 Yong-Heng Huang
Yong-Heng Huang Jin-Shui Lin
Jin-Shui Lin Jin-Cheng Ma
Jin-Cheng Ma Hai-Hong Wang
Hai-Hong Wang- 1Guangdong Provincial Key Laboratory of Protein Function and Regulation in Agricultural Organisms, College of Life Sciences, South China Agricultural University, Guangzhou, China
- 2Shaanxi Engineering and Technological Research Center for Conversation and Utilization of Regional Biological Resources, College of Life Sciences, Yan’an University, Yan’an, China
Pseudomonas aeruginosa is extremely resistant to triclosan. Previous studies have shown that P. aeruginosa encodes a triclosan-resistant enoyl-acyl-carrier protein reductase (ENR), FabV, and that deletion of fabV causes P. aeruginosa to be extremely sensitive to triclosan. In this report, we complemented a P. aeruginosa fabV deletion strain with several triclosan-resistant ENR encoding genes, including Vibrio cholerae fabV, Bacillus subtilis fabL and Enterococcus faecalis fabK. All complemented strains restored triclosan resistance to the level of the wild-type strain, which confirmed that triclosan-resistant ENR allows P. aeruginosa to be extremely resistant to triclosan. Moreover, fabV exhibits pleiotropic effects. Deletion of fabV led P. aeruginosa to show attenuated swarming motility, decreased rhamnolipid, pyoverdine and acyl-homoserine lactones (AHLs) production. Complementation of the fabV mutant with any one ENR encoding gene could restore these features to some extent, in comparison with the wild-type strain. Furthermore, we found that addition of exogenous AHLs could restore the fabV mutant strain to swarm on semisolid plates and to produce more virulence factors than the fabV mutant strain. These findings indicate that deletion of fabV reduced the activity of ENR in P. aeruginosa, decreased fatty acid synthesis, and subsequently depressed the production of AHLs and other virulence factors, which finally may led to a reduction in the pathogenicity of P. aeruginosa. Therefore, fabV should be an ideal target for the control of P. aeruginosa infectivity.
Introduction
Pseudomonas aeruginosa is an aerobic Gram-negative bacterium, which is widespread in the terrestrial environment (Driscoll et al., 2007; Lee and Zhang, 2015). As an important human pathogen, P. aeruginosa is responsible for a myriad of infections of the human body and is also a leading cause of mortality and morbidity in patients with cystic fibrosis (CF; Driscoll et al., 2007; Willcox, 2007; Kerr and Snelling, 2009). These infections are hard to eradicate because P. aeruginosa has developed strong resistance to most conventional antibiotics. The problem is further become complicated by the ability of the pathogen to form a biofilm matrix, which provides bacterial cells with a protected environment and allows them to withstand various stresses including antibiotics (Driscoll et al., 2007; Lee and Zhang, 2015). The breadth of difficult-to-treat P. aeruginosa-related infections makes the development of new anti-pseudomonas drugs a challenging priority. Fatty acid synthesis (FAS) is a vital metabolic pathway central to both mammals and bacteria (Campbell and Cronan, 2001; White et al., 2004; Zhang and Rock, 2008). Therefore, the divergence between mammalian and bacterial FAS pathway makes bacterial FAS an attractive target for the development of new antimicrobial agents (Campbell and Cronan, 2001; Heath et al., 2001; White et al., 2004).
Enoyl-acyl-carrier protein reductase (ENR) is a vital enzyme in the bacterial fatty acid synthetic pathway, which catalyzes the last reduction of trans-2-acyl-ACP (an enoyl-ACP) to the fully saturated acyl-ACP species in the fatty acid elongation cycle (Massengo-Tiasse and Cronan, 2009) (Figure 1). Unlike most enzymes involved in type II fatty acid synthesis, ENRs display extensive sequence and structural diversity among bacteria. Based on the differing sensitivities of bacteria to triclosan, a biocide used in hand soaps and a large variety of other everyday products, four distinct ENR isozymes, FabI (Heath et al., 2001), FabL (Heath et al., 2000), FabK (Heath and Rock, 2000; Marrakchi et al., 2003) and FabV (Massengo-Tiasse and Cronan, 2008), have been identified. FabI has been shown to be the site of action of triclosan (Massengo-Tiasse and Cronan, 2009), while FabL, FabV and FabK are all triclosan-resistant ENRs. The fatty acid biosynthetic pathway of P. aeruginosa has been investigated extensively (Heath and Rock, 1995; Hoang and Schweizer, 1997; Hoang et al., 2002; Zhu et al., 2010; Yuan et al., 2012a,b) (Figure 1). In addition to FabI, P. aeruginosa also encodes a second ENR, FabV (Zhu et al., 2010).
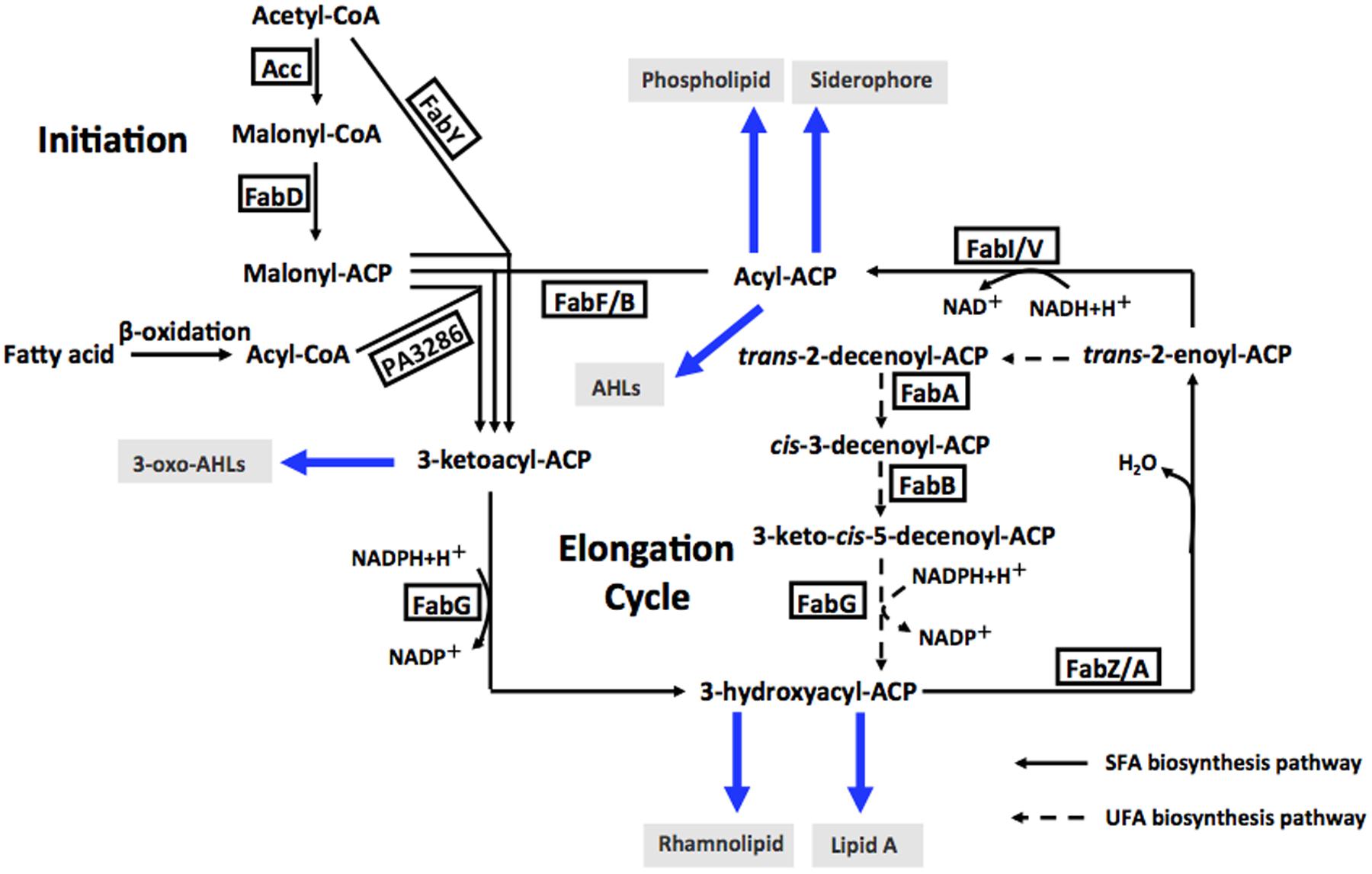
FIGURE 1. Fatty acid biosynthesis in P. aeruginosa, and acyl-ACPs as acyl donors in cellular metabolism and AHL synthesis. Abbreviations: Acc, acetyl-CoA carboxylase; FabD, malonyl-CoA:ACP transacylase; FabY, 3-ketoacyl ACP synthase III; PA3286, a new3-ketoacyl ACP synthase III; FabG, 3-ketoacyl-ACP reductase; FabZ, 3-hydroxyacyl-ACP dehydratase; FabA, 3-hydroxydecanoyl-ACP dehydratase/isomerase; FabF, 3-ketoacyl-ACP synthase II; FabB, 3-ketoacyl-ACP synthase I; FabI and FabV, enoyl-ACP reductase; AHLs, acylhomoserine lactones; SFA, saturated fatty acid; UFA, unsaturated fatty acid.
Pseudomonas aeruginosa is extremely resistant to triclosan: the minimum inhibitory concentration (MIC) of triclosan for the wild-type strain is above 2000 μg/ml (Zhu et al., 2010). But deletion of fabV from the genome causes P. aeruginosa to become extremely sensitive to triclosan (>2,000-fold more sensitive than the wild-type strain). Therefore, it has been suggested that FabV is responsible for the inherent triclosan resistance of P. aeruginosa (Zhu et al., 2010). However, there is no more evidence to support this view.
In P. aeruginosa, FabV also exhibits pleiotropic effects (Bai et al., 2007, 2011; Mou et al., 2011). The fabV (formerly named pfm) gene was first identified to be required for swimming motility, and it was suggested that FabV was involved in energy metabolism, which is critical for the rotation of the flagellum in P. aeruginosa (Bai et al., 2007). Further studies showed that fabV was involved in bacterial protein secretion and bacterial adherence, and mutation of fabV caused P. aeruginosa to produce significantly fewer QS signal molecules and reduced the pathogenicity and virulence in a murine model of acute lung infection. Thus, fabV was suggested to be an important target for the control of P. aeruginosa infectivity (Bai et al., 2011; Mou et al., 2011). However, the mechanism of involvement of FabV in the pathogenicity of P. aeruginosa is still not well studied.
In P. aeruginosa, FAS not only supplies the precursors for phospholipid and lipopolysaccharide synthesis, but also shunt the intermediates to produce siderophores, fatty acid-dependent quorum-sensing signals (PQS and homoserine lactones) and rhamnolipids (Hoang et al., 2002). Thus, we hypothesized that deletion of fabV would reduce the activity of ENR in P. aeruginosa, and subsequently reduce FAS, and then depress the production of QS signals and other virulence factors, and finally would lead to reduced pathogenicity in P. aeruginosa.
In this study, we provided evidence to demonstrate that it is FabV that confers triclosan resistance on P. aeruginosa by complementation of a fabV deletion mutant with Vibrio cholerae fabV, Enterococcus faecalis fabK and Bacillus subtilis fabL. We also confirmed our hypothesis by assaying the fatty acid synthetic rate of the fabV mutant, by measurement of the production of several end products that are related to FAS in the fabV mutant, and by addition of exogenous AHLs to restore the phenotype of the fabV mutant.
Materials and Methods
The supply sources were as follows: fatty acids (FAs), triclosan, rhamnose, N-butyryl-homoserine lactone (C4-HSL), N-3-oxo-dodecanoyl-homoserine lactone (3-oxo-C12-HSL), NADH and antibiotics were from Sigma-Aldrich; Takara Biotechnology Co. provided molecular biology reagents; and Bio-Rad provided the Quick Start Bradford dye reagent. All other reagents were of the highest available quality.
Bacterial Strains, Plasmids, and Growth Media
The strains and plasmids used in this study are listed in Supplementary Table S1. The E. coli K-12 strain DH5α was used in this study for gene cloning. The P. aeruginosa strains used in this study were the wild-type strain PAO1, fabV deletion strain PAO170 and fabI deletion strain PAO272. Luria-Bertani (LB) medium was used as the enriched medium for E. coli and P. aeruginosa growth. Antibiotics were used at the following concentrations (in micrograms per milliliter): sodium ampicillin, 100 (for E. coli); kanamycin sulfate, 30 (for E. coli) or 100 (for P. aeruginosa); gentamicin, 10 (for E. coli) or 100 (for P. aeruginosa); and triclosan, 3 (for P. aeruginosa). Isopropyl-β-D-thiogalactoside (IPTG) was used at a final concentration of 1 mmol/L.
Recombinant DNA Techniques and Construction of Plasmids
Pseudomonas aeruginosa fabI (PafabI) and fabV (PafabV), V. cholerae fabV (VcfabV), E. faecalis fabK (EnfabK) and B. subtilis fabL (BsfabL) were amplified from genomic DNA obtained from wild-type strains of P. aeruginosa, V. cholerae, E. faecalis and B. subtilis, respectively. The primers are listed in Supplementary Table S2. The PCR was performed with Pfu DNA polymerase and the products were inserted into T-vector pMD19, to give plasmids pYH1 (PafabI), pYH2 (PafabV), pYH3 (VcfabV), pYH4 (EnfabK) and pYH5 (BsfabL), respectively. All the genes in the T-vectors were confirmed by sequencing, performed by Shanghai Sangon, Inc. The fab genes in T-vector were digested with NdeI and HindIII, and the appropriate fragments were isolated and ligated into pSRK-Km (Khan et al., 2008) to create plasmids pSRK-PI, pSRK-PV, pSRK-VV, pSRK-EK and pSRK-BL, respectively. All these plasmids were introduced into the P. aeruginosa PAO170 strain via electroporation to give strains: PI170 (PAO170/pSRK-PI), PV170 (PAO170/ pSRK-PV), VV170 (PAO170/pSRK-VV), EK170 (PAO170/pSRK-EK) and BL170 (PAO170/ pSRK-BL), respectively.
Analysis of Fatty Acid Composition of Bacteria
The cellular lipid assay was adapted from that of Stead (1989). Briefly, cultures of the P. aeruginosa strains were grown at 37°C in LB medium overnight. Cells were then harvested from 10 ml aliquots of the cultures and washed with fresh LB medium at room temperature. Cellular lipids were saponified by the addition of 1 ml NaOH in methanol solution (NaOH 45 g; methanol 150 ml; water 150 ml). The samples were placed in a boiling water bath for 30 min. The tubes were vortexed before and once during boiling. FAs were methylated by the addition of 2 ml of 6 mol/L HCl in methanol (325 ml 11.6 mol/L HCl, plus 275 ml methanol). The samples were heated at 80°C for 10 min and immediately cooled to below 20°C. The fatty acid methyl esters were extracted three times with 1.25 ml petroleum ether. The samples were dried under a stream of nitrogen in a fume hood. The esters were analyzed by gas chromatography-mass spectrometry (GC-MS) as described previously (Feng et al., 2015; Mao et al., 2016). The data are presented as percentages of the total FAs and represent the mean ± standard error for three independent determinations.
Enoyl-ACP Reductase Assays
Cell-free extracts of P. aeruginosa were prepared from early stationary phase growing cultures (optical density at 600 nm of 0.8 to 1.0). Cells grown in LB medium at 37°C were harvested by centrifugation and were then suspended in lysis buffer (0.1 mol/L sodium phosphate, pH 7.5, 5 mmol/L β-mercaptoethanol, 1 mmol/L EDTA). The cell lysates were prepared by passing the cell suspensions three times through a French pressure cell. Cell debris was removed by ultracentrifugation for 1 h at 260,000 × g, and the supernatants were dialyzed against lysis buffer for 24 h and saved as cell extracts. The ENR activity of cell-free extracts was determined by using trans-2-decenoyl-ACP as the substrate, which was done by monitoring spectrophotometrically the decrease in the absorbance at 340 nm using an NADH extinction coefficient of 6,220 mol/L-1. The trans-2-decenoyl-ACP was prepared by using a previously published procedure (Zhu et al., 2010). The reaction mixtures for activity assays contained 150 μmol/L NADH, 10 ng of the cell-free extracts, 100 μmol/L trans-2-decenoyl-ACP, and 0.1 mol/L LiCl in 0.1 mol/L sodium phosphate buffer (pH 7.0).
Motility Assays
The swarming, swimming, and twitching motilities of P. aeruginosa were investigated using the following media: (1) swim plate [1% tryptone, 0.5% NaCl, 0.3% agar (Difco Bacto Agar)], (2) swarm plate (0.45% tryptone, 0.13% yeast extract, 0.22% NaCl, 0.5% glucose, 0.5% agar), and (3) twitch plate (1% tryptone, 0.5% yeast extract, 0.5% NaCl, 1% agar). The agar media were air-dried for 5–10 min before use. For the swimming and swarming assays, plates were point-inoculated with bacteria from an overnight culture using a sterile toothpick on the surface, and the plates were incubated at 30°C for 24–48 h. For the twitching motility assay, the cells were stabbed into the bottom of twitching plates using a toothpick and incubated at 37°C for 24 h. The motility was assessed by examining the circle around the inoculation site formed by the growing bacteria.
Rhamnolipid Assay
For the rhamnolipid assay, P. aeruginosa strains were cultured to stationary phase in PPGAS (120 mmol/L Tris-HCl, pH 7.2, 20 mmol/L NH4Cl, 20 mmol/L KCl, 1.6 mmol/L MgSO4, 1% tryptone and 0.5% glucose) at 30°C. The supernatants were first adjusted to pH 2.0 with 1 mol/L HCl, and 1 ml of these supernatant samples was extracted twice with an equal volume of diethyl ether, followed by vacuum drying. The concentration of rhamnolipid was determined by measuring the concentration of rhamnose with the sulfuric acid-anthrone reagent (0.2% anthrone, 85% sulfuric acid) method, using rhamnose sugar as the standard, at 620 nm.
Siderophore Secretion Assay
Chrome azurol S (CAS)-LB agar plates were used for the siderophore secretion assay: 10 ml sterilized 10X CAS solution (1 mmol/L chrome azurol S, 2 mmol/L cetyltrimethylammonium bromide, 500 μmol/L FeCl3⋅6H2O) was mixed with 100 ml LB agar to prepare CAS-LB agar plates. The plates were dried for 1 h at room temperature prior to inoculation, following which 10 μl of overnight P. aeruginosa culture was drop inoculated onto a large CAS-LB agar plate (ø 200 mm). Transparent circles were observed after 48 h of incubation at 37°C.
Extraction and Assay of Quorum-Sensing Signal Molecules
Cultures were grown in LB medium for 24 h at 37°C with shaking (250 rpm) to stationary phase. Supernatants were harvested by centrifugation (12,000 × g, 5 min at room temperature), and 5 ml of supernatant for each sample was extracted using an equal volume of ethyl acetate. The organic phase was concentrated to dryness. For the N-butyryl-L-homoserine lactone (C4-HSL) assay, 10 μl of each sample resolved in 50 μl ethyl acetate was absorbed by a ø 0.5 cm filter paper and then tiled on an LB agar plate, which was newly covered with Chromobacterium violaceum CV026. Purple circles were observed after overnight incubation at 30°C. To detect N-3-oxo-dodecanoyl-L-homoserine lactone (3-oxo-C12-HSL) production, the dry extracts were resolved in 50 μl acetonitrile and 10 μl of each sample was analyzed with HPLC according to the previously published procedure (Ortori et al., 2007). In addition, 10 mmol/L pure 3-oxo-C12-HSL (Sigema) acetonitrile solution was used as a reference.
Pyocyanin Quantitation Assay
The pyocyanin assay is based on the absorbance of pyocyanin at 520 nm in acidic solution. A 5-ml sample of P. aeruginosa culture grown in LB was extracted with 3 ml of chloroform and then re-extracted into 1 ml of 0.2 mmol/L HCl to give a pink to deep red solution. The absorbance of this solution was measured at 520 nm. Concentrations, expressed as micrograms of pyocyanin produced per milliliter of culture supernatant, were determined by multiplying the optical density at 520 nm by 17.072.
LasA Protease Activity Assay
LasA protease activity was determined by measuring the ability of P. aeruginosa culture supernatants to lyse boiled Staphylococcus aureus cells. A 30 ml overnight culture of S. aureus was placed in a boiling water bath for 10 min and then centrifuged for 10 min at 10,000 × g. The resulting pellet was resuspended in 10 mmol/L Na2HPO4 (pH 7.5) and adjusted to an OD600 of 0.9. A 100 μL aliquot of P. aeruginosa supernatant was then added to 900 μL S. aureus suspension, and the OD600 was determined after 5, 10, 15, 20, 25, 30, 35, 40, 45, 60, 75, 90, and 105 min.
Statistical Analyses
Analysis of variance for experimental datasets was performed using JMP soft ware, version 5.0 (SAS Institute Inc., Cary, NC, USA). Significant effects of treatment were determined by the F value. When a significant F test was obtained, separation of means was accomplished by Fisher’s protected LSD (least significant difference) at P ≤ 0.05.
Results
FabV is Responsible for to the Resistance of P. aeruginosa to Triclosan
In order to confirm the role of FabV in P. aeruginosa resistance to triclosan, we tested the sensitivity to triclosan of PAO170 (ΔfabV) carried pSRK-PI (carries P. aeruginosa fabI), pSRK-PV (carries P. aeruginosa fabV) and pSRK-VV (carries V. cholerae fabV), respectively. As expected, the derivative of strain PAO170 carrying the vector plasmid (pSRK-Km) was very sensitive to triclosan (the triclosan MIC was about 1 μg/ml; Figure 2 and Table 1). In contrast, expression of PaFabI, PaFabV, and VcFabV proteins from pSRK-Km-derived plasmids increased triclosan resistance. However, the MIC of strain PAO170 (ΔfabV) carrying plasmid pSRK-PI (5 μg/ml) was much lower than that of strain PAO170 (ΔfabV) carrying plasmid pSRK-PV or pSRK-VV (above 2,000 μg/ml), which was the same as that of the wild-type strain PAO1 (Figure 2 and Table 1). These findings suggest that the sensitivity of mutant strain PAO170 (ΔfabV) to triclosan was due to its lack of triclosan-resistant ENR. To investigate this point further, we also introduced pSRK-EK (carries E. faecalis fabK) and pSRK-BL into P. aeruginosa fabV mutant strain PAO170, respectively. The test of sensitivity to triclosan showed that strain PAO170 (ΔfabV) carrying both plasmids was highly resistant to triclosan; the triclosan MIC was above 2,000 μg/ml (Supplementary Figure S1 and Table 1). These data confirmed that FabV, a triclosan-resistant ENR, leads to resistance of P. aeruginosa to triclosan.
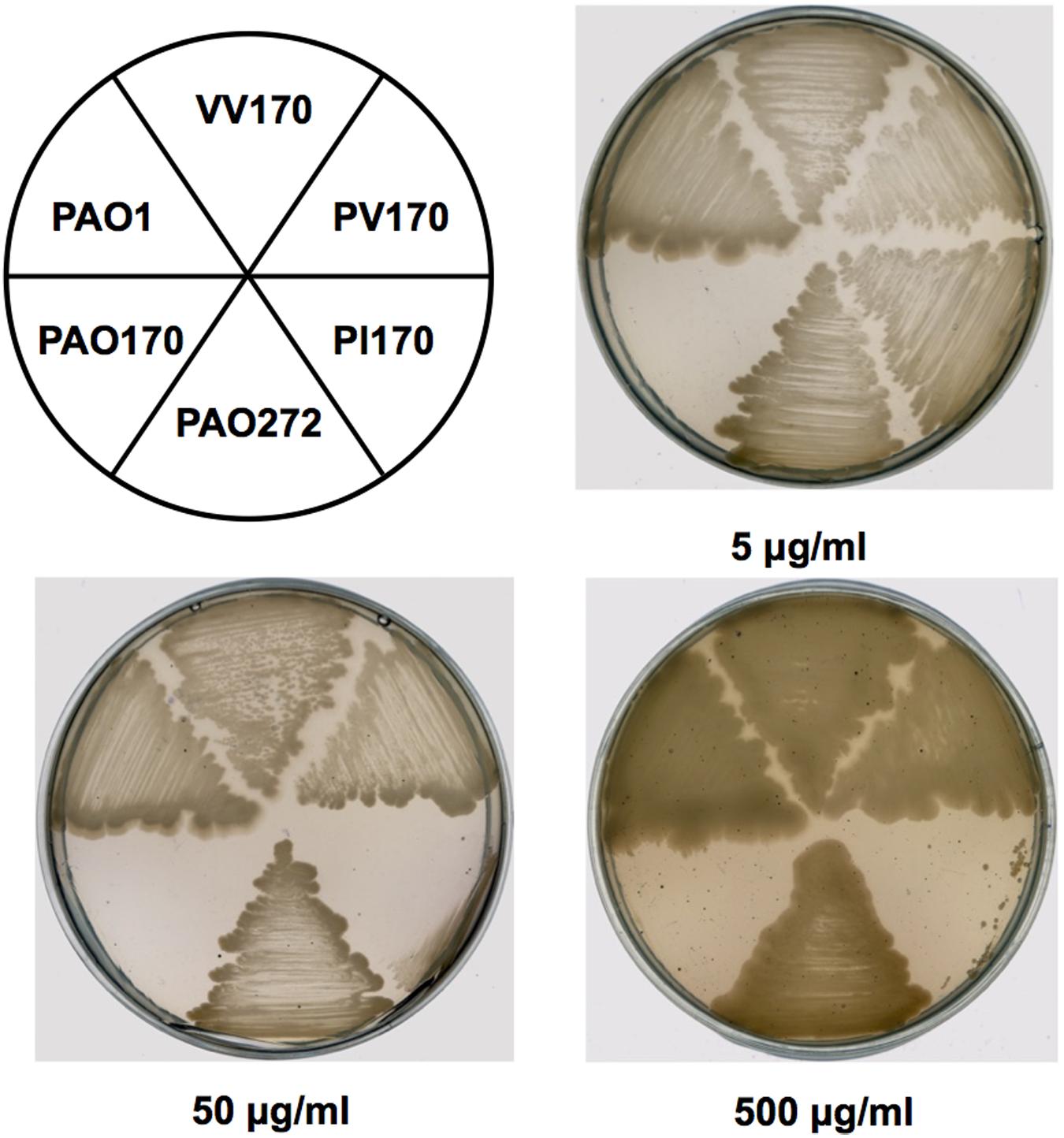
FIGURE 2. Growth inhibition of P. aeruginosa strains by triclosan. The concentration of triclosan is shown below each plate. PAO1 indicates P. aeruginosa wild-type strain PAO1; PAO170 indicates P. aeruginosa fabV deletion strain; PAO272 indicates P. aeruginosa fabI deletion strain; PI170 indicates strain PAO170 carrying plasmid pSRK-PI; PV170 indicates strain PAO170 carrying plasmid pSRK-PV; VV170 indicates strain PAO170 carrying plasmid pSRK-VV.
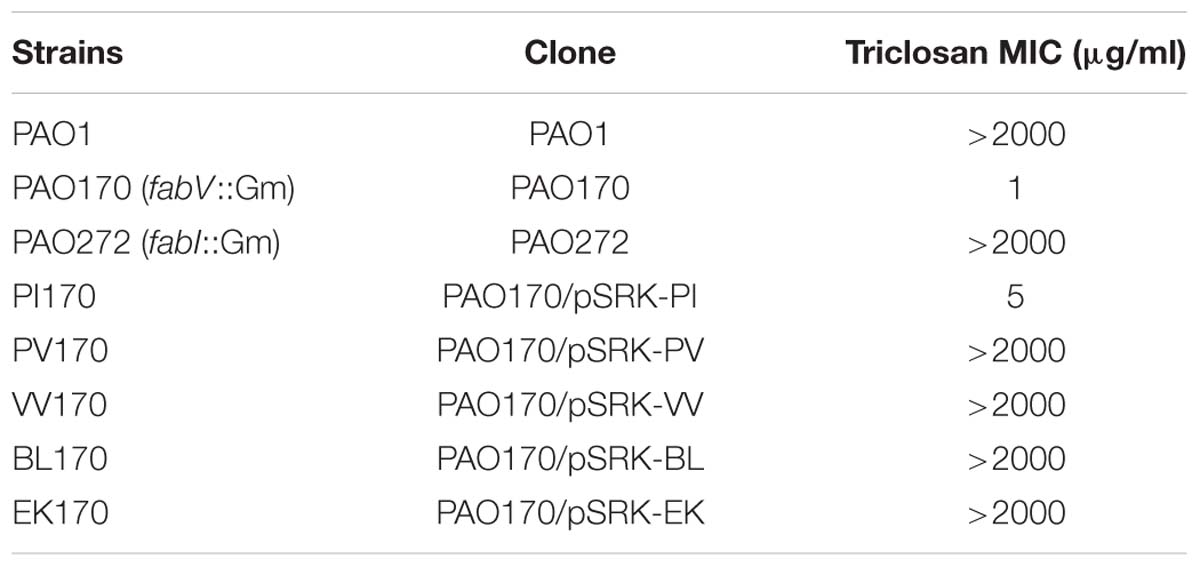
TABLE 1. Triclosan resistance of P. aeruginosa strains.
Deletion of fabV Attenuated P. aeruginosa Swarming Motility
Motility is strongly associated with the pathogenesis of P. aeruginosa. The bacterium exhibits movement on surfaces by utilizing three types of motility: swimming, swarming, and twitching (Inoue et al., 2008; Overhage et al., 2008). Figure 3 shows the colony spreading patterns of these types of motility in P. aeruginosa mutant strains. The twitching pattern of mutant strain PAO272 (ΔfabI) was almost the same as that of wild-type strain PAO1, whereas strain PAO170 (ΔfabV) formed a much smaller twitching pattern than wild-type strain PAO1 (Figure 3A), which suggests that deletion of the fabI gene had no effect on twitching motility of P. aeruginosa, but deletion of fabV inhibited P. aeruginosa twitching motility.
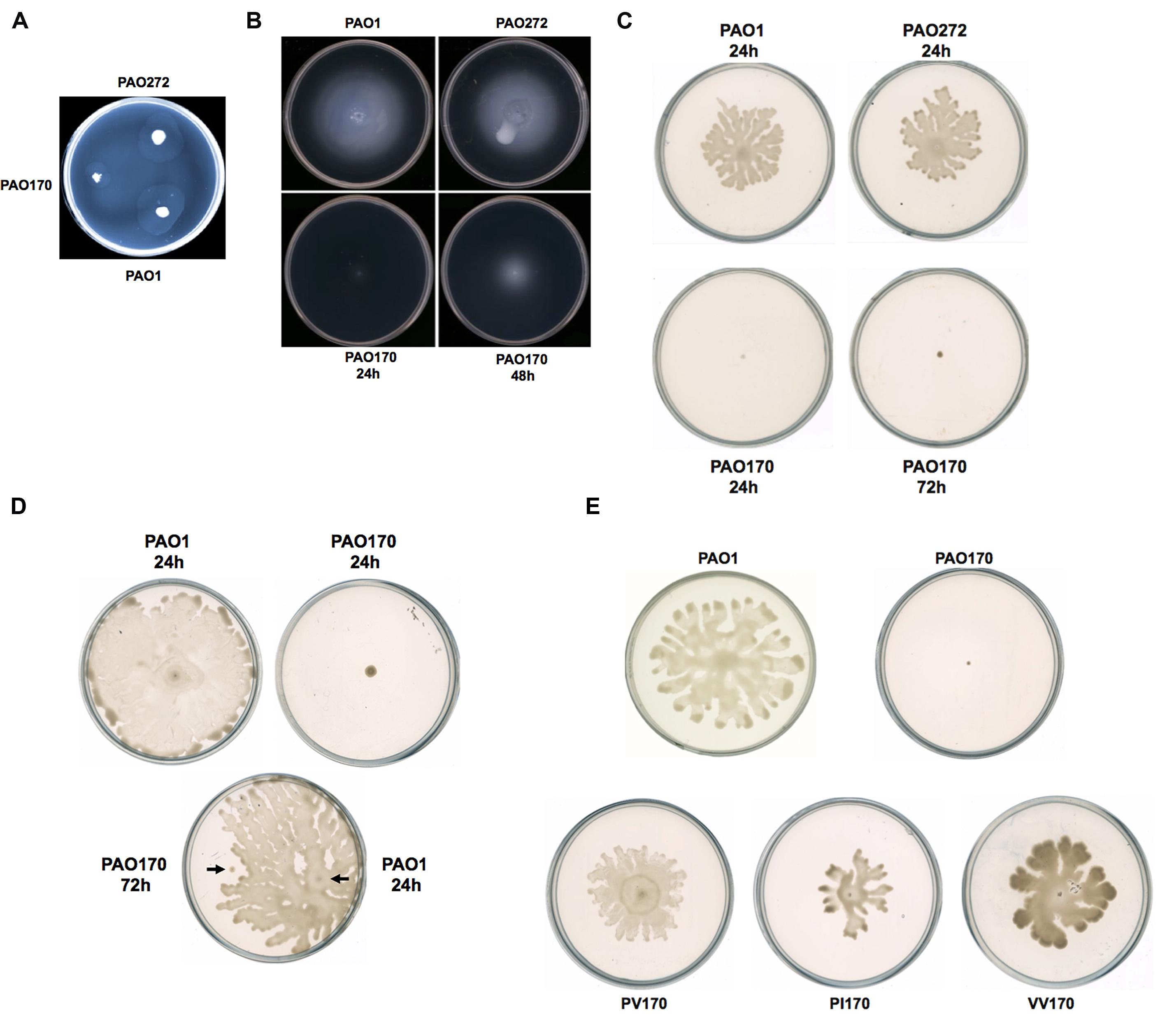
FIGURE 3. Twitching, swimming, and swarming motility of P. aeruginosa strains. (A) Twitching motility patterns of P. aeruginosa strains after incubation at 37°C for 24 h. (B) Swimming motility patterns of P. aeruginosa strains after incubation at 30°C for 24–48 h. (C) Swarming motility patterns of P. aeruginosa strains after incubation at 30°C for 24–72 h. (D) Swarming motility patterns of P. aeruginosa PAO170 grown on a swarming plate containing 0.4% agar. (E) Swarming motility patterns of P. aeruginosa PAO170 carrying ENR encoding genes.
Mutant strain PAO170 (ΔfabV) did not form a swimming pattern after 24 h of incubation on swimming assay plates at 30°C. However, when the incubation time was extended to 48 h, it was able to swim and form a typical swimming pattern, although the average swimming diameter of strain PAO170 (ΔfabV) was much smaller than that of the wild-type strain and the fabI mutant strain PAO272 (ΔfabI; Figure 3B). This result was not consistent with a previous study, in which Bai et al. (2007) reported that mini-Mu insertion in fabV caused the PA68 strain of P. aeruginosa to lose swimming motility. However, it is important to note that, when Bai et al. (2007) carried out the swimming assay for the fabV strain, they incubated the mutant strain for only 16 h on a swimming assay plate. Thus, we speculate that mutation of fabV did not inhibit P. aeruginosa swimming motility directly, but led to slow growth and therefore caused it to form the swimming pattern late. We tested the growth of strain PAO170 (ΔfabV) in swimming medium and confirmed that it grew more slowly than wild-type strain PAO1 (data not shown). Meanwhile, deletion of fabI had no effect on swimming motility of P. aeruginosa because, like wild-type strain PAO1, the mutant strain PAO272 (ΔfabI) was able to form a normal swimming pattern on the swimming assay plate after 24 h of incubation (Figure 3B).
The swarming motility of P. aeruginosa strains was also tested on semisolid plates (containing 0.5% agar) at 30°C (Figure 3C). Although both the fabI mutant strain PAO272 (ΔfabI) and the wild-type strain swarmed normally on semisolid plates after 24 h of incubation, the fabV mutant strain PAO170 failed to form a swarming pattern, even when the incubation time was extended to 48 h (Figure 3C). In swarming medium, the fabV mutant strain grew more slowly than wild-type strain PAO1 (data not shown). Therefore, to test whether the deficiency of swarming motility was due to the weak growth of the fabV mutant strain, we repeated the swarming assay on semisolid plates containing 0.4% agar. As expected, after 24 h of incubation the wild-type strain PAO1 formed obvious swarms on semisolid plates containing 0.4% agar, while the fabV mutant strain failed to form a swarming pattern (Figure 3D). Subsequently, using a toothpick, we inoculated wild-type strain PAO1 on a semisolid plate on which the fabV mutant strain had been cultured for 48 h. After a further 24 h of incubation, the PAO1 strain formed a swarming pattern, but the fabV strain still did not swarm (Figure 3D). These data indicate that it was disruption of fabV that caused P. aeruginosa to lose swarming motility. To test this hypothesis, we checked the swarming motility of complementary strain PAO170/pSRK-PV on a semisolid plate (containing 0.5% agar). Like wild-type strain PAO1, strain PAO170/pSRK-PV was able to form a typical swarming pattern after 24 h of incubation on the semisolid plate (Figure 3E). Moreover, it was interesting that not only P. aeruginosa fabV but also P. aeruginosa fabI could restore normal swarming motility to the fabV mutant (Figure 3E). We also tested whether V. cholerae fabV, E. faecalis fabK and B. subtilis fabL complemented the swarming motility of the fabV mutant. The data showed that all these ENR encoding genes were able to restore swarming motility to the fabV mutant (Figure 3E and Supplementary Figure S2). These results suggest that ENR is required for swarming motility in P. aeruginosa.
The Activity of ENR is Essential to P. aeruginosa Growth
It has been reported that in defined minimal medium the fabV mutant strain PAO170 was viable, but its generation time (about 118 min) was longer than that of the wild-type strain PAO1 (about 40 min; Zhu et al., 2010). To determine whether nutrient conditions affect the growth of strain PAO170, its growth in LB medium was examined. The growth of stain PAO170 in LB medium was significantly slower than that of wild-type strain PAO1, confirming that nutrient conditions did not improve the growth of strain PAO170 (Figure 4A). Complementation of strain PAO170 with plasmid pSRK-PV increased the growth of PAO170 to a certain extent, but not to the level of the wild-type strain (Figure 4A). Plasmid pSRK-PI or pSRK-VV also allowed strain PAO170 to grow to the same level as pSRK-PV (Figure 4A). These results indicated that deletion of PafabV from the genome caused P. aeruginosa to grow slowly and that ENR encoding genes could restore growth of the fabV mutant to a certain extent. We also investigated the growth of PAO170 carrying E. faecalis fabK or B. subtilis fabL encoding plasmids and obtained similar results (Figure 4B). To investigate further possible reasons for the weak growth of strain PAO170, the fatty acid compositions of strain PAO170 were determined by GC-MS. Although the mutant strain PAO170 contained the same fatty acid species as the wild-type strain, PAO170 produced more 3-hydroxy fatty acids (3-HFAs) and fewer FAs (Table 2). The ratio of FAs to 3-HFAs in strain PAO170 was 2.58 and was lower than that of the wild-type strain (3.87), which showed significant difference (P < 0.05) between mutant strain PA170 and wild-type strain PAO1. Complementation of strain PAO170 with plasmid pSRK-PI or pSRK-VV restored the ability of strain PAO170 to produce the wild-type strain’s level of 3-HFAs and FAs. The ratio of FAs to 3-HFAs in strain PAO170 carrying pSRK-PI or pSRK-VV was 3.70 or 3.61, respectively. Moreover, strain PAO170 carrying pSRK-PV produced more FAs and fewer 3-HFAs than the wild-type strain. The ratio of FAs to 3-HFAs in strain PAO170 carrying pSRK-PV was 4.57 (Table 2), and there was highly significant difference between strain PV170 and PAO170 (P < 0.01). It is noteworthy that mutant strain PAO170 carrying pSRK-PV, pSRK-PI or pSRK-VV grew to the same level. This indicated that a change in fatty acid composition caused by deletion of fabV was not the main reason for weak growth in P. aeruginosa fabV mutant. Another possibility was that deletion of fabV led to decreased activity of ENR, which was a contributing factor in the lower growth rate of PAO170. We measured the activity of ENR in cell-free extract of PAO170 by using trans-2-decenoyl-ACP as a substrate and monitoring the decrease in NADH absorbance at 340 nm. The ENR activity (in μmol/min per mg extract protein) in cell-free extract of PAO170 was 0.175 ± 0.006, and was lower than that in the cell-free extract of wild-type strain PAO1 (0.478 ± 0.032). The ENR activity in strain PAO170 carrying plasmids pSRK-PI, pSRK-PV and pSRK-VV was 0.290 ± 0.001, 0.292 ± 0.004 and 0.300 ± 0.006, respectively. Although the ENR activity in strain PAO170 carrying plasmids with genes encoding foreign ENR was increased, it was still lower than that in wild-type strain PAO1. We also detected the ENR activity in cell-free extracts of PAO170 carrying pSRK-EK or pSRK-BL encoding plasmids. The ENR activity in both strains was increased, but did not reach the level of that in the wild-type strain either (data not shown). Therefore, we suggest that the level of activity of ENR, rather than the PafabV gene or protein product, is essential to the growth of P. aeruginosa.
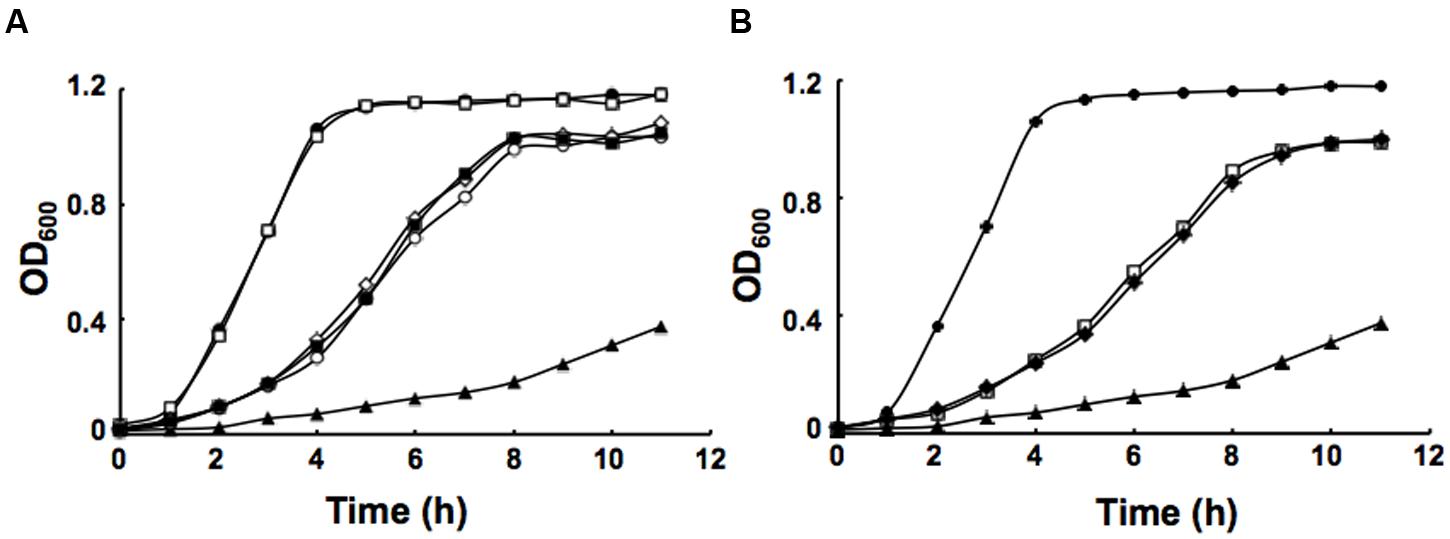
FIGURE 4. Growth of P. aeruginosa strains in LB medium. (A) Growth of the strains PAO1 (filled circle), PAO272 (empty square), PAO170 (filled triangle), PI170 (empty circle), PV170 (filled square) and VV170 (empty diamond). (B) Growth of the strains PAO1 (filled circle), EK170 (empty square), BL170 (filled diamond) and PAO170 (filled triangle). All cultures were grown at 37°C.
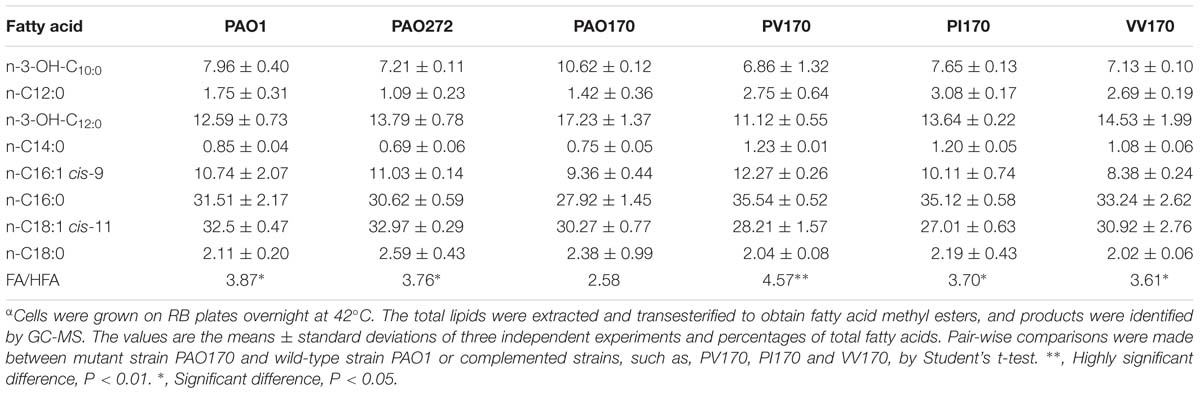
TABLE 2. Fatty acid composition of total lipid extracts from the P. aeruginosaα.
Deletion of fabV Muted the Production of Several Exo-products
Given that deletion of fabV decreased the activity of the ENR of P. aeruginosa significantly, decreased production of several exo-products, which are synthesized using the intermediates in the fatty acid synthetic pathway, would be expected in PAO170 mutant strains. We first looked at levels of rhamnolipids produced by P. aeruginosa strains. Rhamnolipids are secreted surfactant glycolipids assembled by rhamnosyltransferase using L-rhamnose and 3-hydroxydecanoyl-ACP from the FAS pathway (Deziel et al., 2003; Zhu and Rock, 2008). Rhamnolipid quantification by colorimetric detection of rhamnose showed a twofold decrease in the fabV mutant PAO170, while the amount of rhamnolipid produced by complemented strain PAO170/pSRK-PV was up to 80% of that of the wild-type strain PAO1 (Figure 5A).
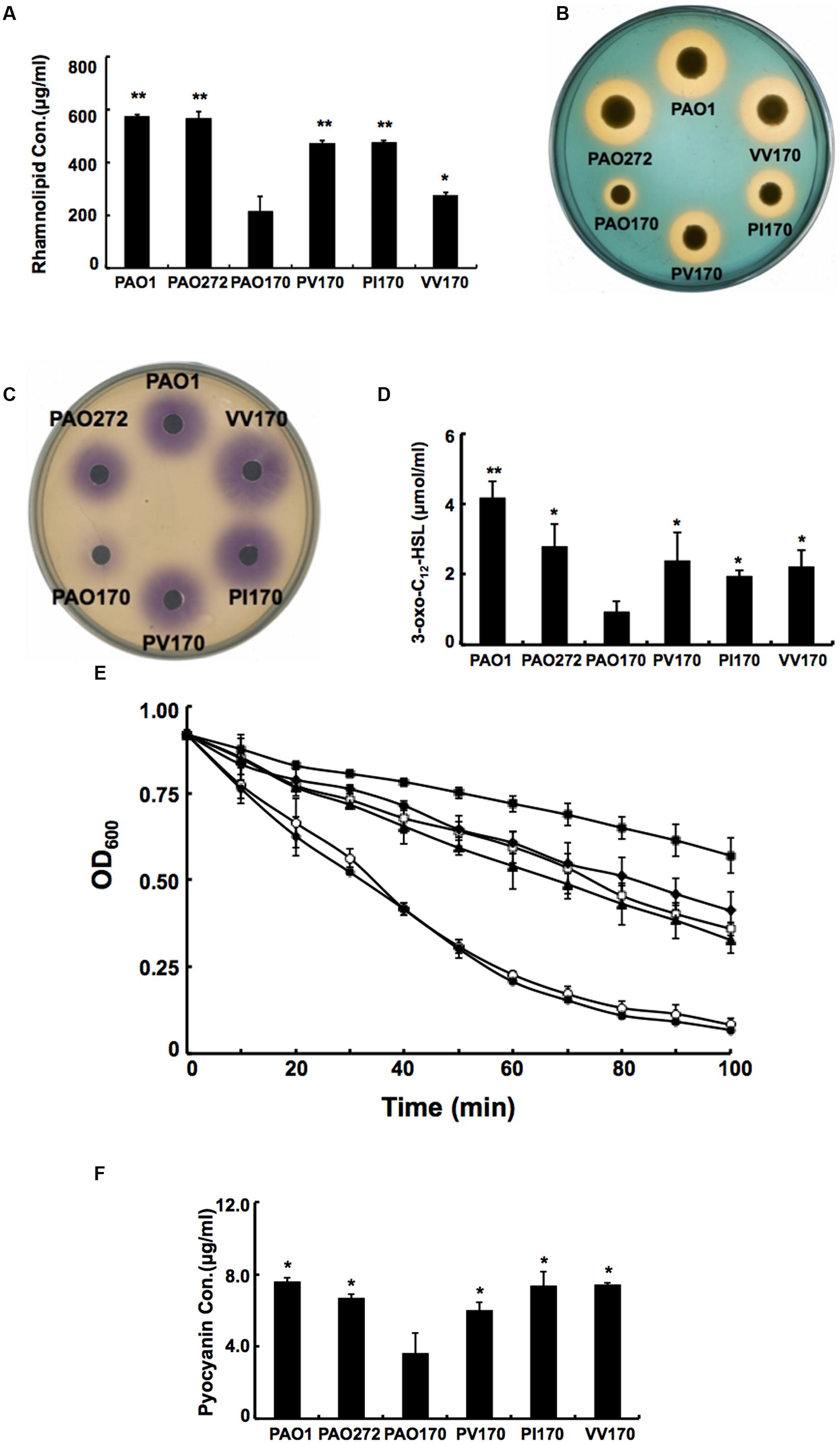
FIGURE 5. Characterization of several exo-products produced by P. aeruginosa strains. (A) Secretion of rhamnolipids by P. aeruginosa strains. (B) Siderophore secretion by P. aeruginosa strains. (C) The C4-HSL produced by P. aeruginosa strains. (D) The 3-oxo-C12-HSL produced by P. aeruginosa strains. (E) LasA protease activities in culture supernatants of P. aeruginosa strains. Filled circle indicates strain PAO1; empty diamond indicates strain PAO272; filled triangle indicates strain PV170; empty square indicates strain VV170; filled diamond indicates strain PI170 and filled square indicates strain PAO170. (F) Pyocyanin in culture supernatants of P. aeruginosa strains. Data are the mean ± standard deviation of triplicate measurements. Pair-wise comparisons were made between mutant strain PAO170 and wild-type strain PAO1 or complemented strains, such as, PV170, PI170 and VV170, by Student’s t-test. ∗∗, Highly significant difference, P < 0.01. ∗, Significant difference, P < 0.05.
Pyoverdine is the dominant siderophore of P. aeruginosa and is assembled from tetradecanoyl-ACP (Drake and Gulick, 2011; Hannauer et al., 2012). The siderophore produced by P. aeruginosa strains was examined on LB-CAS indicator plates. Yellow-orange halos around the cultures indicate where the siderophores have sequestered Fe3+ away from the blue CAS-Fe3+ complex. The halo produced by the PAO170 mutant strain was much smaller than that of the wild-type strain: its average diameter was only a quarter of that of the wild-type strain. After complementation with plasmid pSRK-PV, the average diameter of the yellow-orange halo of the PAO170 mutant strain increased to one half that of the wild-type strain (Figure 5B).
The rhl and las QS signal molecules, N-butanoyl-L-homoserine lactone (C4-HSL) and N-(3-oxododecanoyl)-L-homoserine lactone (3-oxo-C12-HSL), are synthesized from S-adenosylmethionine and butanoyl-ACP or 3-oxododecanoyl-ACP, respectively (Parsek et al., 1999; Hoang et al., 2002). Both acyl-ACPs are intermediates in the FAS pathway. Therefore, we also measured the levels of both QS signal molecules produced by P. aeruginosa strains. The rhl and las QS signals were extracted from culture supernatants of P. aeruginosa strains. The acyl-HSLs were detected using an agar overlay of a C. violaceum reporter strain CV026, which produces a purple halo in response to acyl-HSLs. The purple halo around PAO170 mutant strain was weak and small in comparison to that of the wild-type strain (Figure 5C). Production of acyl-HSLs was increased to wild-type levels by complementation with strain PAO170/pSRK-PV. The 3-oxo-acyl-HSL was detected using HPLC. The level of 3-oxo-acyl-HSL showed a threefold decrease in the fabV mutant in comparison to that of the wild-type strain and there was highly significant difference between strain PV170 and PAO170 (P < 0.01). Complemented strain PAO170/pSRK-PV produced more 3-oxo-acyl-HSL than the fabV mutant, but not to the level of the wild-type strain (Figure 5D).
The PQS signal molecules also utilize 3-ketoacyl medium-chain fatty acid metabolites to synthesize 2-heptyl-3-hydroxy-4-quinolone (PQS; Girard and Bloemberg, 2008; Williams and Camara, 2009). We thus analyzed the levels of PQS in supernatant extract in P. aeruginosa strains. The level of PQS was reduced to a trace in the supernatant extract when fabV was deleted. In contrast, PQS production was markedly enhanced in the complemented strain (data not shown).
We also complemented the PAO170 mutant with other ENR encoding genes, such as P. aeruginosa fabI or V. cholerae fabV, and all complemented strains produced more rhamnolipids, pyoverdine, AHLs and PQS than the fabV mutant (Figures 5A–D).
Addition of Exogenous AHLs Restored the Ability of the fabV Strain to Produce Virulence Factors
The QS systems, including rhl, las and PQS, regulate the ability of P. aeruginosa to produce virulence factors (e.g., LasA/LasB and alkaline proteases, phospholipases, lipases, exotoxin A, rhamnolipid, pyocyanin, and others; Girard and Bloemberg, 2008; Williams and Camara, 2009). Thus, reduction of QS signal molecules should decrease production of these virulence factors. To confirm this, we tested the production of LasA and pyocyanin by P. aeruginosa strains. The data showed that the activity of LasA in PAO170 mutant was much lower than that in the wild-type strain, and the amount of pyocyanin produced by PAO170 strain was about 50% of that produced by the wild-type strain (Figures 5E,F). However, complementation of PAO170 mutant strain with ENR genes increased the activity of LasA and the amount of pyocyanin (Figures 5E,F).
Based on the above study, it would be expected that, on addition of QS signal molecules to cultures of P. aeruginosa strains, production of some virulence factors that are regulated by the QS system would be restored. First, we added C4-HSL or 3-oxo-C12-HSL to swarming plates and tested the swarming motility of the fabV mutant strain. Exogenous addition of both QS signals to the swarming plate restored the ability of the fabV mutant to form a typical swarming pattern after 24 h of incubation, although the pattern was still smaller than that of the wild type (Figure 6A). We also examined production of pyocyanin, rhamnolipid and LasA protease by PAO170 mutant strain after exogenous addition of QS signals. The data showed that, although not up to the levels of the wild-type strain, the production of all these virulence factors was increased to a certain extent (Figures 6B,C). These findings indicated that deletion of fabV reduced the activity of ENR in P. aeruginosa, subsequently depressed the production of QS signals and finally led to reduced virulence factors in P. aeruginosa.
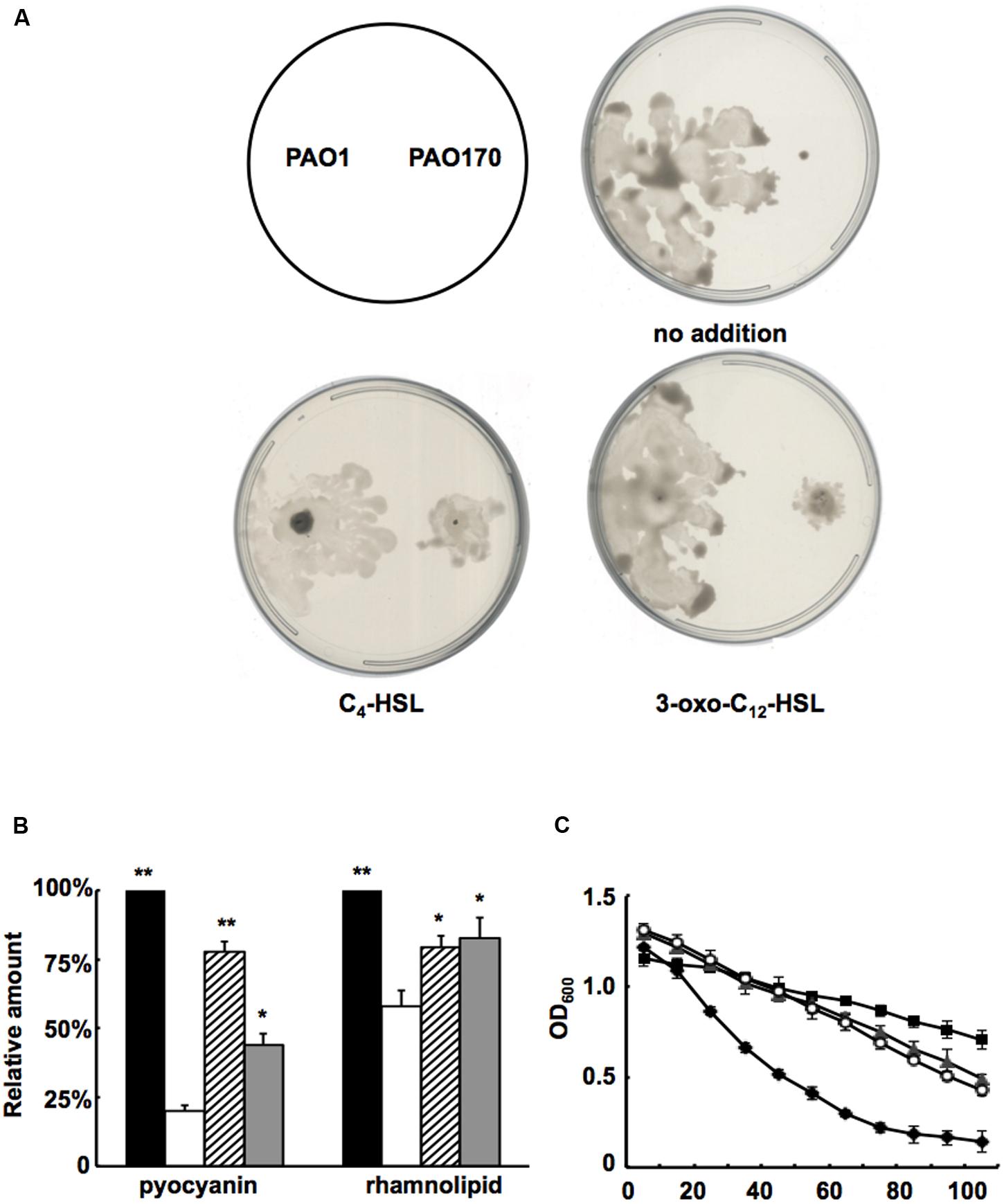
FIGURE 6. Effect of exogenous AHLs on swarming motility and production of several virulence factors by the P. aeruginosa fabV deletion strain. (A) Effect of exogenous AHLs on swarming motility of the P. aeruginosa fabV deletion strain. (B) Effect of exogenous AHLs on pyocyanin and rhamnolipid secretion by the P. aeruginosa fabV deletion strain. Black bar indicates the culture of strain PAO1; white bar indicates the culture of strain PAO170; hatched bar indicates the culture of strain PAO170 after addition of C4-HSL; gray bar indicates the culture of strain PA0170 after addition of 3-oxo-C12-HSL. Data are the mean ± standard deviation of triplicate measurements. Pair-wise comparisons were made between mutant strain PAO170 and wild-type strain PAO1 by Student’s t-test. ∗∗, Highly significant difference, P < 0.01. ∗, Significant difference, P < 0.05. (C) LasA protease activities in culture supernatants of P. aeruginosa strains after addition of exogenous AHLs. Filled diamond indicates the culture of strain PAO1; empty circle indicates the culture of strain PA170 after addition of C4-HSL; filled triangle indicates the culture of strain PV170 after addition of 3-oxo-C12-HSL; filled square indicates the culture of strain PA170.
Discussion
Complementation with any one of the known triclosan-resistant ENR encoding genes, such as V. cholerae fabV, E. faecalis fabK, B. subtilis fabL or P. aeruginosa fabV, restored triclosan resistance in the fabV deleted strain to the level of the wild-type strain, which confirms that FabV is responsible for the inherent triclosan resistance of P. aeruginosa. This confirms that a triclosan-resistant ENR may confer high resistance to triclosan on a bacterium. E. faecalis has a triclosan-resistant ENR encoding gene, fabK, and is resistant to triclosan (the MIC of triclosan is 10 μg/ml), but it has been demonstrated that the E. faecalis FabK does play a role in the inherent triclosan resistance of this bacterium (Zhu et al., 2013). Moreover, S. pneumoniae, B. subtilis and V. cholerae also possesses triclosan-resistant ENR encoding genes (fabK, fabL and fabV; Heath et al., 2000; Marrakchi et al., 2003; Massengo-Tiasse and Cronan, 2008), and expression of S. pneumoniae fabK, B. subtilis fabL or V. cholerae fabV greatly increased the resistance of E. coli to triclosan, but the MIC of triclosan for S. pneumoniae, B. subtilis and V. cholerae is only about 2–4 μg/ml, which is much lower than that of P. aeruginosa. This suggests that triclosan may target a cellular process unrelated to fatty acid synthesis in these bacteria.
The ENRs reduce trans-2-enoyl-ACPs to the fully saturated ACP species in the last step of the elongation cycle in the synthesis of bacterial FAs (White et al., 2004). In E. coli, there is a single, NADH-dependent ENR isoform, FabI, which reduces all of the enoyl intermediates of the pathway and is essential to cell growth and survival. Physiological studies have shown that E. coli FabI plays a determining role in completing rounds of fatty acid elongation and is feedback inhibited by long-chain ACPs (Heath and Rock, 1995). Unlike E. coli, B. subtilis, E. faecalis and P. aeruginosa have two ENRs (Heath et al., 2000; Zhu et al., 2010, 2013), which can be either of the same or of different protein families. However, although both ENR isozymes in these bacteria catalyze reduction of trans-2-enoyl-ACPs to the saturated ACP species, the physiological rationale for this duplication of enzyme activity has been poorly explored. Recently, the functions of both FabI and FabK in E. faecalis were characterized and the results showed that each enzyme has a discrete physiological role. FabI is essential for E. faecalis growth and plays the key role in the fatty acid synthetic pathway, whereas deletion of FabK does not affect growth of E. faecalis and FabK only modulates the composition of phospholipid acyl chains (Zhu et al., 2013). P. aeruginosa possesses FabI and FabV, two ENR isozymes. Although neither the fabI nor the fabV gene is essential to P. aeruginosa, the deletion of fabV produced more 3-hydroxy fatty acids and fewer fatty acids, and this strain grew much more slowly than the wild-type strain. Moreover, fabV deletion attenuated swarming motility, decreased production of rhamnolipid, pyoverdine and AHLs, and reduced virulence factors. All these results suggest that FabV is the main ENR of P. aeruginosa. However, mutation of fabI did not affect P. aeruginosa growth or the fatty acid profiles. This indicates that, unlike FabK in E. faecalis, FabI does not play a role in modulating fatty acid composition at least under the growth conditions we have tested. However, the fabI mutant showed reduced ENR activity in cell extracts and produced fewer AHLs (Hoang and Schweizer, 1999; Zhu et al., 2010). Thus, we speculate that fabI may be required to maintain sufficient ENR activity in P. aeruginosa.
Pseudomonas aeruginosa is a gram-negative bacterium and produces lipid A in its outer membrane (Pier, 2007). When we determined the total cell fatty acid composition in P. aeruginosa strains by GC-MS, we found that deletion of fabV led P. aeruginosa to produce more 3-hydroxydecanoic and 3-hydroxydodecanoic acids, which are the main components of lipid A of P. aeruginosa. This deletion of fabV changed the production of lipid A in P. aeruginosa. However, we also demonstrated that increasing 3-hydroxy fatty acids was not the main reason for the weak growth of P. aeruginosa. In the bacterial fatty acid synthetic pathway, the ENRs are responsible for pulling trans-2-enoyl-ACPs to the fully saturated ACP species. However, the conversion of 3-hydroxyacyl-ACPs to trans-2-enoyl-ACPs is catalyzed by 3-hydroxyacyl-ACP dehydrase (FabA and FabZ) and is an equilibrium reaction (Heath and Rock, 1995). Thus, larger amounts of 3-hydroxy fatty acids may accumulate in a cell when the level of P. aeruginosa ENRs is decreased.
The three dominant QS signal molecules from the las, rhl, and PQS systems, all of which contain fatty acid moieties, together form a complex, cell density-dependent regulatory circuit, which not only regulates the production of many exo-products in P. aeruginosa, but also is involved in bacterial motility (Girard and Bloemberg, 2008; Williams and Camara, 2009). The deletion of fabV led P. aeruginosa to reduce the production of QS signal molecules and many exo-products, and caused P. aeruginosa to fail to swarm on semisolid plates. However, exogenous supplementation of AHLs, C4-HSL or 3-oxo-C12-HSL increased the production of pyocyanin, rhamnolipid and LasA protease in the fabV mutant, and restored fabV mutant swarming on semisolid plates. These findings indicated that deletion of fabV caused decreased production of AHL signals, attenuated QS systems, and subsequently reduced the virulence factors of P. aeruginosa.
Deletion of fabV attenuated not only growth but also QS and many virulence factors. Therefore, inhibition of FabV in P. aeruginosa is a compelling strategy for use in the development of new antimicrobial agents.
Author Contributions
Y-HH cloned ENR genes, constructed several ENRs expression vectors, did complementation experiments with ENRs expression vectors, and carried out biochemical studies. J-SL constructed fabV mutants and tested the growth of mutants in LB medium. J-CM carried out experiments on the pathogenesis of P. aeruginosa. H-HW conceived of the study, and participated in its design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from the Natural Science Foundation of Guangdong Province (2015A030312005), the Natural Science Foundation of Guangdong Province (2014A030313455), and Startup Foundation for Docotors of Yan’an University (YDBK2016-11).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2016.01903/full#supplementary-material
References
Bai, F., Li, Y., Xu, H., Xia, H., Yin, T., Yao, H., et al. (2007). Identification and functional characterization of pfm, a novel gene involved in swimming motility of Pseudomonas aeruginosa. Gene 401, 19–27. doi: 10.1016/j.gene.2007.06.019
Bai, F., Xu, H., Zhang, Q., Qi, X., Mou, R., Bai, G., et al. (2011). Functional characterization of pfm in protein secretion and lung infection of Pseudomonas aeruginosa. Can. J. Microbiol. 57, 829–837. doi: 10.1139/w11-075
Campbell, J. W., and Cronan, J. E. Jr. (2001). Bacterial fatty acid biosynthesis: targets for antibacterial drug discovery. Annu. Rev. Microbiol. 55, 305–332. doi: 10.1146/annurev.micro.55.1.305
Deziel, E., Lepine, F., Milot, S., and Villemur, R. (2003). rhlA is required for the production of a novel biosurfactant promoting swarming motility in Pseudomonas aeruginosa: 3-(3-hydroxyalkanoyloxy)alkanoic acids (HAAs), the precursors of rhamnolipids. Microbiology 149, 2005–2013. doi: 10.1099/mic.0.26154-0
Drake, E. J., and Gulick, A. M. (2011). Structural characterization and high-throughput screening of inhibitors of PvdQ, an NTN hydrolase involved in pyoverdine synthesis. ACS Chem. Biol. 6, 1277–1286. doi: 10.1021/cb2002973
Driscoll, J. A., Brody, S. L., and Kollef, M. H. (2007). The epidemiology, pathogenesis and treatment of Pseudomonas aeruginosa infections. Drugs 67, 351–368. doi: 10.2165/00003495-200767030-00003
Feng, S. X., Ma, J. C., Yang, J., Hu, Z., Zhu, L., Bi, H. K., et al. (2015). Ralstonia solanacearum fatty acid composition is determined by interaction of two 3-ketoacyl-acyl carrier protein reductases encoded on separate replicons. BMC Microbiol. 15:e333. doi: 10.1186/s12866-015-0554-x
Girard, G., and Bloemberg, G. V. (2008). Central role of quorum sensing in regulating the production of pathogenicity factors in Pseudomonas aeruginosa. Future Microbiol. 3, 97–106. doi: 10.2217/17460913.3.1.97
Hannauer, M., Schafer, M., Hoegy, F., Gizzi, P., Wehrung, P., Mislin, G. L., et al. (2012). Biosynthesis of the pyoverdine siderophore of Pseudomonas aeruginosa involves precursors with a myristic or a myristoleic acid chain. FEBS Lett. 586, 96–101. doi: 10.1016/j.febslet.2011.12.004
Heath, R. J., and Rock, C. O. (1995). Enoyl-acyl carrier protein reductase (fabI) plays a determinant role in completing cycles of fatty acid elongation in Escherichia coli. J. Biol. Chem. 270, 26538–26542. doi: 10.1074/jbc.270.44.26538
Heath, R. J., and Rock, C. O. (2000). A triclosan-resistant bacterial enzyme. Nature 406, 145–146. doi: 10.1038/35022656
Heath, R. J., Su, N., Murphy, C. K., and Rock, C. O. (2000). The enoyl-[acyl-carrier-protein] reductases FabI and FabL from Bacillus subtilis. J. Biol. Chem. 275, 40128–40133. doi: 10.1074/jbc.M005611200
Heath, R. J., White, S. W., and Rock, C. O. (2001). Lipid biosynthesis as a target for antibacterial agents. Prog. Lipid Res. 40, 467–497. doi: 10.1016/S0163-7827(01)00012-1
Hoang, T. T., and Schweizer, H. P. (1997). Fatty acid biosynthesis in Pseudomonas aeruginosa: cloning and characterization of the fabAB operon encoding beta-hydroxyacyl-acyl carrier protein dehydratase (FabA) and beta-ketoacyl-acyl carrier protein synthase I (FabB). J. Bacteriol. 179, 5326–5332. doi: 10.1128/jb.179.17.5326-5332.1997
Hoang, T. T., and Schweizer, H. P. (1999). Characterization of Pseudomonas aeruginosa enoyl-acyl carrier protein reductase (FabI): a target for the antimicrobial triclosan and its role in acylated homoserine lactone synthesis. J. Bacteriol. 181, 5489–5497.
Hoang, T. T., Sullivan, S. A., Cusick, J. K., and Schweizer, H. P. (2002). Beta-ketoacyl acyl carrier protein reductase (FabG) activity of the fatty acid biosynthetic pathway is a determining factor of 3-oxo-homoserine lactone acyl chain lengths. Microbiology 148, 3849–3856. doi: 10.1099/00221287-148-12-3849
Inoue, T., Shingaki, R., and Fukui, K. (2008). Inhibition of swarming motility of Pseudomonas aeruginosa by branched-chain fatty acids. FEMS Microbiol. Lett. 281, 81–86. doi: 10.1111/j.1574-6968.2008.01089.x
Kerr, K. G., and Snelling, A. M. (2009). Pseudomonas aeruginosa: a formidable and ever-present adversary. J. Hosp. Infect. 73, 338–344. doi: 10.1016/j.jhin.2009.04.020
Khan, S. R., Gaines, J., Roop, R. M. II, and Farrand, S. K. (2008). Broad-host-range expression vectors with tightly regulated promoters and their use to examine the influence of TraR and TraM expression on Ti plasmid quorum sensing. Appl. Environ. Microbiol. 74, 5053–5062. doi: 10.1128/AEM.01098-08
Lee, J., and Zhang, L. (2015). The hierarchy quorum sensing network in Pseudomonas aeruginosa. Protein Cell 6, 26–41. doi: 10.1007/s13238-014-0100-x
Mao, Y. H., Li, F., Ma, J. C., Hu, Z., and Wang, H. H. (2016). Sinorhizobium meliloti functionally replaces 3-Oxoacyl-Acyl carrier protein reductase (FabG) by overexpressing NodG during fatty acid synthesis. Mol. Plant Microbe Interact. 29, 458–467. doi: 10.1094/MPMI-07-15-0148-R
Marrakchi, H., Dewolf, W. E. Jr., Quinn, C., West, J., Polizzi, B. J., So, C. Y., et al. (2003). Characterization of Streptococcus pneumoniae enoyl-(acyl-carrier protein) reductase (FabK). Biochem. J. 370, 1055–1062. doi: 10.1042/bj20021699
Massengo-Tiasse, R. P., and Cronan, J. E. (2008). Vibrio cholerae FabV defines a new class of enoyl-acyl carrier protein reductase. J. Biol. Chem. 283, 1308–1316. doi: 10.1074/jbc.M708171200
Massengo-Tiasse, R. P., and Cronan, J. E. (2009). Diversity in enoyl-acyl carrier protein reductases. Cell. Mol. Life Sci. 66, 1507–1517. doi: 10.1007/s00018-009-8704-7
Mou, R., Bai, F., Duan, Q., Wang, X., Xu, H., Bai, Y., et al. (2011). Mutation of pfm affects the adherence of Pseudomonas aeruginosa to host cells and the quorum sensing system. FEMS Microbiol. Lett. 324, 173–180. doi: 10.1111/j.1574-6968.2011.02401.x
Ortori, C. A., Atkinson, S., Chhabra, S. R., Camara, M., Williams, P., and Barrett, D. A. (2007). Comprehensive profiling of N-acylhomoserine lactones produced by Yersinia pseudotuberculosis using liquid chromatography coupled to hybrid quadrupole-linear ion trap mass spectrometry. Anal. Bioanal. Chem. 387, 497–511. doi: 10.1007/s00216-006-0710-0
Overhage, J., Bains, M., Brazas, M. D., and Hancock, R. E. (2008). Swarming of Pseudomonas aeruginosa is a complex adaptation leading to increased production of virulence factors and antibiotic resistance. J. Bacteriol. 190, 2671–2679. doi: 10.1128/JB.01659-07
Parsek, M. R., Val, D. L., Hanzelka, B. L., Cronan, J. E. Jr., and Greenberg, E. P. (1999). Acyl homoserine-lactone quorum-sensing signal generation. Proc. Natl. Acad. Sci. U.S.A. 96, 4360–4365. doi: 10.1073/pnas.96.8.4360
Pier, G. B. (2007). Pseudomonas aeruginosa lipopolysaccharide: a major virulence factor, initiator of inflammation and target for effective immunity. Int. J. Med. Microbiol. 297, 277–295. doi: 10.1016/j.ijmm.2007.03.012
Stead, D. E. (1989). Grouping of Xanthomonas campestris pathovars of cereals and grasses by fatty acid profiling. EPPO Bull. 19, 57–68. doi: 10.1111/j.1365-2338.1989.tb00129.x
White, S. W., Zheng, J., Zhang, Y. M., and Rock, C. O. (2004). The structural biology of type II fatty acid biosynthesis. Annu. Rev. Biochem. 74, 791–831. doi: 10.1146/annurev.biochem.74.082803.133524
Willcox, M. D. (2007). Pseudomonas aeruginosa infection and inflammation during contact lens wear: a review. Optom. Vis. Sci. 84, 273–278. doi: 10.1097/OPX.0b013e3180439c3e
Williams, P., and Camara, M. (2009). Quorum sensing and environmental adaptation in Pseudomonas aeruginosa: a tale of regulatory networks and multifunctional signal molecules. Curr. Opin. Microbiol. 12, 182–191. doi: 10.1016/j.mib.2009.01.005
Yuan, Y., Leeds, J. A., and Meredith, T. C. (2012a). Pseudomonas aeruginosa directly shunts β-oxidation degradation intermediates into de novo fatty acid biosynthesis. J. Bacteriol. 194, 5185–5196. doi: 10.1128/JB.00860-12
Yuan, Y., Sachdeva, M., Leeds, J. A., and Meredith, T. C. (2012b). Fatty acid biosynthesis in Pseudomonas aeruginosa is initiated by the FabY class of β-ketoacyl acyl carrier protein synthases. J. Bacteriol. 194, 5171–5184. doi: 10.1128/JB.00792-12
Zhang, Y. M., and Rock, C. O. (2008). Membrane lipid homeostasis in bacteria. Nat. Rev. Microbiol. 6, 222–233. doi: 10.1038/nrmicro1839
Zhu, K., and Rock, C. O. (2008). RhlA converts beta-hydroxyacyl-acyl carrier protein intermediates in fatty acid synthesis to the beta-hydroxydecanoyl-beta-hydroxydecanoate component of rhamnolipids in Pseudomonas aeruginosa. J. Bacteriol. 190, 3147–3154. doi: 10.1128/JB.00080-08
Zhu, L., Bi, H., Ma, J., Hu, Z., Zhang, W., Cronan, J. E., et al. (2013). The two functional enoyl-acyl carrier protein reductases of Enterococcus faecalis do not mediate triclosan resistance. mBio 4:e613–e613. doi: 10.1128/mBio.00613-13
Keywords: Pseudomonas aeruginosa, enoyl-acyl-carrier protein reductase, triclosan, acylhomoserine lactones, swarming
Citation: Huang Y - H, Lin J - S, Ma J - C and Wang H - H (2016) Functional Characterization of Triclosan-Resistant Enoyl-acyl-carrier Protein Reductase (FabV) in Pseudomonas aeruginosa. Front. Microbiol. 7:1903. doi: 10.3389/fmicb.2016.01903
Received: 31 August 2016; Accepted: 14 November 2016;
Published: 29 November 2016.
Edited by:
Dongsheng Zhou, Beijing Institute of Microbiology and Epidemiology, ChinaReviewed by:
Åsa Sjöling, Karolinska Institutet, SwedenHongxia Wang, University of Alabama at Birmingham, USA
Copyright © 2016 Huang, Lin, Ma and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hai-Hong Wang, d2FuZ2hoMzZAc2NhdS5lZHUuY24=
†These authors have contributed equally to this work.