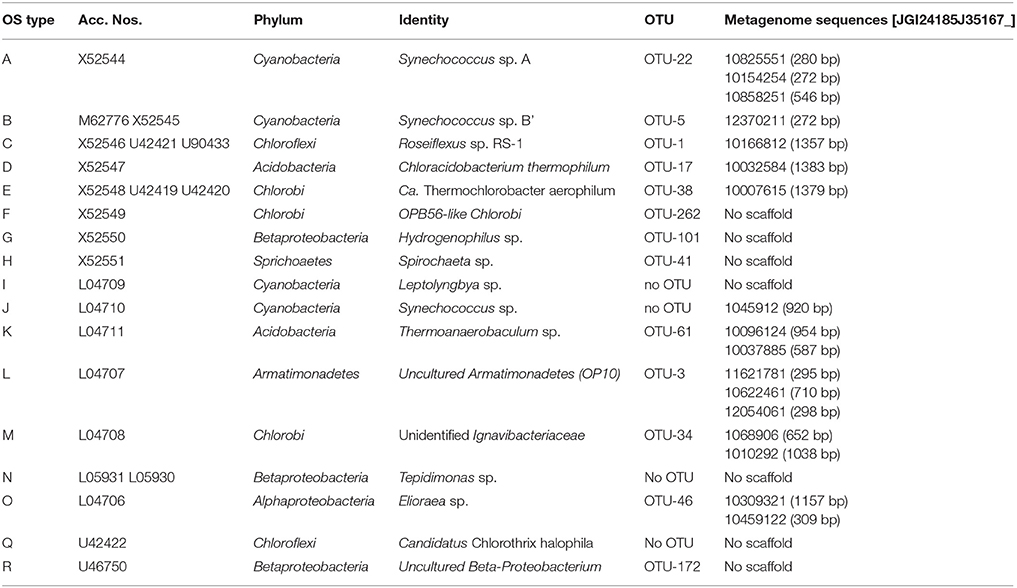
Table 4. OS-type sequences from previous studies (Ward et al., 1990, 1992; Weller et al., 1992; Ferris et al., 1996b, 1997; Ferris and Ward, 1997) and the corresponding sequences obtained in this study.
16S rRNA Gene Amplicons (iTags)
Sequencing of partial 16S rRNA genes resulted in 139,326 total and 30,861 dereplicated (i.e., unique) reads after quality control. Abundance values of dereplicated reads varied between 1 and 30,285, with an average of 5.4 reads per sequence.
Diversity Based on OTUs
The 16S rRNA gene amplicon reads clustered into 317 OTUs of ≥97% nt identity, with abundances between 1 and 68,369 reads per OTU (Table S1). The community was characterized by a low degree of evenness (Figure 3A). The majority of the OTUs were present in low abundance; only 15 OTUs (5% of the taxa) were represented by 1,000 or more reads (Figure 3B). Due to the high number of singleton sequences, the estimated richness based on Chao1 (Schao1 = Sobs+ (no. of singletons2)/(2* no. of doubletons) (Chao, 1984) was rather high, Chao1 = 369.74; a lower value of Chao1 = 220.9 was obtained in a previous study (Klatt et al., 2013a). In contrast, the Simpson's Reciprocal Index () obtained in this study is considerably lower than in previous studies (3.85 in this study vs. 37.5; Klatt et al., 2013a), reflecting the low evenness and strong dominance of only a few OTUs in the amplicon study. While an identity cut-off of 97% for rRNA gene sequences is often used to demarcate species (Stackebrandt and Goebel, 1994; Schloss and Handelsman, 2005; Koeppel and Wu, 2013), this is an arbitrary value that does not necessarily correlate with any species definition. Here, we refer to OTUs as “taxa,” use the term “populations” mainly for dereplicated iTag sequences, and discuss our understanding of the bacterial species concept in Section “Discussion.”
Most Abundant Taxa Based on OTUs
When considering OTU sequences based on 97% nt sequence identity, 15 OTUs were identified with >1,000 reads each, varying in abundance between 1,008 and 68,369 reads (Table 1). These are considered to represent highly abundant taxa and thus are likely to represent key members of the Mushroom Spring undermat community. However, the threshold of 1,000 reads was arbitrarily chosen and does not necessarily correlate with activity or ecological importance. We will focus the discussion on the “very abundant” taxa listed in Table 1, but will also include selected “abundant” and “less abundant” OTUs with read abundances of ≥100 and less, respectively (Table S1).
The 16S rRNA gene amplicons of the microbial undermat community were dominated by sequences derived from Roseiflexus spp. (Figure 2A, OTU-1, 49%) with the second most abundant sequences belonging to a Pseudothermotoga sp. (OTU-2, 10%). An unidentified Armatimonadetes (formerly known as OP10) bacterium (OTU-3), a member of the Aquificae (OTU-4), as well as the sequences derived from member of the Cyanobacteria each represented ~4% of the sequences (Table 1). On the basis of psaA sequences the cyanobacterial sequences can be classified as belonging to ecotype populations of Synechococcus detected in the upper green layer of the mat and are considered likely to arise from buried surface populations that are not expected to represent metabolically active constituents of the undermat community. The sixth most abundant OTU was identified as a phototrophic member of the phylum Chloroflexi, which had previously been detected in the upper green layer using metagenome analysis and identified as the first phototrophic “Anaerolineae-like” Chloroflexi; it has provisionally been named “Ca. Roseilinea gracile” (Klatt et al., 2011, 2013b; Tank et al., in press). Additional abundant OTUs were affiliated with the Atribacteria (OP9) Nitrospirae, Planctomycetes and several phototrophic and non-phototrophic members of the phylum Chloroflexi (Table 1). Three of the fifteen most abundant OTU sequences from the undermat amplicon study represented sequences obtained from the mats of Octopus Spring in previous 16S rRNA gene surveys (OS-B: Synechococcus sp. Type B; OS-C: Roseiflexus sp. RS-1; and OS-L: Armatimonadetes member OTU-3) (Table 4, Ward et al., 1990, 1992; Ferris et al., 1996a; van der Meer et al., 2010).
Most Abundant Populations Based on Dereplicated iTag Sequences
Seventeen dereplicated iTag sequences, representing members of the nine most abundant OTUs, were each detected more than 1,000 times, and in total represent more than half of all iTag reads recovered in this study (Table 2, Figure 2B). These sequences probably correspond to the most abundant “populations” (in contrast to “taxa” for OTUs). Five of these very abundant dereplicated iTag sequences belong to a single OTU representing the most abundant taxon, Roseiflexus spp. (Figure 2B, Table 2 OTU-1, MSunder_iTags-1, 2, 4, 9, and 15). The third most abundant sequence (MSunder_iTag-3), as well as two additional abundant, dereplicated sequences (MSunder_iTags-12 and 14), were representatives of Pseudothermotoga spp. (OTU-2), the second most abundant taxon. The Armatimonadetes (OTU-3) and a member of the phylum Aquificae (OTU-4) contained two slightly different, highly abundant dereplicated iTag sequences each, whereas the other OTUs (OTUs 5–9) had only one very abundant dereplicated iTag sequence. With regard to the single dereplicated iTag sequences, cyanobacteria derived from the green upper layer of the mat community are represented by the eleventh most abundant iTag sequence, and thus the ten most abundant dereplicated iTag sequences (representing eight OTUs) are considered to represent the most abundant populations in the undermat community (MSunder_iTag-1 through MSunder_iTag-10; Table 2).
Microdiversity
We used different methods to assess the degree of sequence heterogeneity and microdiversity within the microbial undermat community. Based on the number of different dereplicated iTag sequences within one 97% OTU, a high degree of diversity was indicated, especially for the most abundant OTU, Roseiflexus spp. We detected 6,193 total dereplicated iTag sequences, 24 of which had >100 reads (Table 1). A similar microdiversity was identified by the oligotyping approach, and was also suggested by a high number of very similar but non-identical clone sequences obtained in a previous study (Klatt et al., 2013a; Figure 5A, and Figure S2, Table S2). Based on ten distinct nucleotide positions, 246 different oligotypes were identified, of which 55 were represented by >10 reads, 23 by >100 reads and nine by > 1,000 reads in the combined dataset (which consisted of ~39,000 upper green layer reads and 75,000 undermat reads). The total “purity scores” of 0.95 and 0.86 for >100 and >10 reads, respectively, indicates a good separation for the highly abundant oligotypes, but also implies further low abundance oligotypes in the samples. Differences in diversity and abundance of oligotypes between the upper green layer and the undermat were detected, e.g., for the most abundant Roseiflexus-oligotypes (Table S2, Figure S2). In general the undermat is more diverse. The upper green layer for example contains a lower number of highly abundant oligotypes (six oligotypes >1% of all Roseiflexus sequences), whereas the undermat is more diverse with nine oligotypes >1% (Table S2, Figure S2). Notably, the most abundant oligotypes are present in both samples in similar abundances. One oligotype dominates both datasets (48% in the upper layer vs. 54% in the undermat). The second most abundant oligotype “CTCTACGGGC” is more abundant in the upper layer (32 vs. 20% of the reads), whereas the third is more abundant in the undermat (9 vs. 6%, Table S2). In general the undermat is more diverse and some oligotypes show distinct differences. For example, the difference in the entropy figures from upper green layer and undermat after separate analyses (considerably lower entropy at pos. 104 and 109 in upper layer; Figures S2B,C) are indicative of a lower abundance of two oligotypes in the upper layer, namely “CCCCGCGTGC” (2.13% in undermat, 0.19% in upper layer) and “CCCCGCGGGC” (1.02 vs. 0.21%) (Table S2).
A high degree of microdiversity was also indicated for other OTUs obtained in this study, e.g., OTU-3 (Armatimonadetes member, OS type L) and OTU-5 (Synechococcus spp.) Overall, the twelve most abundant OTUs also exhibited the highest number of unique amplicon sequences, indicating a correlation between microdiversity and sequencing depth (Table 1). However, the number of abundant dereplicated sequences, i.e., putative ecotypes did not show the self-correlation with sequencing depth, but correlated with the metagenome assembly quality; a high microdiversity was suggested to be interfering with the sequence assembly. Very few contigs with >5 kb length were assembled for the OTUs with the highest microdiversity (OTU-1 and OTU-3).
Metagenome Sequencing
One full lane of Illumina HiSeq sequencing led to 176,741,874 quality-passed reads. 169,595,919 (96%) of these reads were assembled into a 232-Mb metagenome comprising 315,154 total contigs with a maximum scaffold length of 158 kb and a N/L50 value of 32,529/1.24 kb, which defines the number of fragments at or above the Length50 cutoff. There were 13,766 contigs >2.5 kb, 5,362 contigs >5 kb, and 1,665 >10 kb. Contigs >50 kb (n = 38) accounted for 1.14% of all assembled sequence data.
Metagenome Bins
Binning of the metagenome contigs based on tetranucleotide frequency patterns resulted in 36 clusters (Table 3, Figure 4). An additional bin, representing OTU-3 from the iTag study of the undermat, was obtained from an cyanobacterial enrichment culture metagenome (Olsen et al., 2015). Thus, 37 partial genomes, 26 of which contained ≥1 Mb of sequence information, were found by this method (Table 3). Twenty-six of the bins were identified taxonomically, and 22 could be affiliated with abundant OTUs. A specific cut-off with regard to taxonomic levels or sequence threshold cannot be given for the represented populations. However, previous studies, as well as joint binning of the sequences from the presented study with reference genomes, suggest that genomes derived from bacterial populations with 16S rRNA gene sequences identities of ≥96% do not separate into distinct bins (data not shown; Klatt et al., 2011). In this study, the cyanobacterial genomes of Synechococcus Types A and B' (97% 16S rRNA nt identity), and within the Chloroflexi, Roseiflexus castenholzii and Roseiflexus sp. RS-1 (95.6% 16S rRNA nt identity) as well as Chloroflexus aurantiacus J-10-fl and Chloroflexus sp. MS-G (95.7% 16S rRNA nt identity) genomes clustered in single bins containing sequences of both genomes, respectively. All other included Chloroflexi reference genomes (<94% 16S rRNA nt identity) clustered in separate but sometimes adjacent bins. The occurrence of several metagenomic bins affiliated with the Chloroflexi as well as the separate clustering of the included Chloroflexi reference genomes, provides an estimate of the ability of this approach to discriminate and resolve among different members of the same phylum. Based on these observations, as well as 16S rRNA OTU similarities found in this study displaying values of either <95% or >96.8% nt identity, we expect genomes of populations sharing <95% 16S rRNA sequence identity to be represented by distinct metagenomic bins, whereas OTUs of >96.8% similarity would probably be represented by a single partial genome (i.e., metagenomic bin).
Overview of Phyla and Taxa Detected in the Mushroom Spring Undermat
In the following paragraphs we will describe selected taxa from each phylum detected in the undermat community based on combined information of iTag and metagenomic sequence data. The phyla and members thereof are presented in the order of abundance, starting with the most abundant phylum and the most abundant member, respectively. Taxonomic identification was always based on the longest 16S rRNA sequence available, in conjunction with phylogenetic marker genes. Information on additional taxa and phyla can be found in the phylogenetic trees and the Supplemental Materials (Figure 5, and Figure S1, Table S1). Phylogenetic analyses based on 16S rRNA sequences extracted from metagenomic data identified >50 members of 20 different phyla (Figure 5 and Figure S1), most of which could also be affiliated with iTag sequences obtained in the amplicon study.
Chloroflexi
Members of the phylum Chloroflexi were the most diverse group of organisms present in the microbial undermat community. Overall, 41 OTUs were affiliated with the phylum Chloroflexi (Table S1), and twelve Chloroflexi sequences were identified phylogenetically (Figure 5A). Five of the fifteen most abundant OTUs (>1,000 reads), as well as four abundant OTUs with ≥100 reads, were identified as members of the Chloroflexi (Table S1, Figure 5A). Based on the metagenomic information for these taxa, four out of five very abundant Chloroflexi are chlorophototrophic members of this phylum (OTUs-1, 6, 11, and 15; see Figure 5A), while one is a putative chemoheterotroph (OTU-9). Three additional abundant OTUs also are associated with putatively chemoheterotrophic members of this phylum (OTUs 23, 31, and 39). Thirty-two less abundant OTUs were also affiliated with the phylum Chloroflexi (Table S1, Figure 5A).
Binning of the assembled metagenomic data yielded only a very small partial genome for Roseiflexus spp., the most abundant and most diverse OTU in the undermat (Bin-1; Figures 2, 3, 5, Tables 1–3 and Table S1). Bin-1 did not contain any phylogenetic marker genes but was identified by high nucleotide sequence identities (92 ± 5%; range 79–100%) to the Roseiflexus sp. RS-1 genome (CP000686, 5.8 Mb, van der Meer et al., 2010). The Roseiflexus sp. RS-1 genome recruited 23,534 contigs from the metagenome (≥85% nt identity and ≥75 coverage), of which 13,329 contigs showed sequence identity of ≥95%. Only 12 of those contigs were >5 kb in length, sharing a minimum of 94.52% nt identity with the Roseiflexus sp. RS-1 genome sequence. Roseiflexus sp. RS-1 is a filamentous anoxygenic phototroph that synthesizes bacteriochlorophyll (BChl) a but not BChl c. It was previously isolated from Mushroom Spring and was affiliated with OS Type C sequences obtained in early molecular studies (Ward et al., 1990; Ferris et al., 1996b, 1997; Ferris and Ward, 1997). In addition to BChl a-containing photosynthetic reaction centers, the genome of this organism encodes xanthorhodopsin, which was also detected in the undermat metagenome (RoseRS_2966, GenBank Acc. no. ABQ91330.1; JGI24185J3567_10248071), and indicates a possible additional use of light energy (Choi et al., 2014). The small number of long contigs affiliated with this OTU, in combination with the broad coverage range from 31 × to 1,557 ×, reflects a high microdiversity as well as the high abundance of the core genome sequences.
A 1,364-bp partial 16S rRNA sequence identified OTU-6 as a member of the Chloroflexi, which is most closely related to uncultured members in streamer biofilm-producing communities in YNP hot springs (Table 3; Meyer-Dombard et al., 2011). It represents an uncultured chlorophototrophic Anaerolineae-like organism, which was also identified in the upper green layer of the Mushroom Spring microbial mat in a previous metagenomic analysis (Klatt et al., 2011). Despite the absence of a 16S rRNA gene, Bin-6 was identified to represent OTU-6 based on 93 ± 5.6% average nt identity to Cluster 6 from the upper layer metagenome (Klatt et al., 2011), which did contain a ribosomal RNA sequence with 98% identity to OTU-6, as well as 99% sequence identity to a 16S rRNA sequence detected in the metagenome of this study. When first reported by Klatt et al. (2011), this uncultured organism was identified as “Anaerolineae-like,” with Anaerolinea thermophila strain UNI-1 being its closest cultivated and described relative (85% nt identity, Sekiguchi et al., 2003). At the time of this writing [February 2016], a BLAST search identified Thermanaerothrix daxensis strain GNS-1T (Grégoire et al., 2011) and Thermomarinilinea lacunofontalis strain SW7 (Nunoura et al., 2013) as the closest isolated relatives with a 16S rRNA sequence identity value of 87% (Table 1). Phylogenetic analysis based on the full-length 16S rRNA sequences supports a phylogenetic affiliation to the Anaerolineales as well as a more distant relationship to known chlorophototrophic Chloroflexi (Figure 5A). Genes annotated within this metagenomic bin suggest that, like Roseiflexus spp., this anoxygenic chlorophototroph has the potential to produce BChl a but probably doesn't contain BChl c or chlorosomes, although it does possess a putative xanthorhodopsin-like gene (Klatt et al., 2011). Thin short filaments possibly representing this Anaerolineae-like phototrophic Chloroflexi, tentatively named “Ca. Roseilinea gracile” (Tank et al., in press), have been observed in fresh mat samples and enrichment cultures. They exhibit BChl a but not BChl c autofluorescence.
OTU-09 is represented by Bin-9 and was also identified as being derived from a member of a cluster of uncultured Chloroflexi within the Anaerolineae (Figure 5A). However, based on the absence of photosynthesis-related genes in the corresponding metagenomic bin and the absence of unassigned photosynthesis-related genes in the remaining unbinned contigs, the organisms corresponding to OTU-09 are not predicted to be chlorophototrophs.
A close relative of Chloroflexus sp. strain MS-G, a chlorophototrophic member of the Chloroflexi that was previously isolated from this mat (Thiel et al., 2014b), is represented by OTU-11 and Bin-11 in this study. Like strain MS-G, OTU-11 is predicted to be an anoxygenic phototroph containing type-2 (quinone-type) photosynthetic reaction centers, light-harvesting complex 1 and chlorosomes based on a metagenomic bin of 3.1 Mb, with an average read coverage of 30 × (Bin-11, Table 3). The bin contained 21 phylogenetic marker genes, all of which share amino acid sequence identity values of 98.7 to 100% with sequences from Chloroflexus sp. MS-G (Table 3). The organism representing OTU-11/Bin-11 and strain MS-G share 98.3% 16S rRNA and 94 ± 6% overall genomic nucleotide sequence identity, respectively.
A third anoxygenic phototrophic Chloroflexi is represented by OTU-15 and Bin-15. Phylogenetic analysis and BLAST search results indicate this organism to be only distantly related to other chlorophototrophic Chloroflexus spp., displaying 90–91% 16S rRNA sequence identity to Oscillochloris trichoides, Chloroflexus aurantiacus J-10-fl and “Candidatus Chloroploca asiatica.” The organism associated with these sequences presumably represents a novel genus of chlorophototrophic Chloroflexi within the family Chloroflexaceae (Figure 5A). Based on the conserved signature indels that are specific for different groups within the Chloroflexi as described by Gupta et al. (2013), this filamentous anoxygenic phototroph is affiliated with the proposed order of “green nonsulfur bacteria,” Chloroflexales, suborder Chloroflexineae, but is distinct from all known members of the genera Chloroflexus and Oscillochloris. The functional gene content of the associated metagenome bin (Bin-15) indicates that this organism has the capacity to synthesize BChls a and c. A filamentous BChl a- and BChl c-producing isolate similar to Oscillochloris sp. has been obtained in enrichment cultures, and tentatively named “Candidatus Chloranaerofilum corporosum” (Tank et al., in press).
Thermomicrobium roseum, phylum Chloroflexi, which had previously been isolated from the mats (Jackson et al., 1973), was detected in the metagenome in this study and a previous 16S rRNA cloning study (Klatt et al., 2013a), but T. roseum was only present in low numbers based on the analysis of iTag amplicons (OTU-74, 44 reads, Table S1, Figure 5A).
Thermotogae
Only two OTUs, OTU-2, and OTU-107, were identified as members of the phylum Thermotogae by the RDP classifier (Table S1). OTU-2 represents the second most abundant species-level iTag sequence and the corresponding metagenomic 16S rRNA sequence is 99% identical to that of Pseudothermotoga hypogea, formerly known as Thermotoga hypogea (Fardeau et al., 1997; Bhandari and Gupta, 2014). Bin-2 sequences, which represent this Pseudothermotoga sp. OTU-2 mat member (Table 3), show high similarities (98–100% aa sequence identities) to sequences obtained from a previous metagenomic study by Klatt et al. (2013a; IMG/M OID 2015219002), and form a single cluster with the genome sequence of Pseudothermotoga hypogea DSM 11164 in the metagenome binning analysis, which indicates the high similarity of these two genomes. OTU-107 shares 99% nt sequence identity to Fervidobacterium pennivorans strain DSM 9078 as well as to Fervidobacterium sp. isolated from YNP (Sullivan et al., unpublished, AY151268) but is represented by only 20 reads (Table S1, Figure S1). In addition, several sequences were affiliated with group EM3, which has tentatively been placed in the Thermotogae (Reysenbach et al., 2000) (Table S1, Figure S1). OTU-10 was misidentified as a member of the Chlorobi by RDP classifier, but actually represents the most abundant EM3 population and shares highest similarities with hot spring clones OPB88 (AF027006, Hugenholtz et al., 1998b) and OPS2 (AF018187, Graber et al., unpublished) from YNP with 99 and 98% 16S rRNA nt identity, respectively. Bin-10 representing this OTU was identified based on the presence of a matching 16S rRNA gene (Table 3). Phylogenetic affiliations of the phylogenetic marker genes were uncertain with most of the sequences only being assigned to the kingdom (“bacteria”) and phylum level (“Bacteroidetes,” “Chlorobi,” “Deinococcus-Thermus,” “Chloroflexi,” or “Thermotogae,” respectively), which indicates a high degree of novelty for this uncultured organism. Sequences similar to the ones in this metagenomic bin have previously been detected in the oxic upper green layer of the mat community (Klatt et al., 2011). The sequences formed unidentified Cluster 8 in the previous study, which were associated with an uncultivated, putatively heterotrophic bacterium. Bin-10 and Cluster 8 sequences formed a single bin when included in the analysis. A BLASTn comparison revealed an average nucleotide identity of 97 ± 3% between sequences of the previous cluster and the sequences in the bin from this study.
Armatimonadetes (OP10)
Uncultivated members of the Candidate phylum OP10, now named Armatimonadetes (Tamaki et al., 2011; Lee et al., 2013), were first detected in Obsidian Pool in YNP (Hugenholtz et al., 1998b). The undermat community at Mushroom Spring also contains a considerable diversity of members of this phylum. Two of the most highly abundant OTUs, OTUs 3, and 12, were identified as members of the Armatimonadetes. In addition, two abundant (OTUs 18 and 33) and nine less abundant iTag OTUs were identified as members of this phylum (Table S1). Partial genomes were identified for OTUs-3, 12, and 18 (Table 3, Figure S1).
Despite the high abundance of Armatimonadetes member OTU-3 sequences in the amplicon study and the presence of a partial 16S rRNA sequence with high coverage (951 ×; JGI24185J35167_1062246), no corresponding bin was obtained in the undermat metagenome. Serendipitously, a highly similar organism (99% 16S rRNA sequence identity) was identified as a chemoheterotrophic contaminant in a cyanobacterial enrichment culture obtained from these mats in the Ward laboratory at Montana State University (unpublished data). A partial genome of this enrichment contaminant was obtained by binning the assembled contigs of the corresponding enrichment culture metagenome (Bin-3, Table 3). This enrichment partial genome recruited 17,252 sequences (a total of 11 Mb of sequence data) from the undermat metagenome displaying 90.5 ± 7.5% nt id (covering min. 80% of the metagenome scaffold). OTU-3 amplicon sequences were also detected in the upper green layer in lower numbers (4.5 vs. 0.8% relative abundance; Table S1) and a partial genome of this organism was also detected as an unidentified heterotroph Cluster 7 in the upper layer metagenome (Klatt et al., 2011). The partial genome of the upper layer displayed similar identity values of 90.3 ± 7.5% to the enrichment culture metagenome bin and 94.6 ± 5.3% to sequences in the undermat metagenome, and formed a single ESOM bin with the partial genome obtained from the enrichment culture (data not shown). OTU-3 was phylogenetically identified as belonging to the “OS-L clade” within the uncharacterized group 7 of the phylum Armatimonadetes (Lee et al., 2013) (Figure S1). Clade OS-L is named after the first sequence of this clade, OS Type L, obtained from a DGGE study of enrichment cultures from microbial mats in Octopus Spring (Ward et al., 1992), with which the 16S rRNA genes in both Bin-3 from the enrichment culture and the undermat metagenome share 98% nt identity (L04707). So far, no isolated representative has been reported for this phylogenetic group. The presence of all 31 bacterial phylogenetic marker genes in the bin suggests that it contains a nearly complete genome (Table 3). Genes encoded in the partial genome, in combination with its occurrence in an enrichment with oxygenic cyanobacteria, indicates that this organism probably exhibits an aerobic or microaerobic lifestyle, similar to the other isolated members of the Armatimonadetes (Lee et al., 2011; Tamaki et al., 2011; Im et al., 2012). A considerable microdiversity was suggested by the presence of nine abundant iTag sequences (Table 1) as well as the diversity of partial, flagellum-associated genes affiliated with this organism, which were present on short contigs in the metagenome. Additionally, thirteen closely related 16S rRNA sequences were derived from a previous undermat 16S rRNA cloning study (Klatt et al., 2013a). These sequences show high identity values (>97%) to the OTU-3 sequence as well as to each other (assembly based on 97% nt sequence identity, Figure S1A) and also reflect a high microdiversity of these organisms. Similar to the situation found for Roseiflexus spp. (see above), the high microdiversity suggested for this taxon probably caused assembly difficulties, which may explain why no metagenomic bin was recovered directly from the undermat metagenome.
Aquificae
Of four OTUs identified as belonging to members of the Aquificae (Table S1), only OTU-4 was detected in significant numbers (Table S1). The corresponding 1,434-bp rRNA metagenomic sequence is 99% nt identical to clone sequences previously obtained from YNP hot spring habitats (Thermocrinis sp. clone YNP_SBC_BP2A_B2, HM448202, Meyer-Dombard et al., 2011), as well as to the YNP isolate Thermocrinis sp. P2L2B (AJ320219, Eder and Huber, 2002). The closest described relative is Thermocrinis ruber DSM 23557, which was isolated from Octopus Spring and which has a 16S rRNA sequence that shares 97% nt identity to the one found in this study (Huber et al., 1998) (Figure S1). Correlating to the high microdiversity detected for this OTU (Table 1), only a small partial genome was identified in the binning analysis of the metagenome (Bin-4, Table 3). The presence of at least two closely related populations in the undermat community is indicated by two highly similar (96% amino acid identity), Thermocrinis-like soxB genes; these genes are located on three individual scaffolds in the metagenome, each [gene-1, ~270 × coverage: JGI24185J35167_10446912, JGI24185J35167_10438521, JGI24185J35167_10819822; gene-2, ~70 × coverage: JGI24 185J35167_10446972, JGI24185J35167_10438611, JGI24185J35167_10820392], which also suggests problems with sequence assembly that could be related to microdiversity.
Cyanobacteria
The two major photoautotrophic primary producers of the upper green layer, Synechococcus spp. Type A and Type B', were also abundant members of the undermat by iTag analysis (OTUs 5 and 22, Table 1, Table S1). Seventeen additional but less abundant iTag OTUs (each ≤ 25 reads, representing <0.05% of the total iTag sequences) were assigned to cyanobacteria (Table S1). At the temperature sampled in this study (60°C), members of Synechococcus sp. Type B' (OS Type B', Table 4) are the predominant organisms (Klatt et al., 2011; Liu et al., 2011) and were also detected in this study (OTU-5, Bin-5, Table 3). Synechococcus sp. A (OS Type A, Table 4) sequences were detected in lower abundance (OTU-22, Table S1). The small size of Bin-5 (Table 3) reflects a low number of long and well-assembled contigs (68 contigs, 5,005–12,792 bp; 18 × to 96 × coverage) in comparison to a total of 3,353 contigs identified as having their origins in members of the Cyanobacteria in the metagenome (440 to 12,792 bp). Local BLASTn analysis and reference guided assembly using the genome sequence of Synechococcus sp. Type B' as query (applying a 95% nt identity threshold) identified 4,898 contigs as belonging to these organisms. The low assembly quality is indicative of high microdiversity as indicated by the presence of seven abundant iTag sequences (Table 1). Recent studies have found that a high number of ecotype populations occur within this cyanobacterial population, displaying variations in gene content and sequence as well as differences in gene arrangement (Becraft et al., 2011; Olsen et al., 2015). Genome sequences of several ecotypes isolated from the dominant cyanobacteria from Mushroom Spring are now available, and these provide comprehensive insights into the physiological and metabolic capacities of the oxygenic chlorophototrophs in the mat (Bhaya et al., 2007; Nowack et al., 2015; Olsen et al., 2015).
Atribacteria (OP-9/JS1)
The phylum Atribacteria, formerly known as Candidate phylum OP-9/JS1, exhibited low diversity. Of two OTUs identified as belonging to members of this phylum, only OTU-7 was detected in significant numbers in the iTag analysis (Table S1, Figure S1). OTU-7 represented 2.4% of all iTag reads and was represented by only a single abundant dereplicated iTag sequence (Table 1). Bin-7 contained a partial genome of this uncultured bacterium, as identified by the full-length 16S rRNA sequence which shared 99% and 98% sequence identity to Atribacteria clones OPB72 and TP29 obtained from hot springs in YNP and Tibet, respectively (Hugenholtz et al., 1998b; Lau et al., 2009). The affiliated metagenomic bin indicates an anaerobic, fermentative lifestyle for this member of the Atribacteria (data not shown), which is similar to properties deduced from single-cell genome sequences previously obtained from members of the Atribacteria (Dodsworth et al., 2013; Nobu et al., 2016).
Nitrospirae
iTag analysis identified seven Nitrospirae OTUs in the undermat community, of which only one, OTU-8, was abundant (Table S1). Bin-8 was assigned to this Thermodesulfovibrio sp.-like mat member based on presence of the corresponding 16S rRNA sequence (Figure 4, Table 3). OTU-8 represented ~2.0% (3,283 reads) of all iTag sequences (Table 1), and the full 16S rRNA sequence was most closely related to a clone sequence obtained from geothermal groundwater (99%, clone: SMD-B01, NCBI acc. no. AB477993, Kimura et al., 2010) and to Thermodesulfovibrio yellowstonii strain DSM 11347, as the closest isolated relative (96%, NCBI acc. no. CP001147, Henry et al., 1994; Bhatnagar et al., 2015). Bin-8 contained scaffolds with coverage values ranging from 29 to 135, which possibly reflects two different populations with different abundances. This was also suggested by the different read numbers of two abundant, dereplicated iTag sequences (OTU-8, iTag-10, 1,721 reads; and iTag-28, 602 reads; Table S1). The partial genome suggests sulfate-reducing metabolism for this organism, similar to T. yellowstoneii, which was isolated from thermal vent water in Yellowstone Lake, Wyoming, USA (Henry et al., 1994; Bhatnagar et al., 2015). The dsrAB gene sequences associated with dissimilatory sulfate-reduction of this uncultured organism have previously been detected in the Mushroom Spring microbial mat, and the corresponding Thermodesulfovibrio-like organism was associated with the sulfate reduction activity measured in the mat (Dillon et al., 2007). OTU-8 has been detected in both the upper and lower parts of the mat (Table 5, Table S1), possibly indicating that these organisms are not restricted to the undermat; this is further supported by the finding of Thermodesulfovibrio-like sequences also in the green upper layer metagenome in a previous study (Klatt et al., 2011).
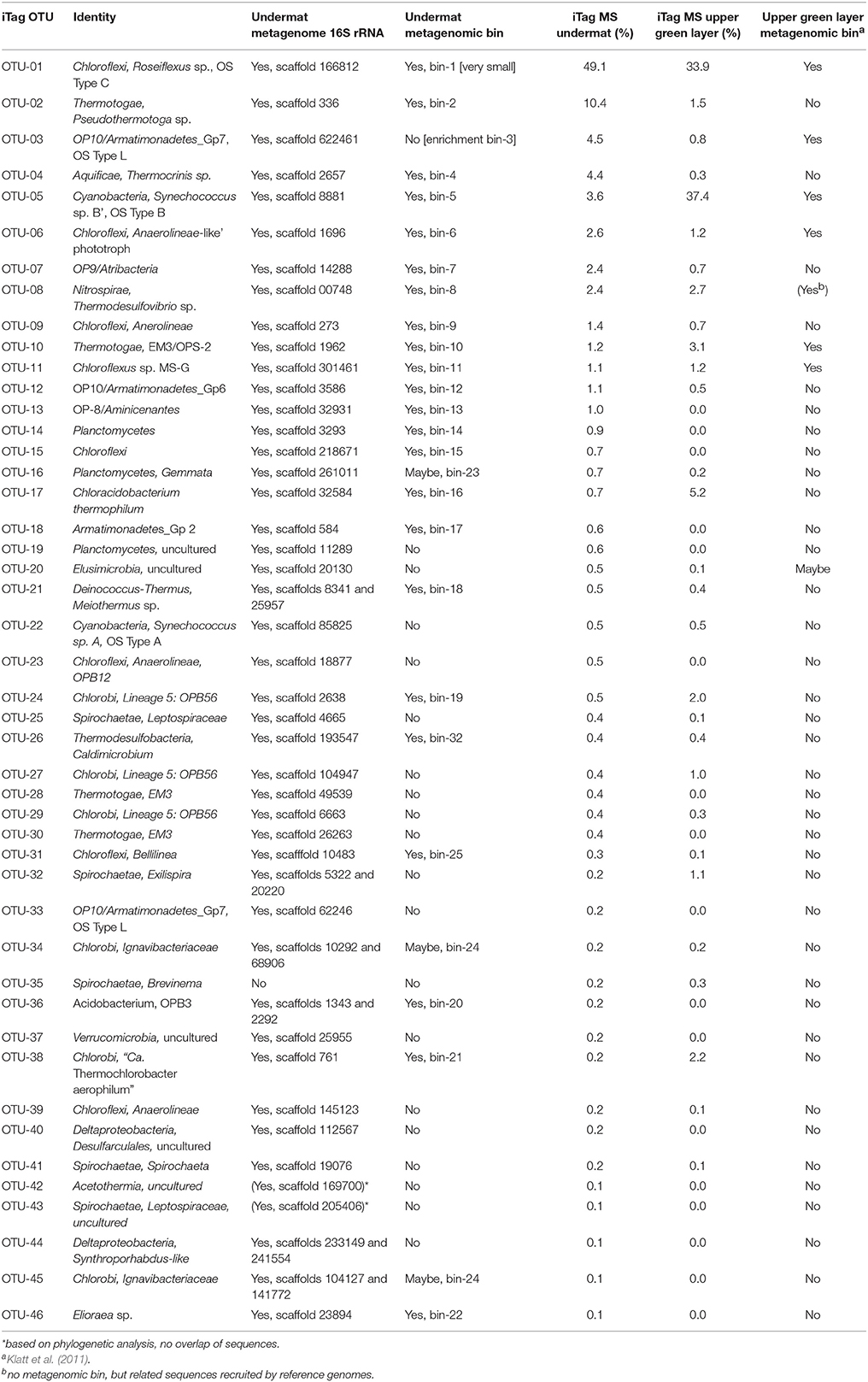
Table 5. Overview of community composition detected by the different methods used in this study (iTag, metagenome 16S rRNA, metagenome binning) and relative abundances in undermat and upper layer iTag sequencing study.
Aminicenantes (OP8)
The Aminicenantes (Candidate phylum OP8) was represented by only a single taxon, OTU-13, and its corresponding metagenomic Bin-13, which contains a 1,497-bp 16S rRNA gene sequence (Table 1, Table S1). Notably, OTU-13 amplicon sequences were found exclusively in the undermat community (Table S1). Although the iTag sequence shared 99% nt identity to the uncultured Aminicenantes bacterium clone OPB95 obtained from a Yellowstone hot spring (AF027060, Hugenholtz et al., 1998b), the full-length sequence showed only 95% nt identity to that sequence. No isolated bacterium shares more than 88% nt identity with this uncultured organism. 16S rRNA gene sequence surveys indicated that members of the Aminicenantes are ubiquitously present in many different habitats and across many environmental parameters (temperature, salinity, and oxygen tension) (Farag et al., 2014). They usually represent only a small fraction (<1%) of microbial communities, but have been found to be more abundant in anoxic environments (Farag et al., 2014).
Planctomycetes
Five abundant iTag OTUs were identified as belonging to members of the phylum Planctomycetes (Table S1), the very abundant OTU-14 (1,260 reads), as well as four less abundant OTUs (OTUs-16, 19, 49, and 51, Table S1). Twelve additional Planctomycetes sequences were found in very low abundance (Table S1).
Bin-14 contained a partial genome for Planctomycetes member OTU-14 and was identified based on the corresponding full-length 16S rRNA sequence as well as nineteen phylogenetic marker genes (Table 3, Figure S1). An uncultured hot spring-associated bacterium from a neutral 61°C geothermal hot-spring mat in Tibet, clone TP5, was identified as closest relative (EF205581, 99%, Lau et al., 2009). The microaerophilic, facultatively anaerobic, thermophilic Planctomycetes strain, Thermogutta terrifontis strain R1T (KC867694, Slobodkina et al., 2014), with 90% sequence identity, is the most closely related isolated relative (Table 1). Based on the number of phylogenetic marker genes present in the metagenome bin, and because of the large sizes of available Planctomycetes genomes (3.8–9.7 Mb for those in JGI/IMG as of December 2015), we expect the 1.87-Mb bin to represent no more than 60% of the genome. The presence of the iTag sequences for this OTU almost exclusively in the undermat sample (a single read was found in iTag analysis of upper green layer; Table 5, Table S1) suggests that this organism lives exclusively in the orange-colored undermat and possibly in its deeper regions below 3 mm, where mainly anoxic conditions occur and persist (Nübel et al., 2002; Jensen et al., 2011).
Bin-23 was also identified as derived from a member of the Planctomycetes, but could not be directly affiliated with any iTag sequence(s) due to absence of an rRNA sequence in the bin (Table 3).
Acidobacteria
Thirteen OTUs representing four different members of the Acidobacteria were identified in the Mushroom Spring undermat community, and two of them were abundant with >100 reads (Table S1). OTU-17 was a member of group 4 of the Acidobacteria and was identified as Cab. aerophilum (Tank and Bryant, 2015a,b). Bin-16 (Table 3) contained a partial genome for this unique microaerophilic, chlorophototrophic member of the phylum Acidobacteria, which was first identified in the phototrophic mats of Mushroom and Octopus Spring and corresponds to the OS Type D sequences from earlier studies (Ward et al., 1990, 1992; Bryant et al., 2007; Tank and Bryant, 2015a,b).
OTU-36, as well as the less abundant OTU-72, were members of Acidobacteria group 3 and were identified as Solibacter-like organisms. Bin-20 was associated with OTU-36 by the presence of a 16S rRNA-containing scaffold as well as by the presence of six phylogenetic marker genes (Table 3). All six phylogenetic marker genes indicated an affiliation with the Acidobacteria and four of them specifically with the candidate species, “Ca. Solibacter usitatus” (Challacombe et al., 2011). Phylogenetic analysis supported the affiliation and placed the sequence in subgroup 3 of the Acidobacteria, closely related to Yellowstone clone OPB3 (98%, AF027004, Hugenholtz et al., 1998b) and “Ca. Solibacter usitatus” Ellin6076 as the closest named relative (Table 1, Figure S1). The low number of phylogenetic marker genes indicates that this member of the Acidobacteria has a large genome, only a part of which is included in the metagenomic bin. This correlates well with the fact that “Ca. Solibacter usitatus” Ellin6076 has an exceptionally large, 9.97-Mb genome (Challacombe et al., 2011).
The fourth member of the phylum Acidobacteria corresponded to a less abundant OTU (OTU-61, 70 reads = 0.1%) and was represented by two partial 16S rRNA sequences in the metagenome. These sequences and the represented uncultured organisms were affiliated with OS Type K sequences from previous studies (Table 4, Ward et al., 1992; Weller et al., 1992).
Proteobacteria
Four abundant OTUs were affiliated with the phylum Proteobacteria by the RDP classifier, one of which was misidentified as Proteobacteria and rather represents a Brevinema-like member of the Spriochaeta (OTU-35), two of which were Deltaproteobacteria (OTUs-40 and 44), and one of which was an Alphaproteobacterium (OTU-46). Twenty-nine additional, low-abundance OTUs were affiliated with Proteobacteria by RDP classifier (Table S1). Sequences for 16S rRNAs of two Alpha-, two Beta- and one Delta-Proteobacteria were found in the metagenome (Figure S1C). The abundant deltaproteobacterial sequence (OTU-44) was closely affiliated to a sequence obtained in a previous metagenome study (Figure S1C, Klatt et al., 2013a). Although the Deltaproteobacteria are commonly known to include members with sulfate-reducing metabolism, and sulfate-reduction has been shown in the microbial mat at Mushroom Spring (Dillon et al., 2007), deltaproteobacterial dsrAB genes were not identified in this nor any previous study. No metagenomic bin was affiliated with a Deltaproteobacterium.
The abundant Alphaproteobacterium (OTU-46) was identified as an Elioraea sp. within the Rhodospirilliales, which corresponds to OS Type O obtained in previous studies (Figure S1C, Table 4, Ward et al., 1992). The corresponding partial genome (Bin-22, Figure 4, Table 3) as well as the genome for the closest relative, Elioraea tepidiphila DSM 17972 (NCBI acc. no. NZ_KB899965.1), contain genes for anoxygenic photosynthesis. Although chlorophototrophy has not been described for Elioraea tepidiphila (Albuquerque et al., 2008), the ability to synthesize BChl a is predicted for the OTU-46 population in the undermat community. A BChl a containing strain, “Candidatus Elioraea thermophilum,” was isolated from the mat, which shares 99.8% and 99.2% sequence identity with the 16S rRNA sequences from the metagenome and amplicon study, respectively (Figure S1C, Tank et al., in press). A low abundance Alphaproteobacterium sequence (OTU-121, 16 reads) was identified as belonging to a Roseomonas/Rhodovarius-like organism, for which an isolate has been obtained from Mushroom Spring and which has tentatively been named “Candidatus Roseovibrio tepidum” (Figure S1C, Tank et al., in press). The isolate exhibits BChl a autofluorescence suggesting a phototrophic lifestyle, which is further strengthened by the presence of low coverage, unidentified alphaproteobacterial pufLM sequences in the metagenome (scaffold JGI24185J35167_1024732, genes 2 and 3, 20 × coverage). Only a single described Roseomonas sp., R. aestuarii, has been reported to produce BChl a, but no pufLM sequences are available for that isolate (Venkata Ramana et al., 2010). Furthermore, two low-abundance OTUs (OTUs-101 and 154) showed the same phylogenetic affiliation (Hydrogenophilius sp., Betaproteobacteria) as OS type G from previous studies (Ward et al., 1990, 1992). The OS Type R sequence (NCBI acc. no. U46750, unpublished) represented an unidentified Betaproteobacterium and a similar, low-abundance iTag sequence (OTU-172) was detected in this study (Table 4, Figure S1C).
Bacteroidetes-Chlorobi
The RDP classifier identified twenty and eight different OTU sequences belonging to members of the phyla Chlorobi and Bacteroidetes, respectively. Seven OTUs affiliated with the Chlorobi were abundant with read numbers >100, and one was very abundant with >1,000 reads (Table S1). However, the most abundant “Chlorobi” sequence (OTU-10) was mis-classified and represents an Thermotogae/EM3 group member (see above, Table 1, Figure 5B). The other abundant Chlorobi sequences were affiliated with the proposed family Thermochlorobacteriaceae (OTU-38) (Liu et al., 2012), “Chlorobi lineage 5” = “OPB56 group” (OTUs 24, 27, and 29) (Iino et al., 2010; Hiras et al., 2015) and “Chlorobi lineage 2” = “SM1H02 group” (OTUs 34 and 45) (Iino et al., 2010; http://www.arb-silva.de/browser/ssu-121/AY555793, named after clone SM1H02, Genbank acc. no. AF445702). Bin-19 (Table 3) was identified as a partial genome representing OTU-24, a representative of OPB56, a subgroup of the Chlorobi with predicted chemoheterotrophic lifestyle that was first detected in YNP (Hugenholtz et al., 1998b; Hiras et al., 2015, Table 3). A low abundance OTU in the OPB56, OTU-262, was identified as a probable representative of the OS Type F sequences from previous studies (Table 4, Ward et al., 1990, 1992). The first aerobic, phototrophic member of the Chlorobi, “Ca. Tcb. aerophilum,” which belongs to the proposed family Thermochlorobacteriaceae and was identified in the upper green layer of the microbial mat by previous metagenomic analyses (Liu et al., 2012), is represented by OTU-38 (Table 1), and was identified as OS Type E in previous studies (Ward et al., 1990, 1992; Ferris et al., 1996b). Bin-21 is derived from this novel phototroph (Table 3) and supports its characterization as a chlorophototroph that synthesizes type-1 reaction centers and chlorosomes, similar to cultivated relatives among the green sulfur bacteria, but which is otherwise very different physiologically. “Ca. Tcb. aerophilum” is proposed to be an aerobic photoheterotroph that cannot oxidize sulfur compounds, cannot fix N2, and does not fix CO2 (Liu et al., 2012).
Bin-24 (Table 3) does not contain a 16S rRNA sequence, but was affiliated with a putative member of the Bacteroidetes-Chlorobi group based on phylogenetic marker genes. It is most closely related to heterotrophic members of the Chlorobi, in the family Ignavibacteriaceae (Liu et al., 2012; Kadnikov et al., 2013) and is presumably affiliated with OTUs-34 or 45 in the Chlorobi Lineage 5/group SM1H02 (Figure 5B). All genes needed for dissimilatory sulfate reduction are present in the partial genome and indicate that this organism is putatively the first sulfate-reducing member of the Bacteroidetes-Chlorobi group. These results will be described in detail elsewhere (Thiel et al., in preparation). The OS Type M sequences obtained in previous studies (Ward et al., 1992) are affiliated with OTU-34 as well as with two partial 16S rRNA sequences from the metagenome (Table 4) within the SM1H02 (Chlorobi Lineage 2) group.
Only low abundance OTUs were affiliated with the Bacteroidetes (Table S1). Many of them were closely related to clone sequences obtained in a previous undermat study, and some also represented partial 16S rRNA sequences from the metagenome (Figure 5B, Klatt et al., 2013a). Schleiferia thermophila, a strain of which has been isolated from Octopus Spring microbial mats (Thiel et al., 2014a), was not detected in this study.
Deinococcus-Thermus/Thermi
Of two different members of the phylum Thermi identified in this study, only Meiothermus sp. was abundant in the undermat community (OTU-21, 656 reads), whereas sequences of Thermus spp. were only present in low numbers in the iTag study (Table S1, Figure S1C). Members of both genera have been isolated from these mat communities (Brock and Freeze, 1969; Ward et al., 1997; Thiel et al., 2015). OTU-21 was identified as a relative of Meiothermus ruber, a member of which, strain A, has previously been isolated from an enrichment culture originally obtained from the microbial mats at Octopus Spring and whose genome has been sequenced (Thiel et al., 2015). Tetranucleotide frequency-based binning of contigs >10 kb led to a 1.3-Mb partial genome (Bin-18, Table 3) for this moderately thermophilic, aerobic, and heterotrophic bacterium. The Meiothermus sp. 16S rRNA sequences obtained from the metagenome share 96.7% nt sequence identity with M. ruber strains A and DSM1279T. Sequences of Bin-18 shared 84.5% (±4.5%) with the M. ruber strain A genome and 84.2% (±4.5%) with M. ruber DSM1279T. Although the (partial) genome sequences of the isolate and the metagenome bin clusters overlap, some separation was visible when the sequences of both organisms were included in the binning analyses (data not shown).
Archaea
Although methanogenesis has been demonstrated in several mats of alkaline siliceous hot springs, including Mushroom Spring (Ward, 1978; Sandbeck and Ward, 1982), and methane has been shown to accumulate in the water above the Mushroom Spring mat in darkness (Kim et al., 2015), iTag sequencing only identified a few partial 16S rRNA sequences as potentially derived from methanogenic Archaea (OTUs-143, 151, 162, 192, and 244; ≤ 11 reads = ≤ 0.01%, Table S1). Phylogenetic analysis confirmed affiliation to the Euryarchaeota for four of them (OTUs-143, 151, 162, and 192, Figure S1A) and three of the sequences were detected in a cloning experiment from a previous study (Klatt et al., 2013a); thus, methanogenic archaea seem to be present in the mat over time, although in very low abundance. One OTU, OTU-151 with 10 reads but no representative sequence in the metagenome, shows high similarity (99% nt id) with the 16S rRNA sequence of the methanogenic archaeon Methanothermobacter thermoautotrophicus, strains of which have been isolated from these mats previously (former Methanobacter thermoautotrophicum; Sandbeck and Ward, 1982). Further, a single, low coverage mcrA gene encoding a methyl-coenzyme M reductase alpha subunit was present in the metagenome (JGI24185J35167_11200021, 7 × coverage) possibly indicating methanogenic metabolism in at least one of the archaeal mat members. Two slightly more abundant 16S rRNA sequences affiliated with ammonia-oxidizing Archaea were detected (Table S1). One (OTU-60, 72 reads) was related to “Candidatus Nitrosocaldus yellowstonii,” which was also identified in an enrichment culture from Octopus Spring mat in previous studies (De La Torre et al., 2008). The other, OTU-67 represents a member of a putatively novel archaeal phylum/division, related to “Candidatus Caldiarchaeum subterranum” (Nunoura et al., 2011). Another less abundant iTag sequence, similar to that of an archaeal 16S rRNA sequence recovered from the undermat metagenome previously (Klatt et al., 2013a), was also detected in the iTag analysis (OTU-125, 15 reads), but not in the metagenome of this study (Figure S1A). None of the metagenomic bins could be identified as belonging to Archaea, and only a few contigs with low coverage values, showed high identities to known archaeal sequences. Thus, our metagenomic and 16S rRNA gene amplicon studies indicate a very low abundance of Archaea, of which sequences related to ammonia-oxidizing Archaea seem to be more abundant than possible methanogenic Archaea. The low abundance of archaeal sequences is consistent with the low relative abundance of archaeal lipids in previous studies, which had been discussed to be related to the energy flows through the trophic structure of the community (Ward et al., 1989).
Firmicutes
Although Anoxybacillus spp. are common members of cyanobacterial enrichment cultures from these environments (e.g., Nowack, 2014; Olsen et al., 2015; Tank and Bryant, 2015b), no evidence for this organism was found in the metagenome nor the iTag analysis. Twenty-four OTUs were classified as belonging to members of the Firmicutes, of which two (OTUs-251 and 255) were predicted to be Bacillus sp.; however, they shared highest sequence similarity to the type strains of Syntrophothermus lipocalidus and Acetomicrobium faecale (both clostridia). None of the 16S rRNA genes retrieved from the metagenome could be affiliated with the Firmicutes. In addition, none of the metagenomic scaffolds were affiliated with Anoxybacillus spp. No sequence from an Anoxybacillus sp. was identified by BLASTn analysis of the metagenome using the partial genome sequence obtained from the Anoxybacillus sp. MT isolated from an enrichment culture from Octopus Spring (Thiel et al. in prep), nor the “phylogenetic distribution of genes by BLAST percent identities” tool implemented in the JGI/IMG website.
Discussion
In this study we analyzed the orange undermat of the microbial mat community at 60°C in Mushroom Spring YNP by 16S rRNA gene amplicon and metagenomic sequencing. Only eight major organismal populations were identified in the upper green layer by genomic, metagenomic and metatranscriptomic analysis (Klatt et al., 2011; Liu et al., 2011). A higher diversity had been speculated to occur in the undermat community (Klatt et al., 2013a). In this study the undermat was found to be a highly diverse but uneven bacterial community, which could be related to the trophic structure associated with mat-decomposing organisms, as hypothesized to explain the variable abundances of lipid biomarkers (Ward et al., 1989) and 16S rRNA sequences (Ward et al., 1998). Out of 317 OTUs, the 15 most abundant ones represent 87% of all iTag sequences, and the single most abundant OTU comprises nearly half of all iTag reads. More than 44 abundant taxa, as defined by read numbers of >100 in the iTag analysis, were detected in the orange-colored undermat at Mushroom Spring. The phylum Chloroflexi displayed the highest diversity with nine abundant and 41 total taxon-specific 16S rRNA sequences (OTUs) found. All of the taxa found in the upper mat by Klatt et al. (2011) were also identified in the undermat.
In this study we analyzed the composition and diversity of the microbial community based on 16S rRNA gene sequences, which cannot easily be translated into species populations. However, relatively high 16S rRNA sequence diversity was found in this study, not only on the OTU level but particularly within the dereplicated iTags, which suggests that this microbial mat community is not simple. Previous observations that closely related cyanobacterial 16S rRNA sequences were differently distributed along environmental gradients (Ferris and Ward, 1997; Ramsing et al., 2000) prompted consideration of the Stable Ecotype Model of species and speciation (Cohan and Perry, 2007), which postulates that some microorganisms exist as ecological species occupying distinct niches (Ward, 1998; Ward and Cohan, 2005). Studies with more rapidly evolving protein-encoding loci led to the prediction of numerous ecotypes with identical or nearly identical 16S rRNA sequences (Ferris et al., 2003; Becraft et al., 2011, 2015; Melendrez et al., 2011). The existence of temperature- and light-adapted Synechococcus ecotypes has been demonstrated by obtaining representative strains and studying their temperature and light preferences as well as their genomes, (Allewalt et al., 2006; Nowack et al., 2015; Olsen et al., 2015). A similar microdiversity and existence of putative ecotypes is suggested by this study for members of the undermat community, and in particular for Roseiflexus spp., the most dominant member in the undermat. The presence of unique 16S rRNA genotypes in the undermat (this study) and at different temperatures (Ferris and Ward, 1997), supports this inference. In addition to the high diversity of OTUs within the phylum Chloroflexi, a high microdiversity was found for Roseiflexus spp. by the presence of 24 abundant and a total of 6,193 dereplicated Roseiflexus sp. iTag sequences, which is further supported by a preliminary analysis of pufLM amplicon sequence data (J. Wood and D. Ward, unpublished data).
The microbial mat as a living and active biological system has been shown to be constantly growing (Doemel and Brock, 1977). In this study we observed phototrophic taxa known from the upper layer in the undermat. Analyses of psaA sequences sampled in this metagenomic study suggest that the Synechococcus populations observed match species found in the upper mat and thus likely occur in the undermat as a consequence of burial. In contrast, similar analyses of pufLM sequences as well as oligotyping suggest that Roseiflexus populations in the undermat are a mixture of those found in the upper green mat layers and those uniquely found in the undermat (Table 5, Figure S2, Wood et al., unpublished). The detection of identical dereplicated iTag and oligotype sequences in both layers might indicate burial. However, the detection of oligotypes and dereplicated iTag sequences with higher relative abundance in the undermat strongly suggests the existence of putative ecotypes specifically adapted to niches in the undermat. Further it is important to note that specifically adapted ecotypes can be so closely related that they have the identical 16S rRNA gene sequence, and can only be detected using more rapidly evolving genes (Becraft et al., 2011, 2015). For other organisms, a greater relative abundance, or exclusive presence in the lower part of the mat, is indicated by the relative number of 16S rRNA gene amplicon reads between the upper layer and undermat samples. For example, Pseudothermotoga spp. OTU-2, Armatimonadetes member OTU-3, Thermocrinis spp. OTU-4, Chloroflexi members OTU-6, 9, and 15, as well as the Atribacteria member OTU-7, the Aminicenantes member OTU-13, and Planctomycetes member OTU-14, are found in much higher relative abundance in the undermat (Table 5, Table S1). Future transcriptomic studies will assess which of the detected populations correspond to the highest transcriptional activities based on gene expression. The presence of aerobic, microaerobic and anaerobic organisms detected in this study indicate a possible layered distribution along the steep and fluctuating oxygen gradient and shows that some oxygen is available during the day below a depth of 2 mm in the microbial mat, as previously suggested by microelectrode measurements (Revsbech and Ward, 1984; Nübel et al., 2002; Jensen et al., 2011). Whereas aerobic bacteria and facultative anaerobes are expected to live in the transition zone adjacent to the upper green layer, abundant anaerobic members of the undermat community, e.g., Pseudothermotoga sp. OTU-2 and Atribacteria member OTU-7 can be expected to be active members mainly in the community below a depth of 3 mm, where anoxic conditions are expected to persist throughout the day (Nübel et al., 2002; Becraft et al., 2011; Jensen et al., 2011). Despite the anaerobic lifestyle of sulfate reduction, Thermodesulfovibrio sp. OTU-8 was detected in higher abundance in the upper layer, which might indicate some degree of oxygen tolerance and diel activity patterns, i.e., primary sulfate-reducing activity under anoxic conditions in the afternoon or at night as measured by Dillon et al. (2007). An Aminicenantes (OP8) member (OTU-13), a Planctomycetes member (OTU-14) and an Oscillochloris-like chlorophototrophic member of the Chloroflexi, “Ca. Chloranaerofilum corporosum” (OTU-15) (Tank et al., in press) were exclusively detected in the undermat by iTag analysis, which suggests that they have an anaerobic lifestyle in the deeper layers of the undermat. However, “Ca. Chloranaerofilum corporosum” is expected to be a phototroph, and only a limited amount of light reaches deep into the undermat. Thus, a layered structure of the microbial community, as has been demonstrated in the upper green layer (Ramsing et al., 2000; Becraft et al., 2011), can only be hypothesized for the undermat at this time. Further studies are needed to determine the distribution of the members of the undermat community.
All seven chlorophototrophs identified in previous genomic and metagenomic studies of the upper green layer were also present in the undermat metagenome (Table 5; Klatt et al., 2011; Liu et al., 2011). Roseiflexus spp. and “Candidatus Roseilinea gracile” showed higher relative abundance in the undermat, whereas the other phototrophs are present in lower relative abundance in comparison to the upper green layer of the mat (Table 5, Table S1). Three additional phototrophic bacteria were detected in the microbial mat for the first time in this study (“Candiatus Chloranaerofilum corporosum” OTU-15, as well as two phototrophic Alphaproteobacteria, “Candidatus Elioraea thermophila” OTU-46, and “Candidatus Roseovibrio tepidum” OTU-121; Tank et al., in press). A total of sixteen phototrophic bacterial taxa representing six different phyla have now been detected in the Mushroom Spring microbial mat (Tank et al., in press). Additionally, the discovery of multiple organisms with genes encoding xanthorhodopsin raises new questions about the role of retinal-based phototrophy (retinalophototrophy; Bryant and Frigaard, 2006) or signaling in the undermat. This will be addressed in more detail elsewhere (Thiel et al., in preparation). The unidentified Cluster 8 previously detected in the upper layer metagenome was identified again here as OTU-10, an organism affiliated with the group EM3, which has tentatively been placed in the phylum Thermotogae (Reysenbach et al., 1994; Klatt et al., 2013a). The second unidentified heterotroph previously detected in the upper layer metagenome, Cluster 7 (Klatt et al., 2011), was identified as an Armatimonadetes member OTU-3. Due to a high microdiversity of this organism in the microbial mat sample, identification was only possible by a serendipitous finding of a closely related organism in an enrichment culture.
Conclusions
In this study we analyzed the community composition and diversity of the orange-colored undermat of Mushroom Spring, an alkaline hot spring in YNP (WY, USA) by 16S rRNA gene amplicon and metagenomic analyses. Despite a long history of research on the microbial mats at Mushroom and Octopus Springs (Brock, 1967; Ward et al., 1998, 2012; Kim et al., 2015), these mats still harbor the potential for many novel discoveries. Members of the genus Roseiflexus dominated a fairly diverse but uneven microbial community, and metagenomic analysis identified several novel organisms with unusual traits. Many unidentified 16S rRNA sequences recovered from these environments in previous studies were detected and phylogenetically identified. Other organisms, which have been cultured from either Mushroom or Octopus Spring, were not detected, once again illustrating the inherent bias of untargeted cultivation experiments. A more detailed analysis of the metagenome, focusing on the metabolic potential of the mat members and their putative interactions, will be published elsewhere (Thiel et al., in preparation). Studies of microbial ecology, diversity, species evolution and interspecies interactions are still subjects of ongoing research with many open questions to be addressed. Comparisons of species in both upper and lower mat and a diel-transcriptomic analysis that will hopefully reveal gene expression activity within the undermat community that will allow us to distinguish between active and inactive members of the community defined in this study, and should provide information on the temporal pattern of gene expression in the undermat. Depth-dependent distributions of OTU populations that may represent putative ecotypes will also be addressed in future studies.
Accession Numbers
16S rRNA gene sequences of iTag OTUs as well as assembled clone sequences have been deposited in GenBank (Acc. nos. KU860141–KU860455 [iTag OTUs]; KX213895–KX214032 [clone OTUs]). Complete metagenome data are available in the Integrated Microbial Genomes with Microbiome Samples (IMG/M, https://img.jgi.doe.gov/) database, taxon object IDs 3300002493, 3300005452 and 2015219002.
Author Contributions
VT conducted sequence analysis after assembly for both amplicon and metagenome sequences, including phylogenetic analysis and phylogenetic marker genes analysis of metagenome bins. JW conducted initial tetranucleotide binning analyses, reference targeted mapping studies and contributed to discussion and manuscript. Sampling and DNA extraction from the hot-spring microbial mat and enrichment cultures was conducted by MO, who also wrote corresponding sections in the manuscript and contributed to the discussion of results. MT isolated and identified all cultures mentioned in the manuscript, contributed to writing the manuscript and discussing the results. CK conducted 16S rRNA cloning and sequencing from undermat samples from a previous time point, analyzed those sequences and contributed to manuscript and discussion. Sequencing, quality check, assembly and dereplication of amplicon and the metagenome was conducted by JGI staff. DW and DB planned the experiments, acquired funding, organized and led field excursions and provided scientific infrastructure. VT, DW, and DB wrote the manuscript.
Funding
This study was partly funded by the Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences of the Department of Energy through Grant DE-FG02-94ER20137. DB and DW additionally acknowledge support from the NASA Exobiology program (NX09AM87G). This work was also partly supported by the U. S. Department of Energy (DOE), Office of Biological and Environmental Research (BER), as part of BER's Genomic Science Program 395 (GSP). This contribution originates from the GSP Foundational Scientific Focus Area (FSFA) at the Pacific Northwest National Laboratory (PNNL) under a subcontract to DB. The nucleotide sequencing was performed as part of a Community Sequencing Program (Project CSP-411) and was performed by the U.S. Department of Energy Joint Genome Institute, which is supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank all of the JGI staff members who contributed to obtaining the sequence data. The materials used in this study were collected under permit #YELL-SCI-0129 held by DW and administered under the authority of Yellowstone National Park. The authors especially thank Christie Hendrix and Stacey Gunther for their advice and assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2016.00919
Figure S1. Phylogenetic tree based on 16S rRNA gene sequences showing the phylogenetic relationship between sequences obtained from the Mushroom Spring microbial undermat community and cultivated and uncultivated relatives in the phyla Armatimonadetes OP10, Aminicenantes OP8, Nitrospirae, Thermodesulfobacteria, Microgenomates OP11, Aquificae, SM2F11 and Archaea (A), Planctomycetes, Verrucomicrobia, Spirochaeta, Acidobacteria, Atribacteria OP9/JS1, Elusimicrobia and Cyanobacteria (B), and Proteobacteria, Thermotogae/EM3 and Thermi (C). The trees were generated based on the Maximum Likelihood method using the phyML software included in the ARB package. Percentage numbers on nodes refer to 100 bootstrap pseudoreplicates conducted. Only values >50% are shown. Bold sequences were obtained from Mushroom or Octopus Spring in this or previous studies. Red bold labels indicate sequences obtained in this study. Blue bold labels indicate “OS type” sequences from previous studies. OTU numbers shown refer to the most abundant OTU represented by the sequence. Only sequences with length >1,000 bp were used for phylogenetic calculations. Sequence length <1,000 bp are given in (gray) in the labels and corresponding sequences were added using the Parsimony method without changing tree topology.
Figure S2. Entropy distribution in Roseiflexus-like 16S rRNA gene amplicon sequences in the combined dataset (A), and the undermat (B), and upper layer (C). The difference in the entropy figures from upper layer (C) and undermat (B) analyzed separately, specifically the considerably lower entropy at pos. 104 and 109 in upper layer, are indicative of a lower abundance of two oligotypes in the upper layer, namely “CCCCGCGTGC” (2.13% in undermat, 0.19% in upper layer) and “CCCCGCGGGC” (1.02 vs. 0.21%) (Table S2).
Table S1. OTUs obtained from 16S rRNA V4 iTag sequencing. Read numbers, relative abundance and number of total and abundant dereplicated iTag sequences are stated. Classification are based on RDP classifier.
Table S2. Read counts and relative abundance of the 23 most abundant Roseiflexus-like oligotypes in undermat and upper layer as determined in the combined amplicon dataset (>100 reads total).
References
Albuquerque, L., Rainey, F. A., Nobre, M. F., and da Costa, M. S. (2008). Elioraea tepidiphila gen. nov., sp. nov., a slightly thermophilic member of the Alphaproteobacteria. Int. J. Syst. Evol. Microbiol. 58, 773–778. doi: 10.1099/ijs.0.65294-0
Allewalt, J. P., Bateson, M. M., Revsbech, N. P., Slack, K., and Ward, D. M. (2006). Effect of temperature and light on growth of and photosynthesis by Synechococcus isolates typical of those predominating in the Octopus Spring microbial mat community of Yellowstone National Park. Appl. Environ. Microbiol. 72, 544–550. doi: 10.1128/AEM.72.1.544
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Amann, R. I., Ludwig, W., and Schleifer, K. H. (1995). Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev. 59, 143–169.
Anderson, K. L., Tayne, T. A., and Ward, D. M. (1987). Formation and fate of fermentation products in hot spring cyanobacterial mats. Appl. Environ. Microbiol. 53, 2343–2352.
Aziz, R. K., Bartels, D., Best, A. A., DeJongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75
Bateson, M. M., and Ward, D. M. (1988). Photoexcretion and fate of glycolate in a hot spring cyanobacterial mat. Appl. Environ. Microbiol. 54, 1738–1743.
Bauld, J., and Brock, T. D. (1973). Ecological studies of Chloroflexis, a gliding photosynthetic bacterium. Arch. Mikrobiol. 92, 267–284. doi: 10.1007/BF00409281
Becraft, E. D., Cohan, F. M., Kuehl, M., Jensen, S. I., and Ward, D. M. (2011). Fine-scale distribution patterns of Synechococcus ecological diversity in the microbial mat of Mushroom Spring, Yellowstone National Park. Appl. Env. Microbiol. 77, 7689–7697. doi: 10.1128/AEM.05927-11
Becraft, E. D., Wood, J. M., Rusch, D. B., Kühl, M., Jensen, S. I., Bryant, D. A., et al. (2015). The molecular dimension of microbial species: 1. Ecological distinctions among, and homogeneity within, putative ecotypes of Synechococcus inhabiting the cyanobacterial mat of Mushroom Spring, Yellowstone National Park. Front. Microbiol. 6:590. doi: 10.3389/fmicb.2015.00590
Bhandari, V., and Gupta, R. S. (2014). Molecular signatures for the phylum (class) Thermotogae and a proposal for its division into three orders (Thermotogales, Kosmotogales ord. nov. and Petrotogales ord. nov.) containing four families (Thermotogaceae, Fervidobacteriaceae fam. nov., Kosmotogaceae fam. nov. and Petrotogaceae fam. nov.) and a new genus Pseudothermotoga gen. nov. with five new combinations. Antonie van Leeuwenhoek 105, 143–168. doi: 10.1007/s10482-013-0062-7
Bhatnagar, S., Badger, J. H., Madupu, R., Khouri, H. M., Connor, E. M. O., Robb, F. T., et al. (2015). Genome sequence of the sulfate-reducing thermophilic bacterium Thermodesulfovibrio yellowstonii strain DSM 11347T (Phylum Nitrospirae). Genome Announc. 3, e01489–e01414. doi: 10.1128/genomeA.01489-14
Bhaya, D., Grossman, A. R., Steunou, A.-S., Khuri, N., Cohan, F. M., Hamamura, N., et al. (2007). Population level functional diversity in a microbial community revealed by comparative genomic and metagenomic analyses. ISME J. 1, 703–713. doi: 10.1038/ismej.2007.46
Boomer, S. M., Lodge, D. P., Dutton, B. E., and Pierson, B. (2002). Molecular characterization of novel red green nonsulfur bacteria from five distinct hot spring communities in Yellowstone National Park. Appl. Environ. Microbiol. 68, 346–355. doi: 10.1128/AEM.68.1.346-355.2002
Boomer, S. M., Pierson, B. K., Austinhirst, R., and Castenholz, R. W. (2000). Characterization of novel bacteriochlorophyll a-containing red filaments from alkaline hot springs in Yellowstone National Park. Arch. Microbiol. 174, 152–161. doi: 10.1007/s002030000189
Brock, T. D., and Freeze, H. (1969). Thermus aquaticus gen. nov. and sp. nov., a non-sporulating extreme thermophile. J. Bacteriol. 98, 289–297.
Bryant, D. A., Costas, A. M. G., Maresca, J. A., Chew, A. G. M., Klatt, C. G., Bateson, M. M., et al. (2007). “Candidatus Chloracidobacterium thermophilum”: an aerobic phototrophic Acidobacterium. Science 317, 523–526. doi: 10.1126/science.1143236
Bryant, D. A., and Frigaard, N.-U. (2006). Prokaryotic photosynthesis and phototrophy illuminated. Trends Microbiol. 14, 488–496. doi: 10.1016/j.tim.2006.09.001
Caporaso, J. G., Bittinger, K., Bushman, F. D., et al. (2010b). PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26, 266–267. doi: 10.1093/bioinformatics/btp636
Caporaso, J. G., Kuczynski, J., Stombaugh, J., et al. (2010a). QUIME allows analysis of high-throughput community sequencing data. Nat Methods 7, 335–336. doi: 10.1038/nmeth.f.303.QIIME
Castenholz, R. W. (1969). Thermophilic blue-green algae and the thermal environment. Bacteriol. Rev. 33, 476–504.
Challacombe, J. F., Eichorst, S. A., Hauser, L., Land, M., Xie, G., and Kuske, C. R. (2011). Biological consequences of ancient gene acquisition and duplication in the large genome of “Candidatus Solibacter usitatus” Ellin6076. PLoS ONE 6:e24882. doi: 10.1371/journal.pone.0024882
Chao, A. (1984). Nonparametric estimation of the number of classes in a population. Scand. J. Stat. 11, 265–270.
Choi, A. R., Shi, L., Brown, L. S., and Jung, K.-H. (2014). Cyanobacterial light-driven proton pump, Gloeobacter rhodopsin: complementarity between rhodopsin-based energy production and photosynthesis. PLoS ONE 9:e110643. doi: 10.1371/journal.pone.0110643
Cohan, F. M., and Perry, E. B. (2007). A systematics for discovering the fundamental units of bacterial diversity. Curr. Biol. 17, 373–386. doi: 10.1016/j.cub.2007.03.032
Cole, J. R., Wang, Q., Cardenas, E., Fish, J., Chai, B., Farris, R. J., et al. (2009). The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucl. Acids Res. 37, D141–D145. doi: 10.1093/nar/gkn879
De La Torre, J. R., Walker, C. B., Ingalls, A. E., Könneke, M., and Stahl, D. A. (2008). Cultivation of a thermophilic ammonia oxidizing archaeon synthesizing crenarchaeol. Environ. Microbiol. 10, 810–818. doi: 10.1111/j.1462-2920.2007.01506.x
Dick, G. J., Andersson, A. F., Baker, B. J., Simmons, S. L., Thomas, B. C., Yelton, A. P., et al. (2009). Community-wide analysis of microbial genome sequence signatures. Genome Biol. 10, R85. doi: 10.1186/gb-2009-10-8-r85
Dillon, J. G., Fishbain, S., Miller, S. R., Bebout, B. M., Habicht, K. S., Webb, S. M., et al. (2007). High rates of sulfate reduction in a low-sulfate hot spring microbial mat are driven by a low level of diversity of sulfate-respiring microorganisms. Appl. Environ. Microbiol. 73, 5218–5226. doi: 10.1128/AEM.00357-07
Dodsworth, J. A., Blainey, P. C., Murugapiran, S. K., Wesley, D., Ross, C. A., Tringe, S. G., et al. (2013). Single-cell and metagenomic analyses indicate a fermentative and saccharolytic lifestyle for member of the OP9 lineage. Nat Commun. 4, 1854. doi: 10.1038/ncomms2884
Doemel, W. N., and Brock, T. D. (1977). Structure, growth, and decomposition of laminated algal-bacterial mats in alkaline hot springs. Appl. Environ. Microbiol. 34, 433–452.
Eder, W., and Huber, R. (2002). New isolates and physiological properties of the Aquificales and description of Thermocrinis albus sp. nov. Extremophiles 6, 309–318. doi: 10.1007/s00792-001-0259-y
Farag, I. F., Davis, J. P., Youssef, N. H., and Elshahed, M. S. (2014). Global patterns of abundance, diversity and community structure of the Aminicenantes (Candidate phylum OP8). PLoS ONE 9:e92139. doi: 10.1371/journal.pone.0092139
Fardeau, M.-L., Ollivier, B., Patel, B. K. C., Magot, M., Thomas, P., and Rimbault, A. (1997). Thermotoga hypogea sp. nov., a xylanolytic, thermophilic bacterium from an oil-producing well. Int. J. Syst. Bacteriol. 47, 1013–1019.
Ferris, M. J., Kopczynski, E. D., Bateson, M. M., and Ward, D. M. (1996a). Enrichment culture and microscopy conceal diverse thermophilic Synechococcus populations in a single hot spring mat habitat. Microbiology 62, 1045–1050.
Ferris, M. J., Kuehl, M., Wieland, A., and Ward, D. M. (2003). Cyanobacterial ecotypes in different optical microenvironments of a 68°C hot spring mat community revealed by 16S-23S rRNA internal transcribed spacer region variation. Appl. Environ. Microbiol. 69, 2893–2898. doi: 10.1128/AEM.69.5.2893
Ferris, M. J., Muyzer, G., and Ward, D. M. (1996b). Denaturing gradient gel electrophoresis profiles of 16S rRNA-defined populations inhabiting a hot spring microbial mat community. Appl. Environ. Microbiol. 62, 340–346.
Ferris, M. J., Nold, S. C., Revsbech, N. P., and Ward, D. M. (1997). Population structure and physiological changes within a hot spring microbial mat community following disturbance. Appl. Environ. Microbiol. 63, 1367–1374.
Ferris, M. J., and Ward, D. M. (1997). Seasonal distributions of dominant 16S rRNA-defined populations in a hot spring microbial mat examined by denaturing gradient gel electrophoresis. Appl. Environ. Microbiol. 63, 1375–1381.
Garcia Costas, A. M., Liu, Z., Tomsho, L. P., Schuster, S. C., Ward, D. M., and Bryant, D. A. (2012a). Complete genome of “Candidatus Chloracidobacterium thermophilum”, a chlorophyll-based photoheterotroph belonging to the phylum Acidobacteria. Environ. Microbiol. 14, 177–190. doi: 10.1111/j.1462-2920.2011.02592.x
Garcia Costas, A. M., Tsukatani, Y., Rijpstra, W. I. C., Schouten, S., Welander, P. V., Summons, R. E., et al. (2012b). Identification of the bacteriochlorophylls, carotenoids, quinones, lipids, and hopanoids of “Candidatus Chloracidobacterium thermophilum.” J. Bacteriol. 194, 1158–1168. doi: 10.1128/JB.06421-11
Grégoire, P., Fardeau, M.-L., Joseph, M., Guasco, S., Hamaide, F., Biasutti, S., et al. (2011). Isolation and characterization of Thermanaerothrix daxensis gen. nov., sp. nov., a thermophilic anaerobic bacterium pertaining to the phylum Chloroflexi, isolated from a deep hot aquifer in the Aquitaine Basin. Syst. Appl. Microbiol. 34, 494–497. doi: 10.1016/j.syapm.2011.02.004
Gupta, R., Chander, P., and George, S. (2013). Phylogenetic framework and molecular signatures for the class Chloroflexi and its different clades: proposal for division of the class Chloroflexi class. nov. into the suborder Chloroflexineae subord. nov., consisting of the emended family Oscillochloridaceae and the family Chloroflexaceae fam. nov., and the suborder Roseiflexinae subord. nov., containing the family Roseiflexaceae fam. nov. Antonie van Leeuwenhoek 103, 99–119. doi: 10.1007/s10482-012-9790-3
Henry, E. A., Devereux, R., Maki, J. S., Gilmour, C. C., Woese, C. R., Mandelco, L., et al. (1994). Characterization of a new thermophilic sulfate-reducing bacterium Thermodesulfovibrio yellowstoneii, gen. nov. and sp. nov.: its phylogenetic relationship to Thermodesulfobacterium commune and their origins deep within the bacterial domain. Arch. Microbiol. 161, 62–69.
Hiras, J., Wu, Y.-W., Eichorst, S. A., Simmons, B. A., and Singer, S. W. (2015). Refining the phylum Chlorobi by resolving the phylogeny and metabolic potential of the representative of a deeply branching, uncultivated lineage. ISME J. 9, 1751–7370. doi: 10.1038/ismej.2015.158
Huber, R., Eder, W., Heldwein, S., Huber, H., Rachel, R., Karl, O., et al. (1998). Thermocrinis ruber gen. nov., sp. nov., a pink-filament-forming hyperthermophilic bacterium isolated from Yellowstone National Park. Appl. Environ. Microbiol. 64, 3576–3583.
Hugenholtz, P., Goebel, B. M., and Pace, N. R. (1998a). Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. J. Bacteriol. 180, 4765–4774.
Hugenholtz, P., and Pace, N. R. (1996). Identifying microbial diversity in the natural environment: a molecular phylogenetic approach. Trends Biotechnol. 14, 190–197. doi: 10.1016/0167-7799(96)10025-1
Hugenholtz, P., Pitulle, C., Hershberger, K. L., and Pace, N. R. (1998b). Novel division level bacterial diversity in a Yellowstone hot spring. J. Bacteriol. 180, 366–376.
Iino, T., Mori, K., Uchino, Y., Nakagawa, T., Harayama, S., and Suzuki, K.-I. (2010). Ignavibacterium album gen. nov., sp. nov., a moderately thermophilic anaerobic bacterium isolated from microbial mats at a terrestrial hot spring and proposal of Ignavibacteria classis nov., for a novel lineage at the periphery of green sulfur bacteria. Int. J. Syst. Evol. Microbiol. 69, 1376–1382. doi: 10.1099/ijs.0.012484-0
Im, W.-T., Hu, Z.-Y., Kim, K.-H., Rhee, S.-K., Meng, H., Lee, S.-T., et al. (2012). Description of Fimbriimonas ginsengisoli gen. nov., sp. nov. within the Fimbriimonadia class nov., of the phylum Armatimonadetes. Antonie Van Leeuwenhoek 102, 307–317. doi: 10.1007/s10482-012-9739-6
Jackson, T. J., Ramaley, R. F., and Meinschein, W. G. (1973). Thermomicrobium, a new genus of extremely thermophilic bacteria. Int. J. Syst. Bacteriol. 23, 28–36. doi: 10.1099/00207713-23-1-28
Jensen, S. I., Steunou, A.-S., Bhaya, D., Kühl, M., and Grossman, A. R. (2011). In situ dynamics of O2, pH and cyanobacterial transcripts associated with CCM, photosynthesis and detoxification of ROS. ISME J. 5, 317–328. doi: 10.1038/ismej.2010.131
Kadnikov, V. V., Mardanov, A. V., Podosokorskaya, O. A., Gavrilov, S. N., Kublanov, I. V., Beletsky, A. V., et al. (2013). Genomic analysis of Melioribacter roseus, facultatively anaerobic organotrophic bacterium representing a novel deep lineage within Bacteriodetes/Chlorobi group. PLoS ONE 8:e53047. doi: 10.1371/journal.pone.0053047
Kerepesi, C., Bánky, D., and Grolmusz, V. (2014). AmphoraNet: the webserver implementation of the AMPHORA2 metagenomic workflow suite. Gene 533, 538–540. doi: 10.1016/j.gene.2013.10.015
Kim, Y.-M., Nowack, S., Olsen, M. T., Becraft, E. D., Wood, J. M., Thiel, V., et al. (2015). Diel metabolomics analysis of a hot spring chlorophototrophic microbial mat leads to new hypotheses of community member metabolisms. Front. Microbiol. 6:209. doi: 10.3389/fmicb.2015.00209
Kimura, H., Nashimoto, H., Shimizu, M., Hattori, S., Yamada, K., Koba, K., et al. (2010). Microbial methane production in deep aquifer associated with the accretionary prism in Japan. ISME J. 4, 531–541. doi: 10.1038/ismej.2009.132
Klatt, C. G., Bryant, D. A., and Ward, D. M. (2007). Comparative genomics provides evidence for the 3-hydroxypropionate autotrophic pathway in filamentous anoxygenic phototrophic bacteria and in hot spring microbial mats. Environ. Microbiol. 9, 2067–2078. doi: 10.1111/j.1462-2920.2007.01323.x
Klatt, C. G., Inskeep, W. P., Herrgard, M. J., Jay, Z. J., Rusch, D. B., Tringe, S. G., et al. (2013a). Community structure and function of high-temperature chlorophototrophic microbial mats inhabiting diverse geothermal environments. Front. Microbiol. 4:106. doi: 10.3389/fmicb.2013.00106
Klatt, C. G., Liu, Z., Ludwig, M., Kühl, M., Jensen, S. I., Bryant, D. A., et al. (2013b). Temporal metatranscriptomic patterning in phototrophic Chloroflexi inhabiting a microbial mat in a geothermal spring. ISME J. 7, 1775–1789. doi: 10.1038/ismej.2013.52
Klatt, C. G., Wood, J. M., Rusch, D. B., Bateson, M. M., Hamamura, N., Heidelberg, J. F., et al. (2011). Community ecology of hot spring cyanobacterial mats: predominant populations and their functional potential. ISME J. 5, 1262–1278. doi: 10.1038/ismej.2011.73
Koeppel, A. F., and Wu, M. (2013). Surprisingly extensive mixed phylogenetic and ecological signals among bacterial Operational Taxonomic Units. Nucl. Acids Res. 41, 5175–5188. doi: 10.1093/nar/gkt241
Lau, M. C. Y., Aitchison, J. C., and Pointing, S. B. (2009). Bacterial community composition in thermophilic microbial mats from five hot springs in central Tibet. Extremophiles 13, 139–149. doi: 10.1007/s00792-008-0205-3
Lee, K. C.-Y., Dunfield, P. F., Morgan, X. C., Crowe, M. A., Houghton, K. M., Vyssotski, M., et al. (2011). Chthonomonas calidirosea gen. nov., sp. nov., an aerobic, pigmented, thermophilic micro-organism of a novel bacterial class, Chthonomonadetes classis nov., of the newly described phylum Armatimonadetes originally designated candidate candiate division OP1. Int. J. Syst. Evol. Microbiol. 61, 2482–2490. doi: 10.1099/ijs.0.027235-0
Lee, K. C.-Y., Herbold, W., Dunfield, P. F., Morgan, X. C., McDonald, I. R., and Stott, M. B. (2013). Phylogenetic delineation of the novel phylum Armatimonadetes (former candidate division OP10) and definition of two novel candidate divisions. Appl. Environ. Microbiol. 79, 2484–2487. doi: 10.1128/AEM.03333-12
Liu, Z., Klatt, C. G., Ludwig, M., Rusch, D. B., Jensen, S. I., Kühl, M., et al. (2012). “Candidatus Thermochlorobacter aerophilum:” an aerobic chlorophotoheterotrophic member of the phylum Chlorobi defined by metagenomics and metatranscriptomics. ISME J. 6, 1869–1882. doi: 10.1038/ismej.2012.24
Liu, Z., Klatt, C. G., Wood, J. M., Rusch, D. B., Ludwig, M., Wittekindt, N., et al. (2011). Metatranscriptomic analyses of chlorophototrophs of a hot-spring microbial mat. ISME J. 5, 1279–1290. doi: 10.1038/ismej.2011.37
Ludwig, W., Strunk, O., Westram, R., Richter, L., Meier, H., Yadhukumar, et al. (2004). ARB: a software environment for sequence data. Nucl. Acids Res. 32, 1363–1371. doi: 10.1093/nar/gkh293
Markowitz, V. M., Chen, I. M. A., Chu, K., Szeto, E., Palaniappan, K., Pillay, M., et al. (2014). IMG/M 4 version of the integrated metagenome comparative analysis system. Nucleic Acids Res. 42, 568–573. doi: 10.1093/nar/gkt919
Melendrez, M. C., Lange, R. K., Cohan, F. M., and Ward, D. M. (2011). Influence of molecular resolution on sequence-based discovery of ecological diversity among Synechococcus populations in an alkaline siliceous hot spring microbial mat. Appl. Environ. Microbiol. 77, 1359–1367. doi: 10.1128/AEM.02032-10
Meyer-Dombard, D. R., Swingley, W., Raymond, J., Havig, J., Shock, E. L., and Summons, R. E. (2011). Hydrothermal ecotones and streamer biofilm communities in the Lower Geyser Basin, Yellowstone National Park. Environ. Microbiol. 13, 2216–2231. doi: 10.1111/j.1462-2920.2011.02476.x
Nobu, M. K., Dodsworth, J. A., Murugapiran, S. K., Rinke, C., Gies, E. A., Webster, G., et al. (2016). Phylogeny and physiology of candidate phylum “Atribacteria” (OP9/JS1) inferred from cultivation-independent genomics. ISME J. 10, 273–286. doi: 10.1038/ismej.2015.97
Nold, S. C., and Ward, D. M. (1996). Photosynthate partitioning and fermentation in hot spring microbial mat communities. Appl. Environ. Microbiol. 62, 4598–4607.
Nowack, S., Olsen, M. T., Schaible, G. A., Becraft, E. D., Shen, G., Klapper, I., et al. (2015). The molecular dimension of microbial species: 2. Synechococcus strains representative of putative ecotypes inhabiting different depths in the Mushroom Spring microbial mat exhibit different adaptive and acclimative responses to light. Front. Microbiol. 6:626. doi: 10.3389/fmicb.2015.00626
Nowack, S. P. (2014). Niche Character in a Temporally Varying Environment. Ph. D. thesis, Montana State University. doi: 10.1017/CBO9781107415324.004
Nübel, U., Bateson, M. M., Vandieken, V., Kühl, M., and Ward, D. M. (2002). Microscopic examination of distribution and phenotypic properties of phylogenetically diverse Chloroflexaceae-related bacteria in hot spring microbial mats. Appl. Environ. Microbiol. 68, 4593–4603. doi: 10.1128/AEM.68.9.4593
Nunoura, T., Hirai, M., Miyazaki, M., Kazama, H., Makita, H., Hirayama, H., et al. (2013). Isolation and characterization of a thermophilic, obligately anaerobic and heterotrophic marine Chloroflexi bacterium from a Chloroflexi-dominated microbial community associated with a Japanese shallow hydrothermal system, and proposal for Thermomarinilinea lacunofontalis gen. nov., sp. nov. Microbes Environ. 28, 228–235. doi: 10.1264/jsme2.ME12193
Nunoura, T., Takaki, Y., Kakuta, J., Nishi, S., Sugahara, J., Kazama, H., et al. (2011). Insights into the evolution of Archaea and eukaryotic protein modifier systems revealed by the genome of a novel archaeal group. Nucl. Acids Res. 39, 3204–3223. doi: 10.1093/nar/gkq1228
Olsen, G. J., Lane, D. J., Giovannoni, S. J., Pace, N. R., and Stahl, D. A. (1986). Microbial ecology and evolution: a ribosomal RNA approach. Annu. Rev. Microbiol. 40, 337–365.
Olsen, M. T., Nowack, S., Wood, J. M., Becraft, E. D., LaButti, K., Lipzen, A., et al. (2015). The molecular dimension of microbial species: 3. Comparative genomics of Synechococcus strains with different light responses and in situ diel transcription patterns of associated putative ecotypes in the Mushroom Spring microbial mat. Front. Microbiol. 6:604. doi: 10.3389/fmicb.2015.00604
Overbeek, R., Olson, R., Pusch, G. D., Olsen, G. J., Davis, J. J., Disz, T., et al. (2014). The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 42, 1–9. doi: 10.1093/nar/gkt1226
Pace, N. R. (2009). Mapping the tree of life: progress and prospects. Microbiol. Mol. Biol. Rev. 73, 565–576. doi: 10.1128/MMBR.00033-09
Ramsing, N. B., Ferris, M. J., and Ward, D. M. (2000). Highly ordered vertical structure of Synechococcus populations within the one-millimeter-thick photic zone of a hot spring cyanobacterial mat. Appl. Environ. Microbiol. 66, 1038–1049. doi: 10.1128/AEM.66.3.1038-1049.2000
Revsbech, N. P., and Ward, D. M. (1984). Microelectrode studies of interstitial water chemistry and photosynthetic activity in a hot spring microbial mat. Appl. Environ. Microbiol. 48, 270–275.
Reysenbach, A., Longnecker, K., and Kirshtein, J. (2000). Novel bacterial and archaeal lineages from an in situ growth chamber deployed at a Mid-Atlantic Ridge hydrothermal vent. Appl. Environ. Microbiol. 66, 3798–3806. doi: 10.1128/AEM.66.9.3798-3806.2000
Reysenbach, A., Wickham, G. S., and Pace, N. R. (1994). Phylogenetic analysis of the hyperthermophilic pink filament community in Octopus Spring, Yellowstone National Park. Appl. Environ. Microbiol. 60, 2113–2119.
Sandbeck, K. A., and Ward, D. M. (1981). Fate of immediate methane precursors in low-sulfate, hot-spring algal-bacterial mats. Appl. Environ. Microbiol. 41, 775–782.
Sandbeck, K. A., and Ward, D. M. (1982). Temperature adaptations in the terminal processes of anaerobic decomposition of yellowstone national park and icelandic hot spring microbial mats. Appl. Environ. Microbiol. 44, 844–851.
Schloss, P. D., and Handelsman, J. (2005). Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl. Environ. Microbiol. 71, 1501–1506. doi: 10.1128/AEM.71.3.1501
Sekiguchi, Y., Yamada, T., Hanada, S., Ohashi, A., Harada, H., and Kamagata, Y. (2003). Anaerolinea thermophila gen. nov., sp. nov. and Caldilinea aerophila gen. nov., sp. nov., novel filamentous thermophiles that represent a previously uncultured lineage of the domain Bacteria at the subphylum level. Int. J. Syst. Evol. Microbiol. 53, 1843–1851. doi: 10.1099/ijs.0.02699-0
Slobodkina, G. B., Kovaleva, O. L., Miroshnichenko, M. L., Slobodkin, A. I., Kolganova, T. V., Novikov, A. A., et al. (2014). Thermogutta terrifontis gen. nov., sp. nov. and Thermogutta hypogea sp. nov., thermophilic anaerobic representatives of the phylum Planctomycetes. Int. J. Syst. Evol. Microbiol. 65, 760–765. doi: 10.1099/ijs.0.000009
Stackebrandt, E., and Goebel, B. M. (1994). Taxonomic Note: a place for DNA-DNA reassociation and 16S rRNA sequence analysis in the present species definition in bacteriology. Int. J. Syst. Bacteriol. 44, 846–849. doi: 10.1099/00207713-44-4-846
Steunou, A.-S., Jensen, S. I., Brecht, E., Becraft, E. D., Bateson, M. M., Kilian, O., et al. (2008). Regulation of nif gene expression and the energetics of N2 fixation over the diel cycle in a hot spring microbial mat. ISME J. 2, 364–378. doi: 10.1038/ismej.2007.117
Tamaki, H., Tanaka, Y., Matsuzawa, H., Muramatsu, M., Meng, X.-Y., Hanada, S., et al. (2011). Armatimonas rosea gen. nov., sp. nov., of a novel bacterial phylum, Armatimonadetes phyl. nov., formally called the candidate phylum OP10. Int. J. Syst. Evol. Microbiol. 61, 1442–1447. doi: 10.1099/ijs.0.025643-0
Tank, M., and Bryant, D. A. (2015a). Chloracidobacterium thermophilum gen. nov., sp. nov.: an anoxygenic microaerophilic chlorophotoheterotrophic acidobacterium. Int. J. Syst. Evol. Microbiol. 65, 1426–1430. doi: 10.1099/ijs.0.000113
Tank, M., and Bryant, D. A. (2015b). Nutrient requirements and growth physiology of the photoheterotrophic acidobacterium, Chloracidobacterium thermophilum. Front. Microbiol. 6:226. doi: 10.3389/fmicb.2015.00226
Tank, M., Thiel, V., Ward, D. M., and Bryant, D. A. (in press). “A panoply of phototrophs: a photomicrographic overview of chlorophototrophs found in the microbial mats of alkaline siliceous hot springs in Yellowstone National Park, WY, USA,” in Modern Topics in the Phototrophic Prokaryotes: Environmental Applied Aspects, ed P. C. Hallenbeck (Berlin: Springer).
Thiel, V., Hamilton, T. L., Tomsho, L. P., Burhans, R., Gay, S. E., Ramaley, R. F., et al. (2014a). Draft genome sequence of the moderately thermophilic bacterium Schleiferia thermophila strain Yellowstone (Bacteroidetes). Genome Announc. 2, e00860–e00814. doi: 10.1128/genomeA.00860-14
Thiel, V., Hamilton, T. L., Tomsho, L. P., Burhans, R., Gay, S. E., Schuster, S. C., et al. (2014b). Draft genome sequence of a sulfide-oxidizing, autotrophic filamentous anoxygenic phototrophic bacterium, Chloroflexus sp. strain MS-G (Chloroflexi). Genome Announc. 2, e00872–e00814. doi: 10.1128/genomeA.00872-14
Thiel, V., Tomsho, L. P., Burhans, R., Gay, S. E., Schuster, S. C., Ward, D. M., et al. (2015). Draft genome sequence of the Deinococcus-Thermus bacterium Meiothermus ruber strain A. Genome Announc. 3, e00202–e00215. doi: 10.1128/genomeA.00202-15
van der Meer, M. T. J., Klatt, C. G., Wood, J., Bryant, D. A., Bateson, M. M., Lammerts, L., et al. (2010). Cultivation and genomic, nutritional, and lipid biomarker characterization of Roseiflexus strains closely related to predominant in situ populations inhabiting Yellowstone hot spring microbial mats. J. Bacteriol. 192, 3033–3042. doi: 10.1128/JB.01610-09
van der Meer, M. T. J., Schouten, S., Damsté, J. S. S., and Ward, D. M. (2007). Impact of carbon metabolism on 13C signatures of cyanobacteria and green non-sulfur-like bacteria inhabiting a microbial mat from an alkaline siliceous hot spring in Yellowstone National Park (USA). Environ. Microbiol. 9, 482–491. doi: 10.1111/j.1462-2920.2006.01165.x
van der Meer, M. T. J., Schouten, S., Jaap, S., Damsté, S., Leeuw, J. W., De Ward, D. M., et al. (2003). Compound-specific isotopic fractionation patterns suggest different carbon metabolisms among Chloroflexus-like bacteria in hot-spring microbial mats. Appl. Environ. Microbiol. 69, 6000–6006. doi: 10.1128/AEM.69.10.6000-6006.2003
van der Meer, M. T. J., Schouten, S., Mary, M., Nübel, U., Wieland, A., Kühl, M., et al. (2005). Diel variations in carbon metabolism by green nonsulfur-like bacteria in alkaline siliceous hot spring microbial mats from Yellowstone National Park. Appl. Environ. Microbiol. 71, 3978–3986. doi: 10.1128/AEM.71.7.3978-3986.2005
Venkata Ramana, V., Sasikala, C., Takaichi, S., and Ramana, C. V. (2010). Roseomonas aestuarii sp. nov., a bacteriochlorophyll-a containing alphaproteobacterium isolated from an estuarine habitat of India. Syst. Appl. Microbiol. 33, 198–203. doi: 10.1016/j.syapm.2009.09.004
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/AEM.00062-07
Ward, D. M. (1978). Thermophilic methanogenesis in a hot-spring algal-bacterial mat (71 to 30 degrees C). Appl. Environ. Microbiol. 35, 1019–1026.
Ward, D. M., Bateson, M. M., Weller, R., and Ruff-Roberts, A. L. (1992). “Ribosomal RNA analysis of microorganisms as they occur in nature,” in Advances in Microbial Ecology, ed K. C. Marshall (New York, NY: Springer US), 219–286.
Ward, D. M., Brassell, S. C., and Eglinton, G. (1985). Archaebacterial lipids in hot-spring microbial mats. Nature 318, 162–163. doi: 10.1038/314731a0
Ward, D. M., Castenholz, R. W., and Miller, S. R. (2012). “Ecology of cyanobacteria II: cyanobacteria in geothermal habitats,” in Ecology of Cyanobacteria II, ed B. A. Whitton (Dordrecht: Springer), 39–63.
Ward, D. M., and Cohan, F. M. (2005). “Microbial diversity in hot spring cyanobacterial mats: pattern and prediction,” in Geothermal Biology and Geochemistry in Yellowstone National Park, eds W. P. Inskeep and T. McDermott (Bozeman: Thermal Biology Institute), 185–202.
Ward, D. M., Ferris, M. J., Nold, S. C., and Bateson, M. M. (1998). A natural view of microbial biodiversity within hot spring cyanobacterial mat communities. Microbiol. Mol. Biol. Rev. 62, 1353–1370.
Ward, D. M., Santegoeds, C. M., Nold, S. C., Ramsing, N. B., Ferris, M. J., and Bateson, M. M. (1997). Biodiversity within hot spring microbial mat communities: molecular monitoring of enrichment cultures. Antonie van Leeuwenhoek 71, 143–150. doi: 10.1023/A:1000131426164
Ward, D. M. J., Shiea, J., Zeng, Y. B., Dobson, G., Brassell, S., and Eglinton, G. (1989). “Lipid biochemical markers and the composition of microbial mats,” in Microbial Mats: Physiological Ecology of Benthic Microbial Communities, eds Y. Cohen and E. Rosenburg (Washington, DC: American Society For Microbiology), 439–454.
Ward, D. M., Weller, R., and Bateson, M. M. (1990). 16S rRNA sequences reveal numerous uncultured microorganisms in a natural community. Nature 346, 183–187. doi: 10.1038/346183a0
Weller, R., Bateson, M. M., Heimbuch, B. K., Kopczynski, E. D., and Ward, D. M. (1992). Uncultivated cyanobacteria, Chloroflexus-like inhabitants, and spirochete-like inhabitants of a hot spring microbial mat. Appl. Environ. Microbiol. 58, 3964–3969.
Keywords: hot spring, microbial community, microbial diversity, extreme environments, phototrophic bacteria
Citation: Thiel V, Wood JM, Olsen MT, Tank M, Klatt CG, Ward DM and Bryant DA (2016) The Dark Side of the Mushroom Spring Microbial Mat: Life in the Shadow of Chlorophototrophs. I. Microbial Diversity Based on 16S rRNA Gene Amplicons and Metagenomic Sequencing. Front. Microbiol. 7:919. doi: 10.3389/fmicb.2016.00919
Received: 07 March 2016; Accepted: 27 May 2016;
Published: 17 June 2016.
Edited by:
Anna-Louise Reysenbach, Portland State University, USAReviewed by:
Charles K. Lee, University of Waikato, New ZealandAnirban Chakraborty, University of Calgary, Canada
Wesley Douglas Swingley, Northern Illinois University, USA
Copyright © 2016 Thiel, Wood, Olsen, Tank, Klatt, Ward and Bryant. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vera Thiel, dnRoaWVsQHRtdS5hYy5qcA==;
Donald A. Bryant, ZGFiMTRAcHN1LmVkdQ==
†Present Address: Vera Thiel and Marcus Tank, Department of Biological Sciences, Tokyo Metropolitan University, Hachioji, Japan