 Tomas Erban1
Tomas Erban1 Pavel B. Klimov2,3
Pavel B. Klimov2,3 Jaroslav Smrz4Thomas W. Phillips5Marta Nesvorna1Jan Kopecky1
Jaroslav Smrz4Thomas W. Phillips5Marta Nesvorna1Jan Kopecky1 Jan Hubert1*
Jan Hubert1*- 1Biologically Active Substances in Crop Protection, Crop Research Institute, Prague, Czech Republic
- 2Department of Ecology and Evolutionary Biology, University of Michigan, Ann Arbor, MI, USA
- 3Faculty of Biology, Tyumen State University, Tyumen, Russia
- 4Department of Zoology, Faculty of Science, Charles University in Prague, Prague, Czech Republic
- 5Department of Entomology, Kansas State University, Manhattan, KS, USA
Background: Tyrophagus putrescentiae colonizes different human-related habitats and feeds on various post-harvest foods. The microbiota acquired by these mites can influence the nutritional plasticity in different populations. We compared the bacterial communities of five populations of T. putrescentiae and one mixed population of T. putrescentiae and T. fanetzhangorum collected from different habitats.
Material: The bacterial communities of the six mite populations from different habitats and diets were compared by Sanger sequencing of cloned 16S rRNA obtained from amplification with universal eubacterial primers and using bacterial taxon-specific primers on the samples of adults/juveniles or eggs. Microscopic techniques were used to localize bacteria in food boli and mite bodies. The morphological determination of the mite populations was confirmed by analyses of CO1 and ITS fragment genes.
Results: The following symbiotic bacteria were found in compared mite populations: Wolbachia (two populations), Cardinium (five populations), Bartonella-like (five populations), Blattabacterium-like symbiont (three populations), and Solitalea-like (six populations). From 35 identified OTUs97, only Solitalea was identified in all populations. The next most frequent and abundant sequences were Bacillus, Moraxella, Staphylococcus, Kocuria, and Microbacterium. We suggest that some bacterial species may occasionally be ingested with food. The bacteriocytes were observed in some individuals in all mite populations. Bacteria were not visualized in food boli by staining, but bacteria were found by histological means in ovaria of Wolbachia-infested populations.
Conclusion: The presence of Blattabacterium-like, Cardinium, Wolbachia, and Solitalea-like in the eggs of T. putrescentiae indicates mother to offspring (vertical) transmission. Results of this study indicate that diet and habitats influence not only the ingested bacteria but also the symbiotic bacteria of T. putrescentiae.
Introduction
Mites, like insects, derive nutritive advantages from persistent associations with microorganisms (Van Asselt, 1999; Dillon and Dillon, 2004; Douglas, 2015). Microorganisms associated with insects can synthetize various nutrients, provide essential amino acids and contribute to digestive processes (Dillon and Dillon, 2004; Douglas, 2009). Domestic mites are inhabitants of human-related habitats such as homes, carpets, beds, and stored food (Spieksma, 1997). Although these mites are commensals on the trophic level, due to allergen production and vectoring their microorganisms are of medical importance (Colloff, 2009). It is hypothesized that mites invaded human-related environments through two different routes: house dust mites (HDMs; e.g., Dermatophagoides pteronyssinus and D. farinae) likely shifted from a parasitic lifestyle back to a commensal life style (Klimov and OConnor, 2013), while ancestors of stored product mites (SPMs; e.g., Acarus siro, Lepidoglyphus destructor, Tyrophagus putrescentiae) were free-living and invaded human houses via the nests of birds and small mammals (OConnor, 1979, 1982).
Tyrophagus putrescentiae (Schrank, 1781) has been reported from agricultural soils (Smrz and Jungova, 1989), commercial bumblebee colonies (Rozej et al., 2012), and the nests of birds and small mammals (Solarz et al., 1999). This mite is very common in human-created habitats such as dust in urban environments, medical and laboratory facilities, farms, the food industry (Franz et al., 1997; Solarz et al., 2007), and in fungal and insect cultures in laboratories (Duek et al., 2001). The most typical food sources of T. putrescentiae are protein and fat-rich substances such as grain germ, nuts, sunflower, oil rape seeds, cheese, ham, and dry dog food (Zakhvatkin, 1959; Robertson, 1961; Hughes, 1976; Garcia, 2004; Palyvos et al., 2008; Erban et al., 2015, 2016; Rybanska et al., 2015). Our broad hypothesis in work reported here is that successful colonization of such a wide range of habitats is facilitated by an inherently broad food plasticity in T. putrescentiae that is likely due to symbiotic microbes.
In general, the ancestors of stored-product mites were fungivorous (OConnor, 1979, 1982). Fungivory is well-documented for T. putrescentiae (Smrz and Catska, 1987; Hubert et al., 2004; Nesvorna et al., 2012). The mite can nutritionally benefit from interactions with bacteria or both fungi and bacteria. T. putrescentiae produces bacteriolytic enzymes which hydrolyze the cell walls of gram positive bacteria (Erban and Hubert, 2008). T. putrescentiae has been found to host bacterial communities in the gut, parenchymal tissues and reproductive tract with various interactions (Hubert et al., 2012a; Kopecky et al., 2014a,b). T. putrescentiae is associated with bacterial parasites or symbionts (Cardinium and Wolbachia) inhabiting the reproductive tract and fat body (Kopecky et al., 2013; Brown and Lloyd, 2015). Another association was described for neutral and alkaline proteases, and exo-chitinase producing Bacillus cereus in T. putrescentiae. B. cereus was presented in all samples and the exoenzymes can interact to utilization of the food sources for T. putrescentiae; however, the addition of B. cereus to the diet led to a substantial suppression of mite population growth (Erban et al., 2016). In contrast, another bacterium, Micrococcus lysodeikticus, had no significant influence on the population growth of T. putrescentiae (Erban and Hubert, 2008). However, those associated bacteria affect the fitness of mites; therefore, their indirect effects on habitat colonization or diet utilization by mites are expected.
The above facts suggest that interactions of mites with microorganisms are important for adaptation to a nutritional food source in human-made habitats as well as in soil. A diet switches from plant-derived food to various fungal species caused changes in the bacterial community associated with the gut and parenchymal tissues of T. putrescentiae (Smrz, 2003; Hubert et al., 2012b) and induced bacteriocytes (sometimes called as extraintestinal bacterial bodies or bacteriome; Smrz and Catska, 1989; Smrz, 2003; Smrz and Soukalova, 2008). It is likely that T. putrescentiae acquires bacteria with chitinolytic activity (i.e., B. cereus and Serratia marcescens) to digest chitin from fungal cell walls or mite bodies (Smrz et al., 1991; Smrz and Catska, 2010; Erban et al., 2016). The bacteria producing exo-chitinases can contribute to degradation of mite exuviae, mite bodies or food boluses consisting of chitin, including the peritrophic membrane (Erban et al., 2016).
In laboratory experiments adding antibiotics to the diet did not eliminate the bacteria in T. putrescentiae; Kocuria and Bacillus were still present in surface cleaned mite body homogenates (Kopecky et al., 2014b). Our recent study showed a population-specific density-dependent growth of T. putrescentiae (Rybanska et al., 2015) and also indicated the possibility that T. putrescentiae populations may differ in acquired bacteria (Smrz and Catska, 2010), suggesting a possible habitat influence on mite internal bacteria.
Here, we compare bacterial community of six distinct populations of T. putrescentiae. The comparison is based on Sanger sequencing of bacterial 16S rRNA genes from adults/juveniles. Because we found diverse symbiotic bacterial community in these populations, we also focused on identification of these symbiotic bacteria in the eggs by bacterial taxa specific primers. Finally, we compared sex ratios and guanine contents in T. putrescentiae populations in relation to the presence of symbiotic bacteria. Limitations associated with the small amount of starting wild material do not allow reliable intra-population comparison of bacterial communities because prolonged mite culture maintenance under standardized laboratory conditions can change bacterial communities.
Materials and Methods
Mites
The bacterial communities of three field and three laboratory populations of Tyrophagus putrescentiae (Schrank, 1781) were compared (Table 1). Morphological determination of populations was performed by under a compound microscope using characters described in Klimov and OConnor (2009a,b). In addition, molecular markers were used for subsequent characterization of the populations. All these T. putrescentiae populations were placed into IWAKI 25 cm2 surface area 70 mL tissue-culture flasks (IWAKI flasks; Cat. No. 3100-025; Sterilin, Newport, UK) with their original food (see Table 1), separately. The flasks were placed into Secador desiccator cabinets (Bel-Art Products, Pequannock, NJ, USA) and incubated under controlled conditions at 25 ± 1°C and 85% RH in darkness for up to 3 months; then the mites were sampled.
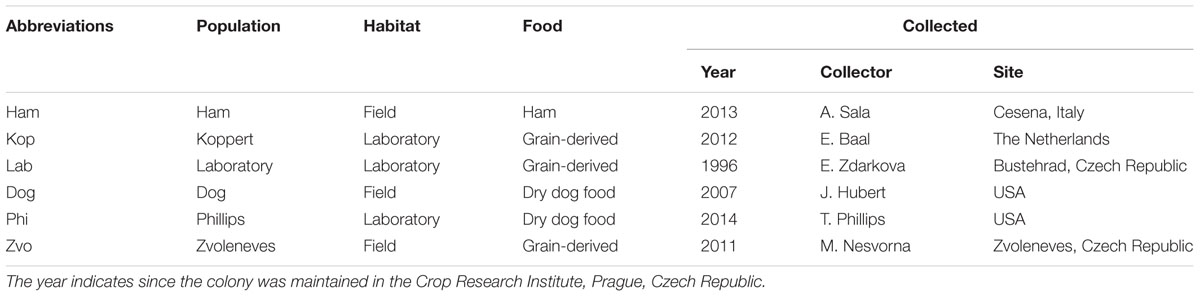
TABLE 1. Sampled populations of Tyrophagus putrescentiae and their habitats at the time of collection.
Tyrophagus putrescentiae mites from field populations were reared for up to 1–2 months in the laboratory. During this time the mite populations multiplied to a level at which sampling of many individuals was possible. For DNA extraction, mites were collected and separated from food particles using a fine-tipped artist’s paint brush, transferred into Eppendorf tubes, and weighed using a microbalance (MS Mettler-Toledo, Greifensee, Switzerland) to obtain 0.05 ± 0.01 g wet weight samples in triplicates per populations. The mean of fresh weight of T. putrescentiae individuals is about 8 μg, so every sample contains ca 6,300 individuals. The Eppendorf tubes with collected mites were filled with 80% ethanol and stored in a refrigerator at 4°C until DNA extraction. Each population of T. putrescentiae was processed in triplicate. For the comparison of the bacterial community of the laboratory population (Table 1) we used previously published sequences of bacteria from laboratory strains (Hubert et al., 2012a,b; Kopecky et al., 2014b). For microanatomical samples, the mites of approximately 100–500 individuals were fixed in modified Bouin-Dubosque-Brasil fluid according to Smrz (1989).
We adopted a method of Stepien and Rodriguez (1973) for eggs accumulation and extraction. The food with mites from rearing flasks was placed on mesh with a size of 176 μm under the water surface. All of the mesh used was polyamide fiber (Silk & Progress, Brnenec, Czech Republic). The females deposited eggs on the water surface after 48 h (Hubert et al., under review). The water was collected and filtrated though the mesh manifold (Stepien and Rodriguez, 1973) using a vacuum pump. The mesh sizes in the manifold were in decreasing order: 411, 300, 206, 176, 139, 109, 86, 42 μm diameter. Next, the eggs were cleaned with ddH20, Tween® 20 (Cat No. P9416, Sigma-Aldrich), bleach, and 80% ethanol (Hubert et al., under review). The eggs were captured at 86 and 42 μm mesh and removed by pipetting into Eppendorf tubes and stored in 80% ethanol. Each sample consisted of 50 eggs in triplicate per population.
Sample Homogenization and DNA Extraction
Prior to DNA extraction, ethanol was replaced, in sequence, by bleach, and then washed three times with sterile phosphate-buffered saline (PBST: 3.2 mM Na2HPO4, 0.5 mM KH2PO4, 1.3 mM KCl, and 135 mM NaCl) with 0.05% w/w Tween® 20 detergent (Cat No. P9416, Sigma-Aldrich, St. Louis, MO, USA) to remove surface microflora. Samples with a total volume of 100 μL PBST were homogenized in a Radnoti tissue grinder (Cat. No. 440613; Monrovia, CA, USA). DNA from the homogenates was extracted using a Wizard® SV Genomic DNA Purification System (Cat No. A2361, Promega) according to the manufacturer’s instructions. Extracted DNA was stored in a freezer at -20°C until the analyses were performed.
Molecular Markers for Identification of T. putrescentiae Populations
The primers (Table 2) spanning a portion of the 5.8S ribosomal DNA (rDNA), the full-length ITS2 region, and a part of the 28S rRNA and primers for the central part of the CO1 region of T. putrescentiae were used to confirm species determinations for mites used in this research (Yang et al., 2011). Amplifications were performed in a C1000 Thermal Cycler (Bio-Rad, Hercules, CA, USA). A total volume of 25 μl polymerase chain reaction (PCR) reaction mixture contained 200 μM dNTPs, 3 mM MgCl2, forward and reverse primers (100 nM each), 0.5 unit Taq polymerase (all from Promega), and 5–30 ng of template DNA that included mite genomic DNA (for reaction conditions, see Yang et al., 2011). The amplicons were done for every sample. The resulting PCR products were visualized by agarose gel electrophoresis. Because we used samples at the population level, amplicons were purified with Wizard® SV Gel and the PCR product clean-up system Kit (Cat No. A9281, Promega) and cloned using pGEM®-T Easy Vector (Cat No. A1380 Promega). We usually selected four clones per sample, i.e., 12 clones per population. Selected clones were sequenced by Macrogen (Seoul, South Korea). The obtained sequences were assembled with CodonCode Aligner, version 5.1.5 (CodonCode Corporation, Dedham, MA, USA).
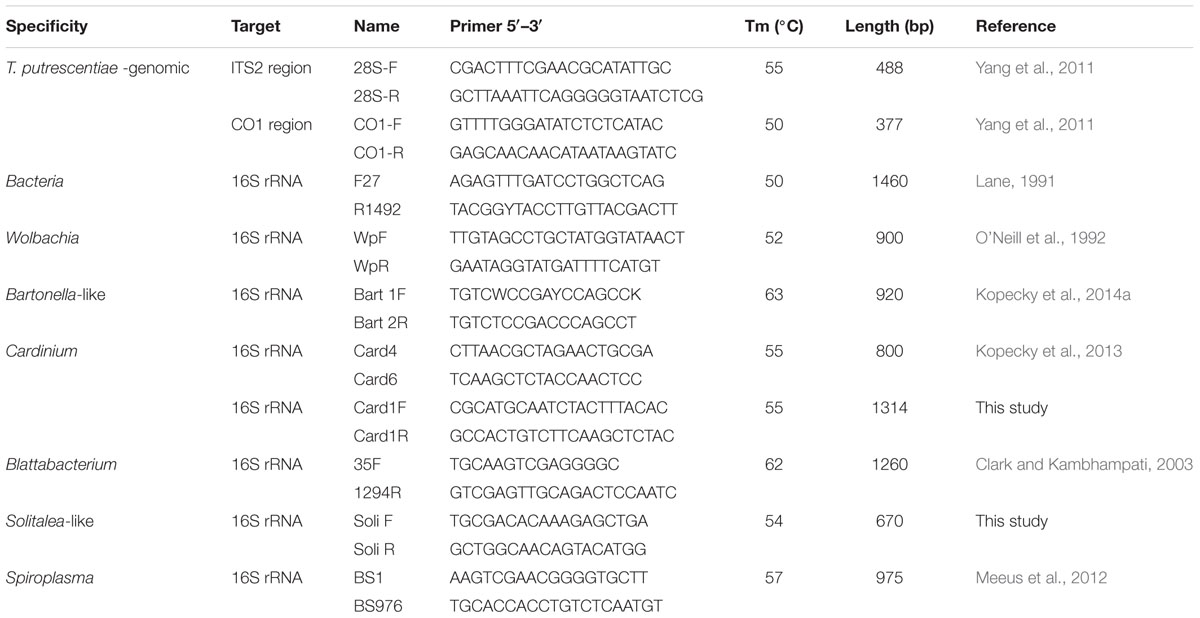
TABLE 2. Primers used for characterization of Tyrophagus putrescentiae populations and detection of bacterial community.
Description of the Bacterial Community
Polymerase chain reaction amplification of 16S rRNA gene fragments was used to characterize the bacterial community using universal 27F/1492R primers (Barbieri et al., 2001). The PCR was done for every sample with the exception of laboratory population of T. putrescentiae. For the latter population, PCR conditions were same as was described above with the exception of annealing temperature (Table 2). The PCR products were cloned using the same protocol as above and 12 colonies were usually selected per sample (i.e., 30 per population) and sequenced in Macrogen. The same laboratory population of T. putrescentiae was analyzed by the same design on the same diet in our previous studies (Hubert et al., 2012a,b; Kopecky et al., 2014b) and we used the clones to characterize the bacterial community of T. putrescentiae laboratory population.
The presence of Cardinium, Wolbachia, Bartonella-like bacteria, Solitalea-like bacteria and Blattabacterium-like bacteria was determined by using taxon-specific primers (see Table 2). As a positive control, we used DNA samples of mites where bacteria had previously been identified using the same methodology (Kopecky et al., 2013, 2014a). The negative control was the double-distilled water used for the PCR master mix preparation. The reaction conditions were the same as described for the molecular markers used for identification of T. putrescentiae populations; the amplification conditions are specified in Table 2. All samples were tested in triplicate. PCR product of the expected size was considered as positive sample. One positive sample was considered as positive presence of bacteria in the population. A negative sample was a sample with no product detected by specific primers. The primer specificity was checked by sequencing of the randomly selected amplicons according to protocol described above.
16S rRNA Sequences
The 16S rRNA sequences were obtained by cloning of amplicons of universal eubacterial primers 27F/1492R (Table 2). The obtained sequences were assembled with CodonCode Aligner and the chimeras were removed using the Mallard and Pintail software (Ashelford et al., 2005, 2006). Altogether, 262 sequences obtained in this study and 176 sequences from previous studies (Hubert et al., 2012a,b; Kopecky et al., 2014b). The sequences were analyzed in MOTHUR v.1.36.0 software (Schloss et al., 2009). The sequences were aligned to Silva reference database (Quast et al., 2013), filtered and clustered and then analyzed and assigned to the operational taxonomic units (OTUs) defined at a distance level of 0.03. The sequences were identified using the Ribosomal Database Project’s (RDP) naive Bayesian rRNA classifier as training set No. 14 (Wang et al., 2007). The representative sequences for individual OTUs97 were compared to the sequences in GenBank using nucleotide Blastn (Altschul et al., 1997).
Phylogenetic Analysis
We conducted phylogenetic analyses on sequences of 16S rRNA, ITS-2 and CO1 of Flavobacteriales symbionts and Wolbachia. The references sequences originated from GenBank or RDP. Alignments of partial 16S rRNA gene sequences were performed using the SILVA Incremental Aligner v.1.2.11 (Pruesse et al., 2012). For the analysis of phylogenetic relationships, the best-fit model of nucleotide substitution was selected using jModelTest v.2.1.7 (Guindon and Gascuel, 2003; Darriba et al., 2012). Based on the selection, the general time reversible model with a proportion of invariable sites and gamma distribution (GTR+G+I), was employed to infer phylogenies in a Bayesian framework in PhyloBayes-MPI, v.1.4e (Lartillot et al., 2009) and the maximum likelihood framework in PhyML v.3.0 (Guindon et al., 2010). Phylograms were visualized in MEGA 6 (Tamura et al., 2007).
Microanatomical Analyses
Sections
Mites were fixed in modified Bouin-Dubosque fluid (Smrz, 1989) for 3 days and then transferred to paraffin. The fixation fluid was replaced by 100% isopropyl alcohol for 12 h (two times), isopropyl alcohol/methyl benzoate (1/1 v/v) for 12 h (two times), methyl benzoate for 12 h (two times), benzene for 2 h, benzene/paraffin (1/1 v/v) for 12 h at 48°C, and paraffin for 12 h (two times) at 56°C (Hubert et al., 1999). Mites were transferred from containers to Peel-A-Way® embedding molds (Polysciences, Eppelheim, Germany) and embedded in Paraplast Plus® (Cat No. 39602004, Leica Biosystems, Nussloch, Germany) at 56°C. Paraffin blocks were sectioned to 4–6 μm sections on Microm HM 200 ErgoStar Microtome (Carl Zeiss, Jena, Germany).
Staining
The slides with sections were placed into different staining solutions equally. The sections were stained by Masson’s triple stain combined with PAS (periodic acid and Schiff agent) and Mann-Dominic and Ziehl–Neelsen staining for bacterial visualization.
Observations
At least 15 specimens per population were observed using an Axioskop compound microscope equipped with a digital camera and Axiovision software (Carl Zeiss).
Visualization in Microscopic Slides
The mites were mounted on permanent slides in Liquide de-Swan medium (distilled water, 20 mL; gum arabic, 15 g; chloral hydrate 50 g and glucose 3 g, glacial acetic acid 5 mL; Kramar, 1953). The sexing of adults and a quantitative estimate of guanine granulae were performed using a compound microscope. Due to the transparency of the mite body, the granulae were visible without staining (Figure 9). At least 100 specimens per one-sex determination and 30 per guanine quantification were observed. The semiquantitative categories were as follows: (0) no guanine granules, (1) low number, the granules filled less than 25% of the hysterosoma, (2) intermediate – granules filled between 25 and 50% of the hysterosoma, and (3) massive, granules filled more than 50% of the hysterosoma (Figure 9).
Statistical Analyses
The similarity of bacterial community in populations was based on analyses of 16S rRNA clones’ library. The shared file was constructed in MOTHUR. The diversity indexes, rarefaction and the comparison of population using Principal coordinate analyses with Euclidian and Jaccard distances were calculated in PAST 3 software (Hammer et al., 2001). The heatmap was constructed in XLSTAT (Addinsoft, New York, NY, USA) using filtering by standard deviation and reduction of low abundant OTUs. The data describing sex ratio and guanine contents of mite bodies were tested by a chi-square test with Bonferroni correction in STATISTIX 9 software (Analytical Software, Tallahassee, FL, USA).
Results
The Comparison of CO1 and ITS in T. putrescentiae Populations
The taxonomic comparison based on Bayesian analyses of CO1 showed that observed T. putrescentiae populations (i.e., Laboratory, Dog, Koppert, and Phillips) clustered together with the known CO1 sequences from GenBank. The exception occurred with the field populations from Zvoleneves, which formed two separate clusters: (i) four sequences (Z 1, 2, 4, 5) formed one cluster with A. siro and (ii) the next sequences clustered to Tyrophagus similis (Figure 1). Using a diagnostic CO1 dataset of 25 identified species of Tyrophagus (PBK, unpublished), we matched these four sequences with Tyrophagus fanetzhangorum. Seven other sequences clustered with T. putrescentiae, based on both GenBank data and our Tyrophagus dataset. The Bayesian analyses of ITS confirm the previous classification for the laboratory populations, Dog, Koppert, and Phillips with T. putrescentiae and similar to the CO1 data, the field Zvoleneves population (eight sequences) clustered with T. fanetzhangorum (Figure 2).
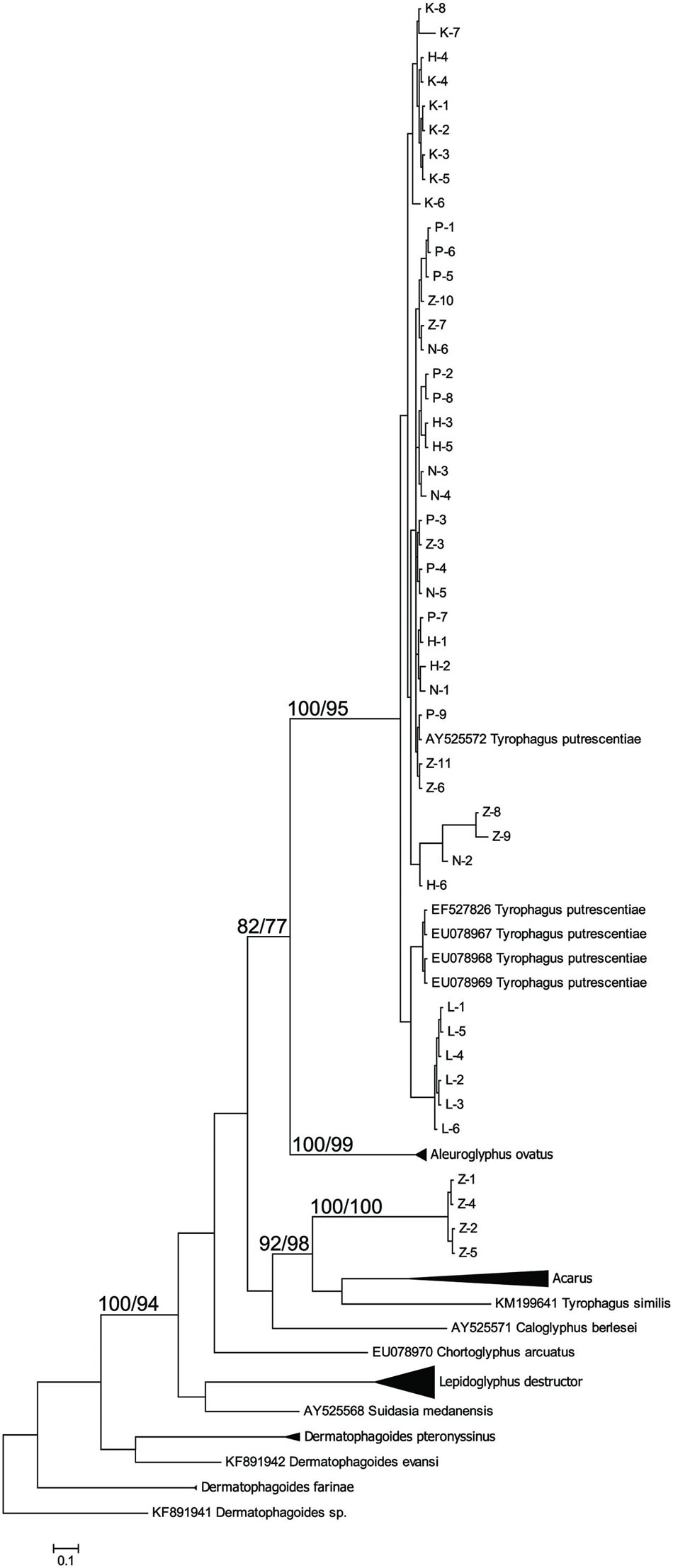
FIGURE 1. Phylogenetic analysis of the CO1 clones of Tyrophagus putrescentiae with the reference sequences from other related astigmatid mites (Webster et al., 2004; Dermauw et al., 2009; Klimov and OConnor, 2009a,b; Bochkov et al., 2014; Ge et al., 2014; Sun et al., 2014). The phylogeny was inferred by Bayesian analysis of 46 partial CO1 gene sequences from observed populations, i.e., L, laboratory; K, Koppert; P, Phillips; D, Dog; H, Ham; and Z, field Zvoleneves. Branch lengths correspond to the mean posterior estimates of evolutionary distances (scale bar: 0.5). Branch labels indicate the Bayesian posterior probabilities and bootstrap support values from maximum likelihood analysis.
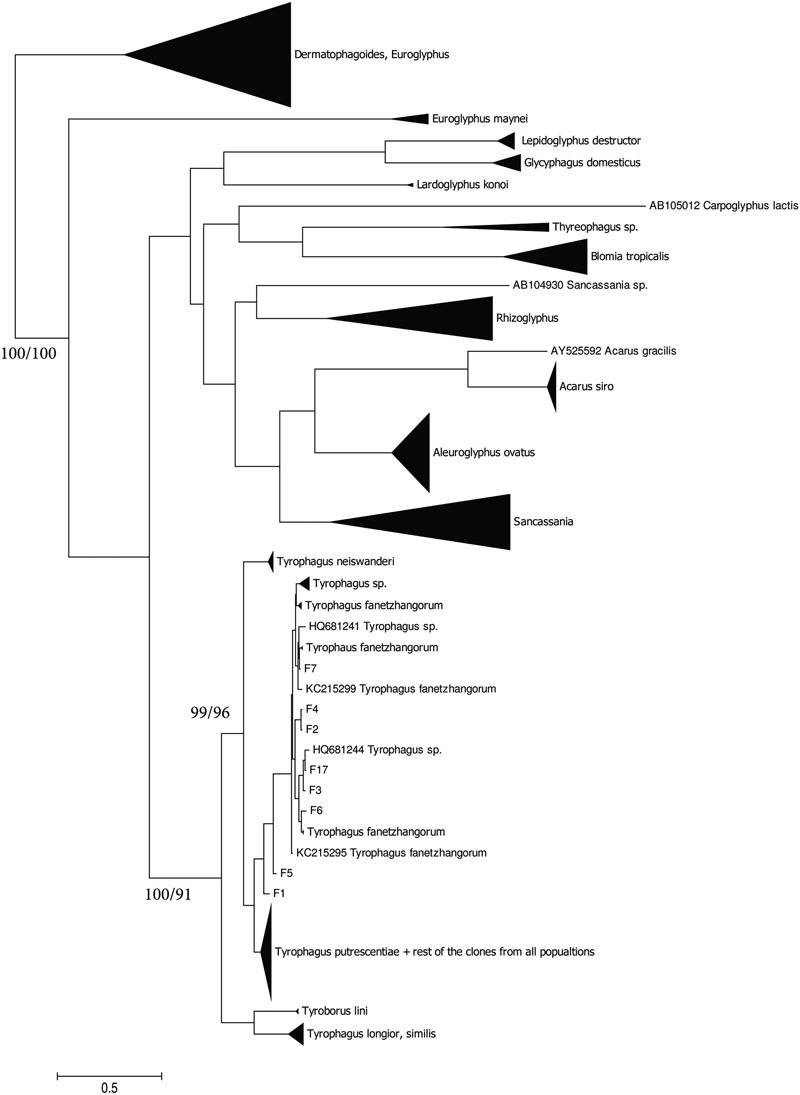
FIGURE 2. Phylogenetic analysis of ITS clones of T. putrescentiae with the reference sequences from other related astigmatid mites (Noge et al., 2005; Liu et al., 2006; Klimov and OConnor, 2008, 2013; Yang et al., 2011; Beroiz et al., 2014). The phylogeny was inferred by Bayesian analysis of 46 partial ITS gene sequences from the observed populations, i.e., L, laboratory; K, Koppert; P, Phillips; D, Dog; H, Ham; and Z, field Zvoleneves. Branch lengths correspond to the mean posterior estimates of evolutionary distances (scale bar: 0.1). Branch labels indicate Bayesian posterior probabilities and supporting bootstrap values from maximum likelihood analysis.
Flavobacteriales Symbiont (Blattabacterium-Like)
The Blattabacterium symbiont was amplified using specific primers designed for identification of Blattabacterium in cockroaches. The symbiont was present in mite populations from dog food only as indicated by 16S rRNA sequences from eubacterial primers, as well as the amplicons of 16S rRNA fragments obtained by specific Blattabacterium primers. A Bayesian analysis of the obtained sequences showed that the sequences clustered as a monophyletic lineage outside Blattabacterium (Clark and Kambhampati, 2003), Candidatus Brownia rhizoecola (Gruwell et al., 2010), C. Uzinora diaspidicola (Gruwell et al., 2007), and C. Sulcia muelleri (Moran et al., 2005; Figure 3).
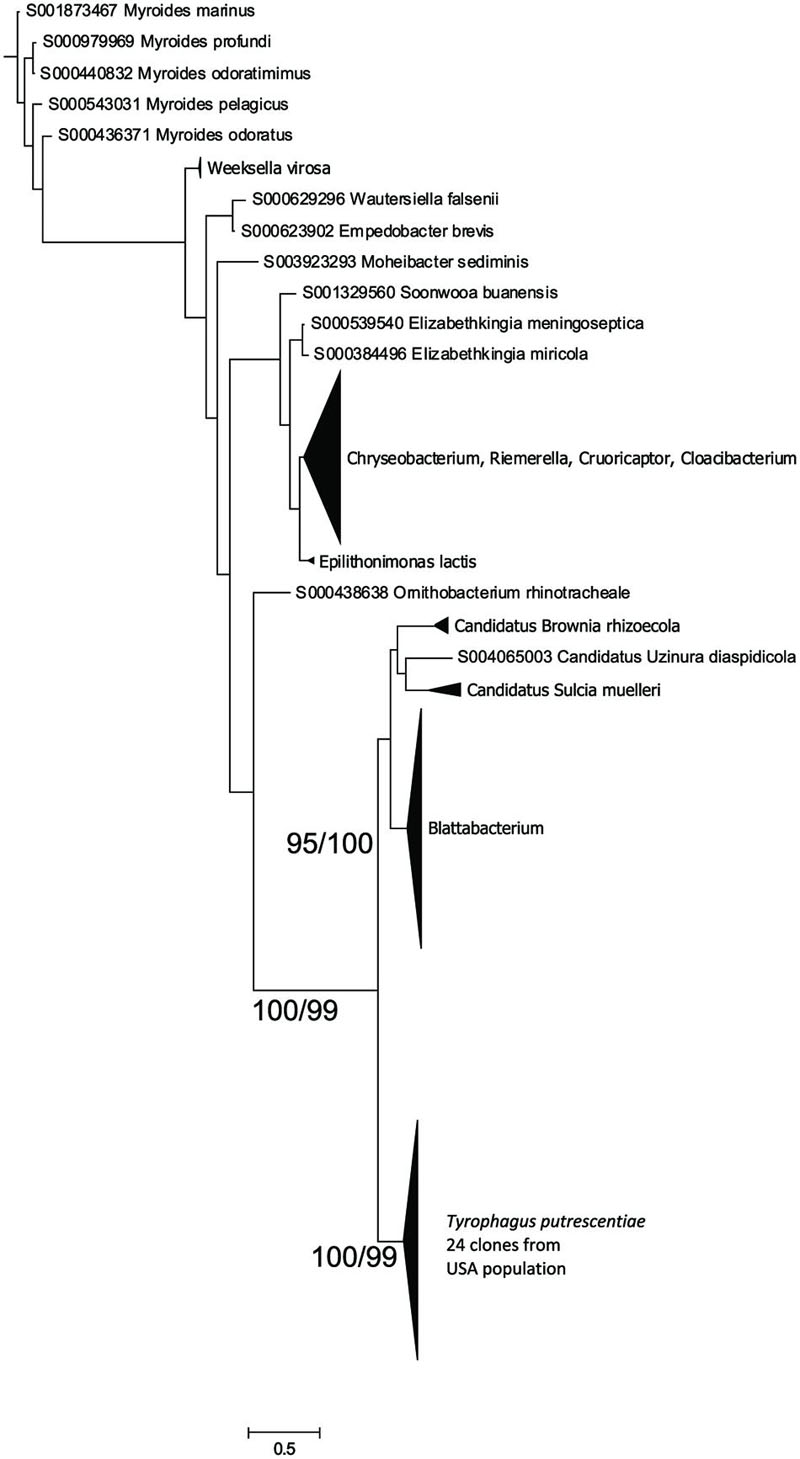
FIGURE 3. Phylogenetic analysis of the Blattabacterium-like symbiont clones obtained from the Dog population of T. putrescentiae. The phylogeny was inferred by a Bayesian analysis of 24 partial 16S rRNA gene sequences with the reference sequences from RDP, i.e., 126 sequences of Blattabacterium, C. Brownia, and C. Sulcia, and the sequences of 398 type strains representing the order Flavobacteriales. Branch lengths correspond to the mean posterior estimates of evolutionary distances (scale bar: 0.5). Branch labels indicate Bayesian posterior probabilities and bootstrap support values from maximum likelihood analysis. The phylogram was rooted using Bacteroides fragilis sequence NR074784 as an outgroup.
Wolbachia
The sequences of Wolbachia were found in the clones of amplicons obtained by the clones form Wolbachia specific primers (WpF/WpR in Table 2) amplicons, i.e., 9 and 10 from Dog and Phillips populations, respectively. A Bayesian analysis of the obtained sequences and sequences in RDP showed that the T. putrescentiae sequences formed separate cluster (Figure 4). Our analyses showed more clusters of the sequences. The analyses differentiated the sequences from the insects and nematodes, with a few exceptions. The sequences were closer to clusters of nematodes and aphids. The sister group was formed by Wolbachia sequences from nematode Radopholus similis (Haegeman et al., 2009) and mite Torotrogla cardueli (Prostigmata: Syringophilidae; Glowska et al., 2015).
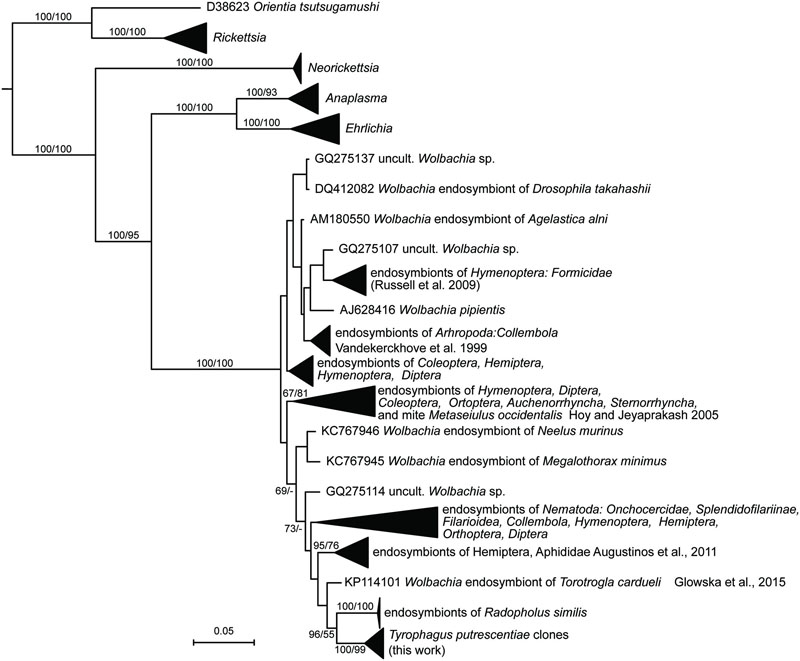
FIGURE 4. Phylogenetic analysis of the Wolbachia clones obtained from the Dog and Phillips populations of T. putrescentiae. The phylogeny was inferred by a Bayesian analysis of 19 partial 16S rRNA gene sequences cloned from T. putrescentiae with the reference sequences from RDP, 242 known Wolbachia endosymbionts (Vandekerckhove et al., 1999; Hoy and Jeyaprakash, 2005; Haegeman et al., 2009; Russell et al., 2009; Augustinos et al., 2011; Glowska et al., 2015) and 27 type strains of the order Rickettsiales. Branch lengths correspond to the mean posterior estimates of evolutionary distances (scale bar: 0.05). Branch labels indicate Bayesian posterior probabilities and bootstrap support values for maximum likelihood analysis. The phylogram was rooted using Rhizobium oryzae sequence EU056823 as an outgroup.
Comparison of Bacterial Community Based on 16S rRNA Gene
Obtained 16S rRNA sequences were deposited in GenBank (Accession Numbers: KX022128–KX022390) and combined to 176 sequences of the laboratory T. putrescentiae population which are available in GenBank (Accession Numbers: JN236405–JN236431; JX001234–JX001344, KJ635082–KJ635148; Hubert et al., 2012a,b; Kopecky et al., 2014b). All the sequences originated from amplification and cloning the amplicons from universal eubacterial primers. Altogether 42 OTUs97 were distinguished (Supplementary Table S1). The diversity of bacterial community was similar for all populations of T. putrescentiae with exception of Zvoleneves population (Figures 5A,B). The bacterial community of mite population was formed from known/suspected symbionts or parasites: Bartonella-like bacteria (OTU97 1), Wolbachia (OTU97 4), Cardinium (OTU97 6), Blattabacterium-like symbiont (OTU97 7), Solitalea-like bacteria (OTU97 8; Figure 5C). The next most frequent and abundant OTUs97 were Bacillus (OTU97 2), Moraxella (OTU97 3), Staphylococcus (OTU97 5), Kocuria (OTU97 9), and Microbacterium (OTU97 10; Supplementary Table S1).
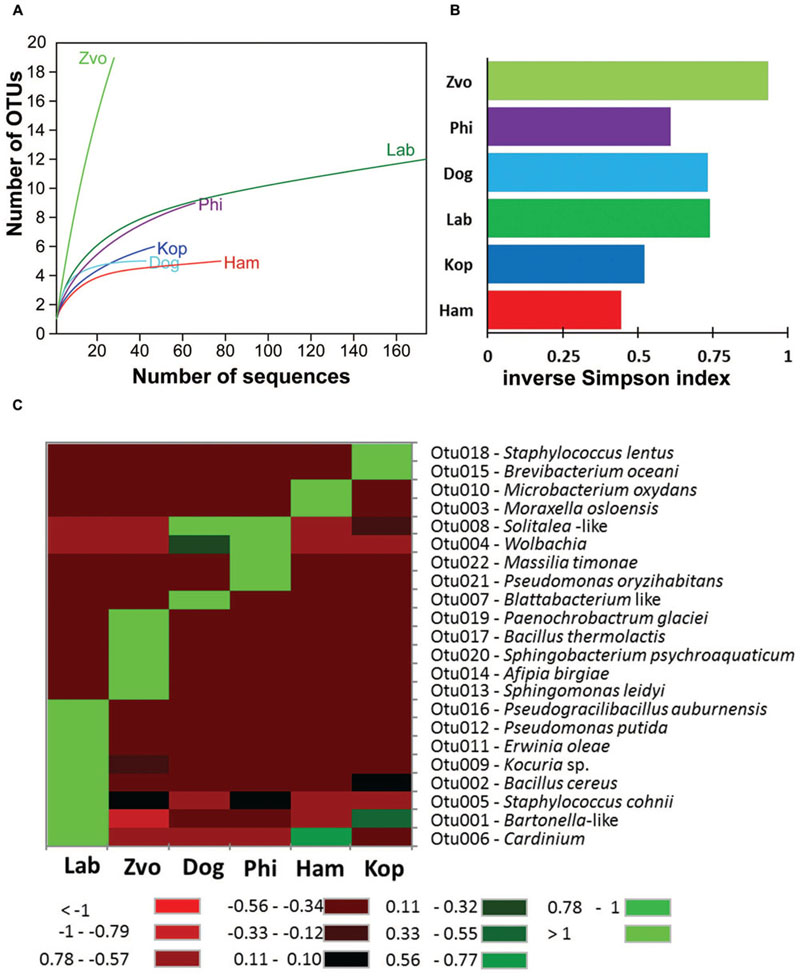
FIGURE 5. The diversity and composition of bacterial communities in T. putrescentiae populations based on Sanger sequencing of the 16S rRNA gene clones, the amplicons originated from eubacterial primers (F24/R1492): (A) rarefaction analyses, (B) inverse Simpson diversity index, (C) heat map. Abbreviations for the T. putrescentiae populations are listed in Table 1.
The populations differed in observed bacterial community as indicated by the 16S rRNA library. Principal coordinate analyses using Euclidian data matrix showed that bacterial communities of Phillips, and Dog mite populations were similar, while Zvoleneves, Laboratory and Ham populations were formed from different bacteria (Figure 6A). The first principal axis explained 60% and the second axis explained 23% of variation in the dataset. When Jaccard data matrix was calculated, the bacterial community was similar in Zvoleneves, Dog and Phillips populations, while Laboratory and Ham populations were different (Figure 6B). The first axis explained 59% and second axis explained 29% of variation in the data set. Composition between populations is reported in Figure 6C.
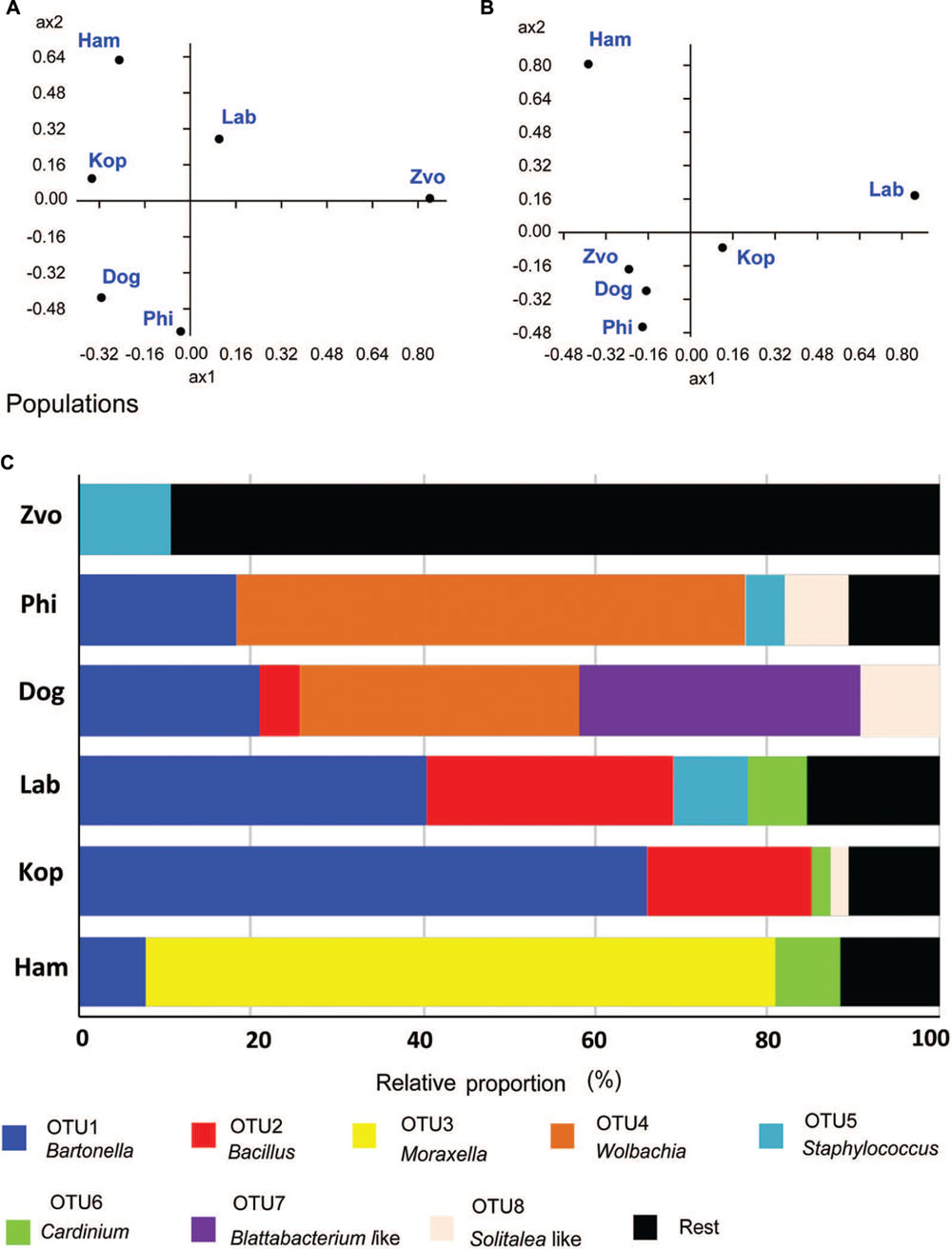
FIGURE 6. Comparison of bacterial communities in T. putrescentiae populations based on Sanger sequencing of the 16S rRNA gene clones, the amplicons originated from eubacterial primers (F24/R1492): (A,B) principal coordinate analyses of bacterial communities in examined mite populations; (A) based on Euclidian distance; (B) based on Jacquard similarity index; (C) relative proportions of cloned bacterial sequences in our 16S rRNA library from different populations of T. putrescentiae. Abbreviations for the T. putrescentiae populations are listed in Table 1.
To confirm the distribution of bacterial taxa obtained from the analyses of cloned sequences from eubacterial (F24/R1492) amplicons, the samples were analyzed by taxa specific primers. The presence/absence of selected taxa of symbiotic/parasitic bacteria was confirmed by specific primers in the samples of adults and in the eggs (Table 3).
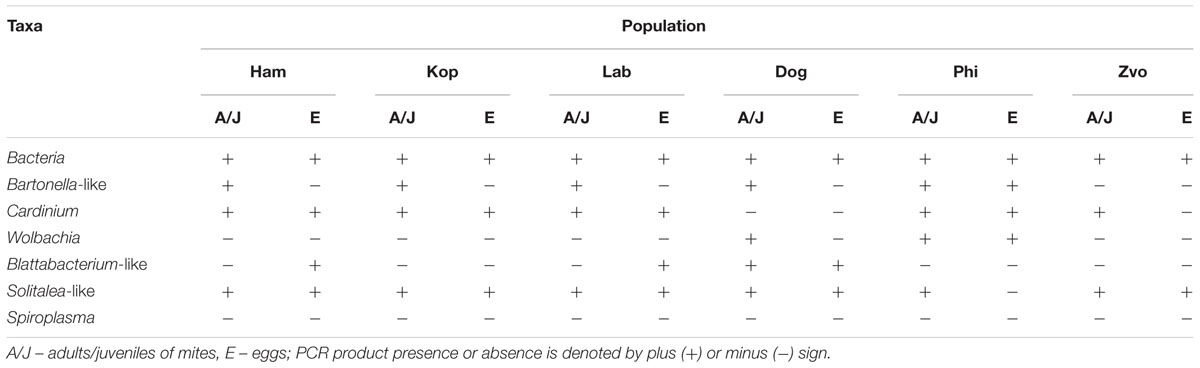
TABLE 3. The presence or absence of amplicons of bacterial 16S rRNA generated with universal and taxon-specific primers in different populations of Tyrophagus putrescentiae.
The highest number of OTUs was found in the Zvoleneves population, while the lowest was in the Phillips population. Generally, no OTU97 (i.e., sequences with 3% of dissimilarity) was shared by all the populations. But when taxa specific primers were used the presence of Solitalea was confirmed in all populations including the eggs. The exception was Phillips T. putrescentiae population, when the Solitalea was not detected in the eggs (Table 3). Bartonella (OTU97 1) had the highest relative numbers in the Koppert and laboratory strains, but Bartonella was present in the Phillips, Dog and Ham populations as well. The amplicon from specific primers confirms the Bartonella in all populations except field Zvoleneves population. Bartonella was not found in the eggs except of the Phillips T. putrescentiae population. Wolbachia (OTU97 4) had high relative numbers in the Dog and Phillips populations. The specific primers confirm Wolbachia in adults/juveniles of Dog and Phillips populations. However, in the eggs, Wolbachia was found only in Dog T. putrescentiae population (Table 3). Bacillus (OTU97 2) formed bacterial communities in the Laboratory and Koppert populations. Moraxella (OTU97 3) formed bacterial communities in the Ham population only and the Blattabacterium-like symbiont (OTU97 7) in the Dog population only. However, the specific primers confirm Blattabacterium-like symbiont in both adults/juveniles of T. putrescentiae Dog population and in the eggs of laboratory T. putrescentiae population (Table 3). Cardinium (OTU97 6) was found in the clones of 16S rRNA of Ham, Koppert and Laboratory T. putrescentiae population. However, the presence was confirmed in all populations by specific primers with exception of Dog population and in the majority of populations was detected in the eggs.
Microanatomical Description of Associated Bacteria
Bacteria were present on the mite surfaces, in bacteryocites, reproductive tracts and salivary glands (Figure 7D). The bacteria were not identified in food boli. The observed food boli (Figure 7A) contained concentrated mucoid substances or unidentified food fragments (Figures 7B,C). However, the mites ingested bacteria randomly, as indicated by the presence of bacteria in the foregut. In the Phillips and Dog populations we found bacteria inside ovaria (Figure 7E).
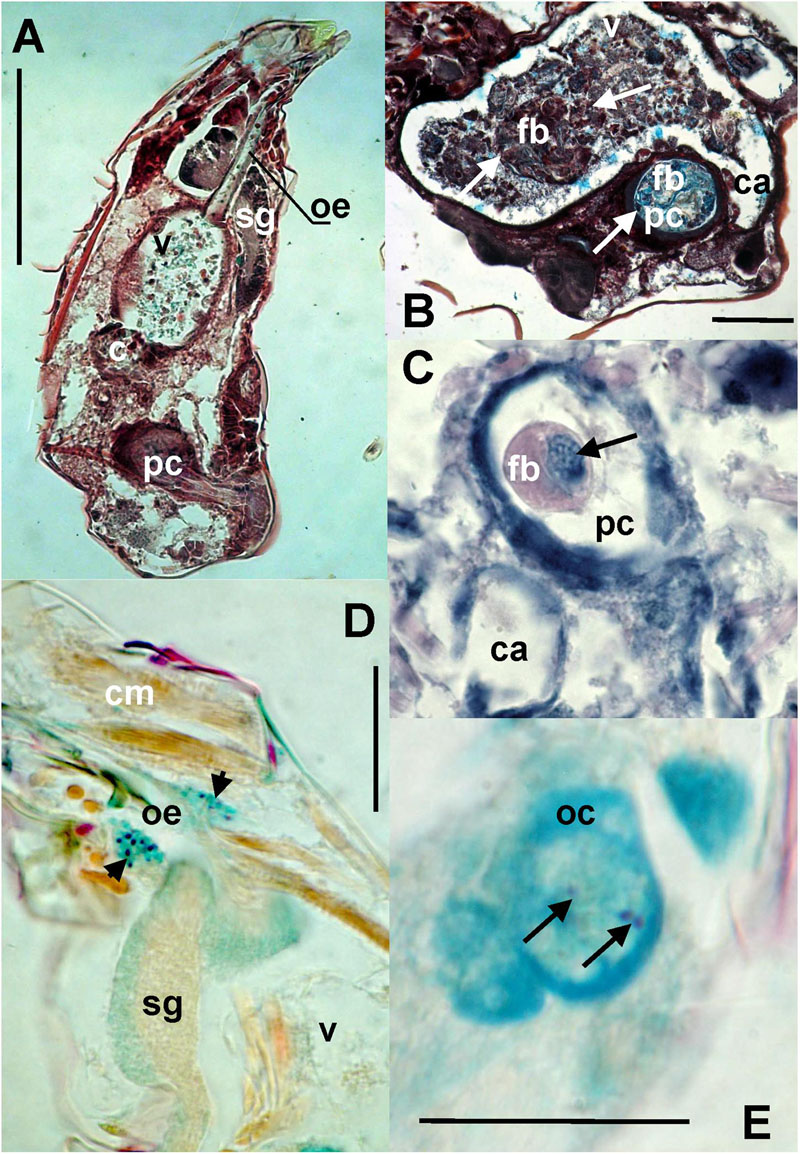
FIGURE 7. The images show: (A) total view of the digestive tract of T. putrescentiae; (B) total view of the ventriculus and post-colon with ingested food; arrows point to food boli; (C) detail view of the post-colon of specimens with food boli formed from mucoid substances and fragments of diet (arrow); (D) detail view of the salivary glands with stained bacterial cells (arrows); (E) detail view of oocyte with stained bacterial cells (arrows). Staining: (A,B) Masson’s triple stain, (C) Ziehl–Neelsen, (D,E) Mann Dominici; Scales: (A,B) 100 μm, (D,E) 25 μm. c, colon; ca, caecum; cm, cheliceral muscles; fb, food bolus; oc, oocyte; oe, esophagus; pc, post-colon; sg, synganglion; v, ventriculus.
The bacteriocytes (Figure 8A) were found in some adults in all observed populations. The bacteria were localized in fat tissues and were of various sizes covering up to one third of the histological sections (Figure 8C). The bacteria in the bacteriocytes were formed by spherical (Figures 8B,D,E) particles with different staining compared to the rest of tissues; we identified these particles as bacteria. In addition, rod-shaped particles were found in the bacteriocytes in the laboratory population of T. putrescentiae (Figure 8F).
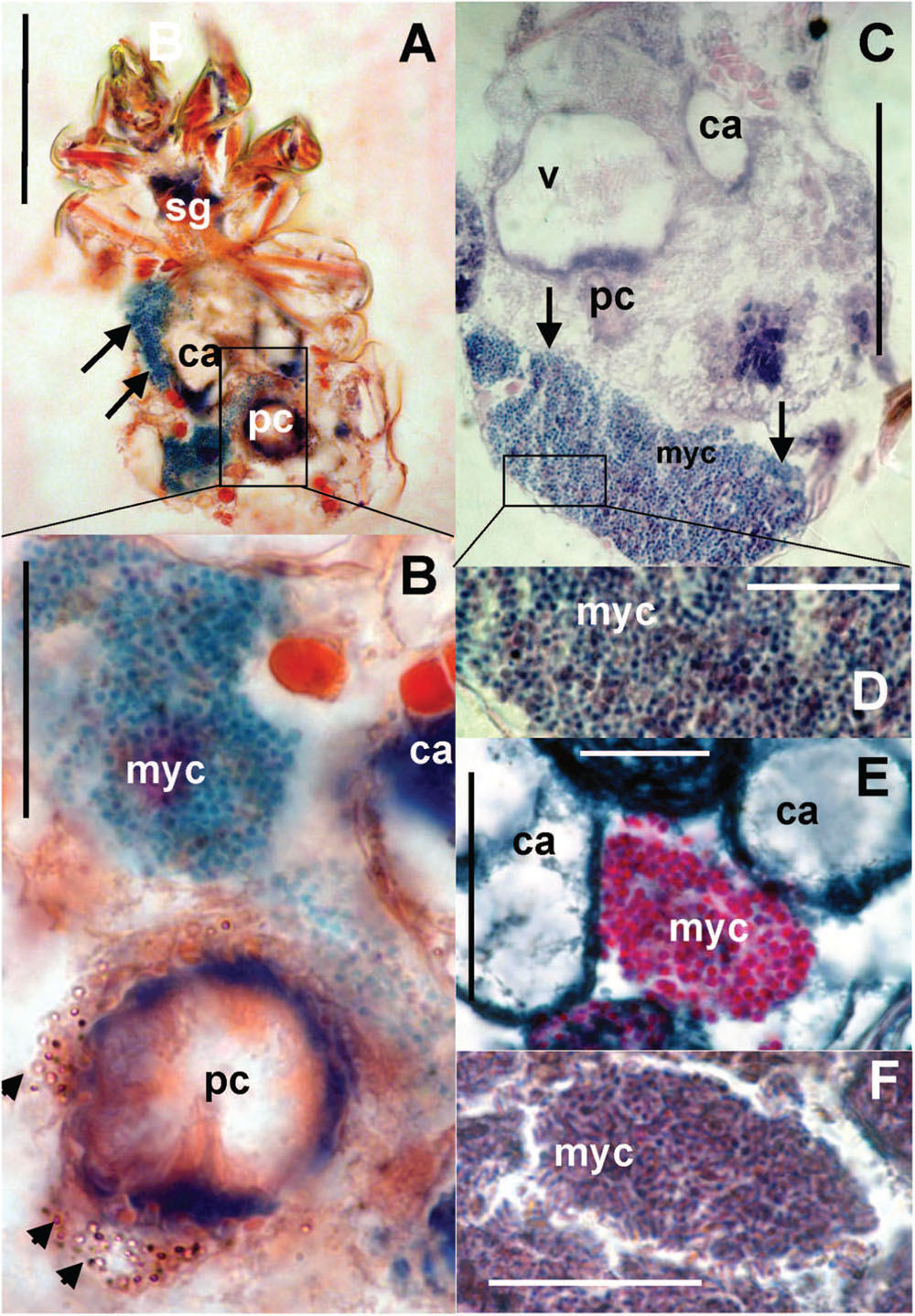
FIGURE 8. Histological sections of T. putrescentiae: (A) Total view of T. putrescentiae with bacteriocyte (arrows); (B) details of the previous image; the arrows point to guanine crystals near the post-colon; (C) sagittal section of the mite body with bacteriocyte (arrows); (E) details of the previous image with spherical bacteria; (F) localization of bacteriocytes with spherical bacteria, (F) details of the bacteriocyte with rod-shaped bacteria. Staining: (A,B) Mann Dominici, (C,D) Ziehl–Neelsen, (E,F) Masson’s triple stain; Scales. (A,C) 100 μm, (D–F) 25 μm. ca, caecum; fb, food bolus; myc, bacteriocyte; pc, post-colon; sg, synganglion.
The guanin granulae filled hysterosoma of mites with different intensity (Figure 9). The populations significantly differed (Chi-square = 119.37, P < 0.001) in the guanine contents in their hysterosoma (Figure 10A). The differences were between Phillips population with prevailing low guanine contents and the rest of populations with high guanine contents (Figure 9).
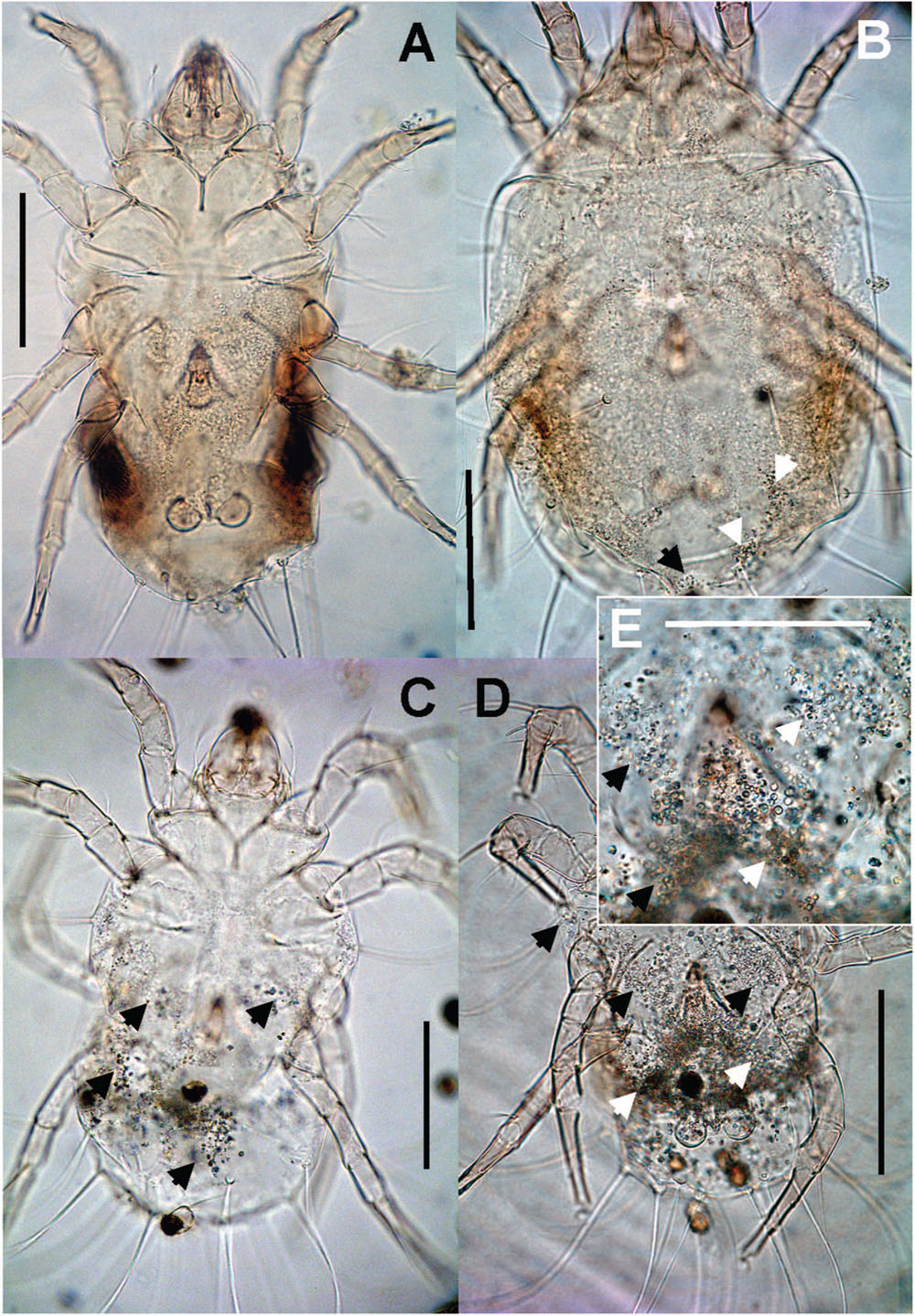
FIGURE 9. Guanine deposits in T. putrescentiae (mite are mounted on permanent microscopic slides): (A) no guanine granules; (B) a low number, the granules fill less than 25% of the hysterosoma; (C) intermediate, the granules fill 25–50% of the hysterosoma; (D) massive, the granules fill more than 50% of the hysterosoma, (E) detail of the previous image. Scales: (A–D) 100 μm, (E) 25 μm.
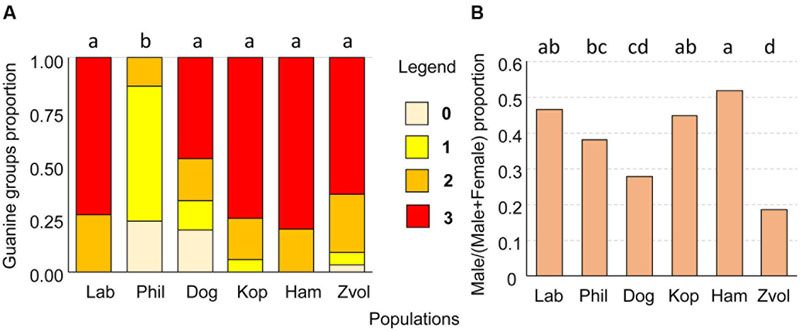
FIGURE 10. (A) The proportion of guanine waste product granules in T. putrescentiae; (B) comparison of sex ratios, male/(male+female) in examined populations of T. putrescentiae. Statistically significant differences (α = 0.05) in multiple comparisons are indicated by different letters. Abbreviations for the T. putrescentiae populations are listed in Table 1. (0) no guanine granules, (1) low number, granules fill less than 25% of the hysterosoma, (2) intermediate, granules fill between 25 and 50% of the hysterosoma, and (3) massive, granules fill more than 50% of the hysterosoma.
Estimation of Sex Ratio
Males were present in all studied populations. As estimated under a compound microscope using slide-mounted mites, proportions male/(male+female) differed among the T. putrescentiae populations (Chi-square = 72.63, P < 0.001) formed following groups: (i) proportion about 0.5: Laboratory, Koppert, and Ham; (ii) female prevailing populations: Phillips, Dog, and Zvoleneves populations (Figure 10B).
Discussion
We found significant differences in bacterial communities among various T. putrescentiae populations using Sanger sequencing of cloned bacterial 16S rRNA amplicons. Because of the complex taxonomy of T. putrescentiae and a possibility of the presence of cryptic species, we inferred single-gene phylogenetic trees aimed at species identification and analysis of sequences available in GenBank. We sequenced two genes most commonly used for Tyrophagus, CO1 and ITS-2. Both generated phylogenetic trees showed the presence of two distinct species, T. putrescentiae and T. fanetzhangorum. T. putrescentiae was present in five populations except. Zvoleneves population was a mixure of T. fanetzhangorum and T. putrescentiae. These analyses also allowed classification of previously unidentified GenBank sequences (Figures 1 and 2). We did not conduct a combined analysis of the two genes because many sequences from GenBank are available for one but not for both of these genes. We suggest that in the case of Zvoleneves population might be the results of bacterial community influenced by an additional factor, that is, the presence of T. fanetzhangorum having its own unique bacterial community.
The differences in bacterial communities confirm the previous results: there is clear variation in bacterial communities among different populations of mites, and this variation may be due to environmental factors such as differences in the diet, micro-habitat or simply the geographic sources, as shown previously for Rhizoglyphus robini (Zindel et al., 2013) and Carpoglpyhus lactis (Hubert et al., 2015). The bacterial community of T. putrescentiae was formed mainly by symbiotic or parasitic bacteria, and contained previously identified bacteria including Bartonella-like, Cardinium (Kopecky et al., 2013, 2014b), and Wolbachia (Brown and Lloyd, 2015). A Blattabacterium-like symbiont and Solitalea-like bacteria were newly identified for T. putrescentiae. The presence of Cardinium, Wolbachia, Solitalea-like bacteria and Blattabacterium-like bacteria in the eggs of some of the compared T. putrescentiae populations indicate symbiotic or parasitic association between these bacteria and T. putrescentiae. It also indicates mother to offspring (vertical) transmission via eggs of these bacteria. In one case (Phillips population), we found Bartonella in the eggs; however, in four T. putrescentiae populations, Bartonella was not observed in the eggs, suggesting that Bartonella is not vertically transmitted.
The next group of identified bacteria, i.e., Bacillus, Moraxella, Staphylococcus, Kocuria, and Microbacterium could have originated from ingested diet (Hubert et al., 2012a,b; Kopecky et al., 2014a,b). We suggest that ingested bacteria serve as source of nutrients for the mites whether by direct food source (Erban and Hubert, 2008) and/or that that they predigest the food with exo-enzymes (Erban et al., 2016).
The interesting finding is the presence of Solitalea-like bacteria, which we identified previously in cloned 16S rRNA sequences from a laboratory strain of the grain mite A. siro (Hubert et al., 2012a,b). However, one sequence of Pedobacter (Solitalea-like) was found in a Chinese T. putrescentiae population from edible fungi by Qu et al. (2015). Solitalea-like bacteria were detected in this study in all the T. putrescentiae populations. Recently, we detected Solitalea-like bacteria in the reproductive tract and parenchymal tissues of A. siro (Hubert et al., under review). The bacteria were also present in the eggs (Hubert et al., under review). Given these findings, we suggest that, similarly to Cardinium, this Solitalea-like bacteria might be either symbiotic or parasitic.
Wolbachia are well-known bacteria in insects and mites (Glowska et al., 2015); however, only a few such studies are available for Astigmata. A recent study demonstrated that T. putrescentiae mites feeding on Drosophila corpses, including Wolbachia-infected corpses, are possible vectors of Wolbachia (Brown and Lloyd, 2015). In this study, we found that the Wolbachia sequences from T. putrescentiae are more similar to Wolbachia in quill mites Torotrogla cardueli (Acari: Syringophilidae; Glowska et al., 2015), aphids (Augustinos et al., 2011), and nematodes (Haegeman et al., 2009). We also found that the Wolbachia sequences from Dog and Phillips T. putrescentiae populations clustered altogether suggesting their high similarity. Wolbachia manipulates sexual reproductions in mites, causing cytoplasmic incompatibility and feminization of males (Breeuwer and Jacobs, 1996). The results for sex ratio observed in both populations of the T. putrescentiae where we identified Wolbachia indicated that males were present. At the population level, T. putrescentiae is infested by at least two bacteria with the known ability to manipulate sexual reproduction in their hosts, i.e., Cardinium and Wolbachia. However, our sampling method did not provide information about the distribution of bacteria among individual mites. We localized the bacteria in oocytes and the bacteria were not observed in all individuals. This means that the infection spreads in a mite population with some dynamics. The described horizontal transfer of Wolbachia from Drosophila to T. putrescentiae (Brown and Lloyd, 2015) might suggest a possibility for mite-to-mite transfer also in our system. However, infection in other species of astigmatid mites in laboratory populations was not observed (Kopecky et al., 2014a), and the results obtained here using Wolbachia-specific primers were similar (see Table 2). In addition, in the populations infected with Wolbachia, a lower proportion of mite males was observed.
Blattabacterium is a member of Flavobacteriales (Bacteroidetes), which are obligate mutualistic endosymbiotic bacteria living in the fat bodies of cockroaches (Sabree et al., 2009). The association between cockroaches and this bacterium allows cockroaches to successfully subsist on nitrogen-poor diets and exploit nitrogenous waste (Sabree et al., 2009). Other related taxa are symbionts of sap-feeding insects, i.e., Hemiptera: Diaspididae, Cicadellidae, Coccoidea (Moran et al., 2005; Gruwell et al., 2007; Rosenblueth et al., 2012). Here, we found a related bacterium in the population found on dog food. Dog food is designed for carnivorous mammals and contains high fat and nitrogen, which makes it very suitable for the rapid growth and development of T. putrescentiae (Rybanska et al., 2015). Acquiring such a bacterium for a T. putrescentiae population that infests dry dog food seems to be beneficial to T. putrescentiae, but the exact nature of mite and bacterium interactions is unknown.
The Blattabacterium and Bartonella-relative taxa are known to influence nitrogen metabolisms of their hosts (van Borm et al., 2002; Sabree et al., 2009). Because we found such bacteria in T. putrescentiae, we focused on the comparison of guanine contents in specimens in the analyzed populations. We hypothesized that Blattabacterium-like bacteria reduce nitrogen waste in the parenchymal tissue of T. putrescentiae in the same way as suggested for cockroaches (Sabree et al., 2009). Guanine is the waste product of nitrogenous metabolisms (Levinson et al., 1991a). It is present in feces and has a kairomone function as was observed in the model species A. siro (Levinson et al., 1991b). T. putrescentiae has the ability to accumulate guanine in the parenchymal tissues (Smrz and Catska, 1989). It was documented that T. putrescentiae on nitrogenous-rich diets, such as some fungi, accumulate guanine in the fat tissues (Smrz and Catska, 1989; Smrz, 2003). Massive feeding on a fungal-type diet leads to an irreversible accumulation of guanine called “white body syndrome,” in which guanine crystals form in large numbers in fat tissues and suppress the internal organ functions of mites, leading to a damage (Smrz and Catska, 1989). But the categorization of guanine granules in mite bodies did not confirm this suggestion, because low guanine contents were observed in Phillips not in Dog populations. The high guanine content in our populations of T. putrescentiae is a possible result of nitrogen un-balanced diets when the mites are not able to eliminate nitrogenous waste (Smrz, 2003).
Bacteriocytes were formed by bacteria in mite specimens that consume a fungal diet (Smrz and Catska, 1989). It was suggested that bacteria can enter from the gut to the bacteriocytes in the fat tissues and participate in chitin digestion (Smrz and Trelova, 1995; Smrz, 2003). In this study, we found bacteriocytes in specimens from all populations, but bacteriocytes were not present in all specimens. We did not identify bacteria forming bacteriocytes, although only Solitalea was identified in all populations. However, previously we observed Solitalea-like bacteria in A. siro, and bacteriocytes were not present (Hubert et al., 2012a).
We hypothesize that the habitat and food plasticity exhibited by T. putrescentiae is mediated by different bacterial communities associated with mite populations. Of the 35 identified OTUs, only Solitalea was shared by all populations, indicating that different populations of T. putrescentiae differ in their bacterial communities. Previously, large changes in bacterial communities were observed after a diet switch in T. putrescentiae. However, those differences were due to bacterial taxa ingested with the diet, i.e., Bacillaceae (Bacillus, Lysinibacillus, Oceanobacillus, and Virgibacillus) and Micrococcales (Kocuria and Brevibacterium; Hubert et al., 2012b; Kopecky et al., 2014b). This was also supported by observations of a field strain of T. putrescentiae in laboratory experiments on fungal diets, when Alcaligenes faecalis, Agrobacterium sp., Serratia marcescens, and Achromobacter sp. were identified by plating and cultivation of bacteria obtained from the homogenates of T. putrescentiae specimens that had consumed the fungi (Smrz et al., 1991; Smrz and Soukalova, 2008; Smrz and Catska, 2010). Here, we observed changes in T. putrescentiae populations in the presence and infestation rates of Bacillus, Staphylococcus, Kocuria, and Moraxella as indicated by sequencing of cloned 16S rRNA sequences. Among these, Moraxella was associated with a mite population feeding on ham. Bacteria forming food boli were not observed in the studied populations of T. putrescentiae. One explanation is that these bacteria are ingested in low numbers along with fragments of fungal mycelium or plant debris.
From a practical point of view, mites are important pests, and mite-caused damage increases as the population density increases. It is well-known that different food sources influence enzyme physiology of mites resulting in differences in population growths (Smrz and Catska, 1987; Erban and Hubert, 2008, 2010; Erban et al., 2009, 2016; Nesvorna et al., 2012; Rybanska et al., 2015). The results of this study indicate that diet and habitats influence not only the ingested spectrum of bacteria but also the symbiotic and parasitic taxa. These two components of the bacterial community can affect both mite fitness and population growth, which result in variability in the growth of various populations, causing higher interspecies variability than variability among species (Bowman, 1984). Therefore, the results of this study are also important for understanding nutritional biology of mites.
Author Contributions
JH, TE, PK, JS, and TP: Scientific writing; JK: bioinformatics; MN: experiments, molecular biology, JH, JS and TE: experimental design, PK: taxonomy and interpretation, JH, and JS: microanatomy.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are obligated to Zuzana Kucerova for morphological determination of the field populations of TP, to Andrea Sala and Elmer van Baal for TP samples, and to Martin Markovic for technical help and Ondrej Ledvinka for advice to statistical analyses. JH, TE, and MN were supported by project number GA15-09038S of the Czech Grant Agency (GA CR), JH, TE, MN, and JK were supported by the project of the Ministry of Agriculture of the Czech Republic RO0415. PK was supported by Ministry of Education and Science of the Russian Federation (grant 16-14-10109 for bioinformatics and No 6.1933.2014/K project code 1933 for laboratory work), Coordenacao de Aperfeicoamento de Pessoal de Nivel Superior (CAPES) Ciencia sem Fronteiras (Brazil; PVE 88881.064989/2014-01 to Almir Pepato and PK), and the Russian Foundation for Basic Research (No 15-04-05185-a to PK, and No 15-04-02706-a to Sergey Ermilov and PK). Several confirmatory sequences were independently generated by PK in the Genomic Diversity Laboratory of the University of Michigan Museum of Zoology.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2016.01046
References
Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Ashelford, K. E., Chuzhanova, N. A., Fry, J. C., Jones, A. J., and Weightman, A. J. (2005). At least 1 in 20 16S rRNA sequence records currently held in public repositories is estimated to contain substantial anomalies. Appl. Environ. Microbiol. 71, 7724–7736. doi: 10.1128/AEM.71.12.7724-7736.2005
Ashelford, K. E., Chuzhanova, N. A., Fry, J. C., Jones, A. J., and Weightman, A. J. (2006). New screening software shows that most recent large 16S rRNA gene clone libraries contain chimeras. Appl. Environ. Microbiol. 72, 5734–5741. doi: 10.1128/AEM.00556-06
Augustinos, A. A., Santos-Garcia, D., Dionyssopoulou, E., Moreira, M., Papapanagiotou, A., Scarvelakis, M., et al. (2011). Detection and characterization of Wolbachia infections in natural populations of aphids: is the hidden diversity fully unraveled? PLoS ONE 6:e28695. doi: 10.1371/journal.pone.0028695
Barbieri, E., Paster, B. J., Hughes, D., Zurek, L., Moser, D. P., Teske, A., et al. (2001). Phylogenetic characterization of epibiotic bacteria in the accessory nidamental gland and egg capsules of the squid Loligo pealei (Cephalopoda: Loliginidae). Environ. Microbiol. 3, 151–167. doi: 10.1046/j.1462-2920.2001.00172.x
Beroiz, B., Couso-Ferrer, F., Ortego, F., Chamorro, M. J., Arteaga, C., Lombardero, M., et al. (2014). Mite species identification in the production of allergenic extracts for clinical use and in environmental samples by ribosomal DNA amplification. Med. Vet. Entomol. 28, 287–296. doi: 10.1111/mve.12052
Bochkov, A. V., Klimov, P. B., Hestvik, G., and Saveljev, A. P. (2014). Integrated Bayesian species delimitation and morphological diagnostics of chorioptic mange mites (Acariformes: Psoroptidae: Chorioptes). Parasitol. Res. 113, 2603–2627. doi: 10.1007/s00436-014-3914-9
Bowman, C. E. (1984). “Comparative enzymology of economically important astigmatid mites,” in Acarology VI, Vol. 2, eds D. A. Griffiths and C. E. Bowman (Chichester: Ellis Horwood), 993–1001.
Breeuwer, J. A. J., and Jacobs, G. (1996). Wolbachia: intracellular manipulators of mite reproduction. Exp. Appl. Acarol. 20, 421–434. doi: 10.1007/BF00053306
Brown, A. N., and Lloyd, V. K. (2015). Evidence for horizontal transfer of Wolbachia by a Drosophila mite. Exp. Appl. Acarol. 66, 301–311. doi: 10.1007/s10493-015-9918-z
Clark, J. W., and Kambhampati, S. (2003). Phylogenetic analysis of Blattabacterium, endosymbiotic bacteria from the wood roach, Cryptocercus (Blattodea: Cryptocercidae), including a description of three new species. Mol. Phylogenet. Evol. 26, 82–88. doi: 10.1016/S1055-7903(02)00330-5
Darriba, D., Taboada, G. L., Doallo, R., and Posada, D. (2012). jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 9, 772–772. doi: 10.1038/nmeth.2109
Dermauw, W., Van Leeuwen, T., Vanholme, B., and Tirry, L. (2009). The complete mitochondrial genome of the house dust mite Dermatophagoides pteronyssinus (Trouessart): a novel gene arrangement among arthropods. BMC Genomics 10:107. doi: 10.1186/1471-2164-10-107
Dillon, R. J., and Dillon, V. M. (2004). The gut bacteria of insects: nonpathogenic interactions. Annu. Rev. Entomol. 49, 71–92. doi: 10.1146/annurev.ento.49.061802.123416
Douglas, A. E. (2009). The microbial dimension in insect nutritional ecology. Funct. Ecol. 23, 38–47. doi: 10.1111/j.1365-2435.2008.01442.x
Douglas, A. E. (2015). Multiorganismal insects: diversity and function of resident microorganisms. Annu. Rev. Entomol. 60, 17–34. doi: 10.1146/annurev-ento-010814-020822
Duek, L., Kaufman, G., Palevsky, E., and Berdicevsky, I. (2001). Mites in fungal cultures. Mycoses 44, 390–394. doi: 10.1046/j.1439-0507.2001.00684.x
Erban, T., Erbanova, M., Nesvorna, M., and Hubert, J. (2009). The importance of starch and sucrose digestion in nutritive biology of synanthropic acaridid mites: alpha-amylases and alpha-glucosidases are suitable targets for inhibitor-based strategies of mite control. Arch. Insect Biochem. Physiol. 71, 139–158. doi: 10.1002/arch.20312
Erban, T., and Hubert, J. (2008). Digestive function of lysozyme in synanthropic acaridid mites enables utilization of bacteria as a food source. Exp. Appl. Acarol. 44, 199–212. doi: 10.1007/s10493-008-9138-x
Erban, T., and Hubert, J. (2010). Comparative analyses of proteolytic activities in seven species of synanthropic acaridid mites. Arch. Insect Biochem. Physiol. 75, 187–206. doi: 10.1002/arch.20388
Erban, T., Rybanska, D., Harant, K., Hortova, B., and Hubert, J. (2016). Feces derived allergens of Tyrophagus putrescentiae reared on dried dog food and evidence of the strong nutritional interaction between the mite and Bacillus cereus producing protease bacillolysins and exo-chitinases. Front. Physiol. 7:53. doi: 10.3389/fphys.2016.00053
Erban, T., Rybanska, D., and Hubert, J. (2015). Population growth of the generalist mite Tyrophagus putrescentiae (Acari: Acaridida) following adaptation to high- or low-fat and high- or low-protein diets and the effect of dietary switch. Environ. Entomol. 44, 1599–1604. doi: 10.1093/ee/nvv129
Franz, J.-T., Masuch, G., Musken, H., and Bergmann, K.-C. (1997). Mite fauna of German farms. Allergy 52, 1233–1237. doi: 10.1111/j.1398-9995.1997.tb02529.x
Garcia, N. (2004). Efforts to control mites on Iberian ham by physical methods. Exp. Appl. Acarol. 32, 41–50. doi: 10.1023/B:APPA.0000018165.80420.c9
Ge, M.-K., Sun, E.-T., Jia, C.-N., Kong, D.-D., and Jiang, Y.-X. (2014). Genetic diversity and differentiation of Lepidoglyphus destructor (Acari: Glycyphagidae) inferred from inter-simple sequence repeat (ISSR) fingerprinting. Syst. Appl. Acarol. 19, 491–498. doi: 10.11158/saa.19.4.12
Glowska, E., Dragun-Damian, A., Dabert, M., and Gerth, M. (2015). New Wolbachia supergroups detected in quill mites (Acari: Syringophilidae). Infect. Genet. Evol. 30, 140–146. doi: 10.1016/j.meegid.2014.12.019
Gruwell, M. E., Hardy, N. B., Gullan, P. J., and Dittmar, K. (2010). Evolutionary relationships among primary endosymbionts of the mealybug subfamily Phenacoccinae (Hemiptera: Coccoidea: Pseudococcidae). Appl. Environ. Microbiol. 76, 7521–7525. doi: 10.1128/AEM.01354-10
Gruwell, M. E., Morse, G. E., and Normark, B. B. (2007). Phylogenetic congruence of armored scale insects (Hemiptera: Diaspididae) and their primary endosymbionts from the phylum Bacteroidetes. Mol. Phylogenet. Evol. 44, 267–280. doi: 10.1016/j.ympev.2007.01.014
Guindon, S., Dufayard, J.-F., Lefort, V., Anisimova, M., Hordijk, W., and Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Guindon, S., and Gascuel, O. (2003). A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52, 696–704. doi: 10.1080/10635150390235520
Haegeman, A., Vanholme, B., Jacob, J., Vandekerckhove, T. T., Claeys, M., Borgonie, G., et al. (2009). An endosymbiotic bacterium in a plant-parasitic nematode: member of a new Wolbachia supergroup. Int. J. Parasitol. 39, 1045–1054. doi: 10.1016/j.ijpara.2009.01.006
Hammer, O., Harper, D. A. T., and Ryan, P. D. (2001). PAST: Paleontological Statistics Software Package for Education and Data Analysis. Palaeontol. Electron. 4:4. Available at: http://palaeo-electronica.org/2001_1/past/issue1_01.htm [accessed June 10, 2016].
Hoy, M. A., and Jeyaprakash, A. (2005). Microbial diversity in the predatory mite Metaseiulus occidentalis (Acari: Phytoseiidae) and its prey, Tetranychus urticae (Acari: Tetranychidae). Biol. Control 32, 427–441. doi: 10.1016/j.biocontrol.2004.12.012
Hubert, J., Kopecky, J., Perotti, M. A., Nesvorna, M., Braig, H. R., Sagova-Mareckova, M., et al. (2012a). Detection and identification of species-specific bacteria associated with synanthropic mites. Microb. Ecol. 63, 919–928. doi: 10.1007/s00248-011-9969-6
Hubert, J., Nesvorna, M., Kopecky, J., Sagova-Mareckova, M., and Poltronieri, P. (2015). Carpoglyphus lactis (Acari: Astigmata) from various dried fruits differed in associated micro-organisms. J. Appl. Microbiol. 118, 470–484. doi: 10.1111/jam.12714
Hubert, J., Nesvorna, M., Sagova-Mareckova, M., and Kopecky, J. (2012b). Shift of bacterial community in synanthropic mite Tyrophagus putrescentiae induced by Fusarium fungal diet. PLoS ONE 7:e48429. doi: 10.1371/journal.pone.0048429
Hubert, J., Stejskal, V., Munzbergova, Z., Kubatova, A., Vanova, M., and Zdarkova, E. (2004). Mites and fungi in heavily infested stores in the Czech Republic. J. Econ. Entomol. 97, 2144–2153. doi: 10.1093/jee/97.6.2144
Hubert, J., Sustr, V., and Smrz, J. (1999). Feeding of the oribatid mite Scheloribates laevigatus (Acari: Oribatida) in laboratory experiments. Pedobiologia 43, 328–339.
Hughes, A. M. (1976). The Mites of Stored Food and Houses Volume 9 of Technical bulletin (Great Britain. Ministry of Agriculture, Fisheries and Food), 2nd Edn. London: Her Majesty’s Stationery Office.
Klimov, P. B., and OConnor, B. (2013). Is permanent parasitism reversible?—critical evidence from early evolution of house dust mites. Syst. Biol. 62, 411–423. doi: 10.1093/sysbio/syt008
Klimov, P. B., and OConnor, B. M. (2008). Origin and higher-level relationships of psoroptidian mites (Acari: Astigmata: Psoroptidia): evidence from three nuclear genes. Mol. Phylogenet. Evol. 47, 1135–1156. doi: 10.1016/j.ympev.2007.12.025
Klimov, P. B., and OConnor, B. M. (2009a). Conservation of the name Tyrophagus putrescentiae, a medically and economically important mite species (Acari: Acaridae). Int. J. Acarol. 35, 95–114. doi: 10.1080/01647950902902587
Klimov, P. B., and OConnor, B. M. (2009b). Improved tRNA prediction in the American house dust mite reveals widespread occurrence of extremely short minimal tRNAs in acariform mites. BMC Genomics 10:598. doi: 10.1186/1471-2164-10-598
Kopecky, J., Nesvorna, M., and Hubert, J. (2014a). Bartonella-like bacteria carried by domestic mite species. Exp. Appl. Acarol. 64, 21–32. doi: 10.1007/s10493-014-9811-1
Kopecky, J., Nesvorna, M., Mareckova-Sagova, M., and Hubert, J. (2014b). The effect of antibiotics on associated bacterial community of stored product mites. PLoS ONE 9:e112919. doi: 10.1371/journal.pone.0112919
Kopecky, J., Perotti, M. A., Nesvorna, M., Erban, T., and Hubert, J. (2013). Cardinium endosymbionts are widespread in synanthropic mite species (Acari: Astigmata). J. Invertebr. Pathol. 112, 20–23. doi: 10.1016/j.jip.2012.11.001
Kramar, J. (1953). The contribution to microscopic preparation of Arthropods. [Prispevek k mikroskopicke preparaci clenovcu.]. Ceskoslov. Biol. 2, 57–58.
Lane, D. J. (1991). “16S/23S rRNA sequencing,” in Nucleic Acid Techniques in Bacterial Systematics, eds E. Stackebrandt and M. Goodfellow (New York, NY, USA: John Wiley and Sons), 115–175.
Lartillot, N., Lepage, T., and Blanquart, S. (2009). PhyloBayes 3: a Bayesian software package for phylogenetic reconstruction and molecular dating. Bioinformatics 25, 2286–2288. doi: 10.1093/bioinformatics/btp368
Levinson, H. Z., Levinson, A. R., and Muller, K. (1991a). Functional adaption of two nitrogenous waste products in evoking attraction and aggregation of flour mites (Acarus siro L.). Anz. Schadlingskd. Pfl. Umwelt. 64, 55–60. doi: 10.1007/bf01909743
Levinson, H. Z., Levinson, A. R., and Muller, K. (1991b). The adaptive function of ammonia and guanine in the biocoenotic association between Ascomycetes and flour mites (Acarus siro L.). Naturwissenschaften 78, 174–176. doi: 10.1007/bf01136207
Liu, Y. C., Chang, S. C., Chen, W. H., and Shu, W. B. (2006). The application of single-step nested multiplex polymerase chain reaction for the identification of Rhizoglyphus echinopus, R. robini and R. setosus simultaneously. Plant Prot. Bull. 48, 101–116.
Meeus, I., Vercruysse, V., and Smagghe, G. (2012). Molecular detection of Spiroplasma apis and Spiroplasma melliferum in bees. J. Invertebr. Pathol. 109, 172–174. doi: 10.1016/j.jip.2011.11.006
Moran, N. A., Tran, P., and Gerardo, N. M. (2005). Symbiosis and insect diversification: an ancient symbiont of sap-feeding insects from the bacterial phylum Bacteroidetes. Appl. Environ. Microbiol. 71, 8802–8810. doi: 10.1128/AEM.71.12.8802-8810.2005
Nesvorna, M., Gabrielova, L., and Hubert, J. (2012). Suitability of a range of Fusarium species to sustain populations of three stored product mite species (Acari: Astigmata). J. Stored Prod. Res. 48, 37–45. doi: 10.1016/j.jspr.2011.08.006
Noge, K., Mori, N., Tanaka, C., Nishida, R., Tsuda, M., and Kuwahara, Y. (2005). Identification of astigmatid mites using the second internal transcribed spacer (ITS2) region and its application for phylogenetic study. Exp. Appl. Acarol. 35, 29–46. doi: 10.1007/s10493-004-1953-0
OConnor, B. M. (1979). “Evolutionary origins of astigmatid mites inhabiting stored products,” in Proceedings of the V International Congress of Acarology: Recent Advances in Acarology, Volume 1, August 6–12, 1978, Michigan State University, East Lansing, ed. G. J. Rodriguez (New York, NY: Academic Press), 273–278. doi: 10.1016/b978-0-12-592201-2.50038-5
OConnor, B. M. (1982). Evolutionary ecology of astigmatid mites. Annu. Rev. Entomol. 27, 385–409. doi: 10.1146/annurev.en.27.010182.002125
O’Neill, S. L., Giordano, R., Colbert, A. M., Karr, T. L., and Robertson, H. M. (1992). 16S rRNA phylogenetic analysis of the bacterial endosymbionts associated with cytoplasmic incompatibility in insects. Proc. Natl. Acad. Sci. U.S.A. 89, 2699–2702. doi: 10.1073/pnas.89.7.2699
Palyvos, N. E., Emmanouel, N. G., and Saitanis, C. J. (2008). Mites associated with stored products in Greece. Exp. Appl. Acarol. 44, 213–226. doi: 10.1007/s10493-008-9145-y
Pruesse, E., Peplies, J., and Glockner, F. O. (2012). SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28, 1823–1829. doi: 10.1093/bioinformatics/bts252
Qu, S.-X., Li, H.-P., Ma, L., Hou, L.-J., Lin, J.-S., Song, J.-D., et al. (2015). Effects of different edible mushroom hosts on the development, reproduction and bacterial community of Tyrophagus putrescentiae (Schrank). J. Stored Prod. Res. 61, 70–75. doi: 10.1016/j.jspr.2014.12.003
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
Robertson, P. L. (1961). A morphological study of variation in Tyrophagus (Acarina), with particular reference to populations infesting cheese. Bull. Entomol. Res. 52, 501–529. doi: 10.1017/s0007485300055565
Rosenblueth, M., Sayavedra, L., Samano-Sanchez, H., Roth, A., and Martinez-Romero, E. (2012). Evolutionary relationships of flavobacterial and enterobacterial endosymbionts with their scale insect hosts (Hemiptera: Coccoidea). J. Evol. Biol. 25, 2357–2368. doi: 10.1111/j.1420-9101.2012.02611.x
Rozej, E., Witalinski, W., Szentgyorgyi, H., Wantuch, M., Moron, D., and Woyciechowski, M. (2012). Mite species inhabiting commercial bumblebee (Bombus terrestris) nests in Polish greenhouses. Exp. Appl. Acarol. 56, 271–282. doi: 10.1007/s10493-012-9510-8
Russell, J. A., Moreau, C. S., Goldman-Huertas, B., Fujiwara, M., Lohman, D. J., and Pierce, N. E. (2009). Bacterial gut symbionts are tightly linked with the evolution of herbivory in ants. Proc. Natl. Acad. Sci. U.S.A. 106, 21236–21241. doi: 10.1073/pnas.0907926106
Rybanska, D., Hubert, J., Markovic, M., and Erban, T. (2015). Dry dog food integrity and mite strain influence the density-dependent growth of the stored-product mite Tyrophagus putrescentiae (Acari: Acaridida). J. Econ. Entomol. 109, 454–460. doi: 10.1093/jee/tov298
Sabree, Z. L., Kambhampati, S., and Moran, N. A. (2009). Nitrogen recycling and nutritional provisioning by Blattabacterium, the cockroach endosymbiont. Proc. Natl. Acad. Sci. U.S.A. 106, 19521–19526. doi: 10.1073/pnas.0907504106
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Smrz, J. (1989). Internal anatomy of Hypochthonius rufulus (Acari: Oribatida). J. Morphol. 200, 215–230. doi: 10.1002/jmor.1052000210
Smrz, J. (2003). Microanatomical and biological aspects of bacterial associations in Tyrophagus putrescentiae (Acari: Acaridida). Exp. Appl. Acarol. 31, 105–113. doi: 10.1023/b:appa.0000005111.05959.d6
Smrz, J., and Catska, V. (1987). Food selection of the field population of Tyrophagus putrescentiae (Schrank) (Acari. Acarida). J. Appl. Entomol. 104, 329–335. doi: 10.1111/j.1439-0418.1987.tb00533.x
Smrz, J., and Catska, V. (1989). The effect of the consumption of some soil fungi on the internal microanatomy of the mite Tyrophagus putrescentiae (Schrank) (Acari. Acaridida). Acta Univ. Carol. Biol. 33, 81–93.
Smrz, J., and Catska, V. (2010). Mycophagous mites and their internal associated bacteria cooperate to digest chitin in soil. Symbiosis 52, 33–40. doi: 10.1007/s13199-010-0099-6
Smrz, J., and Jungova, E. (1989). The ecology of a field population of Tyrophagus putrescentiae (Acari. Acaridida). Pedobiologia 33, 183–192.
Smrz, J., and Soukalova, H. (2008). “Mycophagous mites (Acari: Oribatida and Acaridida) and their cooperation with chitinolytic bacteria,” in Proceedings of the Sixth European Congress: Integrative Acarology, 21-25 July 2008, Montpellier, eds M. Bertrand, S. Kreiter, K. D. McCoy, A. Migeon, M. Navajas, M.-S. Tixier, et al. (Montpellier: European Association of Acarologists (EURAAC)), 374–377.
Smrz, J., Svobodova, J., and Catska, V. (1991). Synergetic participation of Tyrophagus putrescentiae (Schrank) (Acari; Acaridida) and its associated bacteria on the destruction of some soil micromycetes. J. Appl. Entomol. 111, 206–210. doi: 10.1111/j.1439-0418.1991.tb00312.x
Smrz, J., and Trelova, M. (1995). The association of bacteria and some soil mites (Acari: Oribatida and Acaridida). Acta Zool. Fenn. 196, 120–123.
Solarz, K., Senczuk, L., Maniurka, H., Cichecka, E., and Peszke, M. (2007). Comparisons of the allergenic mite prevalence in dwellings and certain outdoor environments of the Upper Silesia (southwest Poland). Int. J. Hyg. Environ. Health 210, 715–724. doi: 10.1016/j.ijheh.2006.11.007
Solarz, K., Szilman, P., and Szilman, E. (1999). “Allergenic mites associated with bird nests in Poland (Astigmata: Pyroglyphidae, Acaridae, Glycyphagidae),” in Proceedings of the 3rd Symposium of the European Association of Acarologists: Ecology and Evolution of the Acari, Series Entomologica, Vol. 55, 1–5 July 1996, Amsterdam, eds J. Bruin, L. P. S. van der Geest, and M. W. Sabelis (Boston, MA: Kluwer Academic Publishers), 651–656. doi: 10.1007/978-94-017-1343-6_56
Spieksma, F. T. M. (1997). Domestic mites from an acarologic perspective. Allergy 52, 360–368. doi: 10.1111/j.1398-9995.1997.tb01012.x
Stepien, Z., and Rodriguez, J. G. (1973). Collecting large quantities of acarid mites. Ann. Entomol. Soc. Am. 66, 478–480. doi: 10.1093/aesa/66.2.478
Sun, E.-T., Li, C.-P., Nie, L.-W., and Jiang, Y.-X. (2014). The complete mitochondrial genome of the brown leg mite, Aleuroglyphus ovatus (Acari: Sarcoptiformes): evaluation of largest non-coding region and unique tRNAs. Exp. Appl. Acarol. 64, 141–157. doi: 10.1007/s10493-014-9816-9
Tamura, K., Dudley, J., Nei, M., and Kumar, S. (2007). MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol. Biol. Evol. 24, 1596–1599. doi: 10.1093/molbev/msm092
Van Asselt, L. (1999). Interactions between domestic mites and fungi. Indoor Built Environ. 8, 216–220. doi: 10.1159/000024644
van Borm, S., Buschinger, A., Boomsma, J. J., and Billen, J. (2002). Tetraponera ants have gut symbionts related to nitrogen-fixing root-nodule bacteria. Proc. Biol. Sci. 269, 2023–2027. doi: 10.1098/rspb.2002.2101
Vandekerckhove, T. T. M., Watteyne, S., Willems, A., Swings, J. G., Mertens, J., and Gillis, M. (1999). Phylogenetic analysis of the 16S rDNA of the cytoplasmic bacterium Wolbachia from the novel host Folsomia candida (Hexapoda, Collembola) and its implications for wolbachial taxonomy. FEMS Microbiol. Lett. 180, 279–286. doi: 10.1111/j.1574-6968.1999.tb08807.x
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/AEM.00062-07
Webster, L. M. I., Thomas, R. H., and McCormack, G. P. (2004). Molecular systematics of Acarus siro s. lat., a complex of stored food pests. Mol. Phylogenet. Evol. 32, 817–822. doi: 10.1016/j.ympev.2004.04.005
Yang, B., Cai, J., and Cheng, X. (2011). Identification of astigmatid mites using ITS2 and COI regions. Parasitol. Res. 108, 497–503. doi: 10.1007/s00436-010-2153-y
Zakhvatkin, A. A. (1959). A Translation of Fauna of U.S.S.R. Arachnoidea Tyroglyphoidea (Acari), Vol. VI, No. 1, eds and trans. A. Ratcliffe and A. M. Hughes (Washington, DC: The American Institute of Biological Sciences).
Keywords: Tyrophagus putrescentiae, bacteria, symbiont, feeding, Blattabacterium, Wolbachia, 16S rRNA
Citation: Erban T, Klimov PB, Smrz J, Phillips TW, Nesvorna M, Kopecky J and Hubert J (2016) Populations of Stored Product Mite Tyrophagus putrescentiae Differ in Their Bacterial Communities. Front. Microbiol. 7:1046. doi: 10.3389/fmicb.2016.01046
Received: 27 February 2016; Accepted: 22 June 2016;
Published: 12 July 2016.
Edited by:
Thomas Carl Bosch, Kiel University, GermanyReviewed by:
Stephan Schmitz-Esser, Iowa State University, USAAndreas Vilcinskas, University of Giessen, Germany
Copyright © 2016 Erban, Klimov, Smrz, Phillips, Nesvorna, Kopecky and Hubert. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jan Hubert, aHViZXJ0QHZ1cnYuY3o=