 Peter Deines
Peter Deines Thomas C. G. Bosch
Thomas C. G. Bosch- Zoological Institute and Interdisciplinary Research Center, Kiel Life Science, Christian-Albrechts-Universität zu Kiel, Kiel, Germany
Animals are home to complex microbial communities, which are shaped through interactions within the community, interactions with the host, and through environmental factors. The advent of high-throughput sequencing methods has led to novel insights in changing patterns of community composition and structure. However, deciphering the different types of interactions among community members, with their hosts and their interplay with their environment is still a challenge of major proportion. The emerging fields of synthetic microbial ecology and community systems biology have the potential to decrypt these complex relationships. Studying host-associated microbiota across multiple spatial and temporal scales will bridge the gap between individual microorganism studies and large-scale whole community surveys. Here, we discuss the unique potential of Hydra as an emerging experimental model in microbiome research. Through in vivo, in vitro, and in silico approaches the interaction structure of host-associated microbial communities and the effects of the host on the microbiota and its interactions can be disentangled. Research in the model system Hydra can unify disciplines from molecular genetics to ecology, opening up the opportunity to discover fundamental rules that govern microbiome community stability.
Introduction
Microbes sustain life on this planet as they perform not only important ecosystem functions but also inhabit all organisms. The entirety of a host with its associated microbial community, including viruses and cellular microbes is called the “metaorganism” or “holobiont” (Bosch and McFall-Ngai, 2011; Bosch and Miller, 2016). The existence of such a unifying term indicates the significance of the microbial community for understanding the biology of any host. These host-associated microbial communities (microbiomes) live on host surfaces, are associated with different tissues, and can reside inter- and intracellularly (Huttenhower et al., 2012; Kostic et al., 2013). Host-associated microbial communities are dynamic, changing throughout the hosts’ life, and are not passive players but actively engage in host development, metabolism, immunity, and health as found in established model systems, like corals, worms, insects, mice, and Hydra (Ley et al., 2008; Fraune and Bosch, 2010; Fukuda et al., 2011; Naik et al., 2012; Lee and Brey, 2013; Sommer and Bäckhed, 2013; McFall-Ngai, 2014; Thompson et al., 2015). How microbes increase the host’s stress tolerance and modulate its niche breath are active fields of research (Mueller and Sachs, 2015). Despite the evident importance of the microbiome in affecting host fitness (in a Darwinian sense), insights into the underlying mechanisms are still lacking.
Transitioning from Descriptive to Predictive Microbiome Research
Recent technological advances (e.g., high-throughput sequencing, proteomics, metabolomics) and expanded efforts through a large number of human and environmental microbiome initiatives (summarized in Stulberg et al., 2016) have led to great progress in characterizing the composition of host-, habitat-, or ecosystem-associated microbial communities (Huttenhower et al., 2012; Gilbert et al., 2014; Hacquard et al., 2015; Sunagawa et al., 2015). A recent assessment of US microbiome research, for example, highlights the importance of microbiome studies for tackling current world problems, such as food production, human, and ecosystem health (Stulberg et al., 2016). Yet for achieving this, microbiome research needs to shift from a descriptive to a more predictive science (Alivisatos et al., 2015), where the ecology of these highly diverse communities is addressed. Central for the ability to predict and manage the function of host-associated microbial communities is the knowledge about the factors determining their dynamics and stability. The concept of a core microbiome (taxa or functional core) has been very helpful in addressing the stability of this core and how it changes with age, diet, geographic location, time, or other factors. Recent findings from human microbiome research suggest that a core microbiome can be defined at the functional rather than the taxonomic level (Lloyd-Price et al., 2016). Yet, what constitutes a core still remains elusive and depends on the question of interest. In Hydra, microbial communities of wild caught and domesticated animals have been found to be surprisingly similar and to share a core microbiota at the taxonomic level (Fraune and Bosch, 2007). It is likely that the microbiome (like microbial communities associated with abiotic environments) is affected by various extrinsic and intrinsic factors, e.g., temperature, pH, resource availability, microbe–microbe interactions, but also by interactions with the host. While a number of studies exist on the interactions between host and the microbiome, comparatively little is known on the interactions between microbes within the microbiome (including the virome) and on how these impact the metaorganism.
Within-microbiome interactions can be driven by diverse features such as metabolism, social traits (production of public goods), or environmental factors, like spatial organization (Kim et al., 2008; Nadell et al., 2010; Mitri and Foster, 2013; Großkopf and Soyer, 2014). Six different interaction patterns between members of different species can be distinguished (Lidicker, 1979) (see Figure 1 for potential interactions within the metaorganism exemplified in Hydra).
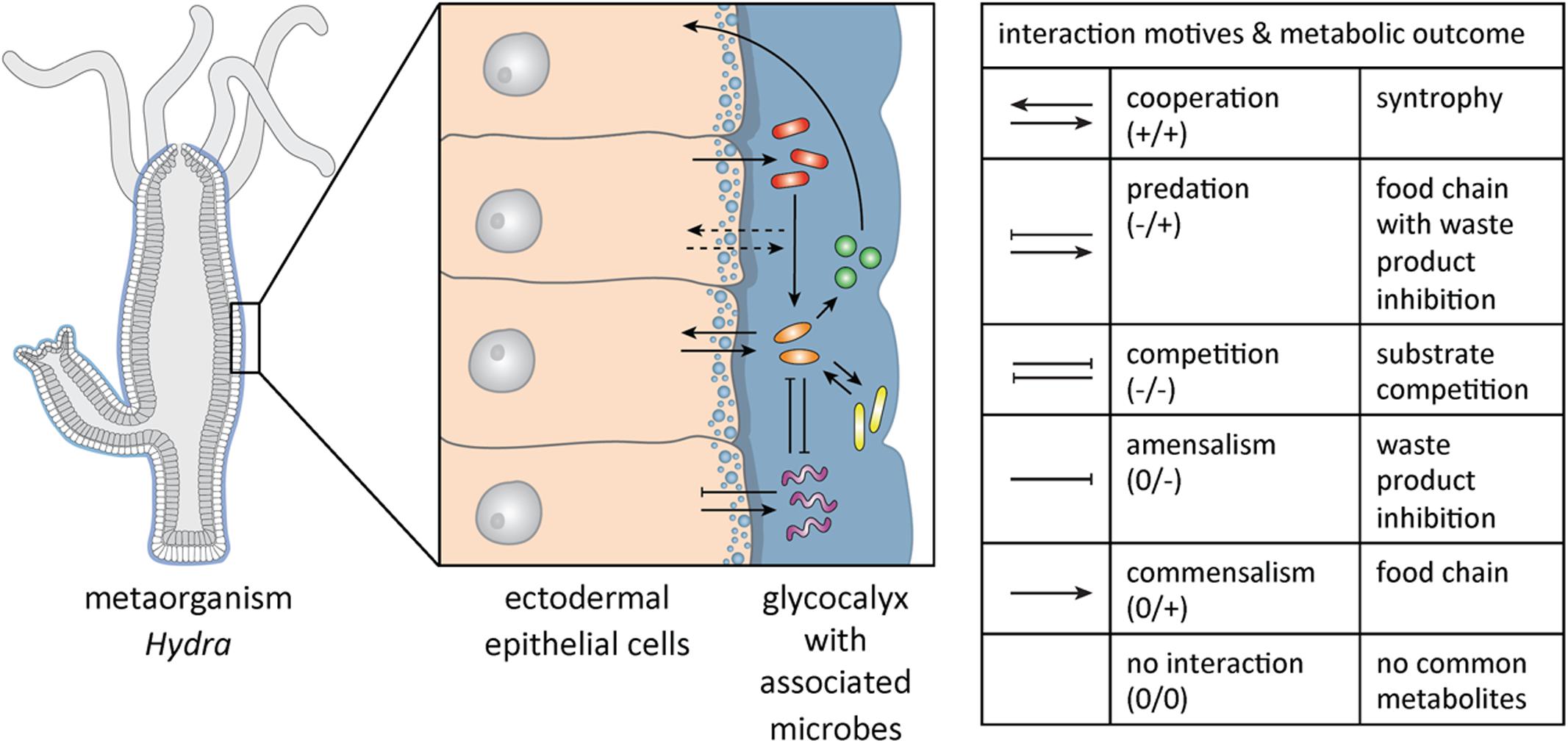
FIGURE 1. Complex microbial community interactions in Hydra. Schematic drawing of Hydra, with a magnified section of the ectodermal epithelial cells that are covered with a glycocalyx and the associated microbial community. In this complex system microbes may interact with each other in multiple ways as depicted by arrows. For the species involved interactions can have a positive (+), a negative (-) or no impact (0). A summary of the possible ecological interactions and their meaning in metabolic terms are summarized in the table (modified from West et al., 2007; Faust and Raes, 2012; Großkopf and Soyer, 2014). It is likely that interactions between specific microbes and the host can be modulated by metabolic or physical signals but that the host also directly affects the interaction between microbial species (dashed arrows) via epithelial selection.
Microbe–Microbe Interactions: Challenges and Future Research
For disentangling the interactions in microbial communities, it is essential to understand the importance of the two key motifs, cooperation and competition. This has been well studied in environmental communities, e.g., by Foster and Bell (2012), where species from two aquatic environments were isolated. By analyzing pair-wise interactions between the culturable microbes it was observed that the majority of interactions were competitive (Foster and Bell, 2012). This pattern is confirmed by results from other studies, which suggest that at least 86% of interactions between strains or species are competitive (reviewed in Mitri and Foster, 2013). This is in contrast to what had been assumed for host-associated microbial communities until very recently. Cooperative interactions among community members were predicted to be the major driving force for a productive and stable microbiome, e.g., in the human gut (Bäckhed et al., 2005; Van den Abbeele et al., 2011). This view was challenged by a seminal study that integrated ecological network analysis and recent data from mammalian microbiomes (Coyte et al., 2015). Whereas a cooperative microbial network was indeed found to be very productive in the short term, competition between the different microbes was identified as the driver of long-term microbiome stability, and so as a benefit for the host. The counter-intuitive result that cooperation between microbial species is destabilizing is based on positive feedback loops that will lead to runaway effects (Coyte et al., 2015). Unconstrained cooperation is predicted to lead to an ever-increasing abundance of the cooperating groups of species, which in turn can result in the collapse of competing populations and eventually in the destabilization of the whole community. Competitive interactions between microbes are in contrast thought to help stabilize the community (McNally and Brown, 2016).
Another factor facilitating the stability of its microbiome is the host itself (Coyte et al., 2015). Several mechanisms have been identified by which a host may be able to suppress the positive feedback between cooperating species and weaken their interaction: (i) regulation through the immune response dependent on the density of the different microbial species, (ii) spatial segregation reducing between-species contact, and (iii) provision of carbon sources via epithelial feeding minimizing cross-feeding between microbes. This study also implies that for understanding and manipulating the host microbiome, close attention needs to be paid to the strength and nature of the ecological interactions between the different microbial species (Coyte et al., 2015), for which experimental data is still scarce.
Although impressive advances have been made with the help of high-throughput sequencing techniques in describing the (highly) diverse species compositions of host-associated communities [such as the ‘Human Microbiome Project’ (Huttenhower et al., 2012)], little data is currently available on ecological interactions within the microbiome (Coyte et al., 2015). One central aspect for approaching a predictive understanding of microbial community function and dynamics will be the integration of theory and experiments (Widder et al., 2016). For generating data that can feed into mathematical models, ‘model’ microbial communities need to be identified that can be manipulated and where theoretical predictions can be tested.
Hydra and Its Microbiome as an Experimental Model
We here propose to extend the utilization of the metaorganism Hydra beyond the fields of developmental biology (Bosch, 2007), stem cell research (Bosch, 2009; Bosch et al., 2010), immunity (Bosch, 2013, 2014), aging (Nebel and Bosch, 2012; Boehm et al., 2013), and animal–microbe interactions (Bosch, 2013, 2014) as a model organism for studying interactions within the animal-associated microbiota and on how that affects the host and vice versa (Figure 1).
Hydra is a cnidarian, which in contrast to other model systems for host-associated microbiota, is phylogenetically basal. Hydra’s phylogenetic position thus provides the benefit of a very simple body plan with a limited number of cells and a basal immune and nervous system. Its tube-like body is akin to the vertebrate intestine (Bosch, 2012) and changes in Hydra’s epithelial homeostasis lead to significant changes in the microbial community (Fraune et al., 2009). Hydra possesses a species-specific and core microbiome of low complexity (Fraune and Bosch, 2007; Bosch, 2013), from which the most dominant microbes can individually be cultured under laboratory conditions (Bosch, 2013; Fraune et al., 2014). The observation of distinct and reproducible colonization patterns of Hydra during its developmental life cycle suggests that host factors are involved in shaping the microbial composition (Franzenburg et al., 2013a). Antimicrobial peptides have been identified as prominent effector molecules of the innate immune system that drive epithelial selection (Augustin et al., 2009; Bosch et al., 2009; Franzenburg et al., 2013b). Yet it is unlikely that the host has the ability to control each microbial phylotype individually.
Another key tool for studying interactions between the host and its microbiota is the creation of germ-free or gnotobiotic animals, which has been achieved in the Hydra system (Franzenburg et al., 2012). This in combination with its transparency, its fast life cycle, the ease of its cultivation and clonal propagation, and the rich pool of knowledge on features important for characterizing, understanding, but also manipulating Hydra as a host (for details see Bosch, 2014), makes it a perfect system for ‘deconstructing’ a metaorganism and its interactions. Hydra as a basal eumetazoan thus not only allows us to gain insight into the early evolution of host–microbe interactions but also into the ecological interactions within low complexity microbiomes. Dissections of the ecological interactions at play are key for understanding microbiome dynamics, which is the prerequisite for the ability to reconstructing and restoring a ‘healthy microbiome’.
For disentangling microbial interactions within the Hydra microbiome, we propose an integrated approach based on constructing synthetic communities of various complexities in vivo and in vitro that can be compared to the in situ community and to single genotypes (Figure 2). Contrasting identical microbial communities through in vivo and in vitro experiments will offer valuable clues to the extent of host effects on microbe–microbe interactions and ultimately their fitness, and vice versa of microbial effects on the fitness of the host. Classical co-culture experiments (Mitri and Foster, 2013) can easily be performed in the Hydra system. Here the community is deconstructed to its individual components and fitness of each microbial species within the synthetic communities of various compositions compared to its fitness when grown singly. Negative interactions [such as competition (net negative effect)] result in reduced growth of the co-culture compared to the sum of the species (mono-culture) yields. If additive growth equals the sum of mono-culture growth, the net effect is neutral, so species do not interact. If the net effect is positive, i.e., the combined growth is greater than the additive growth, interactions are cooperative. This approach has been used for species assemblages with a manageable number of species (e.g., Fiegna et al., 2015). With the ability to culture most of the microbial phylotypes in Hydra in vitro (Bosch, 2013), the interaction between individual members and the role of individual microbes within the community can be disentangled.
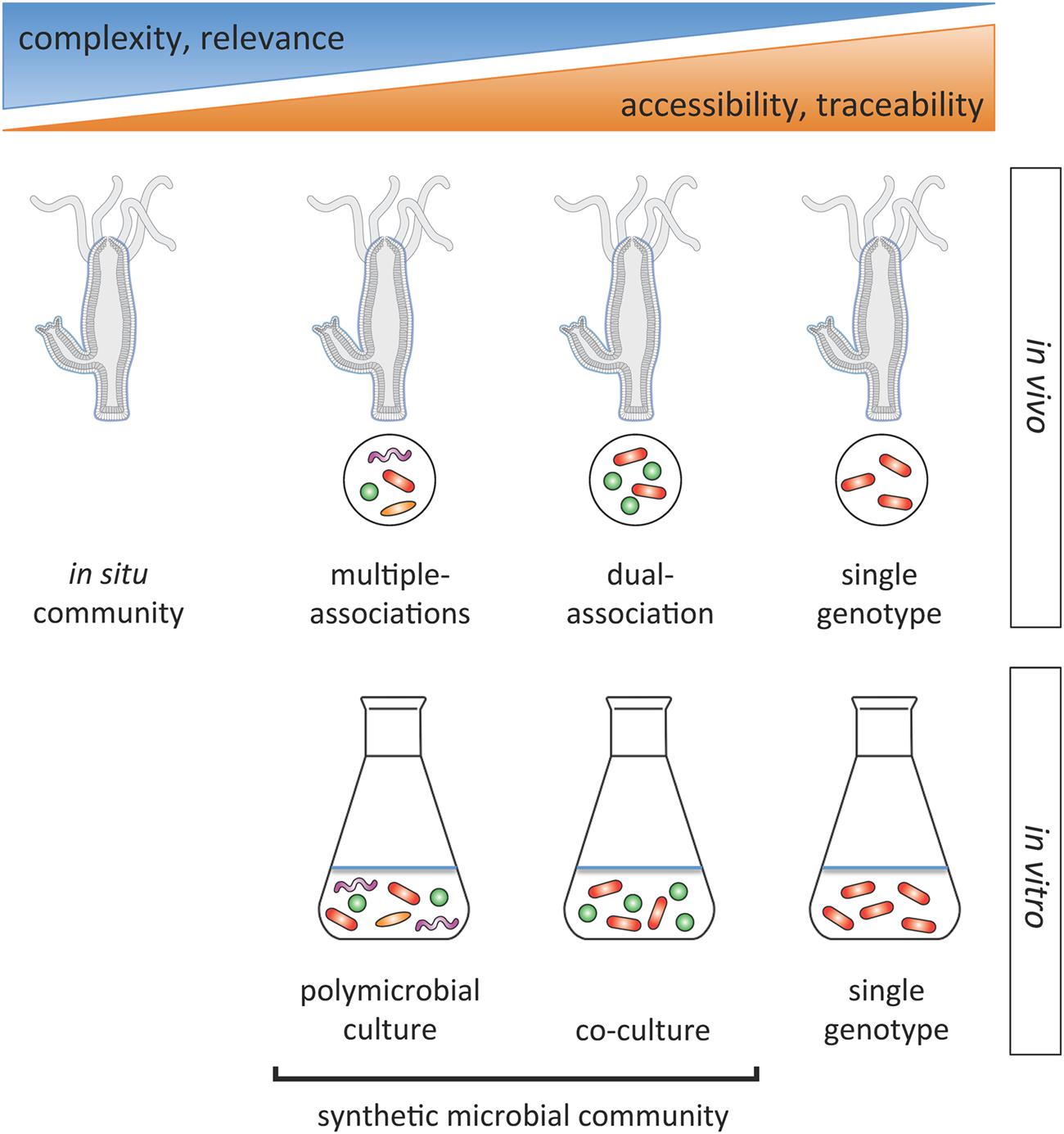
FIGURE 2. Hydra’s experimental potential for studying interactions within the microbiome. Overview of the spectrum of microbial community study systems for unraveling interactions in the relatively simple Hydra microbiome. The integration of in vivo and in vitro experiments offers the opportunity to capture clues on how the host might affect microbe–microbe interactions and vice versa. The opportunity to study inter- and intra-specific interactions at different resolutions will help to unravel general principles of microbial interactions in microbiomes.
Co-culture experiments will provide information on the interactions between the different microbes but also on the interactions with the host (Figure 1), which can be implemented into simple theoretical models that can in turn be extended to providing predictions for more complex microbial communities, such as the mouse or human microbiome. The value of such an approach in gaining new insights into the function of the metaorganism has been showcased by a recent study, where Hydra’s microbiome was found to provide protection from infection with the fungus Fusarium sp., which was only able to infect germ-free animals or the ones with a reduced microbial community (Fraune et al., 2014). Data from these experiments fed into theoretical models, which confirmed the findings using a game theory approach, but also indicated that more experiments are needed, as the findings could not fully be explained by pairwise interactions between the microbial species (Li et al., 2015). Such integration of mathematical predictive models with experimental data is essential for advancing the understanding of the function and dynamics of microbial communities (Widder et al., 2016).
For gaining information on the mechanistic underpinning of specific interactions, co-culture experiments can be complemented with gene knockout experiments (Bernstein et al., 2012; Nakashima and Miyazaki, 2014; Khare and Tavazoie, 2015) in which a target gene is deleted in one of the strains and the nature of the interaction in the co-culture compared to the one with the gene still present. Rakoff-Nahoum et al. (2016) used such an in vitro approach in combination with gnotobiotic mouse experiments to test for the evolution of cooperation within the mammalian gut microbiota. Further, there is evidence that not only genetic changes might be necessary for the establishment and maintenance of interspecies interactions but also that changes of the transcriptome can solely be sufficient (reviewed in Tan et al., 2015). The opportunity of analyzing the mechanistic (genetic) underpinning of the interactions among microbes is possible in the Hydra metaorganism but can moreover be extended to the host as not only the bacteria can be genetically manipulated, but also transgenic Hydra polys can be generated by embryo microinjection (Wittlieb et al., 2006). Another aspect that is advantageous in this system is the ability to create a ‘static’ host that doesn’t coevolve with the microbiome as Hydra can reproduce asexually via budding, and so create identical copies of itself. The microbiome in contrast can artificially be manipulated and selected upon, e.g., with the goal of improving host performance. This host-mediated microbiome engineering (recently reviewed in Mueller and Sachs, 2015) is discussed as one of the new research frontiers in medicine and agriculture. Microbiome engineering also allows the studying of the emergence of new interactions between community members and how this affects the productivity and long-term stability of the microbiome and also on how it impacts on host fitness (Mueller and Sachs, 2015). This is especially of interest in times of rapid environmental change, where shifted interactions within the microbiome can function as a buffer against environmental effects on the host.
Using host-mediated microbiome engineering, another layer of complexity can be added by including the virome. A recent study revealed that viral communities in Hydra are species-specific (Grasis et al., 2014) but their role in establishing and maintaining the microbiome or affecting species interactions has to be still resolved (Bosch et al., 2015; Bosch and Miller, 2016).
Decoding the connection between metabolites, microbes, and the host is yet another exciting frontier in metaorganism research (Dorrestein et al., 2014). This is highly relevant in the light of recent evidence that microbially produced metabolites influence different organ systems, such as microbe-brain connections, bone metabolism, or immune functions (Cryan and Dinan, 2012; Dorrestein et al., 2014; Charles et al., 2015; Eisthen and Theis, 2016). Multi-omics approaches in which the metabolome, the microbiome, and the host immune system are assessed simultaneously are feasible in the Hydra model and will help to decipher this new connection. The metabolite exchange between the different microbial players in an interaction can be addressed in an experimental set up, where a conditioned- or spent-medium approach allows to control metabolite production and consumption (Ponomarova and Patil, 2015). The method is based on a cell-free culture filtrate of a donor species, which is added to a recipient microbial culture to assess the activity of the secretome (Vetsigian et al., 2011; Rakoff-Nahoum et al., 2014). In addition to classical experiments in liquid culture media, other co-culture systems and technologies are currently used, such as microfluidics, petri dishes, solid support systems, bioreactors, and transwell systems (reviewed in detail in Goers et al., 2014).
An alternative approach for inferring cooperative and competitive relationships between the different community members (but not the underlying mechanisms) is based on the availability of high-throughput sequence data, where co-occurrence data and correlation patterns are analyzed (Faust and Raes, 2012). A positive relationship can be assumed, when two species co-occur or show a similar abundance pattern over multiple samples; a negative one is predicted, when they show mutual exclusion (Faust and Raes, 2012). With this information co-occurrence interaction networks can be constructed, enabling the identification of keystone species that stabilize a community, or predicting the (in)stability of communities to environmental change (Faust et al., 2012; Berry and Widder, 2014; Widder et al., 2014).
Another set of models based on whole-genome sequence data (or at least drafts) focuses on metabolic dependencies in microbial communities (Klitgord and Segrè, 2010; Mahadevan and Henson, 2012). Based on the success of systems biology in developing in silico predictive capabilities for individual species, community systems biology (CoSy) attempts to determine and predict the interactions among multiple species (Zengler and Palsson, 2012; Tan et al., 2015). The role of community systems biology in helping to unravel modes of interactions in complex communities has recently been highlighted by Zengler and Palsson (2012), and specifically for the human microbiome by Manor et al. (2014). Genome-scale metabolic modeling has been instrumental for the reconstruction of the genotype to phenotype relationship for single organisms (Borenstein et al., 2008; Zengler and Palsson, 2012). The advancement of these frameworks and techniques for multispecies metabolic models is a very promising route for the prediction of complex relationships in multispecies systems (such as the human microbiome) in the near future (Borenstein, 2012; Manor et al., 2014). Such an approach can be applied to the Hydra system due to its comparatively simple microbiome. Hypotheses can be generated based on genomic data for subsequent tests in the laboratory as most of the bacterial phylotypes identified can be cultured in vitro.
The ease of working with the Hydra system in the laboratory allows that experiments can be performed under well-controlled conditions, that can easily be replicated, perturbed, and sampled at different time scales. Hydra shares many ancestral genes with humans that have been lost in Drosophila and Caenorhabditis (Chapman et al., 2010; Hemmrich et al., 2012), so insights into host–microbe–microbe relationships might unravel general principles that are also relevant to humans and their microbiota. The Hydra system provides an excellent bridge between the simplicity of synthetic communities and the mouse model. As important features of the host, e.g., the host immune response, are difficult to recapitulate in vitro, in vivo experiments in combination with an in silico approach will close the knowledge gap from microbiome composition to ecological interactions within these communities.
Author Contributions
PD and TB jointly contributed to conception and writing of the manuscript. Both have approved it for publication.
Funding
The work related to this perspective received funding from the European Union’s Framework Programme for Research and Innovation Horizon 2020 (2014–2020) under the Marie Skłodowska-Curie Grant Agreement No. 655914 and from the DAAD (Deutscher Akademischer Austausch Dienst). Work in the Bosch laboratory is supported in part by grants from the Deutsche Forschungsgemeinschaft (DFG), the Collaborative Research Center (CRC) 1182 (“Origin and Function of Metaorganisms”) and grants from the DFG Cluster of Excellence Programme “Inflammation at Interfaces”.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Katrin Hammerschmidt and the three referees for helpful comments on an earlier version of the manuscript and Katja Schröder for the graphical design of the Hydra polyp. TB gratefully appreciates support from the Canadian Institute for Advanced Research (CIFAR).
References
Alivisatos, A. P., Blaser, M. J., Brodie, E. L., Chun, M., Dangl, J. L., Donohue, T. J., et al. (2015). A unified initiative to harness earth’s microbiomes. Science 350, 507–508. doi: 10.1126/science.aac8480
Augustin, R., Anton-Erxleben, F., Jungnickel, S., Hemmrich, G., Spudy, B., Podschun, R., et al. (2009). Activity of the novel peptide arminin against multiresistant human pathogens shows the considerable potential of phylogenetically ancient organisms as drug sources. Antimicrob. Agents Chemother. 53, 5245–5250. doi: 10.1128/AAC.00826-09
Bäckhed, F., Ley, R. E., Sonnenburg, J. L., Peterson, D. A., and Gordon, J. I. (2005). Host-bacterial mutualism in the human intestine. Science 307, 1915–1920. doi: 10.1126/science.1104816
Bernstein, H. C., Paulson, S. D., and Carlson, R. P. (2012). Synthetic Escherichia coli consortia engineered for syntrophy demonstrate enhanced biomass productivity. J. Biotechnol. 157, 159–166. doi: 10.1016/j.jbiotec.2011.10.001
Berry, D., and Widder, S. (2014). Deciphering microbial interactions and detecting keystone species with co-occurrence networks. Front. Microbiol. 5:219. doi: 10.3389/fmicb.2014.00219
Boehm, A.-M., Rosenstiel, P., and Bosch, T. C. G. (2013). Stem cells and aging from a quasi-immortal point of view. Bioessays 35, 994–1003. doi: 10.1002/bies.201300075
Borenstein, E. (2012). Computational systems biology and in silico modeling of the human microbiome. Brief. Bioinform. 13, 769–780. doi: 10.1093/bib/bbs022
Borenstein, E., Kupiec, M., Feldman, M. W., and Ruppin, E. (2008). Large-scale reconstruction and phylogenetic analysis of metabolic environments. Proc. Natl. Acad. Sci. U.S.A. 105, 14482–14487. doi: 10.1073/pnas.0806162105
Bosch, T. C. G. (2007). Why polyps regenerate and we don’t: towards a cellular and molecular framework for Hydra regeneration. Dev. Biol. 303, 421–433. doi: 10.1016/j.ydbio.2006.12.012
Bosch, T. C. G. (2009). Hydra and the evolution of stem cells. Bioessays 31, 478–486. doi: 10.1002/bies.200800183
Bosch, T. C. G. (2012). Understanding complex host-microbe interactions in Hydra. Gut. Microbes 3, 345–351. doi: 10.4161/gmic.20660
Bosch, T. C. G. (2013). Cnidarian-microbe interactions and the origin of innate immunity in metazoans. Annu. Rev. Microbiol. 67, 499–518. doi: 10.1146/annurev-micro-092412-155626
Bosch, T. C. G. (2014). Rethinking the role of immunity: lessons from Hydra. Trends Immunol. 35, 495–502. doi: 10.1016/j.it.2014.07.008
Bosch, T. C. G., Anton-Erxleben, F., Hemmrich, G., and Khalturin, K. (2010). The Hydra polyp: nothing but an active stem cell community. Dev. Growth Differ. 52, 15–25. doi: 10.1111/j.1440-169X.2009.01143.x
Bosch, T. C. G., Augustin, R., Anton-Erxleben, F., Fraune, S., Hemmrich, G., Zill, H., et al. (2009). Uncovering the evolutionary history of innate immunity: the simple metazoan Hydra uses epithelial cells for host defence. Dev. Comp. Immunol. 33, 559–569. doi: 10.1016/j.dci.2008.10.004
Bosch, T. C. G., Grasis, J. A., and Lachnit, T. (2015). Microbial ecology in Hydra: why viruses matter. J. Microbiol. 53, 193–200. doi: 10.1007/s12275-015-4695-2
Bosch, T. C. G., and McFall-Ngai, M. J. (2011). Metaorganisms as the new frontier. Zoology 114, 185–190. doi: 10.1016/j.zool.2011.04.001
Bosch, T. C. G., and Miller, D. J. (2016). The Holobiont Imperative. Vienna: Springer, doi: 10.1007/978-3-7091-1896-2
Chapman, J. A., Kirkness, E. F., Simakov, O., Hampson, S. E., Mitros, T., Weinmaier, T., et al. (2010). The dynamic genome of Hydra. Nature 464, 592–596. doi: 10.1038/nature08830
Charles, J. F., Ermann, J., and Aliprantis, A. O. (2015). The intestinal microbiome and skeletal fitness: connecting bugs and bones. Clin. Immunol. 159, 163–169. doi: 10.1016/j.clim.2015.03.019
Coyte, K. Z., Schluter, J., and Foster, K. R. (2015). The ecology of the microbiome: networks, competition, and stability. Science 350, 663–666. doi: 10.1126/science.aad2602
Cryan, J. F., and Dinan, T. G. (2012). Mind-altering microorganisms: the impact of the gut microbiota on brain and behaviour. Nat. Rev. Neurosci. 13, 701–712. doi: 10.1038/nrn3346
Dorrestein, P. C., Mazmanian, S. K., and Knight, R. (2014). Finding the missing links among metabolites, microbes, and the host. Immunity 40, 824–832. doi: 10.1016/j.immuni.2014.05.015
Eisthen, H. L., and Theis, K. R. (2016). Animal-microbe interactions and the evolution of nervous systems. Philos. Trans. R. Soc. Lond. B Biol. Sci. 371, 20150052. doi: 10.1098/rstb.2015.0052
Faust, K., and Raes, J. (2012). Microbial interactions: from networks to models. Nat. Rev. Microbiol. 10, 538–550. doi: 10.1038/nrmicro2832
Faust, K., Sathirapongsasuti, J. F., Izard, J., Segata, N., Gevers, D., Raes, J., et al. (2012). Microbial co-occurrence relationships in the human microbiome. PLoS Comput. Biol. 8:e1002606. doi: 10.1371/journal.pcbi.1002606
Fiegna, F., Moreno-Letelier, A., Bell, T., and Barraclough, T. G. (2015). Evolution of species interactions determines microbial community productivity in new environments. ISME J. 9, 1235–1245. doi: 10.1038/ismej.2014.215
Foster, K. R., and Bell, T. (2012). Competition, not cooperation, dominates interactions among culturable microbial species. Curr. Biol. 22, 1845–1850. doi: 10.1016/j.cub.2012.08.005
Franzenburg, S., Fraune, S., Altrock, P. M., Künzel, S., Baines, J. F., Traulsen, A., et al. (2013a). Bacterial colonization of Hydra hatchlings follows a robust temporal pattern. ISME J. 7, 781–790. doi: 10.1038/ismej.2012.156
Franzenburg, S., Walter, J., Künzel, S., Wang, J., Baines, J. F., Bosch, T. C. G., et al. (2013b). Distinct antimicrobial peptide expression determines host species-specific bacterial associations. Proc. Natl. Acad. Sci. U.S.A. 110, E3730–E3738. doi: 10.1073/pnas.1304960110
Franzenburg, S., Fraune, S., Künzel, S., Baines, J. F., Domazet-Loso, T., and Bosch, T. C. G. (2012). MyD88-deficient Hydra reveal an ancient function of TLR signaling in sensing bacterial colonizers. Proc. Natl. Acad. Sci. U.S.A. 109, 19374–19379. doi: 10.1073/pnas.1213110109
Fraune, S., Abe, Y., and Bosch, T. C. G. (2009). Disturbing epithelial homeostasis in the metazoan Hydra leads to drastic changes in associated microbiota. Environ. Microbiol. 11, 2361–2369. doi: 10.1111/j.1462-2920.2009.01963.x
Fraune, S., Anton-Erxleben, F., Augustin, R., Franzenburg, S., Knop, M., Schröder, K., et al. (2014). Bacteria-bacteria interactions within the microbiota of the ancestral metazoan Hydra contribute to fungal resistance. ISME J. 9, 1543–1556. doi: 10.1038/ismej.2014.239
Fraune, S., and Bosch, T. C. G. (2007). Long-term maintenance of species-specific bacterial microbiota in the basal metazoan Hydra. Proc. Natl. Acad. Sci. USA 104, 13146–13151. doi: 10.1073/pnas.0703375104
Fraune, S., and Bosch, T. C. G. (2010). Why bacteria matter in animal development and evolution. Bioessays 32, 571–580. doi: 10.1002/bies.200900192
Fukuda, S., Toh, H., Hase, K., Oshima, K., Nakanishi, Y., Yoshimura, K., et al. (2011). Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nature 469, 543–547. doi: 10.1038/nature09646
Gilbert, J. A., Jansson, J. K., and Knight, R. (2014). The earth microbiome project: successes and aspirations. BMC Biol. 12:69. doi: 10.1186/s12915-014-0069-1
Goers, L., Freemont, P., and Polizzi, K. M. (2014). Co-culture systems and technologies: taking synthetic biology to the next level. J. R. Soc. Interface 11, 20140065–20140065. doi: 10.1098/rsif.2014.0065
Grasis, J. A., Lachnit, T., Anton-Erxleben, F., Lim, Y. W., Schmieder, R., Fraune, S., et al. (2014). Species-specific viromes in the ancestral holobiont Hydra. PLoS ONE 9:e109952. doi: 10.1371/journal.pone.0109952
Großkopf, T., and Soyer, O. S. (2014). Synthetic microbial communities. Curr. Opin. Microbiol. 18, 72–77. doi: 10.1016/j.mib.2014.02.002
Hacquard, S., Garrido-Oter, R., González, A., Spaepen, S., Ackermann, G., Lebeis, S., et al. (2015). Microbiota and host nutrition across plant and animal kingdoms. Cell Host Microbe 17, 603–616. doi: 10.1016/j.chom.2015.04.009
Hemmrich, G., Khalturin, K., Boehm, A.-M., Puchert, M., Anton-Erxleben, F., Wittlieb, J., et al. (2012). Molecular signatures of the three stem cell lineages in Hydra and the emergence of stem cell function at the base of multicellularity. Mol. Biol. Evol. 29, 3267–3280. doi: 10.1093/molbev/mss134
Huttenhower, C., Gevers, D., Knight, R., Abubucker, S., Badger, J. H., Chinwalla, A. T., et al. (2012). Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214. doi: 10.1038/nature11234
Khare, A., and Tavazoie, S. (2015). Multifactorial competition and resistance in a two-species bacterial system. PLoS Genet. 11:e1005715. doi: 10.1371/journal.pgen.1005715
Kim, H. J., Boedicker, J. Q., Choi, J. W., and Ismagilov, R. F. (2008). Defined spatial structure stabilizes a synthetic multispecies bacterial community. Proc. Natl. Acad. Sci. U.S.A. 105, 18188–18193. doi: 10.1073/pnas.0807935105
Klitgord, N., and Segrè, D. (2010). Environments that induce synthetic microbial ecosystems. PLoS Comput. Biol. 6:e1001002. doi: 10.1371/journal.pcbi.1001002
Kostic, A. D., Howitt, M. R., and Garrett, W. S. (2013). Exploring host-microbiota interactions in animal models and humans. Gene. Dev. 27, 701–718. doi: 10.1101/gad.212522.112
Lee, W.-J., and Brey, P. T. (2013). How microbiomes influence metazoan development: insights from history and Drosophila modeling of gut-microbe interactions. Annu. Rev. Cell Dev. Biol 29, 571–592. doi: 10.1146/annurev-cellbio-101512–122333
Ley, R. E., Lozupone, C. A., Hamady, M., Knight, R., and Gordon, J. I. (2008). Worlds within worlds: evolution of the vertebrate gut microbiota. Nat. Rev. Microbiol. 6, 776–788. doi: 10.1038/nrmicro1978
Li, X.-Y., Pietschke, C., Fraune, S., Altrock, P. M., Bosch, T. C. G., and Traulsen, A. (2015). Which games are growing bacterial populations playing? J. R. Soc. Interface 12, 20150122. doi: 10.1098/rsif.2015.0121
Lidicker, W. Z. (1979). A clarification of interactions in ecological systems. BioScience 29, 475–477. doi: 10.2307/1307540
Lloyd-Price, J., Abu-Ali, G., and Huttenhower, C. (2016). The healthy human microbiome. Genome Med. 8, 51. doi: 10.1186/s13073-016-0307-y
Mahadevan, R., and Henson, M. A. (2012). Genome-based modeling and design of metabolic interactions in microbial communities. Comput. Struct. Biotechnol. J. 3, e201210008. doi: 10.5936/csbj.201210008
Manor, O., Levy, R., and Borenstein, E. (2014). Mapping the inner workings of the microbiome: genomic- and metagenomic-based study of metabolism and metabolic interactions in the human microbiome. Cell Metab. 20, 742–752. doi: 10.1016/j.cmet.2014.07.021
McFall-Ngai, M. (2014). Divining the essence of symbiosis: insights from the squid-vibrio model. PLoS Biol. 12:e1001783. doi: 10.1371/journal.pbio.1001783
McNally, L., and Brown, S. P. (2016). Microbiome: ecology of stable gut communities. Nat. Microbiol. 1, 1–2. doi: 10.1038/nmicrobiol.2015.16
Mitri, S., and Foster, K. R. (2013). The genotypic view of social interactions in microbial communities. Annu. Rev. Genet. 47, 247–273. doi: 10.1146/annurev-genet-111212-133307
Mueller, U. G., and Sachs, J. L. (2015). Engineering microbiomes to improve plant and animal health. Trends Microbiol. 23, 606–617. doi: 10.1016/j.tim.2015.07.009
Nadell, C. D., Foster, K. R., and Xavier, J. B. (2010). Emergence of spatial structure in cell groups and the evolution of cooperation. PLoS Comput. Biol. 6:e1000716. doi: 10.1371/journal.pcbi.1000716
Naik, S., Bouladoux, N., Wilhelm, C., Molloy, M. J., Salcedo, R., Kastenmuller, W., et al. (2012). Compartmentalized control of skin immunity by resident commensals. Science 337, 1115–1119. doi: 10.1126/science.1225152
Nakashima, N., and Miyazaki, K. (2014). Bacterial cellular engineering by genome editing and gene silencing. Int. J. Mol. Sci. 15, 2773–2793. doi: 10.3390/ijms15022773
Nebel, A., and Bosch, T. C. G. (2012). Evolution of human longevity: lessons from Hydra. Aging 4, 1–2. doi: 10.18632/aging.100510
Ponomarova, O., and Patil, K. R. (2015). Metabolic interactions in microbial communities: untangling the Gordian knot. Curr. Opin. Microbiol. 27, 37–44. doi: 10.1016/j.mib.2015.06.014
Rakoff-Nahoum, S., Coyne, M. J., and Comstock, L. E. (2014). An ecological network of polysaccharide utilization among human intestinal symbionts. Curr. Biol. 24, 40–49. doi: 10.1016/j.cub.2013.10.077
Rakoff-Nahoum, S., Foster, K. R., and Comstock, L. E. (2016). The evolution of cooperation within the gut microbiota. Nature 533, 255–259. doi: 10.1038/nature17626
Sommer, F., and Bäckhed, F. (2013). The gut microbiota - masters of host development and physiology. Nat. Rev. Micorbiol. 11, 227–238. doi: 10.1038/nrmicro2974
Stulberg, E., Fravel, D., Proctor, L. M., Murray, D. M., LoTempio, J., Chrisey, L., et al. (2016). An assessment of US microbiome research. Nat. Microbiol. 1:15015. doi: 10.1038/nmicrobiol.2015.15
Sunagawa, S., Coelho, L. P., Chaffron, S., Kultima, J. R., Labadie, K., Salazar, G., et al. (2015). Ocean plankton. Structure and function of the global ocean microbiome. Science 348, 1261359–1261359. doi: 10.1126/science.1261359
Tan, J., Zuniga, C., and Zengler, K. (2015). Unraveling interactions in microbial communities - from co-cultures to microbiomes. J. Microbiol. 53, 295–305. doi: 10.1007/s12275-015-5060-1
Thompson, J. R., Rivera, H. E., Closek, C. J., and Medina, M. (2015). Microbes in the coral holobiont: partners through evolution, development, and ecological interactions. Front. Cell Infect. Micorbiol. 4:176. doi: 10.3389/fcimb.2014.00176/abstract
Van den Abbeele, P., Van de Wiele, T., Verstraete, W., and Possemiers, S. (2011). The host selects mucosal and luminal associations of coevolved gut microorganisms: a novel concept. FEMS Microbiol. Rev. 35, 681–704. doi: 10.1111/j.1574-6976.2011.00270.x
Vetsigian, K., Jajoo, R., and Kishony, R. (2011). Structure and evolution of Streptomyces interaction networks in soil and in silico. PLoS Biol. 9:e1001184. doi: 10.1371/journal.pbio.1001184
West, S. A., Griffin, A. S., and Gardner, A. (2007). Social semantics: altruism, cooperation, mutualism, strong reciprocity and group selection. J. Evol. Biol. 20, 415–432. doi: 10.1111/j.1420-9101.2006.01258.x
Widder, S., Allen, R. J., Pfeiffer, T., Curtis, T. P., Wiuf, C., Sloan, W. T., et al. (2016). Challenges in microbial ecology: building predictive understanding of community function and dynamics. ISME J. doi: 10.1038/ismej.2016.45 [Epub ahead of print].
Widder, S., Besemer, K., Singer, G. A., Ceola, S., Bertuzzo, E., Quince, C., et al. (2014). Fluvial network organization imprints on microbial co-occurrence networks. Proc. Natl. Acad. Sci. U.S.A. 111, 12799–12804. doi: 10.1073/pnas.1411723111
Wittlieb, J., Khalturin, K., Lohmann, J. U., Anton-Erxleben, F., and Bosch, T. C. G. (2006). Transgenic Hydra allow in vivo tracking of individual stem cells during morphogenesis. Proc. Natl. Acad. Sci. U.S.A. 103, 6208–6211. doi: 10.1073/pnas.0510163103
Keywords: microbiome, microbial interactions, synthetic communities, model system, systems biology, modeling
Citation: Deines P and Bosch TCG (2016) Transitioning from Microbiome Composition to Microbial Community Interactions: The Potential of the Metaorganism Hydra as an Experimental Model. Front. Microbiol. 7:1610. doi: 10.3389/fmicb.2016.01610
Received: 28 June 2016; Accepted: 26 September 2016;
Published: 13 October 2016.
Edited by:
Monica Medina, Pennsylvania State University, USAReviewed by:
Janelle Renee Thompson, Massachusetts Institute of Technology, USAOded Yarden, Hebrew University of Jerusalem, Israel
Cynthia B. Silveira, San Diego State University, USA
Copyright © 2016 Deines and Bosch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter Deines, cGRlaW5lc0B6b29sb2dpZS51bmkta2llbC5kZQ==