 Yanmei Zhang1,2,3†
Yanmei Zhang1,2,3† Fuju Zhao1†
Fuju Zhao1† Mimi Kong4†
Mimi Kong4† Shiwen Wang1†Li Nan4
Shiwen Wang1†Li Nan4 Binjie Hu1
Binjie Hu1 Michal A. Olszewski5
Michal A. Olszewski5 Yingxin Miao1Danian Ji6Wenrong Jiang1Yi Fang1Jinghao Zhang1Fei Chen1Ping Xiang6*Yong Wu4*
Yingxin Miao1Danian Ji6Wenrong Jiang1Yi Fang1Jinghao Zhang1Fei Chen1Ping Xiang6*Yong Wu4* Hu Zhao1,2,3*
Hu Zhao1,2,3*- 1Department of Laboratory Medicine, Huadong Hospital affiliated to Fudan University, Shanghai, China
- 2Key Laboratory of Clinical Geriatric Medicine, Shanghai, China
- 3Research Center on Aging and Medicine, Fudan University, Shanghai, China
- 4Ningbo HEALTH Gene Technologies Co., Ltd, Ningbo, China
- 5Division of Pulmonary and Critical Care Medicine, Department of Internal Medicine, University of Michigan Health System and Research Service, VA Ann Arbor Healthcare System, Ann Arbor, MI, USA
- 6Digestive Endoscopic Center, Huadong Hospital affiliated to Fudan University, Shanghai, China
Helicobacter pylori (H. pylori) infection is closely related to various gastroduodenal diseases. Virulence factors and bacterial load of H. pylori are associated with clinical outcomes, and drug-resistance severely impacts the clinical efficacy of eradication treatment. Existing detection methods are low-throughput, time-consuming and labor intensive. Therefore, a rapid and high-throughput method is needed for clinical diagnosis, treatment, and monitoring for H. pylori. High-throughput Multiplex Genetic Detection System (HMGS) assay was established to simultaneously detect and analyze a set of genes for H. pylori identification, quantification, virulence, and drug resistance by optimizing the singlet-PCR and multiple primers assay. Twenty-one pairs of chimeric primers consisted of conserved and specific gene sequences of H. pylori tagged with universal sequence at the 5′ end were designed. Singlet-PCR assay and multiple primers assay were developed to optimize the HMGS. The specificity of HMGS assay was evaluated using standard H. pylori strains and bacterial controls. Six clinical isolates with known genetic background of target genes were detected to assess the accuracy of HMGS assay. Artificial mixed pathogen DNA templates were used to evaluate the ability to distinguish mixed infections using HMGS assay. Furthermore, gastric biopsy specimens with corresponding isolated strains were used to assess the capability of HMGS assay in detecting biopsy specimens directly. HMGS assay was specific for H. pylori identification. HMGS assay for H. pylori target genes detection were completely consistent with the corresponding genetic background. Mixed infection with different drug-resistant isolates of H. pylori could be distinguished by HMGS assay. HMGS assay could efficiently diagnose H. pylori infection in gastric biopsy specimens directly. HMGS assay is a rapid and high throughput method for the simultaneous identification and quantification of H. pylori, analysis of virulence and drug resistance in both isolated strains and biopsy specimens. It could also be used to distinguish the mixed infection with different resistant genotype strains. Furthermore, HMGS could detect H. pylori infection in gastric biopsy specimens directly.
Introduction
Helicobacter pylori (H. pylori), a micro-aerobic Gram-negative bacteria, is closely associated with a variety of gastroduodenal diseases, such as chronic superficial gastritis (CSG), chronic atrophic gastritis (CAG), peptic ulcer diseases (PUD), and gastric carcinoma (GC) (Venerito et al., 2016). More than half of the adult population worldwide was infected by this pathogen and the most serious incidence of infection was observed in East Asia (de Martel et al., 2013). The analysis of different virulence, gene-mutation related resistance and the bacterial load have been revealed to be very important for effective clinical diagnosis, treatment, and monitoring of H. pylori infection (Kusters et al., 2006; Proença-Modena et al., 2009; Yamaoka, 2010).
Currently, various detection methods are available in clinical practice, such as culture, histopathology, urea breath test (UBT), rapid urease test (RUT), serology, and PCR, but each has some disadvantages (Wang et al., 2015). To date, no single method incorporates simultaneous analysis of strain-specific features including identification, virulence characteristics, drug-resistance, quantification, and evaluation of multi-strain infections (Shukla et al., 2011). Thus, a rapid, accurate, and high-content quantitative assay is required to directly assist clinical diagnosis in tissue and improve the effectiveness of diagnosis and antibiotic treatment.
Previously, our team has developed a genetic analysis method using 16S rRNA and ureC for H. pylori identification and quantification (Zhou et al., 2015). Here, we further explored and developed a new multiple genes detection assay. In this study, total 21 pairs of chimeric gene specific primers were designed to establish a high-throughput multiple genetic detection system (HMGS) for simultaneous bacterial diagnosis and identification of virulence and resistance genes. The specificity of HMGS assay was assessed using the standard H. pylori strains and bacterial controls. Six clinical isolates with known genetic background of target genes were detected to assess the accuracy of HMGS assay. Artificially mixed DNA from different wild type and mutant resistant isolates of H. pylori was used to evaluate the ability to distinguish mixed infections. Furthermore, gastric biopsy specimens with the corresponding isolated strains were used to evaluate the capacity of HMGS assay in detecting biopsy specimens directly.
Materials and Methods
Ethics Statement
The study has been approved by Ethics Committee for human studies of Huadong Hospital. The Ethics Approval Number: [2013]-077.
Bacterial Strains and Clinical Specimens
The standard strains H. pylori ATCC43504, Escherichia coli (E. coli) ATCC8739, Pseudomonas aeruginosa (P. aeruginosa) ATCC35218, and Acinetobacter baumannii (A. baumannii) ATCC19606 were purchased from Shanghai Municipal Center for Disease Control & Prevention. Six clinical strains of H. pylori with known genetic background and Campylobacter jejuni (C. jejuni), Klebsiellar pneumonia (K. pneumonia), Stenotrophomonas maltophilia (S. maltophilia) were isolated and confirmed by their specific and conserved species genes. Two clinical antrum tissue specimens were obtained from patients who were diagnosed as H. pylori positive by C14 expiration test and one uninfected antrum tissue from a healthy control in the Huadong Hospital, Shanghai, China.
Culture and DNA extraction
Each antrum biopsy specimen was placed in 0.9% sterile saline solution kept at 4°C and sent to the microbiology laboratory within 4 h. The biopsy specimens were homogenized using TissueLyser (Jingxin Co., Ltd., Shanghai, China) and plated on Columbia agar (OXOID Microbiology Products, Thermo Fisher Scientific Inc., Waltharm, MA, USA) supplemented with medium containing 8% sterile defibrinated sheep blood (Shunwei Biotech Co., Ltd., Shanghai, China) and 0.5% selective antibiotics (Sigma-Aldrich, USA) supplement. The cultures were kept under microaerophilic conditions (5%O2, 10%CO2, 85%N2) at 35°C for 3-7 days. The isolates were frozen in glycerin broth at -80°C and the remained homogenated specimens were kept in -80°C for DNA extraction. Total DNA from the standard strains, clinical isolates and H. pylori positive biopsy specimens was extracted with a bacterial genomic DNA extraction kit (Tiangen Biotech Co., Ltd., Beijing, China) following the manufacturer’s instruction. The extracts were eluted with 100 μL of DNase/RNase-free H2O (ddH2O). Concentration of each extract was determined using a ThermoNano drop 2000 spectrophotometer (Thermo Fisher Scientific Inc., Waltharm, MA, USA). The extracts were stored at -20°C for further analysis.
Primer Design
The primer set for HMGS assay consisted of 21 pairs of chimeric primers, including identification gene 16S rRNA, 10 main virulence-associated genotypes cagA, vacA s1, vacA s2, vacA m1, vacA m2, iceA1, iceA2, luxS, dupA, and oipA, four drug resistance genes 23S rRNA (A2143G), rdxA (C148T), gyrA(C261A/G), and pbp1A (A1777G), quantification gene of ureC, and human tissue internal control beta-globin gene which induced to normalize the specific gene expression. In addition, it also has one pair of universal primers. All chimeric primers consisted of a gene-specific sequence tagged with a universal sequence at the 5′ end to ensure the specific and equivalent amplification. The forward universal primers were Cy5-labeled at the 5′ end of the sequence. Hundreds of sequences for each target from different region of the world, especially for those from Asia were downloaded from the National Center of Biotechnology Information (NCBI) and analyzed using Vector NTI to identify the homolog regions. The primers were designed in highly conserved regions using DNA star software (DNASTAR Inc., Madison, WI, USA) and Primer Premier 5.0 software (Premier Biosoft International, Palo Alto, CA, USA). The specificity of each single pair of primers was verified by conventional PCR and sequencing. These gene-specific primers were designed and optimized following several main principles: homogeneity, the length of amplification product is 150-350 bp, at least three bases difference between each amplicon, no significant dimers between different primers, and no non-specific products by each pair of gene-specific primers (Robertson and Walsh-Weller, 1998; Bruce, 2003). The primer sequences (Sunny Biotech Co., Ltd), the size of the resulting amplicons, and their target genes were listed in Table 1.
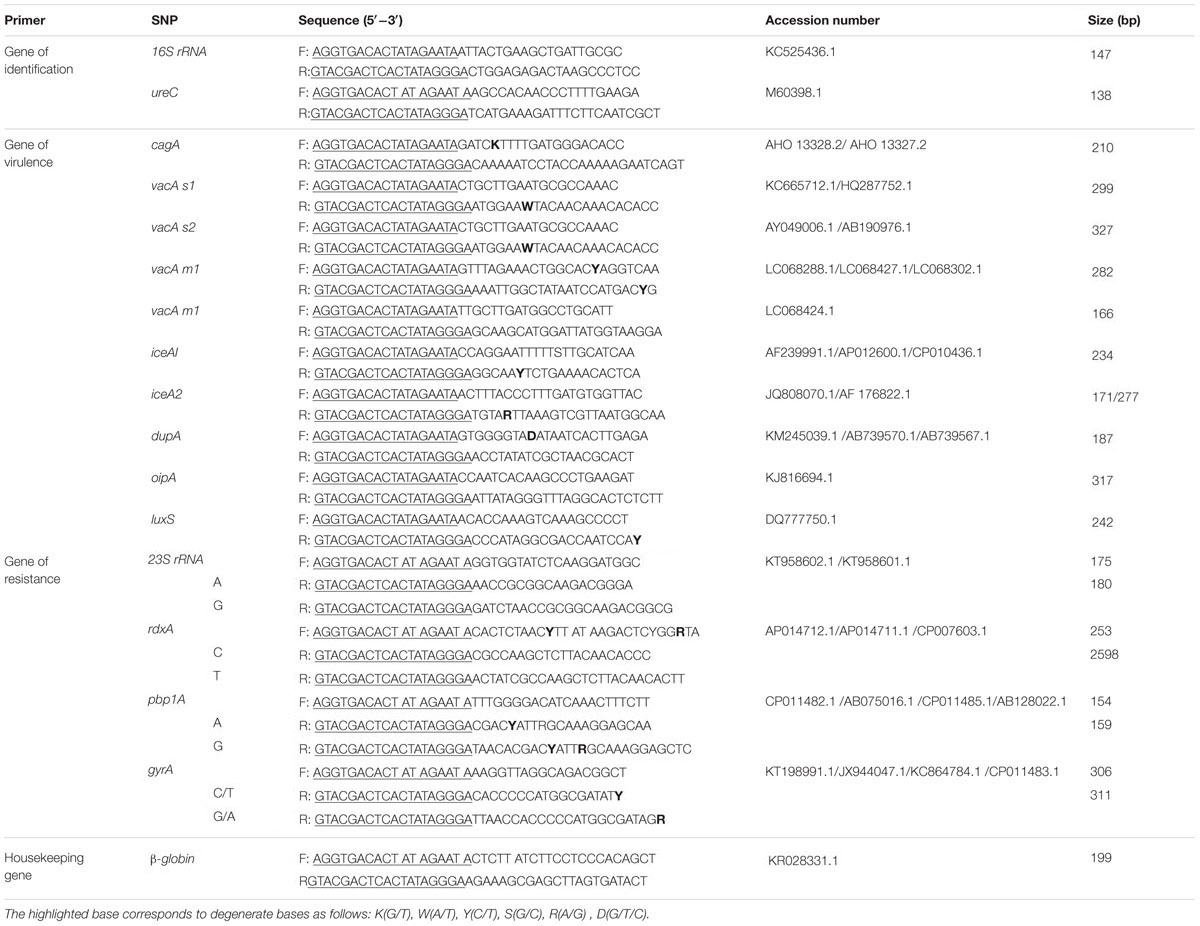
TABLE 1. Primer sequences and product size in HMGS.
Singlet-PCR Assay
The singlet-PCR assay was conducted using DNA templates verified by sequencing to evaluate the specificity of each pair of pathogen-specific primers and to ascertain the actual fragment size of each amplicon. The singlet-PCR assay was developed as follows: The singlet-PCR assay contained 0.5 μL of DNA, 12.5 μL of 2× buffer (Ex Taq polymerase, Mg2+, dNTP), 20 μM each of the forward universal primer and reverse universal primer, and 1 μM each of the forward chimeric primer and reverse chimeric primer. ddH2O was added to the PCR reaction to a final volume of 25 μL. The PCR mixture was incubated at 95°C for 5 min, followed by two steps with different annealing temperatures: step one, 12 PCR cycles of 30 s at 95°C, 30 s at 60°C, and 25 s at 72°C; step two, 20 cycles at 95°C for 30 s, 50°C for 30 s, and 72°C for 25 s; 5 min at 72°C, before then being held at 4°C in a thermal cycler. After amplification, 1 μL of PCR product that was diluted 10-fold was added to a 38.75 μL of specimens loading solution, along with 0.25 μL of DNA size standard-400 (GenomeLab GeXP Start Kit; Beckman Coulter, USA). Finally, a drop of mineral oil was added after blending. The GeXP Genetic Analysis System (Beckman Coulter, USA) was then used to analyze PCR products based on size separation using high-resolution capillary gel electrophoresis. The peak height for each PCR products was reported in the electropherogram and the reaction was considered positive when the dye signal was greater than 2,000 relative fluorescence units (rfu). ddH2O was used as a negative control throughout the assay.
Establishment and Optimization of HMGS
The multiple primers and reaction factors were further optimized in one HMGS assay reaction. The multiplex quantitative PCR was performed in a mixture containing MgCl2 solution, PCR buffer, forward universal primers, reverse universal primers, Taq polymerase, DNA template, and ddH2O. In details, the reaction system was optimized as follows: All primers were added to the reaction system with the same concentration and the volume. After amplification, PCR products were prepared for fragment analysis using GeXP Genetic Analysis System following manufacture suggested protocols. One microliter of PCR product was added to 30 μL of specimens loading solution along with 0.5 μL of DNA Size Standard 400 (AB Sciex, Inc., USA). The mixture was added to a 96-well plate and was loaded onto a GeXP Genetic Analysis System for capillary electrophoresis and fragment separation. The peaks were initially analyzed based on the sizes of the appropriate amplified products. The peak height for each gene was reported in the electropherogram and the dye signal intensity was measured by fluorescence spectrophotometry in rfu (relative fluorescence unit). Then, the concentrations of primers were adjusted to optimize signal intensity for each target to be detected from 2,000 to 175,000 rfu. If a signal peak was too high, the corresponding concentration would be reduced until the peak intensity was moderate. If a signal peak was too low or not detectable, the primer concentration would be increased to enhance the signal intensity. Additionally, the annealing temperature was optimized through temperature gradient descent method (chimeric primers from 50 to 65°C). Meanwhile, other factors of reaction conditions were also optimized. The main optimized principle was to keep all amplicons with similar amplification efficiency and presenting the gene-specific target amplicon without cross-amplification. Here, the ratio and concentration of primers were critical. Meanwhile, other parameters of the reaction conditions such as Tm value, reaction time and temperature were also important and were systematically optimized. HMGS protocols referred to the instruction of GeXP and previously reported paper (Zhou et al., 2015).
Artificial Mixture
To mimic clinical phenomenon in which patients infected different drug-resistant types of H. pylori simultaneously, four pairs of isolated strains from biopsy specimens with known genetic background, with mutation sites in 23S rRNA (A2143G), rdxA (C148T), gyrA (C261A/G), and pbp1A (A1777G) were chosen, and DNA from the wild type and mutant resistant strains were artificially mixed together in equal concentrations in HMGS assay. In addition, DNA extracts from clarithromycin resistant isolated strain with mutation site in 23S rRNA (23S rRNA_G) and from the wild type strain (23S rRNA_A) were chosen to mix together using following template ratio: 1:8, 1:4, 1:2,1:1, 2:1, 4:1, and 8:1. HMGS assay was performed using the mixed DNA.
Specificity, Sensitivity, and Accuracy of HMGS Assay
To determine if optimized HMGS maintained high specificity for H. pylori detection, HMGS assay readout of a standard H. pylori stain (ATCC43504), ddH2O, and six control bacterial species were tested in HMGS platform. Our control bacterial species included: E. coli, P. aeruginosa, A. baumannii, S. maltophilia, K. pneumoniae, as well as C. jejuni which has very similar growth characteristics with H. pylori. To further confirm the specificity of HMGS in complicated environment, we have mixed six control bacterial species C. jejuni, E. coli, P. aeruginosa, A. baumannii, S. maltophilia, and K. pneumoniae DNA (0.8 ng/μL of each bacterial specie) with and without H. pylori DNA (0.4 ng/μL of each bacterial specie) in PBS and then detected by HMGS assay. The sensitivity of 21 primer pairs HMGS was conducted by eight serial double dilutions of H. pylori DNA in PBS from the concentration of 0.4 ng/μL to 3.125 × 10-3 ng/μL The HMGS limit of detection was set by the criteria that the all H. pylori specific signals would read above 2,000 rfu. Total six clinical isolates with known genetic background of target genes verified by Sanger sequencing and three antrum biopsy specimens (two cases from the patients with H. pylori infection and one from uninfected healthy control) were used to verify the accuracy of HMGS.
Statistical Analysis
Statistical analysis of the linear correlation between the peak areas and the template ratios of wild type and mutant drug resistant isolates was carried out using SPSS version 17.0 software.
Results
Individual Primers Selected for HMGS Were Verified by Singlet-PCR Assay
The amplicon sizes for the target genes were as follows (Figures 1A–T): identification gene16S rRNA: 147 bp; quantification gene ureC: 138 bp; internal control gene beta-globin: 199 bp; 10 important virulence genes cagA: 210 bp, dupA: 187 bp, luxS: 242 bp, vacA s1: 299 bp, oipA: 317 bp, vacA m1: 282 bp, vacA m2: 166bp, iceA1: 234 bp, and iceA2: 171bp/277 bp; four main resistance genes 23S rRNA(A2143G): 175 bp/180 bp, rdxA(C148T): 253 bp/258 bp, pbp1A(A1777G): 154 bp/159 bp, and gyrA(C261A/G):306 bp/311 bp, respectively. The result showed that all target genes were specifically amplified without unintended products by their corresponding primers.
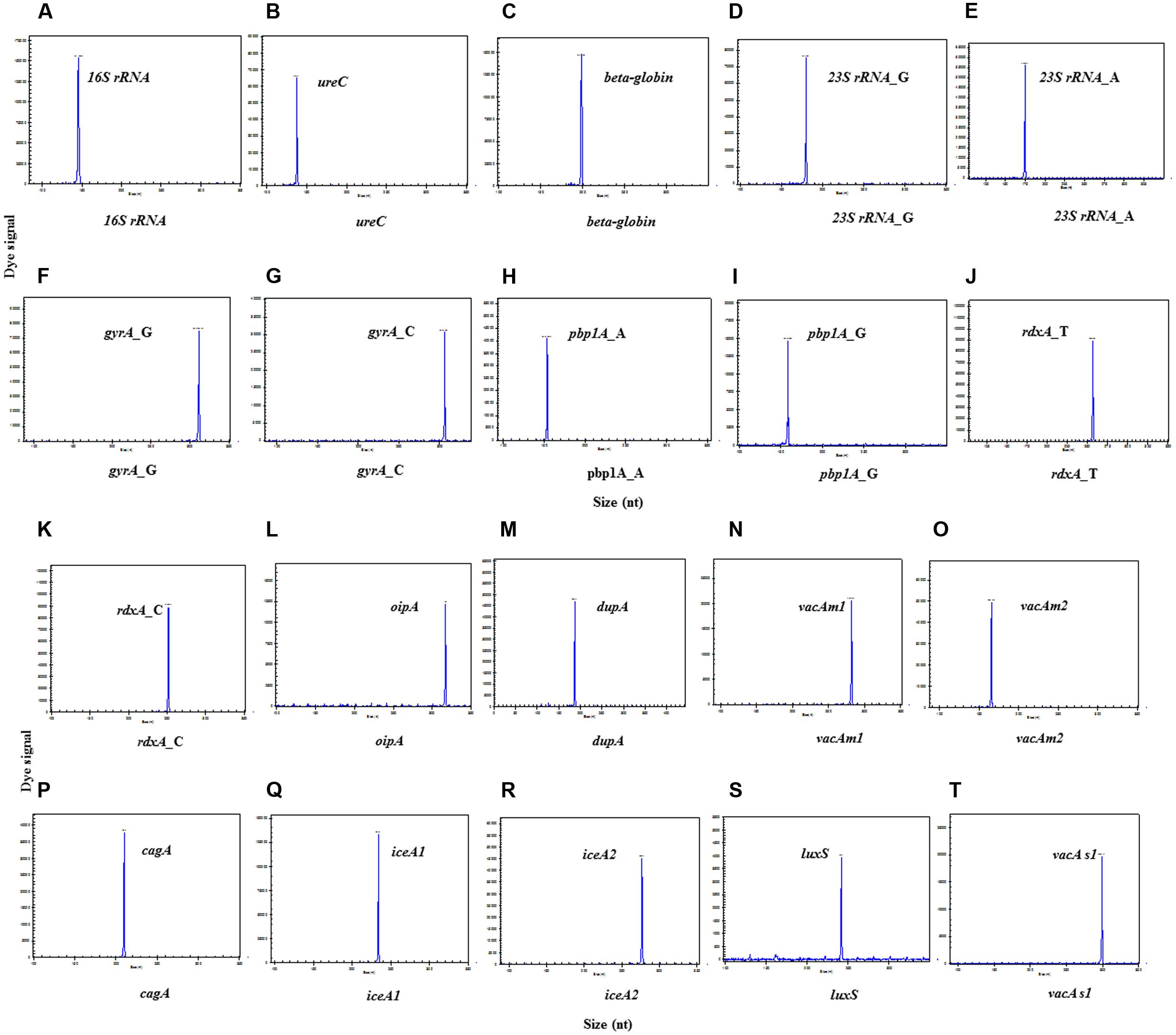
FIGURE 1. Singlet-PCR assay. The X-axis indicates the actual PCR product size, and the Y-axis indicates the dye signal. (A-T) Showed the results of the amplification of genes, 16S rRNA, ureC, beta-globin, 23S rRNA_G, 23S rRNA_A, gyrA_G, gyrA_C, pbp1A gyrA_A, pbp1A gyrA_G, rdxA_T,, rdxA_C,oipA, dupA, vacA m1, vacA m2, cagA, iceA1, iceA2, luxS, and vacA s1, respectively. Note that all gene targets were specifically amplified without nonspecific amplification by singlet-PCR assay.
The Optimization of HMGS Conditions Allowed for Efficient Use of Multiplex Assay with High-Resolution Using Isolates
The intensity of amplification signals for different targets varied up to 100-fold when all primers were used at the same concentration (Figure 2A). Therefore, the primer concentrations for genes with saturated peaks, such as 16S rRNA and ureC, were reduced to reach a moderate signal levels. On the other hand, the primer concentrations for genes with very low peaks, such as dupA, gyrA, and iceA1, were increased two- to threefold to significantly enhance the signal levels. The overall signal intensity for all targets was moderate to the middle range in HMGS assay after optimization (Figure 2B).
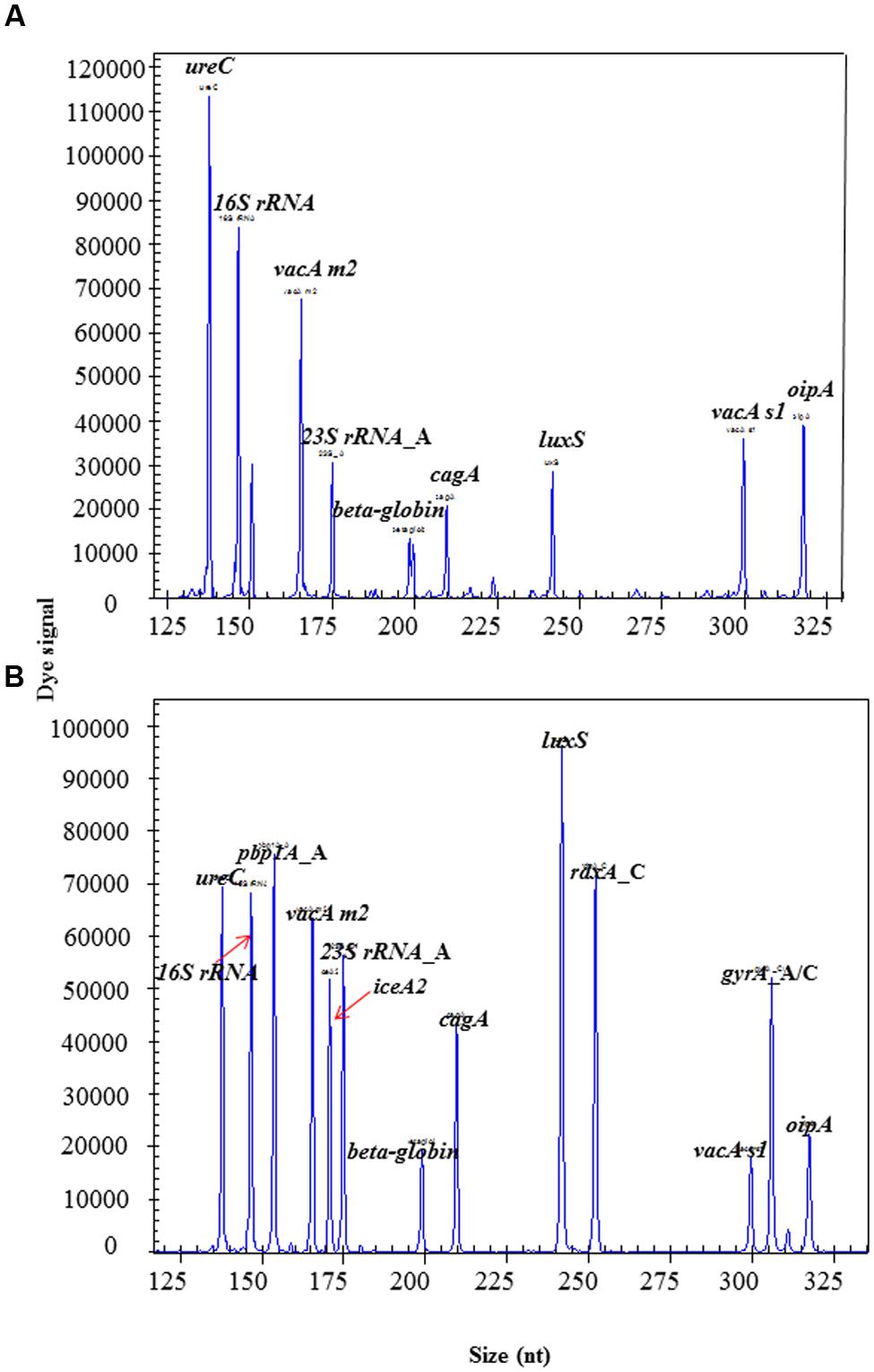
FIGURE 2. The optimization of HMGS assay for H. pylori detection. (A) The HMGS assay result showed specific gene from left to right was as follow: ureC, 16S rRNA, vacA m2, 23S rRNA_A, beta-globin, cagA, luxS, vacA s1, and oipA. (B) The targets genes from left to right are as follow: ureC, 16S rRNA, pbp1A, vacA m2, 23S rRNA_A, iceA2, beta-globin, cagA, luxS, rdxA_C, vacA s1, gyrA_C, and oipA. Note that the specific genes pbp1A, iceA2, and gyrA_C showed up and all the intensity of amplification signals for target genes moderated to the middle range after optimization.
HMGS Assay is Specific for H. pylori Identification
The H. pylori standard strain ATCC43504 showed specific amplification signals of H. pylori identification genes 16S rRNA and ureC (Figure 3A), while other six control bacterial species including C. jejuni, E. coli, P. aeruginosa, A. baumannii, S. maltophilia, K. pneumoniae, and ddH2O did not produce any signals that would indicate non-specific amplification (Figures 3B–F). Furthermore, the control bacteria mix did not display any H. pylori specific signals, while the control mix with H. pylori showed all the specific signals (Figures 3H,I). The above results demonstrated that the newly established and optimized HMGS assay was highly specific for H. pylori identification. The limit of sensitivity for HMGS simultaneous detection of all genes using 21 primer pairs was 6.25 × 10-3 ng/μL while over 80% of genes were still above the detection limit at 3.125 × 10-3 ng/μL dilution of H. pylori DNA diluted in PBS.
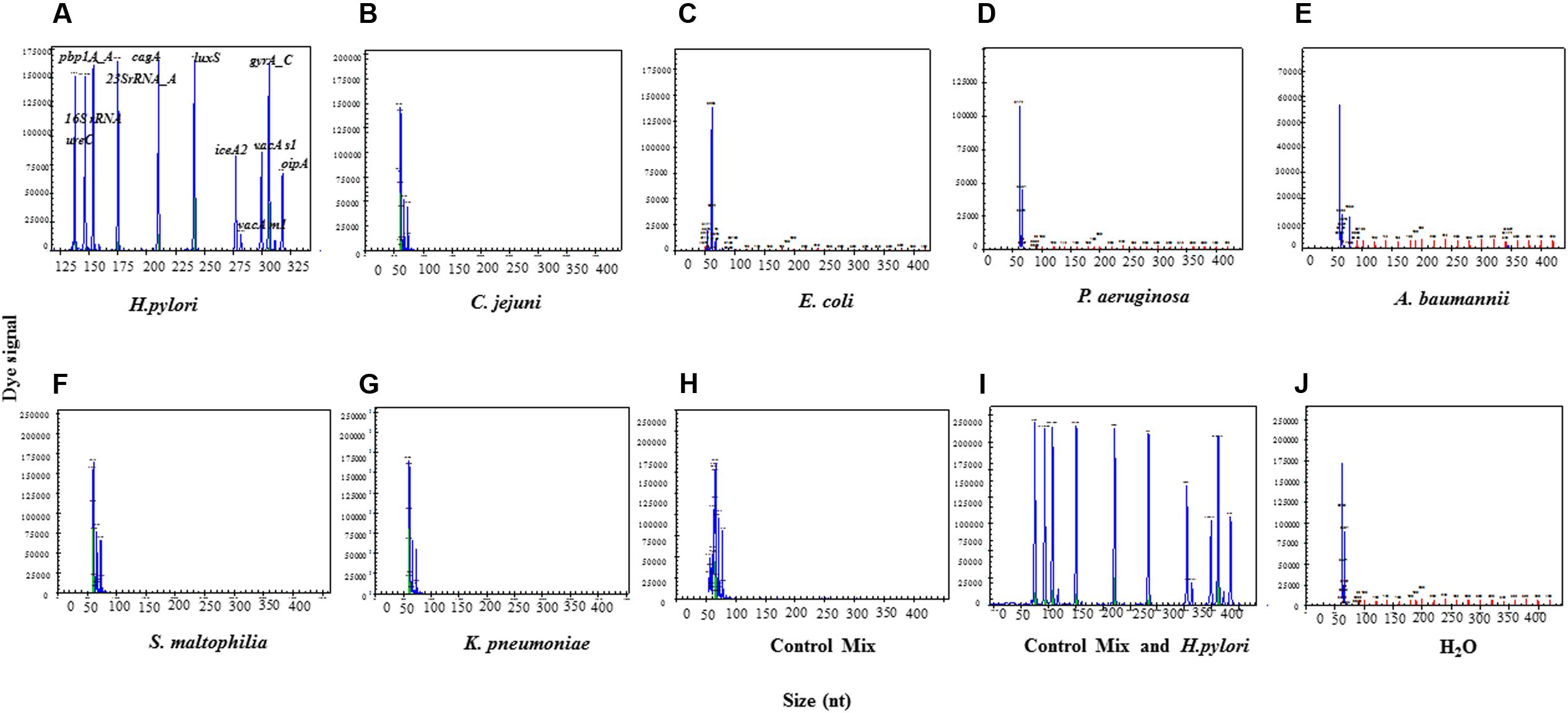
FIGURE 3. The specificity of the HMGS assay. (A) The standard H. pylori strain (ATCC43504) was used as positive control. (B) C. jejuni, (C) E. coli, (D) P. aeruginosa, (E) A. baumannii, (F) S. maltophilia, (G) K. pneumoniae, (H) control mix, (I) control mix and H. pylori, and (J) ddH2O were used as controls. 16S rRNA had a specific peak at 146 bp and ureC had a specific peak at 137 bp for standard H. pylori strain. No specific peak was found in all negative controls. The peaks in (B) to (H) and (J) around 60 bp were primer dimers, excluded according to the standards provided by the manufacturer.
HMGS Can Simultaneously Detect Virulence and Drug Resistance Genes of H. pylori
Six clinical isolates with known genetic background of target genes were used to evaluate the accuracy of HMGS assay. All the specific amplification signals were observed including identification gene 16S rRNA, quantification gene ureC, internal control beta-globin, 10 main virulence-associated genes cagA, vacA s1, vacA s2, vacA m1, vacA m2, iceA1, iceA2, luxS, dupA, and oipA, and four drug resistance genes pbp1A, gyrA, 23SrRNA, rdxA (Figure 4). These results demonstrated that the optimized HMGS assay for target genes of H. pylori were completely consistent with the corresponding genetic background.
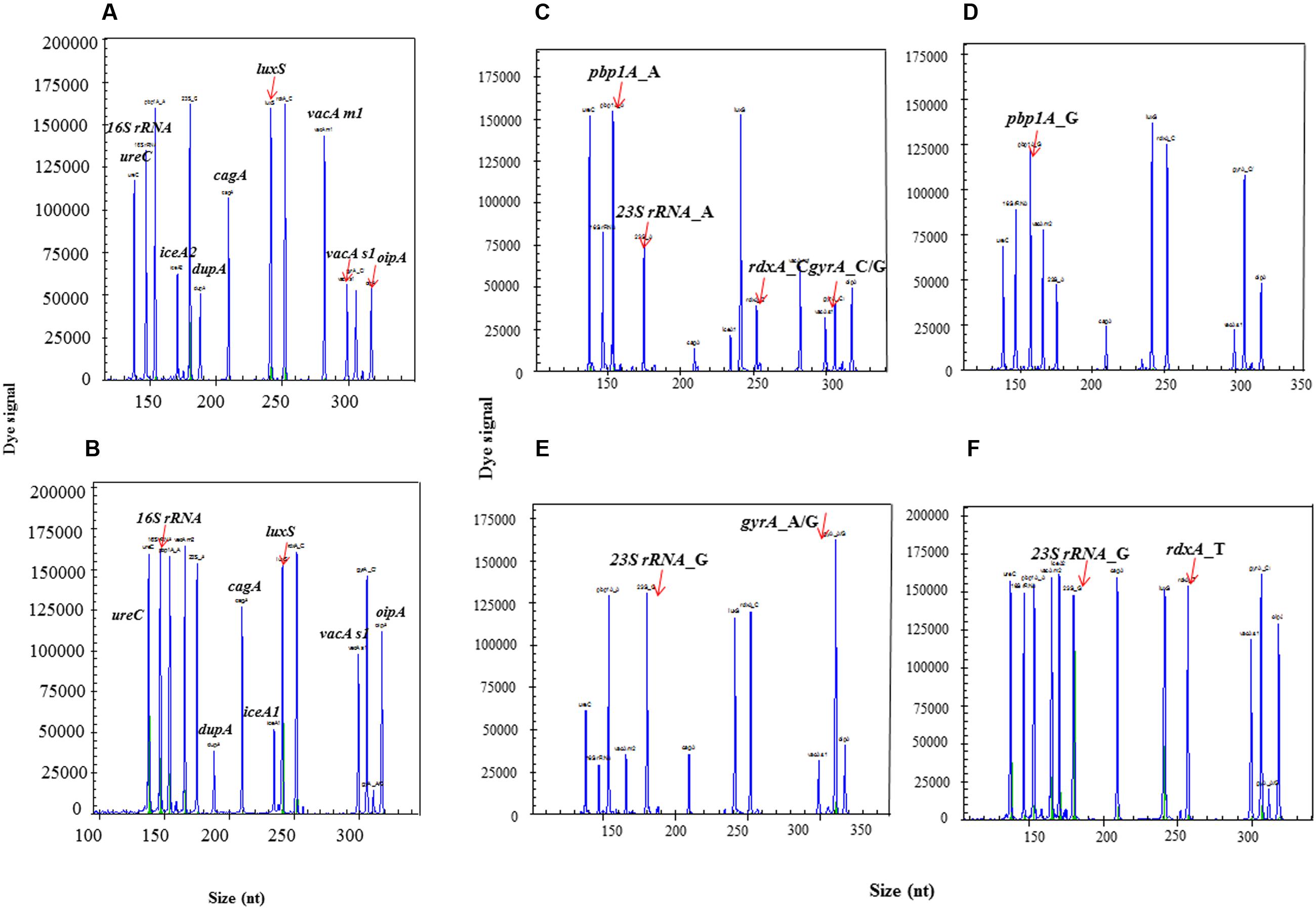
FIGURE 4. The detection of identification, virulence and resistance genes by HMGS assay. All the target genes could be efficiently detected by HMGS, including identification genes 16S rRNA and ureC (A-F), virulence genes cagA, dupA, luxS, vacA s1, oipA, vacA m1, and iceA2 (A), cagA, dupA, luxS, vacA s1, oipA, and iceA1 (B), resistance genes pbp1A (A1777G) (C,D), 23S rRNA (A2143G) (C,E), gyrA(C261A/G) (C,E), and rdxA (C148T) (C,F). Note that different isolates showed at the individual panels expressed distinct sets of identification, virulence and resistance genes, clearly identified by HMGS.
HMGS Assay Allowed for Detecting Mixed Infection with Drug Resistant Mutant and Wild Type Isolates in a Wide Range of Proportions
The specific and distinct amplification signals for mixture templates with wild type and mutant of H. pylori were shown in Figure 5. These data demonstrated that HMGS assay could successfully detect and differentiate mixed infection with wild type and mutant resistant genes of H. pylori isolates. We perform additional test to further explore the possibility to detect the relative contribution of wild type strain and resistant mutant in mixed infections. DNA templates from CLA-resistant mutant clinical isolate (23S rRNA_G) and wild type isolate (23S rRNA_A) were prepared and amplified using following ratio: 1:8, 1:4, 1:2,1:1, 2:1, 4:1, and 8:1 per reaction in the HMGS assay. The relative magnitude of signals from the wild type and mutant clinical strains corresponded to the ratio of the mixed template. A near-identity (R2= 0.987) linear correlation between the peak areas and the template ratios has been found. These results showed that wild type and mutant isolates could be accurately quantify by HMGS assay at relatively broad range of proportions of the mixed infection strains (Figure 6).
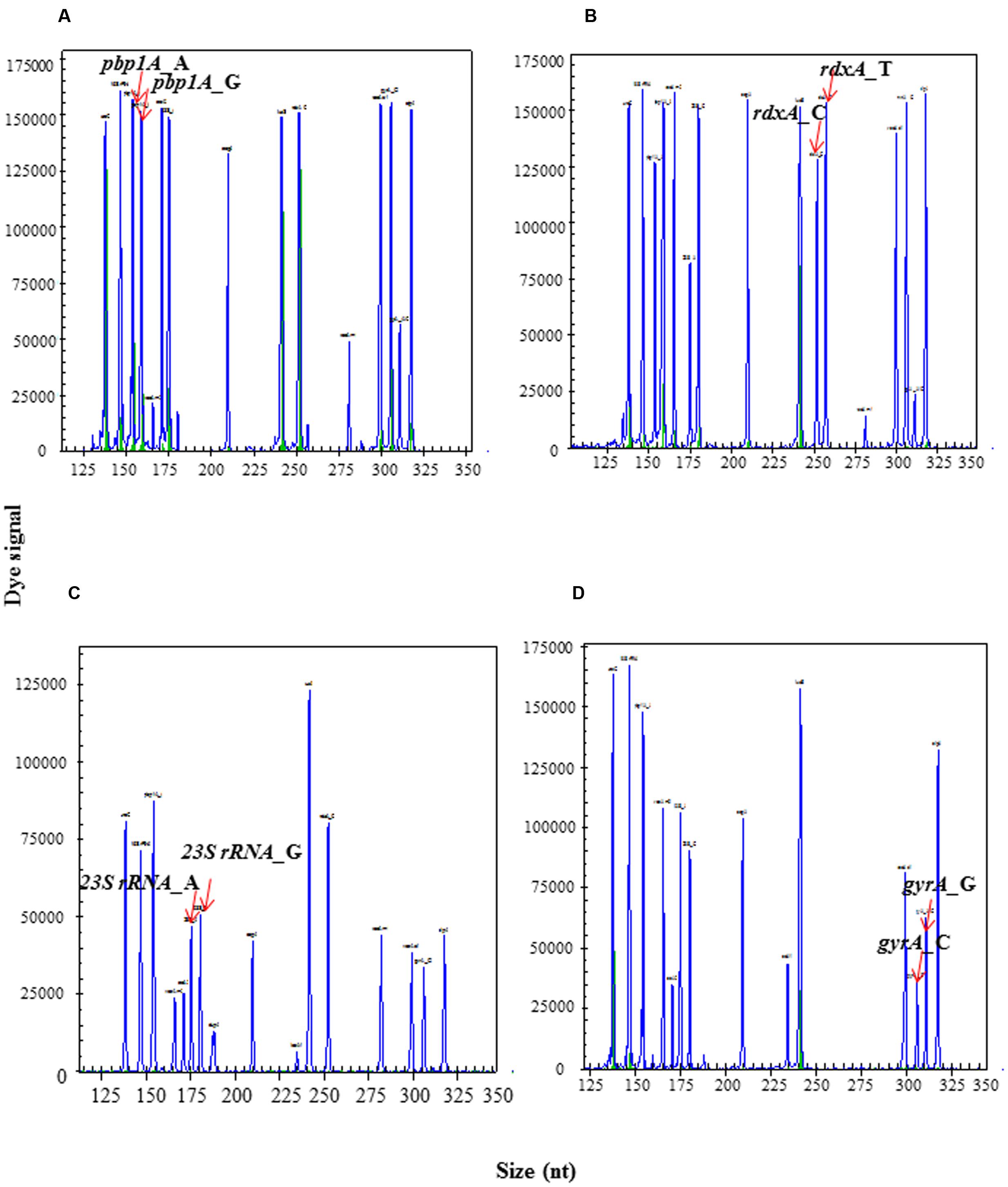
FIGURE 5. Artificial mixture of wild type and resistant mutant isolates detected by HMGS assay. The multiplex assay was carried out with artificial mixed wild type and resistant mutant templates for (A) pbp1A(A1777G), (B) rdxA(C148T), (C) 23SrRNA (A2143G), and (D) gyrA(C261A/G), respectively. Note that mixed infection of wild type and mutant isolates could be accurately distinguished at a relatively broad range of proportions by HMGS assay.
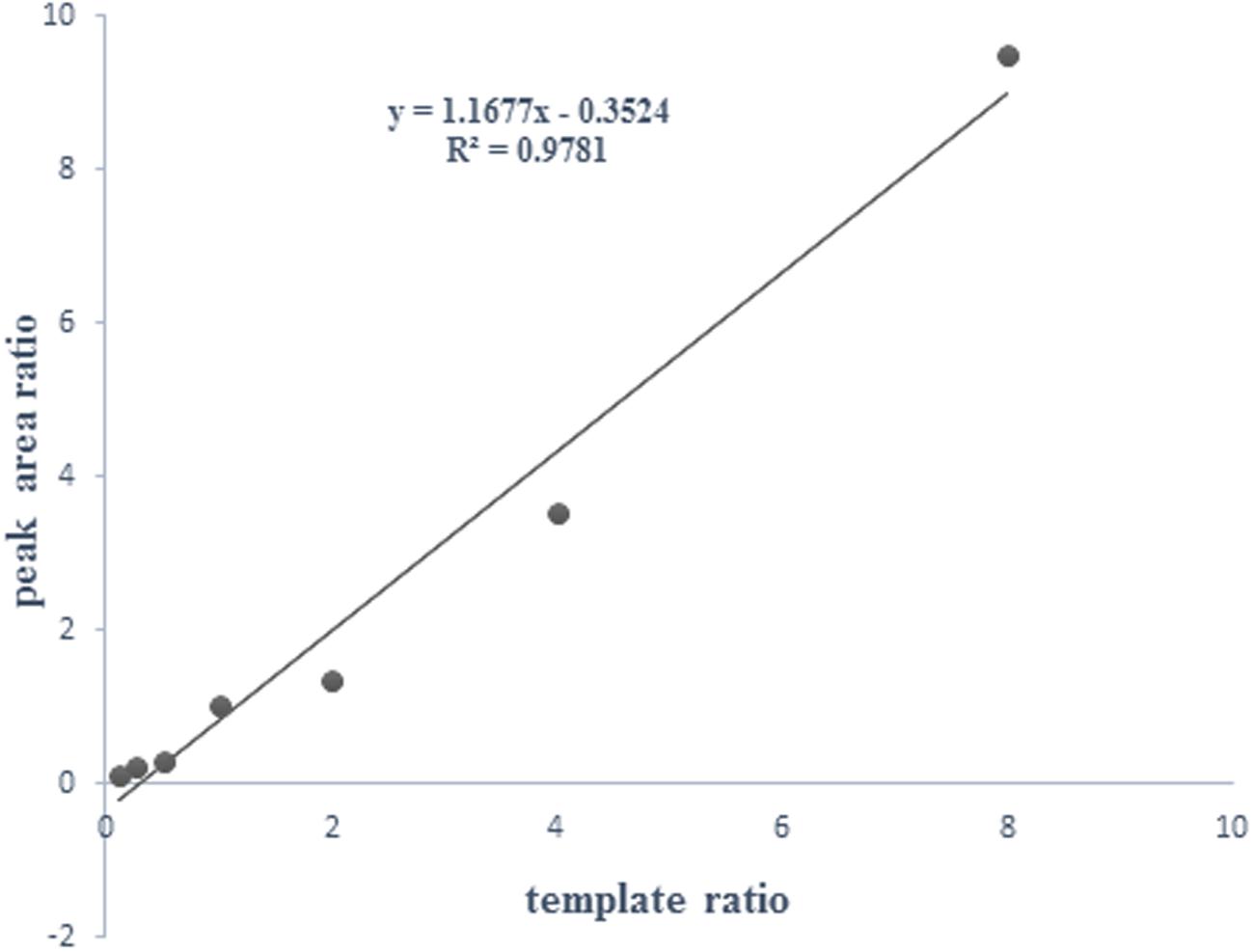
FIGURE 6. Correlation between the peak areas and the template ratios of the wild type and mutant drug resistant isolates. A near-identity (R2= 0.987) linear correlation between the peak areas and the template ratios showed that wild type and mutant isolates could be accurately quantify by HMGS assay at relatively broad range of proportions of the mixed infection.
Optimization of HMGS Conditions Allowed for Efficiently Detecting of H. pylori Directly in Gastric Biopsy Specimens
Wild type and mutant resistant gene readouts of HMGS assay in biopsy specimens were completely consistent with their corresponding isolated strains. The peaks of gyrA_C were simultaneously showed in the wild type gastric biopsy specimen as well as in its corresponding LEV-susceptible isolates (Figures 7A,B), and the peaks of gyrA_A/G were showed in the mutant gastric biopsy specimens and its corresponding LEV-resistant isolates (Figures 7C,D). Meanwhile, all H. pylori specific peaks and human internal control gene beta-globin were detected in two infected biopsy specimens (Figures 7A,C), while only beta-globin was detected in uninfected biopsy specimens (Figure 7E). Thus, these results showed an excellent concordance between gastric biopsy specimens and corresponding isolated strains, documenting that HMGS assay could efficiently diagnose H. pylori infection and analyze the virulence and drug resistance in gastric biopsy specimens directly.
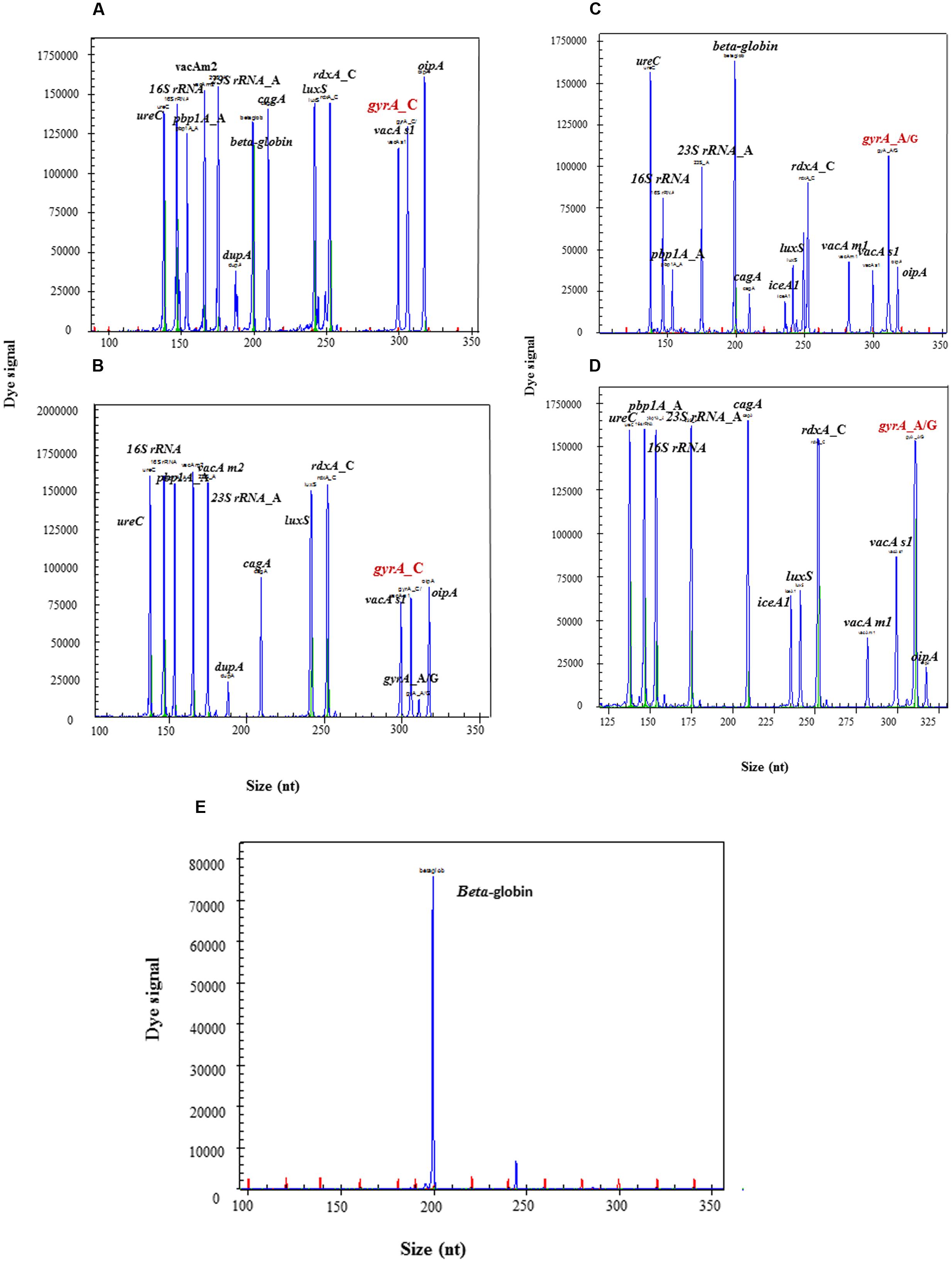
FIGURE 7. Detection of antrum biopsy specimens and corresponding strains by HMGS assay. The expected amplification peaks appeared in the wild type gyrA_C biopsy specimen (A) and its corresponding LEV-susceptible isolated strain (B). The expected amplification peaks appeared in the mutant gyrA_A/G biopsy specimen (C) and its corresponding LEV-resistant isolated strain (D). Only beta-globin peak appeared in uninfected biopsy specimen (E).
Discussion
Helicobacter pylori is an important pathogen causing major gastroduodenal disease for which rapid non-culture based identification assays are needed (Graham, 1998). Virulence factors contribute to the development of severe complications in H. pylori infections and become predictors for risk of gastric atrophy, intestinal metaplasia and other severe clinical outcomes (Shiota et al., 2013). The severity of clinical manifestations is associated with bacterial load (Kusters et al., 2006). Meanwhile, the growing H. pylori drug resistance in Asia countries severely influences the efficacy of eradication treatment, which required drug resistance analysis before treatment (Katelaris, 2009; Cammarota et al., 2012; Alfizah et al., 2014; Smith et al., 2014). Therefore, it is important to analyze the virulence, bacterial load and resistance profiles during the identification of H. pylori. The traditional methods such as histology, culture, RUT, UBT as well as serology are available for the detection of H. pylori, and each method has its usefulness and limitations in different clinical situations (Hirschl and Makristathis, 2007; Khalifehgholi et al., 2013; Wang et al., 2015). The published guidance proposed molecular methods, like PCR sequencing, real-time (RT-PCR) PCR, and dual-priming oligonucleotide (DPO-PCR) should be introduced as alternatives for conventional H. pylori detection (Lehours et al., 2011; Malfertheiner et al., 2012) due to their high sensitivity and accuracy compared with conventional methods. However, these molecular methods are relatively expensive and could not detect multiple genes at a time (Lee et al., 2014; Wang et al., 2015). At present, there were no existing molecular methods that could simultaneously identify and quantify H. pylori, analyze the virulence and drug-resistance genes in single-infection and mixed infection condition. Thus, it is urgent to explore a rapid, effective, and high-throughput molecular technique to diagnose, treat, and monitor H. pylori infection. Previously, our team has established a genetic method using 16S rRNA and ureC for H. pylori identification and quantification (Zhou et al., 2015). The sensitivity, specificity, positive predictive value (PPV) and negative predictive value (NPV) were comparable to other conventional methods, such as culture, RUT and histopathology. In this study, we incorporated this readout into a newly developed and optimized HMGS assay, introducing 21 newly designed primer pairs to simultaneously detect the identification gene, 10 virulence genes, 4 drug resistance genes, quantification genes ureC of H. pylori and internal control gene beta-globin of human tissue to normalize the specific gene expression.
Currently, some identification methods for H. pylori such as culture, histology, and RUT are available in clinical diagnosis but each of these methods has some limitations (Wang et al., 2015). The culture of gastric mucosa biopsy specimens is generally considered as the gold standard for the identification of H. pylori. However, culture is time consuming (Hachem et al., 1995; Cuchi et al., 2002), and its sensitivity and specificity are restricted with the technical difficulties, incubation environment and low bacterial loads (Cover, 2012). Histopathology has high sensitivity and specificity but the choice of staining methods and the bacterial load could directly impact the histological assessment (Christensen et al., 1992; Atherton et al., 1996; El-Zimaity et al., 1996). Besides, RUT is also susceptible to bacteria loads (Leodolter et al., 2001). In present study, we simultaneously detected the identification gene, virulence and resistance genes in six clinical isolated strains with known genetic background (Figure 3) and the results showed an excellent concordance with sequencing. In addition, specificity of the HMGS assay was examined using the standard H. pylori strain (ATCC43504), and six bacterial controls. The result showed that only standard H. pylori strains had specific amplification signals in 16S rRNA and ureC genes at the size of 146 and 137 bp. However, neither H. pylori identification genes nor any of the other H. pylori-specific amplification signals appeared in HMGS conducted on control bacteria and negative control of ddH2O specimens. This result demonstrated that the optimized HMGS assay was highly specific to detect H. pylori (Figure 6). Therefore, this novel HMGS assay is a highly rapid, specific and effective technique compared with culture, histopathology and RUT.
Virulence factors have been reported to contribute to the development of H. pylori related-diseases and assist to diagnose in H. pylori infection (Proença-Modena et al., 2009). The specific functions of the virulence-associated factors of H. pylori is complicated, and its outcome likely affected by different combinations of variable regions of the virulence genes and influence of “local factors” in each studies (Shiota et al., 2013; Yamaoka and Graham, 2014; Sgouras et al., 2015). For example, there are variations in the functional vacA gene signal regions (s1 and s2) and middle regions (m1 and m2 in different H. pylori strains. Individuals infected with s1 or m1 H. pylori strains have an increased risk compared with individuals infected with s2 or m2 strain. Additionally, “i” region plays a role in the development of peptic ulcer in people in Iraq and Italy (Yamaoka, 2010); however, in a study of the patients from East and Southeast Asia, association between the region and disease has not been detected (Shiota et al., 2013). The immunoassay could be used to detect the virulence factors of H. pylori. However, immunoassay is relatively low-throughput in a single reaction. Especially, there is a window phase between the timing of H. pylori infection and antibody producing, which may lead to the high false negative rate (FNR) in clinical diagnosis of H. pylori infection. By using of HMGS assay, we here detected and analyzed 10 main virulence factors simultaneously (Figure 4). While understanding interactions between hosts and microbes expressing various combinations of virulence gene is very complex and will require an amount of data and system biology approach to analyze, we anticipate that our rapid, accurate, and high-throughput multiple virulence gene analysis will be a useful tool in gathering such information for further analysis. We believe that such information will enable us to identify the high-risk groups among carriers of H. pylori.
The most common antibiotics for H. pylori treatment are clarithromycin (CLA), metronidazole (MTZ), amoxicillin (AMX), and levofloxacin (LEV) according to the guidelines (Wolle and Malfertheiner, 2007; Gao et al., 2010; Yu et al., 2011; Malfertheiner et al., 2012). However, the growing drug resistance has seriously impacted the efficacy of eradication treatment, which emphasized the need of resistance analysis before antibiotic treatment. Conventional detection methods for H. pylori drug resistance are culture-dependent, time-consuming, low-throughput and could not distinguish mixed infection. H. pylori drug resistance was related to specific gene mutations (Gerrits et al., 2006; Tanih and Ndip, 2013). In our study, the four wild-type genes and their main gene mutation sites of 23S rRNA (A2143G), rdxA (C148T), gyrA (C261A/G), and pbp1A (A1777G) were selected for HMGS to assess CLA, MTZ, LEV, and AMX resistance. We demonstrated that the HMGS assay performed on isolated strains was fast, effective and accurate for simultaneous analysis for drug resistance genes (Figure 4). Notably, the possibility of mixed infection with different drug resistant H. pylori strains increases the difficulty of differential diagnosis and individual eradication therapy. Here, we explored HMGS performance in detecting mixed infection using the artificial mixture with the wild type and mutant resistant genes, which showed the specific and distinct amplification signals for wild type and mutant type of H. pylori (Figure 5). Additionally, a linear correlation between the amplification trend of wild and mutant strains were analyzed (R2= 0.987), indicating that HMGS had accurately established the ratio in a board range of mixture proportions between drug resistant mutant isolate and the wild type H. pylori (Figure 6). Therefore, mixed infection with wild and mutant isolates could be detected and quantified by HMGS assay and predominant bacteria could be distinguished in mixed infections. We anticipated that border use of HMGS assay would improve the clinical accurate diagnosis, treatment and monitoring.
The severity of the clinical manifestations of the H. pylori infection was also associated with bacterial load (Kusters et al., 2006). The most regular method used for H. pylori quantification is RT-PCR. However, the selection of probes could easily influence the sensitivity and reproducibility of the result (He et al., 2002; Mikula et al., 2003). Moreover, RT-PCR could not amplify the target and internal control genes in the same reaction. Here, we selected the ureC as the quantification genes for its single copy. Meanwhile, beta-globin gene was introduced as internal control of human tissue that could normalize the expression of specific genes of H. pylori. Therefore, HMGS had a good prospect for the clinical diagnosis and treatment monitoring of H. pylori.
After optimizing HMGS assay in detecting H. pylori, as well as analyze the virulence and drug-resistance genes in isolated strains, we further explored the clinical utility of the HMGS assay for H. pylori diagnosis with 3 gastric biopsy specimens. Our results showed the readouts of HMGS assay performed on biopsy specimens were completely in agreement with the results of clinical diagnosis. Only human internal control beta-globin appeared in uninfected biopsy specimen (Figure 7E), while both beta-globin and the specific peaks of H. pylori appeared in the infected biopsy specimens (Figures 7A,C). Besides, the specific peaks in biopsy specimens were completely consistent with that in their corresponding isolated strains which had been confirmed by sequencing (Figures 4A–D). Considering the limitations of conventional detection methods for H. pylori, HMGS assay may efficiently skip the culture step, shorten the detection time and decrease the FNR. Based on these advantages, HMGS assay would identify H. pylori and detect virulence and drug resistance directly using biopsy specimens.
Conclusion
In conclusion, we explored and optimized HMGS assay in detecting and analyzing a set of genes for H. pylori identification, quantification, virulence, and drug resistance in both isolated strains and biopsy specimens. Also, HMGS assay could detect mixed infections and differentiate the predominant bacteria in artificial mixed infection strains. While present study has focused on technological details and demonstration of possible clinical applications of HMGS, we believe that broad validation of its full potential will result from future studies conducted in a lager cohort of patients, possibly, at multiple study centers. We anticipate that HMGS will improve the clinical diagnosis, treatment and monitoring of H. pylori infection.
Author Contributions
YZ, FZ, MK, SW contributed to this work equally. YZ, PX, YW, and HZ designed the experiments. YZ, FZ, SW, OM, and HZ wrote the manuscript.
Funding
This work was supported by Shanghai Science and Technology Committee “Lead project” (grant no: 14411962800 and 16411968000), Shanghai Science and Technology Committee “Natural Science Foundation” (grant no: 14ZR1413100), Shanghai Health Bureau “Key research projects” (grant no: 20134008), Shanghai Shenkang Hospital Development Center “New frontier technology joint research project” (grant no: SHDC12013123, SHDC12015107, and SHDC22014003), Ministry of Science and Technology “The National High Technology Research and Development Program of China (863 Program)” (grant no: 2015AA021107-019). Dr. Olszewski was supported by VA Merit Grant (grant no: I01BX000656).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful to Prof. Zhijun Bao (Department of Gastroenterology, Gerontology Institute of Shanghai affiliated to Huadong Hospital, Huadong Hospital affiliated to Fudan University, Shanghai, China) for instruction, and grateful to Dr. Jun Zhou and Dr. Renxiang Huang (Department of Endoscopy, Huadong Hospital, Fudan University, Shanghai, China) for providing gastric mucosa biopsy samples.
References
Alfizah, H., Norazah, A., Hamizah, R., and Ramelah, M. (2014). Resistotype of Helicobacter pylori isolates: the impact on eradication outcome. J. Med. Microbiol. 63, 703–709. doi: 10.1099/jmm.0.069781-0
Atherton, J. C., Tham, K. T., Peek, R. J., Cover, T. L., and Blaser, M. J. (1996). Density of Helicobacter pylori infection in vivo as assessed by quantitativeculture and histology. J. Infect. Dis. 174, 552–556. doi: 10.1093/infdis/174.3.552
Bruce, A. (2003). White Polymerase Chain Reaction (PCR): design and optimization of reactions. Methods Mol. Biol. 226, 89–99.
Cammarota, G., Sanguinetti, M., Gallo, A., and Posteraro, B. (2012). Review article: biofilm formation by Helicobacter pylori as a target for eradication of resistant infection. Aliment. Pharmacol. Ther. 36, 222–230. doi: 10.1111/j.1365-2036.2012.05165.x
Christensen, A. H., Gjørup, T., Hilden, J., Fenger, C., Henriksen, B., Vyberg, M., et al. (1992). Observer homogeneity inthe histologic diagnosis of Helicobacter pylori. Latent class analysis,kappa coefficient, and repeat frequency. Scand. J. Gastroenterol. 27, 933–939. doi: 10.3109/00365529209000166
Cover, T. L. (2012). Perspectives on methodology for in vitro culture of Helicobacter pylori. Methods Mol. Biol. 921, 11–15.
Cuchi, E., Forne, M., and Quintana, S. (2002). Comparison of two transportmedia and threeulture media for primary isolation of Helicobacterpylori from gastric biopsies. Clin. Microbiol. Infect. 8, 609–610. doi: 10.1046/j.1469-0691.2002.00454.x
de Martel, C., Forman, D., and Plummer, M. (2013). Gastric cancer: epidemiology and risk factors. Gastroenterol. Clin. North Am. 42, 219–240. doi: 10.1016/j.gtc.2013.01.003
El-Zimaity, H. M., Graham, D. Y., Al-Assi, M. T., Malaty, H., Karttunen, T. J., Graham, D. P., et al. (1996). Interobserver variationin the histopathological assessment of Helicobacter pylorigastritis. Hum. Pathol. 27, 35–41. doi: 10.1016/S0046-8177(96)90135-5
Gao, W., Cheng, H., Hu, F., Li, J., Wang, L., Yang, G., et al. (2010). The evolution of Helicobacter pylori antibiotics resistance over 10 years in Beijing. China. Helicobacter 15, 460–466. doi: 10.1111/j.1523-5378.2010.00788.x
Gerrits, M. M., van Vliet, A. H., Kuipers, E. J., and Kusters, J. G. (2006). Helicobacter pylori and antimicrobial resistance: molecular mechanisms and clinical implications. Lancet Infect. Dis. 6, 699–709. doi: 10.1016/S1473-3099(06)70627-2
Graham, D. Y. (1998). Antibiotic resistance in Helicobacter pylori: implications for therapy. Gastroenterology 115, 1272–1277. doi: 10.1016/S0016-5085(98)70100-3
Hachem, C. Y., Clarridge, J. E., Evans, D. G., and Graham, D. Y. (1995). Comparisonof agar based media for primary isolation of Helicobacter pylori. J. Clin. Pathol. 48, 714–716.
He, Q., Wang, J., Osato, M., and Lachman, L. B. (2002). Real-Time quantitative PCR for detection of Helicobacter pylori. J. Clin. Microbiol. 40, 3720–3728. doi: 10.1128/JCM.40.10.3720-3728.2002
Hirschl, A. M., and Makristathis, A. (2007). Methods to detect Helicobacter pylori: from culture to molecular biology. Helicobacter 12(Suppl. 2), 6–11. doi: 10.1111/j.1523-5378.2007.00560.x
Katelaris, P. H. (2009). Helicobacter pylori: antibiotic resistance and treatment options. J. Gastroenterol. Hepatol. 24, 1155–1157. doi: 10.1111/j.1440-1746.2009.05911.x
Khalifehgholi, M., Shamsipour, F., Ajhdarkosh, H., Ebrahimi Daryani, N., Pourmand, M. R., Hosseini, M., et al. (2013). Comparison of five diagnostic methods for Helicobacter pylori. Iran. J. Microbiol. 5, 396–401.
Kusters, J. G., Van Vliet, A. H. M., and Kuipers, E. J. (2006). Pathogenesis of Helicobacter pylori infection. Clin. Microbiol. Rev. 19, 449–490. doi: 10.1128/CMR.00054-05
Lee, J. W., Kim, N., Nam, R. H., Park, J. H., Choi, Y. J., and Kim, J. M. (2014). GenoType HelicoDR test in the determination of antimicrobial resistance of Helicobacter pylori in Korea. Scand. J. Gastroenterol. 49, 1058–1067. doi: 10.3109/00365521.2014.894117
Lehours, P., Siffré, E., and Mégraud, F. (2011). DPO multiplex PCR as an alternative to culture and susceptibility testing to detect Helicobacter pylori and its resistance to clarithromycin. BMC Gastroenterol. 11:112. doi: 10.1186/1471-230X-11-112
Leodolter, A., Wolle, K., and Malfertheiner, P. (2001). Current standards in the diagnosis of Helicobacter pylori infection. Dig. Dis. 19, 116–122. doi: 10.1159/000050665
Malfertheiner, P., Megraud, F., O’Morain, C. A., Atherton, J., Axon, A. T., Bazzoli, F., et al. (2012). Management of Helicobacter pylori infection–the Maastricht IV/ Florence consensus report. Gut 61, 646–664. doi: 10.1136/gutjnl-2012-302084
Mikula, M., Dzwonek, A., Jagusztyn-Krynicka, K., and Ostrowski, J. (2003). Quantitative detection for low levels of Helicobacter pylori infection in experimentally infected mice by real-time PCR. J. Microbiol. Methods 55, 351–359. doi: 10.1016/S0167-7012(03)00166-0
Proença-Modena, J. L., Acrani, G. O., and Brocchi, M. (2009). Helicobacter pylori: phenotypes, genotypes and virulence genes. Future Microbiol. 4, 223–240. doi: 10.2217/17460913.4.2.223
Robertson, J. M., and Walsh-Weller, J. (1998). An introduction to PCR primer design and optimization of amplification reactions. Methods Mol. Biol. 98, 121–154.
Sgouras, D. N., Trang, T. T., and Yamaoka, Y. (2015). Pathogenesis of Helicobacter pylori infection. Helicobacter. 20(Suppl. 1), 8–16. doi: 10.1111/hel.12251
Shiota, S., Suzuki, R., and Yamaoka, Y. (2013). The significance of virulence factors in Helicobacter pylori. J. Digest. Dis. 14, 341–349. doi: 10.1111/1751-2980.12054
Shukla, S. K., Prasad, K. N., Tripathi, A., Ghoshal, U. C., Krishnani, N., and Nuzhat, H. (2011). Quantitation of Helicobacter pylori ureC gene and its comparison with different diagnostic techniques and gastric histopathology. J. Microbiol. Methods 86, 231–237. doi: 10.1016/j.mimet.2011.05.012
Smith, S. M., O’Morain, C., and McNamara, D. (2014). Antimicrobial susceptibility testing for Helicobacter pylori in times of increasing antibiotic resistance. World J. Gastroenterol. 20, 9912–9921. doi: 10.3748/wjg.v20.i29.9912
Tanih, N. F., and Ndip, R. N. (2013). Molecular detection of antibiotic resistance in South African isolates of Helicobacter pylori. Gastroenterol. Res. Pract. 2013:259457. doi: 10.1155/2013/259457
Venerito, M., Goni, E., and Malfertheiner, P. (2016). Helicobacter pylori screening: options and challenges. Expert Rev. Gastroenterol. Hepatol. 10, 497–503. doi: 10.1586/17474124.2016.1126507
Wang, Y. K., Kuo, F. C., Liu, C. J., Wu, M. C., Shih, H. Y., Wang, S. S., et al. (2015). Diagnosis of Helicobacter pylori infection: current options and developments. World J. Gastroenterol. 21, 11221–11235. doi: 10.3748/wjg.v21.i40.11221
Wolle, K., and Malfertheiner, P. (2007). Treatment of Helicobacter pylori. Best Pract. Res. Clin. Gastroenterol. 21, 315–324. doi: 10.1016/j.bpg.2006.11.001
Yamaoka, Y. (2010). Mechanisms of disease: Helicobacter pylori virulence factors. Nat. Rev. Gastroenterol. Hepatol. 7, 629–641.
Yamaoka, Y., and Graham, D. Y. (2014). Helicobacter pylori virulence and cancer pathogenesis. Future Oncol. 10, 1487–1500. doi: 10.2217/fon.14.29
Yu, C., Li, L., Chen, W., Jiao, Y., Yang, N., Yang, E., et al. (2011). Levofloxacin susceptibility testing for Helicobacter pylori in China: comparison of E-test and disk diffusion method. Helicobacter 16, 119–123. doi: 10.1111/j.1523-5378.2011.00820.x
Keywords: Helicobacter pylori, high-throughput multiplex genetic detection system, identification, quantification, virulence, resistance genes, mixed infection
Citation: Zhang Y, Zhao F, Kong M, Wang S, Nan L, Hu B, Olszewski MA, Miao Y, Ji D, Jiang W, Fang Y, Zhang J, Chen F, Xiang P, Wu Y and Zhao H (2016) Validation of a High-Throughput Multiplex Genetic Detection System for Helicobacter pylori Identification, Quantification, Virulence, and Resistance Analysis. Front. Microbiol. 7:1401. doi: 10.3389/fmicb.2016.01401
Received: 18 April 2016; Accepted: 24 August 2016;
Published: 07 September 2016.
Edited by:
Manuela Caniça, National Institute of Health Dr. Ricardo Jorge, PortugalReviewed by:
Abdel Rahman Zueter, Universiti Sains Malaysia, MalaysiaYan Sun, University of Pennsylvania, USA
Copyright © 2016 Zhang, Zhao, Kong, Wang, Nan, Hu, Olszewski, Miao, Ji, Jiang, Fang, Zhang, Chen, Xiang, Wu and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hu Zhao, aHViZXJ0emhhb0AxNjMuY29t Yong Wu, eW9uZy53dUBoZWFsdGhnZW5ldGVjaC5jb20= Ping Xiang, eGlhbmdwaW5nODEzQDEyNi5jb20=
†These authors have contributed equally to this work.