 Sherry L. Seston1‡
Sherry L. Seston1‡ Roxanne A. Beinart2†‡
Roxanne A. Beinart2†‡ Neha Sarode3
Neha Sarode3 Abigail C. Shockey4
Abigail C. Shockey4 Piyush Ranjan3Sangita Ganesh3
Piyush Ranjan3Sangita Ganesh3 Peter R. Girguis2
Peter R. Girguis2 Frank J. Stewart3*
Frank J. Stewart3*- 1Department of Biology, Alverno College, Milwaukee, WI, USA
- 2Department of Organismic and Evolutionary Biology, Harvard University, Cambridge, MA, USA
- 3School of Biology, Georgia Institute of Technology, Atlanta, GA, USA
- 4Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, Madison, WI, USA
Endosymbioses between animals and chemoautotrophic bacteria are ubiquitous at hydrothermal vents. These environments are distinguished by high physico-chemical variability, yet we know little about how these symbioses respond to environmental fluctuations. We therefore examined how the γ-proteobacterial symbionts of the vent snail Ifremeria nautilei respond to changes in sulfur geochemistry. Via shipboard high-pressure incubations, we subjected snails to 105 μM hydrogen sulfide (LS), 350 μM hydrogen sulfide (HS), 300 μM thiosulfate (TS) and seawater without any added inorganic electron donor (ND). While transcript levels of sulfur oxidation genes were largely consistent across treatments, HS and TS treatments stimulated genes for denitrification, nitrogen assimilation, and CO2 fixation, coincident with previously reported enhanced rates of inorganic carbon incorporation and sulfur oxidation in these treatments. Transcripts for genes mediating oxidative damage were enriched in the ND and LS treatments, potentially due to a reduction in O2 scavenging when electron donors were scarce. Oxidative TCA cycle gene transcripts were also more abundant in ND and LS treatments, suggesting that I. nautilei symbionts may be mixotrophic when inorganic electron donors are limiting. These data reveal the extent to which I. nautilei symbionts respond to changes in sulfur concentration and species, and, interpreted alongside coupled biochemical metabolic rates, identify gene targets whose expression patterns may be predictive of holobiont physiology in environmental samples.
Introduction
Hydrothermal vents are dynamic ecosystems where the vigorous emission of hot, chemically reduced fluid from the seafloor into the surrounding seawater results in a temporally and spatially variable physico-chemical environment. The dominant animals in these habitats live in partnership with bacterial symbionts that oxidize chemical reductants from the venting fluid, including hydrogen sulfide, methane, and hydrogen (Tivey, 2007), as well as thiosulfate, an intermediate oxidation product of sulfide that accumulates at some vents (Gru et al., 1998; Waite et al., 2008; Gartman et al., 2011). The symbionts oxidize these reduced compounds to fuel carbon fixation (chemoautotrophy), which provides primary nutrition for themselves and their animal hosts (Cavanaugh et al., 2006; Dubilier et al., 2008).
To acquire the chemical substrates needed for symbiont metabolism, symbiotic vent animals live where they can access both emitted fluid and oxygenated seawater (Stewart et al., 2005). Vent symbioses thrive in mixing zones where fluid turbulence causes conditions to change rapidly over small spatial scales such that adjacent animals may experience vastly different chemical and physical environments (Johnson et al., 1986). Sulfur chemistry in these zones can be particularly dynamic, with concentrations of hydrogen sulfide and derivatives of sulfide oxidation (e.g., thiosulfate) varying temporally and in proximity to vent sites (Gru et al., 1998; Mullaugh et al., 2008; Waite et al., 2008; Gartman et al., 2011). Furthermore, venting can halt, either transiently or permanently, due to changes in volcanic activity or the path of fluid flow (Butterfield et al., 1997). The dynamic nature of hydrothermal vents thus exposes organisms to fluctuating concentrations of diverse reductants and oxidants (Shank et al., 1998), and may drive variation in the metabolism and growth dynamics of symbiotic partners (Duperron et al., 2007; Dubilier et al., 2008; Robidart et al., 2011).
Descriptive and experimental studies over the past 30 years have enabled a broad understanding of the chemoautotrophic metabolism of vent symbionts. Physiological experiments with intact animal hosts (e.g., Childress et al., 1986, 1991; Girguis et al., 2000; Girguis and Childress, 2006; Nyholm et al., 2008; Petersen et al., 2011; Goffredi et al., 2014; Beinart et al., 2015) and excised symbionts (Belkin et al., 1986; Fisher et al., 1987; Wilmot and Vetter, 1990; Childress et al., 1991; Nelson et al., 1995) have established that vent symbionts can fix inorganic carbon via the oxidation of hydrogen sulfide, thiosulfate, hydrogen and/or methane. Separately, analyses of symbiont gene content (Kuwahara et al., 2007; Newton et al., 2007; Robidart et al., 2008; Nakagawa et al., 2014), gene and protein expression (Markert et al., 2007, 2011; Nyholm et al., 2008; Robidart et al., 2011; Gardebrecht et al., 2012; Wendeberg et al., 2012; Sanders et al., 2013), and enzyme activity (Felbeck, 1981; Stein et al., 1988; Robinson et al., 1998) from vent symbioses have clarified the pathways that symbionts employ for these metabolisms.
While it has been speculated that shifts in chemical availability are coupled to rapid physiological responses by the host, the symbiont, or both (Girguis and Childress, 2006), inferences made from freshly collected animals reveal at best a general picture of the conditions experienced in situ, making it challenging to identify the relationships between environmental conditions, symbiont/host physiology, and shifts in the metabolic coupling of the symbiont and host. A greater understanding of how the environment shapes the physiology of vent symbionts requires controlled experiments that couple measurements of metabolic rates with analyses of gene expression. Accordingly, we conducted high-pressure respirometric experiments on the symbiotic vent snail Ifremeria nautilei to characterize changes in symbiont gene expression (metatranscriptomes) under varying regimes of reduced sulfur species and availability.
I. nautilei lives in mixing zones around hydrothermal vents in the southwestern Pacific, deriving its nutrition from intracellular gill symbionts that can oxidize both sulfide and thiosulfate to fuel autotrophy (thioautotrophy; Beinart et al., 2015). Previous characterization of the I. nautilei gill symbiont community revealed associations with a lineage of γ-proteobacterial sulfur oxidizers from the Order Chromatiales (Urakawa et al., 2005; Suzuki et al., 2006). Methanotrophic bacteria may also be present (Gal'chenko et al., 1992; Borowski et al., 2002), although their representation across host populations is inconsistent (Windoffer and Giere, 1997; Suzuki et al., 2006) and molecular characterization of I. nautilei methanotrophs is lacking (Petersen and Dubilier, 2009). We herein present 16S rRNA gene and metatranscriptome data elucidating symbiont community composition and metabolism over a range of environmentally relevant conditions: (i) no electron donor (ND), (ii) 105 μM hydrogen sulfide (LS), (iii) 350 μM hydrogen sulfide (HS), and (iv) 300 μM thiosulfate (TS). These experiments clarify pathways of sulfur, nitrogen, and carbon metabolism in the Ifremeria-associated bacterial community and provide the first molecular evidence for the activity of methanotrophic bacteria in the gill-associated community. The results are derived from experiments designed to study the effects of geochemical dynamics on sulfur oxidation and carbon fixation rates (Beinart et al., 2015). An analysis of symbiont transcripts in light of the observed metabolic rates reveals how I. nautilei symbionts sustain chemoautotrophic activity at low sulfide concentrations, but exhibit a marked change in their physiology as sulfur availability increases.
Materials and Methods
Animal Collection and Experimental Incubations
As described in Beinart et al. (2015) I. nautilei were collected and incubated in four separate experiments that were performed to compare symbiont metabolism and gene expression when exposed to 105 μM hydrogen sulfide (low sulfide, LS), 300 hydrogen sulfide μM (high sulfide, HS), 300 thiosulfate μM (TS), or no exogenous electron donor (no donor, ND). Snails were collected from the vent fields ABE (HS and TS) or Tu'i Malila (LS and ND) at the Eastern Lau Spreading Center by the remotely operated vehicle JASON II during expedition TM-235 in 2009 (Table 1). Snails were brought to the ship in insulated containers and kept in 4°C seawater. Individuals responsive to the touch were either immediately preserved as described below or were transferred to flow-through, 2.68 L titanium aquaria in a high-pressure respirometry system (HPRS) that maintains the animals at 3000 psi. All treatments had five snails per aquaria, except the LS treatment, which had only four individuals. Snails in the HPRS were acclimated for 8 h prior to the start of the treatment incubations, during which time snails were exposed to input water comprised of filtered seawater amended with sodium nitrate (NaNO3, final concentration 40 μM) bubbled with carbon dioxide, oxygen, and nitrogen to achieve concentrations of 4 mM, >300 μM, and 400 μM, respectively. Snails used for the HS, TS, and LS treatments were acclimated without an electron donor, whereas those used in the ND treatment were acclimated with 300 μM sulfide, achieved by bubbling input water with gaseous hydrogen sulfide (5% H2S/95% N2). The addition of sulfide during ND snail acclimation was done to avoid overly stressed or starved individuals at the end of the long experimental period. Following acclimation, input water for TS, HS, and LS treatments were amended with either 300 μM sodium thiosulfate (NaS2O3) or bubbled with gaseous hydrogen sulfide (5% H2S/95% N2) to achieve target concentrations, whereas ND treatments received no energy source. For the duration of each experiment (between 27 and 40 h, Table 2), the concentration of hydrogen sulfide, thiosulfate, and polysulfide was monitored in the input water and effluent from the aquaria via an inline voltammetric microelectrode, allowing for the calculation of mass-specific rates of sulfur metabolism. Oxygen (O2) was detectable in the outflow of all vessels during the experiments, as measured by an oxygen optode (Golden Scientific), however, we were unable to quantify exact oxygen concentrations due to problems with calibration. Though the total length of each experiment varied due to logistical constraints, the sulfur metabolism and/or respiration of the animals in all experiments reached steady state after at least 10 h into the experiment. In addition, the incorporation of inorganic carbon (i.e., carbon fixation) by snail symbionts was measured by amending input seawater with isotopically labeled sodium carbonate (Na13CO3; 99.9% atom percent; Icon Services). A full description of the experimental system, conditions, measurements, and analysis can be found in Beinart et al. (2015).
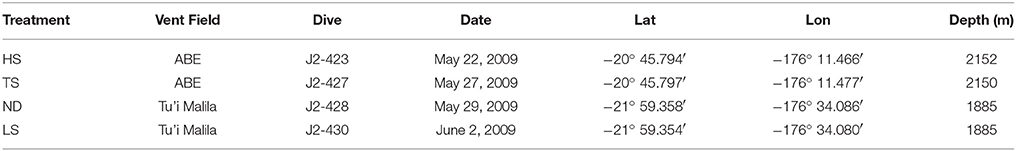
Table 1. Date and location of experimental animal collection.
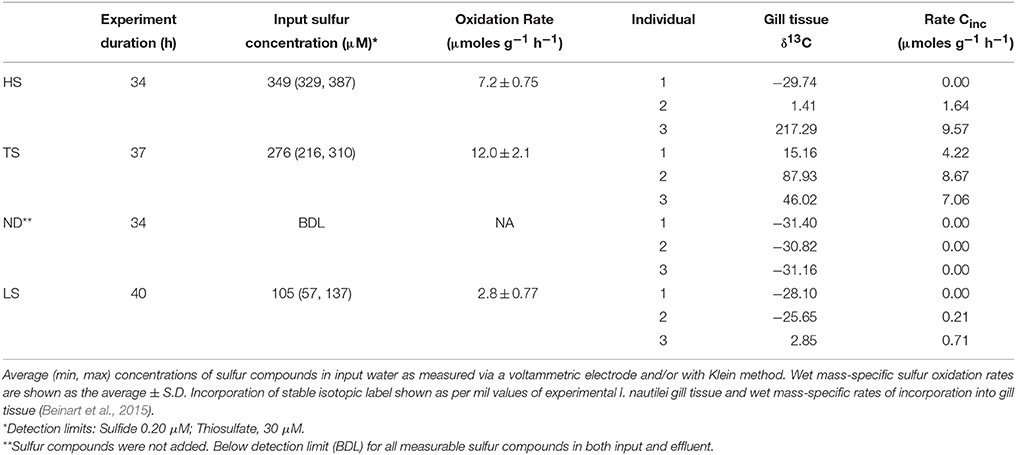
Table 2. Net sulfur uptake (oxidation) and carbon incorporation by Ifremeria during experiments.
Nucleic Acid Extraction
Immediately after collection from the vent field or at the conclusion of each experiment, animals were confirmed to be alive via responsiveness to touch, and then quickly excised from their shells. Subsamples of gill tissue from each individual were promptly homogenized with a Tissue Tearor and preserved in Trizol™ (LifeTechnologies) and stored at −80°C. RNA was extracted via the manufacturer's protocol. DNA from the same samples was back-extracted from the resulting interphase with a buffer consisting of 4 M guanine thiocyanate, 50 mM sodium citrate, and 1 M Tris (free base). The DNA was then extracted again with chloroform isoamyl alcohol and precipitated with isopropanol. The resulting DNA pellets were washed twice with 75% ethanol and air-dried. DNA pellets were resuspended in 8 mM sodium hydroxide, adjusted to pH 7–8 with 0.1 M HEPES and amended with 1 mM EDTA.
16S rRNA Gene Amplification, Sequencing, and Analysis
High-throughput sequencing of dual-indexed PCR amplicons encompassing the V4 hypervariable region of the 16S rRNA gene was used to assess the identity and relative abundance of symbionts in I. nautilei gill tissue. Amplicons were synthesized using Platinum PCR supermix (Life Technologies) with V4-specific primers F515 and R806 (Caporaso et al., 2010). Both forward and reverse primers were barcoded and appended with Illumina-specific adapters as per instructions by Kozich et al. (2013). Thermal cycling conditions were: initial denaturation at 94°C (3 min), followed by 30 cycles of denaturation at 94°C (45 s), primer annealing at 55°C (45 s), and primer extension at 72°C (90 s), followed by final extension at 72°C for 10 min. Amplicons were analyzed by agarose gel electrophoresis to verify size (~400 bp) and purified using Diffinity RapidTip2 PCR purification tips. Barcoded and Illumina adaptor-appended amplicons for each sample were pooled at equimolar concentrations and sequenced on an Illumina MiSeq (software v.2.4.0.4) using a 500 cycle MiSeq reagent kit, with 5% PhiX added to increase sequence diversity.
Demultiplexed amplicon read pairs were quality trimmed with Trim Galore! (Babraham Bioinformatics), using a base Phred33 score threshold of Q25 and a minimum length cutoff of 100 bp. High quality paired reads were then merged using the software FLASH (Magoč and Salzberg, 2011). Merged reads were analyzed using the software pipeline QIIME v1.8.0 (Caporaso et al., 2010). Reads were first screened for chimeras using QIIME's identify_chimeric_seqs.py script with usearch61. Non-chimeric sequences were clustered into Operational Taxonomic Units (OTUs) at 97% sequence similarity using the open reference OTU picking protocol with the script pick_open_reference_otus.py. Taxonomy was assigned to representative OTUs from each cluster using the Greengenes reference database (Aug 2013 release; DeSantis et al., 2006). The core set of QIIME diversity analyses was performed using the workflow script core_diversity_analyses.py at an even sampling depth of 19 978 sequences. One representative sequence from the 5 most abundant OTUs was aligned with related proteobacterial sequences using MUSCLE in MEGA 5.2.2 (Edgar, 2004). Phylogenetic analysis of the aligned representative sequences for each OTU was performed to further clarify the identity of host-associated 16S rRNA gene sequences. A maximum-likelihood tree was constructed using 252 unambiguously aligned nucleotide positions in webPRANK (Löytynoja and Goldman, 2010), and included sequences from diverse chemoautotrophic symbionts and free-living marine bacteria found in Genbank. Bootstrap analysis (1000 replicates) was performed to assess node confidence.
Metatranscriptome Library Preparation, Sequencing, and Analysis
Analysis of protein-coding genes and transcripts followed that of Stewart et al. (2012). Aliquots (50 ng) of total RNA were used to prepare cDNA libraries using the ScriptSeq™ v2 RNA-Seq Library Preparation Kit (Epicenter, Madison, WI, USA) according to manufacturer instructions. Briefly, cDNA was synthesized from fragmented total RNA (rRNA was not removed) using reverse transcriptase and amplified and barcoded using ScriptSeq Index PCR Primers (Epicenter) to generate single-indexed cDNA libraries. cDNA libraries were pooled and sequenced on an Illumina MiSeq using a 500 cycle kit. Barcoded sequences were de-multiplexed and filtered to remove low-quality reads (Phred score <25). Paired end sequences were merged using custom scripts incorporating the FASTX toolkit (http://hannonlab.cshl.edu/fastx_toolkit/index.html) and USEARCH algorithm (Edgar, 2010), with criteria of minimum 10% overlap and 95% nucleotide identity within the overlapping region
Prokaryotic and eukaryotic ribosomal RNA (rRNA) reads were separated from total RNA reads with riboPicker (Schmieder et al., 2012) using the comprehensive non-redundant rRNA database (2012 update) as reference. Reads with at least 75% nucleotide identity and 50% query coverage were considered a match to a ribosomal sequence. Reads identified as rRNA were submitted to MG-RAST (Meyer et al., 2008) to analyze overall rRNA taxonomic distribution. A subset of 100 000 rRNA reads from each sample were also analyzed in QIIME v 1.8 as described above.
Non-rRNA reads from each sample were processed with PRINSEQ Lite 0.20.4 (Schmieder and Edwards, 2011) to remove exact duplicate sequences (reads with 100% identity and equal length). Non-rRNA reads were annotated using BLASTX against the NCBI-nr database of non-redundant protein sequences (as of November 2013). BLASTX results were examined in MEGAN5 (build 4.1; Huson et al., 2011) and matches to bacterial and archaeal genes above a bit score of 50 were retained and classified according to functional categories based on the SEED subsystems ontology (Overbeek et al., 2005). Reads annotated as unassigned or eukaryotic were not included in further analyses. Each metatranscriptome was normalized to the sample with the smallest number of bacterial non-rRNA reads (Snail TS-1: 11 654 reads) in MEGAN5 by randomly subsampling 11 654 reads from each metatranscriptome 1000 times. The average count for each SEED subsystem category rounded to the nearest integer was used in all subsequent analyses. Differential expression of SEED Subsystem categories and proteins was analyzed with the baySeq R package v 2.6 (Hardcastle and Kelly, 2010). The heat map and distance clustering of SEED subsystems and samples was computed using heatmap.2 within the gplots R package (Warnes et al., 2013). To validate the results obtained in MEGAN and explore expression of particular genes of interest, BLASTX results (bit score > 50) were manually examined via keyword searches based on NCBI-nr annotations independent of MEGAN, as in (Canfield et al., 2010). Manual searches focused on enzymes of the Calvin Benson Bassham (CBB) cycle, reductive tricarboxylic acid (rTCA) cycle, methane oxidation, and nitrogen and sulfur metabolism in bacteria (Klotz and Stein, 2008) including: pyrophosphate-dependent phosphofructokinase, membrane-bound proton-translocating pyrophosphatase, ATP citrate lyase, 2-oxoglutarate oxidoreductase, fumarate reductase, SoxABCDHWXYZ proteins, Sulfide:quinone (oxido)reductase (Sqr), sulfide dehydrogenase (Fcc), APS reductase (AprAB), sulfate adenylytransferase (Sat), DsrABCEFHJKLMOPNR proteins, heterodisulfide reductase, methane monooxygenase, methanol dehydrogenase, pyrroloquinoline quinone synthase, 3-hexulose-6-phosphate synthase, and 3-hexulose-6-phosphate isomerase.
Amplicon and metatranscriptome sequences are available through NCBI under BioProject ID PRJNA288684.
Results and Discussion
Metabolic Activity
Ifremeria nautilei individuals were collected in four separate dives from four locations within the ABE and Tu'i Malila vent fields of the Eastern Lau Spreading Center (Table 1 and Figure S1). All individuals were subjected to an 8-h acclimation period in pressurized aquaria before the treatment incubations. Snails used for the HS, TS, and LS treatments were acclimated without an electron donor, whereas those used in the ND treatment were acclimated with 300 μM sulfide. After acclimation, treatment conditions were established in the aquaria and remained constant for 27–40 h. The same snails that were used for rate measurements were used for subsequent symbiont community composition and metatranscriptome analysis. Sulfur oxidation and carbon fixation rates for all individuals were reported in Beinart et al. (2015) and are summarized here for convenience in interpreting the gene expression patterns. Sulfur metabolism rates were measured by assessing the net flux of sulfide, thiosulfate, and polysulfide through high-pressure aquaria under each treatment. I. nautilei demonstrated net uptake (oxidation) of sulfide or thiosulfate when these compounds were supplied across all tested conditions (Table 2). Mass-specific sulfur oxidation rates were highest in I. nautilei exposed to 300 μM of thiosulfate (TS), with the next highest rates in the HS followed by the LS treatments (Table S2). The measurement of net sulfur flux through the aquaria also revealed the excretion of polysulfides by I. nautilei when exposed to the HS treatment (Beinart et al., 2015). Oxygen (O2) was detectable in HPRS outflow of all treatments during the experiments, indicating that complete anaerobiosis was not established in any treatment.
Symbiont carbon fixation was observed in treatments exposed to both sulfide and thiosulfate (Table 2). In contrast to sulfur metabolism rates, which were based on the collective uptake by all experimental individuals (i.e., sulfide disappearance from aquaria) and therefore reflected an average metabolic rate across individuals per treatment, carbon fixation rates were measured by the incorporation of stable isotope labeled (13C) inorganic carbon into individual snails. Compared to symbiont-free foot tissue (average δ13C −28.0 ‰± 0.95 s.d; Beinart et al., 2015), elevated carbon isotopic compositions were detected in the gills of I. nautilei individuals exposed to HS, LS, and TS (Table 2), whereas carbon incorporation was not observed in any individuals in the no electron donor (ND) treatment. Although mass-specific incorporation rates were relatively consistent among individuals in the TS treatment, rates differed widely among individuals within the HS and LS treatments. Notably, an individual in the HS treatment demonstrated the highest rate across all treatments, while other HS individuals showed low or undetectable carbon incorporation (Table 2). This pattern, seen also in the LS treatment, reflects the position of the individuals in the aquaria, with the most productive individuals closest to the incoming water, suggesting that metabolism by downstream individuals was limited by the activity of those upstream (Beinart et al., 2015).
Symbiont Taxonomic Composition—16S rRNA Gene Amplicons
At the conclusion of each incubation, gills of three host individuals were immediately sampled to assess symbiont phylogenetic affiliation and gene transcription profiles. Illumina sequencing of 16S rRNA gene (DNA) amplicons revealed that all 12 experimental I. nautilei hosted highly related communities of symbionts (Figure 1). A single OTU (97% similarity cluster) within the order Chromatiales of the γ-proteobacteria was dominant across all individuals, accounting for 90.1% of all sequences (Figure 1). A representative sequence from this OTU clustered phylogenetically with the known sulfur-oxidizing I. nautilei symbiont, differing from published sequences by 1-2 nucleotides (<0.8%; Figure 2).
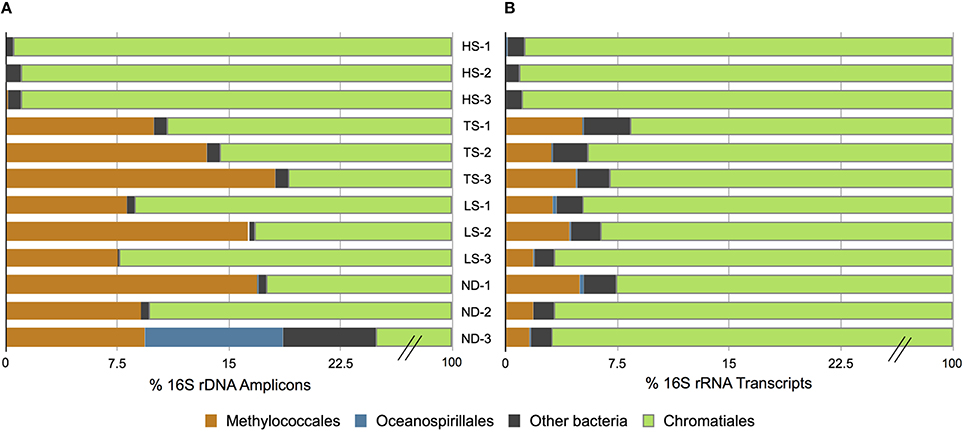
Figure 1. Distribution of amplified 16S rRNA genes (A) and 16S rRNA transcripts (B) in Ifremeria nautilei samples from experimental shipboard incubations.

Figure 2. Maximum-likelihood phylogenetic tree of the top 5 most abundant 16S rRNA gene OTUs. The 5 OTUs shown here accounted for 98% of all 781 662 reads analyzed. The tree is based on 252 unambiguously aligned nucleotide positions. Two ε-proteobacterial Alviniconcha hessleri symbionts are used as the outgroup. The numbers to the left of each node are bootstrap values obtained from 1000 replicates; only values greater than 50% are indicated.
Amplicon sequencing also detected putative methanotrophic bacteria in association with I. nautilei used in the treatments, although at much lower abundance than the dominant Chromatiales symbionts. Sequences related to the Methylococcaceae, a γ-proteobacterial family of methanotrophic bacteria, represented 7.7% of all amplicons (all individuals combined) and three distinct OTUs (Figure 2). These OTUs were abundant in the TS, LS, and ND incubations (up to 16.8% of amplicons from a single host), but represented <0.02% of sequences from individuals in the HS incubation (Figure 1, and discussed in more detail below). The Methylococcaceae OTUs did not cluster with the methanotrophic symbionts of other animals, but rather formed a separate cluster with a cloned 16S rRNA sequence from a shallow hydrothermal vent system (Hirayama et al., 2007; Figure 2). These findings represent the first published DNA sequences identifying a γ-proteobacterial methanotrophic symbiont in I. nautilei. Previous electron microscopy studies have observed multiple bacterial morphotypes in the gill cells of some I. nautilei individuals, including a low abundance morphotype with stacked cytoplasmic membranes characteristic of type I methanotrophs (Gal'chenko et al., 1992; Borowski et al., 2002). However, prior molecular analyses detected only the Chromatiales symbiont (Windoffer and Giere, 1997; Borowski et al., 2002; Urakawa et al., 2005; Suzuki et al., 2006).
The fifth most abundant OTU recovered from the amplicon library was affiliated with the genus Endozoicomonas of the order Oceanospirillales, and accounted for 0.5% of all sequences (Figure 2). Endozoicomonas bacteria are aerobic heterotrophs that appear commonly in association with marine invertebrates (Dubilier et al., 2008). There is evidence that Oceanospirillales are intranuclear parasites that cause host cell death in symbiotic bathymodiolin mussels (Zielinski et al., 2009). Intracellular Oceanospirillales have also been observed in the hydrothermal vent gastropod Alviniconcha that co-occurs with I. nautilei in the Lau Basin, although the nature of the association is not clear (Beinart et al., 2014).
To investigate whether differences in symbiont diversity resulted from incubation conditions or from in situ variability among the sampled populations, we extended the 16S rRNA gene analysis to include 10 snails collected from the same vent fields as the experimental animals. This analysis revealed a similar dominance of the single Chromatiales OTU, but also considerable variation in the relative abundance of Methylococcaceae among host individuals (Figure 3). Notably, Methylococcaceae OTUs composed < 0.05% of the symbiont population in snails from the ABE vent field collected during the same dive as snails HS1-3 used in the HS treatment, while the symbiont population within snails from the Tu'i Malila vent field contained 5–17% Methylococcaceae. These results suggest that the low proportion of Methylococcaceae in the HS 16S rRNA gene datasets may reflect environmental variation among symbiont populations rather than an effect of incubation conditions (Figure 1). Our results, from both experimental and environmental specimens, highlight the potential for local variation in the proportional abundances of Chromatiales and Methylococcaceae symbionts in I. nautilei. Additional studies are necessary to confirm whether such variation is linked with environmental sulfide and methane availability, as shown for other dual chemosynthetic symbioses (Duperron et al., 2011).
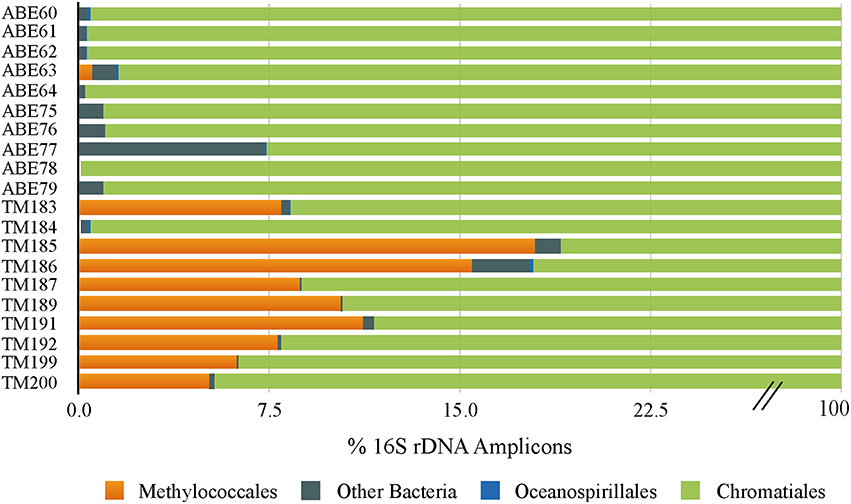
Figure 3. Bar graph showing the distribution of 16S rRNA OTUs in Ifremeria nautilei from ABE and Tui Malila vent fields.
Metatranscriptome Characteristics and Taxonomic Composition
Each metatranscriptome was derived from a single snail and consisted of 1 to 5 million high quality paired sequence reads representing both symbiont and host RNA (Table 3). Metatranscriptomes were generated from the same 12 individuals (3 from each treatment) that were used in the metabolic experiments and phylogenetic analyses. Of the reads generated, 69–77% represented rRNA, while 5–9% represented mRNA transcripts with matches to protein-coding genes in the NCBI nr database. The remaining 16–24% of the reads not identified as either rRNA or mRNA may represent unidentified protein-coding genes or enzymatic and regulatory RNA. Approximately 51–73% of mRNA transcripts identified in MEGAN were eukaryotic in origin, with only 8–12% of these transcripts successfully assigned a functional annotation. The single most abundant transcript identified in all 12 metatranscriptomes (2–7% of all identified protein-coding transcripts) matched the cytochrome oxidase subunit I gene of I. nautilei with > 99% identity, confirming the species identity of the host individuals. Other abundant eukaryotic transcripts identified in all host individuals regardless of treatment included cytochrome oxidase subunit II, tRNAs, NADH dehydrogenase complex I, fumarate reductase, cytochrome bc1 complex, and ATP synthase subunits, suggesting that all snails were metabolically active at the time of sampling and aerobic conditions were maintained throughout all experiments.
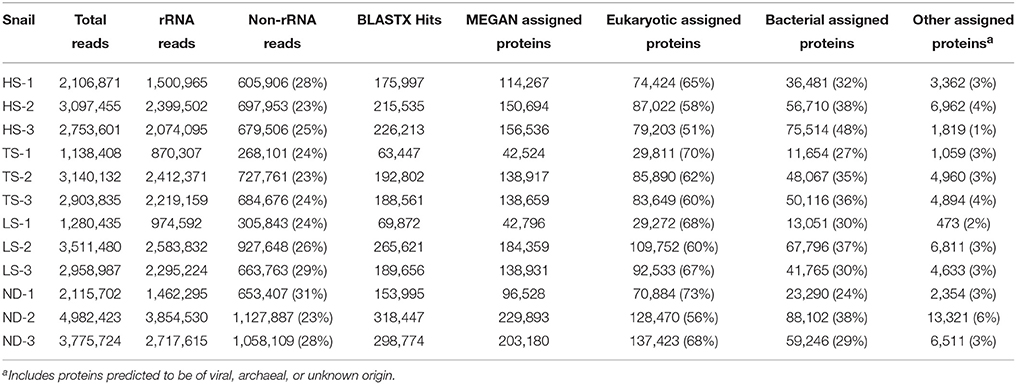
Table 3. Metatranscriptome sequence characteristics.
Bacterial 16S rRNA transcripts composed 19–53% of the total small subunit (SSU) rRNA reads (16S + host 18S) in each metatranscriptome. Interestingly, 16S rRNA represented a higher proportion of total SSU transcripts in metatranscriptomes from the HS treatments (average 45 ± 7%) compared to those from the other treatments (average 26 ± 6%; Table 3). This difference may reflect variation in either symbiont density or activity. This is consistent with the aforementioned metabolic data, and suggests that at elevated sulfide concentrations the bacterial symbiont population is more active. The vast majority of 16S rRNA transcripts (91.6–99.1% in all metatranscriptomes) matched sequences related to the Chromatiales (Figure 1). Consistent with the 16S gene amplicon results, a proportion of 16S rRNA transcripts (1.6–5.1%) from the TS, LS, and ND metatranscriptomes were classified as Methylococcaceae, whereas Methylococcaceae represented < 0.02% of total 16S transcripts in the three HS transcriptomes (Figure 1). In all specimens, the proportion of Methylococcaceae sequences in the 16S rRNA datasets derived from metatranscriptomes was approximately 1/4 the proportion observed in the DNA-amplified 16S gene datasets. This consistently lower proportion in the transcript data could indicate a low cellular RNA to DNA ratio, suggesting that Methylococcaceae bacteria were less metabolically active than the sulfur-oxidizing population during the incubations. This is consistent with the assumption that all of the incubations lacked appreciable methane or other C1 compounds that methylotrophs typically metabolize (neither methane nor any other C1 compounds were added to these incubations). Alternatively, the discrepancy between the DNA and RNA data could result from PCR bias and uneven amplification of different templates. Finally, consistent with the amplicon data, a minor fraction (0.2–0.5%) of 16S rRNA transcripts in each metatranscriptome matched sequences of the Oceanospirillales order (Figure 1).
Differential Gene Expression
To detect differential expression, protein-coding transcripts were assigned to functional SEED subsystems (Level 1 and 2) and proteins (Level 4) in MEGAN5 (Huson et al., 2011). The baySeq R package was then used to determine which model (treatment grouping) best explained subsystem and protein expression patterns (Table 4). Differential expression patterns were best explained by dividing the metatranscriptomes into two groups based on the concentration of electron donor in the incubation experiments. Grouping transcriptomes according to high electron donor (HD: HS and TS treatments) vs. low electron donor (LD: LS and ND treatments) conditions explained an estimated 39% of Level 1 subsystem differential expression, compared to 24% when transcriptomes were grouped according to the four individual treatments (Table 4). This trend was also observed when transcripts were assigned to Level 3 SEED subsystems and functional proteins (Table 4 and Tables S1, S2). Other models, including one comparing the thiosulfate treatment to the sulfide-containing treatments, explained less than 20% of variation at all subsystem levels. The availability of electron donor, and not donor compound type, therefore appears to be the strongest driver of symbiont differential gene expression in these experiments.
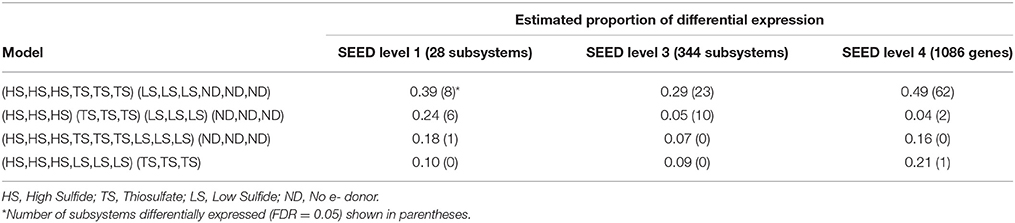
Table 4. Estimated proportion of differential expression based on posterior likelihoods of differential expression among treatments.
While our data indicate electron donor availability as the major driver of symbiont transcriptional variation, it is possible that factors independent of treatment conditions also affected the observed transcription patterns. First, some genes may be constitutively transcribed at uniform levels among treatments. Second, both the host and symbionts may produce storage molecules that could—over the duration of the incubations—dampen the transcriptional and physiological response to our treatments. For example, some chemoautotrophic symbionts produce elemental sulfur that can be stored and may be used as energy to drive carbon fixation when exogenous reductants are absent or low (Vetter, 1985; Wilmot and Vetter, 1990; Windoffer and Giere, 1997; Vetter and Fry, 1998; Pflugfelder et al., 2005). The presence of such molecules could enable symbionts to maintain thioautotrophic growth and associated gene expression patterns without exogenous reduced sulfur. It is possible that the snails used here may have contained varying amounts of storage compounds due to differences in sulfide availability at the collection sites. Finally, the half-life of mRNA varies widely, generally from minutes to hours (Rauhut and Klug, 1999; Deana and Belasco, 2005), raising the possibility of a decoupling between transcript profiles and the inferred physiological outcomes linked to transcribed genes. However, the long duration during which snails were maintained in HPRS (8 h acclimation + 27–40 h incubation) makes it unlikely that our results reflect transcriptional variation due to differences in in situ conditions at the collection sites.
Eight of the 28 Level 1 SEED subsystems differed significantly in expression (FDR < 0.05, Table 4 and Table S1). Sample clustering based on transcript representation in these 8 categories grouped high electron donor (HD) samples separate from low electron donor (LD) samples (Figure 4). In agreement with the higher carbon fixation rates measured directly in HD samples, transcripts associated with nitrogen metabolism, respiration, and nucleosides/nucleotides were more abundant in metatranscriptomes from HD treatments. Closer inspection of differentially expressed genes in these categories indicated that the availability of reduced sulfur compounds increased the representation of transcripts involved in nitrogen assimilation, ATP synthesis, cytochrome oxidase biogenesis, and dissimilatory nitrate reduction (Table S2). In contrast, LD incubations were enriched in transcripts involved in stress response, protein metabolism, phages/prophages/transposable elements/plasmids, clustering-based subsystems, and iron acquisition/metabolism (Figure 4).
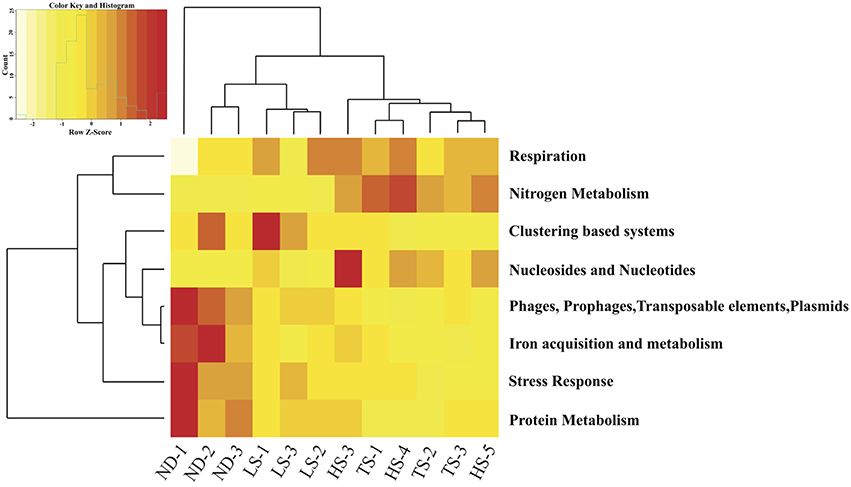
Figure 4. Heat map displaying hierarchical distance clustering of the eight SEED Subsystems differential expressed under HD vs. LD conditions based on normalized transcript abundance. Each row corresponds to a SEED subsystem, and each column to an individual experimental Ifremeria nautilei. Relative expression levels are indicated by colors shown in the scale at the top left. The dendrogram at the top shows the clustering of the individual snail samples. The dendrogram at left demonstrates the clustering of the SEED Subsystems.
It should be noted that many proteins are listed in multiple SEED subsystems. For example, the chaperone protein DnaK is found in both protein metabolism and stress response subsystems. Other proteins are listed in seemingly unrelated subsystems, as is the case for the Fe-S cluster scaffold protein SufB, which is included in the phages/prophages/transposable elements/plasmids subsystem. Thus, drawing definitive conclusions from Level 1 SEED subsystem expression is difficult. Below, we use the results of the differential expression analysis of SEED functional proteins combined with supplemental manual queries of BLASTX results to describe in detail the expression patterns for key processes of symbiont physiology, especially those identified as differentially expressed in baySeq. Based on the 16S rRNA gene and transcript patterns described above, the vast majority of bacterial transcripts described are likely derived from the sulfur-oxidizing symbiont population. However, in the absence of genome data, we are not able to definitively localize all transcripts to a particular symbiont phylotype.
S Metabolism
Sulfur oxidation genes typical of chemoautotrophic γ-proteobacteria were transcribed in all individuals (Figure 5). The canonical sulfur oxidation pathway in chemoautotrophic γ-proteobacteria is believed to start with the incomplete oxidation of sulfide or thiosulfate to elemental sulfur in the periplasm by the SoxABXYZ proteins (Ghosh and Dam, 2009). Through manual searches of BLASTX results (bit score > 50), we identified transcripts matching the soxABHWXYZ genes (Table S4). Typical of γ-proteobacterial sulfur oxidizers, transcripts corresponding to the SoxCD proteins, which are necessary for complete oxidation of sulfide to sulfate by the Sox system, were not found, which may indicate that I. nautilei symbionts form elemental sulfur in the periplasm (Ghosh and Dam, 2009). SoxH, SoxK, and the thioredoxin SoxW were also detected in all metatranscriptomes and are commonly found in γ-proteobacterial sulfur oxidizers, but their roles in sulfide oxidation are unclear. In addition, manual searches of BLASTX results detected sulfide:quinone (oxido)reductase (Sqr) and sulfide dehydrogenase (Fcc) transcripts, both of which oxidize sulfide to polysulfides in the periplasm, although only Sqr has been shown to be necessary for sulfide oxidation (Dahl and Friedrich, 2008). The abundance of sqr, fcc, and soxABHWXYZ transcripts was relatively uniform in all metatranscriptomes, demonstrating no differential expression based on metabolic activity or the concentration or type of electron donor (Tables S3, S4).
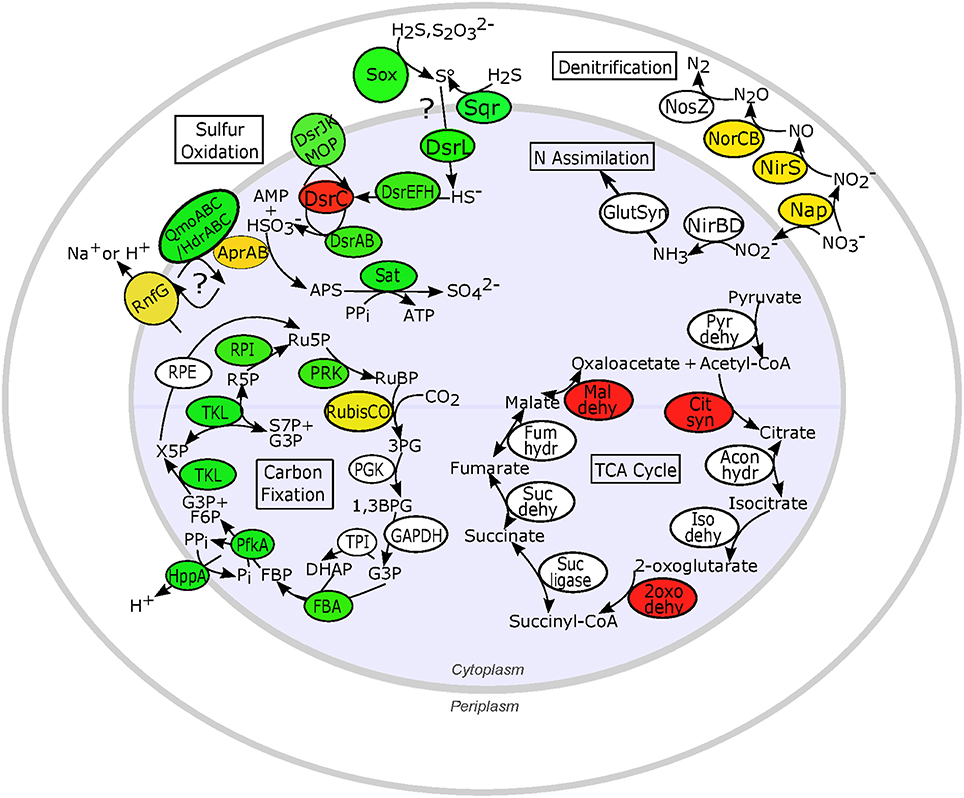
Figure 5. Expression patterns for key proteins involved in proposed sulfur oxidation, nitrogen reduction and assimilation, and carbon metabolism pathways in chemosynthetic Ifremeria nautilei symbionts. Proteins and complexes are colored based on transcript abundance in snail metatranscriptomes: green, constitutive expression (transcripts detected in all metatranscriptomes); yellow, HD expression > LD expression (FDR < 0.05); red, LD expression > HD expression (FDR < 0.05); white, sporadic or low expression (transcripts detected in fewer than 9 of the 12 metatranscriptomes); only proteins for which transcripts were detected are shown. Sulfur oxidation: Sox, Sox multi enzyme complex; AprAB, adenylylsulfate reductase; DsrL, Sulfur oxidation-associated protein DsrL; DsrEFH, putative sulfurtransferase complex; DsrC, putative bacterial heterodisulfide; DsrAB, reverse-type dissimilatory sulfite reductase; DsrJKMOP, sulfite reduction-associated complex DsrMKJOP; QmoABC/HdrABC, putative quinone-interacting membrane-bound oxidoreductase; RnfG, Rnf electron transport complex; Sat, sulfur adenylyltransferase; Sqr, sulfide quinone (oxido)reductase. N metabolism: Nap, periplasmic nitrate reductase; NirS, membrane-bound respiratory nitrite reductase; NirBD, NADH-dependent siroheme nitrite reductase; NorCB, nitric oxide reductase; NosZ, nitrous oxide reductase; GlutSyn, glutamine synthetase + glutamate synthase. TCA cycle: Pyr dehy, pyruvate dehydrogenase; Cit syn, citrate synthase; Acon hydr, Aconitate hydratase; Iso dehy, Isocitrate dehydrogenase; 2oxo dehy, 2-oxoglutarate dehydrogenase; Suc ligase, Succinyl-CoA ligase; Suc dehy, Succinate dehydrogenase; Fum hydr, Fumarate hydratase class I; Mal dehy, Malate dehydrogenase. Carbon fixation: RubisCO, Ribulose bisphosphate carboxylase; PGK, Phosphoglycerate kinase; GAPDH, NAD-dependent glyceraldehyde-3-phosphate dehydrogenase; TPI, Triosephosphate isomerase; FBA, Fructose bisphosphate aldolase; PfkA, Reversible pyrophosphate-dependent phosphofructokinase; HppA, membrane-bound proton-translocating pyrophosphatase; TKL, Transketolase; RPE, Ribulose-phosphate-3-epimerase; RPI, Ribose-5-phosphate isomerase A; PRK, Phosphoribulokinase; RuBP, Ribose-1,5-bisphosphate; 3PG, 3-phosphoglycerate; G3P, Glyceraldehyde-3-phosphate; DHAP, Dihydroxyacetone phosphate; FBP, Fructose-1,6-bisphosphate; F6P, Fructose-6-phosphate; X5P, Xylose-5-phosphate; Ru5P, Ribulose-5-phosphate; R5P, Ribose-5-phosphate; S7P, Sedoheptulose-7-phosphate; PPi, inorganic pyrophosphate; Pi, inorganic phosphate.
Following the production of elemental sulfur, sulfur-oxidizing γ-proteobacteria use the reverse dissimilatory sulfite reduction pathway (rDSR) to produce sulfite, followed by sulfite oxidation to sulfate by APS reductase (AprAB) and sulfate adenylytransferase (Sat; Dahl et al., 2005). Both MEGAN SEED protein classification and manual searches of BLASTX results showed that DSR genes dsrABCEFHJKLMOPNR were similarly represented in all metatranscriptomes (Tables S3, S4 and Figure 5). Transcription of aprAB, encoding the subunits of APS reductase, was correlated with sulfur metabolism rates (R2 = 0.99), and aprAB was the only sulfur metabolism gene identified by the baySeq model as significantly up-regulated in HD incubations (FDR = 0.006; Table S2). Multiple studies of free-living and endosymbiotic dissimilatory sulfur-metabolizing bacteria have found that AprAB expression is high in relation to other proteins (>6% of total proteome; Markert et al., 2007, 2011) and correlated with the energy state of the cell (Zhang et al., 2006; Pereira et al., 2008).
Transcription of the genes encoding the quinone-interacting membrane-bound oxidoreductase QmoABC complex, which is proposed to transfer electrons from AprAB to the periplasmic quinone pool, was sporadic among treatments (Figure 5). However, both SEED identifications and manual searches showed that all metatranscriptomes contained abundant sequences encoding heterodisulfide reductase HdrABC, which may be homologous to the QmoABC complex (Ramos et al., 2012). Indeed, genomic analysis of Thiolapillus brandeum strain Hiromi 1, a free-living strain closely related to I. nautilei sulfur-oxidizing symbionts, identified a chimeric cluster of qmoAB-hdrBC genes (Nunoura et al., 2014) and metagenomic/proteomic analysis of Riftia pachyptila γ-proteobacterial symbionts found abundant proteins annotated as heterodisulfide reductases with high similarity to Chlorobium tepidum QmoAB proteins (Markert et al., 2011). Thus, differentiation of these homologous protein complexes is problematic. In addition, the nature of electron transfer from AprAB to the periplasm is unclear and may involve heterodisulfide reductase and flavin-based electron bifurcation in order to conserve energy (Ramos et al., 2012). Evidence for expression of multiple proteins of the Rnf electron transport complex, which could couple with HdrABC to generate an ion motive force and thus ATP (Biegel et al., 2011; Ramos et al., 2012; Buckel and Thauer, 2013), was also observed in all symbiont metatranscriptomes (Figure 5 and Tables S3, S4), with transcripts for RnfG significantly more abundant in HD vs. LD incubations (FDR = 0.04; Table S2).
In summary, we observed expression of the full suite of expected sulfur oxidation genes in I. nautilei symbiont metatranscriptomes; however, expression of most sulfur oxidation genes, excluding AprAB, was not impacted by electron donor concentration or type. This result could be explained by several factors, including (i) stability and persistence of sulfur-oxidation transcripts produced prior to our experiments (i.e., during in situ activity), (ii) oxidation of elemental sulfur molecules stored by the symbionts to maintain thioautotrophic growth in the absence of sufficient exogenous reduced sulfur, or (iii) constitutive expression of most symbiont sulfur oxidation genes. Of these, the latter option may be most likely given that the predicted half-life of most mRNA molecules is considerably shorter than the duration of our experiments (Rauhut and Klug, 1999), and that there is no evidence that I. nautilei produce sulfur storage granules (Windoffer and Giere, 1997; Borowski et al., 2002). Rather, it has been suggested that indispensable metabolic pathways in bacteria are often constitutively expressed and are not repressible (Salmon et al., 2003, 2005). Thus, if I. nautilei symbionts are obligate sulfur oxidizers and cannot oxidize other electron donors, it is not surprising that sulfur oxidation genes are transcribed constitutively. Here, APS reductase (AprAB) and RnfG were the only sulfur oxidation genes whose transcription increased in response to reduced sulfur, suggesting that these proteins are tightly regulated by environmental conditions and are important for electron transport and energy conservation when either sulfide or thiosulfate is abundant. Indeed, other studies have also found that the transcription of AprAB is relatively responsive to environmental conditions compared to that of other sulfur oxidation genes (Zhang et al., 2006; Markert et al., 2007; Wendeberg et al., 2012). AprAB may therefore be a particularly good indicator of sulfur oxidation activity.
Carbon Metabolism
Previous work has shown that most γ-proteobacterial chemoautotrophic symbionts fix carbon via the Calvin-Benson-Bassham (CBB) cycle (Woyke et al., 2006; Duperron et al., 2007; Markert et al., 2007; Kleiner et al., 2012a; Sanders et al., 2013). Both SEED classification in MEGAN and manual searches of BLASTX data detected transcripts corresponding to all CBB cycle enzymes, although expression levels depended on the enzyme and incubation conditions (Figure 5 and Tables S3, S4). Furthermore, transcripts encoding the three key enzymes of the rTCA cycle (ATP citrate lyase, 2-oxoglutarate oxidoreductase, and fumarate reductase) were not detected, indicating that I. nautilei symbionts use only the CBB cycle for carbon fixation. Based on SEED classifications, form II Ribulose-1,5-bisphosphate carboxylase/oxygenase (RubisCO) was the most highly transcribed CBB cycle gene and the only CBB gene significantly more abundant in HD incubations (Table S2 and Figure 5). RubisCO transcript abundances and average 13C incorporation rates were both substantially higher in HD vs. LD incubations, although individual values were only moderately correlated (R2 = 0.24), suggesting that post-transcriptional regulation, redox poise, or other factors may control activity of the RubisCO enzyme and carbon incorporation rates (Beinart et al., 2015; Table 2). One individual in particular, snail HS3, exhibited a relative high abundance of RubisCO transcripts, but did not incorporate appreciable levels of 13C.
Form II RubisCO (CbbM), the only form detected in our data, is sensitive to oxygen and is hypothesized to function best in environments with low O2 and high CO2 concentrations (Tabita et al., 2008). However, it is thought that cbbM expression is unlikely to be directly regulated by CO2 concentration but may instead be regulated by redox conditions (Badger and Bek, 2008; Tabita et al., 2008; Alfreider et al., 2012). Our data are consistent with the hypothesis that cbbM transcription is induced when oxygen is low, and suppressed when oxygen concentrations are high, such as likely to be the case in the LD treatments if reduced access to electron donor decreases the symbionts' requirement for oxygen. Thus, transcription levels may be tightly linked to redox conditions experienced by the symbionts. Further investigation on the regulation of RubisCO in symbionts is needed to understand how this critical enzyme is regulated and optimized for conditions within the host.
Transcripts matching phosphoribulokinase, fructose-bisphosphate aldolase, transketolase, and ribulose-5-phosphate isomerase were detected in all 12 metatranscriptomes, whereas transcripts for other CBB cycle enzymes were detected at lower levels and more sporadically among datasets (Table S3). As has been reported for symbionts of deep-sea tubeworms (Riftia pachyptila), clams (Calyptogena magnifica), and shallow water gutless marine worms, we did not detect transcription of the genes encoding sedoheptulose-1,7-bisphosphatase and fructose-1,6-bisphosphatase, which are necessary to regenerate ribulose-1,5-bisphosphate (Markert et al., 2007; Newton et al., 2007; Kleiner et al., 2012b). However, manual searches of BLASTX results detected transcription of genes for reversible pyrophosphate-dependent phosphofructokinase (PfkA) and a membrane-bound proton-translocating pyrophosphatase (HppA), which together have been proposed to replace sedoheptulose-1,7-bisphosphatase and fructose-1,6-bisphosphatase in some chemolithotrophic symbionts (Table S4). Using reversible PfkA in combination with HppA in the CBB cycle reduces the net ATP consumption of carbon fixation as the membrane-bound proton-translocating pyrophosphatase establishes a proton gradient across the cytoplasmic membrane, allowing for ATP generation by ATP synthase (Reshetnikov et al., 2008; Kleiner et al., 2012a,b). In support of this hypothesis, PfkA enzyme activity was detected in strain Hiromi 1, a strain closely related to I. nautilei sulfur-oxidizing symbionts, pfkA is conserved in all complete genomes of sulfur-oxidizing γ-proteobacteria available in public databases, and pfkA:hppA are co-localized in the genomes of many free-living and symbiotic chemoautotrophic bacteria (Kleiner et al., 2012a,b; Nunoura et al., 2014).
Evidence for expression of bacterial oxidative TCA enzymes was observed in all metatranscriptomes, and transcripts corresponding to citrate synthase, 2-oxoglutarate dehydrogenase, and malate dehydrogenase were significantly more abundant in LD vs. HD treatments based on the baySeq differential expression analysis (Table S2). In addition, transcripts encoding two enzymes involved in the biosynthesis of lipoyl cofactors (octanoate-[acyl-carrier-protein]-protein-N-octanoyltransferase and lipoate synthase) essential for the activation of oxidative TCA cycle enzymes and a long chain fatty acid coenzyme A ligase involved in activation of fatty acid breakdown were also significantly enriched in LD metatranscriptomes (Table S2).
Activation of fatty acids and expression of oxidative TCA cycle enzymes when electron donors are limiting could provide a number of advantages for symbiont survival. Incorporation of fatty acids and other organic carbon through the TCA cycle is less energy intensive than fixing inorganic carbon and also produces NADPH, which presumably would be in short supply when electron donors are not available (Nunoura et al., 2014). However, net heterotrophic growth requires uptake of organic compounds, and we did not find evidence for the expression of genes involved in the transport of organic compounds into the cell (Kleiner et al., 2012b). Alternatively, increased expression of TCA cycle enzymes could be driven by the oxidizing conditions in the LD incubations, as has been observed in multiple studies in Escherichia coli (Salmon et al., 2003, 2005; Shalel-Levanon et al., 2005; Toya et al., 2012).
It is not clear from our data whether I. nautilei symbionts are performing net heterotrophy or are using internal carbon molecules such as fatty acids and glycogen as a source of carbon and reducing equivalents when electron donors are limited. Free-living strains related to I. nautilei symbionts have been observed to incorporate complex organic carbon sources such as fumarate, formate, citrate, pyruvate, and peptone (Sievert and Vetriani, 2012; Nunoura et al., 2014). Our data raise the possibility that I. nautilei symbionts are mixotrophic, fixing inorganic carbon when reduced sulfur is available, but switching to chemoheterotrophy when energy supplies are limiting.
Nitrogen Metabolism
Inorganic nitrogen compounds are generally abundant at hydrothermal vents, while organic nitrogen compounds are scarce (Johnson et al., 1986). Vent fluids may contain ammonium, which can be assimilated by many microorganisms. However, nitrate is typically more abundant and can be used as both an oxidant for energy generation (respiration) and a primary nitrogen source for assimilation. Both processes begin with nitrate reduction to nitrite, but proceed through different enzymatic pathways (Potter et al., 1999; Klotz and Stein, 2008). The next step in nitrogen assimilation is the cytoplasmic reduction of nitrite to ammonia by the complex NirBD and assimilation by glutamine synthetase/glutamate synthase or asparagine synthase. In the dissimilatory pathway, nitrite may be further reduced to nitrogen gas (denitrification) in three steps: nitrite reduction by NirS or NirK, nitric oxide reduction by the NorCB complex, and finally nitrous oxide reduction by NosZ. Alternatively, nitrite can be reduced to ammonium by Nrf to generate energy in a process of dissimilatory nitrate reduction to ammonia (Potter et al., 1999; Klotz and Stein, 2008).
Metatranscriptomes from HD incubations were significantly enriched in transcripts encoding proteins of both assimilatory and dissimilatory (respiratory) nitrate reduction and ammonium assimilation compared to LD treatments (Figure 5 and Table S2). Interestingly, no transcripts corresponding to the membrane-bound dissimilatory Nar or cytoplasmic assimilatory Nas nitrate reductases were identified. We only found transcripts encoding the periplasmic nitrate reductase NapCFGH proteins, which may be used in either assimilation or respiration, and in some cases may serve as an electron shunt when electron flow through the aerobic respiratory chain is restricted due to low oxygen concentrations (Potter et al., 1999; Klotz and Stein, 2008). The relative abundance of Nap transcripts was on average over 20X higher in HD compared to LD conditions (Table S3). Transcripts encoding a complete suite of respiratory denitrification [nirS, norCB, and nosZ] and cytoplasmic nitrogen assimilation [nirBD, glutamine synthetase-glutamate synthase] enzymes were also significantly enriched in HD metatranscriptomes (Table S2). These results suggest that when sufficient electron donor is available I. nautilei symbionts reduce nitrate to nitrite in the periplasm and subsequently use it for respiration (denitrification) in the periplasm or transport the nitrite to the cytoplasm for assimilation (Figure 5). Metatranscriptomic analysis of sulfur-oxidizing symbionts of the vent snail Alviniconcha found evidence for the same nitrate assimilation and denitrification pathways reported here (Sanders et al., 2013). Furthermore, a recent study showed that a close free-living relative of the I. nautilei symbiont coupled complete denitrification to sulfur oxidation and reached higher cell densities with nitrate as electron acceptor compared to oxygen (Nunoura et al., 2014).
Using the periplasmic Nap complex for nitrate reduction provides considerable adaptability in nitrogen metabolism and respiration in response to dynamic nitrogen concentrations and redox conditions. After reduction of nitrate to nitrite in the periplasm, nitrite can be transported into the cytoplasm only when it is needed for assimilation, keeping cytoplasmic nitrite concentrations low and thus avoiding nitrite toxicity (Rowe et al., 1979). Additionally, unlike the respiratory Nar proteins, the Nap complex is not inhibited by oxygen (Potter et al., 1999). Consequently, the Nap complex can be expressed under aerobic conditions, potentially allowing symbionts to use nitrate as an electron acceptor and avoid competition with the host for oxygen (Sanders et al., 2013). Our results confirm that sulfur-oxidizing symbionts tightly regulate the expression of nitrogen assimilation and denitrification genes in response to reduced sulfur and oxygen availability.
Stress Response
Symbiont populations in LD incubations express genes that are consistent with increased oxidative stress (Chen et al., 2009). Of 19 protein-coding genes identified as significantly more abundant in LD incubations using the baySeq model, 10 were related to the bacterial stress response (Table S2). In bacteria, a highly conserved “heat-shock” stress response occurs in response to heat, starvation, radiation, and oxidative agents (Neidhardt et al., 1984). The heat-shock response involves production of a set of heat-shock proteins, many of which are molecular chaperones involved in transit across membranes, targeted proteolysis, and polypeptide folding (Genevaux et al., 2007). Stress-induced transcription of heat-shock genes is typically controlled by binding of the sigma factor RpoH to RNA polymerase (Bukau, 1993). Based on SEED protein classification in MEGAN, metatranscriptomes from LD incubations contained 9X more RpoH-related transcripts than those from HD incubations, indicating that the heat-shock response was induced in symbionts without sufficient electron donor in the presence of oxygen (Table S2).
In accordance with an increase in RpoH, transcripts encoding proteins in the DnaK chaperone system and the Clp/Hsp100 family of ATP-dependent protein remodeling machines also increased in LD incubations, notably representing the most abundant transcripts in ND datasets. The DnaK chaperone system is composed of DnaK, DnaJ, and GrpE, which work together with Clp/Hsp100 family proteins to catalyze protein disaggregation and refolding or protein degradation during physiologic stress (Dougan et al., 2002). Here, DnaK, DnaJ, and GrpE transcripts were significantly enriched in LD incubations, although DnaJ and GrpE transcripts were 10 to 100X less abundant than DnaK (Tables S2, S3). Transcripts encoding ClpA and ClpX, as well as FtsH subunits and peptidases associated with ATP-dependent protein degradation (Dougan et al., 2002), were also significantly enriched under LD conditions (Table S2). Other subunits associated with the Clp/Hsp100 family, such as ClpB and ClpS, were highly expressed in LD incubations, but were not significantly differentially expressed under the criteria of the baySeq model (Table S3).
Transcription of genes involved in iron regulation and 4Fe-4S cluster biosynthesis were also significantly more abundant under LD conditions, potentially due to the importance of iron homeostasis for cell survival during oxidative stress (Table S2). Ferrous iron reacts with the hydrogen peroxide present during oxidative stress to form damaging peroxide radicals. The rate of oxidative damage is therefore elevated when cytoplasmic iron concentrations are high (Imlay, 2013). Transcripts related to the MerR family of metalloregulators, which detect and respond to reactive metals and changes in redox conditions, were significantly enriched in LD metatranscriptomes. These included transcripts encoding the oxidative stress metalloregulator SoxR, which is known to induce expression of multiple proteins that mitigate cellular damage, including superoxide dismutase and the regulatory Fur protein (Brown et al., 2003; Imlay, 2013). Both the Fur protein, which suppresses iron import and utilization, and superoxide dismutase, were significantly more abundant in LD incubations (Table S2), suggesting that I. nautilei symbionts without sufficient electron donating compounds were attempting to limit iron uptake and neutralize superoxide to avoid oxidative damage.
Proteins containing 4Fe-4S centers and mononuclear iron also are very susceptible to damage from reactive oxygen species (Imlay, 2013). Transcripts from two Fe-S cluster assembly pathways, the Isc (iron sulfur cluster) system and the Suf (sulfur formation) system, were detected in all snail metatranscriptomes (Table S3). The Suf system has been found to operate specifically to protect and assemble Fe-S clusters during oxidative stress (Ayala-Castro et al., 2008). In accordance with this suggested function, transcripts encoding a key scaffold protein (SufB) in the Suf system were on average 20X more abundant in LD vs. HD incubations (Table S2). LD transcriptomes were also significantly enriched in transcripts for the DNA repair enzyme RecA (Imlay, 2013) and the stress-induced morphogene BolA (Santos et al., 1999). Together, these results indicate significant oxidative stress in symbiont populations without sufficient electron donor, suggesting that I. nautilei cannot shield its symbionts from fluctuations in redox conditions that may negatively impact symbiont survival.
Methane (C1) Metabolism
Despite a presumed lack of exogenous methane in these experiments, we detected transcripts indicating that methanotroph-related I. nautilei symbionts were active during the incubations. Phylogenetic analysis of 16S rRNA transcripts and amplified rRNA genes placed the methanotroph-related I. nautilei symbionts in a monophyletic group within the γ-proteobacterial family Methylococcaceae (Figure 2). Methylococcaceae are type I methanotrophs typically characterized by the use of the ribulose monophosphate (RuMP) pathway for incorporation of C1 carbon into biomass and a membrane-associated particulate methane monooxygenase (pMMO) that catalyzes the oxidation of methane to methanol (Chistoserdova, 2011). Manual searches of BLASTX files revealed transcripts matching the pMMO genes pmoCAB, accounting for 0.1-1.5% of non-rRNA reads in the TS, LS, and ND datasets (Table S4). Recovered pMMO sequences shared >85% amino acid similarity to pMMO proteins from Methylomicrobium album, Methylomicrobium buryatense, and Methylomarinum vadi, supporting the phylogenetic placement of these symbionts within the Methylococcaceae, but not specifically related to methanotrophic symbionts of deep-sea bathymodiolin mussels. No transcripts indicative of sMMO were detected (Hanson and Hanson, 1996; Chistoserdova, 2011). PmoC-related reads represented the most abundant methanotroph-related functional gene transcript observed in the TS, LS, and ND metatranscriptomes, and were 5–10X more abundant than PmoA or PmoB transcripts. Two previous transcriptomic studies of alpha-proteobacterial methanotroph pure cultures observed that the pmoCAB operon is the most highly expressed operon in the genome when sufficient copper is present, with pmoC gene expression 6–7X higher than pmoA/B (Matsen et al., 2013; Vorobev et al., 2014).
Evidence for the further oxidation and assimilation of methane was also detected, although transcripts from these pathways were one to two orders of magnitude less abundant than pMMO-related transcripts (Table S4). Transcripts encoding methanol dehydrogenase proteins (MDH), which catalyze the oxidation of methanol to formaldehyde (Chistoserdova, 2011), were present in three of the nine methanotroph-containing metatranscriptomes. In addition, six metatranscriptomes contained transcripts matching methanotroph-related genes for the biosynthesis of the methanol dehydrogenase cofactor pyrroloquinoline quinone. Expression of unique genes for formaldehyde oxidation and assimilation through the RuMP pathway (3-hexulose-6-phosphate synthase and 3-hexulose-6-phosphate isomerase) were also detected in six of the nine methanotroph-containing metatranscriptomes (Table S4).
Collectively, these results confirm the transcriptional activity of a methanotrophy pathway in I. nautilei symbionts. The presence of methanotrophy-related transcripts despite a lack of C1 compounds in these experiments is surprising as no methane was introduced into these experiments, and the chance of methane being present in any of the injected mixed gases is very low. Our data may therefore be explained by a constitutive transcription of methanotrophy genes, or a relatively high stability of methanotrophy transcripts. Indeed, both constitutive expression and high transcript stability have been reported for the pmoCAB operon (Lieberman and Rosenzweig, 2004; Deana and Belasco, 2005; Chen et al., 2008; Wendeberg et al., 2012; Matsen et al., 2013). Alternatively, putatively methanotrophic I. nautilei symbionts could be growing on substrates other than methane, such as methylated sulfur compounds or even multi-carbon substrates. However, this explanation seems unlikely, as all Methylococcaceae characterized to date are obligate methanotrophs (Chistoserdova, 2011). The functional contributions of I. nautilei methanotrophs to holobiont fitness remain to be determined.
Concluding Remarks
This study of deep-sea vent snails is unique in that it examined the transcriptional responses of bacterial symbionts under well-controlled environmental conditions. These results show that while I. nautilei symbionts can use either sulfide or thiosulfate for energy during chemoautotrophic growth (as observed in Beinart et al., 2015), the transition between these substrates appears to have surprisingly little effect on symbiont transcription patterns. Periplasmic nitrate reduction appears critical at higher sulfide or thiosulfate conditions, likely reflecting a dependence on nitrate respiration at elevated sulfur species concentrations, as well as enhanced nitrogen assimilation to meet potentially elevated growth rates.
Notably, when sulfide is low or absent, there are marked and unexpected changes in symbiont transcription. First, I. nautilei symbionts express genes that are consistent with mixotrophy, suggesting the possibility of transitioning to heterotrophic growth when energy for CO2 fixation is limiting. The origin and composition of the organic carbon for symbiont heterotrophy is unknown in I. nautilei, but could involve recycling of organics leaked from host cells or the catabolism of internally stored carbon (Kleiner et al., 2012b). Second, these data suggest that at lower sulfide concentrations I. nautilei symbionts may experience oxidative stress. We posit that the symbionts may be experiencing a reduction in reactive oxygen species scavenging when electron donors are scarce, or alternatively may be exposed to elevated reactive oxygen species concentrations caused by stressed hosts. While the causal factor remains unclear, it is important to note that these conditions are comparable to those found in situ, especially around lower flow diffuse vents. Third, a striking commonality among many of the differentially expressed genes identified in this study, including form II RubisCO, oxidative TCA cycle enzymes, and genes involved in iron uptake, is that redox potential is implicated in their regulation based on studies in other bacteria; thus environmental redox conditions may control many processes in I. nautilei symbionts.
Our results also highlight the variability of I. nautilei symbiont community composition across the Lau Basin vent system and provide the first molecular evidence for the presence and activity of methanotrophic symbionts in Ifremeria. The persistence of methanotrophy transcripts in our incubations, despite the lack of a clear methane source, may reflect an adaptation to the dynamic vent environment, with the Ifremeria-associated bacterial community being poised to rapidly consume methane when this energy source becomes available.
Together these data reveal that vent geochemical dynamics affect a broad range of metabolic systems and subsystems, such as carbon, nitrogen and sulfur metabolism. Such variation will in turn govern the extent to which the holobiont's (host + symbiont) activity influences environmental geochemistry. Finally, the apparent response to low or no sulfide was quite striking, and future studies should further examine the extent to which oxidative stress influences symbiont function in situ. It should be noted that the relative abundances of transcripts, for example those encoding RubisCO, AprAB, or stress proteins, may be reasonably accurate indicators of substrate conditions and symbiont metabolic state. Other loci, however, appear less transcriptionally responsive to environmental change and their expression patterns should be interpreted with caution. In the future, similar controlled experiments that couple measurements of chemical flux (e.g., carbon fixation, sulfur and methane oxidation) to biomolecule abundances are necessary for determining which molecular patterns accurately reflect symbiont physiology, and therefore the extent to which meta-omic datasets can be used for predictive models of symbiont contributions to holobiont fitness.
Author Contributions
RB and PG conceived and designed the study, performed on-board experiments, and extracted nucleic acids. SS and FS designed the sequencing and analysis approach. SS, NS, SG, and AS performed sequencing reactions and sequence processing. SS, NS, SG, and PR analyzed the data. SS and RB wrote the paper with contributions from FS, PG, and NS.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by National Science Foundation Grants OCE-0732369 (to PG), DGE-1144152 (to RB), and (1151698 to FS) and the Alfred P. Sloan Foundation (grant RC944 to FS). Thanks to Josh Parris and members of the DiChristina Laboratory at Georgia Tech for assistance in the laboratory and helpful discussions.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2016.01074
Figure S1. Maps showing the vent fields ABE and Tu'i Malila at the Eastern Lau Spreading Center where individual snails used in the experimental treatments were collected.
Table S1. List of differentially expressed SEED Level 1 subsystems including normalized transcript abundance and false discovery rate (FDR) values.
Table S2. List of differentially expressed SEED Level functional proteins as annotated in MEGAN5 including normalized transcript abundance and false discovery rate (FDR) values.
Table S3. Normalized abundance of transcripts in all 12 metatranscriptomes corresponding to SEED subsystem functional proteins annotated in MEGAN5.
Table S4. Abundance of specific transcripts of interest (see Materials and Methods) in all 12 metatranscriptomes from manual searches of processed BLASTX results (bit score >50) normalized to 100 000 non-rRNA reads per metatranscriptome.
References
Alfreider, A., Schirmer, M., and Vogt, C. (2012). Diversity and expression of different forms of rubisco genes in polluted groundwater under different redox conditions. FEMS Microbiol. Ecol. 79, 649–660. doi: 10.1111/j.1574-6941.2011.01246.x
Ayala-Castro, C., Saini, A., and Outten, F. W. (2008). Fe-S cluster assembly pathways in bacteria. Microbiol. Mol. Biol. Rev. 72, 110–125. doi: 10.1128/MMBR.00034-07
Badger, M. R., and Bek, E. J. (2008). Multiple RubisCO forms in proteobacteria: their functional significance in relation to CO2 acquisition by the CBB cycle. J. Exp. Bot. 59, 1525–1541. doi: 10.1093/jxb/erm297
Beinart, R. A., Gartman, A., Sanders, J. G., Luther, G. W., and Girguis, P. R. (2015). The uptake and excretion of partially oxidized sulfur expands the repertoire of energy resources metabolized by hydrothermal vent symbioses. Proc. R. Soc. Lond. B Biol. Sci. 282:20142811. doi: 10.1098/rspb.2014.2811
Beinart, R. A., Nyholm, S. V., Dubilier, N., and Girguis, P. R. (2014). Intracellular Oceanospirillales inhabit the gills of the hydrothermal vent snail Alviniconcha with chemosynthetic, γ-Proteobacterial symbionts. Environ. Microbiol. Rep. 6, 656–664. doi: 10.1111/1758-2229.12183
Belkin, S., Nelson, D. C., and Jannasch, H. W. (1986). Symbiotic assimilation of CO2 in two hydrothermal vent animals, the mussel Bathymodiolus thermophilus and the tube worm Riftia pachyptila. Biol. Bull. 170, 110–121. doi: 10.2307/1541384
Biegel, E., Schmidt, S., Gonzalez, J. M., and Müller, V. (2011). Biochemistry, evolution and physiological function of the Rnf complex, a novel ion-motive electron transport complex in prokaryotes. Cell Mol. Life Sci. 68, 613–634. doi: 10.1007/s00018-010-0555-8
Borowski, C., Giere, O., Krieger, J., Amann, R., and Dubilier, N. (2002). New aspects of the symbiosis in the provannid snail Ifremeria nautilei from the North Fiji Back Arc Basin. Cahiers Biol. Mar. 43, 321–324.
Brown, N. L., Stoyanov, J. V., Kidd, S. P., and Hobman, J. L. (2003). The MerR family of transcriptional regulators. FEMS Microbiol. Rev. 27, 145–163. doi: 10.1016/S0168-6445(03)00051-2
Buckel, W., and Thauer, R. K. (2013). Energy conservation via electron bifurcating ferredoxin reduction and proton/Na+ translocating ferredoxin oxidation. Biochim. Biophys. Acta 1827, 94–113. doi: 10.1016/j.bbabio.2012.07.002
Bukau, B. (1993). Regulation of the Escherichia coli heat-shock response. Mol. Microbiol. 9, 671–680. doi: 10.1111/j.1365-2958.1993.tb01727.x
Butterfield, D. A., Jonasson, I. R., Massoth, G. J., Feely, R. A., Roe, K. K., Embley, R. E., et al. (1997). Seafloor eruptions and evolution of hydrothermal fluid chemistry. Philos. Trans. R. Soc. Lond. A 355, 369–386. doi: 10.1098/rsta.1997.0013
Canfield, D. E., Stewart, F. J., Thamdrup, B., De Brabandere, L., Dalsgaard, T., Delong, E. F., et al. (2010). A cryptic sulfur cycle in oxygen-minimum-zone waters off the chilean coast. Science 330, 1375–1378. doi: 10.1126/science.1196889
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Cavanaugh, C., Mckiness, Z., Newton, I. G., and Stewart, F. (2006). “Marine chemosynthetic symbioses,” in The Prokaryotes, eds M. Dworkin, S. Falkow, E. Rosenberg, K.-H. Schleifer, and E. Stackebrandt (New York, NY: Springer), 475–507.
Chen, Y., Azad, M. B., and Gibson, S. B. (2009). Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Diff. 16, 1040–1052. doi: 10.1038/cdd.2009.49
Chen, Y., Dumont, M. G., Neufeld, J. D., Bodrossy, L., Stralis-Pavese, N., McNamara, N. P., et al. (2008). Revealing the uncultivated majority: combining DNA stable-isotope probing, multiple displacement amplification and metagenomic analyses of uncultivated Methylocystis in acidic peatlands. Environ. Microbiol. 10, 2609–2622. doi: 10.1111/j.1462-2920.2008.01683.x
Childress, J. J., Fisher, C. R., Brooks, J. M., Kennicutt, M. C., Bidigare, R., and Anderson, A. E. (1986). A methanotrophic marine molluscan (Bivalvia, Mytilidae) symbiosis: mussels fueled by gas. Science 233, 1306–1308. doi: 10.1126/science.233.4770.1306
Childress, J. J., Fisher, C. R., Favuzzi, J. A., and Sanders, N. K. (1991). Sulfide and carbon dioxide uptake by the hydrothermal vent clam, Calyptogena magnifica, and its chemoautotrophic symbionts. Physiol. Zool. 64, 1444–1470. doi: 10.1086/physzool.64.6.30158224
Chistoserdova, L. (2011). Modularity of methylotrophy, revisited. Environ. Microbiol. 13, 2603–2622. doi: 10.1111/j.1462-2920.2011.02464.x
Dahl, C., Engels, S., Pott-Sperling, A. S., Schulte, A., Sander, J., Lübbe, Y., et al. (2005). Novel genes of the Dsr gene cluster and evidence for close interaction of Dsr proteins during sulfur oxidation in the phototrophic sulfur bacterium Allochromatium vinosum. J. Bacteriol. 187, 1392–1404. doi: 10.1128/JB.187.4.1392-1404.2005
Deana, A., and Belasco, J. G. (2005). Lost in translation: the influence of ribosomes on bacterial mRNA decay. Genes Dev. 19, 2526–2533. doi: 10.1101/gad.1348805
DeSantis, T. Z., Hugenholtz, P., Larsen, N., Rojas, M., Brodie, E. L., Keller, K., et al. (2006). Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72, 5069–5072. doi: 10.1128/AEM.03006-05
Dougan, D. A., Mogk, A., and Bukau, B. (2002). Protein folding and degradation in bacteria: to degrade or not to degrade? That is the question. Cell Mol. Life Sci. 59, 1607–1616. doi: 10.1007/PL00012487
Dubilier, N., Bergin, C., and Lott, C. (2008). Symbiotic diversity in marine animals: the art of harnessing chemosynthesis. Nat. Rev. Microbiol. 6, 725–740. doi: 10.1038/nrmicro1992
Duperron, S., Guezi, H., Gaudron, S. M., Pop Ristova, P., Wenzhöfer, F., and Boetius, A. (2011). Relative abundances of methane- and sulphur-oxidising symbionts in the gills of a cold seep mussel and link to their potential energy sources. Geobiology 9, 481–491. doi: 10.1111/j.1472-4669.2011.00300.x
Duperron, S., Sibuet, M., MacGregor, B. J., Kuypers, M. M., Fisher, C. R., and Dubilier, N. (2007). Diversity, relative abundance, and metabolic potential of bacterial endosymbionts in three Bathymodiolus mussel species from cold seeps in the Gulf of Mexico. Environ. Microbiol. 9, 1423–1438. doi: 10.1111/j.1462-2920.2007.01259.x
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucelic Acid Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Felbeck, H. (1981). Chemoautotrophic potential of the hydrothermal vent tube worm, Riftia pachyptila Jones (Vestimentifera). Science 213, 336–338. doi: 10.1126/science.213.4505.336
Fisher, C. R., Childress, J. J., Oremland, R. S., and Bidigare, R. R. (1987). The importance of methane and thiosulfate in the metabolism of the bacterial symbionts of two deep-sea mussels. Mar. Biol. 96, 59–71. doi: 10.1007/BF00394838
Gal'chenko, V. F., Pimenov, N. V., Lein, A. Y., Galkin, S. V., Miller, Y. M., and Ivanov, M. V. (1992). Mixotrophic type of feeding of Olgaconcha tufaribeck (gastropoda: Prosobranchia) from the active hydrothermal field of the Manus Basin (Bismark Sea). Dok. Biol. Sci. 323, 125–129.
Gardebrecht, A., Markert, S., Sievert, S. M., Felbeck, H., Thürmer, A., Albrecht, D., et al. (2012). Physiological homogeneity among the endosymbionts of Riftia pachyptila and Tevnia jerichonana revealed by proteogenomics. ISME J. 6, 766–776. doi: 10.1038/ismej.2011.137
Gartman, A., Yucel, M., Madison, A. S., Chu, D. W., Ma, S., Janzen, C. P., et al. (2011). Sulfide oxidation across diffuse flow zones of hydrothermal vents. Aquatic Geochem. 17, 583–601. doi: 10.1007/s10498-011-9136-1
Genevaux, P., Georgopoulos, C., and Kelly, W. L. (2007). The Hsp70 chaperone machines of Escherichia coli: a paradigm for the repartition of chaperone functions. Mol. Microbiol. 66, 840–857. doi: 10.1111/j.1365-2958.2007.05961.x
Ghosh, W., and Dam, B. (2009). Biochemistry and molecular biology of lithotrophic sulfur oxidation by taxonomically and ecologically diverse bacteria and archaea. FEMS Microbiol. Rev. 33, 999–1043. doi: 10.1111/j.1574-6976.2009.00187.x
Girguis, P. R., and Childress, J. J. (2006). Metabolite uptake, stoichiometry and chemoautotrophic function of the hydrothermal vent tubeworm Riftia pachyptila: responses to environmental variations in substrate concentrations and temperature. J. Exp. Biol. 209, 3516–3528. doi: 10.1242/jeb.02404
Girguis, P. R., Lee, R. W., Desaulniers, N., Childress, J. J., Pospesel, M., Felbeck, H., et al. (2000). Fate of nitrate acquired by the tubeworm Riftia pachyptila. Appl. Environ. Microbiol. 66, 2783–2790. doi: 10.1128/AEM.66.7.2783-2790.2000
Goffredi, S. K., Yi, H., Zhang, Q., Klann, J. E., Struve, I. A., Vrijenhoek, R. C., et al. (2014). Genomic versatility and functional variation between two dominant heterotrophic symbionts of deep-sea osedax worms. ISME J. 8, 908–924. doi: 10.1038/ismej.2013.201
Gru, C., Legoff, H., Narcon, S., Sarradin, P.-M., Caprais, J.-C., and Lallier, F. H. (1998). Determination of reduced sulfur compounds by high-performance liquid chromatography in hydrothermal seawater and body fluids from Riftia pachyptila. Analyst 123, 1289–1293. doi: 10.1039/a800032h
Hardcastle, T. J., and Kelly, K. A. (2010). Bayseq: empirical bayesian methods for identifying differential expression in sequence count data. BMC Bioinformatics 11:422. doi: 10.1186/1471-2105-11-422
Hirayama, H., Sunamura, M., Takai, K., Nunoura, T., Noguchi, T., Oida, H., et al. (2007). Culture-dependent and -independent characterization of microbial communities associated with a shallow submarine hydrothermal system occurring within a coral reef off Taketomi Island, Japan. Appl. Environ. Microbiol. 73, 7642–7656. doi: 10.1128/AEM.01258-07
Huson, D. H., Mitra, S., Weber, N., Ruscheweyh, H., and Schuster, S. C. (2011). Integrative analysis of environmental sequences using MEGAN4. Genome Res. 21, 1552–1560. doi: 10.1101/gr.120618.111
Imlay, J. A. (2013). The molecular mechanisms and physiological consequences of oxidative stress: lessons from a model bacterium. Nat. Rev. Microbiol. 11, 443–454. doi: 10.1038/nrmicro3032
Johnson, K. S., Beehler, C. L., Sakamoto-Arnold, C. M., and Childress, J. J. (1986). In situ measurements of chemical distributions in a deep-sea hydrothermal vent field. Science 231, 1139–1141. doi: 10.1126/science.231.4742.1139
Kleiner, M., Petersen, J. M., and Dubilier, N. (2012a). Convergent and divergent evolution of metabolism in sulfur-oxidizing symbionts and the role of horizontal gene transfer. Curr. Opin. Microbiol. 15, 621–631. doi: 10.1016/j.mib.2012.09.003
Kleiner, M., Wentrup, C., Lott, C., Teeling, H., Wetzel, S., Young, J., et al. (2012b). Metaproteomics of a gutless marine worm and its symbiotic microbial community reveal unusual pathways for carbon and energy use. Proc. Natl. Acad. Sci. U.S.A. 109, 7148–7149. doi: 10.1073/pnas.1121198109
Klotz, M. G., and Stein, L. Y. (2008). Nitrifier genomics and evolution of the nitrogen cycle. FEMS Microbiol. Lett. 278, 146–156. doi: 10.1111/j.1574-6968.2007.00970.x
Kozich, J. J., Westcott, S. L., Baxter, N. T., Highlander, S. K., and Schloss, P. D. (2013). Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 79, 5112–5120. doi: 10.1128/AEM.01043-13
Kuwahara, H., Yoshida, T., Takaki, Y., Shimamura, S., Nishi, S., Harada, M., et al. (2007). Reduced genome of the thioautotrophic intracellular symbiont in a deep-sea clam,Calyptogena okutanii. Curr. Biol. 17, 881–886. doi: 10.1016/j.cub.2007.04.039
Lieberman, R. L., and Rosenzweig, A. C. (2004). Biological methane oxidation: regulation, biochemistry, and active site structure of particulate methane monooxygenase. Crit. Rev. Biochem. Mol. Biol. 39, 147–164. doi: 10.1080/10409230490475507
Löytynoja, A., and Goldman, N. (2010). Webprank: a phylogeny-aware multiple sequence aligner with interactive alignment browser. BMC Bioinformatics 11:579. doi: 10.1186/1471-2105-11-579
Magoč, T., and Salzberg, S. L. (2011). Flash: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
Markert, S., Arndt, C., Felbeck, H., Becher, D., Sievert, S. M., Hügler, M., et al. (2007). Physiological proteomics of the uncultured endosymbiont of riftia pachyptila. Science 315, 247–250. doi: 10.1126/science.1132913
Markert, S., Gardebrecht, A., Felbeck, H., Sievert, S. M., Klose, J., Becher, D., et al. (2011). Status quo in physiological proteomics of the uncultured Riftia pachyptila endosymbiont. Proteomics 11, 3106–3117. doi: 10.1002/pmic.201100059
Matsen, J. B., Yang, S., Stein, L. Y., Beck, D., and Kalyuzhnaya, M. G. (2013). Global molecular analyses of methane metabolism in methanotrophic alphaproteobacterium, Methylosinus trichosporium OB3B. Part I: transcriptomic study. Front. Microbiol. 4:40. doi: 10.3389/fmicb.2013.00040
Meyer, F., Paarmann, D., D'souza, M., Olson, R., Glass, E., and Kubal, M. (2008). The metagenomics RAST server - a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics 9:386. doi: 10.1186/1471-2105-9-386
Mullaugh, K. M., Luther, G. W., Ma, S., Moore, T. S., Yucel, M., Becker, E. L., et al. (2008). Voltammetric (micro)electrodes for the in situ study of Fe2+ oxidation kinetics in hot springs and S2Oproduction at hydrothermal vents. Electroanalysis 20, 280–290. doi: 10.1002/elan.200704056
Nakagawa, S., Shimamura, S., Takaki, Y., Suzuki, Y., Murakami, S. I., Watanabe, T., et al. (2014). Allying with armored snails: the complete genome of gammaproteobacterial endosymbiont. ISME J. 8, 40–51. doi: 10.1038/ismej.2013.131
Neidhardt, F. C., VanBogelen, R. A., and Vaughn, V. (1984). The genetics and regulation of heat-shock proteins. Annu. Rev. Genet. 18, 295–329. doi: 10.1146/annurev.ge.18.120184.001455
Nelson, D. C., Hagen, K. D., and Edwards, D. B. (1995). The gill symbiont of the hydrothermal vent mussel Bathymodiolus thermophilus is a physchrophilic, chemoautotrophic, sulfur bacterium. Mar. Biol. 121, 487–495. doi: 10.1007/BF00349457
Newton, I. L. G., Woyke, T., Auchtung, T. A., Dilly, G. F., Dutton, R. J., Fisher, M. C., et al. (2007). The Calyptogena magnifica chemoautotrophic symbiont genome. Science 315, 998–1000. doi: 10.1126/science.1138438
Nunoura, T., Takaki, Y., Kazama, H., Kakuta, J., Shimamura, S., Makita, H., et al. (2014). Physiological and genomic features of a novel sulfur-oxidizing gammaproteobacterium belonging to a previously uncultivated symbiotic lineage isolated from a hydrothermal vent. PLoS ONE 9:e104959. doi: 10.1371/journal.pone.0104959
Nyholm, S. V., Robidart, J., and Girguis, P. R. (2008). Coupling metabolite flux to transcriptomics: insights into the molecular mechanisms underlying primary productivity by the hydrothermal vent tubeworm Ridgeia piscesae. Biol. Bull. 214, 255–265. doi: 10.2307/25470667
Overbeek, R., Begley, T., Butler, R. M., Choudhuri, J. V., Chuang, H. Y., Cohoon, M., et al. (2005). The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucelic Acids Res. 33, 5691–5702. doi: 10.1093/nar/gki866
Pereira, P. M., He, Q., Valente, F. M., Xavier, A. V., Zhou, J., Pereira, I. A., et al. (2008). Energy metabolism in Desulfovibrio vulgaris Hildenborough: insights from transcriptome analysis. Anton. Van Leeuw. 93, 347–362. doi: 10.1007/s10482-007-9212-0
Petersen, J. M., and Dubilier, N. (2009). Methanotrophic symbioses in marine invertebrates. Environ. Microbiol. Rep. 1, 319–335. doi: 10.1111/j.1758-2229.2009.00081.x
Petersen, J. M., Zielinski, F. U., Pape, T., Seifert, R., Moraru, C., Amann, R., et al. (2011). Hydrogen is an energy source for hydrothermal vent symbioses. Nature 476, 176–180. doi: 10.1038/nature10325
Pflugfelder, B., Fisher, C. R., and Bright, M. (2005). The color of the trophosome: elemental sulfur distribution in the endosymbionts of Riftia pachyptila (Vestimentifera; Siboglinidae). Mar. Biol. 146, 895–901. doi: 10.1007/s00227-004-1500-x
Potter, L. C., Millington, P., Griffiths, L., Thomas, G. H., and Cole, J. A. (1999). Competition between Eschericia coli strains expressing either a periplasmic or a membrane-bound nitrate reductase: does Nap confer a selective advantage during nitrate-limited growth? Biochem. J. 344, 77–84.
Ramos, A. R., Keller, K. L., Wall, J. D., and Pereira, I. A. (2012). The membrane QmoABC complex interacts directly with the dissimilatory adenosine 5′-phosphosulfate reductase in sulfate reducing bacteria. Front. Microbiol. 3:137. doi: 10.3389/fmicb.2012.00137
Rauhut, R., and Klug, G. (1999). mRNA degradation in bacteria. FEMS Microbiol. Rev. 23, 353–370. doi: 10.1111/j.1574-6976.1999.tb00404.x
Reshetnikov, A. S., Rozova, O. N., Khmelenina, V. N., Mustakhimov, I. I., Beschastny, A. P., Murrell, J. C., et al. (2008). Characterization of the pyrophosphate-dependent 6-phosphofructokinase from Methylococcus capsulatus Bath. FEMS Microbiol. Lett. 288, 202–210. doi: 10.1111/j.1574-6968.2008.01366.x
Robidart, J. C., Bench, S. R., Feldman, R. A., Novoradovsky, A., Podell, S. B., Gaasterland, T., et al. (2008). Metabolic versatility of the Riftia pachyptila endosymbiont revealed through metagenomics. Environ. Microbiol. 10, 727–737. doi: 10.1111/j.1462-2920.2007.01496.x
Robidart, J. C., Roque, A., Song, P., and Girguis, P. R. (2011). Linking hydrothermal geochemistry to organismal physiology: physiological versatility in Riftia pachyptila from sedimented and basalt-hosted vents. PLoS ONE 6:e21692. doi: 10.1371/journal.pone.0021692
Robinson, J., Polz, M., Fiala-Medioni, A., and Cavanaugh, C. (1998). Physiological and immunological evidence for two distinct C1-utilizing pathways in Bathymodiolus puteoserpentis (Bivalvia: Mytilidae), a dual endosymbiotic mussel from the mid-atlantic ridge. Mar. Biol. 132, 625–633. doi: 10.1007/s002270050427
Rowe, J. J., Yarbrough, J. M., Rake, J. B., and Eagon, R. G. (1979). Nitrite inhibition of aerobic bacteria. Curr. Microbiol. 2, 51–54. doi: 10.1007/BF02601735
Salmon, K. A., Hung, S. P., Mekjian, K., Baldi, P., Hatfield, G. W., and Gunsalus, R. P. (2003). Global gene expression profiling in Escherichia coli K12 the effects of oxygen availability and Fnr. J. Biol. Chem. 278, 29837–29855. doi: 10.1074/jbc.M213060200
Salmon, K. A., Hung, S. P., Steffen, N. R., Krupp, R., Baldi, P., Hatfield, G. W., et al. (2005). Global gene expression profiling in Escherichia coli K12 effects of oxygen availability and ArcA. J. Biol. Chem. 280, 15084–15096. doi: 10.1074/jbc.M414030200
Sanders, J. G., Beinart, R. A., Stewart, F. J., Delong, E. F., and Girguis, P. R. (2013). Metatranscriptomics reveal differences in in situ energy and nitrogen metabolism among hydrothermal vent snail symbionts. ISME J. 7, 1556–1567. doi: 10.1038/ismej.2013.45
Santos, J. M., Freire, P., Vicente, M., and Arraiano, C. M. (1999). The stationary-phase morphogene bolA from Escherichia coli is induced by stress during early stages of growth. Mol. Microbiol. 32, 789–798. doi: 10.1046/j.1365-2958.1999.01397.x
Schmieder, R., and Edwards, R. (2011). Quality control and preprocessing of metagenomic datasets. Bioinformatics 27, 863–864. doi: 10.1093/bioinformatics/btr026
Schmieder, R., Lim, Y. W., and Edwards, R. (2012). Identification and removal of ribosomal RNA sequences from metatranscriptomes. Bioinformatics 28, 433–435. doi: 10.1093/bioinformatics/btr669
Shalel-Levanon, S., San, K. Y., and Bennett, G. N. (2005). Effect of oxygen, and ArcA and Fnr regulators on the expression of genes related to the electron transfer chain and the TCA cycle in Escherichia coli. Metab. Eng. 7, 364–374. doi: 10.1016/j.ymben.2005.07.001
Shank, T. M., Fornari, D. J., Von Damm, K. L., Lilley, M. D., Haymon, R. M., and Lutz, R. A. (1998). Temporal and spatial patterns of biological community development at nascent deep-sea hydrothermal vents (9°50′n, East Pacific Rise). Deep Sea Res Part II Topical Stud. Oceanogr. 45, 465–515. doi: 10.1016/S0967-0645(97)00089-1
Sievert, S. M., and Vetriani, C. (2012). Chemoautotrophy at deep-sea vents: past, present, and future. Oceanography 25, 218–233. doi: 10.5670/oceanog.2012.21
Stein, J. L., Cary, S. C., Hessler, R. R., Ohta, S., Vetter, R. D., Childress, J. J., et al. (1988). Chemoautotrophic symbiosis in a hydrothermal vent gastropod. Biol. Bull. 174, 373–378. doi: 10.2307/1541963
Stewart, F. J., Newton, I. L. G., and Cavanaugh, C. M. (2005). Chemosynthetic endosymbioses: adaptations to oxic-anoxic interfaces. Trends Microbiol. 13, 439–448. doi: 10.1016/j.tim.2005.07.007
Stewart, F. J., Ulloa, O., and DeLong, E. F. (2012). Microbial metatranscriptomics in a permanent marine oxygen minimum zone. Environ. Microbiol. 14, 23–40. doi: 10.1111/j.1462-2920.2010.02400.x
Suzuki, Y., Kojima, S., Watanabe, H., Suzuki, M., Tsuchida, S., Nunoura, T., et al. (2006). Single host and symbiont lineages of hydrothermal-vent gastropods Ifremeria nautilei (Provannidae): biogeography and evolution. Mar. Ecol. Prog. Ser. 315, 167–175. doi: 10.3354/meps315167
Tabita, F. R., Hanson, T. E., Satagopan, S., Witte, B. H., and Kreel, N. E. (2008). Phylogenetic and evolutionary relationships of RubisCO and the RubisCO-like proteins and the functional lessons provided by diverse molecular forms. Philos. Trans. R. Soc. Lond. B Biol. Sci. 363, 2629–2640. doi: 10.1098/rstb.2008.0023
Tivey, M. K. (2007). Generation of seafloor hydrothermal vent fluids and associated mineral deposits. Oceanography 20, 50–65. doi: 10.5670/oceanog.2007.80
Toya, Y., Nakahigashi, K., Tomita, M., and Shimizu, K. (2012). Metabolic regulation analysis of ArcA mutant Eschericia coli under nitrate conditions using different levels of omics data. Mol. Biosyst. 8, 2593–2604. doi: 10.1039/c2mb25069a
Urakawa, H., Dubilier, N., Fujiwara, Y., Cunningham, D. E., Kojima, S., and Stahl, D. A. (2005). Hydrothermal vent gastropods from the same family (Provannidae) harbour ε- and γ-proteobacteria endosymbionts. Environ. Microbiol. 7, 750–754. doi: 10.1111/j.1462-2920.2005.00753.x
Vetter, R. D. (1985). Elemental sulfur in the gills of three species of clams containing chemoautotrophic symbiotic bacteria, A possible inorganic energy storage compound. Mar. Biol. 88, 33–42. doi: 10.1007/BF00393041
Vetter, R. D., and Fry, B. (1998). Sulfur contents and sulfur-isotope compositions of thiotrophic symbioses in bivalve molluscs and vestimentiferan worms. Mar. Biol. 132, 453–460. doi: 10.1007/s002270050411
Vorobev, A., Jagadevan, S., Jain, S., Anantharaman, K., Dick, G. J., Vuilleumier, S., et al. (2014). Genomic and transcriptomic analyses of the facultative methanotroph Methylocystis sp. Strain SB2 grown on methane or ethanol. Appl. Environ. Microbiol. 80, 3044–3052. doi: 10.1128/AEM.00218-14
Waite, T. J., Moore, T. S., Childress, J. J., Hsu-Kim, H., Mullaugh, K. M., Nuzzio, D. B., et al. (2008). Variation in sulfur speciation with shellfish presence at a lau basin diffuse flow vent site. J. Shellfish Res. 27, 163–168. doi: 10.2983/0730-8000(2008)27[163:VISSWS]2.0.CO;2
Warnes, G. R., Bolker, B., Bonebakker, L., Gentleman, R., Huber, W., Liaw, A., et al. (2013). Gplots: Various R programming Tools for Plotting Data. R Package Version 2.12. 11. Available online at: https://cran.r-project.org/web/packages/gplots/index.html
Wendeberg, A., Zielinski, F. U., Borowski, C., and Dubilier, N. (2012). Expression patterns of mRNAs for methanotrophy and thiotrophy in symbionts of the hydrothermal vent mussel Bathymodiolus puteoserpentis. ISME J. 6, 104–112. doi: 10.1038/ismej.2011.81
Wilmot, D. B. Jr., and Vetter, R. D. (1990). The bacterial symbiont from the hydrothermal vent tubeworm Riftia pachyptila is a sulfide specialist. Mar. Biol. 106, 273–283. doi: 10.1007/BF01314811
Windoffer, R., and Giere, O. (1997). Symbiosis of the hydrothermal vent gastropod Ifremeria nautilei (Provannidae) with endobacteria - structural analyses and ecological considerations. Biol. Bull. 193, 381–392. doi: 10.2307/1542940
Woyke, T., Teeling, H., Ivanova, N. N., Huntemann, M., Richter, M., Gloeckner, F. O., et al. (2006). Symbiosis insights through metagenomic analysis of a microbial consortium. Nature 443, 950–955. doi: 10.1038/nature05192
Zhang, W., Culley, D. E., Scholten, J. C., Hogan, M., Vitiritti, L., and Brockman, F. J. (2006). Global transcriptomic analysis of Desulfovibrio vulgaris on different electron donors. Anton. Van Leeuw. 89, 221–237. doi: 10.1007/s10482-005-9024-z
Keywords: Ifremeria nautilei, chemoautotroph, endosymbiont, methanotrophic bacteria, Sulfur oxidizers, metatranscriptomics, deep sea vents
Citation: Seston SL, Beinart RA, Sarode N, Shockey AC, Ranjan P, Ganesh S, Girguis PR and Stewart FJ (2016) Metatranscriptional Response of Chemoautotrophic Ifremeria nautilei Endosymbionts to Differing Sulfur Regimes. Front. Microbiol. 7:1074. doi: 10.3389/fmicb.2016.01074
Received: 25 April 2016; Accepted: 27 June 2016;
Published: 19 July 2016.
Edited by:
Shana Goffredi, Occidental College, USAReviewed by:
Satoshi Nakagawa, Kyoto University, JapanSébastien Duperron, Université Pierre et Marie Curie, France
Copyright © 2016 Seston, Beinart, Sarode, Shockey, Ranjan, Ganesh, Girguis and Stewart. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Frank J. Stewart, ZnJhbmsuc3Rld2FydEBiaW9sb2d5LmdhdGVjaC5lZHU=
†Present Address: Roxanne A. Beinart, Department of Geology and Geophysics, Woods Hole Oceanographic Institution, Woods Hole, MA, USA
‡ These authors have contributed equally to this work.