 Aimi Hanafi1
Aimi Hanafi1 Woon Ching Lee1
Woon Ching Lee1 Mun Fai Loke1,2Xinsheng Teh1Ain Shaari1Mojdeh Dinarvand1
Mun Fai Loke1,2Xinsheng Teh1Ain Shaari1Mojdeh Dinarvand1 Philippe Lehours3,4Francis Mégraud3,4Alex Hwong Ruey Leow5
Philippe Lehours3,4Francis Mégraud3,4Alex Hwong Ruey Leow5 Jamuna Vadivelu1*
Jamuna Vadivelu1* Khean Lee Goh5
Khean Lee Goh5- 1Department of Medical Microbiology, Faculty of Medicine, University of Malaya, Kuala Lumpur, Malaysia
- 2Department of Microbiology and Immunology, Yong Loo Lin School of Medicine, National University of Singapore, Singapore, Singapore
- 3Laboratoire de Bactériologie, Université de Bordeaux, Bordeaux, France
- 4Institut National de la Santé et de la Recherche Médicale U853, Bordeaux, France
- 5Department of Medicine, Faculty of Medicine, University of Malaya, Kuala Lumpur, Malaysia
Antibiotic resistance in bacteria incurs fitness cost, but compensatory mechanisms may ameliorate the cost and sustain the resistance even under antibiotics-free conditions. The aim of this study was to determine compensatory mechanisms of antibiotic resistance in H. pylori. Five strains of levofloxacin-sensitive H. pylori were induced in vitro to develop resistance. In addition, four pairs of metronidazole-sensitive and -resistant H. pylori strains were isolated from patients carrying dual H. pylori populations that consist of both sensitive and resistant phenotypes. Growth rate, virulence and biofilm-forming ability of the sensitive and resistant strains were compared to determine effects of compensatory response. Proteome profiles of paired sensitive and resistant strains were analyzed by liquid chromatography/mass spectrophotometry (LC/MS). Although there were no significant differences in growth rate between sensitive and resistant pairs, bacterial virulence (in terms of abilities to induce apoptosis and form biofilm) differs from pair to pair. These findings demonstrate the complex and strain-specific phenotypic changes in compensation for antibiotics resistance. Compensation for in vitro induced levofloxacin resistance involving mutations of gyrA and gyrB was functionally random. Furthermore, higher protein translation and non-functional protein degradation capabilities in naturally-occuring dual population metronidazole sensitive-resistant strains may be a possible alternative mechanism underlying resistance to metronidazole without mutations in rdxA and frxA. This may explain the lack of mutations in target genes in ~10% of metronidazole resistant strains.
Introduction
Helicobacter pylori is a common bacterial pathogen that colonize the human stomach and is related to incidence of gastric cancer and peptic ulcer diseases (Parsonnet et al., 1991; Dhar et al., 2003). H. pylori infection can often be successfully eradicated with antibiotics (Heo and Jeon, 2014). However, the increasing prevalence of antibiotic resistance in H. pylori is a cause of concern as this is one of the most important causes of therapy failure (Graham and Fischbach, 2010). The prevalence of H. pylori antibiotic resistance has been associated with extensive use of antibiotics within a population (Megraud and Lehours, 2007; de Francesco et al., 2010).
The antibiotics used to treat H. pylori infection were mainly amoxicillin, clarithromycin, and metronidazole; these would be administered for 10–14 days in combination with an anti-secretory drug to increase the pH (Lind et al., 1999). Current recommendations for H. pylori treatment include the first line therapy, which is standard triple therapy consisting a combination of proton pump inhibitors (PPI), clarithromycin, and amoxicillin or metronidazole; the second line therapy will be used in the case of treatment failure, in which bismuth-based quadruple therapy or levofloxacin-containing triple therapy are recommended (Malfertheiner et al., 2007).
Levofloxacin, a fluoroquinolone, was shown to have eradicated H. pylori effectively (Cammarota et al., 2000). Fluoroquinolones generally target chromosome replication and in particular, DNA gyrase, which allows DNA unraveling before replication. However, the prevalence of levofloxacin resistance in H. pylori has been increasing worldwide (de Francesco et al., 2010); with resistance rates at 14.1% in Europe (Megraud et al., 2013), 20.6% in southeast region of China (Su et al., 2013), and 18.4% in Vietnam (Binh et al., 2013). This resistance has been associated with the point mutations occurring at positions Asn87 and Asp91 of the quinolone resistance determining region (QRDR) within gyrA gene (Miyachi et al., 2006; Rozen et al., 2007; Lee et al., 2011). Other mutations that have also been linked to levofloxacin resistance include mutations at positions Ala88, Ala97, and Met191 of gyrA and Phe438, Asp481, and Arg484 of gyrB (Miyachi et al., 2006; Liu et al., 2011; Teh et al., 2014).
Metronidazole, a nitroimidazole, acts as a biocidal agent by its interaction with a nitroreductase homolog, RdxA. Reduction of metronidazole results in the formation of DNA-damaging and mutagenic products (Sisson et al., 2000). Mutations in rdxA were shown to be the cause of H. pylori resistance to metronidazole (Goodwin et al., 1998). Inactivation of rdxA reduces the effect of nitroreductases, which comes with the decrease of conversion of metronidazole into hydroxylamine that damages bacterial DNA (Olekhnovich et al., 2009). Mutation in another gene, frxA, encoding for NADH flavin oxidoreductase, was also implicated in H. pylori metronidazole resistance (Kwon et al., 2000). FrxA, another nitroreductase of H. pylori, may also activate metronidazole bactericidal action although the overall effects of frxA mutation are still being investigated (Justino et al., 2014). Mutations, such as frameshift, missense, premature truncations, deletions, and insertions within rdxA and frxA genes, are associated with metronidazole resistance (Kwon et al., 2000; Teh et al., 2014; Binh et al., 2015). In an earlier study, it was shown that 4/37 (10.8%) of the metronidazole resistant H. pylori strains from Malaysia could not be attributed to mutations in rdxA and/or fdxA (Teh et al., 2014). Thus, H. pylori may become resistant to the antibiotic via other mechanisms.
Despite the studies on the resistance-related genes, the consequences of mutations on the physiological state of H. pylori are poorly understood. The impact of mutation in antibiotic resistance has been studied in vitro in Streptococcus pneumoniae for levofloxacin resistance; different resistance-encoding genes were shown to cost different levels of fitness (Rozen et al., 2007). Björkholm et al. (2001) studied the biological cost of mutation in response to clarithromycin resistance in H. pylori; compensatory mutation has been suggested to affect bacterial fitness. However, the effects of mutations that result in levofloxacin and metronidazole resistance have not been studied in H. pylori. Furthermore, antibiotic resistance-related mutations may affect bacterial virulence and survival. Implications of mutations in H. pylori virulence proteins involved in apoptosis (Oldani et al., 2009) and biofilm formation (Cole et al., 2004) have been studied. Maintenance of plasmids and mutated virulence genes cost energy (Martínez and Baquero, 2002). Therefore, in the absence of antibiotic selective pressure, higher level of virulence may still be maintained for the survival of bacteria could be attributed to the development of compensatory mutations (Martínez and Baquero, 2002). Analysis of H. pylori proteome may reveal patterns of compensatory mutations in response to metronidazole and fluoroquinolone resistance. An objective of this study was to determine the proteome of resistant H. pylori in response to amelioration of fitness cost as part of a compensatory response.
Materials and Methods
Induction of Levofloxacin Resistant Strains
H. pylori strains sensitive to levofloxacin were incubated at 37°C in a microaerophilic atmosphere of 10% CO2. They were inoculated into BHI broth with a series of levofloxacin concentrations (0.0156, 0.0313, 0.0625, 0.125, 0.25, 0.5, 1, 2, 4, 8) in a 96-well plate. After each passage, the cells would be exposed to increased concentration of levofloxacin. For example, the cells grew in the broth with 0.0156 μg/ml levofloxacin would be inoculated to the broth with 0.0313 μg/ml in the next growth passage. The bacterial suspensions were transferred to antibiotics-free chocolate agar to determine the minimum inhibitory concentration (MIC) of levofloxacin after each exposure. Stable induced-resistant strains would be grown on chocolate agar supplemented with levofloxacin to confirm resistance. The identity between resistant strains and their corresponding parental sensitive strains before induction were verified by random amplification of polymorphic DNA- polymerase chain reaction (RAPD-PCR) typing using primers 1254 and 1281 as described by Akopyanz et al. (1992) with the following modifications. The reaction mix (Thermo Fisher Scientific, USA) consisted of 1 × Taq buffer with KCl, 0.4 mM of each deoxynucleotide triphosphates (dNTPs), 3 mM MgCl2, 1 U Taq DNA polymerase, 0.2 μM of RAPD primer, 10 ng genomic DNA in a volume of 25 μl. The conditions for PCR amplification were denaturation at 95°C for 3 min, followed by 35 cycles of 95°C for 1 min, 38°C (for primer 1254) or 32°C (for primer 1281) for 1 min, 72°C for 2 min; and then a final extension at 72°C for 5 min.
Screening for Dual Population Metronidazole-Sensitive and Resistant Strains
For in vitro susceptibility testing of the H. pylori strains, a suspension equal to the McFarland tube no. 3 was prepared for each strain in BHI broth supplemented with 2% β-cyclodextrin and 0.4% yeast extract. The cell suspension was inoculated by confluent swabbing of the surface of non-selective chocolate agar with the adjusted inoculum suspensions. Metronidazole discs were laid on the agar surface. The plates were then be incubated at 37°C under microaerophilic conditions. The breakpoint zone diameter was recorded after 48–72 h of incubation. Colonies that grew within clear zones were isolated and grown on metronidazole-supplemented chocolate agar to confirm resistance. Colonies that grew around the clear zones were isolated and grown on non-selective and metronidazole-supplemented chocolate agar to confirm susceptibility. Resistant and susceptible isolates of the same strain from the antibiotics screening were selected for dual population study. Genomic DNA of H. pylori was extracted for PCR amplification and sequencing of gyrA, gyrB, rdxA, and frxA, which were performed as described by Teh et al. (2014).
H. pylori Strains and Growth
The H. pylori UM137, UM171, UM229, UM233, UM276 which consist of levofloxacin-sensitive and induced-resistant variant of each strain, and UM163, UM303, UM400, UM443 which consist of metronidazole-sensitive and naturally-occuring resistant variants of each strain were used in this study. UM276 was also resistant to metronidazole and UM229 was additionally resistant to clarithromycin. The 18 strains of H. pylori were categorized into sensitive and resistant groups based on their resistance to metronidazole, levofloxacin, clarithromycin, and multi-drug resistance (Figure 1). The H. pylori strains were cultured on chocolate agar for 3 days in microaerobic atmosphere at 37°C and the cells were harvested for protein extraction.

Figure 1. H. pylori strains according to sensitivity or resistance to metronidazolea, levofloxacin, clarithromycinb, and combined (multi-drugc) resistance. aUM276 and UM276L (levofloxacin-sensitive and resistant), UM163M, UM303M, UM400M, UM443M have naturally-occuring resistance to metronidazole. bUM229 and UM229L (levofloxacin-sensitive and resistant) has naturally-occuring resistance to clarithromycin. cResistant to metronidazole, levofloxacin, and/or clarithromycin.
Optical Density and Viable Count
H. pylori strains exhibiting dual phenotypes (sensitive and resistant) were incubated respectively in BHI broth supplemented with 10% fetal bovine serum and 0.4% yeast extract in a microaerobic atmosphere at 37°C. At t = 0 h, the optical density (OD) of the H. pylori suspensions was standardized to be at the same level at 600 nm by spectrophotometry. Samples were collected every 24 h over 7 days. The OD600 of the cell suspension was measured by spectrophotometry and viable count was performed to compare their growth curves; a serial dilution of the incubated samples was performed and the number of colony-forming units (CFU) was assessed after each time interval.
Apoptosis Assay
AGS cells, a gastric epithelial cell line, were infected in vitro with H. pylori susceptible- and its corresponding resistant-strains at a multiplicity of infection (MOI) of 50:1 bacteria to cell (Menaker et al., 2004). H. pylori was removed and the infected AGS cells were harvested at time intervals of t = 4, 8, and 12 h. The cells were stained with Annexin V Alexa Fluor® 488 and propidium iodide according to the instructions in Tali® apoptosis kit (Thermo Fisher Scientific). Annexin V-positive and propidium iodide-negative cells were considered as apoptotic cells (Sawai and Domae, 2011). The levels of apoptosis induced by the antibiotics-resistant H. pylori strain and its parental susceptible strain were compared using Tali® image-based cytometer (Thermo Fisher Scientific). Apoptosis induction is an indicator of the H. pylori virulence activity (Cho et al., 2003).
Biofilm Measurement
Various strains of H. pylori cultured on chocolate agar for 3 days were harvested and incubated for another 3 days in BHI broth supplemented with 2% β-cyclodextrin and 0.4% yeast extract. The total bacterial population for the cultures was standardized at the same OD600 value. A volume of 2 ml of each bacterial suspension was inoculated into each well of a 24-well plate. The growth of biofilm was observed after every 24 h over 7 days of incubation at 37°C in 10% CO2; the bacterial suspension was aspirated and 0.1% crystal violet was added into the well. The plate was gently agitated for 30 min and the crystal violet was removed. The crystal violet-treated wells were washed with distilled water and the stained biofilm was destained with 19:1 ethanol-acetic acid. The destaining solution was collected and its absorbance was measured at OD600. The amount of biofilm produced by antibiotic-resistant H. pylori strain and its sensitive strain was compared. The ability to form biofilm indicates the ability to survive against the activity of biocidal agents (Yonezawa et al., 2010).
Protein Extraction and Mass Spectrometry
H. pylori cell pellets were lysed and its protein was extracted using Norgen's Proteospin™ total protein purification kit (Norgen Biotek Corporation, Canada). Cells were resuspended in 50 μl lysis buffer and centrifuged at 14,000 × g for 2 min. The supernatant was transferred into a filter column fitted in an elution tube, and centrifuged at 14,000 × g for 1 min. One microliter of protease inhibitor (Halt Protease and Phosphatase Inhibitor; Thermo Fisher Scientific) was added to the tube. Protein concentrations were determined by Bradford assay (Bio-Rad, USA). Each tube of protein sample was reduced with a volume of 10 mM dithiothreitol (DTT; Bio-Rad), and alkylated in the dark with a volume of 20 mM iodoacetamide (IAA; Bio-Rad). The samples were incubated at room temperature for 30 min and added with Pierce™ trypsin protease (Thermo Scientific) to digest the proteins at 1:50 trypsin:protein. The samples were then incubated at 4°C for 30 min, and subsequently at 37°C for overnight. The extracted protein was then treated for liquid chromatography mass spectrophotometry according to a previous study (Chan et al., 2015).
Protein Profiling and Identification
Raw data was exported and sequencing was performed using PEAKS software (version 7.5; Bioinformatics Solution Inc., ON, Canada). The reference proteome database was H. pylori J99. Results were refined by using a false discovery rate (FDR) of 1%, and 1 unique peptide according to metronidazole- and levofloxacin-sensitive and resistant strain groups (Supplementary Material).
Proteins common to all strains in each group were selected (levofloxacin-sensitive, levofloxacin-resistant, metronidazole-sensitive, metronidazole-resistant) for protein expression analysis. Protein-protein interaction networks were analyzed by using STRING 10 (Szklarczyk et al., 2015). Proteins which were differentially expressed between sensitive and resistant groups were further analyzed for their functions in molecular and biological functions from UniProt database (The Uniprot Consortium, 2008) and pathways from KEGG database (Kanehisa et al., 2014), and possible roles in compensatory response.
Independence Test of Resistance and Genes
The up- or down-regulation of all expressed proteins in each strain was scored, and the difference between antibiotic-sensitive and -resistant strain groups was determined in terms of fold-expression. Proteins with the difference in scores of between 0.5- and 2 fold-difference were eliminated. The independence test of association between antibiotics resistance and the genes corresponding to the expressed proteins was determined by Fisher's exact test which was performed using SPSS (version 20; SPSS Inc, Chicago, USA). The antibiotics resistance in this test refers to the resistance against levofloxacin, metronidazole, clarithromycin or the combination resistance of any of the antibiotics (levofloxacin and clarithromycin, or levofloxacin, and metronidazole).
Results and Discussion
The emergence of resistant bacteria as a result of adaptability to antibiotics has no doubt been an emerging concern for human health. In turn, resistant bacteria presumably gained mutations that could ameliorate the reduced fitness as a result of its adaptation to antibiotics. These mutations were considered to be involved in compensatory mechanism that compensates for the fitness lost due to resistance (Handel et al., 2006). Compensatory mutations could occurred in regions of the genome due to selective pressure to compensate for the deleterious effects of the initial resistance mutations (Maisnier-Patin and Andersson, 2004). These mutations may be involved in any particular molecular or biological functions. A proteomic study on metronidazole-resistant H. pylori had examined metabolic changes in the bacteria which reported down- and up-regulation of various proteins including a protein with reductase activity (McAtee et al., 2001). The protein composition of the antibiotics resistant H. pylori may reveal the biological pathways involved in a compensatory response.
Functional Characteristics
Based on MIC, strains were considered to be levofloxacin-sensitive (<1 μg/ml) or resistant (≥1 μg/ml; Chisholm and Owen, 2009); or metronidazole-sensitive (<8 μg/ml) or resistant (≥8 μg/ml; Osato et al., 2001). Five levofloxacin-sensitive H. pylori strains with MIC ≤ 0.125 μg/ml were successfully induced in vitro to become resistant with MIC > 32 μg/ml, while four pairs of naturally-occurring metronidazole-resistant H. pylori strains with MIC > 256 μg/ml and metronidazole-sensitive strains with MIC ≤ 0.75 μg/ml were isolated for this study (Table 1). These four pairs of metronidazole-sensitive and -resistant strains were isolated after screening through 170 H. pylori positive gastric tissue biopsies collected from patients seen at the Endoscopy Unit (University of Malaya Medical Centre, Kuala Lumpur, Malaysia) during the period from July 2011 to June 2014. All pairs of sensitive and resistant strains for both antibiotics were verified to be identical by RAPD-PCR genotyping (Figure 2).
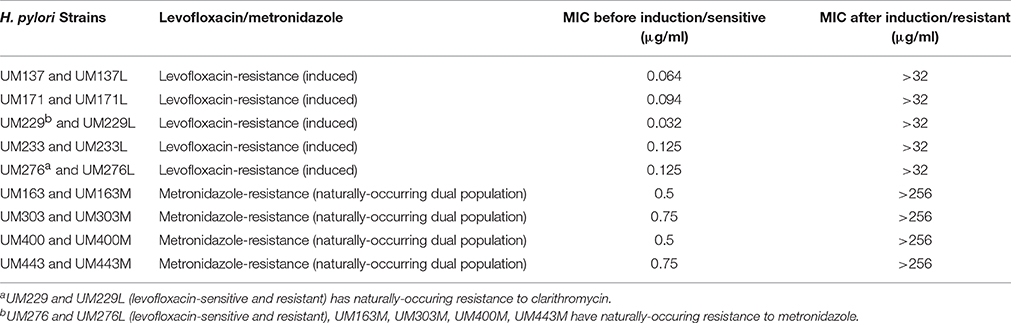
Table 1. MICs of H. pylori strains before and after levofloxacin-resistance in vitro induction (UM137, UM171, UM229, UM233, and UM276) and naturally-occuring metronidazole-sensitive and resistant H. pylori strains (UM163, UM303, UM400, UM443).
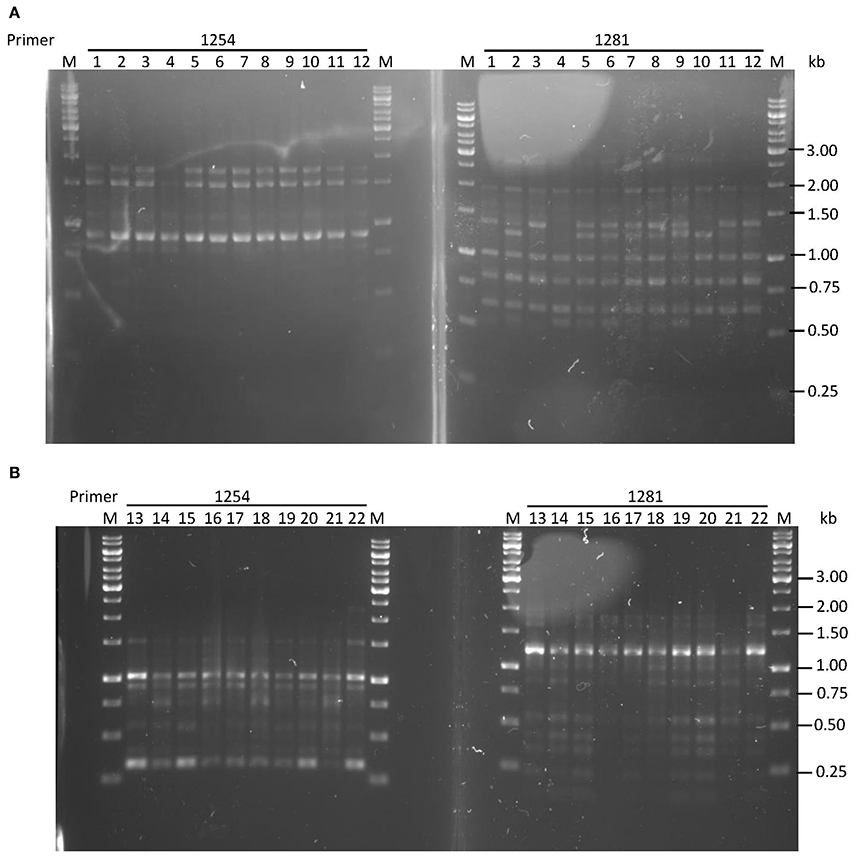
Figure 2. RAPD-PCR typing of (A) levofloxacin-induced H. pylori UM137 strains 1–8 sensitive, 9–12 resistant (B) metronidazole dual population of H. pylori UM163 strains 13–19 sensitive, 20–22 resistant. Primers used were 1254 and 1281, and M: 1 kb DNA ladder.
Determination of growth rate and generation time is often used to measure fitness costs associated with antibiotic resistance (Pope et al., 2010). The growth curves of these five pairs of levofloxacin-sensitive and resistant, and four pairs of metronidazole-sensitive and resistant H. pylori strains were not significantly different within pairs (p ≥ 0.05; Figure 3). Thus, change in antibiotics resistance status did not reduce the growth fitness of the resistant strains compared to its sensitive counterparts. However, a limitation of growth curve is that the competitive fitness of sensitive strains over its resistant counterparts could not be assessed.
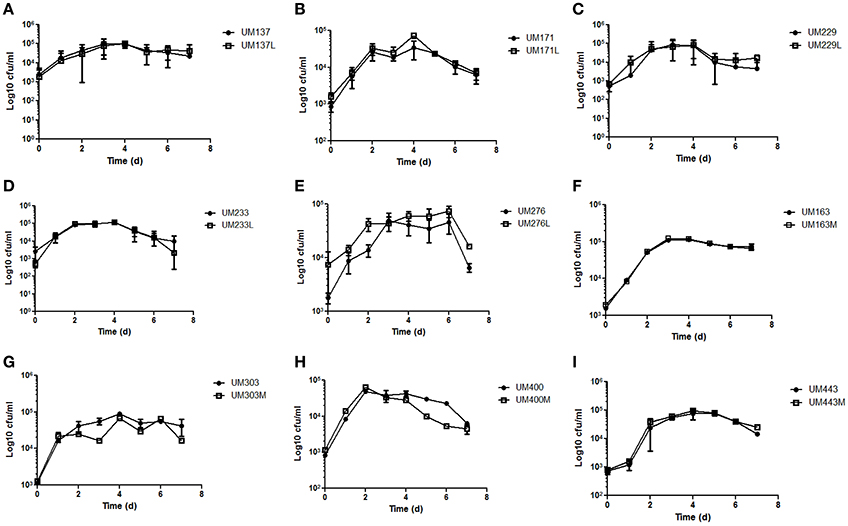
Figure 3. Growth curves of (A) UM137 (B) UM171 (C) UM229 (D) UM233 (E) UM276 (F) UM163 (G) UM303 (H) UM400 (I) UM443 H. pylori strains. Data represent mean ± SEM (n = 3).
Among the nine pairs of sensitive and resistant strains, only three pairs of dual strains showed significant change in their ability to induce apoptosis in AGS cells within pairs (p < 0.05). The level of apoptotic AGS cells induced by H. pylori infection post-12 h showed a decrease in the metronidazole resistant strains, UM163M and UM303M, and an increase in the resistant strain, UM400M, compared to their sensitive counterparts (Table 2). In contrast, none of the levofloxacin resistant strains showed significant change in ability to induce apoptosis compared to their sensitive counterparts. Uninfected cells acted as negative control while staurosporine-treated AGS cells act as the positive control (four-fold increase in apoptosis compared to untreated AGS cells).
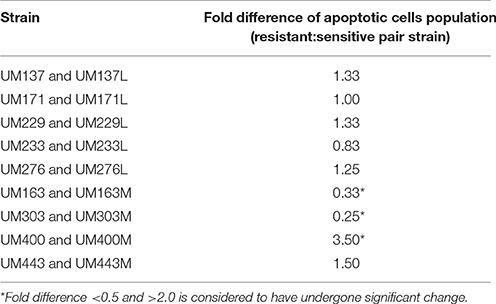
Table 2. Fold differences of the apoptotic AGS cells population induced by H. pylori from the ratio of resistant and sensitive pair strains, post-12 h of infection.
At population level, biofilm formation is a multicellular strategy for survival, and indirectly increases overall bacterial survival fitness. Bacterial biofilm presents as a physiological barrier at stationary growth phase against antibiotics whereby viable cells may persist due to the impenetrable structural matrix, the equilibrium of live and dead cells, or the regulation of genes expression (Normark and Normark, 2002). A majority of these strains were low-level biofilm formers, except for UM137, UM163, UM171, and UM443, and fold changes between the resistant and sensitive strains in the low-level biofilm forming pairs were not significantly different (p ≥ 0.05) (Table 3). Among the high-level biofilm forming strains, UM137L showed a significant decrease in biofilm formed. On the other hand, UM443M showed no significant increase of the biofilm formed over 7 days (p ≥ 0.05), while the biofilm formed by UM163M and UM171L were decreased compared to UM163 and UM171 respectively; the fold change did not reach statistical significance (p ≥ 0.05).
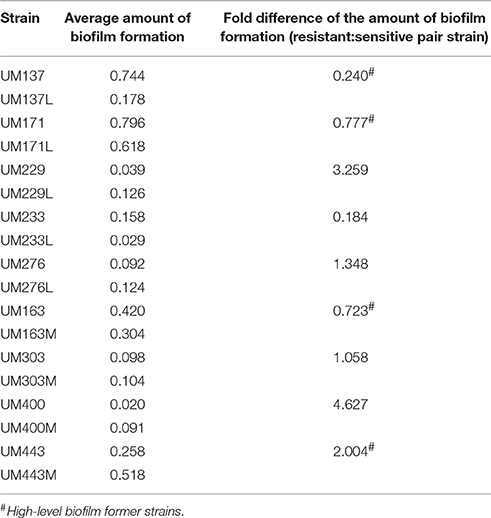
Table 3. Average biofilm formation of H. pylori strains over 7 days of growth and the fold difference between resistant and sensitive pair strains.
Molecular Characteristics
GyrA mutations occurred in the QRDR of the induced levofloxacin-resistant strains at N87K for UM137L and UM276L, A88V for UM233L, and D91N for UM171L and UM229L, D155N for UM137L and UM233L whereas substitutions S429T and R484K occurred in gyrB of UM233L (Figure 4). On the other hand, no specific mutations occurred in naturally-occuring metronidazole resistant strains, except for A40V in frxA of UM443M. Thus, in vitro induction of levofloxacin resistance could be explained by known gyrA and/or gyrB mutations while the naturally-occurring dual population metronidazole resistance could be caused by mutations in other genes or may involve other mechanisms.

Figure 4. Amino acid sequences of levofloxacin resistance-associated gene regions: (A) gyrA, (B) gyrB, and metronidazole resistance-associated gene regions: (C) rdxA, (D) frxA of H. pylori strains. gyrA and gyrB sequences were compared to UM038 strain (positive gyrA mutation), and rdxA and frxA sequences were compared to a reference strain, 26695. Positions of expected mutations associated with levofloxacin are in box outlines, and positions of variations of mutation in either antibiotic-resistant strains are in gray boxes.
Inter-Gene or Intra-Molecular Protein Interactions
In order to explore the underlying compensatory mechanisms adopted by antibiotic resistant H. pylori to maintain their overall fitness, protein profiling on the nine pairs of sensitive and resistant H. pylori strains was performed. All expressed proteins with significant association with levofloxacin, metronidazole, clarithromycin, and/or any combination of them (levofloxacin and clarithromycin, or levofloxacin, and metronidazole) were identified using Fisher's Exact test with p < 0.05 considered to be significant (Table 4).
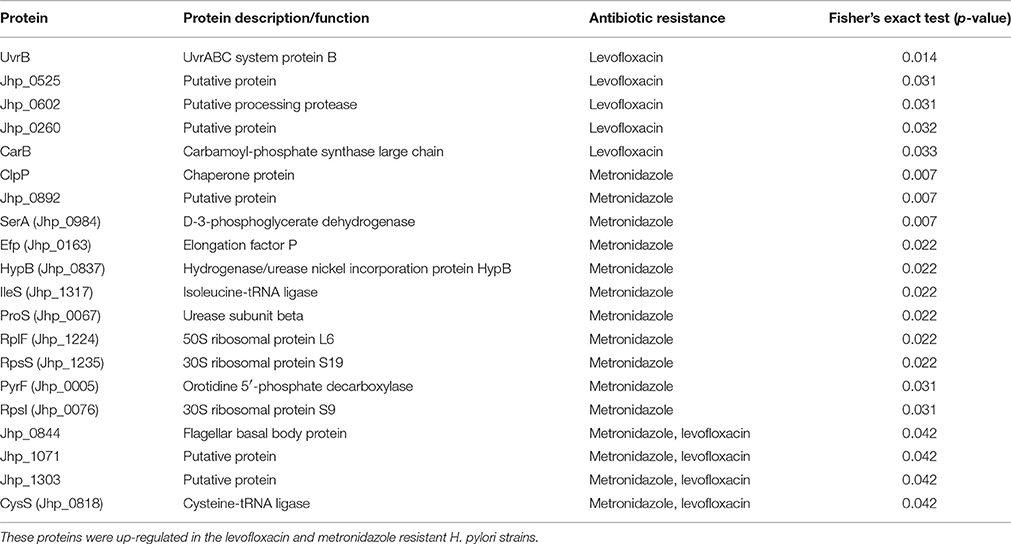
Table 4. Proteins of significant association with resistance to metronidazole, levofloxacin, and/or clarithromycin in all of the 18 H. pylori strains tested using Fisher's exact test (2-tailed, p < 0.05 is considered significant).
From the prediction of possible protein-protein interactions by STRING, changes in protein expression profiles of metronidazole-resistant strains appeared to be functionally related compared to levofloxacin-resistant strains (Figure 5). Metronidazole resistance associated proteins are involved in translation, ATP binding, ligase activity, aminoacyl-tRNA editing activity, rRNA binding, or structural constituent of ribosome (Table 5). These up-regulated proteins may either represent an alternative mechanism of metronidazole resistance in a dual population environment without genetic mutations in rdxA and frxA and/ or as a compensatory mechanism underlying metronidazole resistance. The alteration in protein profile may explain the dual population phenomenon whereby some bacterial cells were more resistant to antibiotics than others despite having similar genetic makeup.
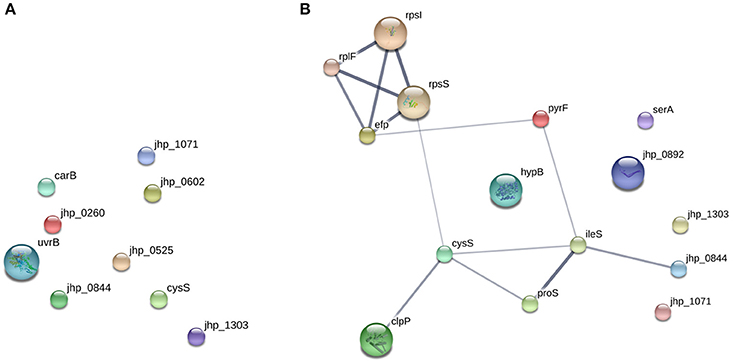
Figure 5. STRING protein-protein interaction network of proteins from PEAKS refined data. The gene names correspond with the proteins that were recovered from (A) levofloxacin and (B) metronidazole-sensitive and resistant strains.
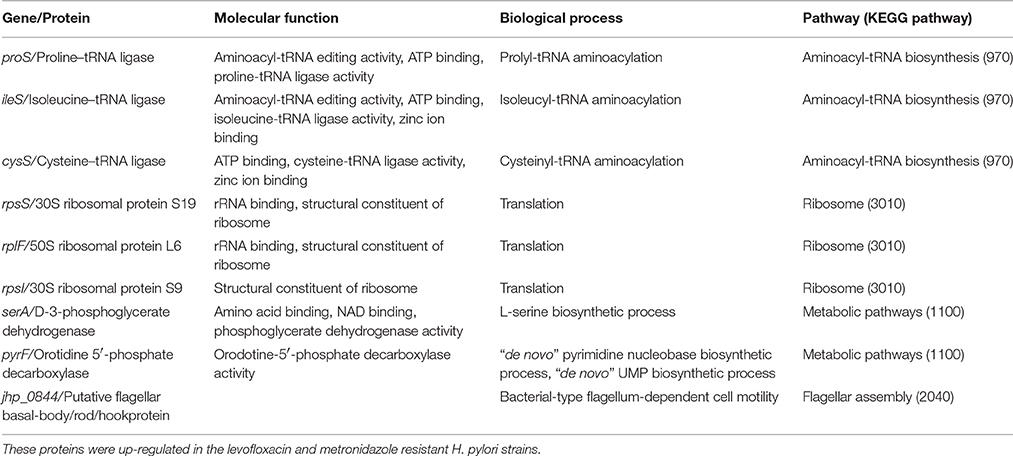
Table 5. Proteins with differential expression among nine pairs of levofloxacin and metronidazole-resistant and sensitive strains.
It has been suggested that the presence of different proteins in strains with different antibiotic resistance may be attributed to epistasis (Salverda et al., 2011). The effect of compensatory response against antibiotic resistance may be due to the interactions between the primary resistance genes and the secondary mutated genes (Moore et al., 2000; Baker et al., 2013). In addition, the association of genes with the sensitive-resistant groups of different antibiotic groups may be due to the effect of either positive or negative epistasis (Schenk et al., 2013). It may also be possible that these compensatory protein responses are in the vicinity of their expressed resistance mutations in their primary or tertiary protein structure (Davis et al., 2009). This may explain the levofloxacin sensitive strains protein expression profile, which seem to be functionally random, as compared to that of the metronidazole resistant strains. Afterall, point mutations in gyrA and gyrB, which are responsible for levofloxacin resistence, also occurred at random positions along the genes. These point mutations at different positions may alter the interaction and functions of these proteins differently.
Protein Translation of Antibiotic Resistant Strains
ProS, IleS, and CysS are aminoacyl-tRNA ligases (also known as aminoacyl-tRNA synthetases) that are involved in the synthesis of aminoacyl-tRNAs, which bind to ribosomes during the translation process (Hendrickson and Schimmel, 2003). In the current study, these aminoacyl-tRNA synthetases were found to be expressed in metronidazole resistant H. pylori strains (except CysS that was also expressed in levofloxacin resistant strains) but not in sensitive strains. In Clostridium difficile, aminoacyl-tRNA synthetases, CysS, and SerS, were highly expressed in bacterial strains that exhibit resistance to metronidazole (Moura et al., 2014). Interestingly, it was also shown that these aminoacyl-tRNA synthetases were not detected the same metronidazole-resistant C. difficle when cultured in the presence of the antibiotic (Moura et al., 2014). In the current study, in order to compare the protein profiles between sensitive and resistant pair of strains, bacteria for proteomics profiling were cultured in the absence of antibiotics.
Ribosomal proteins (RpsS, RplF, and RpsI) were expressed in metronidazole resistant H. pylori strains but not in sensitive strains.
Different expressions of ribosomal proteins have been associated with bacterial fitness (Lind et al., 2010). It has been shown that the alteration of rplF gene (50S ribosomal protein L6) in Escherichia coli mutants interrupted cell proliferation since this protein plays an important role in E. coli 50S subunit assembly (Shigeno et al., 2016). Similarly, down-regulation of RpsI (30S ribosomal protein S9) has been associated with decreasing rate of protein synthesis in E. coli (Dabbs, 1983; Hoang et al., 2004).
Elongation factor P (EFP), which is a prokaryotic protein translation factor required for efficient peptide bond synthesis on 70S ribosomes (Blaha et al., 2009), was also associated with metronidazole resistance in H. pylori further suggesting that protein translation were up-regulated in antibiotic resistant strains. ATP-dependent caseinolytic proteases that acts both as a chaperone and as an ATPase driving the degradation of damaged or mis-made proteins has been demonstrated to play a role in modulation susceptibility to antibiotics causing protein damage and/or oxidative stress (Loughlin et al., 2009). From our results, ClpP was found to be expressed in metronidazole resistant strains indicating that the ability to degrade non-functional proteins is an important part of the mechanism. Thus, we hypothesized that in a dual population environment, bacterial cells with more efficient translation machinery were better at overcoming the action of metronidazole, which targets bacterial DNA. This may represent an alternative mechanism that could be used by H. pylori to counter the actions of metronidazole. Unlike mutations in frxA and rdxA, this alternative mechanism of metronidazole resistance will be harder to detect.
Metabolism of Antibiotic Resistant Strains
SerA (D-3-phosphoglycerate dehydrogenase) is an enzyme that is involved in the conversion of 3-phosphoglycerate into 3-phosphohydroxypyruvate, one of the steps in serine biosynthesis pathway (Shimizu et al., 2008). PyrF (orotidine 5′-phosphate decarboxylase) is known to be an efficient enzyme in the catalyzation of orotidine 5′-monophosphate to uridine 5′monophosphate (Harris et al., 2000) and plays a role in pyrimidine metabolism (Capone et al., 2007). Bacteria has been suggested of undergoing different levels of metabolism when proteins involved in amino acid metabolism are up-regulated (Sauer et al., 2002; Drenkard, 2003). In this study, SerA and PyrF were shown to be associated with metronidazole resistance in H. pylori. Alteration in metabolic pathways of antibiotic resistant strains may be a compensatory response in order to balance the energy production associated with antibiotic resistance. The higher cost of fitness needs to be compensated thus enabling a higher survival rate of the bacteria.
Conclusion
Growth rates of both induced and naturally occurring levofloxacin and metronidazole resistant strains of H. pylori strains were comparable with their respective parental strains indicating that the acquisition of antibiotic resistance was well-compensated with no loss of growth fitness. However, virulence and biofilm forming abilities were altered in certain antibiotic resistant strains compared to their parental strains suggesting that gain or loss of virulence and survival fitness in H. pylori is strain specific reflecting the genetic diversity among H. pylori strains. Our results may also suggest that in a dual population environment, higher protein translation, and non-functional protein degradation capabilities may be used by H. pylori to as an alternative strategy to counter the action of metronidazole in the absence of mutations in frxA and rdxA. In order to maintain energy balance and fitness, metabolic pathways may be altered in compensation. In comparison to metronidazole, compensation for levofloxacin resistance, which involves mutations in gyrA and gyrB, the compensatory mechanism was functionally more random as suggested by their proteomic profiles.
Author Contributions
PL, FM, ML, and KG conceived the work. AH, AS, and MD conducted the levofloxacin resistance induction and AH performed the growth, apoptosis, biofilm formation assays. XT performed the PCR amplification of the resistance-associated genes. ML and AH performed the protein profiling and bioinformatics analysis. AH, WL, ML, AL, and JV contributed to the preparation of the manuscript. All authors participated in the critical review of this manuscript.
Funding
This project was funded by the Ministry of Education, Malaysia through University of Malaya High Impact Research Grant UM.C/625/1/HIR/MoE/CHAN/13/2 (Account No. H-50001-A000032) and University of Malaya Research Grant (UMRG) Reference No. RP016B-13HTM.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2016.02015/full#supplementary-material
References
Akopyanz, N., Bukanov, N. O., Westblom, T. U., Kresovich, S., and Berg, D. E. (1992). DNA diversity among clinical isolates of Helicobacter pylori detected by PCR-based RAPD fingerprinting. Nucleic Acids Res. 20, 5137–5142. doi: 10.1093/nar/20.19.5137
Baker, S., Duy, P. T., Nga, T. V., Dung, T. T. N., Phat, V. V., Chau, T. T. A., et al. (2013). Fitness benefits in fluoroquinolone-resistant Salmonella typhi in the absence of antimicrobial pressure. Elife 2:e01229. doi: 10.7554/eLife.01229
Binh, T. T., Shiota, S., Nguyen, L. T., Ho, D. D., Hoang, H. H., Ta, L., et al. (2013). The incidence of primary antibiotic resistance of Helicobacter pylori in Vietnam. J. Clin. Gastroenterol. 47, 233–238. doi: 10.1097/MCG.0b013e3182676e2b
Binh, T. T., Suzuki, R., Trang, T. T., Kwon, D. H., and Yamaoka, Y. (2015). Search for novel candidate mutations for metronidazole resistance in Helicobacter pylori using next-generation sequencing. Antimicrob. Agents Chemother. 59, 2343–2348. doi: 10.1128/AAC.04852-14
Björkholm, B., Sjölund, M., Falk, P. G., Berg, O. G., Engstrand, L., and Andersson, D. I. (2001). Mutation frequency and biological cost of antibiotic resistance in Helicobacter pylori. Proc. Natl. Acad. Sci. U.S.A. 98, 14607–14612. doi: 10.1073/pnas.241517298
Blaha, G., Stanley, R. E., and Steitz, T. A. (2009). Formation of the first peptide bond: the structure of EF-P bound to the 70S ribosome. Science 325, 966–970. doi: 10.1126/science.1175800
Cammarota, G., Cianci, R., Cannizzaro, O., Cuoco, L., Pirozzi, G., Gasbarrini, A., et al. (2000). Efficacy of two one-week rabeprazole/levofloxacin-based triple therapies for Helicobacter pylori infection. Aliment. Pharmacol. Ther. 14, 1339–1343. doi: 10.1046/j.1365-2036.2000.00846.x
Capone, R. F., Ning, Y., Pakulis, N., Alhazzazi, T., and Fenno, J. C. (2007). Characterization of Treponema denticola pyrF encoding orotidine-5′- monophosphate decarboxylase. FEMS Microbiol. Lett. 268, 261–267. doi: 10.1111/j.1574-6968.2006.00589.x
Chan, K. G., Loke, M. F., Ong, B. L., Wong, Y. L., Hong, K. W., Tan, K. H., et al. (2015). Multiphasic strain differentiation of atypical mycobacteria from elephant trunk wash. Peer J. 3:e1367. doi: 10.7717/peerj.1367
Chisholm, S. A., and Owen, R. J. (2009). Frequency and molecular characteristics of ciprofloxacin- and rifampicin-resistant Helicobacter pylori from gastric infections in the UK. J. Med. Microbiol. 58, 1322–1328. doi: 10.1099/jmm.0.011270-0
Cho, S. J., Kang, N. S., Park, S. Y., Kim, B. O., Rhee, D. K., and Pyo, S. (2003). Induction of apoptosis and expression of apoptosis related genes in human epithelial carcinoma cells by Helicobacter pylori VacA toxin. Toxicon 42, 601–611. doi: 10.1016/j.toxicon.2003.08.003
Cole, S. P., Harwood, J., Lee, R., She, R., and Guiney, D. G. (2004). Characterization of monospecies biofilm formation by Helicobacter pylori. J. Bacteriol. 186, 3124–3132. doi: 10.1128/JB.186.10.3124-3132.2004
Dabbs, E. R. (1983). Escherichia coli kasugamycin dependence arising from mutation at the rpsI locus. J. Bacteriol. 153, 709–715.
Davis, B. H., Poon, A. F., and Whitlock, M. C. (2009). Compensatory mutations are repeatable and clustered within proteins. Proc. Biol. Sci. 276, 1823–1827. doi: 10.1098/rspb.2008.1846
De Francesco, V., Giorgio, F., Hassan, C., Manes, G., Vannella, L., Panella, C., et al. (2010). Worldwide, H. pylori antibiotic resistance: a systematic review. J. Gastrointest. Liver Dis. 19, 409–414. doi: 10.4292/WJGPT.v3.i4.68
Dhar, S. K., Soni, R. K., Das, B. K., and Mukhopadhyay, G. (2003). Molecular mechanism of action of major Helicobacter pylori virulence factors. Mol. Cell. Biochem. 253, 207–215. doi: 10.1023/A:1026051530512
Drenkard, E. (2003). Antimicrobial resistance of Pseudomonas aeruginosa biofilms. Microbes Infect. 5, 213–219. doi: 10.1016/j.micinf.2003.08.009
Goodwin, A., Kersulyte, D., Sisson, G., Veldhuyzen van Zanten, S. J., Berg, D. E., and Hoffman, P. S. (1998). Metronidazole resistance in Helicobacter pylori is due to null mutations in a gene (rdxA) that encodes an oxygen-insensitive NADPH nitroreductase. Mol. Microbiol. 28, 383–393. doi: 10.1046/j.1365-2958.1998.00806.x
Graham, D. Y., and Fischbach, L. (2010). Helicobacter pylori treatment in the era of increasing antibiotic resistance. Gut 59, 1143–1153. doi: 10.1136/gut.2009.192757
Handel, A., Regoes, R. R., and Antia, R. (2006). The role of compensatory mutations in the emergence of drug resistance. PLoS Comput. Biol. 2:e137. doi: 10.1371/journal.pcbi.0020137
Harris, P., Navarro Poulsen, J. C., Jensen, K. F., and Larsen, S. (2000). Structural basis for the catalytic mechanism of a proficient enzyme: orotidine 5′-monophosphate decarboxylase. Biochemistry 39, 4217–4224. doi: 10.1021/bi992952r
Hendrickson, T. L., and Schimmel, P. (2003). “Transfer RNA-dependent amino acid discrimination by aminoacyl-tRNA synthetases,” in Translation Mechanisms, eds J. Lapointe and L. Brakier-Gingras (Georgetown, TX: Kluwer Academic Plenum Publishers), 34–64.
Heo, J., and Jeon, S. W. (2014). Optimal treatment strategy for Helicobacter pylori: era of antibiotic resistance. World J. Gastroenterol. 20, 5654–5659. doi: 10.3748/wjg.v20.i19.5654
Hoang, L., Fredrick, K., and Noller, H. F. (2004). Creating ribosomes with an all-RNA 30S subunit P site. Proc. Natl. Acad. Sci. U.S.A. 101, 12439–12443. doi: 10.1073/pnas.0405227101
Justino, M. C., Parente, M. R., Boneca, I. G., and Saraiva, L. M. (2014). FrxA is an s-nitrosoglutathione reductase enzyme that contributes to Helicobacter pylori pathogenicity. FEBS J. 281, 4495–4505. doi: 10.1111/febs.12958
Kanehisa, M., Goto, S., Sato, Y., Kawashima, M., Furumichi, M., and Tanabe, M. (2014). Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 42, D199–D205. doi: 10.1093/nar/gkt1076
Kwon, D. H., Kato, M., El-Zaatari, F. A., Osato, M. S., and Graham, D. Y. (2000). Frame-shift mutations in NAD(P)H flavin oxidoreductase encoding gene (frxA) from metronidazole resistant Helicobacter pylori ATCC43504 and its involvement in metronidazole resistance. FEMS Microbiol. Lett. 188, 197–202. doi: 10.1111/j.1574-6968.2000.tb09193.x
Lee, J. W., Kim, N., Nam, R. H., Park, J. H., Kim, J. M., Jung, H. C., et al. (2011). Mutations of Helicobacter pylori associated with fluoroquinolone resistance in Korea. Helicobacter 16, 301–310. doi: 10.1111/j.1523-5378.2011.00840.x
Lind, P. A., Berg, O. G., and Andersson, D. I. (2010). Mutational robustness of ribosomal protein genes. Science 330, 825–827. doi: 10.1126/science.1194617
Lind, T., Mégraud, F., Unge, P., Bayerdörffer, E., O'morain, C., Spiller, R., et al. (1999). The MACH2 study: role of omeprazole in eradication of Helicobacter pylori with 1-week triple therapies. Gastroenterology 116, 248–253. doi: 10.1016/S0016-5085(99)70119-8
Liu, G., Xu, X., He, L., Ding, Z., Gu, Y., Zhang, J., et al. (2011). Primary antibiotic resistance of Helicobacter pylori isolated from Beijing children. Helicobacter 16, 356–362. doi: 10.1111/j.1523-5378.2011.00856.x
Loughlin, M. F., Arandhara, V., Okolie, C., Aldsworth, T. G., and Jenks, P. J. (2009). Helicobacter pylori mutants defective in the clpP ATP-dependant protease and the chaperone clpA display reduced macrophage and murine survival. Microb. Pathog. 46, 53–57. doi: 10.1016/j.micpath.2008.10.004
Maisnier-Patin, S., and Andersson, D. I. (2004). Adaptation to the deleterious effects of antimicrobial drug resistance mutations by compensatory evolution. Res. Microbiol. 155, 360–369. doi: 10.1016/j.resmic.2004.01.019
Malfertheiner, P., Megraud, F., O'Morain, C., Bazzoli, F., El-Omar, E., Graham, D., et al. (2007). Current concepts in the management of Helicobacter pylori infection: the Maastricht III consensus report. Gut 56, 772–781. doi: 10.1136/gut.2006.101634
Martínez, J. L., and Baquero, F. (2002). Interactions among strategies associated with bacterial infection: pathogenicity, epidemicity, and antibiotic resistance. Clin. Microbiol. Rev. 15, 647–679. doi: 10.1128/CMR.15.4.647-679.2002
McAtee, C. P., Hoffman, P. S., and Berg, D. E. (2001). Identification of differentially regulated proteins in metronidozole resistant Helicobacter pylori by proteome techniques. Proteomics 1, 516–521. doi: 10.1002/1615-9861(200104)1:4<516::AID-PROT516>3.0.CO;2-O
Megraud, F., Coenen, S., Versporten, A., Kist, M., Lopez-Brea, M., Hirschl, A. M., et al. (2013). Helicobacter pylori resistance to antibiotics in Europe and its relationship to antibiotic consumption. Gut 62, 34–42. doi: 10.1136/gutjnl-2012-302254
Mégraud, F., and Lehours, P. (2007). Helicobacter pylori detection and antimicrobial susceptibility testing. Clin. Microbiol. Rev. 20, 280–322. doi: 10.1128/CMR.00033-06
Menaker, R. J., Ceponis, P. J., and Jones, N. L. (2004). Helicobacter pylori induces apoptosis of macrophages in association with alterations in the mitochondrial pathway. Infect. Immun. 72, 2889–2898. doi: 10.1128/IAI.72.5.2889-2898.2004
Miyachi, H., Miki, I., Aoyama, N., Shirasaka, D., Matsumoto, Y., Toyoda, M., et al. (2006). Primary levofloxacin resistance and gyrA/B mutations among Helicobacter pylori in japan. Helicobacter 11, 243–249. doi: 10.1111/j.1523-5378.2006.00415.x
Moore, F. B., Rozen, D. E., and Lenski, R. E. (2000). Pervasive compensatory adaptation in Escherichia coli. Proc. Biol. Sci. 267, 515–522. doi: 10.1098/rspb.2000.1030
Moura, I., Monot, M., Tani, C., Spigaglia, P., Barbanti, F., Norais, N., et al. (2014). Multidisciplinary analysis of a nontoxigenic Clostridium difficile strain with stable resistance to metronidazole. Antimicrob. Agents Chemother. 58, 4957–4960. doi: 10.1128/AAC.02350-14
Normark, B. H., and Normark, S. (2002). Evolution and spread of antibiotic resistance. J. Intern. Med. 252, 91–106. doi: 10.1046/j.1365-2796.2002.01026.x
Oldani, A., Cormont, M., Hofman, V., Chiozzi, V., Oregioni, O., Canonici, A., et al. (2009). Helicobacter pylori counteracts the apoptotic action of its VacA toxin by injecting the CagA protein into gastric epithelial cells. PLoS Pathog. 5:e1000603. doi: 10.1371/journal.ppat.1000603
Olekhnovich, I. N., Goodwin, A., and Hoffman, P. S. (2009). Characterization of the NAD(P)H oxidase and metronidazole reductase activities of the RdxA nitroreductase of Helicobacter pylori. FEBS J. 276, 3354–3364. doi: 10.1111/j.1742-4658.2009.07060.x
Osato, M. S., Reddy, R., Reddy, S. G., Penland, R. L., and Graham, D. Y. (2001). Comparison of the Etest and the NCCLS-approved agar dilution method to detect metronidazole and clarithromycin resistant Helicobacter pylori. Int. J. Antimicrob. Agents 17, 39–44. doi: 10.1016/S0924-8579(00)00320-4
Parsonnet, J., Friedman, G. D., Vandersteen, D. P., Chang, Y., Vogelman, J. H., Orentreich, N., et al. (1991). Helicobacter pylori infection and the risk of gastric carcinoma. N. Engl. J. Med. 325, 1127–1131. doi: 10.1056/NEJM199110173251603
Pope, C. F., McHugh, T. D., and Gillespie, S. H. (2010). “Methods to determine fitness in bacteria,” in Antibiotic Resistance Protocols, 2nd Edn, eds S. H. Gillespie, and T. D. McHugh (New York NY: Humana Press), 113–121. doi: 10.1007/978-1-60327-279-7_9
Rozen, D. E., McGee, L., Levin, B. R., and Klugman, K. P. (2007). Fitness costs of fluoroquinolone resistance in Streptococcus pneumoniae. Antimicrob. Agents Chemother. 51, 412–416. doi: 10.1128/AAC.01161-06
Salverda, M. L., Dellus, E., Gorter, F. A., Debets, A. J., van der Oost, J., Hoekstra, R. F., et al. (2011). Initial mutations direct alternative pathways of protein evolution. PLoS Genet. 7:e1001321. doi: 10.1371/journal.pgen.1001321
Sauer, K., Camper, A. K., Ehrlich, G. D., Costerton, J. W., and Davies, D. G. (2002). Pseudomonas aeruginosa displays multiple phenotypes during development as a biofilm. J. Bacteriol. 184, 1140–1154. doi: 10.1128/jb.184.4.1140-1154.2002
Sawai, H., and Domae, N. (2011). Discrimination between primary necrosis and apoptosis by necrostatin-1 in Annexin V-positive/propidium iodide-negative cells. Biochem. Biophys. Res. Commun. 411, 569–573. doi: 10.1016/j.bbrc.2011.06.186
Schenk, M. F., Szendro, I. G., Salverda, M. L., Krug, J., and de Visser, J. A. (2013). Patterns of epistasis between beneficial mutations in an antibiotic resistance gene. Mol. Biol. Evol. 30, 1779–1787. doi: 10.1093/molbev/mst096
Shigeno, Y., Uchiumi, T., and Nomura, T. (2016). Involvement of ribosomal protein L6 in assembly of functional 50S ribosomal subunit in Escherichia coli cells. Biochem. Biophys. Res. Commun. 473, 237–242. doi: 10.1016/j.bbrc.2016.03.085
Shimizu, Y., Sakuraba, H., Doi, K., and Ohshima, T. (2008). Molecular and functional characterization of D-3-phosphoglycerate dehydrogenase in the serine biosynthetic pathway of the hyperthermophilic archaeon Sulfolobus tokodaii. Arch. Biochem. Biophys. 470, 120–128. doi: 10.1016/j.abb.2007.11.010
Sisson, G., Jeong, J. Y., Goodwin, A., Bryden, L., Rossler, N., Lim-Morrison, S., et al. (2000). Metronidazole activation is mutagenic and causes DNA fragmentation in Helicobacter pylori and in Escherichia coli containing a cloned H. pylori rdxA(+) (nitroreductase) gene. J. Bacteriol. 182, 5091–5096. doi: 10.1128/JB.182.18.5091-5096.2000
Su, P., Li, Y., Li, H., Zhang, J., Lin, L., Wang, Q., et al. (2013). Antibiotic resistance of Helicobacter pylori isolated in the southeast coastal region of China. Helicobacter 18, 274–279. doi: 10.1111/hel.12046
Szklarczyk, D., Franceschini, A., Wyder, S., Forslund, K., Heller, D., Huerta-Cepas, J., et al. (2015). STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43, D447–D452. doi: 10.1093/nar/gku1003
Teh, X., Khosravi, Y., Lee, W. C., Leow, A. H., Loke, M. F., Vadivelu, J., et al. (2014). Functional and molecular surveillance of Helicobacter pylori antibiotic resistance in Kuala Lumpur. PLoS ONE 9:e101481. doi: 10.1371/journal.pone.0101481
The Uniprot Consortium (2008). The universal protein resource (UniProt). Nucleic Acids Res. 36, D190–D195. doi: 10.1093/nar/gkl929
Keywords: Helicobacter pylori, levofloxacin, metronidazole, bacterial fitness, antibiotic resistance, proteome
Citation: Hanafi A, Lee WC, Loke MF, Teh X, Shaari A, Dinarvand M, Lehours P, Mégraud F, Leow AHR, Vadivelu J and Goh KL (2016) Molecular and Proteomic Analysis of Levofloxacin and Metronidazole Resistant Helicobacter pylori. Front. Microbiol. 7:2015. doi: 10.3389/fmicb.2016.02015
Received: 03 June 2016; Accepted: 01 December 2016;
Published: 15 December 2016.
Edited by:
Patrick Rik Butaye, Ghent University, BelgiumReviewed by:
Santi M. Mandal, Vidyasagar University, IndiaMarie-Cecile Ploy, University of Limoges, France
Copyright © 2016 Hanafi, Lee, Loke, Teh, Shaari, Dinarvand, Lehours, Mégraud, Leow, Vadivelu and Goh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jamuna Vadivelu, amFtdW5hQHVtbWMuZWR1Lm15