 Neng Fei Wang1†
Neng Fei Wang1† Tao Zhang2†
Tao Zhang2† Xiao Yang3
Xiao Yang3 Shuang Wang3
Shuang Wang3 Yong Yu4Long Long Dong5Yu Dong Guo5Yong Xing Ma1Jia Ye Zang1*
Yong Yu4Long Long Dong5Yu Dong Guo5Yong Xing Ma1Jia Ye Zang1*
- 1Key Lab of Marine Bioactive Substances, First Institute of Oceanography, State Oceanic Administration, Qingdao, China
- 2Institute of Medicinal Biotechnology, Chinese Academy of Medical Sciences, Beijing, China
- 3Chemical Engineering Institute, Qingdao University, Qingdao, China
- 4Polar Research Institute of China, Shanghai, China
- 5Department of Bioengineering and Biotechnology, Qingdao University of Science and Technology, Qingdao, China
This study assessed the diversity and composition of bacterial communities within soils and lake sediments from an Arctic lake area (London Island, Svalbard). A total of 2,987 operational taxonomic units were identified by high-throughput sequencing, targeting bacterial 16S rRNA gene. The samples from four sites (three samples in each site) were significantly different in geochemical properties and bacterial community composition. Proteobacteria and Acidobacteria were abundant phyla in the nine soil samples, whereas Proteobacteria and Bacteroidetes were abundant phyla in the three sediment samples. Furthermore, Actinobacteria, Chlorobi, Chloroflexi, Elusimicrobia, Firmicutes, Gemmatimonadetes, Nitrospirae, Planctomycetes, Proteobacteria significantly varied in their abundance among the four sampling sites. Additionally, members of the dominant genera, such as Clostridium, Luteolibacter, Methylibium, Rhodococcus, and Rhodoplanes, were significantly different in their abundance among the four sampling sites. Besides, distance-based redundancy analysis revealed that pH (p < 0.001), water content (p < 0.01), ammonium nitrogen (-N, p < 0.01), silicate silicon (-Si, p < 0.01), nitrite nitrogen (-N, p < 0.05), organic carbon (p < 0.05), and organic nitrogen (p < 0.05) were the most significant factors that correlated with the bacterial community composition. The results suggest soils and sediments from a lake area in the Arctic harbor a high diversity of bacterial communities, which are influenced by many geochemical factors of Arctic environments.
Introduction
Thaw lakes and ponds are major features of the Arctic landscape and can account for up to 90% of the total land surface area in the parts of the Arctic (Yoshikawa and Hinzman, 2003; Smol and Douglas, 2007; Pienitz et al., 2008). Over the past 100 years, the Arctic average temperatures have increased at about twice the global average rate (Solomon, 2007) and one consequence of the rising temperatures in the Arctic is the changes in the distribution and abundance of thaw lakes and ponds (Vincent et al., 2013). In some regions of the High Arctic, a few lakes and ponds are beginning to shrink, and some have even disappeared (Smith et al., 2005). In contrasts, in some regions of Low Arctic or Subarctic, some thaw lakes and ponds are growing in size because of permafrost thaw (Payette et al., 2004). Therefore, lakes and ponds are good sentinels of global climate change (Adrian et al., 2009).
Arctic microbes are sensitive to environmental changes in the Arctic ecosystem (Vincent, 2010) and thereby understanding the Arctic microbial community is of great importance to predict the response of the Arctic ecosystems to climate warming (Blaud et al., 2015a). As the key components of microbes, Bacteria and Archaea within soils or lake sediments may play important roles in driving biogeochemical cycles (e.g., carbon, nitrogen, and phosphorus) in the Arctic lake area. Hitherto, a few studies were carried out to provide a great deal of information on the bacterial diversity in the soils from various area types, such as diesel-contaminated soils (Yergeau et al., 2012), peat soils (Lipson et al., 2013), permafrost-affected soils (Gittel et al., 2014b), buried soils (Gittel et al., 2014a), vegetation-impacted soils (Shi et al., 2015), tundra tussock and shrub soils (Wallenstein et al., 2007). To the best of our knowledge, only two studies have yet been carried out to provide insight to the bacterial communities that inhabit soils or lake sediments from an Arctic lake area in the Canadian High Arctic (Steven et al., 2013; Stoeva et al., 2014). Moreover, the environmental factors which influence the bacterial community composition and function in the Arctic lake area are poorly understood.
Previous studies mainly focused on the diversity of cultured soil bacteria in the Arctic using traditional isolation methods (Master and Mohn, 1998) and conventional DNA-based molecular methods (e.g., DGGE, T-RFLP, Q-PCR, clone libraries) (Neufeld and Mohn, 2005; Wallenstein et al., 2007; Stoeva et al., 2014; Blaud et al., 2015b). Recently, bacterial diversity and community composition in the Arctic soils or lake sediments have been revealed by next-generation sequencing (Campbell et al., 2010; Yergeau et al., 2010, 2012, 2014; Mackelprang et al., 2011; Lipson et al., 2013; Gittel et al., 2014a,b; Kim et al., 2014; Koyama et al., 2014; Schostag et al., 2015; Shi et al., 2015). In the present study, we use high-throughput sequencing to investigate the bacterial communities in the soils and sediments from an Arctic lake area (Svalbard, High Arctic) and address the following questions: (1) what are bacterial diversity and community composition in the Arctic lake area; (2) what are differences in the bacterial taxonomic groups among different sampling sites in the Arctic lake area; (3) what are the key geochemical factors that determine the bacteria community composition in the Arctic lake area?
Materials and Methods
Study Sites and Sample Collection
The studied area was located in the London Island of Konsfjorden, which is on the west coast of Spitsbergen, Svalbard archipelago (Figure 1A). During winter, this shallow lake (~2 m deep) can be frozen to the bottom, whereas during summer, it is supplied by meltwater from the lake ice and accumulated snow on the hillslope. Soils and sediments (about 50 g) were sampled from surface (5 cm) near each other (about 1 m apart) in triplicate at four sites (Hill, Up, Down, and Sedi; Figure 1B). Samples were collected using a sterile shovel and directly put into TWIRL’EM sterile sampling bags (Labplas Inc., Sainte-Julie, QC, Canada). Sampling occurred during China’s Arctic expedition in July 2014. A total of nine soil samples and three lake sediment samples were collected and directly put into sterile plastic sampling bags. Samples were placed in centrifuge tubes at -20°C in the Yellow River Station (China) for about 20 days and then taken to the home laboratory in China by plane. During the flights, samples were conserved in an incubator with frozen ice bags. In the home laboratory, soil samples were frozen at -80°C until nucleic acid extraction.

FIGURE 1. Location (red dot; A) and image (B) of the sampling sites from an Arctic lake area in the present study.
Geochemical Properties of Soils and Lake Sediments
A total of nine geochemical properties were assessed, including pH, water content, organic carbon, organic nitrogen, ammonium nitrogen (-N), silicate silicon (-Si), nitrite nitrogen (-N), phosphate phosphorus (-P), and nitrate nitrogen (-N) (Table 1). Adding 10 ml of distilled water to 4 g of soil/sediment, pH was measured by a pH electrode (PHS-3C, Shanghai REX Instrument Factory, Shanghai, China). Water content (10 g of each sample) was determined as gravimetric weight loss after drying the soil/sediment at 105°C until constant weight. The organic C and organic N were analyzed according to the following procedure (Hu et al., 2012). Samples (10 g of each sample) were dried at 50°C, homogenized and pulverized with an agate mortar. Approximately 0.5 g samples were thoroughly decalcified using 1 M hydrochloric acid. They were then subsequently rinsed with Milli-Q water for complete acid removal and dried overnight at 50°C. The carbonate-free total organic carbon and total organic nitrogen samples were analyzed using an elemental analyzer (EA3000, Euro Vector SpA, Milan, Italy). The amounts of organic C and organic N were indicated by percentage. The other five chemical properties (-Si, -P, -N, -N, and -N; 5 g of each sample) were determined colorimetrically on a nutrient auto-analyzer (QuAAtro, SEAL, Germany; relative standard deviation < 5%; Liu et al., 2016), and the detection limits for -Si, -P, -N, -N, and -N were 0.030, 0.024, 0.015, 0.003, and 0.040 μmol L-1, respectively.
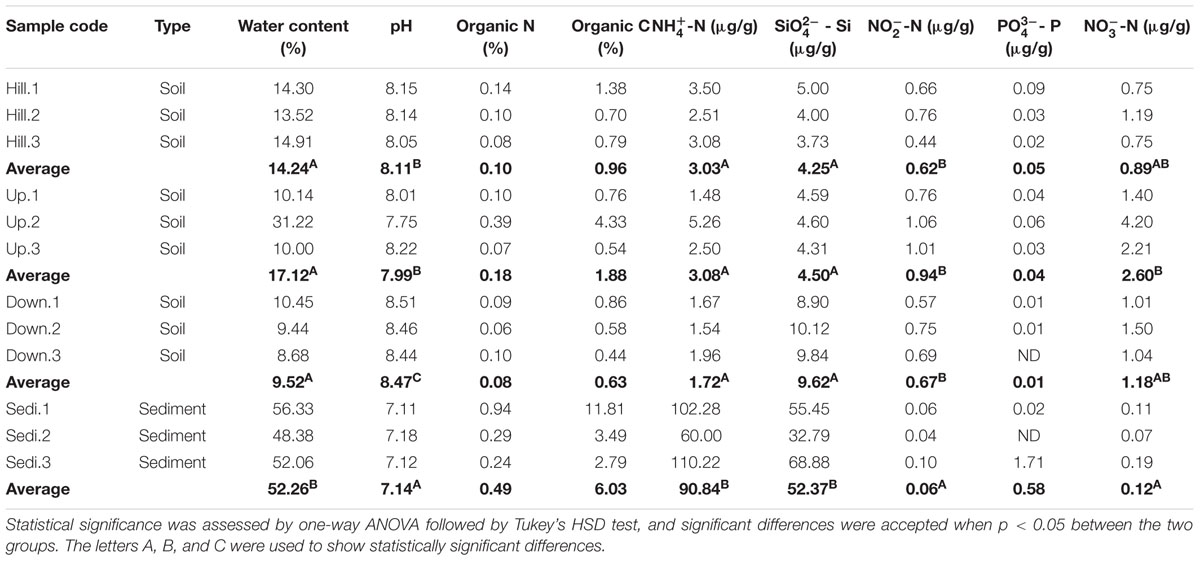
TABLE 1. Geochemical properties of 12 samples investigated in the present study.
DNA Extraction and PCR Amplification
DNA was extracted from an aliquot of 0.25 g soil/sediment from each sample, using a PowerSoil DNA Isolation Kit (MO BIO Laboratories, San Diego, CA, USA) according to the manufacturer’s instructions. The V3 and V4 hypervariable regions of the bacterial 16S ribosomal RNA gene were amplified by PCR (98°C for 1 min, followed by 30 cycles at 98°C for 10 s, 50°C for 30 s, and 72°C for 30 s and a final extension at 72°C for 5 min) using primers 338F 5′-barcode-ACTCCTACGGGAGGCAGCA-3′ and 806R 5′-barcode- GGACTACHVGGGTWTCTAAT-3′, where barcode is an eight base sequence unique to each sample. All PCR reactions were carried out in 30 μL reactions with 15 μL of Phusion High-Fidelity PCR Master Mix (New England Biolabs); 0.2 μM of forward and reverse primers, and about 10 ng template DNA.
PCR Products Quantification, Qualification, and Purification
Mix same volume of 1X loading buffer (contained SYB green) with PCR products and operate electrophoresis on 2% agarose gel for detection. Samples with bright main strip between 400 and 450 bp were chosen for further experiments. PCR products were mixed in equidensity ratios. Then, mixture PCR products were purified with GeneJET Gel Extraction Kit (Thermo Scientific).
Library Preparation and Sequencing
Following manufacturer’s recommendations, sequencing libraries were generated by using NEB Next Ultra DNA Library Prep Kit for Illumina (NEB, USA) and then index codes were added to them. The library quality was assessed on the Qubit 2.0 Fluorometer (Thermo Scientific) and Agilent Bioanalyzer 2100 system. At last, the library was sequenced on an Illumina MiSeq platform and 300 bp paired-end reads were generated. The raw reads were deposited into the NCBI Sequence Read Archive (SRA) database (Accession Number: SRR3186997).
Processing of Sequencing Data
Paired-end reads from the original DNA fragments are merged by using FLASH software (Magoč and Salzberg, 2011), which is designed to merge paired-end reads when there are overlaps between reads1 and reads2. Paired-end reads was assigned to each sample according to the unique barcodes. Raw sequencing data were quality-filtered using QIIME 1.8.0 software (Caporaso et al., 2010) with the following criteria: (i) The 300 bp reads were truncated at any site receiving an average quality score <20 over a 50-bp sliding window, discarding the truncated reads that were shorter than 50 bp; (ii) exact barcode matching, two nucleotide mismatch in primer matching, reads containing ambiguous characters were removed; (iii) only sequences that overlap longer than 10 bp were assembled according to their overlap sequence. Reads which could not be assembled were discarded. Operational taxonomic units (OTUs) were clustered with 97% similarity cutoff using UPARSE (Edgar, 2013); chimeric sequences were identified and removed using UCHIME (Edgar et al., 2011). Singleton OTUs were removed. The taxonomy of 16S rRNA gene sequence was analyzed by RDP Classifier (Wang et al., 2007) against the Silva rRNA gene database1 using confidence threshold of 80%. All samples were normalized at the same sequence depth (12,401 reads) and these OTUs were then used as a foundation for calculating alpha-diversity and beta-diversity metrics using QIIME 1.8.0 software (Caporaso et al., 2010).
Statistical Analyses
Statistical analyses of the alpha-diversity of each soil sample via Chao1, Good’s coverage estimator, and Shannon’s index (H′) were performed using QIIME 1.8.0 software (Caporaso et al., 2010). One-way analysis of variance (ANOVA) followed by Tukey’s HSD (Honest Significant Difference) test was performed for the geochemical properties and the diversity parameters of samples to determine the level of significance using Statistical Package for the Social Sciences software (SPSS) v.17.0. The relationships among the bacterial communities in the 12 samples were analyzed by hierarchical clustering analysis using the R v.3.1.1 statistical software. Analysis of similarities (ANOSIM) test was performed to determine whether the four sampling sites had statistically significantly different bacterial communities by using QIIME v.1.8.0 software (Caporaso et al., 2010). The abundance-based Bray–Curtis similarity coefficient was used to examine the dissimilarity of different samples. The relevance of environmental factors in explaining the distribution patterns of bacterial communities in different samples was analyzed by Bray–Curtis distance-based redundancy analysis (db-RDA) using the R v.3.1.1 statistical software. Monte Carlo permutation test was also performed to examine the relationship between the nine geochemical properties and bacterial community composition in this Arctic lake area. A linear discriminant analysis effect size (LEfSe) method was used to identify the significantly different bacterial groups in different sampling sites (Segata et al., 2011).
Results
Geochemical Properties of Soil and Sediment Samples
The highest values of water content, organic C, organic N, -N, -Si, and -P were observed from the sediment samples (site Sedi), whereas the lowest values of pH, -N, and -N were also detected at the site Sedi. Among the nine geochemical properties, six properties (i.e., water content, pH, -N, -Si, -N, and -N) were significantly different between sediment samples and soil samples. For examples, the water contents of soils were in the range of 8.68 to 31.22 %, where the water contents of sediments ranged from 48.38 to 56.33%; the -N concentrations in the sediments ranged from 60.00 to 110.22 μg/g, which was much higher than those in the soils (1.48–5.26 μg/g); the concentrations of -Si in the sediments were from 32.79 to 68.88 μg/g, which were also much higher than those in soils (3.73–10.12 μg/g). In contrast, the concentrations of -N (0.07–0.19 μg/g) and -N (0.04–0.10 μg/g) in the sediments were lower than those in the soils (-N, 0.75–4.20 μg/g; -N, 0.44–1.06 μg/g). Additionally, the other three factors (i.e., organic C, organic N, and -P) were not significantly different among four samples sites. Specifically, the organic C contents ranged from 0.44 to 11.81%, whereas organic N contents were from 0.06 to 0.94%. The concentrations of -P were 0–1.71 μg/g. Furthermore, eight geochemical properties (i.e., water content, organic C, organic N, -N, -Si, -N, -P, and -N) were not significantly different among three sampling sites of soils (Hill, Up, and Down).
Bacterial Diversity and Community Composition
A total of 148,812 bacterial sequences and 2,987 OTUs (at the 3% evolutionary distance) were identified in the present study. The sequence number of each sample was 12,401, from which 1,022 to 1,308 OTUs were recognized. The Good’s coverage estimator of the OTUs in the samples ranged from 95.33 to 97.52% (Table 2), indicating that the sequences sufficiently covered the diversity of bacterial populations in all the samples. According to the OTU numbers and Shannon’s indices, no significant differences in bacterial richness and diversity were detected among the four sampling sites (Table 2).
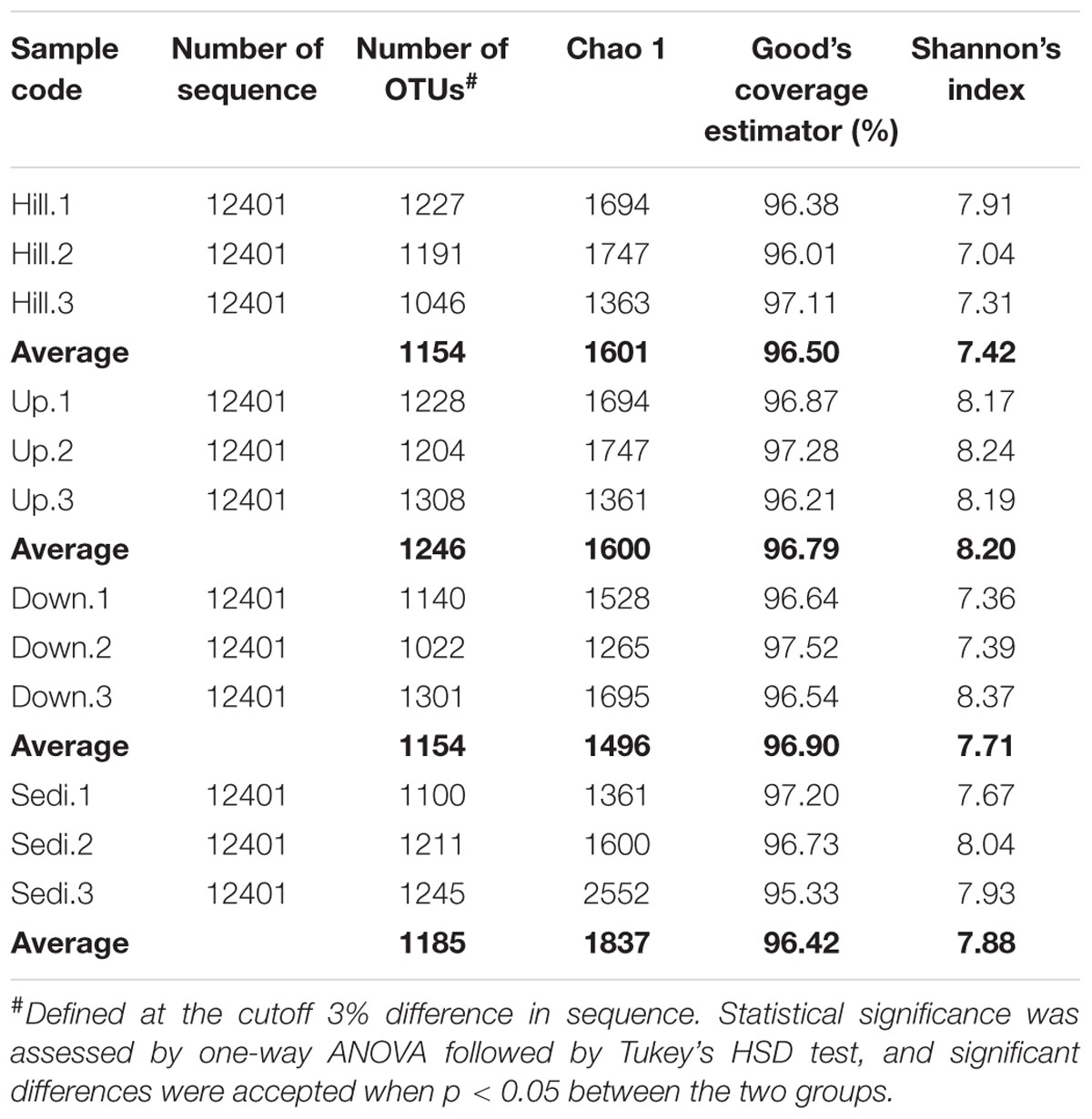
TABLE 2. Summary data for Miseq sequencing data from the 12 samples in the present study.
Sequences affiliated with Proteobacteria, Acidobacteria, Gemmatimonadetes, and Bacteroidetes were common in all the four sites (Figure 2). Proteobacteria and Acidobacteria were abundant phyla in the nine soil samples, whereas Proteobacteria and Bacteroidetes were abundant phyla in the three sediment samples.
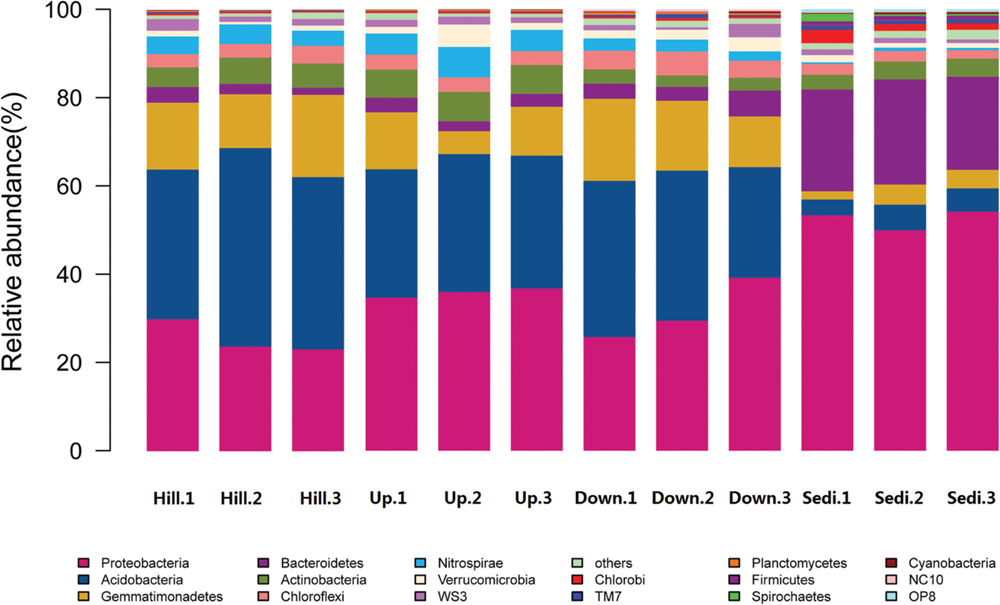
FIGURE 2. The relative abundance of different phyla in the 12 soil/sediment samples in the present study.
The most abundant sequences in Proteobacteria phylum belonged to Betaproteobacteria, followed by Alphaproteobacteria, Deltaproteobacteria, Gammaproteo-bacteria, and Epsilonproteobacteria. Analysis of Proteobacteria revealed dominance of the order Burkholderiales (Class Betaproteobacteria), followed by Sphingomonadales (Class Alphaproteobacteria), Rhizobiales (Class Alphaproteobacteria), Xanthomonadales (Class Gammaproteobacteria), and Syntro-phobacterales (Class Deltaproteobacteria). In Acidobacteria, the most abundant class was Chloracidobacteria. The phylum Bacteroidetes was represented by bacteria belonging to the classes Flavobacteriia, Bacteroidia, and Sphingobacteriia, whereas the abundant order were Bacteroidales (Class Bacteroidia), Sphingobacteriales (Class Sphingobacteriia), and Flavobacteriales (Class Flavobacteriia).
The bacterial genera detected in this study (>1000 reads) included Zymomonas (1635 reads, phylum Proteobacteria), Kaistobacter (1397 reads, phylum Proteobacteria), Methylibium (1340 reads, phylum Proteobacteria), Sulfuricurvum (1321 reads, phylum Proteobacteria), Rhodoplanes (1278 reads, phylum Proteobacteria), Candidatus Solibacter (1141 reads, phylum Acidobacteria), and Syntrophus (1098 reads, phylum Proteobacteria).
The Correlation between Bacterial Communities and Soil/Sediment Geochemical Properties
Operational taxonomic units cluster analysis (Figure 3A) revealed that the 12 samples were clustered into four groups which corresponded to four sampling sites very well. It was shown that nine soil samples from sites Hill, Up, and Down were closely related and they were obviously distinguished from three sediment samples from site Sedi. An ANOSIM test (R = 0.87, p = 0.002) supported that the four sampling sites harbored significantly different bacterial communities (Figure 3B). A Venn diagram demonstrated that OTUs differed among the four sites (Figure 4). The number of site-specific OTUs ranged from 93 (Site Hill) to 361 (Site Sedi). Only 805 in 2,987 OTUs were shared by all four sampling sites.
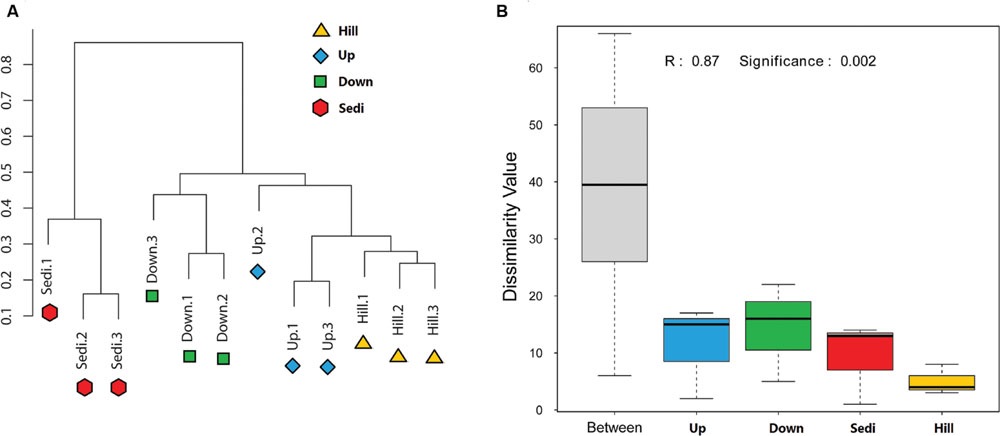
FIGURE 3. (A) Clustering analysis of bacterial communities in the 12 soil/sediment samples based on OTU abundance-based Bray–Curtis similarity coefficients. (B) Analysis of similarities (ANOSIM) of the four sampling sites in the present study.
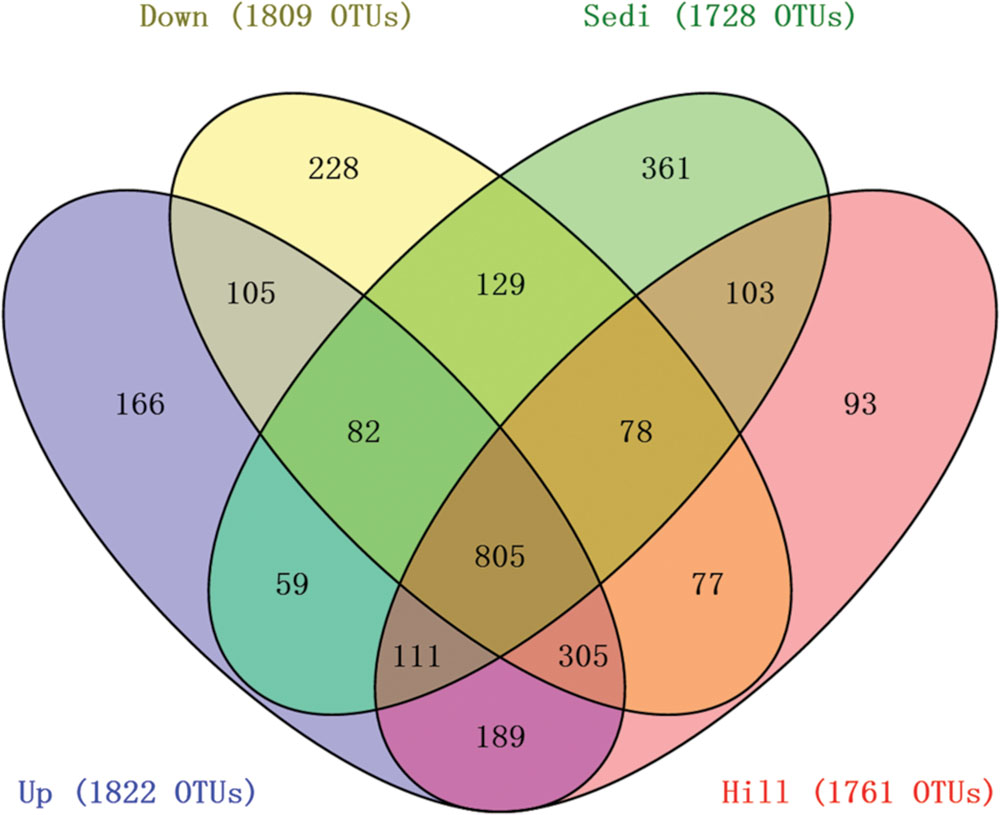
FIGURE 4. A Venn diagram displaying the degree of overlap of bacterial OTUs (at the 3% evolutionary distance) among the four sampling sites.
At different taxonomic ranks, the four sampling sites can be significantly distinguished from each others, as shown in Supplementary Table S1. For examples, at the phylum rank, members of Chlorobi, Firmicutes, Proteobacteria were significantly abundant in the sediment samples (site Sedi); at the class rank, members of Actinobacteria, Anaerolineae, Clostridia, Ignavibacteria, and Verrucomicrobiae were significantly higher in site Sedi than those in other three sites. Similarly, the bacterial community composition in the four sampling sites can be distinguished at ranks of order, family, and genus. For example, sequences of the genera, such as Clostridium, Luteolibacter, Methylibium, Rhodococcus, and Rhodoplanes, were significantly different in their abundance among the four sampling sites. Particularly, members of the genera Clostridium, Dechloromonas, Desulfomicrobium, Haliscomenobacter, Hyphomicrobium, Luteolibacter, Methylibium, Mycobacterium, Nitrospira, and Roseomonas were significantly abundant in the sediment samples (site Sedi; Supplementary Table S1).
Distance-based redundancy analysis (db-RDA; Figure 5) and Monte Carlo permutation test (Table 3) were performed to examine the relationship between the nine geochemical properties and bacterial community composition in the Arctic lake area. It was also shown that six soil samples from sites Hill and Up were closely related and they were obviously distinguished from soil samples from site Down and three sediment samples from site Sedi. The combination of the nine environmental factors showed a significant correlation with bacterial community composition (F = 8.184374, p = 0.001). These factors explained 97.36% of the bacterial community variation, while 2.64% of the variation was not explained by any of the selected nine environmental factors. Among the nine geochemical factors, pH (p < 0.001), water content (p < 0.01), -N (p < 0.01), -Si (p < 0.01), -N (p < 0.05), organic C (p < 0.05), and organic N (p < 0.05) were important factors that correlated with the bacterial community composition in this area. However, the other two environmental factors, including -P and -N, were not significantly correlated with bacteria community composition (Table 3).
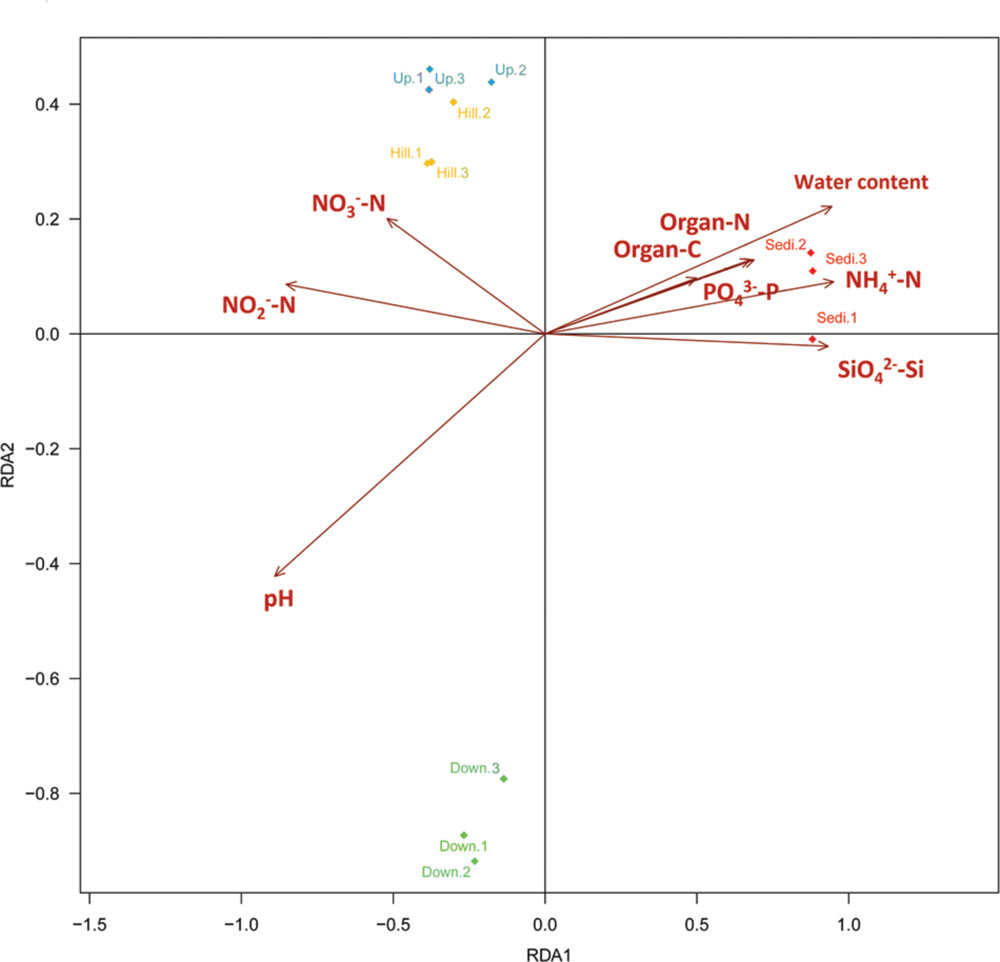
FIGURE 5. Distance-based redundancy analysis to show correlations between the bacterial communities and environmental factors of the 12 samples from the four sampling sites. The arrows represent geochemical factors measured. The 12 samples are labeled with unique sampling code.
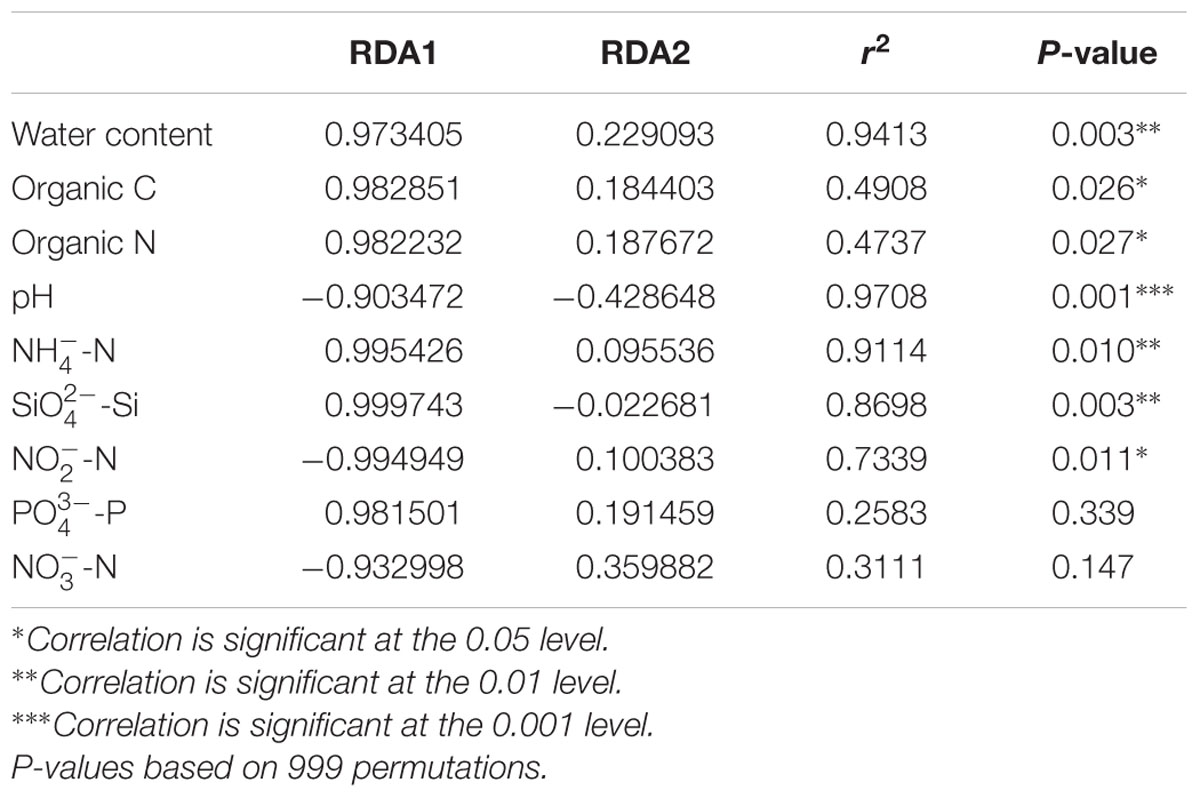
TABLE 3. A Monte Carlo permutation test of relationship between environmental factors and bacterial community composition.
Discussion
In the present study, the high level of Shannon diversity indices (H′ = 7.04–8.37) and the identification of 1022–1308 OTUs suggest the presence of high richness and diversity of bacterial communities in the soils and lake sediments from Arctic lake area. By comparison, using 454 pyrosequencing, Steven et al. (2013) reported a Shannon diversity of 6.4–7.2 and 1801–2264 OTUs for soil bacterial communities in the catchment area of a lake in the Canadian High Arctic. The observed bacterial richness and diversity indices in this study were somewhat similar to those in the lake area of the Canadian High Arctic.
The relative abundances of the sequences affiliated with Proteobacteria, Acidobacteria, Gemmatimonadetes, and Bacteroidetes in the soils and lake sediments (Svalbard, High Arctic), were somewhat different from those previously described for soils in the Arctic using high-throughput sequencing (Steven et al., 2013). Based on the 16S rDNA pyrosequencing data, Acidobacteria, Cyanobacteria, Proteobacteria, Planctomycetes, and Verrucomicrobia were the dominant soil bacterial phyla in the catchment lake area of the Canadian High Arctic (Steven et al., 2013). Bacterial communities in both organic and mineral soils (at Toolik Lake, Alaska, USA) were predominated by Acidobacteria and Proteobacteria, followed by the Actinobacteria and Bacteroidetes (Campbell et al., 2010). In addition, the dominant sequences in the moist acidic tussock tundra soil in a subarctic region (Northwest Alaska) were Acidobacteria, Proteobacteria, and Actinobacteria (Kim et al., 2014). In the soils collected from Adventdalen (Svalbard), the dominant sequences (>5% of total) were Proteobacteria, Actinobacteria, Verrucomicrobia, Acidobacteria, and Gemmatimonadetes (Schostag et al., 2015). These differences of dominant phyla may be due to the geographical distance and differences in soil properties among the different regions. For example, the carbon contents in this study were in the range of 0.44–11.81%, where the carbon contents in the Canadian High Arctic ranged from 4.25 to 18.97%.
Although bacterial diversity and richness indices did not vary much among the four different sites in this study, some specific bacterial groups varied obviously. For example, members of the genera Clostridium, Dechloromonas, Desulfomicrobium, Haliscomenobacter, Hyphomicrobium, Luteolibacter, Methylibium, Mycobacterium, Nitrospira, and Roseomonas were significantly abundant in the lake sediment samples (site Sedi). As significant differences in geochemical properties and bacterial communities were observed between the three soil sites (Hill, Up, Down) and one sediment site (Sedi), the above genera may be specifically associated with the environmental factors in the lake sediments (site Sedi). For example, the carbohydrate-fermenting butyric acid bacteria of the genus Clostridium can act as precursors for sulfate reducers and methanogens and thereby perform an important step of anaerobic decomposition of organic matter in aquatic ecosystems (Gorlenko et al., 1977), whereas Nitrospira is a chemolithoautotrophic nitrite-oxidizing bacterium that is generally found in freshwater (Hovanec et al., 1998). Therefore, the genera Clostridium and Nitrospira may be much abundant in the lake sediments as compared with soils near lake.
The combined nine geochemical factors showed a significant correlation with bacterial community composition in this lake area. In this study, pH (p < 0.001) was the best predictor of soil bacterial community composition. Furthermore, content of water (p < 0.01), -N (p < 0.01), -Si (p < 0.01), -N (p < 0.05), organic C (p < 0.05), and organic N (p < 0.05) showed significant correlation with the bacterial community composition. The above abiotic factors may directly alter bacterial community composition by affecting growth of certain bacteria in soils or lake sediments. In the previous studies, soil pH was found to be the most influential soil properties to determine the bacterial community structure in subarctic tundra soil (Kim et al., 2014; Shi et al., 2015), whereas Steven et al. (2013) reported that soil moisture might influenced the bacterial communities of samples inside and outside of the water tracks in an Arctic lake area. Koyama et al. (2014) found that soil bacterial composition was altered by increased nutrient availability in Arctic tundra soils. Additionally, Shi et al. (2015) found that dissolved organic carbon (DOC), dissolved organic carbon (DON), C/N ratio, concentration, and N mineralization also contributed to soil bacterial community variability in the subarctic region. In this study, more organic N and -N were found in sediment samples compared to soil samples. Interestingly, the anaerobic bacteria Clostridium was also significantly abundant in the lake sediments compared to soil samples, and some Clostridium species were capable of fixing free N2 into nitrogenous and ammonium compound (Kanamori et al., 1989).
In the future, more and more thaw lakes and ponds would shrink and disappear, as climate warming dries out the freshwater mass in the High Arctic (Vincent et al., 2013). Our results provide the possible consequences of the climate change on the soil geochemical properties and bacterial communities in the High Arctic lake area. If the lake’s water level and size would shrink in the future, the lake sediments would be exposed to air and their geochemical properties would also be altered. As a consequence, the sediment bacterial communities in the lake water (as shown in the Site Sedi) would be changed to those in the soils near lake’s water (as shown in Site Down) and may even to those in the soils far from lake’s water (as shown in Site Up and Hill). Additionally, we will measure methane or CO2, which are accumulated on the soils and sediments and eventually liberated to the atmosphere as a consequence of climate warming. Furthermore, little is known about the presence, diversity, and community composition of Archaea, which may play important roles in biogeochemical cycling in the Arctic soils and sediments. All in all, if we want to determine the relationships between climate change and microbial communities in this lake ecosystem, the long-time survey (10–50 years) on the soil/sediment geochemical properties and microbial communities in this lake area would be necessary in the further study.
Author Contributions
NW planned the study, collected samples, conducted lab work. TZ wrote the manuscript and conducted parts of lab work. XY, YY, LD, YG, YM, and SW joined in lab work and laboratory analyses. JZ contributed with planning the project and revising the manuscript. All authors reviewed the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was supported by Chinese polar environment comprehensive investigation & assessment programmers (No. 02-01), the Key Research Program of Shandong Province (No. 2015GSF115004) and Basic Scientific Fund for National Public Research Institutes of China (No. 2015T04).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2016.01170
Footnotes
References
Adrian, R., O’Reilly, C. M., Zagarese, H., Baines, S. B., Hessen, D. O., Keller, W., et al. (2009). Lakes as sentinels of climate change. Limnol. Oceanogr. 54, 2283–2297. doi: 10.4319/lo.2009.54.6_part_2.2283
Blaud, A., Lerch, T. Z., Phoenix, G. K., and Osborn, A. M. (2015a). Arctic soil microbial diversity in a changing world. Res. Microbiol. 166, 796–813. doi: 10.1016/j.resmic.2015.07.013
Blaud, A., Phoenix, G. K., and Osborn, A. M. (2015b). Variation in bacterial, archaeal and fungal community structure and abundance in High Arctic tundra soil. Polar Biol. 38, 1009–1024. doi: 10.1007/s00300-015-1661-8
Campbell, B. J., Polson, S. W., Hanson, T. E., Mack, M. C., and Schuur, E. A. (2010). The effect of nutrient deposition on bacterial communities in Arctic tundra soil. Environ. Microbiol. 12, 1842–1854. doi: 10.1111/j.1462-2920.2010.02189.x
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Edgar, R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998. doi: 10.1038/nmeth.2604
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Gittel, A., Bárta, J., Kohoutová, I., Mikutta, R., Owens, S., Gilbert, J., et al. (2014a). Distinct microbial communities associated with buried soils in the Siberian tundra. ISME J. 8, 841–853. doi: 10.1038/ismej.2013.219
Gittel, A., Bárta, J., Kohoutová, I., Schnecker, J., Wild, B., Čapek, P., et al. (2014b). Site- and horizon-specific patterns of microbial community structure and enzyme activities in permafrost-affected soils of Greenland. Front. Microbiol. 5:541. doi: 10.3389/fmicb.2014.00541
Gorlenko, M. V., Dubinina, G. A., and Kuznetsov, S. I. (1977). Ekologiya Vodnykh Mikroorganizmov [Ecology of Aquatic Microorganisms]. Moscow: Nauka.
Hovanec, T. A., Taylor, L. T., Blakis, A., and DeLong, E. (1998). Nitrospira-like bacteria associated with nitrite oxidation in freshwater aquaria. Appl. Environ. Microbiol. 64, 258–264.
Hu, L. M., Shi, X. F., Yu, Z. G., Lin, T., Wang, H. J., Ma, D. Y., et al. (2012). Distribution of sedimentary organic matter in estuarine-inner shelf regions of the East China Sea: implications for hydrodynamic forces and anthropogenic impact. Mar. Chem. 142, 29–40. doi: 10.1016/j.marchem.2012.08.004
Kanamori, K., Weiss, R. L., and Roberts, J. D. (1989). Ammonia assimilation pathways in nitrogen-fixing Clostridium kluyverii and Clostridium butyricum. J. Bacteriol. 171, 2148–2154.
Kim, H. M., Jung, J. Y., Yergeau, E., Hwang, C. Y., Hinzman, L., Nam, S., et al. (2014). Bacterial community structure and soil properties of a subarctic tundra soil in Council, Alaska. FEMS Microbiol. Ecol. 89, 465–475. doi: 10.1111/1574-6941.12362
Koyama, A., Wallenstein, M. D., Simpson, R. T., and Moore, J. C. (2014). Soil bacterial community composition altered by increased nutrient availability in Arctic tundra soils. Front. Microbiol. 5:516. doi: 10.3389/fmicb.2014.00516
Lipson, D. A., Haggerty, J. M., Srinivas, A., Raab, T. K., Sathe, S., and Dinsdale, E. A. (2013). Metagenomic insights into anaerobic metabolism along an Arctic peat soil profile. PLoS ONE 8:e64659. doi: 10.1371/journal.pone.0064659
Liu, J., Zang, J. Y., Zhao, C. Y., Yu, Z. G., Xu, B. C., Li, J. X., et al. (2016). Phosphorus speciation, transformation, and preservation in the coastal area of Rushan Bay. Sci. Total Environ. 565, 258–270. doi: 10.1016/j.scitotenv.2016.04.177
Mackelprang, R., Waldrop, M. P., DeAngelis, K. M., David, M. M., Chavarria, K. L., Blazewicz, S. J., et al. (2011). Metagenomic analysis of a permafrost microbial community reveals a rapid response to thaw. Nature 480, 368–371. doi: 10.1038/nature10576
Magoč, T., and Salzberg, S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
Master, E. R., and Mohn, W. W. (1998). Psychrotolerant bacteria isolated from Arctic soil that degrade polychlorinated biphenyls at low temperatures. Appl. Environ. Microbiol. 64, 4823–4829.
Neufeld, J. D., and Mohn, W. W. (2005). Unexpectedly high bacterial diversity in Arctic tundra relative to boreal forest soils, revealed by serial analysis of ribosomal sequence tags. Appl. Environ. Microbiol. 71, 5710–5718. doi: 10.1128/AEM.71.10.5710-5718.2005
Payette, S., Delwaide, A., Caccianiga, M., and Beauchemin, M. (2004). Accelerated thawing of subarctic peatland permafrost over the last 50 years. Geophys. Res. Lett. 31:L18208. doi: 10.1029/2004GL020358
Pienitz, R., Doran, P. T., and Lamoureux, S. F. (2008). “Origin and Geomorphology of Lakes in the polar regions,” in Polar Lakes and Rivers—Limnology of Arctic and Antarctic Aquatic Ecosystems, eds W. F. Vincent and J. Laybourn-Parry (Oxford: Oxford University Press), 25–41. doi: 10.1093/acprof:oso/9780199213887.003.0002
Schostag, M., Stibal, M., Jacobsen, C. S., Bælum, J., Taş, N., Elberling, B., et al. (2015). Distinct summer and winter bacterial communities in the active layer of Svalbard permafrost revealed by DNA- and RNA-based analyses. Front. Microbiol. 6:399. doi: 10.3389/fmicb.2015.00399
Segata, N., Izard, J., Waldron, L., Gevers, D., Miropolsky, L., Garrett, W. S., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12:R60. doi: 10.1186/gb-2011-12-6-r60
Shi, Y., Xiang, X., Shen, C., Chu, H., Neufeld, J. D., Walker, V. K., et al. (2015). Vegetation-associated impacts on arctic tundra bacterial and microeukaryotic communities. Appl. Environ. Microbiol. 81, 492–501. doi: 10.1128/AEM.03229-14
Smith, L. C., Sheng, Y., MacDonald, G. M., and Hinzman, L. D. (2005). Disappearing Arctic lakes. Science 308:1429. doi: 10.1126/science.1108142
Smol, J. P., and Douglas, M. S. V. (2007). Crossing the final ecological threshold in high Arctic ponds. Proc. Natl. Acad. Sci. U.S.A. 104, 12395–12397. doi: 10.1073/pnas.0702777104
Solomon, S. (2007). Climate Change 2007-the Physical Science Basis: Working Group I Contribution to the Fourth Assessment Report of the Ipcc, Vol. 4. Cambridge: Cambridge University Press.
Steven, B., Lionard, M., Kuske, C. R., and Vincent, W. F. (2013). High bacterial diversity of biological soil crusts in water tracks over permafrost in the High Arctic polar desert. PLoS ONE 8:e71489. doi: 10.1371/journal.pone.0071489
Stoeva, M. K., Aris-Brosou, S., Chételat, J., Hintelmann, H., Pelletier, P., and Poulain, A. J. (2014). Microbial community structure in lake and wetland sediments from a high Arctic polar desert revealed by targeted transcriptomics. PLoS ONE 9:e89531. doi: 10.1371/journal.pone.0089531
Vincent, W. F. (2010). Microbial ecosystem responses to rapid climate change in the Arctic. ISME J. 4, 1089–1091. doi: 10.1038/ismej.2010.108
Vincent, W. F., Lemay, M., Allard, M., and Wolfe, B. B. (2013). Adapting to permafrost change: a science framework. Eos Trans. Am. Geophys. Union 94, 373–375. doi: 10.1002/2013EO420002
Wallenstein, M. W., McMahon, S., and Schimel, J. (2007). Bacterial and fungal community structure in Arctic tundra tussock and shrub soils. FEMS Microbiol. Ecol. 59, 428–435. doi: 10.1111/j.1574-6941.2006.00260.x
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/AEM.00062-07
Yergeau, E., Hogues, H., Whyte, L. G., and Greer, C. W. (2010). The functional potential of High Arctic permafrost revealed by metagenomic sequencing, qPCR and microarray analyses. ISME J. 4, 1206–1214. doi: 10.1038/ismej.2010.41
Yergeau, E., Sanschagrin, S., Beaumier, D., and Greer, C. W. (2012). Metagenomic analysis of the bioremediation of diesel-contaminated Canadian high arctic soils. PLoS ONE 7:e30058. doi: 10.1371/journal.pone.0030058
Yergeau, E., Sanschagrin, S., Maynard, C., St-Arnaud, M., and Greer, C. W. (2014). Microbial expression profiles in the rhizosphere of willows depend on soil contamination. ISME J. 8, 344–358. doi: 10.1038/ismej.2013.163
Keywords: bacterial diversity and community composition, Arctic lake area, soils and lake sediments, high-throughput sequencing, geochemical factor
Citation: Wang NF, Zhang T, Yang X, Wang S, Yu Y, Dong LL, Guo YD, Ma YX and Zang JY (2016) Diversity and Composition of Bacterial Community in Soils and Lake Sediments from an Arctic Lake Area. Front. Microbiol. 7:1170. doi: 10.3389/fmicb.2016.01170
Received: 28 February 2016; Accepted: 14 July 2016;
Published: 28 July 2016.
Edited by:
Kelly Wrighton, Ohio State University, USAReviewed by:
Antonio Ventosa, University of Seville, SpainTrinity L. Hamilton, University of Cincinnati, USA
Copyright © 2016 Wang, Zhang, Yang, Wang, Yu, Dong, Guo, Ma and Zang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jia Ye Zang, emp5QGZpby5vcmcuY24=
†These authors have contributed equally to this work.