 Xinpeng Tian1†
Xinpeng Tian1† Zhewen Zhang2†
Zhewen Zhang2† Tingting Yang2,3†Meili Chen2Jie Li1Fei Chen2Jin Yang4Wenjie Li4
Tingting Yang2,3†Meili Chen2Jie Li1Fei Chen2Jin Yang4Wenjie Li4 Bing Zhang4
Bing Zhang4 Zhang Zhang2Jiayan Wu2Changsheng Zhang1
Zhang Zhang2Jiayan Wu2Changsheng Zhang1 Lijuan Long1*
Lijuan Long1* Jingfa Xiao2*
Jingfa Xiao2*- 1Key Laboratory of Tropical Marine Bio-resources and Ecology and Guangdong Key Laboratory of Marine Materia Medica, South China Sea Institute of Oceanology – Chinese Academy of Sciences, Guangzhou, China
- 2Key Laboratory of Genome Sciences and Information, Beijing Institute of Genomics – Chinese Academy of Sciences, Beijing, China
- 3University of Chinese Academy of Sciences, Beijing, China
- 4Core Genomic Facility, Beijing Institute of Genomics – Chinese Academy of Sciences, Beijing, China
Over 200 genomes of streptomycete strains that were isolated from various environments are available from the NCBI. However, little is known about the characteristics that are linked to marine adaptation in marine-derived streptomycetes. The particularity and complexity of the marine environment suggest that marine streptomycetes are genetically diverse. Here, we sequenced nine strains from the Streptomyces genus that were isolated from different longitudes, latitudes, and depths of the South China Sea. Then we compared these strains to 22 NCBI downloaded streptomycete strains. Thirty-one streptomycete strains are clearly grouped into a marine-derived subgroup and multiple source subgroup-based phylogenetic tree. The phylogenetic analyses have revealed the dynamic process underlying streptomycete genome evolution, and lateral gene transfer is an important driving force during the process. Pan-genomics analyses have revealed that streptomycetes have an open pan-genome, which reflects the diversity of these streptomycetes and guarantees the species a quick and economical response to diverse environments. Functional and comparative genomics analyses indicate that the marine-derived streptomycetes subgroup possesses some common characteristics of marine adaptation. Our findings have expanded our knowledge of how ocean isolates of streptomycete strains adapt to marine environments. The availability of streptomycete genomes from the South China Sea will be beneficial for further analysis on marine streptomycetes and will enrich the South China Sea’s genetic data sources.
Introduction
The specific environment conditions of the ocean, especially its high hydrostatic pressure, low temperature, and oligotrophic deep-sea (water depths greater than 1,000 m) conditions, have affected marine bacteria in various respects, forcing them to evolve special features to adapt to the unique deep-sea environment (Li et al., 2011; Qin et al., 2011, 2014). In general, the genomes of cultured deep-sea bacteria contain more transposable and phage-related elements and larger intergenic spacers than those of surface bacteria (Ivars-Martinez et al., 2008). Deep-sea bacteria also have more genomic islands than other bacteria that confer specific features, such as drug and heavy metal resistance (Qin et al., 2011). Moreover, genes that are important for cold, pressure, and oligotrophic adaptations, such as membrane unsaturation genes and signal transduction genes, are better represented in deep-sea bacteria than in other bacteria (Eloe et al., 2008; Qin et al., 2011). However, the genetic bases for marine adaptation are still unknown. Considering the high diversity of deep-sea bacteria (Qin et al., 2011), more genomic sequences of representative bacterial strains must be analyzed to characterize the mechanism of marine environmental adaptation.
Pan-genomics provide a new method for studying bacterial species diversity, evolution, adaptability, population structures and others (Medini et al., 2005; Muzzi et al., 2007; Zhang et al., 2011; Qin et al., 2014; Richards et al., 2014; Vernikos et al., 2015; Xiao et al., 2015). With the development of sequencing technology and sharp decreases in sequencing costs and time, more and more bacterial genomes are now openly available. This availability has made pan-genomics analysis possible for many strains at the species/genus level (Qin et al., 2014). A pan-genomic analysis of seven Salmonella strains provides insights into their genomic variations and clarifies their genomic evolution profile (Liang et al., 2012). A study on 11 marine Glaciecola strains revealed that the genus has an open pan-genome and contains some common genomic features related to cold adaptation, such as glycine betaine and exopolysaccharide synthesis genes as well as genes that encode cold-shock proteins and tRNA-dihydrouridine synthase (Qin et al., 2014). Therefore, with the accumulation of bacterial genomes, pan-genomics can be a very good method for exploring the phylogenetic relations and environmental adaptation mechanisms of bacterial genera.
Streptomycetes are a major branch of gram-positive bacteria with G+C contents generally ranging between 66 and 74% (Hopwood, 2006; Kämpfer, 2006). Most of these bacteria prefer neutral to alkaline soils as a natural habitat, and like other soil bacteria, they can also co-exist with earthworms or other arthropods within intestinal tracts or other body parts (Hulcr et al., 2011). Rivers, lakes, and marine environments, and especially the sediments of these environments, are also streptomycete habitats (Kämpfer, 2006). Although, streptomycetes are well-known for producing antibiotics that are used to treat bacterial, mycobacterial, fungal, and parasitic infections, some of them infect plants or humans and cause disease, such as potato scab and human mycetoma (Dunne et al., 1998; Loria, 2007). In comparison with streptomycetes from other environments, marine streptomycetes can produce a variety of novel active materials, and they have some different pathways (Li et al., 2011; Lee et al., 2014; Barakat and Beltagy, 2015) resulting from the uniquely high hydrostatic pressure, low temperature, and oligotrophy of the marine environment. However, the phylogenetic relations and genome diversity of streptomycetes between marine and other environments are still unknown.
In this study, we sequenced nine Streptomyces strains that were isolated from different longitudes, latitudes, and depths of the South China Sea. We compared them with the genomes of 22 NCBI downloaded streptomycete strains. The phylogenetic and pan-genomic analyses of the 31 strains reveal the evolutional relations of streptomycetes from different environments, and they also reveal streptomycete adaptation mechanisms to marine environments.
Materials and Methods
Bacterial Strains
Nine streptomycete strains (which belonged to four species, namely S. abyssalis, S. oceani, S. qinglanensis, and S. nanshensis) were isolated from different longitudes, latitudes, and depths of marine sediments or gorgonians in the South China Sea (detailed information is listed in Supplementary Table S1). Among these strains, S. abyssalis 10389, S. abyssalis 10390, and S. nanshensis 10429 were isolated from unidentified gorgonians, and others were separated from marine sediment. The sediment isolates were all taken from deep-sea environments (deeper than 500 m). Moreover, Streptomycetes oceani 02100 can survive only in medium that is prepared with seawater instead of distilled water, and the other strains can live with or without seawater.
Biomass was obtained by cultivating bacteria in modified ISP 2 broth (at 28°C, 1 week, 150 rpm). The genomic DNA extraction and purification of the representative strains were performed as described by Orsini and Romano-Spica (2001).
In addition, 22 genomes from Streptomyces genera were downloaded from the NCBI ftp (Bacteria and Bacteria_DRAFT parts in http://ftp.ncbi.nlm.nih.gov/genomes). They are all draft genomes except those of S. fulvissimus DSM 40593 and S. griseus NBRC 13350. These strains were separated from different habitats, such as soils, marine areas, plants, insect-associated substrate, humans, and so on (basic information for the 22 strains is listed in Supplementary Table S2).
Genome Sequencing and Assembly
Paired-end and mate-pair libraries with insert sizes of 500 bp, 1–3 and 3–5 kb were constructed for nine streptomycete strains that were isolated from the South China Sea. All the libraries were sequenced using Illumina HiSeq2000 with a sequencing read length of 101 bp. The sequencing data were assembled with SOAPdenovo, version 2.04 (Luo et al., 2012). The total read lengths (after quality filtration) of each library, genome size, and average genome coverage of each strain are listed in Supplementary Table S3. The final genome assembly results are listed in Supplementary Table S4.
Genome Annotation and Analysis
The tRNA genes were predicted by tRNAscan-SE (Lowe and Eddy, 1997). The rRNA genes were identified by RNAmmer (Lagesen et al., 2007), and the coding sequences (CDSs) were found with Prodigal, version 2.60 (Hyatt et al., 2010). The predicted genes were annotated by performing a BLAST (Altschul et al., 1990) search against databases of non-redundant proteins from the NCBI and COG (Tatusov et al., 2001; E-value 1e-5). Genes that were annotated as transporters and hypothetical proteins were selected, and they are regarded as transporter candidates. For these candidates, transporter predictions were made by performing a BLAST search against sequences that were downloaded from the TransportDB (Ren et al., 2007), which is widely used for comparative analyses of transport capabilities in prokaryotes (Ren and Paulsen, 2005, 2007). The metabolic pathways of streptomycetes were determined using the KEGG Automatic Annotation Server (KAAS; Moriya et al., 2007)1 with the bi-directional best hit (BBH) assignment method.
Moreover, the comparison of general genomic features, such as the genome size, G+C content and CDSs, were conducted with R. A principal component analysis (PCA) of these features was performed with R package psych (Revelle, 2015). Clustered regularly interspaced short palindromic repeats (CRISPRs) were found with pilercr1.06 (Edgar, 2007).
Pan-genomics Analyses
All the genes from the 31 streptomycetes strains were delineated into clusters with putative shared homologies by MP method as implemented in the pan-genome analysis pipeline (PGAP) with a 50% cut-off for protein sequence identity. Pan-genome characteristic curves were depicted by PanGP (Zhao et al., 2014) with DG sampling algorithms. The COG functional enrichment of core genes was analyzed by PGAP with the parameter “–function.” Gene clusters that were shared among all strains and contained only single gene copies for each strain were called single copy core genes. The pairwise sequence alignment of all single copy core genes was performed with the BLAST program on the basis of an E-value cut-off of 1e-5.
Single Copy Core Gene Identity Comparison
For this comparison, the single copy genes in the 31 streptomycete strains were first identified. The identities of each pair of genes between two independent strains were then determined by BLAST. A plot of the identities density of all pairs of genes between two independent strains was created with R.
Phylogenetic Analyses
For the phylogenetic analysis, Catenulispora acidiphila DSM 44928 was selected as the outgroup. The gene clusters of the 32 strains were identified by PGAP using the MP method. The single copy core gene sequences from the 32 strains that belonged to one cluster were aligned with MAFFT (Katoh et al., 2002). The recombination for genes in these clusters was assessed using PhiPack [Bruen et al., 2006; which calculates the p-values for three individual methods, namely the neighbor similarity score (NSS), Maxχ2 and Phi] and GENECONV (Padidam et al., 1999). Recombination was inferred for p-values of less than 0.05. Thirty-three gene clusters (3.7%) that showed evidence of recombination for all four methods were removed according to the comparison analyzed by Richards et al. (2014). The rooted maximum likelihood (ML) species tree was constructed from a gene alignment concatenation of the 853 clusters with PhyML v3.0 (Guindon et al., 2010). The phylogenetic analysis was performed with the GTR + I + G substitution model, which was determined to have the best fit for the data when using jmodeltest-2.1.6 (Darriba et al., 2012). The rooted species tree based on 16S rRNA was also constructed with PhyML v3.0. For both phylogenetic trees, branch supports were provided by generating 100 bootstrap replicates.
Gene Gain and Loss
The gene gain/loss on the species tree was assessed by parsimony-based gene-tree species-tree reconciliation as implemented in the AnGST program. The most parsimonious reconciliation was obtained by inferring a minimum set of evolutionary events [gene loss, gene duplication, speciation, lateral gene transfer (LGT), and gene birth or genesis] for a gene tree of every gene cluster containing three or more sequences, when using the default event penalty values of AnGST (LGT = 3.0, duplication = 2.0, loss = 1.0, and speciation = 0). We did not constrain the time-consistent reconciliation, allowing gene transfer events to occur between any two lineages.
The gene trees for all gene clusters containing three or more genes were constructed using PhyML v3.0 with the GTR + I + G substitution model. One hundred bootstrap replicates for every gene tree were provided to AnGST to account for phylogenetic uncertainty in the gene tree. Four gene clusters that contained particularly large number of sequences (more than 900) were excluded from the AnGST analysis. Genes in clusters containing only one gene were considered a birth in every genome. For gene clusters containing two genes, we explained the evolutionary history of these genes by following the method mentioned by Richards et al. (2014).
Results
General Genomic Information on Streptomycetes
Constructing genomic maps of streptomycetes is a great challenge as a result of the high GC contents, linear chromosomes, unstable chromosome structures and large genome sizes of these bacteria. In this study, libraries with various insert fragment lengths were used for genome sequencing and assembly, and the sequencing depths of these nine strains were all approximately 300×. We used two software of Velvet (Zerbino and Birney, 2008) and SOAPdenovo2 (Luo et al., 2012) to obtain optimal assembly results. After the comparison of the N50 scaffolds and Contigs from two software, we chose SOAPdenovo2 to conduct the genome assembly. The scaffold numbers ranged from 22 to 2,310. The scaffold N50 were all above 14 kb (Supplementary Table S4).
The general genome features of the nine newly sequenced streptomycete strains and 22 previously sequenced streptomycete strains are summarized in Table 1. Twenty-two previously sequenced streptomycete strains were selected through the phylogenetic analyses of 136 streptomycete strain genomes (127 strain genomes were downloaded from NCBI and nine strain genomes were sequenced in this study) with Co-phylog (Yi and Jin, 2013; Supplementary Figure S1). The G+C contents of 31 streptomycete strains were all above 70% (70.1–73.1%). The genomes of S. fulvissimus DSM 40593 and S. griseus NBRC 13350 were 7.91 and 8.55 Mb in size, respectively. The others were draft genomes with sizes ranging from 5.93 to 10.27 Mb. The predicted protein CDSs ranged from 5,163 to 9,122. Here, we could conclude that the streptomycete genome size and CDSs number have great variability. The CDSs number was usually consistent with the genome size (R = 0.84; Figure 1A). Most streptomycete strains derived from marine environments had smaller genome sizes, but more than half of them (11/20, and 20 strains are isolated from marine environments) possessed a slightly higher GC content (Figure 1A). Streptomyces sp. CNS606 had the smallest genome size (5.93 Mb) but the highest G+C content (73.1%; Figure 1A). We also tested the continuous distributions of these three general features in marine-derived streptomycete strains and strains from other sources by two-tailed Kolmogorov–Smirnov test. The CDSs numbers and genome sizes had significantly different continuous distributions (with p-values of 0.003 and 0.006, respectively). However, the continuous distributions of the G+C content were not remarkable (p-value 0.676). According to the PCA, the genome size, CDS number and G+C content were divided into two principal components (Figure 1B). The majority of the marine-derived streptomycete strains can be divided from other sourced strains by the first dimension (genome size and CDS number), with ellipses on 95% confidence intervals of environment classifications (PCA1 61% variance, PCA2 34% variance).
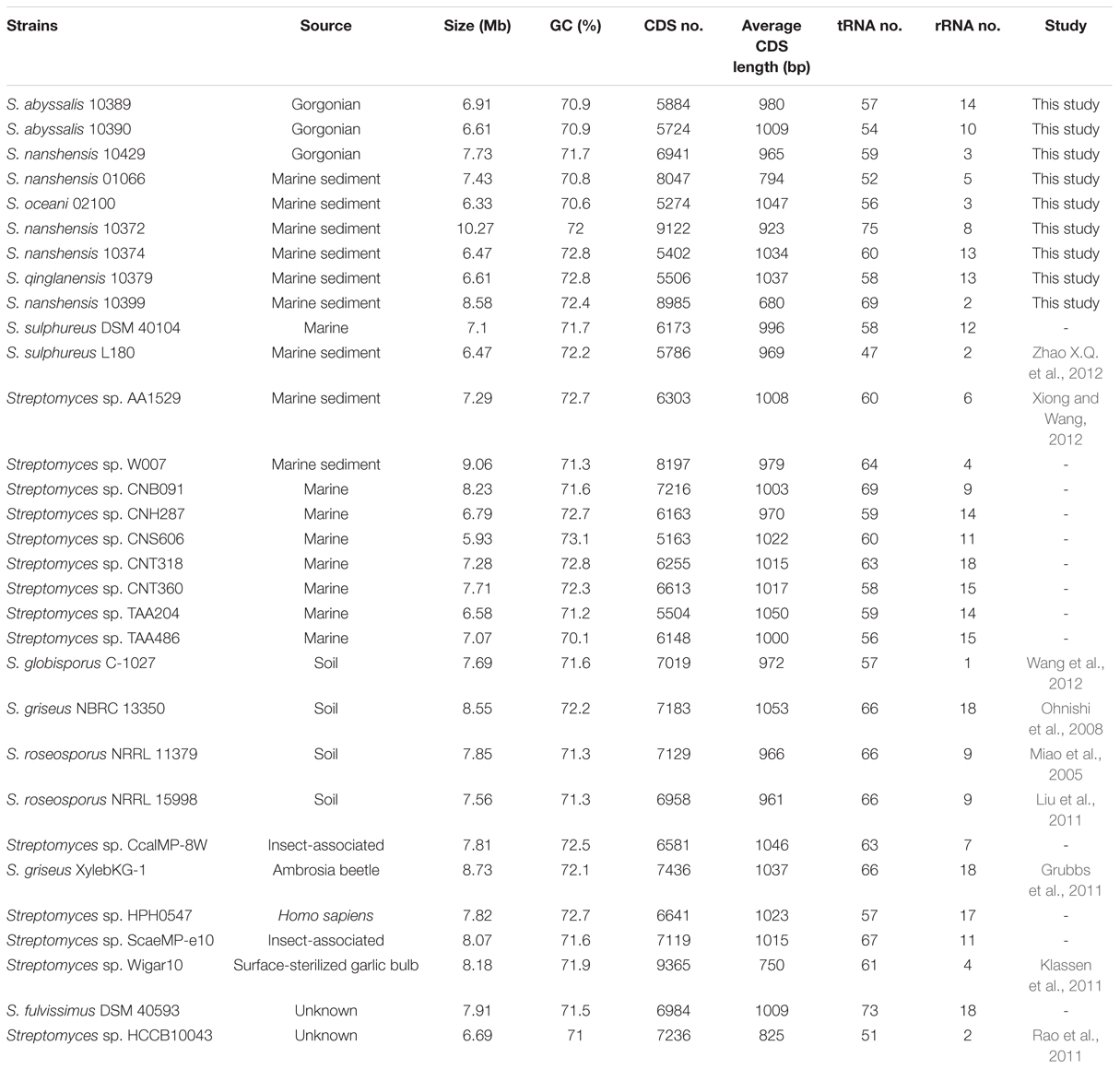
TABLE 1. Genomic information of 31 Streptomyces.
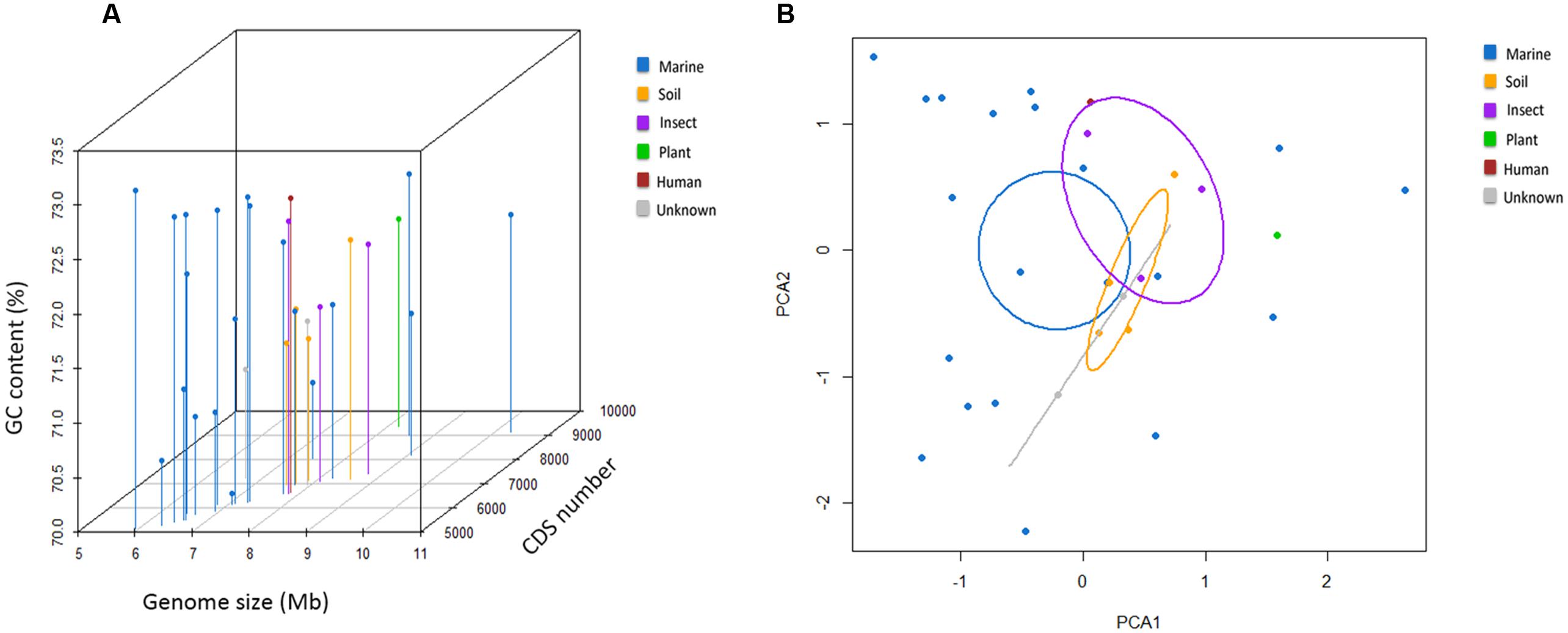
FIGURE 1. General genomic features of 31 streptomycete strains. (A) Three-dimensional plots of the GC content, genome size and CDS number. (B) The principal component analysis (PCA) of the genome size, CDS number and G+C content.
Phylogenetic Analyses of Streptomycetes
To infer the phylogenetic relations of 31 streptomycete strains, we reconstructed phylogenetic trees on the basis of 16S rRNA and single copy core genes with Catenulispora acidiphila DSM 44928 as the outgroup (for details, see Materials and Methods). In general, the two trees shared the same topology. In the phylogenetic tree based on 16S rRNA gene sequences (Figure 2A), 27 strains were primarily grouped into two subgroups, with the exception of Streptomyces sp. HPH0547. The S. sulphureus DSM 40104, Streptomyces sp. CNT360, S. roseosporus NRRL 15998 and Streptomyces sp. CcalMP-8W strains were not included in this tree because 16S rRNA genes were not detected in their draft genomes. However, the internal branches of the tree that were based on 16S rRNA gene sequences had low bootstrap support. Single-gene phylogenies might not always reflect the evolutionary history of a species due to the high degree of LGT (Marri et al., 2006). We also reconstructed a phylogenetic tree using concatenated single copy core genes (Figure 2B). This tree exhibited larger bootstrap scores and higher robustness. According to the tree based on concatenated single copy core genes, 31 streptomycete strains were clearly grouped into two subgroups. One subgroup with 18 strains contains 17 strains isolated from marine environments, and one from human beings is called the marine-derived subgroup. The other subgroup contains 13 strains that were isolated from different environments, such as marine areas, soil, insects, plants, and others, and it is called the multiple sources subgroup. The tree of concatenated single copy core genes suggested that S. nanshensis 10399, S. qinglanensis 10379 and S. nanshensis 10374 are tightly grouped and that S. abyssalis 10389 and S. abyssalis 10390 are sister taxa that are equally closely related to S. nanshensis 01066. This finding may reflect that close, isolated locations are associated with genetic recombination more frequently, which may reduce the divergence of these bacteria. Our analysis suggests that Streptomyces sp. CNH287, S. oceani 02100 and Streptomyces sp. CNS606 form a separate clade. Streptomyces sp. HPH0547 was isolated from Homo sapiens, and it was clustered in the marine-derived group as an exception. This finding may imply that this strain originated in the ocean and that its ancestor was transmitted to humans because of frequent activity. The three marine-derived strains (Streptomyces sp. W007, Streptomyces sp. CNB091, and S. nanshensis 10372) were clustered into a multiple sources subgroup with some soil-derived strains. We boldly conjecture that these three bacterial ancestors were transmitted to the marine environment recently, and they still retain many characteristics from the land.
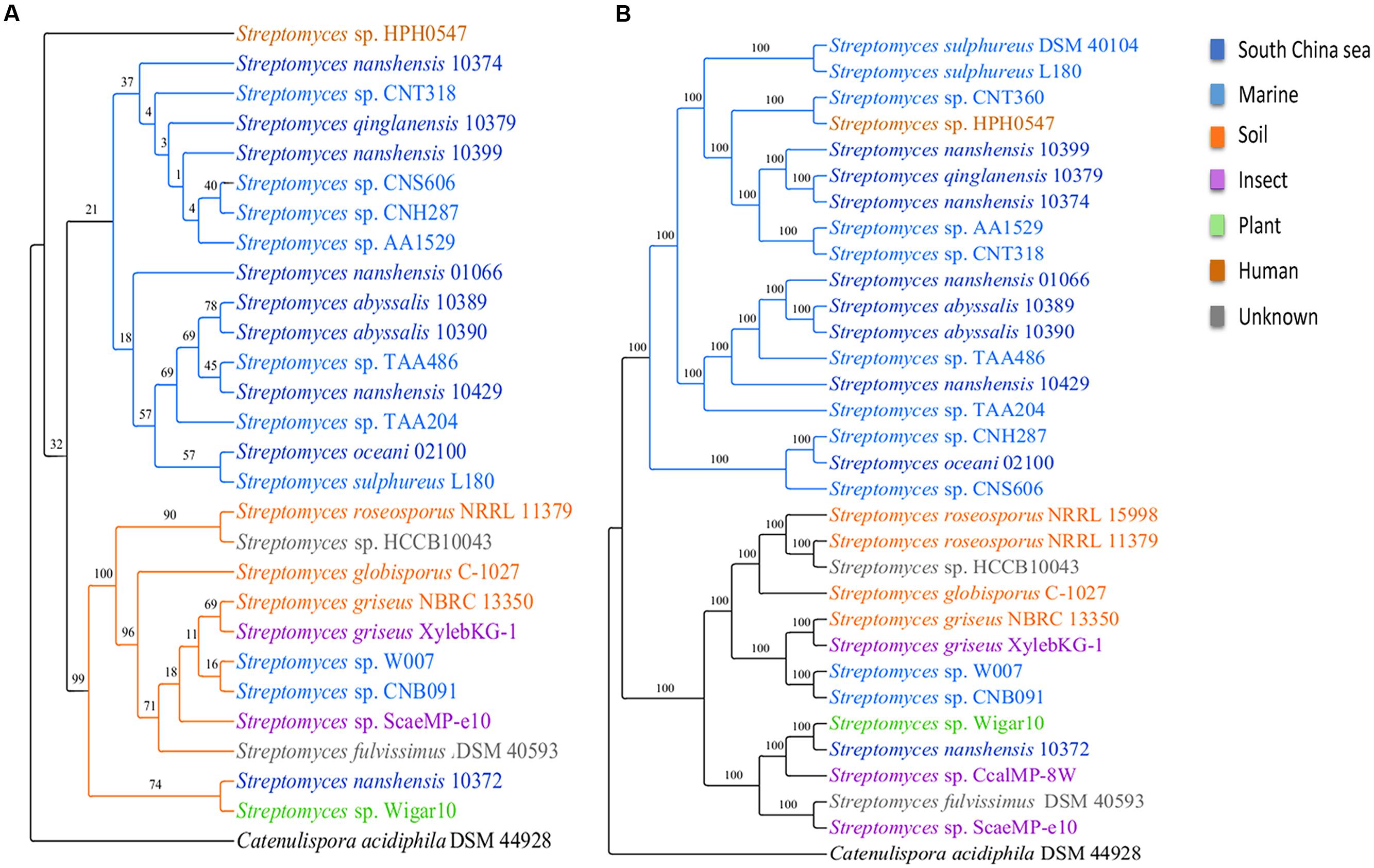
FIGURE 2. Phylogenetic trees for streptomycete strains. The phylogenetic trees were inferred using maximum likelihood on the basis of (A) the 16S rRNA gene and (B) the concatenated alignment of single copy core genes. The numbers on the branches represent the support derived from 100 bootstrap replicates.
Tang et al. (2013) found sharp genetic distinctions among bacteria from closely related lineages. They showed the existence of an abrupt turning point in the sequence divergence between any pair of Salmonella lineages in their comparisons (Tang et al., 2013). We compared the sequence identity levels between the single copy core genes among 31 strains. In the marine-derived subgroup, most of the genes shared 85% of their sequence identity. Because S. nanshensis 10374 and S. qinglanensis 10379, S. abyssalis 10389 and S. abyssalis 10390 exhibited high similarity, there is a small peak at the 100% sequence identity point (Supplementary Figure S2A). In the multiple sources subgroup, the majority of sequence identities among the strains were above 90%, and the peak of the identity distribution was at 95%. Similarly, we also calculated the percentages of genes with 80% or higher shared identity (Supplementary Figure S2B), and more than 90% (Supplementary Figure S2C) of the identities between each pair of genes were found in the single copy core genes of 31 strains. In general, these results suggested that the genomic conservation that presented within the multiple sources subgroup is relatively high.
Recombination plays a key role in bacterial adaptation. We also estimated the relative effects of recombination and mutation (r/m) for each strain through the concatenated alignment of all single copy orthologs with ClonalFrameML (Didelot and Wilson, 2015). The r/m ratio for each branch in the tree ranged from 0.23 on the branch to S. griseus XylebKG-1 to 116.45 on the branch to S. oceani 02100 (Supplementary Table S5). The high r/m ratio of S. oceani 02100 suggested that the overall recombination caused 116 times more substitutions than mutations, confirming the importance of recombination in helping S. oceani 02100 adapt to the variable ocean environment. However, S. griseus XylebKG-1 is an ambrosia beetle-associated actinomycete, and its low r/m ratio suggests long-term co-evolution with its host. The r/m ratios in the marine-derived subgroup were clearly different from those in the multiple sources subgroup (Welch’s two sample t-test, p-values 0.024). This finding showed that most marine-derived streptomycetes had obviously higher r/m ratio than those multiple sources streptomycetes.
Streptomycete Genome Dynamics
To clarify the genome dynamics of streptomycetes, we analyzed the gene gain/loss events that occurred during the evolution of every strain using AnGST (David and Alm, 2011). We identified 29,695 clusters of functional genes containing a total of 210,057 gene copies with using MP method in PGAP (Zhao Y.B. et al., 2012). Among these clusters, 9,293 (31.3%) possessed three or more gene copies, 2,562 (8.6%) included two gene copies (doublets), and 17,840 (60.1%) contained one gene copy (singletons). Using the default penalties, we detected a dynamic pattern of gene gain/loss through the phylogeny. During the process of forming the two subgroups, namely the marine-derived subgroup and the multiple sources subgroup, more (1,466 and 2,632, respectively) gene gain events occurred (Figure 3). During this period, the multiple sources subgroup had more than 79.5% of the genes in comparison with the marine-derived subgroup, which always had a larger genome. Subsequently, some branches of the two subgroups excluding the terminal branches experienced several periods of genome shrinkage (more gene loss than gain). Finally, in recent times, nearly all 31 (27/31) of the strains except for S. nanshensis 10374, S. abyssalis 10390, Streptomyces sp. TAA204, and Streptomyces sp. CcalMP-8W were characterized by much more gains than losses. Phylogenetic analyses of 46 Streptococcus strains by Richards et al. (2014) acquired a similar pattern of genome evolution. Of the proportion of genes gained on the terminal branches, 30.7% were identified as LGT. The majority (60.1%) of the acquired genes were classified as genes that were born on these branches (singletons). However, given that these genes were not present in any other strains, it is possible that they were acquired via LGT from bacteria that were not included in this study. To explore this possibility, each singleton gene was searched for significant sequence matches using BLASTp (E-value cutoff = 1e-5) against the NCBI NR database. A total of 70.2% (12,531) of the singleton genes matched other species, which indicated that over two-thirds of the singletons were actually acquired through LGT. In addition, 94.2% (11,810) of these genes match a streptomycete species. Lefebure et al. (2012) performed a similar AnGST analysis on 15 Streptococcus species, and they showed that these genes account for approximately two-thirds of the genes born on the Streptococcus pyogenes branch. Although, the remaining singleton genes may be born de novo on this branch, it is also possible that homologous genes haven’t yet to be sequenced (Richards et al., 2014). Overall, our findings suggest that a large proportion of the streptomycete pan-genome (all gene clusters) has been involved in LGT. Among all the gene clusters, 60.2% (17,876) were identified as being involved in LGT.
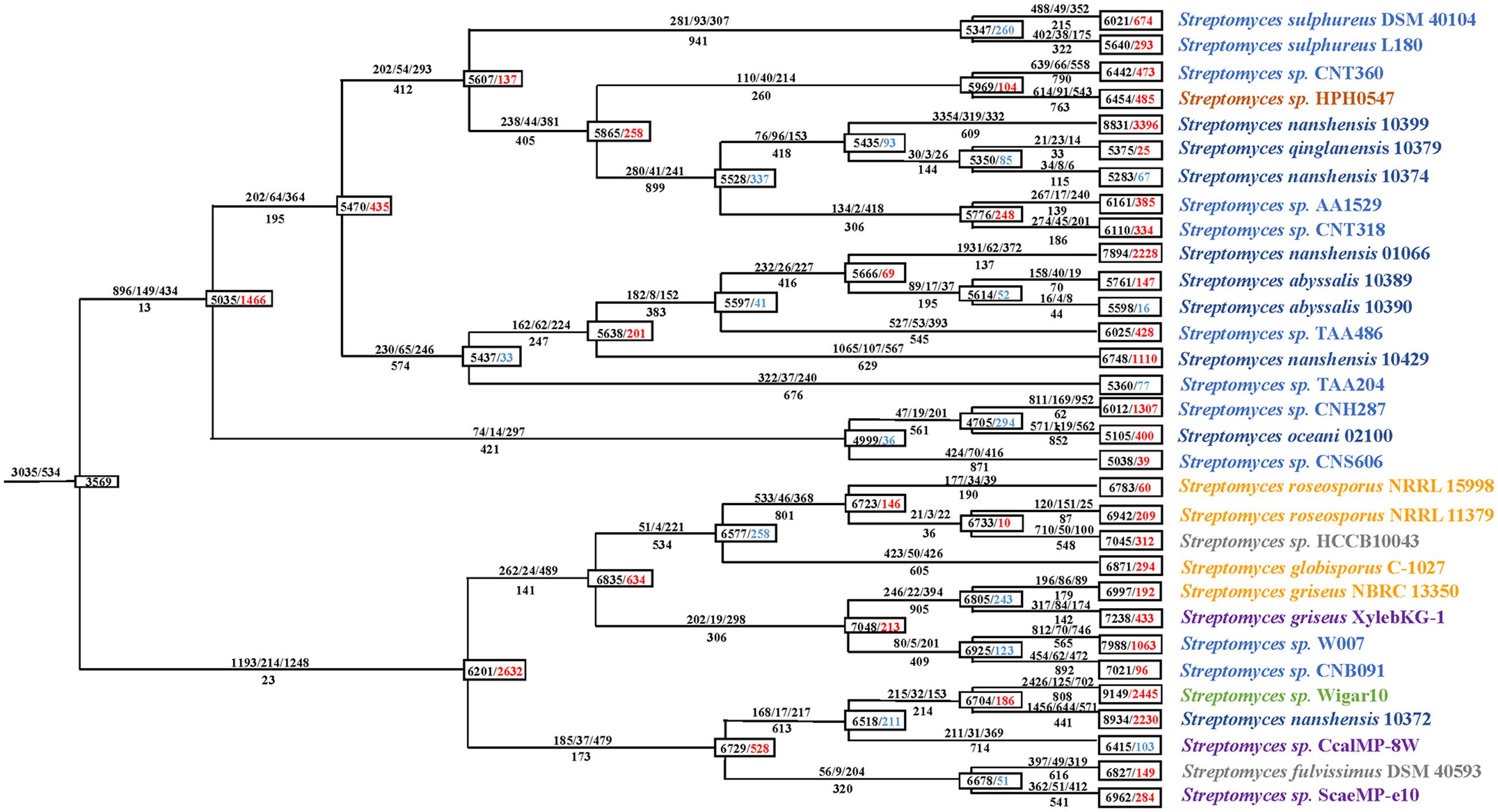
FIGURE 3. Gene gain and loss events during the evolution of 31 streptomycete strains. The numbers in boxes on every node represent the genome size and the overall balance (gain-loss: balance), with black indicating the genome size, red indicating an overall gain and blue indicating an overall loss. The numbers above and below every lineage show the number of birth/duplication/transfer and loss events.
From the perspective of gene gain/loss for the phylogenetic process of individual species, S. oceani 02100 and S. nanshensis 10372 are notable. After the initial expansion period (with a gain of 1,466 genes), S. oceani 02100 underwent two consecutive periods of gene loss (it lost 330 genes in total) and recently obtained 400 genes. In comparison with other strains, the S. oceani 02100 strain obtained fewer genes during evolution, which may have caused it to be unable to live without salt (Tian et al., 2012). S. nanshensis 10372 experienced only one period of streamlining (lost 211 genes) during evolution and recently gained 2,230 genes. These changes all resulted in S. nanshensis 10372s possession of the largest genome size among the nine marine strains.
Core and Pan-Genome Analyses
The pan-genome defines the entire genomic repertoire of a given phylogenetic clade and encodes for all possible lifestyles of its organisms (Vernikos et al., 2015). This genome primarily contains the core genome, dispensable genome and strain-specific genes. The core genome is essential for a bacterium basic lifestyle, and the dispensable genome offers species diversity, environmental adaptation and other characteristics (Tettelin et al., 2008). To understand the genetic composition of the 31 streptomycete strains in the pan-genome insight more thoroughly, we clustered all 210,057 protein CDSs in 31 streptomycete strains with PGAP. All the protein CDSs were clustered into 29,695 orthologs; 2,048 (6.90%) orthologs were identified in 31 strains as the streptomycete core genome (Figure 4A), and 1,272 of them were single-copy. The 9,807 orthologs were identified as dispensable genomes, and 17,840 genes were strain-specific. S. nanshensis 10374 and S. qinglanensis 10379 as well as S. abyssalis 10389 and S. abyssalis 10390 had a smaller number of strain-specific genes because these two pairs of strains had high similarities in the genome. However, S. nanshensis 10399 had the largest number of strain-specific genes, indicating that it has the greatest difference with the other strain genomes. For further analysis, we also identified the core ortholog genes and strain-specific genes in marine-derived and multiple source subgroups (Figures 4B,C). In an attempt to understand the relations between the streptomycete pan-genome size, the core genome number and the strain number, we plotted the pan-genome profile fitted curves of the 31 strains, the marine-derived subgroup and the multiple sources subgroup according to a Heaps’ law model (Tettelin et al., 2005, 2008) with PanGP (Zhao et al., 2014). As shown in Figure 5A, we can intuitively observe that the more genomes we added, the more new ortholog clusters were discovered, implying an open pan-genome of streptomycete strains (Figure 5A, blue curve). The green curve in the same picture, which is fit by an exponential function, indicated that the average number of core genes converged to a relatively constant number of 2,266 (2264–2268, 95% confidence interval). In the marine-derived subgroup and the multiple sources subgroup, the pan-genome profiles were basically similar (Figures 5B,C). We have also conducted a COG function category comparison among core genes from the marine-derived subgroup and the multiple sources subgroup. As shown in Figure 6, the core genomes of these two subgroups both harbor a large proportion of genes pertaining to transcription (K), replication, recombination and repair (L), signal transduction mechanisms (T), cell wall/membrane/envelope biogenesis (M), energy production and conversion (C), carbohydrate transport and metabolism (G), amino acid transport and metabolism (E) and others. The marine-derived subgroup contains a higher proportion of core genes belonging to the COG categories of translation, ribosomal structure and biogenesis (J), and post-translational modification, protein turnover, and chaperones (O). The abundance of these genes might play a role in bacterial adaptations to the low temperature, high pressure, oligotrophic, saline and dark marine environment by ensuring protein synthesis and maintaining protein functional stability. The overrepresentation of COG category J was also observed in the genome of the halophilic archaeon Halococcus hamelinensis (Gudhka et al., 2015). The abundance of COG category O was also consistent with previous research (Kuwahara et al., 2007; Wang et al., 2008).
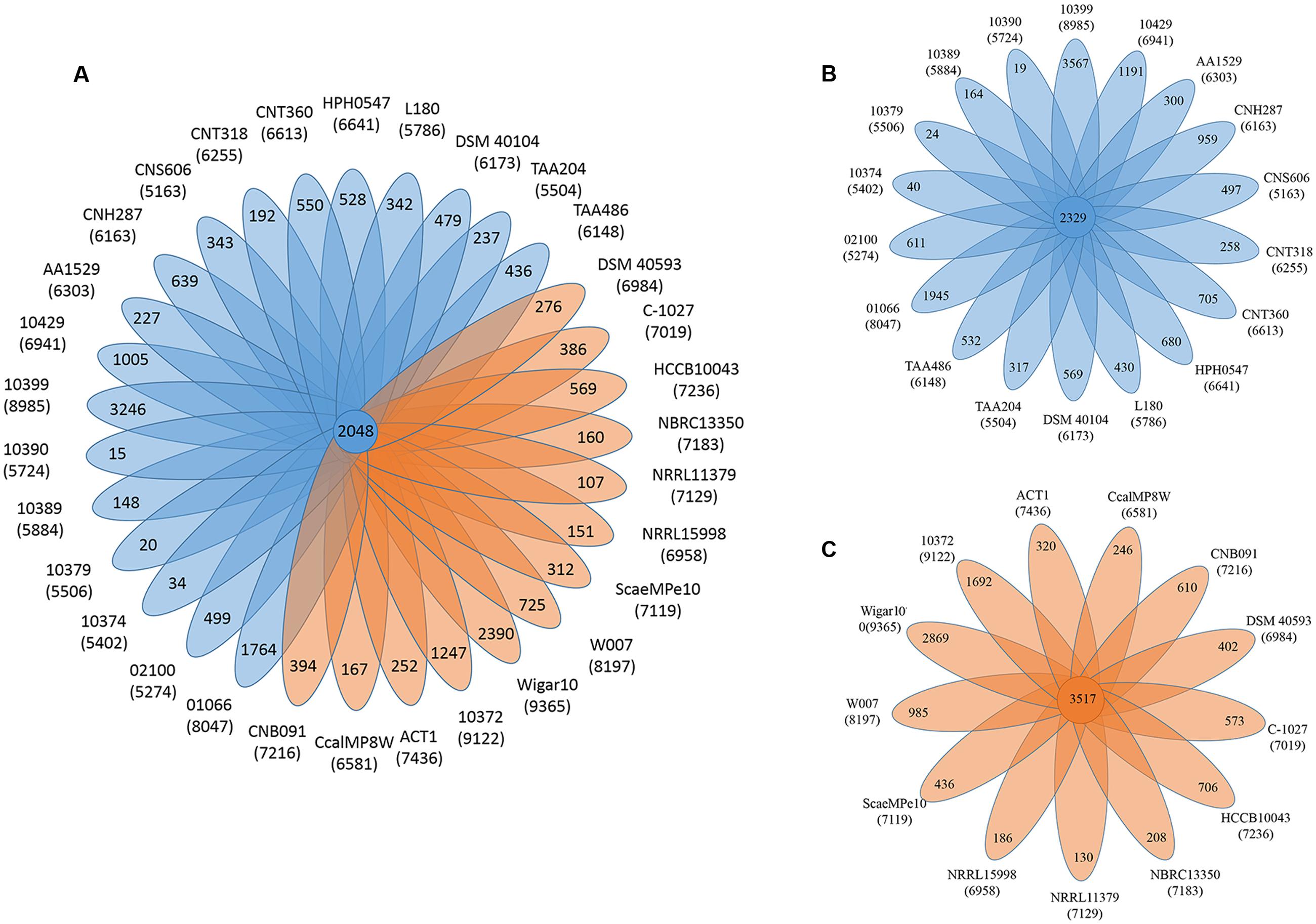
FIGURE 4. The pan-genome of streptomycetes. (A) Flower plots showing the core gene number (in the center) and strain-specific gene number (in the petals) in 31 streptomycete strains. (B) Flower plots showing the core gene number (in the center) and strain-specific gene number (in the petals) in the marine-derived subgroup. (C) Flower plots showing the core gene number (in the center) and strain-specific gene number (in the petals) in the multiple sources subgroup. The numbers under the strain name represent the total number of coding proteins. Blue indicates the marine-derived subgroup and orange indicates the multiple sources subgroup.
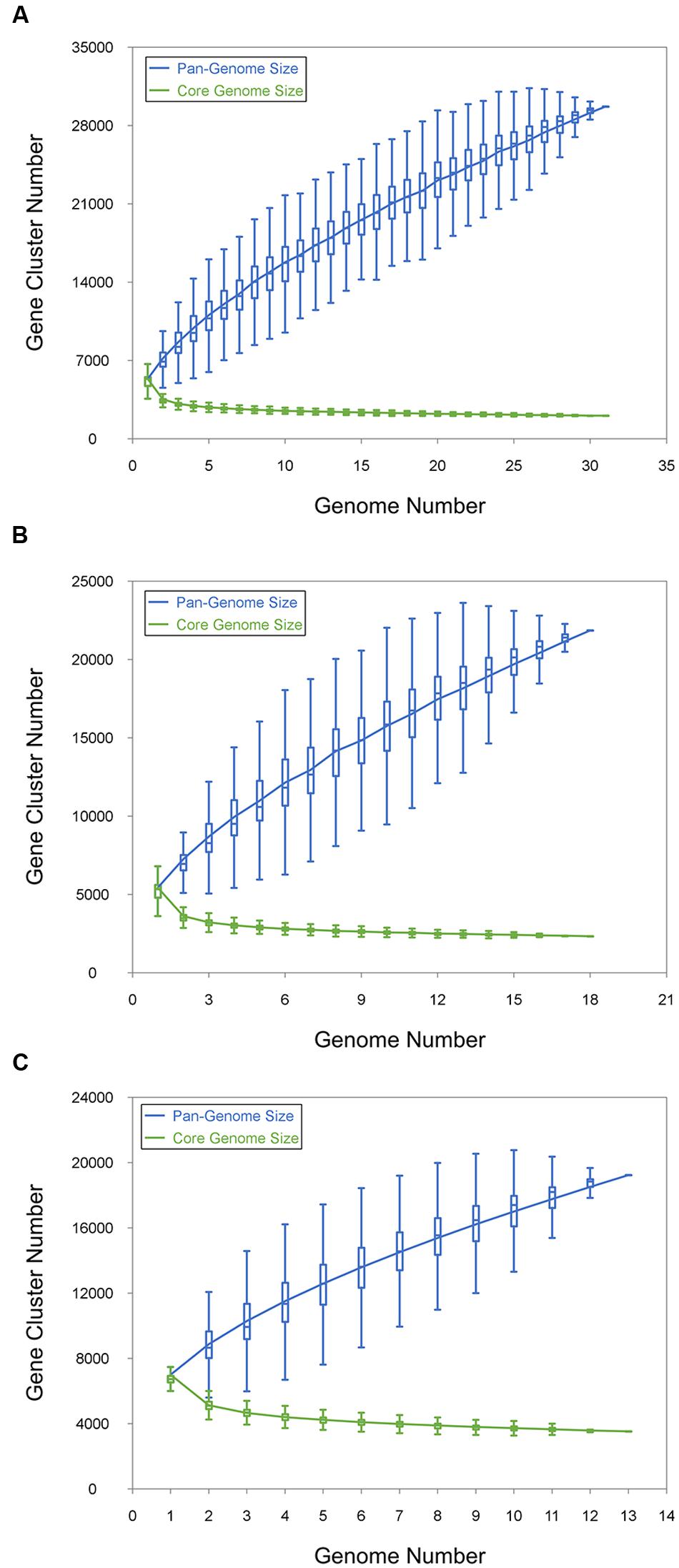
FIGURE 5. Pan- and core genome size prediction. (A) Curves for 31 streptomycete pan-genomes and core genomes. (B) Curves for the pan-genomes and core genomes of 18 marine-derived strains. (C) Curves for the pan-genomes and core genomes of 13 multiple source strains. The blue dots denote the pan-genome size and the green dots denote the core genome size. The connected median values represent the relations between the genome number and gene cluster number.
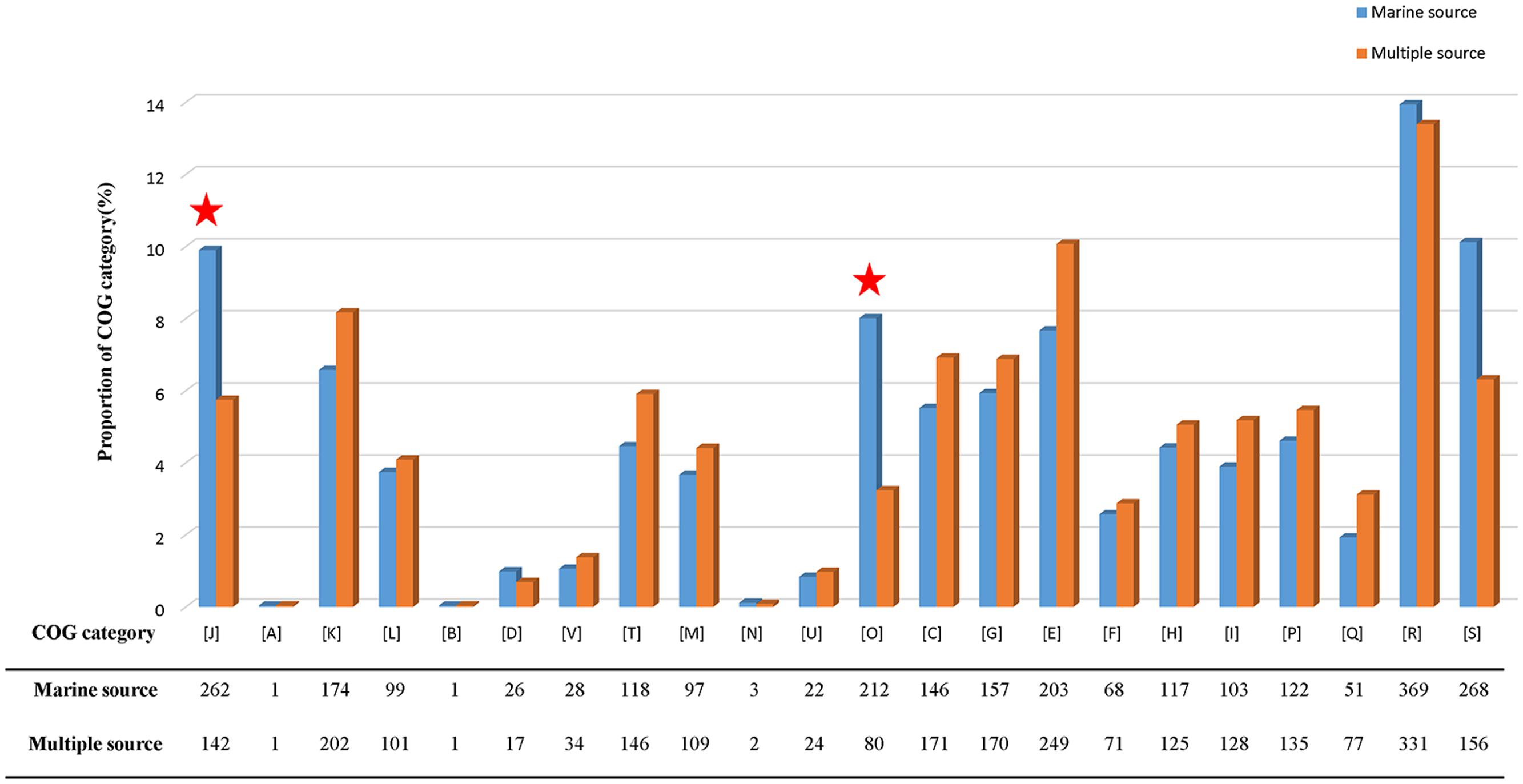
FIGURE 6. COG category percentage of core genes in the marine-derived subgroup and multiple sources subgroup. The COG categories are described as follows: J, translation, ribosomal structure, and biogenesis; A, RNA processing and modification; K, transcription; L, replication, recombination, and repair; B, chromatin structure and dynamics; D, cell cycle control, cell division, and chromosome partitioning; V, defense mechanisms; T, signal transduction mechanisms; M, cell wall/membrane/envelope biogenesis; N, cell motility; U, intracellular trafficking, secretion, and vesicular transport; O, post-translational modification, protein turnover, and chaperones; C, energy production and conversion; G, carbohydrate transport and metabolism; E, amino acid transport and metabolism; F, nucleotide transport and metabolism; H, coenzyme transport and metabolism; I, lipid transport and metabolism; P, inorganic ion transport and metabolism; Q, secondary metabolites biosynthesis, transport and catabolism; R, general function prediction only; and S, function unknown.
Transporters and Various Marine Environment Adaptations
To investigate how streptomycetes interact with environments, we compared how cytoplasmic transport systems differ between the two subgroups. All the strains in both subgroups contain a large number of ATP-binding cassette transporters (ABC transporters) and the major facilitator superfamily (MFS). The transporters in these two families are ubiquitously present in all biological kingdoms. More transporters, such as the solute: sodium symporter (SSS), metal ion (Mn2+) transporter (Nramp), tripartite ATP-independent periplasmic transporters (TRAP-T) and others are found among the strains in the marine-derived subgroup that showed a significant difference (Figure 7A). Some transporters occur specifically in strains from the South China Sea, such as the branched chain amino acid exporter (LIV-E), small conductance mechanosensitive ion channel (MscS) and large conductance mechanosensitive ion channel (MscL; Figure 7A). During the transporter analysis, we found that marine-derived streptomycete strains enriched the K+ transporter (Trk) and (betaine/carnitine/choline transporter) BCCT more than multiple sources did (Supplementary Table S6). The enrichment of Trk may help marine-derived streptomycete strains accumulate K+, which is the primary strategy for many extremophiles that survive in high-osmolality environments (Roberts, 2004). An abundance of BCCT was also observed in several marine Vibrio strains, such as V. vulnificus and V. fischeri (Naughton et al., 2009). BCCT would assist in the accumulation of betaine, carnitine and choline, which are known as compatible solutes. These compatible solutes not only maintain the cellular osmotic balance but also serve as stabilizers of proteins and cell components against the denaturing effects of high ionic strength (Kempf and Bremer, 1998). The transporter analysis also showed that most streptomycete strains that are derived from the South China Sea possess MscS and MscL proteins, except S. oceani 02100, S. nanshensis 10372 and S. nanshensis 10429. When bacteria are moved from marine to freshwater environments (leading to osmotic downshock), the mechanosensitive channels may help the cells reduce their turgor pressure rapidly by releasing cytoplasmic solutes (Berrier et al., 1992). Consistent with previous reports, the MscL protein alleviated cell lysis following a sudden osmotic downshock in the marine halophile Vibrio alginolyticus (Nakamaru et al., 1999).
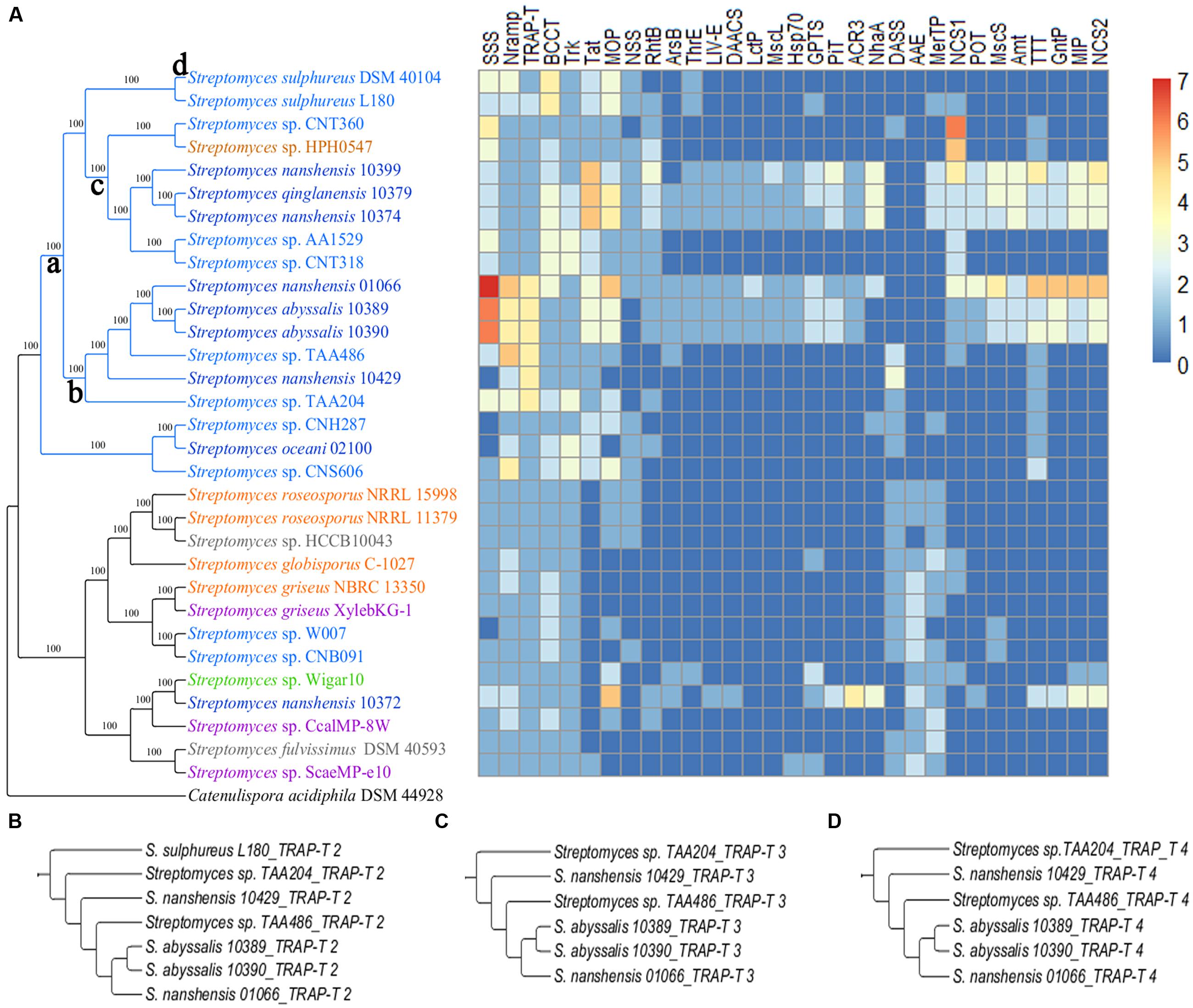
FIGURE 7. (A) A streptomycete species tree and a heatmap showing the distribution of transport protein families. The transport protein families are described as follows: SSS, solute: sodium symporter; Nramp, metal ion (Mn2+-iron) transporter; TRAP-T, tripartite ATP-independent periplasmic transporter; BCCT, betaine/carnitine/choline transporter; Trk, K+ transporter; Tat, twin arginine targeting; MOP, multidrug/oligosaccharidyl-lipid/polysaccharide; NSS, neurotransmitter: sodium symporter; RhtB, resistance to homoserine/threonine; ArsB, arsenite-antimonite; ThrE, threonine/serine exporter; LIV-E, branched chain amino acid exporter; DAACS, dicarboxylate/amino acid: cation (Na+ or H+) symporter; LctP, lactate permease; MscL, large conductance mechanosensitive ion channel; Hsp70, cation channel-forming heat shock protein-70; GPTS, general phosphotransferase system; PiT, the inorganic phosphate transporter; ACR3, arsenical resistance-3; NhaA, Na+: H+ antiporter; DASS, divalent anion: Na+ symporter; AAE, aspartate: alanine exchanger; MerTP, mercuric ion (Hg2+) permease; NCS1, nucleobase: cation symporter-1; POT, proton-dependent oligopeptide transporter; MscS, small conductance mechanosensitive ion channel; Amt, ammonia transporter channel; TTT, tricarboxylate transporter; GntP, gluconate: H+ symporter; MIP, the major intrinsic protein; and NCS2, the nucleobase:cation symporter-2. (B) The gene tree of TRAP-T 2. (C) The gene tree of TRAP-T 3. (D) The gene tree of TRAP-T 4.
Strains of the marine-derived subgroup contained more transporters that were involved in nutrient absorption, such as TRAP-T, SSS, the nucleobase: cation symporter-1 (NSC1), the twin arginine targeting (Tat) family and the neurotransmitter: sodium symporter (NSS). Substrate binding proteins (SBPs) that are anchored in the cytomembrane of gram-positive bacteria have high affinities to the substrate and assist bacteria in taking advantage of low nutrient concentrations. The known SBP-dependent secondary transporters are currently categorized into two families, namely TRAP-T and the tripartite tricarboxylate transporters (TTTs; Mulligan et al., 2011). Although, there is no obvious sequence consistency between TRAP-T, SSS and NSC1, the latter two have the same folding structure and the same transport mechanism as the TRAP-T (Weyand et al., 2008). The Tat family is likely to play a major role in nutrient acquisition from complex sources, permitting streptomycetes growth when more readily used soluble nutrients are not available. The known or predicted Tat substrates in S. coelicolor include a diverse array of hydrolytic enzymes (Chater et al., 2010). Moreover, a comparison of the two-component system indicates that 15 strains of the marine-derived subgroup can assimilate phosphate under phosphate-limiting conditions, and only four strains in the other subgroup have the effective pathway. Furthermore, according to the AnGST results, the gene-tree species-tree reconciliation allows us to infer the evolutionary history of every gene family in an explicit fashion. For the example of TRAP-T, there are four clusters of TRAP-T genes in the data set. The first TRAP-T cluster contained 31 genes, with one for each strain. This cluster was born on the root (Figure 7A) and has been retained throughout evolutionary history. This finding suggests that the TRAP-T 1 gene (TRAP transporter solute receptor, TAXI family) is important to Streptomyces strains. The other three cluster gene trees were shown in Figures 7B–D. For TRAP-T 2 (permease with DctM domain; Figure 7B), it first gained on branch a and then lost on branch c and branch d. For TRAP-T 3 (hypothetical protein with DctQ domain; Figure 7C) and TRAP-T 4 (hypothetical protein with PBP2_TRAP_Siap_TeaA_like domain; Figure 7D), they were all born on branch b. Not surprisingly, S. nanshensis 01066, S. abyssalis 10389, S. abyssalis 10390, Streptomyces sp. TAA486, S. nanshensis 10429, and Streptomyces sp. TAA204 had three protein components of TRAP-T (TRAP-T 1, TRAP-2, and TRAP-T 3), which helps these strains cope with the oligotrophic marine environment.
Most strains that were isolated from the South China Sea also have other transporter families that are involved in taking up nutrients, such as the TTT, the gluconate: H+ symporter (GntP), the dicarboxylate/amino acid: cation (Na+ or H+) symporter (DAACS), the lactate permease (LctP), proton-dependent oligopeptide transporter (POT), the general phosphotransferase system (GPTS) and the ammonia transporter channel (Amt). As mentioned above, the TTT permits streptomycetes to take up nutrients under oligotrophic conditions. The remaining families enable streptomycetes to make use of alternative carbon sources and nitrogen sources and to obtain more energy and thereby increase their competitiveness in response to ecological stress (Kleiner, 1985; Caspari and Urlinger, 1996; Yurgel et al., 2000; Nunez et al., 2002; Govindarajan et al., 2013; Lyons et al., 2014). Additionally, the PhoR-PhoB two-component system and the multiple sugar ABC transporter that are contained in more strains of the marine-derived subgroup may also help these strains take full advantage of nutrients under oligotrophic conditions. The PhoR-PhoB two-component system plays an important role in detecting and responding to changes in the environmental phosphate concentration (Marzan and Shimizu, 2011). Thus, the system facilitates the ability of the marine-derived subgroup strain to regulate its phosphate uptake in a sophisticated manner and survive, even under phosphate-limiting conditions. Most marine-derived strains possessed a complete, putative multiple sugar transport system (ChvE, GguB, GguA). ChvE has been suggested to be involved in sugar binding, sugar utilization and virulence in Agrobacterium tumefaciens (He et al., 2009). Previous research has also indicated that GguA and GguB play a role in sugar utilization. In A. tumefaciens, the chvE-, gguA-, and gguB-encoded products are predicted to constitute a complete binding protein-dependent ABC transporter, which has a wide range of substrates, including L-arabinose, D-fucose, D-galactose, D-glucose, and D-xylose (Zhao and Binns, 2011).
In addition to the above transporter families, strains from the marine-derived subgroup also possess transporters that participate in drug/metabolite transport, metalloid transport, and pH regulation. Members pertaining to resistance-nodulation-cell division (RND), the drug/metabolite transporter (DMT), multidrug/oligosaccharidyl-lipid/polysaccharides (MOPs), and resistance to homoserine/threonine (RhtB) are involved in drug/metabolite or homoserine lactone efflux (Jack et al., 2001; Li et al., 2011), and there are more strains from the marine-derived subgroup than from the multiple sources subgroup. Members of the threonine/serine exporter (ThrE) family contained more strains that were isolated from the South China Sea than other strains. Threonine and serine will be exported when their high concentrations inhibit cell growth (Yen et al., 2002). The proper content of manganese (Mn), which is called a “life guard,” is important for maintaining the normal physiological function of the cell (Li and Tang, 2012). The presence of more members of the metal ion (Mn2+) transporter (Nramp) family in the marine-derived subgroup strains reflects their adaptability to low marine Mn2+ concentrations (Komorsky-Lovric, 1998; Yen et al., 2002). Members of the arsenical resistance-3 (ACR3) and arsenite-antimonite (ArsB) efflux family included more strains that were isolated from the South China Sea, and these proteins pumped As(III) and Sb(III) away to reduce toxicity and enhance survivability (Rosen, 1996; Ostrowski et al., 1999; Hughes, 2002; Fu et al., 2009). Additionally, these strains have more Na+: H+ antiporters (NhaA), which are involved in maintaining the pH homeostasis and exhausting the cell’s Na+ to prevent toxicity from high Na+ concentrations (Padan and Schuldiner, 1994).
Genetic Constitution of CRISPRs
Clustered regularly interspaced short palindromic repeat elements are common in bacteria and archaea. A CRISPR is characterized by direct repeats (DRs) that are separated by similarly sized non-repetitive spacers, and the spacer sequences are related to antiphage functions via an RNA-silencing-like mechanism (Qin et al., 2010, 2014). CRISPRs were detected in 11 marine-derived strains and seven other habitat-origin strains (Table 2). The number of CRISPR locations ranges from one (in S. oceani 02100, Streptomyces sp. HPH0547, and S. griseus XylebKG-1) to 12 (in S. sulphureus L180). Seven strains of the 11 marine streptomycetes contained five or more than five CRISPRs, and the strains that were isolated from other habitats possessed less than three CRISPRs (Table 2). Unlike the other marine bacterium from the genus Glaciecola with a conservative DR in almost all the species (Qin et al., 2014), the DR length in the 18 streptomycete strains varied from 25 to 38 bp, and the DR sequences had low shared identity (Supplementary Table S7), which implies diverse origins for the CRISPRs in the Streptomyces genus. The number of spacers ranged from 2 to 41. The sequences of the spacers exhibited high sequence diversity. Considering the large variety of phages (approximately 5–10 times more than the quantity of bacteria) in the marine environment, it is not surprising that spacer sequences show substantial diversity (Wommack and Colwell, 2000; Qin et al., 2010).
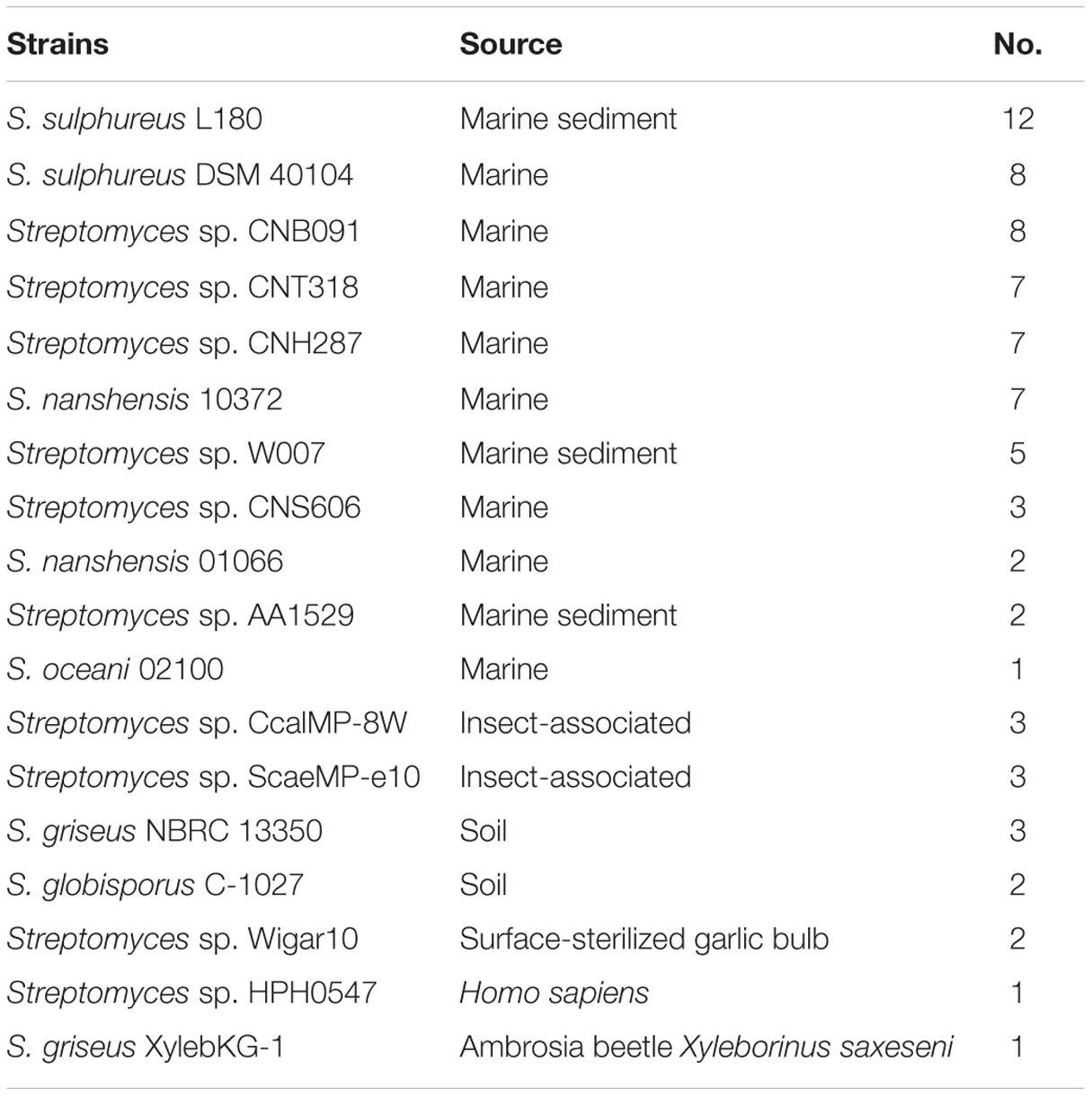
TABLE 2. The number of CRISPRs locations and habitat of every strain.
Discussion
The marine environment is prone to high salinity and oligotrophic conditions. Therefore, it is likely that marine-derived streptomycetes would need to possess various strategies to cope with this type of environment. A phylogenetic analysis, a pan-genomics analysis and a functional genomics analysis were used to reveal the characteristics of marine-derived streptomycetes, which reflected their ability to survive in a marine environment. The phylogeny results may suggest that marine-derived streptomycetes and multiple sources have independent origins. The marine-derived streptomycete strains likely originated from a common ancestor with an ocean origin. Based on the genome dynamics analysis, we concluded that a genomic expansion in streptomycetes during the initial period help the genus adapt to different environments and divide them into subgroups. The genes that are redundant in the new environment are then lost. Finally, most genomes went through a more recent period of genome expansion along with the present day species. Furthermore, most of the genes born on terminal branches were from LGT, which affects a substantial proportion of the streptomycete pan-genome and drives genome expansion. The same pattern of genomic expansion and degradation was also observed by Cuypers and Hogeweg (2012) in a virtual cell model.
The deep sea exhibits extreme conditions, such as high hydrostatic pressures (up to 1,100 bar) and low temperatures (less than 4°C; Jorgensen and Boetius, 2007), which are challenges to marine bacterial survival. Seawater always has a high sodium concentration, which is likely to form a high-osmolality environment. The water in a cell will rapidly flux out along the osmotic gradient when microorganisms are exposed to high-osmolality environments, which may cause a reduction in turgor and the dehydration of the cytoplasm (Kempf and Bremer, 1998). In using a functional genomics analysis as a guide, we found that marine-derived streptomycete strains possessed more Trk and BCCT genes related to high-osmolality environment adaptation. We predicted that the primary strategy for marine-derived streptomycete strain survival is to accumulate K+ by Trk. The abundance of BCCT in marine-derived streptomycete strains may suggest that they accumulate intracellular betaine/carnitine/choline as a major adaptive response to a high-osmolality environment.
Deep-sea sediments are oligotrophic, and particulate organic matter (POM) in sediments is the major nutrient resource for deep-sea bacteria. Every year, most nutrients arrive at the deep sea in pulses, and deep-sea bacteria must therefore respond to this nutrient pulse quickly as well (Qin et al., 2011). In coping with the oligotrophic environment, marine-derived subgroup strains developed more transporters to respond to trace nutrients and absorb a variety of carbon and nitrogen sources to enhance their competitiveness. The tat family helps streptomycete strains from the marine source subgroup hydrolyze complex compounds into soluble nutrients; TRAP-T increases the absorption of nutrients, and SSS, NCS1, and NSS provide effective ways to transfer various nutrients. For most strains that were isolated from the South China Sea, there are also many other transporter families that are involved in taking up nutrients, which were already mentioned above. However, further work is needed to clarify the exact roles of these transporters in marine-derived streptomycete physiology.
The analyses presented here also confirmed the presence of genes that encode drug/metabolite transport, metalloid transport, and pH regulation. Evidently, the toxic effects of high concentrations of sugars, amino acids, other metabolites, and their detrimental analogs have forced bacteria to acquire these efflux systems during evolution (Jack et al., 2001). To ensure bacterial survival, the active efflux of drugs and metabolites is essential (Levy, 1992; Nikaido, 1994). In this study, we found that more drug or metabolite efflux transporters were processed in marine-derived streptomycete strains, such as RND, DMT, MOP, and RhtB. Many of the proteins in the DMT superfamily were found to be pumps for drug or metabolite efflux (Jack et al., 2001). Previous studies suggested that RhtB is involved in the efflux of homoserine and threonine from Escherichia coli (Aleshin et al., 1999), and it also might be involved in quorum sensing (Vicente et al., 2009). These drug or metabolite pumps may help marine-derived streptomycete strains eject intracellular toxic substances, and they are beneficial for survival in the marine environment. In addition, the abundance of CRISPRs in marine isolates may also help marine streptomycetes against phages in the marine environment.
In summary, by comparing the genomes of ocean isolates with those of isolates from other environments, we are beginning to identify some genomic characteristics that enable marine streptomycetes to thrive in complex marine environments. Phylogenetic analyses have revealed the dynamic process of streptomycete genome evolution, and LGT is an important driving force during this process, as already shown by Ochman et al. (2000). Pan-genomic analyses revealed that streptomycetes have an open pan-genome that reflects the diversity of these streptomycete species and their high levels of adaptability to different environments. The functional genomics analyses indicate that marine-derived subgroup streptomycetes possess some characteristics of marine adaptation, such as high hydrostatic pressure, low temperature, and oligotrophic adaptability. To adapt to the high hydrostatic pressure and low temperature conditions, marine-derived subgroup strains accumulate more functional genes that are related to post-translational modification, protein turnover, chaperones, translation, ribosomal structure and biogenesis as well as more BCCT, Trk, MscS, and MscL transporter families. The genomes of most marine-derived subgroup strains harbored a variety of transporters to cope with the oligotrophic environment, and these transporters included TRAP-T, SSS, NCS1, NSS, Tat, TTT, GntP, DAACS, LctP, POT, GPTS, and Amt. Furthermore, more transporters take part in drug/metabolite efflux, metalloid resistance, and pH regulation, thereby adapting to other marine conditions, and they are contained in marine-derived subgroup strains. In sum, marine streptomycetes possess more functional genes and transporters than other streptomycetes to adapt to the cold, hyperosmosis, oligotrophy, and other marine conditions, and marine isolates also have more CRISPRs, which is related to phage resistance.
Data Accessibility
The nine draft genome sequences of Streptomyces were submitted to the NCBI. The accession numbers were JHEJ00000000, LJGS00000000, LJGT00000000, LJGU00000000, LJGV00000000, LJGW00000000, LJGX00000000, LJGY00000000, and LJGZ00000000.
Author Contributions
XT, Zhewen Zhang, and TY contributed equally to this manuscript. MC, FC, Zhang Zhang, JW, CZ, LL, and JX participated in the design and discussion of the research. XT, JL, JY, and BZ carried out the experimental part of the work. Zhewen Zhang and TY carried out the analysis of the data and wrote the manuscript. All authors have read and approved the final manuscript.
Funding
This work was supported by the Key deployment project of the Chinese Academy of Sciences (KSZD-EW-TZ-009), grants from National Programs for High Technology Research and Development (863 Program; 2014AA021503), and grants from the National Science Foundation of China (31471248, 31501024, 41230962, and 41276004).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
The authors thank the Core Genomic Facility at the Beijing Institute of Genomics for their sequencing support.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2016.00998
Footnotes
References
Aleshin, V. V., Zakataeva, N. P., and Livshits, V. A. (1999). A new family of amino-acid-efflux proteins. Trends Biochem. Sci. 24, 133–135. doi: 10.1016/S0968-0004(99)01367-5
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1006/jmbi.1990.9999
Barakat, K. M., and Beltagy, E. A. (2015). Bioactive phthalate from marine Streptomyces ruber EKH2 against virulent fish pathogens. Egypt. J. Aquat. Res. 41, 49–56. doi: 10.1016/j.ejar.2015.03.006
Berrier, C., Coulombe, A., Szabo, I., Zoratti, M., and Ghazi, A. (1992). Gadolinium ion inhibits loss of metabolites induced by osmotic shock and large stretch-activated channels in bacteria. Eur. J. Biochem. 206, 559–565. doi: 10.1111/j.1432-1033.1992.tb16960.x
Bruen, T. C., Philippe, H., and Bryant, D. (2006). A simple and robust statistical test for detecting the presence of recombination. Genetics 172, 2665–2681. doi: 10.1534/genetics.105.048975
Caspari, T., and Urlinger, S. (1996). The activity of the gluconate-H+ symporter of Schizosaccharomyces pombe cells is down-regulated by D-glucose and exogenous cAMP. FEBS Lett. 395, 272–276. doi: 10.1016/0014-5793(96)01052-6
Chater, K. F., Biro, S., Lee, K. J., Palmer, T., and Schrempf, H. (2010). The complex extracellular biology of Streptomyces. FEMS Microbiol. Rev. 34, 171–198. doi: 10.1111/j.1574-6976.2009.00206.x
Cuypers, T. D., and Hogeweg, P. (2012). Virtual genomes in flux: an interplay of neutrality and adaptability explains genome expansion and streamlining. Genome Biol. Evol. 4, 212–229. doi: 10.1093/Gbe/Evr141
Darriba, D., Taboada, G. L., Doallo, R., and Posada, D. (2012). jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 9, 772–772. doi: 10.1038/nmeth.2109
David, L. A., and Alm, E. J. (2011). Rapid evolutionary innovation during an Archaean genetic expansion. Nature 469, 93–96. doi: 10.1038/nature09649
Didelot, X., and Wilson, D. J. (2015). ClonalFrameML: efficient inference of recombination in whole bacterial genomes. PLoS Comput. Biol. 11:e1004041. doi: 10.1371/journal.pcbi.1004041
Dunne, E. F., Burman, W. J., and Wilson, M. L. (1998). Streptomyces pneumonia in a patient with human immunodeficiency virus infection: case report and review of the literature on invasive Streptomyces infections. Clin. Infect. Dis. 27, 93–96. doi: 10.1086/514612
Edgar, R. C. (2007). PILER-CR: fast and accurate identification of CRISPR repeats. BMC Bioinformatics 8:18. doi: 10.1186/1471-2105-8-18
Eloe, E. A., Lauro, F. M., Vogel, R. F., and Bartlett, D. H. (2008). The deep-sea bacterium Photobacterium profundum SS9 utilizes separate flagellar systems for swimming and swarming under high-pressure conditions. Appl. Environ. Microbiol. 74, 6298–6305. doi: 10.1128/AEM.01316-08
Fu, H. L., Meng, Y. L., Ordonez, E., Villadangos, A. F., Bhattacharjee, H., Gil, J. A., et al. (2009). Properties of arsenite efflux permeases (Acr3) from Alkaliphilus metalliredigens and Corynebacterium glutamicum. J. Biol. Chem. 284, 19887–19895. doi: 10.1074/jbc.M109.011882
Govindarajan, S., Elisha, Y., Nevo-Dinur, K., and Amster-Choder, O. (2013). The general phosphotransferase system proteins localize to sites of strong negative curvature in bacterial cells. Mbio 4:e00443-13. doi: 10.1128/mBio.00443-13
Grubbs, K. J., Biedermann, P. H. W., Suen, G., Adams, S. M., Moeller, J. A., Klassen, J. L., et al. (2011). Genome sequence of Streptomyces griseus strain XylebKG-1, an ambrosia beetle-associated actinomycete. J. Bacteriol. 193, 2890–2891. doi: 10.1128/Jb.00330-11
Gudhka, R. K., Neilan, B. A., and Burns, B. P. (2015). Adaptation, ecology, and evolution of the halophilic stromatolite archaeon halococcus hamelinensis inferred through genome analyses. Archaea 2015, 241608. doi: 10.1155/2015/241608
Guindon, S., Dufayard, J. F., Lefort, V., Anisimova, M., Hordijk, W., and Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
He, F. L., Nair, G. R., Soto, C. S., Chang, Y. C., Hsu, L. L., Ronzone, E., et al. (2009). Molecular basis of ChvE function in sugar binding, sugar utilization, and virulence in Agrobacterium tumefaciens. J. Bacteriol. 191, 5802–5813. doi: 10.1128/Jb.00451-09
Hopwood, D. A. (2006). Soil to genomics: the Streptomyces chromosome. Annu. Rev. Genet. 40, 1–23. doi: 10.1146/annurev.genet.40.110405.090639
Hughes, M. F. (2002). Arsenic toxicity and potential mechanisms of action. Toxicol. Lett. 133, 1–16. doi: 10.1016/S0378-4274(02)00084-X
Hulcr, J., Adams, A. S., Raffa, K., Hofstetter, R. W., Klepzig, K. D., and Currie, C. R. (2011). Presence and diversity of Streptomyces in Dendroctonus and sympatric bark beetle galleries across North America. Microb. Ecol. 61, 759–768. doi: 10.1007/s00248-010-9797-0
Hyatt, D., Chen, G. L., LoCascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119
Ivars-Martinez, E., Martin-Cuadrado, A. B., D’Auria, G., Mira, A., Ferriera, S., Johnson, J., et al. (2008). Comparative genomics of two ecotypes of the marine planktonic copiotroph Alteromonas macleodii suggests alternative lifestyles associated with different kinds of particulate organic matter. ISME J. 2, 1194–1212. doi: 10.1038/ismej.2008.74
Jack, D. L., Yang, N. M., and Saier, M. H. (2001). The drug/metabolite transporter superfamily. Eur. J. Biochem. 268, 3620–3639. doi: 10.1046/j.1432-1327.2001.02265.x
Jorgensen, B. B., and Boetius, A. (2007). Feast and famine – microbial life in the deep-sea bed. Nat. Rev. Microbiol. 5, 770–781. doi: 10.1038/Nrmicro1745
Kämpfer, P. (2006). “The family streptomycetaceae, part I: taxonomy,” in The Prokaryotes: A Handbook on the Biology of Bacteria, eds M. Dworkin, S. Falkow, E. Rosenberg, K.-H. Schleifer, and E. Stackebrandt (Berlin: Springer), 538–604.
Katoh, K., Misawa, K., Kuma, K., and Miyata, T. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066. doi: 10.1093/nar/gkf436
Kempf, B., and Bremer, E. (1998). Uptake and synthesis of compatible solutes as microbial stress responses to high-osmolality environments. Arch. Microbiol. 170, 319–330. doi: 10.1007/s002030050649
Klassen, J. L., Adams, S. M., Bramhacharya, S., Giles, S. S., Goodwin, L. A., Woyke, T., et al. (2011). Draft genome sequence of Streptomyces sp strain wigar10, isolated from a surface-sterilized garlic bulb. J. Bacteriol. 193, 6999–7000. doi: 10.1128/Jb.06257-11
Kleiner, D. (1985). Bacterial ammonium transport. FEMS Microbiol. Lett. 32, 87–100. doi: 10.1111/j.1574-6968.1985.tb01185.x
Komorsky-Lovric, S. (1998). A simple method for detection of manganese in marine sediments. Croat. Chem. Acta 71, 263–269. doi: 10.1016/j.talanta.2011.01.025
Kuwahara, H., Yoshida, T., Takaki, Y., Shimamura, S., Nishi, S., Harada, M., et al. (2007). Reduced genome of the thioautotrophic intracellular symbiont in a deep-sea clam, Calyptogena okutanii. Curr. Biol. 17, 881–886. doi: 10.1016/j.cub.2007.04.039
Lagesen, K., Hallin, P., Rodland, E. A., Staerfeldt, H. H., Rognes, T., and Ussery, D. W. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108. doi: 10.1093/nar/gkm160
Lee, S. H., Moon, K., Kim, H., Shin, J., Oh, D. C., and Oh, K. B. (2014). Bahamaolide A from the marine-derived Streptomyces sp. CNQ343 inhibits isocitrate lyase in Candida albicans. Bioorg. Med. Chem. Lett. 24, 4291–4293. doi: 10.1016/j.bmcl.2014.07.021
Lefebure, T., Richards, V. P., Lang, P., Pavinski-Bitar, P., and Stanhope, M. J. (2012). Gene repertoire evolution of Streptococcus pyogenes inferred from phylogenomic analysis with Streptococcus canis and Streptococcus dysgalactiae. PLoS ONE 7:e37607. doi: 10.1371/journal.pone.0037607
Levy, S. B. (1992). Active efflux mechanisms for antimicrobial resistance. Antimicrob. Agents Chemother. 36, 695–703. doi: 10.1128/AAC.36.4.695
Li, F., Jiang, P., Zheng, H., Wang, S., Zhao, G., Qin, S., et al. (2011). Draft genome sequence of the marine bacterium Streptomyces griseoaurantiacus M045, which produces novel manumycin-type antibiotics with a pABA core component. J. Bacteriol. 193, 3417–3418. doi: 10.1128/JB.05053-11
Li, W., and Tang, X. (2012). Manganese trafficking, metabolism and homeostasis. Chin. Bull. Life Sci. 24, 867–880.
Liang, W. L., Zhao, Y. B., Chen, C. X., Cui, X. Y., Yu, J., Xiao, J. F., et al. (2012). Pan-genomic analysis provides insights into the genomic variation and evolution of Salmonella paratyphi A. PLoS ONE 7:e45346. doi: 10.1371/journal.pone.0045346
Liu, W. T., Kersten, R. D., Yang, Y. L., Moore, B. S., and Dorrestein, P. C. (2011). Imaging mass spectrometry and genome mining via short sequence tagging identified the anti-infective agent arylomycin in Streptomyces roseosporus. J. Am. Chem. Soc. 133, 18010–18013. doi: 10.1021/ja2040877
Lowe, T. M., and Eddy, S. R. (1997). tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964. doi: 10.1093/nar/25.5.0955
Luo, R. B., Liu, B. H., Xie, Y. L., Li, Z. Y., Huang, W. H., Yuan, J. Y., et al. (2012). SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1, 18. doi: 10.1186/2047-217x-1-18
Lyons, J. A., Parker, J. L., Solcan, N., Brinth, A., Li, D., Shah, S. T. A., et al. (2014). Structural basis for polyspecificity in the POT family of proton-coupled oligopeptide transporters. EMBO Rep. 15, 886–893. doi: 10.15252/embr.201338403
Marri, P. R., Hao, W., and Golding, G. B. (2006). Gene gain and gene loss in streptococcus: is it driven by habitat? Mol. Biol. Evol. 23, 2379–2391. doi: 10.1093/molbev/msl115
Marzan, L. W., and Shimizu, K. (2011). Metabolic regulation of Escherichia coli and its phoB and phoR genes knockout mutants under phosphate and nitrogen limitations as well as at acidic condition. Microb. Cell Fact. 10, 39. doi: 10.1186/1475-2859-10-39
Medini, D., Donati, C., Tettelin, H., Masignani, V., and Rappuoli, R. (2005). The microbial pan-genome. Curr. Opin. Genet. Dev. 15, 589–594. doi: 10.1016/j.gde.2005.09.006
Miao, V., Coeffet-LeGal, M. F., Brian, P., Brost, R., Penn, J., Whiting, A., et al. (2005). Daptomycin biosynthesis in Streptomyces roseosporus: cloning and analysis of the gene cluster and revision of peptide stereochemistry. Microbiology 151, 1507–1523. doi: 10.1099/mic.0.27757-0
Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A. C., and Kanehisa, M. (2007). KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 35, W182–W185. doi: 10.1093/Nar/Gkm321
Mulligan, C., Fischer, M., and Thomas, G. H. (2011). Tripartite ATP-independent periplasmic (TRAP) transporters in bacteria and archaea. FEMS Microbiol. Rev. 35, 68–86. doi: 10.1111/j.1574-6976.2010.00236.x
Muzzi, A., Masignani, V., and Rappuoli, R. (2007). The pan-genome: towards a knowledge-based discovery of novel targets for vaccines and antibacterials. Drug Discov. Today 12, 429–439. doi: 10.1016/j.drudis.2007.04.008
Nakamaru, Y., Takahashi, Y., Unemoto, T., and Nakamura, T. (1999). Mechanosensitive channel functions to alleviate the cell lysis of marine bacterium, Vibrio alginolyticus, by osmotic downshock. FEBS Lett. 444, 170–172. doi: 10.1016/S0014-5793(99)00054-X
Naughton, L. M., Blumerman, S. L., Carlberg, M., and Boyd, E. F. (2009). Osmoadaptation among Vibrio species and unique genomic features and physiological responses of Vibrio parahaemolyticus. Appl. Environ. Microbiol. 75, 2802–2810. doi: 10.1128/Aem.01698-08
Nikaido, H. (1994). Prevention of drug access to bacterial targets: permeability barriers and active efflux. Science 264, 382–388. doi: 10.1126/science.8153625
Nunez, M. F., Kwon, O., Wilson, T. H., Aguilar, J., Baldoma, L., and Lin, E. C. C. (2002). Transport of L-lactate, D-lactate, and glycolate by the LldP and GlcA membrane carriers of Escherichia coli. Biochem. Biophys. Res. Commun. 290, 824–829. doi: 10.1006/bbrc.2001.6255
Ochman, H., Lawrence, J. G., and Groisman, E. A. (2000). Lateral gene transfer and the nature of bacterial innovation. Nature 405, 299–304. doi: 10.1038/35012500
Ohnishi, Y., Ishikawa, J., Hara, H., Suzuki, H., Ikenoya, M., Ikeda, H., et al. (2008). Genome sequence of the streptomycin-producing microorganism Streptomyces griseus IFO 13350. J. Bacteriol. 190, 4050–4060. doi: 10.1128/Jb.00204-08
Orsini, M., and Romano-Spica, V. (2001). A microwave-based method for nucleic acid isolation from environmental samples. Lett. Appl. Microbiol. 33, 17–20. doi: 10.1046/j.1472-765X.2001.00938.x
Ostrowski, S. R., Wilbur, S., Chou, C. H. S. J., Pohl, H. R., Stevens, Y. W., Allred, P. M., et al. (1999). Agency for toxic substances and disease Registry’s 1997 priority list of hazardous substances. Latent effects-carcinogenesis, neurotoxicology, and developmental deficits in humans and animals. Toxicol. Ind. Health 15, 602–644. doi: 10.1177/074823379901500702
Padan, E., and Schuldiner, S. (1994). Molecular physiology of the Na+/H+ antiporter in Escherichia-coli. J. Exp. Biol. 196, 443–456.
Padidam, M., Sawyer, S., and Fauquet, C. M. (1999). Possible emergence of new geminiviruses by frequent recombination. Virology 265, 218–225. doi: 10.1006/viro.1999.0056
Qin, Q. L., Li, Y., Zhang, Y. J., Zhou, Z. M., Zhang, W. X., Chen, X. L., et al. (2011). Comparative genomics reveals a deep-sea sediment-adapted life style of Pseudoalteromonas sp. SM9913. ISME J. 5, 274–284. doi: 10.1038/ismej.2010.103
Qin, Q. L., Xie, B. B., Yu, Y., Shu, Y. L., Rong, J. C., Zhang, Y. J., et al. (2014). Comparative genomics of the marine bacterial genus Glaciecola reveals the high degree of genomic diversity and genomic characteristic for cold adaptation. Environ. Microbiol. 16, 1642–1653. doi: 10.1111/1462-2920.12318
Qin, Q. L., Zhang, X. Y., Wang, X. M., Liu, G. M., Chen, X. L., Xie, B. B., et al. (2010). The complete genome of Zunongwangia profunda SM-A87 reveals its adaptation to the deep-sea environment and ecological role in sedimentary organic nitrogen degradation. BMC Genomics 11:247. doi: 10.1186/1471-2164-11-247
Rao, M., Li, Q. S., Feng, L., Xia, X., Ruan, L. J., Sheng, X. F., et al. (2011). A new aminopeptidase inhibitor from Streptomyces strain HCCB10043 found by UPLC-MS. Anal. Bioanal. Chem. 401, 699–706. doi: 10.1007/s00216-011-5093-1
Ren, Q., Chen, K., and Paulsen, I. T. (2007). TransportDB: a comprehensive database resource for cytoplasmic membrane transport systems and outer membrane channels. Nucleic Acids Res. 35, D274–D279. doi: 10.1093/nar/gkl925
Ren, Q., and Paulsen, I. T. (2005). Comparative analyses of fundamental differences in membrane transport capabilities in prokaryotes and eukaryotes. PLoS Comput. Biol. 1:e27. doi: 10.1371/journal.pcbi.0010027
Ren, Q., and Paulsen, I. T. (2007). Large-scale comparative genomic analyses of cytoplasmic membrane transport systems in prokaryotes. J. Mol. Microbiol. Biotechnol. 12, 165–179. doi: 10.1159/000099639
Revelle, W. (2015). psych: Procedures for Psychological, Psychometric, and Personality Research. Available at: http://CRAN.R-project.org/package=psych
Richards, V. P., Palmer, S. R., Bitar, P. D. P., Qin, X., Weinstock, G. M., Highlander, S. K., et al. (2014). Phylogenomics and the dynamic genome evolution of the genus Streptococcus. Genome Biol. Evol. 6, 741–753. doi: 10.1093/gbe/evu048
Roberts, M. F. (2004). Osmoadaptation and osmoregulation in archaea: update 2004. Front. Biosci. 9:1999–2019. doi: 10.2741/1366
Rosen, B. P. (1996). Bacterial resistance to heavy metals and metalloids. J. Biol. Inorg. Chem. 1, 273–277. doi: 10.1007/s007750050053
Tang, L., Li, Y., Deng, X., Johnston, R. N., Liu, G. R., and Liu, S. L. (2013). Defining natural species of bacteria: clear-cut genomic boundaries revealed by a turning point in nucleotide sequence divergence. BMC Genomics 14:489. doi: 10.1186/1471-2164-14-489
Tatusov, R. L., Natale, D. A., Garkavtsev, I. V., Tatusova, T. A., Shankavaram, U. T., Rao, B. S., et al. (2001). The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 29, 22–28. doi: 10.1093/nar/29.1.22
Tettelin, H., Masignani, V., Cieslewicz, M. J., Donati, C., Medini, D., Ward, N. L., et al. (2005). Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome.” Proc. Natl. Acad. Sci. U.S.A. 102, 13950–13955. doi: 10.1073/pnas.0506758102
Tettelin, H., Riley, D., Cattuto, C., and Medini, D. (2008). Comparative genomics: the bacterial pan-genome. Curr. Opin. Microbiol. 11, 472–477. doi: 10.1016/j.mib.2008.09.006
Tian, X. P., Xu, Y., Zhang, J., Li, J., Chen, Z., Kim, C. J., et al. (2012). Streptomyces oceani sp nov., a new obligate marine actinomycete isolated from a deep-sea sample of seep authigenic carbonate nodule in South China Sea. Antonie Van Leeuwenhoek 102, 335–343. doi: 10.1007/s10482-012-9743-x
Vernikos, G., Medini, D., Riley, D. R., and Tettelin, H. (2015). Ten years of pan-genome analyses. Curr. Opin. Microbiol. 23, 148–154. doi: 10.1016/j.mib.2014.11.016
Vicente, C. M., Santos-Aberturas, J., Guerra, S. M., Payero, T. D., Martin, J. F., and Aparicio, J. F. (2009). PimT, an amino acid exporter controls polyene production via secretion of the quorum sensing pimaricin-inducer PI-factor in Streptomyces natalensis. Microb. Cell Fact. 8, 33. doi: 10.1186/1475-2859-8-33
Wang, F. P., Wang, J. B., Jian, H. H., Zhang, B., Li, S. K., Wang, F., et al. (2008). Environmental adaptation: genomic analysis of the piezotolerant and psychrotolerant deep-sea iron reducing bacterium Shewanella piezotolerans WP3. PLoS ONE 3:e1937. doi: 10.1371/Journal.Pone.0001937
Wang, L. F., Wang, S. M., He, Q., Yu, T. F., Li, Q. L., and Hong, B. (2012). Draft genome sequence of Streptomyces globisporus C-1027, which produces an antitumor antibiotic consisting of a nine-membered enediyne with a chromoprotein. J. Bacteriol. 194, 4144. doi: 10.1128/Jb.00797-12
Weyand, S., Shimamura, T., Yajima, S., Suzuki, S., Mirza, O., Krusong, K., et al. (2008). Structure and molecular mechanism of a nucleobase-cation-symport-1 family transporter. Science 322, 709–713. doi: 10.1126/science.1164440
Wommack, K. E., and Colwell, R. R. (2000). Virioplankton: viruses in aquatic ecosystems. Microbiol. Mol. Biol. Rev. 64, 69–114. doi: 10.1128/MMBR.64.1.69-114.2000
Xiao, J., Zhang, Z., Wu, J., and Yu, J. (2015). A brief review of software tools for pangenomics. Genomics Proteomics Bioinformatics 13, 73–76. doi: 10.1016/j.gpb.2015.01.007
Xiong, Z. Q., and Wang, Y. (2012). Draft genome sequence of the marine Streptomyces sp strain AA1529, isolated from the Yellow Sea. J. Bacteriol. 194, 5474–5475. doi: 10.1128/Jb.01247-12
Yen, M. R., Tseng, Y. H., Simic, P., Sahm, H., Eggeling, L., and Saier, M. H. (2002). The ubiquitous ThrE family of putative transmembrane amino acid efflux transporters. Res. Microbiol. 153, 19–25. doi: 10.1016/S0923-2508(01)01281-5
Yi, H. G., and Jin, L. (2013). Co-phylog: an assembly-free phylogenomic approach for closely related organisms. Nucleic Acids Res. 41:e75. doi: 10.1093/nar/gkt003
Yurgel, S., Mortimer, M. W., Rogers, K. N., and Kahn, M. L. (2000). New substrates for the dicarboxylate transport system of Sinorhizobium meliloti. J. Bacteriol. 182, 4216–4221. doi: 10.1128/Jb.182.15.4216-4221.2000
Zerbino, D. R., and Birney, E. (2008). Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18, 821–829. doi: 10.1101/gr.074492.107
Zhang, A. D., Yang, M., Hu, P., Wu, J. Y., Chen, B., Hua, Y. F., et al. (2011). Comparative genomic analysis of Streptococcus suis reveals significant genomic diversity among different serotypes. BMC Genomics 12:523. doi: 10.1186/1471-2164-12-523
Zhao, J. L., and Binns, A. N. (2011). Characterization of the mmsAB-araD1 (gguABC) genes of Agrobacterium tumefaciens. J. Bacteriol. 193, 6586–6596. doi: 10.1128/Jb.05790-11
Zhao, X. Q., Geng, X., Chen, C., Chen, L. Y., Jiao, W. C., and Yang, C. (2012). Draft genome sequence of the marine actinomycete Streptomyces sulphureus L180, isolated from marine sediment. J. Bacteriol. 194, 4482–4482. doi: 10.1128/Jb.00900-12
Zhao, Y. B., Jia, X. M., Yang, J. H., Ling, Y. C., Zhang, Z., Yu, J., et al. (2014). PanGP: a tool for quickly analyzing bacterial pan-genome profile. Bioinformatics 30, 1297–1299. doi: 10.1093/bioinformatics/btu017
Keywords: comparative genomics, streptomycete, pan-genomics, genome dynamics, marine environment adaptation
Citation: Tian X, Zhang Z, Yang T, Chen M, Li J, Chen F, Yang J, Li W, Zhang B, Zhang Z, Wu J, Zhang C, Long L and Xiao J (2016) Comparative Genomics Analysis of Streptomyces Species Reveals Their Adaptation to the Marine Environment and Their Diversity at the Genomic Level. Front. Microbiol. 7:998. doi: 10.3389/fmicb.2016.00998
Received: 05 May 2016; Accepted: 13 June 2016;
Published: 27 June 2016.
Edited by:
Feng Gao, Tianjin University, ChinaReviewed by:
Jonathan Badger, National Cancer Institute, USAQin Ma, South Dakota State University, USA
Copyright © 2016 Tian, Zhang, Yang, Chen, Li, Chen, Yang, Li, Zhang, Zhang, Wu, Zhang, Long and Xiao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lijuan Long, bG9uZ2xqQHNjc2lvLmFjLmNu; Jingfa Xiao, xiaojingfa@big.ac.cn
†These authors have contributed equally to this work.