 Patrick Buerger1,2,3*
Patrick Buerger1,2,3* Elisha M. Wood-Charlson4
Elisha M. Wood-Charlson4 Karen D. Weynberg1
Karen D. Weynberg1 Bette L. Willis3,5
Bette L. Willis3,5 Madeleine J. H. van Oppen1,6
Madeleine J. H. van Oppen1,6- 1Australian Institute of Marine Science (AIMS), Townsville, QLD, Australia
- 2Australian Institute of Marine Science, James Cook University (AIMS@JCU), Townsville, QLD, Australia
- 3College of Science and Engineering, James Cook University (JCU), Townsville, QLD, Australia
- 4Center for Microbial Oceanography: Research and Education, University of Hawaii, Honolulu, HI, USA
- 5Australian Research Council (ARC) Centre of Excellence for Coral Reef Studies, College of Science and Engineering, Townsville, QLD, Australia
- 6School of BioSciences, University of Melbourne, Melbourne, VIC, Australia
Understanding how pathogens maintain their virulence is critical to developing tools to mitigate disease in animal populations. We sequenced and assembled the first draft genome of Roseofilum reptotaenium AO1, the dominant cyanobacterium underlying pathogenicity of the virulent coral black band disease (BBD), and analyzed parts of the BBD-associated Geitlerinema sp. BBD_1991 genome in silico. Both cyanobacteria are equipped with an adaptive, heritable clustered regularly interspaced short palindromic repeats (CRISPR)-Cas defense system type I-D and have potential virulence genes located within several prophage regions. The defense system helps to prevent infection by viruses and mobile genetic elements via identification of short fingerprints of the intruding DNA, which are stored as templates in the bacterial genome, in so-called “CRISPRs.” Analysis of CRISPR target sequences (protospacers) revealed an unusually high number of self-targeting spacers in R. reptotaenium AO1 and extraordinary long CRIPSR arrays of up to 260 spacers in Geitlerinema sp. BBD_1991. The self-targeting spacers are unlikely to be a form of autoimmunity; instead these target an incomplete lysogenic bacteriophage. Lysogenic virus induction experiments with mitomycin C and UV light did not reveal an actively replicating virus population in R. reptotaenium AO1 cultures, suggesting that phage functionality is compromised or excision could be blocked by the CRISPR-Cas system. Potential prophages were identified in three regions of R. reptotaenium AO1 and five regions of Geitlerinema sp. BBD_1991, containing putative BBD relevant virulence genes, such as an NAD-dependent epimerase/dehydratase (a homolog in terms of functionality to the third and fourth most expressed gene in BBD), lysozyme/metalloendopeptidases and other lipopolysaccharide modification genes. To date, viruses have not been considered to be a component of the BBD consortium or a contributor to the virulence of R. reptotaenium AO1 and Geitlerinema sp. BBD_1991. We suggest that the presence of virulence genes in potential prophage regions, and the CRISPR-Cas defense systems are evidence of an arms race between the respective cyanobacteria and their bacteriophage predators. The presence of such a defense system likely reduces the number of successful bacteriophage infections and mortality in the cyanobacteria, facilitating the progress of BBD.
Introduction
Diseases have become a major contributor to coral mortality over the last few decades (Willis et al., 2004; Rosenberg et al., 2007; Bourne et al., 2009). Many disease outbreaks can be linked to environmental and biological stressors, including direct human activities (Aeby et al., 2011; Lamb and Willis, 2011; Lamb et al., 2014), elevated seawater temperatures (Bruno et al., 2007; Maynard et al., 2015), and decreasing water quality (reviewed in Sutherland et al., 2004). Environmental perturbations are expected to increase in severity and frequency over time and are predicted to exacerbate the prevalence of coral diseases in the future (Selig et al., 2006; Aeby et al., 2011). While around 20 coral diseases have been described since the early 1970s, our understanding of their causative agents and pathogenesis is still limited (Harvell et al., 2007; Sheridan et al., 2013). Only a profound understanding of disease etiologies and diagnostics will lead to effective and adequate mitigation strategies (Pollock et al., 2011).
Black band disease (BBD) is one of the earliest described coral diseases (Antonius, 1973) and the most widely reported (Green and Bruckner, 2000; Page and Willis, 2006; Barneah et al., 2007; Aeby et al., 2015; Johan et al., 2015). The disease consists of a microbial consortium of cyanobacterial species, sulfate -reducing and -oxidizing bacteria, Alphaproteobacteria, Cytophaga, as well as other heterotrophic bacteria (Cooney et al., 2002; Miller and Richardson, 2011; Sato et al., 2011). The bacteria form a black mat (Figure 1) that is characterized by an anoxic, sulfide gradient towards the coral surface, which is lethal for the underlying coral tissue, and that progresses over the colony surface at rates up to 2 cm per day (reviewed by Sato et al., 2016). In terms of biomass, most abundant in the disease mat is a filamentous cyanobacterium, Roseofilum reptotaenium (Rasoulouniriana) Casamatta (Rasoulouniriana et al., 2009; Casamatta et al., 2012; Buerger et al., 2016). The cyanobacterium is thought to be one of the main pathogens within the BBD microbial consortium because of its ability to penetrate polyp tissues and gastrovascular cavities (Kramarsky-Winter et al., 2014; Richardson et al., 2014), thereby providing a framework for disease development (Sato et al., 2016). It also supplies energy and nutrients to the microbial consortium, possibly through fermentation and photosynthesis in the sulfide-rich BBD environment, as suggested for Geitlerinema sp. BBD_1991 (Den Uyl et al., 2016). However, important questions, such as the direct cause of BBD onset and progression drivers, remain unclear (Sato et al., 2016). Although the role of viruses, in particular bacteriophages, can provide new insights into the drivers and causation of a coral disease (Weynberg et al., 2015), BBD etiology has most commonly been studied by investigating the functions of associated bacteria, while the role of bacteriophages has not been considered.

FIGURE 1. Black band disease (BBD) on the coral Pavona duerdeni. The black band microbial mat is dominated by the cyanobacterium Roseofilum reptotaenium. The band kills the underlying coral tissue and progresses at rates typically in the order of 3–4 mm per day.
Lysogenic bacteriophages integrate into the genome of an infected bacterium and can change the bacterium’s behavior and virulence through the newly introduced genetic material (Brüssow et al., 2004). For example, the pathogenicity of Vibrio cholerae primarily depends on infection by a lysogenic bacteriophage (CTXphi). The bacteriophage transfers genes that encode for one of the primary virulence factors, such as the cholera toxin (CT), and converts V. cholerae from a non-pathogenic to a pathogenic strain (Waldor and Mekalanos, 1996; Faruque and Mekalanos, 2003). Lysogenic conversion may also transform Vibrio coralliilyticus into a coral pathogen triggering the coral disease white syndrome, since some of its virulence factors are found on pathogenicity islands that contain toxin genes homologous to those of the V. cholerae prophage (Weynberg et al., 2015).
In addition to triggering diseases through lysogenic conversion, bacteriophages can also lyse bacteria and mitigate bacterial diseases through their lytic cycle. The lytic ability of phages has been harnessed by humans to treat disease, known as ‘phage therapy’ (Duckworth and Gulig, 2002). Phage therapy has been successfully used to treat human diseases in clinical trials (reviewed in Abedon et al., 2011), mitigate diseases in aquaculture (reviewed in Oliveira et al., 2012), and to treat coral diseases (Efrony et al., 2007; Atad et al., 2012; Cohen et al., 2013). The bacteriophage infects the target host, takes over the cell machinery, replicates itself, lyses the host bacterium, and subsequently spreads its progeny phages, which infect remaining host cells and mitigate the disease over a short period of time (d’Herelle, 1930; Adams, 1959).
Conversely, bacteria can prevent bacteriophage infection by maintaining defense mechanisms, such as ‘clustered regularly interspaced short palindromic repeats’ (CRISPR) associated systems. Short sequences (spacers) that match the foreign target DNA and RNA sequences (protospacers) are stored in repetitive regions, known as CRISPR arrays (reviewed in Makarova et al., 2011a,b). An operon of associated genes codes for proteins (Cas) that detect and cleave the foreign DNA, guided by the transcribed spacers (crRNAs). CRISPR-Cas systems are known to be prevalent in ∼10% of currently sequenced bacterial genomes (Burstein et al., 2016), but seem to be more widespread among cyanobacterial genomes (detected in 68.3%, 86 out of 126 cyanobacterial genomes, Cai et al., 2013).
Evidence of typically high bacteriophage densities in cyanobacterial mats (Hennes et al., 1995; Voorhies et al., 2016), and the generally rapid co-evolution between hosts and their associated viruses in microbial mats (Heidelberg et al., 2009), led us to hypothesize that cyanobacterium-specific bacteriophages are likely to be present in BBD. In this study, we test for the existence of interactions between bacteriophages and the main BBD cyanobacteria, R. reptotaenium and Geitlerinema sp. BBD_1991. We evaluate the likelihood that the cyanobacteria are target of bacteriophage predation, which could influence their virulence and the development of BBD pathogenicity. By sequencing the genome of R. reptotaenium AO1 (Buerger et al., 2016) and retrieving the available genome for Geitlerinema sp. BBD_1991, we analyze the two data sets in silico for prophage integrations, bacteriophage–host interactions, and host defense mechanisms against virus infections. We also tested cultures of R. reptotaenium AO1 for evidence of active lysogenic infection, which was suggested from our genomic results, using mitomycin C and UV treatments.
Materials and Methods
Coral Sampling and Cyanobacteria Culturing
Samples of the coral Pavona duerdeni were collected at Orpheus Island in the central Great Barrier Reef (S 18-34.609; E 146-29.793) in 3–5 m water depth, transported to the Australian Institute of Marine Science (Townsville), and kept in flow-through aquaria until further processing. The main BBD-associated cyanobacterium, R. reptotaenium (Rasoulouniriana) Casamatta, was isolated and cultured as described elsewhere (Buerger et al., 2016). In brief, ∼1 cm2 of the black band mat was removed from an infected coral, inoculated onto an agar plate (0.6% bacteriological agar in autoclaved seawater, enriched with L1 medium at a final 1x concentration), and placed under unidirectional light of 40 PAR in 30°C. After 6 h of incubation, cyanobacteria furthest away from the inoculation spot were transferred to a new agar plate. Incubation steps were repeated three times until cyanobacteria were clean of contaminating bacteria. Non-axenic monocultures (single cyanobacterial genotype with associated bacteria) were obtained by isolating a single cyanobacterium from an agar plate under a stereo-microscope (Wild 143 Heerbrugg, M3Z, Switzerland). Cyanobacteria were grown in 125 mL L1 medium, with a 12 h light cycle until the end of their exponential phase, at which time a few aggregations of filaments were taken for DNA extraction.
DNA-Extraction and Sequencing
DNA was extracted from 50 mg (dry weight) samples of the cyanobacterial biomass (n = 2 samples) with a Mo-Bio Power Plant Pro DNA extraction kit (cat. 13400-50), according to the manufacturer’s recommendations, with the following small modifications. Samples were: (1) bead-beaten in Power Plant Pro kit solution PD1 (450 μl), PD2 (50 μl), and Rnase A (3 μl, 25 mg/ml) for 1 min at max speed with (Fastprep-25 5G, MP Biomedicals); (2) incubated for 1 h at 56°C and 10 min at 65°C with proteinase k (15 μl, 20 mg/ml); and (3) eluted with 2 × 50 μl TE (10 mM Tris/Hcl, pH 8.5, 0.1 mM EDTA). Approximately 2.5 μg of purified DNA (Zymo genomic DNA clean and concentrator) was sent for next generation sequencing with a Truseq library preparation on a MiSeq 2 × 300 V3 (Ramaciotti Centre, University of New South Wales).
Genome Assembly and Annotation
Paired-end sequences were merged with PEAR 0.9.5, using default parameters. Low quality reads (phred score < 33 within 95% of the sequence) identified by Fastx v0.0.13 and below 100 bp were removed. Reads were assembled into contigs with SPAdes 3.5.0, k-mer range = 55, 99, 127 (Bankevich et al., 2012). Contig bins were created based on marker genes, nucleotide composition, and contig abundance (minimum contig length 1000 bp) with MaxBin-1.4.2 (Wu et al., 2014), and taxonomically identified to genus level using Kraken v0.10.5-beta (Wood and Salzberg, 2014). A circular genome view was created with the software CGView (Stothard and Wishart, 2005). Assemblies were annotated using RAST (Rapid Annotations using Subsystems Technology, Overbeek et al., 2014), submitted to NCBI GenBank (accession numbers in results section and Supplementary Table S1) and filtered for genes that were relevant for bacteriophages (weblinks for bioinformatics tools, Supplementary Table S1). An additional data set was retrieved from Geitlerinema sp. BBD_1991 (Den Uyl et al., 2016) and compared to the results from the in silico R. reptotaenium AO1 analyses.
CRISPRs
Clustered regularly interspaced short palindromic repeats (CRISPR), associated Cas genes, and direct repeats were identified within the respective cyanobacterium genome bins of R. reptotaenium AO1 (Buerger et al., 2016) and Geitlerinema sp. BBD_1991 (Den Uyl et al., 2016) with CRISPRfinder, CRISPRdb and CRISPRcompar (Grissa et al., 2007). Only confirmed CRISPR arrays were considered. To assess protospacers and potential self-targeting spacers, CRISPR arrays were compared to: (a) publicly available databases: viral RefSeq, plasmid RefSeq and GenBank-phage accessed through CRISPRtarget online tool, as well as (b) local databases of the assembled genomic bins, by retrieving best possible BLASTn matches with default parameters for short sequences (-gapopen 10, -gapextend 2, -dust no, -reward 1, -penalty -1, -word_size 7, -qcov_hsp_perc 100, Biswas et al., 2013). For both analyses, only protospacers with the CRISPRtarget default minimum matching score of 20 were considered as a possible categorical match (bacteriophage, plasmid, unknown). A less stringent matching score (i.e., 18 and 19) was only considered if protospacer matches were relevant to the respective environment (e.g., cyanophage). Cas gene assignments, self-targeted genes, and open reading frames adjacent to CRISPR arrays were verified against the NCBI nr database to known protein sequences (tBLASTx) using Artemis (version 16, Rutherford et al., 2000).
Prophage
The assembled cyanobacterium genome contigs of R. reptotaenium AO1 (Buerger et al., 2016) and Geitlerinema sp. BBD_1991 (Den Uyl et al., 2016) were analyzed for prophage gene signatures with PHAST (Zhou et al., 2011), PHASTER (Arndt et al., 2016) and VIRsorter (Roux et al., 2015), using default parameters for submission as metagenomic contigs. Although incomplete prophages are reported in this paper, only complete prophage signatures were considered as potentially functional prophages. Annotations of potential prophage regions were checked for unrecognized phage genes, toxicity genes and genes of virulence with Blast2GO against the Swissprot database (Conesa and Götz, 2008) and BLASTp against the NCBI nr database.
Lysogenic Virus Induction
R. reptotaenium AO1 was grown at 30°C with 50–80 μE m-2 s-1 light intensity in 12 well plates until mid-exponential phase (Buerger et al., 2016) and then exposed to a mitomycin C treatment to induce virus replication of potential lysogenic bacteriophages. Mitomycin C was added to the cyanobacterial cultures (exposure time 2 h) in final concentrations of 0.1, 0.5, and 1 μg ml-1 (three wells per concentration, three plate replicates). After the 2-h exposure, mitomycin C-treated medium was replaced by fresh L1 growth medium and plates were maintained at control incubation conditions (Paul and Weinbauer, 2010). In a separate experiment, cyanobacteria were exposed to UVA and UVB light (intensity 2.5 mW/cm2) for 1, 2, 4, 10, 15, 20, and 30 min. After UV exposure, cultures were kept at control incubation conditions. Cyanobacterial growth was monitored for potential lysis with fluorescence readings over time (Synergy H4, Biotek). Fluorescence readings were averaged from an area scan, with 25 reads per well, and transformed into a measure of cyanobacterial biomass (Buerger et al., 2016).
To quantify viral abundance in each treatment well, the abundance of virus-like particles (VLPs) was measured on a flow cytometer (BD FACSVerse) with a 488 nm argon-ion laser (Brussaard et al., 2010). In brief, samples were fixed in the dark at a final concentration of 4% μl glutaraldehyde, incubated for 30 min at 4°C, and stained with 1x SYBR green I (Invitrogen) at 80°C for 10 min. Samples were counted in a dilution series at a low flow rate with approximately 200–400 events per second, same VLP standards as Pollock et al. (2014).
Results
Assembly and Gene Annotation
The sequences retrieved from the R. reptotaenium culture were assembled using the software SPAdes 3.5.0 and resulted in three genomic bins that were submitted for automated annotation with RAST. The three bins were taxonomically identified as cyanobacterium R. reptotaenium AO1 (RAST 564709.3; NCBI GenBank project accession number MLAW00000000), Alphaproteobacterium (RAST 28211.29, and Cytophagaceae sp. (RAST 89373.4). The R. reptotaenium AO1 genome bin matched our expected BBD cyanobacterial target and was chosen for further analyses. The R. reptotaenium AO1 genome bin consisted of 134 contigs, with a total length of 5,826,181 bp (Table 1). The full annotation contained 5,491 features, with 39 possible missing genes (data access, RAST and NCBI GenBank, Supplementary Table S1).
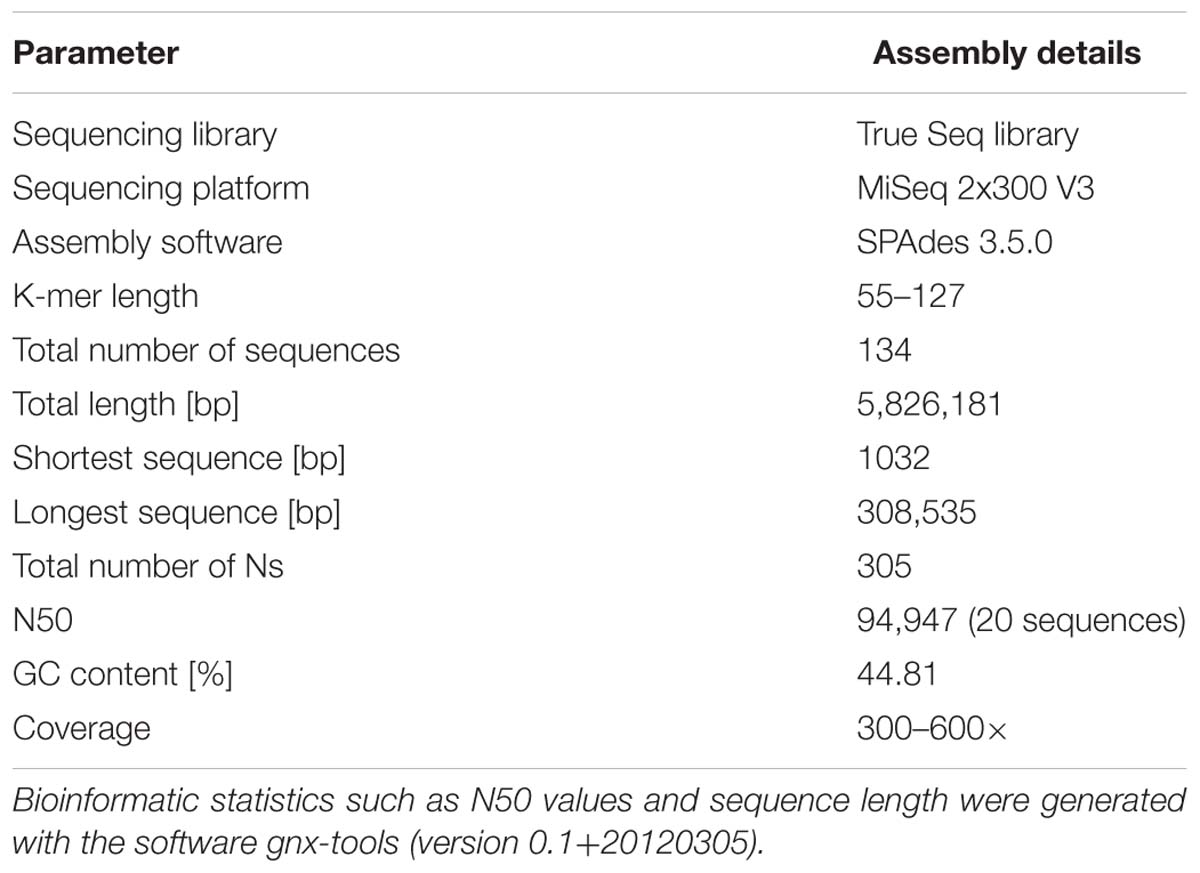
TABLE 1. Assembly details of the draft genome of Roseofilum reptotaenium AO1.
CRISPRs
A CRISPR-Cas immune system type I-D, also known as CASCADE (CRISPR associated complex for antiviral defense), was identified within the R. reptotaenium AO1 genome (Figure 2). CRISPR type I systems are known to target DNA only, not RNA, and require a specific protospacer-adjacent motif (PAM) on the target sequence next to the protospacer to be functional (reviewed in Jiang and Marraffini, 2015). The associated genes, adjacent to CRISPR array #2, were homologs of the known Cas genes, such as Cas3 helicase, Cas10d, Cas7, Cas5, Cas6, Cas2, and Cas1 (respective functions in Supplementary Table S2, modified from Makarova et al., 2011a,b). Compared to other cyanobacterium genomes, the R. reptotaenium AO1 CRISPR-Cas system is of average complexity, with seven array loci and a total of 100 spacers (median of type I systems contain approximately 92 spacers and three CRISPR loci; Cai et al., 2013).
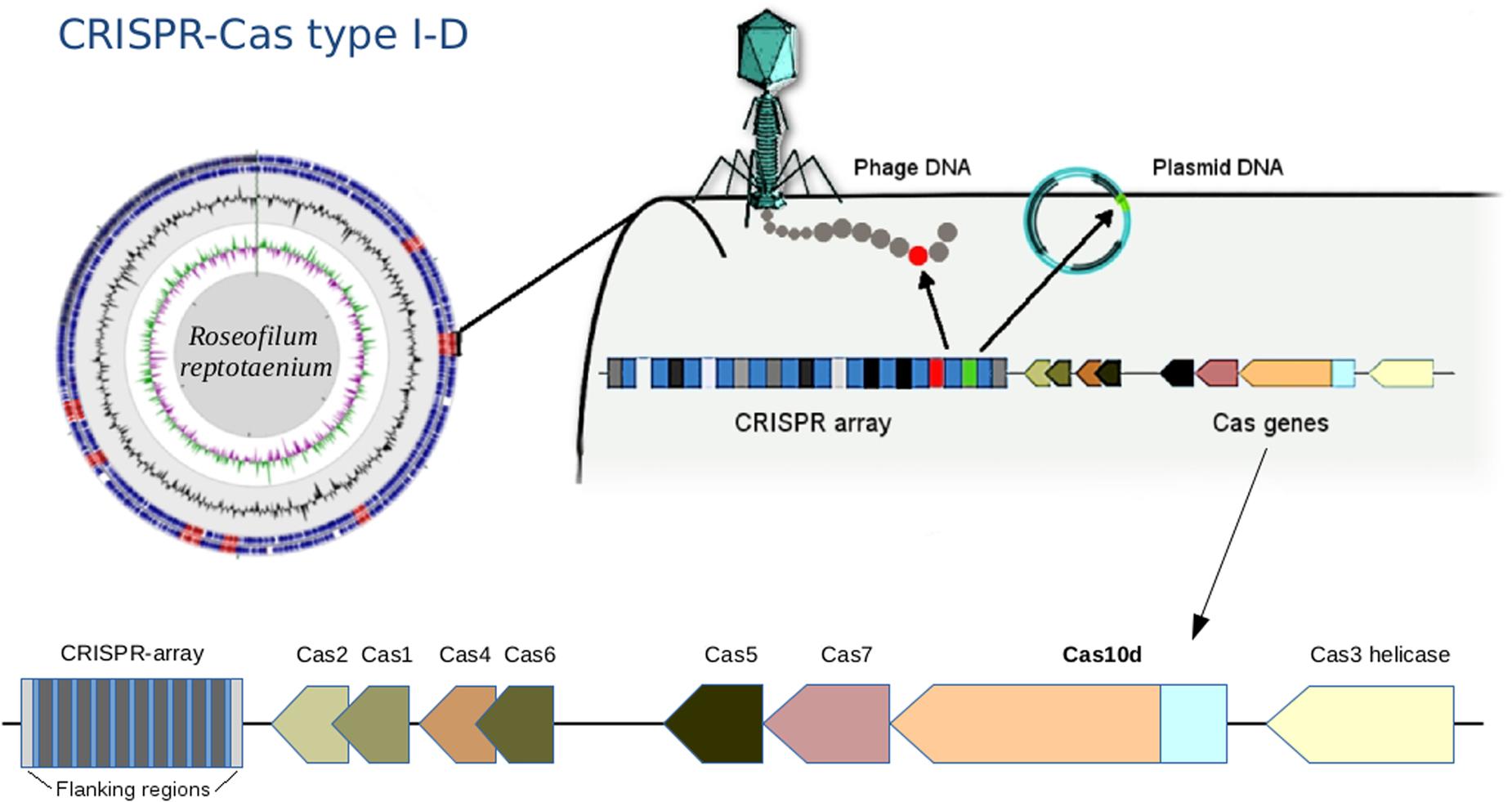
FIGURE 2. Draft genome assembly and illustration of the CRISPR-Cas system in Roseofilum reptotaenium AO1. The adaptive, heritable CRISPR-Cas system defends the cyanobacterium against bacteriophage infections, plasmids and mobile genetic elements. The gene Cas10d is representative of the CRISPR-type I-D, commonly found in other cyanobacteria. CRISPR arrays are marked in red on the genome contigs, displayed in a circular view. The Cas gene arrangement is indicated at the bottom of the figure.
Horizontal gene transfer has been inferred as one of the main methods for distributing parts of the CRISPR-Cas systems among bacteria, based on high sequence similarity of direct repeats (Godde and Bickerton, 2006). In R. reptotaenium AO1, all direct repeats of the CRISPR arrays were 37 bp long (Table 2) and showed high similarity to direct repeats of other cyanobacterial species, such as the marine Rivularia sp. PCC 7116 strain (Supplementary Table S3). The respective CRISPR-flanking regions were found to be specific for R. reptotaenium AO1, with no BLAST homologies to other known sequences.
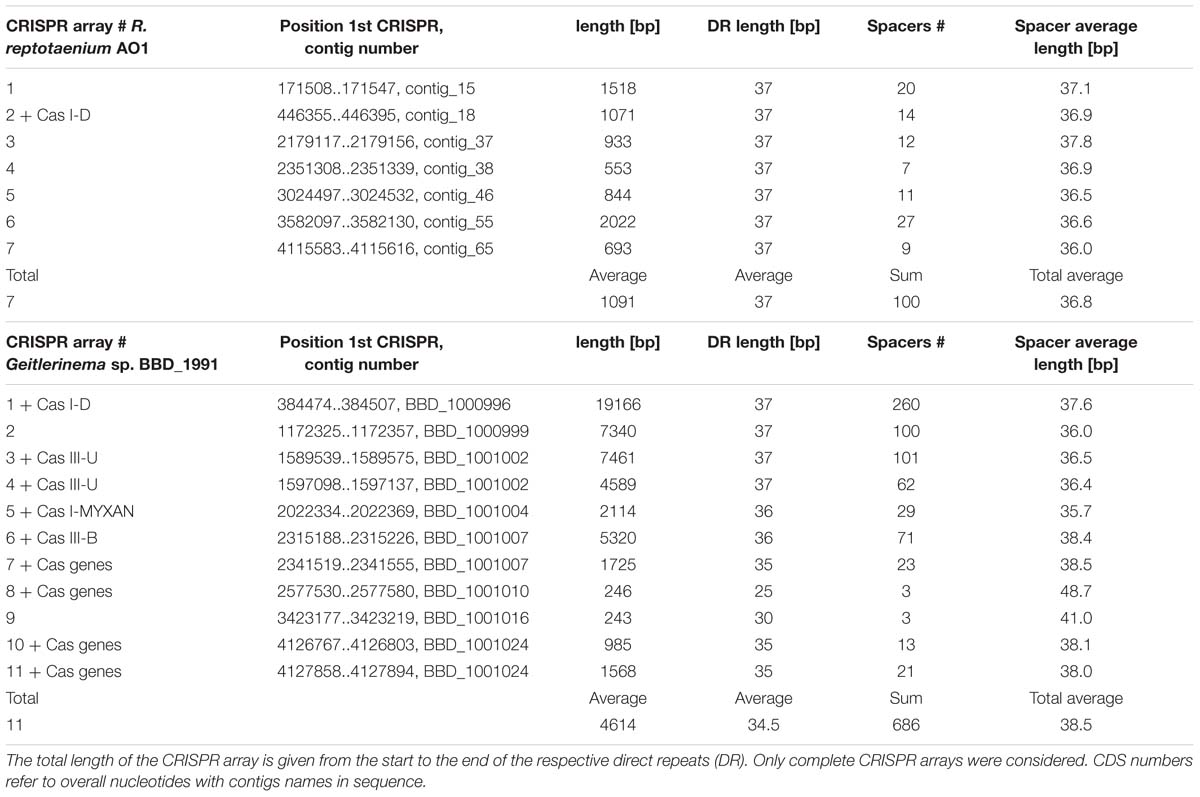
TABLE 2. CRISPR-Cas spacers of R. reptotaenium AO1 and Geitlerinema sp. BBD_1991.
In total, seven CRISPR arrays containing a total of 100 unique spacers were identified in R. reptotaenium AO1. Identical matches (protospacers) to eight spacer sequences were found on one cyanobacterial genome contig (contig 93, cov. 558, length 31,342 bp). The protospacers on contig 93 match spacers in conserved middle parts of the CRISPR arrays (average spacer position: 8.75 out of 15.88, Supplementary Table S4). One of the target regions on contig 93 codes for a DnaB domain-containing helicase (related to a bacteriophage multi-domain, phage_DnaB, BLASTp e-value < 6.28e - 20), while all others code for hypothetical proteins (Supplementary Table S4). Analysis of the assembly’s de Bruijn graphs showed that contig 93 is not circular shaped, but can be connected to other contigs, and is therefore not part of a plasmid or linear extra-chromosomal element (Supplementary Figure S1). Contig 93 was not integrated into other contigs, likely due to multiple connection possibilities, strain variations, or repetitive regions. Although a phage integrase was detected on one of the adjacent contigs (contig 184, pfam00589, e-value < 8.47e - 03, Supplementary Table S5), no other genes related to bacteriophages or mobile genetic elements were uncovered.
Results from protospacer matches to public databases (40% of spacers had potential protospacer matches) showed that protospacers were mostly unrelated to cyanobacteria or the marine environment (Supplementary Table S6), indicating that spacer sequences were novel and not represented in publicly available databases. Protospacer origins were non-redundant and matched to a diversity of plasmids and viruses, with multiple hits to Synechococcus phages, a Cyanothece sp. plasmid, and a Sinorhizobium fredii plasmid (number of hits were 3, 2, and 2, respectively, Supplementary Table S6).
Eleven CRISPR arrays were detected in the genome of Geitlerinema sp. BBD_1991 (Den Uyl et al., 2016), which had an extraordinarily high number of spacers (n = 260), approximately six times as many spacer sequences compared to R. reptotaenium AO1 (Table 2). While most of the target sequences (protospacers) were unknowns (95% ± 6), no identical spacer sequences were identified between the two genomes and also no self-targeting spacers were found in Geitlerinema sp. BBD_1991 (Figure 3; Supplementary Table S6). Cas genes were adjacent to almost all CRISPR arrays of Geitlerinema sp. BBD_1991, whereas R. reptotaenium AO1 had only one CRISPR array with adjacent Cas genes (Table 2; Supplementary Table S2). Several genes representative of different CRISPR-Cas types were detected in Geitlerinema sp. BBD_1991, such as types I-D, II-U, III-B and I-MYXAN. The direct repeat sequences of Geitlerinema sp. BBD_1991 were similar to R. reptotaenium AO1, and closely related to other cyanobacteria species, such as Crinalium epipsammum and Synechococcus sp. (Supplementary Table S3).
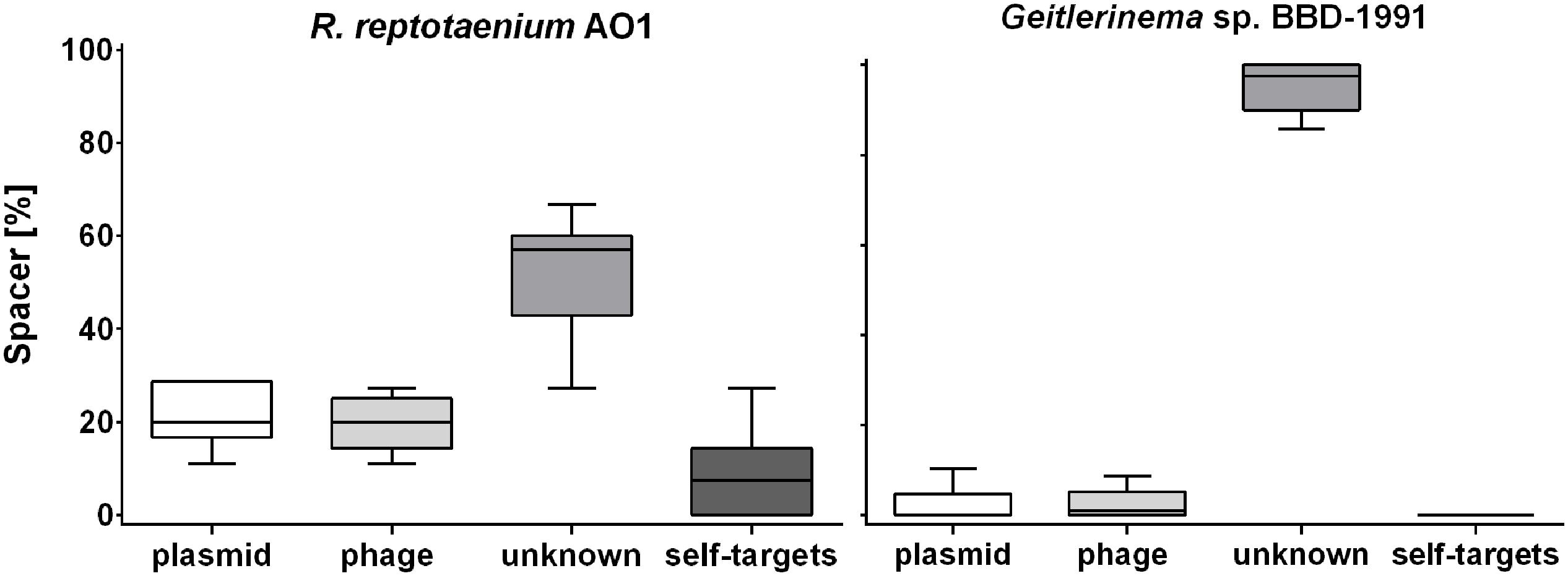
FIGURE 3. Potential CRISPR-Cas spacer targets within BBD. Most of the spacer targets (protospacers) could not be identified by matches to CRISPR sequences in publicly available databases. An unusually high number of protospacers were located within a short region of the R. reptotaenium AO1 genome. Spacer targets in % were calculated for the respective categories within each of the seven CRISPR arrays and visualized as replicates in a boxplot (detailed protospacer Supplementary Table S6). Bar = mean, whiskers = min to max values, box = 25th and 75th percentiles.
Prophage Detection
Three potential prophages were detected in R. reptotaenium AO1 (R1-R3, Table 3; Supplementary Tables S7 and S8). These were classified as incomplete and questionable prophages, because some of the required genes to fully form an assembled virus, such as genes coding for a tail and capsid, were missing from the contigs. It is noteworthy that the tools PHAST, PHASTER, and VIRsorter did not identify prophage signatures in the same regions of the genome, which may suggest that our ability to capture prophage signatures is still limited.
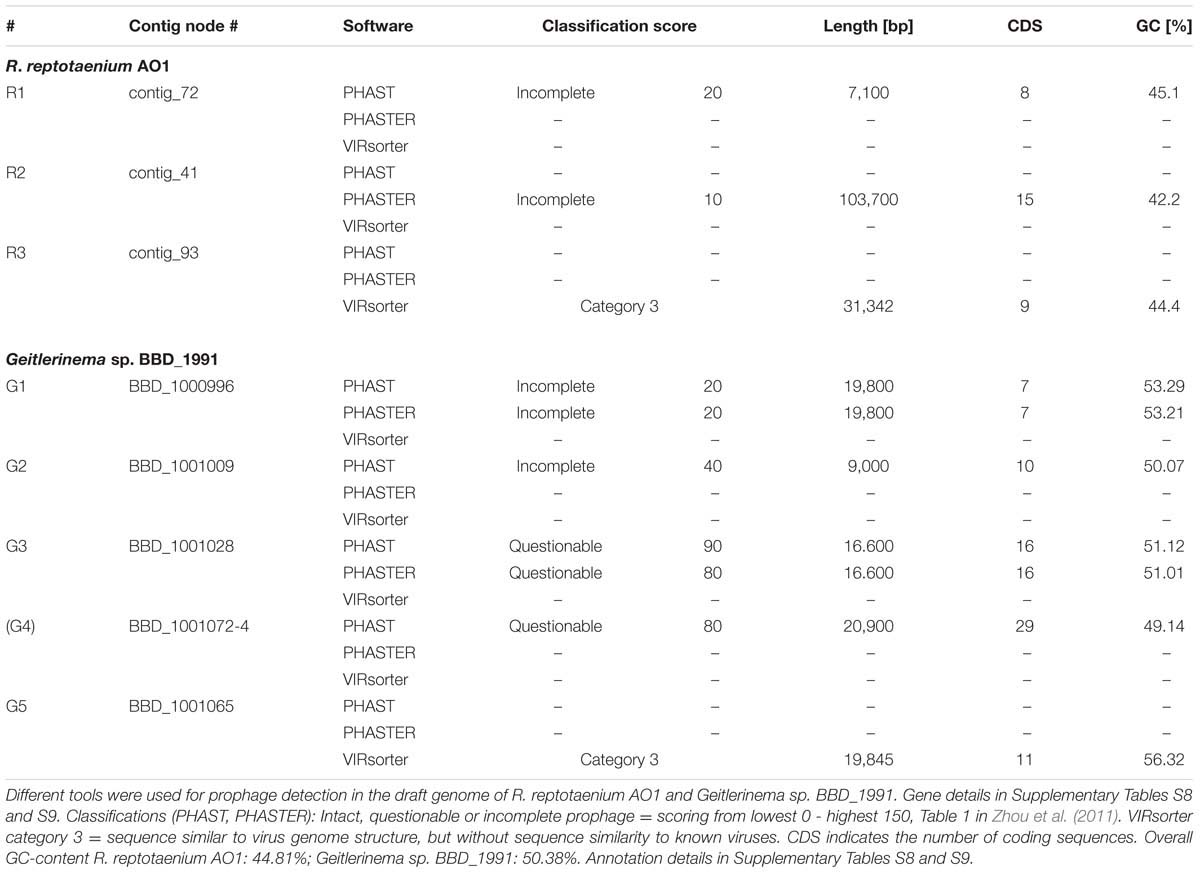
TABLE 3. Details of prophage detection.
The potential prophage regions of the R. reptotaenium AO1 genome carried genes that could be involved in processes other than virion assembly, such as the genes DNA adenine methyltransferase, transketolase, GDP-D-mannose 4,6-dehydratase, D, D-heptose 7-phosphate kinase, phosphoheptose isomerase, and a ADP-L-glycero-D-mannoheptose-6-epimerase (Supplementary Table S8). Some of these associated genes are involved in the non-oxidative pentose phosphate pathway (GDP-D-mannose 4,6-dehydratase), which provides energy during replication of virus when photosynthesis is not present (Shestakov and Karbysheva, 2015). However, potential cyanobacterial virulence factors that were located on prophage regions included lysozyme/metalloendopeptidases (e.g., region R1, e-value 7.00e - 017, Supplementary Table S8), genes potentially involved in lipopolysaccharide production (phosphoheptose isomerase, ADP-L-glycero-D-mannoheptose-6-epimerase and an NAD-dependent epimerase/dehydratase in region R2, e-value 3.37e - 029, Supplementary Table S8). Genes related to bacteriophages functionality were detected by RAST outside potential prophage regions and scattered across the R. reptotaenium AO1 contigs, such as a phage endolysin gene, phage tail protein, a T4-like virus tail tube protein gp19, a phage associated DNA primase, a putative prophage protein, a phage shock protein and a phage integrase (Supplementary Table S7).
By comparison, five potential prophages were detected in Geitlerinema sp. BBD_1991 (Table 3; Supplementary Table S9) (Den Uyl et al., 2016). Prophage_G1 region, recognized by PHAST and PHASTER (not VIRsorter), was the only region flanked by integration sites AttL and AttR, but was missing phage-related assembly genes. Phage-related genes were detected by PHAST and PHASTER mainly in the regions G3, resulting in a high prediction score (scored 90, questionable prophage) including genes such as phage baseplate, tail tube and tail sheath. In addition, the prophage_G4 region stretched over three consecutive contigs (BBD_1001072–BBD_1001074) that were not necessarily connected to each other in the genomic assembly and therefore likely represent a false positive result. Nevertheless, a gap-less assembly may rearrange the respective contig connections into a fully intact prophage. Genes directly located within the Geitlerinema sp. BBD_1991 potential prophage regions (Supplementary Table S9), which are likely to be involved in other processes than phage assembly, include photosystem II components, such as PsbE, PsbF, and PsbJ (prophage_G1), genes that contribute to Fe(II) transport systems, such as iron permease FTR1, to a non-oxidative pentose phosphate pathway, such as the GDP-D-mannose 4,6-dehydratase and other genes, such as a phosphoribosylglycinamide formyltransferase, orotate phosphoribosyltransferase, guanine deaminase and a biotin-(acetyl-CoA-carboxylase) ligase BirA. Potential virulence genes located in Geitlerinema sp. BBD_1991 prophage regions include a lysozyme/metalloendopeptidase (region G3 and G4, see annotations Supplementary Table S1 in Den Uyl et al., 2016), a peptidase S8/S53 (subtilase family protease, PHAST in region G4) and lipopolysaccharide modification genes, such as GDP-L-fucose synthase and glycosyl transferase (PHAST in region G4).
Lysogenic Virus Induction
It was not possible to induce a lysogenic virus from our R. reptotaenium AO1 cultures. Although cyanobacterial cell densities in cultures declined over time in response to the mitomycin C and UV treatments, we did not detect any evidence of bacteriophage replication in our flow cytometry counts (Figure 4). We interpret this to indicate that the mitomycin C and UV treatments caused the cyanobacteria to disintegrate and to reduce their fluorescence compared to controls.
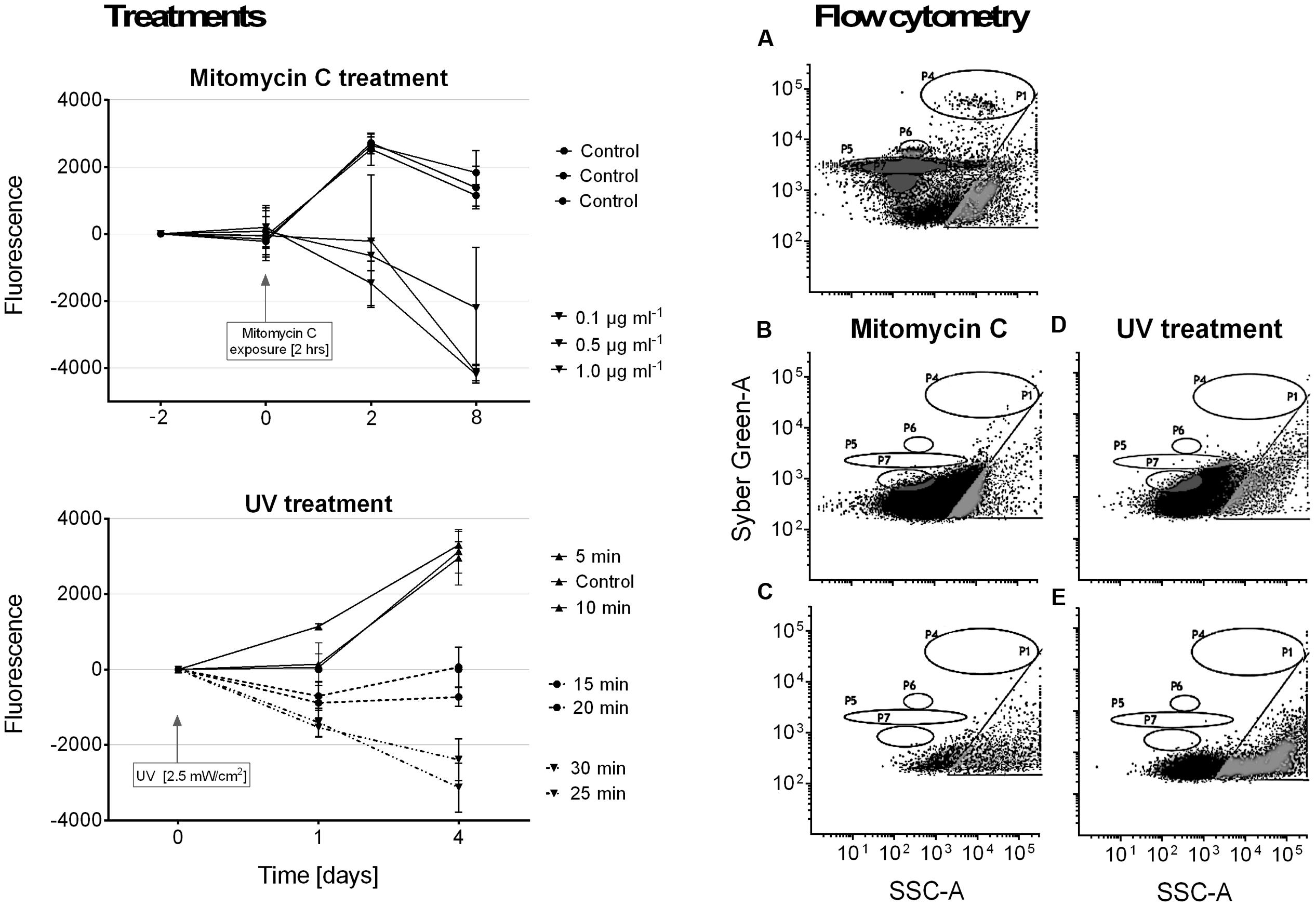
FIGURE 4. Results of lysogenic virus induction experiments using mitomycin C and UV light treatments. Panel on the left shows cyanobacterial growth, measured as fluorescence, in response to the respective treatments. Samples were normalized by calculating differences from starting values. Panels on the right show examples of particle size distributions from flow cytometry measurements after treatment (quantification not included due to no hits in relevant areas). In accordance with measured known references (Pollock et al., 2014), gate P4 = bacteria populations, and gates P5–P7 = virus like particles (VLPs). (A) Reference plot of bacteria and virus populations, (B) 2 h mitomycin C exposure at 1 μg/ml, (C) mitomycin C control measurement, (D) 30 min UV exposure, (E) UV treatment control measurement.
Discussion
Our analyses reveal that the genome of the cyanobacterium R. reptotaenium AO1, the dominant member of the microbial consortium causing BBD in corals, and that of Geitlerinema sp. BBD_1991 are equipped with a CRISPR-Cas adaptive defense system and several prophage regions that contain potential virulence-related genes. CRISPR-Cas systems thrive in environments where there is a high incidence of phage predation (Jiang et al., 2013), and can be multi-functional, ranging from preventing bacteriophage infections, interfering with the uptake of plasmids and mobile genetic elements (Barrangou et al., 2007), to the control of gene expression (Hatoum-Aslan and Marraffini, 2014) and support of DNA repair mechanisms (Babu et al., 2011).
CRISPR-Cas Self-targets
We found that a section of the R. reptotaenium AO1 genome is targeted by an unusually high number of spacer sequences (8 out of 100 spacers, Supplementary Table S6). Although spacer sequences of the CRISPR-Cas systems are usually thought to target foreign genetic material, approximately 1 in 250 spacer sequences can be self-targeting and match to particular regions of the host’s genome (Stern et al., 2010). Typically, a large proportion of these self-targeting spacers are located within the first or second position of a CRISPR array and may be acquired accidentally, resulting in autoimmunity through digestion of the host’s genetic code or inactivation of the CRISPR array (Stern et al., 2010). However, the self-targeting CRISPR-Cas spacers of R. reptotaenium AO1 do not result in autoimmunity and are not inactivated for the following reasons. The presence of the self-targeting spacers is unlikely to be accidental, because they originate from relatively conserved middle parts of the CRISPR arrays (average spacer position: 8.75 out of 15.88, Supplementary Table S4). They target loci that are not randomly distributed across the genome, but located in a narrow region of 31,342 bp (contig 93). The CRISPR arrays are still active, because up to three self-targeting spacers are present on single CRISPR arrays, indicating that they were acquired as separate events. These results suggest a secondary regulatory role of this CRISPR-Cas system in which cleavage of the host genome is prevented, possibly by the lack of PAM recognition sequences on the target regions or through another unknown process.
Contig 93, a Potential Prophage
In R. reptotaenium AO1, contig 93 was detected to be an incomplete prophage by VIRsorter (Table 2, Supplementary Table S8). Self-targeting spacers are known to target environmental lysogenic bacteriophages or prophages of other bacteria, but rarely their own prophage signatures (Touchon and Rocha, 2010; Hargreaves et al., 2014; Briner et al., 2015). The potential prophage is classified as incomplete, because only a single gene (DnaB domain-containing helicase) was detected as having a phage origin (Phage_cluster_71 PFAM-AAA_25, coding for a DNA repair protein), while the contig lacks essential genes for virus replication and assembly, such as capsid, head, or tail genes. It is possible that essential parts of the potential bacteriophage could be spread over multiple assembled contigs. Indeed, according to the assembly de Bruijn graphs (Supplementary Figure S1), contig 93 was separated from other contigs due to multiple possibilities for continued assembly and probably unresolved repetitive regions. However, the only bacteriophage-related gene on adjacent contigs is a phage integrase (contig 184, pfam00589, e-value < 4e–46, Supplementary Table S5), which is required for site-specific DNA excision and integration (Fogg et al., 2014).
The area of R. reptotaenium AO1 contig 93 could resemble a bacteriophage that is still hidden in the genome bin. To test this hypothesis, we attempted to induce a potential hidden prophage with mitomycin C and UV treatments (Figure 4). Although cyanobacterial biomass declined in response to the treatments, the lack of any measurable virus replication events indicates that the observed cell degradation was probably due to the respective treatments, rather than viral lysis. Alternatively, it has been shown that a CRISPR-Cas system of Escherichia coli can prevent prophage induction without killing the host bacterium when both processes, virus replication and CRISPR-Cas defense, are activated at similar time points (Edgar and Qimron, 2010). Although unconfirmed, the authors suggest possible regulation by promoters of cas proteins during a stress response, such as the sigma factor σ32 of E. coli, which could silence CRISPR activity until required in order to prevent prophage induction (Edgar and Qimron, 2010). Consequently, even if contig 93 was part of a prophage, as indicated by the presence of multiple spacers that target the incomplete prophage region, it might not have been possible to induce it.
Functional Role of Potential Prophage Regions
Contig 93 contains predicted coding regions for 39 hypothetical proteins with unknown functions, seven of which match directly to CRISPR-Cas spacers (Supplementary Tables S4 and S6). Other known genes on contig 93 code for a Rec-D like helicase, DnaB helicase, a putative proteinase, and a DNA-damage inducible protein. CRISPR-Cas systems can be multi-functional and involved in processes other than defense against foreign genetic material, such as expression regulation of pathogenicity genes leading to increased virulence (Hatoum-Aslan and Marraffini, 2014; Louwen et al., 2014), biofilm formation (Zegans et al., 2009), and DNA repair (Babu et al., 2011). The CRISPR-Cas1 protein (YgbT) of E. coli is known to interact with Rec repair proteins, which can increase resistance to DNA damage (Babu et al., 2011). In R. reptotaenium AO1, the proteins encoded by contig 93 may be interacting with the CRISPR-Cas system for increased DNA repair. Such increased DNA repair would be beneficial for R. reptotaenium during UV-induced DNA damage, for example in high light environments, a condition that would lead to more rapid BBD progression (Sato et al., 2010). However, further research is required to tease apart alternative hypotheses about the function of the CRISPR-Cas system in R. reptotaenium AO1.
Several genes that represent potential virulence factors were located within other prophage regions of the R. reptotaenium AO1 and Geitlerinema sp. BBD_1991 genomes. These genes are likely involved in lipopolysaccharide production and code for lysozyme/metalloendopeptidases. Although some of the gene annotation differed between RAST and the prophage detection tools (PHAST and PHASTER), their functional roles were consistently associated with virulence factors of other known bacteria. The third and fourth most highly expressed cyanobacterial gene within BBD that encodes for an NAD-dependent epimerase/dehydratase (Arotsker et al., 2016) was homologous in terms of its functionality to a gene located in R. reptotaenium AO1 prophage region_R2 (PHAST annotation, Supplementary Table S8, e-value < 3.37e - 029). NAD-dependent epimerase/dehydratases have been linked to increased virulence in the bacterium Pectobacterium carotovorum causing soft rot disease of vegetables (Islam, 2016). P. carotovorum with an intact NAD-dependent epimerase/dehydratase (wcaG gene) showed increased secretion of virulence associated exoenzymes and caused 21.5–26.7% macerated tissue, while mutants with a disrupted gene caused only 5.8–6.5% tissue damage on vegetables (Islam, 2016). Since the respective NAD-dependent epimerase/dehydratase of P. carotovorum (Islam, 2016) and R. reptotaenium AO1 are homologs to the wcaG gene coding for a GDP-fucose synthetase, their virulence associated exoenzymes are likely to be involved in the production of lipopolysaccharides and colanic acid, both of which are known bacterial virulence factors (Fry et al., 2000). The same respective gene in R. reptotaenium AO1 prophage_R2 region has been annotated by RAST as a rhamnose containing glycans subsystem coding for an UDP-glucose 4-epimerase (EC 5.1.3.2). This particular annotation is also a known virulence factor, which is located within a gene cluster that produces endotoxic lipopolysaccharides in Campylobacter spp. (Fry et al., 2000). A deactivation of the UDP-glucose 4-epimerase coding gene resulted in the expression of incomplete lipopolysaccharides and a virulence reduction (Fry et al., 2000). In addition, both cyanobacterial genomes had other virulence-associated genes in potential prophage regions involved in lipopolysaccharide production (Figure 5). For R. reptotaenium AO1 these were: GDP-D-mannose 4,6-dehydratase (Webb et al., 2004), phosphoheptose isomerase, ADP-L-glycero-D-mannoheptose-6-epimerase and an O-antigen export system (Brooke and Valvano, 1996); and for Geitlerinema sp. BBD_1991: GDP-L-fucose synthase (Mäki and Renkonen, 2004), GDP-D-mannose 4,6-dehydratase (Webb et al., 2004), glycosyl transferase (Davies et al., 2013). In Geitlerinema sp. BBD_1991 some of these genes are located within prophage_G1 region, that contained an integrase (G1_1), recombinase (G1_2) and was flanked by phage attachment sites attL/attR (Figure 5; Supplementary Table S9), known arrangements of genomic and pathogenicity islands (Bellanger et al., 2014). Several studies have suggested that cyanobacterial lipopolysaccharides are less endotoxic than classic lipopolysaccharides (reviewed in Gemma et al., 2016). However, the high expression of NAD-dependent epimerase/dehydratase (Arotsker et al., 2016) leads to the assumption that lipopolysaccharides could play an important role in the virulence of BBD associated cyanobacteria.
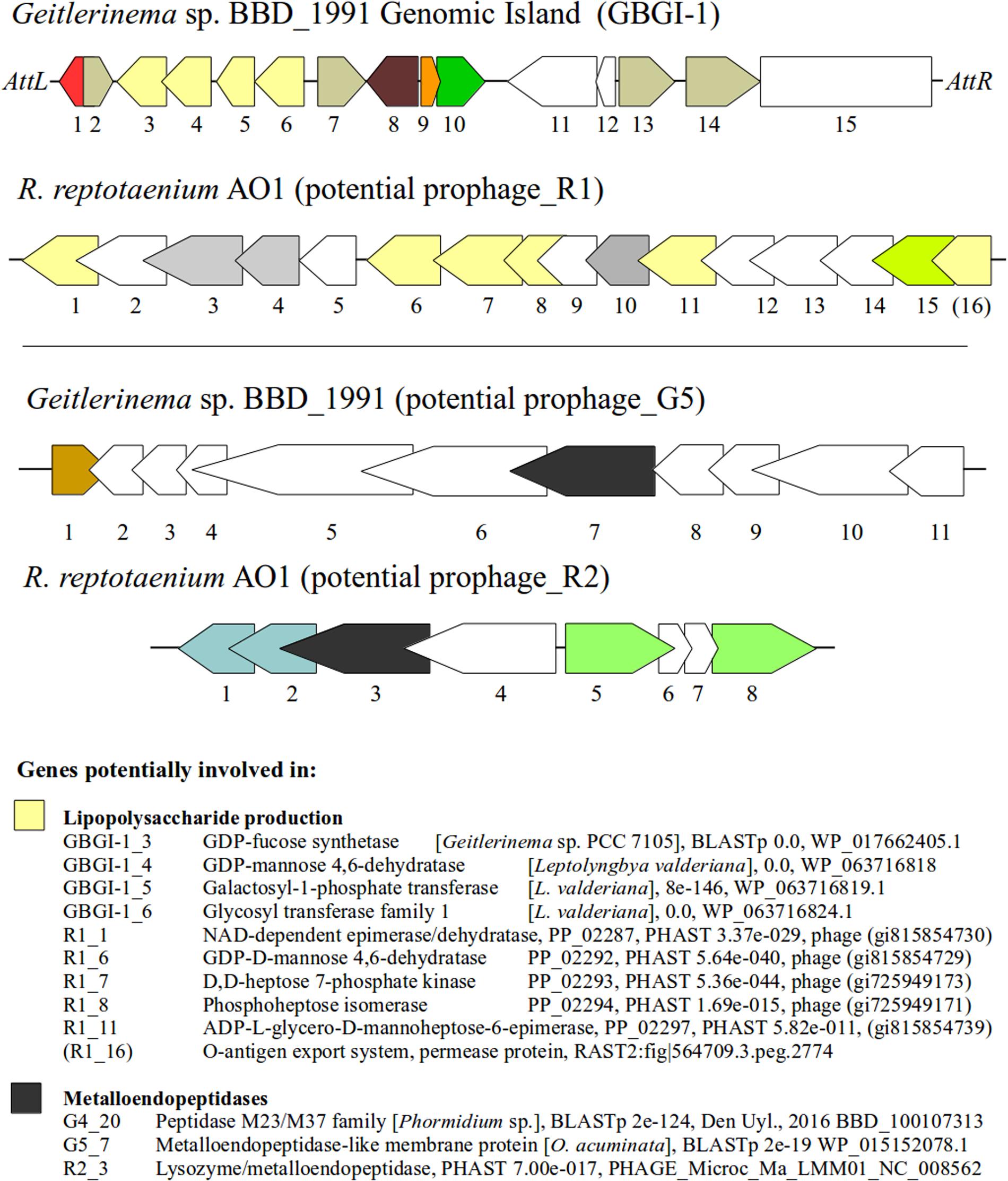
FIGURE 5. Genetic structure of prophage regions and their potential virulence factors. Genes with similar function are color coded, e.g., yellow = genes potentially involved in lipopolysaccharide production, dark gray = metalloendopeptidases, white = hypothetical proteins, other colors = see annotations Supplementary Tables S8 and S9. Genomic island of Geitlerinema sp. BBD_1991 is shown on top with the attachment sites AttL and AttR. BLASTp and PHAST results are given with E-values and protein accession numbers.
Other virulence associated genes coding for lysozyme/metalloendopeptidases (superfamily peptidase M23, zinc metallopeptidases, BLASTp e-value < 2.23e - 36), were present in both genomes (prophage_R1 in Supplementary Table S8; G4 BBD_100107313 and G5 BBD_10010657 in Supplementary Table S9). Although unreported for cyanobacteria, metalloendopeptidases are known virulence factors in a wide range of bacterial pathogens causing tissue damage (Miyoshi and Shinoda, 1997).
We found potential virulence genes that had overlapping functionality in prophage regions of both genomes, R. reptotaenium AO1 and Geitlerinema sp. BBD_1991. To date, viruses have not been considered as a contributor to the virulence of BBD associated bacteria. Our results strongly suggest that genetic material related to lysogenic bacteriophages contributes to the virulence of R. reptotaenium AO1 and Geitlerinema sp. BBD_1991.
Conclusion
Here we show that the cyanobacterium R. reptotaenium AO1, the dominant microbe within the BBD consortium, and Geitlerinema sp. BBD_1991, a less abundant member of the consortium, acquire resistance against bacteriophages by maintaining adaptive, heritable CRISPR-Cas defense systems. It is not surprising to find CRISPR-Cas systems and potential prophage regions in BBD associated cyanobacteria. Microbial mats, such as BBD, can be hot-spots for bacteriophage–host interactions due to the high abundance of bacteria and associated viruses (Heidelberg et al., 2009; Carreira et al., 2015). Cyanobacteria are the most abundant microbes in terms of biomass within the BBD consortium, and therefore a likely target for bacteriophage infections (Thingstad, 2000). Such constant phage predation pressure can result in an arms race and initiate evolution of host defense mechanisms, such as CRISPR-Cas systems (Levin, 2010; Held et al., 2013). CRISPR-Cas systems are not an absolute barrier and can be overcome by rapidly evolving bacteriophages (Andersson and Banfield, 2008), reflected by the multiple potential prophage regions within the R. reptotaenium AO1 and Geitlerinema sp. BBD_1991 genomes. Genes located in potential prophage regions are coding for known virulence factors and indicate that bacteriophages and foreign genetic material are likely contributors to the virulence of R. reptotaenium AO1 and Geitlerinema sp. BBD_1991. Maintaining CRISPR-Cas systems is costly, but crucial for protection against a potentially high number of lytic and lysogenic bacteriophages present in the BBD consortium. While a lytic infection could decrease the abundance and biomass of cyanobacteria in the mat, a lysogenic conversion could introduce new genetic material that might change the phenotypic characteristics of the infected cyanobacteria. Under both scenarios, an infection might alter the cyanobacteria’s functional role within the BBD mat: i.e., increase- or decrease its virulence. The detected CRISPR-Cas systems and potential prophage regions are evidence of a close interaction between bacteriophages and their host and highlight viruses as functional members of the BBD microbial consortium and as possible contributors to the virulence of the BBD-associated cyanobacteria R. reptotaenium AO1 and Geitlerinema sp. BBD_1991.
Ethics Statement
No ethics approval required for corals.
Sampling permit: Great Barrier Reef Marine Park Authority.
Permit number: G14/.1 to: James Cook University, School of Marine and Tropical Biology, QLD 4811.
Author Contributions
PB: Designed the study, performed experiments, data analyses, contributed materials, wrote the manuscript, and reviewed the drafts. EW-C, KW, BW, and MvO: Designed the study, contributed materials, wrote the manuscript, and reviewed the drafts.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Yui Sato and David Bourne for their support in the laboratory and in the field; Patrick Laffy for bioinformatics discussions and the Australian Nectar Research Cloud for access to computational resources. We acknowledge funding from AIMS@JCU (#17625) and the Australian Research Council (Future Fellowship #FT100100088 to MvO and SuperScience Fellowship #FS110200034 to KW).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2016.02077/full#supplementary-material
References
Abedon, S. T., Kuhl, S. J., Blasdel, B. G., and Kutter, E. M. (2011). Phage treatment of human infections. Bacteriophage 1, 66–85. doi: 10.4161/bact.1.2.15845
Aeby, G. S., Williams, G. J., Franklin, E. C., Haapkyla, J., Harvell, C. D., Neale, S., et al. (2011). Growth anomalies on the coral genera Acropora and Porites are strongly associated with host density and human population size across the Indo-Pacific. PLoS ONE 6:e16887. doi: 10.1371/journal.pone.0016887
Aeby, G. S., Work, T. M., Runyon, C. M., Shore-Maggio, A., Ushijima, B., Videau, P., et al. (2015). First record of black band disease in the Hawaiian archipelago: response, outbreak status, virulence, and a method of treatment. PLoS ONE 10:e0120853. doi: 10.1371/journal.pone.0120853
Andersson, A. F., and Banfield, J. F. (2008). Virus population dynamics and acquired virus resistance in natural microbial communities. Science 320, 1047–1050. doi: 10.1126/science.1157358
Antonius, A. (1973). New observations on coral destruction in reefs. 10th Meet. Assoc. Isl. Mar. Lab Carib 10: 3.
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, 1–6. doi: 10.1093/nar/gkw387
Arotsker, L., Kramarsky-Winter, E., Ben-Dov, E., and Kushmaro, A. (2016). Microbial transcriptome profiling of black band disease in a Faviid coral during a seasonal disease peak. Dis. Aquat. Organ. 118, 77–89. doi: 10.3354/dao02952
Atad, I., Zvuloni, A., Loya, Y., and Rosenberg, E. (2012). Phage therapy of the white plague-like disease of Favia favus in the Red Sea. Coral Reefs 31, 665–670. doi: 10.1007/s00338-012-0900-5
Babu, M., Beloglazova, N., Flick, R., Graham, C., Skarina, T., Nocek, B., et al. (2011). A dual function of the CRISPR-Cas system in bacterial antivirus immunity and DNA repair. Mol. Microbiol. 79, 484–502. doi: 10.1111/j.1365-2958.2010.07465.x
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Barneah, O., Ben-Dov, E., Kramarsky-Winter, E., and Kushmaro, A. (2007). Characterization of black band disease in Red Sea stony corals. Environ. Microbiol. 9, 1995–2006. doi: 10.1111/j.1462-2920.2007.01315.x
Barrangou, R., Fremaux, C., Deveau, H., Richards, M., Boyaval, P., Moineau, S., et al. (2007). CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–12. doi: 10.1126/science.1138140
Bellanger, X., Payot, S., Leblond-Bourget, N., and Guédon, G. (2014). Conjugative and mobilizable genomic islands in bacteria: evolution and diversity. FEMS Microbiol. Rev. 38, 720–760. doi: 10.1111/1574-6976.12058
Biswas, A., Gagnon, J. N., Brouns, S. J. J., Fineran, P. C., and Brown, C. M. (2013). CRISPRTarget: bioinformatic prediction and analysis of crRNA targets. RNA Biol. 10, 817–27. doi: 10.4161/rna.24046
Bourne, D. G., Garren, M., Work, T. M., Rosenberg, E., Smith, G. W., and Harvell, C. D. (2009). Microbial disease and the coral holobiont. Trends Microbiol. 17, 554–562. doi: 10.1016/j.tim.2009.09.004
Briner, A. E., Lugli, G. A., Milani, C., Duranti, S., Turroni, F., Gueimonde, M., et al. (2015). Occurrence and diversity of CRISPR-Cas systems in the genus bifidobacterium. PLoS ONE 10:e0133661. doi: 10.1371/journal.pone.0133661
Brooke, J. S., and Valvano, M. A. (1996). Molecular cloning of the Haemophilus influenzae gmhA (lpcA) gene encoding a phosphoheptose isomerase required for lipooligosaccharide biosynthesis. J. Bacteriol. 178, 3339–3341. doi: 10.1128/jb.178.11.3339-3341.1996
Bruno, J. F., Selig, E. R., Casey, K. S., Page, C. A., Willis, B. L., Harvell, C. D., et al. (2007). Thermal stress and coral cover as drivers of coral disease outbreaks. PLoS Biol. 5:1220–1227. doi: 10.1371/journal.pbio.0050124
Brussaard, C. P., Payet, J. P., Winter, C., and Weinbauer, M. G. (2010). Quantification of aquatic viruses by flow cytometry. Man. Aquat. Viral Ecol. 11, 102–107. doi: 10.4319/mave.2010.978-0-9845591-0-7.102
Brüssow, H., Canchaya, C., and Hardt, W. (2004). Phages and the evolution of bacterial pathogens: from genomic rearrangements to lysogenic conversion. Microbiol. Mol. Biol. Rev. 68, 560–602. doi: 10.1128/MMBR.68.3.560
Buerger, P., Alvarez-Roa, C., Weynberg, K. D., Baekelandt, S., and van Oppen, M. J. H. (2016). Genetic, morphological and growth characterisation of a new Roseofilum strain (Oscillatoriales, Cyanobacteria) associated with coral black band disease. PeerJ 4: e2110. doi: 10.7717/peerj.2110
Burstein, D., Sun, C. L., Brown, C. T., Sharon, I., Anantharaman, K., Probst, A. J., et al. (2016). Major bacterial lineages are essentially devoid of CRISPR-Cas viral defence systems. Nat. Commun. 7:10613. doi: 10.1038/ncomms10613
Cai, F., Axen, S. D., and Kerfeld, C. A. (2013). Evidence for the widespread distribution of CRISPR-Cas system in the phylum cyanobacteria. RNA Biol. 10, 687–693. doi: 10.4161/rna.24571
Carreira, C., Piel, T., Staal, M., Stuut, J.-B. W., Middelboe, M., and Brussaard, C. P. D. (2015). Microscale spatial distributions of microbes and viruses in intertidal photosynthetic microbial mats. Springerplus 4:239.
Casamatta, D., Stanic, D., Gantar, M., and Richardson, L. L. (2012). Characterization of Roseofilum reptotaenium (Oscillatoriales, Cyanobacteria) gen. et sp. nov. isolated from Caribbean black band disease. Phycologia 51, 489–499. doi: 10.2216/11-10.1
Cohen, Y., Joseph Pollock, F., Rosenberg, E., and Bourne, D. G. (2013). Phage therapy treatment of the coral pathogen Vibrio coralliilyticus. Microbiologyopen 2, 64–74. doi: 10.1002/mbo3.52
Conesa, A., and Götz, S. (2008). Blast2GO: a comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genomics 2008:619832. doi: 10.1155/2008/619832
Cooney, R. P., Pantos, O., Le Tissier, M. D. A., Barer, M. R., O’Donnell, A. G., and Bythell, J. C. (2002). Characterization of the bacterial consortium associated with black band disease in coral using molecular microbiological techniques. Environ. Microbiol. 4, 401–413. doi: 10.1046/j.1462-2920.2002.00308.x
Davies, M. R., Broadbent, S. E., Harris, S. R., Thomson, N. R., and van der Marjan, W. (2013). Horizontally acquired glycosyltransferase operons drive salmonellae lipopolysaccharide diversity. PLoS ONE 9:e1003568. doi: 10.1371/journal.pgen.1003568
Den Uyl, P. A., Richardson, L. L., Jain, S., and Dick, G. J. (2016). Unraveling the physiological roles of the cyanobacterium Geitlerinema sp. BBD and other black band disease community members through genomic analysis of a mixed culture. PLoS ONE 11:e0157953. doi: 10.1371/journal.pone.0157953
d’Herelle, F. (1930). Elimination du bacteriophage dans les symbioses bacterie- bacteriophage. Compt. Rend. Soc. Biol. 104:1254.
Duckworth, D. H., and Gulig, P. A. (2002). Potential treatment for bacterial infections. Biodrugs 16, 57–62. doi: 10.2165/00063030-200216010-00006
Edgar, R., and Qimron, U. (2010). The Escherichia coli CRISPR system protects from λ lysogenization, lysogens, and prophage induction. J. Bacteriol. 192, 6291–6294. doi: 10.1128/JB.00644-10
Efrony, R., Loya, Y., Bacharach, E., and Rosenberg, E. (2007). Phage therapy of coral disease. Coral Reefs 26, 7–13. doi: 10.1007/s00338-006-0170-1
Faruque, S. M., and Mekalanos, J. J. (2003). Pathogenicity islands and phages in Vibrio cholerae evolution. Trends Microbiol. 11, 505–510. doi: 10.1016/j.tim.2003.09.003
Fogg, P. C. M., Colloms, S., Rosser, S., Stark, M., and Smith, M. C. M. (2014). New applications for phage integrases. J. Mol. Biol. 426, 2703–2716. doi: 10.1016/j.jmb.2014.05.014
Fry, B. N., Feng, S., Chen, Y. Y., Newell, D. G., Coloe, P. J., and Korolik, V. (2000). The galE gene of Campylobacter jejuni is involved in lipopolysaccharide synthesis and virulence. Infect. Immun. 68, 2594–2601. doi: 10.1128/IAI.68.5.2594-2601.2000
Gemma, S., Molteni, M., and Rossetti, C. (2016). Lipopolysaccharides in cyanobacteria: a Brief Overview. Adv. Microbiol. 6, 391–397. doi: 10.4236/aim.2016.65038
Godde, J. S., and Bickerton, A. (2006). The repetitive DNA elements called CRISPRs and their associated genes: evidence of horizontal transfer among prokaryotes. J. Mol. Evol. 62, 718–729. doi: 10.1007/s00239-005-0223-z
Green, E. P., and Bruckner, A. W. (2000). The significance of coral disease epizootiology for coral reef conservation. Biol. Conserv. 96, 347–361. doi: 10.1016/S0006-3207(00)00073-2
Grissa, I., Vergnaud, G., and Pourcel, C. (2007). The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinformatics 8:172. doi: 10.1186/1471-2105-8-172
Hargreaves, K. R., Flores, C. O., Lawley, T. D., and Clokie, M. R. J. (2014). Abundant and diverse clustered regularly interspaced short palindromic repeat spacers in Clostridium difficile strains and prophages target multiple phage types within this pathogen. MBio 5, 1–10. doi: 10.1128/mBio.01045-13
Harvell, D., Jordán-Dahlgren, E., Merkel, S., Rosenberg, E., Raymundo, L., Smith, G., et al. (2007). Coral disease, environmental drivers, and the balance between coral and microbial associates. Oceanography 20, 172–195. doi: 10.5670/oceanog.2007.91
Hatoum-Aslan, A., and Marraffini, L. A. (2014). Impact of CRISPR immunity on the emergence and virulence of bacterial pathogens. Curr. Opin. Microbiol. 17, 82–90. doi: 10.1016/j.mib.2013.12.001
Heidelberg, J. F., Nelson, W. C., Schoenfeld, T., and Bhaya, D. (2009). Germ warfare in a microbial mat community: CRISPRs provide insights into the co-evolution of host and viral genomes. PLoS ONE 4:e4169. doi: 10.1371/journal.pone.0004169
Held, N. L., Childs, L. M., Davison, M., Weitz, J. S., Whitaker, R. J., and Bhaya, D. (2013). “CRISPR-Cas systems to probe ecological diversity and host-viral interactions,” in CRISPR-Cas Systems: RNA-Mediated Adaptive Immunity in Bacteria and Archaea, eds R. Barrangou and J. van der Oost (Berlin: Springer), 221–250. doi: 10.1007/978-3-642-34657-6
Hennes, K. P., Suttle, C. A., Chan, A. M., and Chan, A. M. Y. M. (1995). Fluorescently labeled virus probes show that natural virus populations can control the structure of marine microbial communities. Appl. Environ. Microbiol. 61:3623.
Islam, R. (2016). NAD-Dependent Epimerase/Dehydratase Affects Cell Surface Properties, Virulence and Extracellular Enzyme Production in the Soft Rot Phytopathogen, Pectobacterium Carotovorum. Master of Science thesis, Tennessee State University, Nashville, TN.
Jiang, W., Maniv, I., Arain, F., Wang, Y., Levin, B. R., and Marraffini, L. A. (2013). Dealing with the evolutionary downside of CRISPR immunity: bacteria and beneficial plasmids. PLoS Genet. 9:e1003844. doi: 10.1371/journal.pgen.1003844
Jiang, W., and Marraffini, L. A. (2015). CRISPR-Cas: new tools for genetic manipulations from bacterial immunity systems. Annu. Rev. Microbiol. 69, 209–228. doi: 10.1146/annurev-micro-091014-104441
Johan, O., Bengen, D. G., Zamani, N. P., and Sweet, M. J. (2015). The distribution and abundance of black band disease and white syndrome in Kepulauan Seribu, Indonesia. HAYATI J. Biosci. 22, 105–112. doi: 10.1016/j.hjb.2015.09.001
Kramarsky-Winter, E., Arotsker, L., Rasoulouniriana, D., Siboni, N., Loya, Y., and Kushmaro, A. (2014). The possible role of cyanobacterial filaments in coral black band disease pathology. Microb. Ecol. 67, 177–185. doi: 10.1007/s00248-013-0309-x
Lamb, J. B., True, J. D., Piromvaragorn, S., and Willis, B. L. (2014). Scuba diving damage and intensity of tourist activities increases coral disease prevalence. Biol. Conserv. 178, 88–96. doi: 10.1016/j.biocon.2014.06.027
Lamb, J. B., and Willis, B. L. (2011). Using coral disease prevalence to assess the effects of concentrating tourism activities on offshore reefs in a tropical marine park. Conserv. Biol. 25, 1044–1052. doi: 10.1111/j.1523-1739.2011.01724.x
Levin, B. R. (2010). Nasty viruses, costly plasmids, population dynamics, and the conditions for establishing and maintaining CRISPR-mediated adaptive immunity in bacteria. PLoS Genet. 6:e1001171. doi: 10.1371/journal.pgen.1001171
Louwen, R., Staals, R. H. J., Endtz, H. P., van Baarlen, P., and van der Oost, J. (2014). The role of CRISPR-Cas systems in virulence of pathogenic bacteria. Microbiol. Mol. Biol. Rev 78, 74–88. doi: 10.1128/MMBR.00039-13
Makarova, K. S., Aravind, L., Wolf, Y. I., and Koonin, E. V. (2011a). Unification of Cas protein families and a simple scenario for the origin and evolution of CRISPR-Cas systems. Biol. Direct 6:38. doi: 10.1186/1745-6150-6-38
Makarova, K. S., Haft, D. H., Barrangou, R., Brouns, S. J. J., Charpentier, E., Horvath, P., et al. (2011b). Evolution and classification of the CRISPR-Cas systems. Nat. Rev. Microbiol. 9, 467–477. doi: 10.1038/nrmicro2577
Mäki, M., and Renkonen, R. (2004). Biosynthesis of 6-deoxyhexose glycans in bacteria. Glycobiology 14, 1–15. doi: 10.1093/glycob/cwh040
Maynard, J., van Hooidonk, R., Eakin, C. M., Puotinen, M., Garren, M., Williams, G., et al. (2015). Projections of climate conditions that increase coral disease susceptibility and pathogen abundance and virulence. Nat. Clim. Change 5, 688–694. doi: 10.1038/nclimate.2625
Miller, A. W., and Richardson, L. L. (2011). A meta-analysis of 16S rRNA gene clone libraries from the polymicrobial black band disease of corals. FEMS Microbiol. Ecol. 75, 231–241. doi: 10.1111/j.1574-6941.2010.00991.x
Miyoshi, S., and Shinoda, S. (1997). Bacterial metalloprotease as the toxic factor in infection. J. Toxicol. Toxin Rev. 16, 177–194. doi: 10.3109/15569549709016455
Oliveira, J., Castilho, F., Cunha, A., and Pereira, M. J. (2012). Bacteriophage therapy as a bacterial control strategy in aquaculture. Aquac. Int. 20, 879–910. doi: 10.1007/s10499-012-9515-7
Overbeek, R., Olson, R., Pusch, G. D., Olsen, G. J., Davis, J. J., Disz, T., et al. (2014). The SEED and the rapid annotation of microbial genomes using subsystems technology (RAST). Nucleic Acids Res. 42, D206–D214. doi: 10.1093/nar/gkt1226
Page, C. A., and Willis, B. L. (2006). Distribution, host range and large-scale spatial variability in black band disease prevalence on the Great Barrier Reef, Australia. Dis. Aquat. Organ. 69, 41–51. doi: 10.3354/dao069041
Paul, J. H., and Weinbauer, M. (2010). “Detection of lysogeny in marine environments,” in Manual of Aquatic Viral Ecology, ed. S. W. Wilhelm (Waco, TX: ASLO), 30–33. doi: 10.4319/mave.2010.978-0-9845591-0-7.30
Pollock, F. J., Morris, P. J., Willis, B. L., and Bourne, D. G. (2011). The urgent need for robust coral disease diagnostics. PLoS Pathog. 7:e1002183. doi: 10.1371/journal.ppat.1002183
Pollock, F. J., Wood-Charlson, E. M., van Oppen, M. J. H., Bourne, D. G., Willis, B. L., and Weynberg, K. D. (2014). Abundance and morphology of virus-like particles associated with the coral Acropora hyacinthus differ between healthy and white syndrome-infected states. Mar. Ecol. Prog. Ser. 510, 39–43. doi: 10.3354/meps10927
Rasoulouniriana, D., Siboni, N., Ben-Dov, E., Kramarsky-Winter, E., Loya, Y., and Kushmaro, A. (2009). Pseudoscillatoria coralii gen. nov., sp nov., a cyanobacterium associated with coral black band disease (BBD). Dis. Aquat. Organ. 87, 91–96. doi: 10.3354/dao02089
Richardson, L., Stanić, D., May, A., Brownell, A., Gantar, M., and Campagna, S. (2014). Ecology and physiology of the pathogenic cyanobacterium Roseofilum reptotaenium. Life 4, 968–987. doi: 10.3390/life4040968
Rosenberg, E., Kellogg, C. A., and Rohwer, F. (2007). Coral microbiology. Oceanography 20, 146–154. doi: 10.5670/oceanog.2007.60
Roux, S., Enault, F., Hurwitz, B. L., and Sullivan, M. B. (2015). VirSorter: mining viral signal from microbial genomic data. PeerJ 3:e985. doi: 10.7717/peerj.985
Rutherford, K., Parkhill, J., Crook, J., Horsnell, T., Rice, P., Rajandream, M. A., et al. (2000). Artemis: sequence visualization and annotation. Bioinformatics 16, 944–945. doi: 10.1093/bioinformatics/16.10.944
Sato, Y., Bourne, D. G., and Willis, B. L. (2011). Effects of temperature and light on the progression of black band disease on the reef coral, Montipora hispida. Coral Reefs 30, 753–761. doi: 10.1007/s00338-011-0751-5
Sato, Y., Civiello, M., Bell, S. C., Willis, B. L., and Bourne, D. G. (2016). Integrated approach to understanding the onset and pathogenesis of black band disease in corals. Environ. Microbiol. 18, 752–765. doi: 10.1111/1462-2920.13122
Sato, Y., Willis, B. L., and Bourne, D. G. (2010). Successional changes in bacterial communities during the development of black band disease on the reef coral, Montipora hispida. ISME J. 4, 203–214. doi: 10.1038/ismej.2009.103
Selig, E. R., Harvell, C. D., Bruno, J. F., Willis, B. L., Page, C. A., Casey, K. S., et al. (2006). “Analyzing the relationship between ocean temperature anomalies and coral disease outbreaks at broad spatial scales,” in Coral Reefs and Climate Change: Science and Management, eds J. Phinney, O. Hoegh-Guldberg, J. Kleypas, W. Skirving, and A. Strong (Washington, DC: American Geophysical Union), 111–128. doi: 10.1029/61CE07
Sheridan, C., Kramarsky-Winter, E., Sweet, M., Kushmaro, A., and Costa, M. (2013). Diseases in coral aquaculture: causes, implications and preventions. Aquaculture 39, 124–135. doi: 10.1016/j.aquaculture.2013.02.037
Shestakov, S. V., and Karbysheva, E. A. (2015). The role of viruses in the evolution of cyanobacteria. Med. Hypotheses 135, 115–127.
Stern, A., Keren, L., Wurtzel, O., Amitai, G., and Sorek, R. (2010). Self-targeting by CRISPR: gene regulation or autoimmunity? Trends Genet. 26, 335–340. doi: 10.1016/j.tig.2010.05.008
Stothard, P., and Wishart, D. S. (2005). Circular genome visualization and exploration using CGView. Bioinformatics 21, 537–539. doi: 10.1093/bioinformatics/bti054
Sutherland, K. P., Porter, J. W., and Torres, C. (2004). Disease and immunity in Caribbean and Indo-Pacific zooxanthellate corals. Mar. Ecol. Prog. Ser. 266, 273–302. doi: 10.3354/meps266273
Thingstad, T. F. (2000). Elements of a theory for the mechanisms controlling abundance, diversity, and biogeochemical role of lytic bacterial viruses in aquatic systems. Limnol. Ocean. 45, 1320–1328. doi: 10.4319/lo.2000.45.6.1320
Touchon, M., and Rocha, E. P. C. (2010). The small, slow and specialized CRISPR and anti-CRISPR of Escherichia and Salmonella. PLoS ONE 5:e11126. doi: 10.1371/journal.pone.0011126
Voorhies, A. A., Eisenlord, S. D., Marcus, D. N., Duhaime, M. B., Biddanda, B. A., Cavalcoli, J. D., et al. (2016). Ecological and genetic interactions between cyanobacteria and viruses in a low-oxygen mat community inferred through metagenomics and metatranscriptomics. Environ. Microbiol. 18, 358–371. doi: 10.1111/1462-2920.12756
Waldor, M., and Mekalanos, J. (1996). Lysogenic conversion by a filamentous phage encoding cholera toxin. Science 272, 1910–1914. doi: 10.1126/science.272.5270.1910
Webb, N. A., Mulichak, A. M., Lam, J. S., Rocchetta, H. L., and Garavito, R. M. (2004). Crystal structure of a tetrameric GDP-D-mannose 4, 6-dehydratase from a bacterial GDP-D-rhamnose biosynthetic pathway. Protein Sci. 13, 529–539. doi: 10.1110/ps.03393904.with
Weynberg, K. D., Voolstra, C. R., Neave, M. J., Buerger, P., and van Oppen, M. J. H. (2015). From cholera to corals: viruses as drivers of virulence in a major coral bacterial pathogen. Sci. Rep. 5:17889. doi: 10.1038/srep17889
Willis, B. L., Page, C. A., and Dinsdale, E. A. (2004). “Coral disease on the Great Barrier Reef,” in Coral Health and Disease, eds E. Rosenberg and Y. Loya (New York, NY: Springer), 69–104.
Wood, D. E., and Salzberg, S. L. (2014). Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 15: R46. doi: 10.1186/gb-2014-15-3-r46
Wu, Y.-W., Tang, Y.-H., Tringe, S. G., Simmons, B., and Singer, S. W. (2014). MaxBin: an automated binning method to recover individual genomes from metagenomes using an expectation-maximization algorithm. Microbiome 2: 26. doi: 10.1186/2049-2618-2-26
Zegans, M. E., Wagner, J. C., Cady, K. C., Murphy, D. M., Hammond, J. H., and O’Toole, G. A. (2009). Interaction between bacteriophage DMS3 and host CRISPR region inhibits group behaviors of Pseudomonas aeruginosa. J. Bacteriol. 91, 210–219. doi: 10.1128/JB.00797-08
Keywords: BBD, bacteriophage, viruses, pathogenicity, virulence
Citation: Buerger P, Wood-Charlson EM, Weynberg KD, Willis BL and van Oppen MJH (2016) CRISPR-Cas Defense System and Potential Prophages in Cyanobacteria Associated with the Coral Black Band Disease. Front. Microbiol. 7:2077. doi: 10.3389/fmicb.2016.02077
Received: 27 September 2016; Accepted: 08 December 2016;
Published: 22 December 2016.
Edited by:
Rainer Kurmayer, University of Innsbruck, AustriaReviewed by:
Lucas Stal, Royal Netherlands Institute for Sea Research (NIOZ), NetherlandsJulie L. Meyer, University of Florida, USA
Copyright © 2016 Buerger, Wood-Charlson, Weynberg, Willis and van Oppen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Patrick Buerger, cGF0cmljay5idWVyZ2VyQG15LmpjdS5lZHUuYXU=