 Chung T. Ngo
Chung T. Ngo Sara Romano-Bertrand
Sara Romano-Bertrand Sylvie Manguin
Sylvie Manguin Estelle Jumas-Bilak
Estelle Jumas-Bilak- 1Institut de Recherche pour le Développement France, UMR-MD3, Faculté de Pharmacie, Montpellier, France
- 2National Institute of Veterinary Research, Hanoi, Vietnam
- 3UMR 5569 Hydrosciences, Equipe Pathogènes Hydriques, Santé et Environnements, Faculté de Pharmacie, Université de Montpellier, Montpellier, France
- 4Département d’Hygiène Hospitalière, Centre Hospitalier Universitaire de Montpellier, Montpellier, France
The naturally acquired microbiota of Anopheles can influence vector’s susceptibility to Plasmodium and its capacity to transmit them. Microbiota modification is a new challenge to limit disease transmission but it still needs advanced knowledges on bacterial community in Anopheles, especially in wild and infected specimens from diverse origin and species. Bacterial culture and 16S rRNA gene-PCR associated to Temporal Temperature Gradient Electrophoresis (TTGE) were applied to explore the bacterial diversity in the abdomen of 100 wild specimens (eight Anopheles species) collected in the Binh Phuoc Province, Vietnam. Culture and PCR-TTGE were complementary. The bacterial richness of the mosquito collection encompassed 105 genera belonging to seven phyla, mostly Proteobacteria, Firmicutes, and Actinobacteria. Staphylococcus, Clostridium, and Bacillus in Firmicutes were the most prevalent genera. However, Proteobacteria represented by 57 genera was the most diversified phylum in Anopheles microbiota. The high overall of Anopheles-associated bacteria is confirmed with, to our knowledge, 51 genera described for the first time in Anopheles microbiota. However, the diversity per specimen was low with average diversity index and the average Shannon–Wiener score (H) of 4.843 and 5.569, respectively. The most represented bacterial genera were present in <30% of the specimens. Consequently, the core microbiota share by Anopheles from Binh Phuoc was very narrow, suggesting that Anopheles microbiota was greatly influenced by local environments. The repertory of bacterial genera in two specimens of An. dirus and An. pampanai naturally infected by Plasmodium vivax was also described as preliminary results. Finally, this study completed the repertory of bacteria associated to wild Anopheles. Anopheles associated-bacteria appeared specimen-dependent rather than mosquitoe species- or group-dependent. Their origin and the existence of Anopheles-specific bacterial taxa are discussed.
Introduction
Despite some success in controlling malaria, this disease continues to be a major health burden in many countries around the world with 438,000 deaths reported in 2015, particularly in sub-Saharan Africa recording 395,000 deaths (90%), but also in forested regions of Southeast Asia with 32,000 deaths (7%; WHO, 2015). Until now, vector control has been one of the key elements for controlling this disease based on the use of insecticides, such as DDT, pyrethroids, organophosphates, carbamates (Verhaeghen et al., 2009; Kabula et al., 2013). However, this strategy of insecticide use has many serious side-effects, in particular on human health and the environment (Dabrowski and Balderacchi, 2013; Singleton et al., 2013; Dabrowski et al., 2014), having also a direct impact on the increase of Anopheles resistance to insecticides which has now been reported in 64 countries (WHO, 2012; Chang et al., 2013; Kabula et al., 2013; Tangena et al., 2013). Therefore, new and innovative approaches to control Anopheles vectors for reducing malaria transmission on a more durable and safer manner for human health are, hence, needed.
In Vietnam, malaria remains the most important vector-borne parasitic disease with a strong prevalence in forested regions, in particular along the international borders with Cambodia, where Binh Phuoc Province is located. This province, situated in central-south Vietnam, is considered as having the highest malarial transmission of all (Abe et al., 2009; Tran et al., 2012). The local malaria situation reported both resistance of Plasmodium to anti-plasmodial drugs and Anopheles mosquitoes to the insecticide used in this province (Huong et al., 2001; Nguyen et al., 2003; Thanh et al., 2010; Verhaeghen et al., 2010; Tran et al., 2012). However, there is a lack of information on the distribution and vector competence of the local Anopheles vectors.
The impact of the microbiota on mosquito infection and more specifically on Anopheles resistance to malaria pathogens is showing great potential toward reducing the mosquito vector competence and blocking transmission of several infectious diseases (Dong et al., 2006, 2009, 2011; Blumberg et al., 2013). The influence of the Anopheles microbiota on its vectorial competence to transmit pathogens is a growing field of investigation as demonstrated by the recent increase in studies, to cite a few (Favia et al., 2007; Briones et al., 2008; Dong et al., 2009; Rodrigues et al., 2010; Cirimotich et al., 2011; Blumberg et al., 2013; Gendrin and Christophides, 2013; Dennison et al., 2014; Buck et al., 2016). A previous report on the biodiversity of the abdomen microbiota of 100 wild Anopheles specimens (five species) from Dak Nong Province showed a high taxonomic diversity, including species reported as implicated in the mosquito resistance to Plasmodium infection (Ngo et al., 2015). By the same approach, we present here the bacterial diversity detected in 100 Anopheles specimens of eight species collected from Binh Phuoc, a Province characterized by high malaria endemicity. The Anopheles studied herein included two specimens naturally infected by Plasmodium vivax.
Materials and Methods
Anopheles Samples: Collection and Identification
One hundred Anopheles (out of 486 specimens collected from 4 sites located in Bu Gia Map District, Binh Phuoc Province in central-southern Vietnam (Ngo et al., 2014), which belonged to eight Anopheles species, including Anopheles dirus, An. jeyporiensis, An. maculatus, An. minimus, An. pampanai, An. rampae, An. sawadwongporni, and An. scanloni, were randomly chosen for the present study. These Anopheles specimens were collected between November and December, 2011 (rainy-beginning of the dry season) during 11 consecutive nights (17:00–20:00 h using methods, such as light trap capture and human landing catches indoor and outdoor (Ngo et al., 2014).
Anopheles mosquito identification was morphologically done in the field by sorting out each taxon. Specimens that belonged to the Dirus Complex or the Funestus and Maculatus Groups were individually identified to species level using the appropriate PCR-based assay as described by Walton et al. (1999, 2007) and Garros et al. (2005). Each individual was split into two pieces stored at -80°C until use: (1) head-thorax for species identification and detection of Plasmodium by molecular methods (Ngo et al., 2014); and (2) abdomen for bacterial analysis. One hundred abdomens of wild-caught Anopheles females were used for the subsequent bacterial study by both methods, culture and DNA fingerprint, as described below.
Bacterial Culture, DNA Extraction, and Strains Identification
Anopheles abdomens were surface rinsed twice in sterilized DNA-free water, and each abdomen was thoroughly disrupted using a sterilized tissue crusher device in 150 μl of sterile DNA-free water. Then, 10 μl of this suspension was spread on each prepared culture medium plate: blood sheep agar, R2A and Acetobacter agar. The detailed protocol followed the one published by Ngo et al. (2015). In order to identify strains, 16S rRNA gene was amplified using the Taq DNA polymerase (Go Taq Promega) and the universal primer pair (27f and 1492r) as previously described (Romano-Bertrand et al., 2012). PCR amplifications were checked by DNA electrophoresis in 1.5% agarose gels containing ethidium bromide and visualized under ultraviolet light. Sterile DNA-free water used for mosquito preparations was used in negative control for each series of DNA extraction and PCR. The successfully amplified products were sequenced with primer 27f, on an ABI 3730xl sequencer (Cogenics, Meylan, France). Each sequencing chromatograph was visually inspected and corrected.
Independent-Culture Analysis by 16S PCR-Temporal Temperature Gel Electrophoresis (PCR-TTGE)
Whole DNA was extracted from 100 μl of mosquito abdomen suspension using the Master Pure Gram Positive DNA purification kit as recommended by the supplier (Epicentre Biotechnologies, Madison, WI, USA). The purified and raw DNAs were kept at -80°C before further analyses. Because of low bacterial load in mosquito’s samples, a nested-PCR approach was necessary to obtain a sufficient PCR amplification (Romano-Bertrand et al., 2012). The first PCR amplified almost entirely the 16S rRNA gene using the 27f and 1492r primers as previously described (Romano-Bertrand et al., 2012). Then, products of the first PCR were used as template for the amplification of the V2–V3 hyper variable region using the FastStart high fidelity PCR system (Roche apply science) and the primers HDA1 with GC-clamp and HDA2 (Ogier et al., 2002; Roudiere et al., 2009). Again, sterile DNA-free water was used as negative control for each series of nested-PCR. If positive signal was observed, controls were analyzed in parallel of samples by TTGE migration. TTGE profiles of samples and controls were then compared. Positive controls, consisting on decreasing inoculums of Elizabethkingia anophelis (which is especially retrieved from mosquitos samples), were used to validate the analysis, from the DNA extraction and throughout the whole Temporal Temperature Gel Electrophoresis (TTGE) process. TTGE migration and TTGE bands sequencing were performed as described in Ngo et al. (2015).
Sequence Analysis, Species Identification, and OTU Affiliation
The sequences were analyzed by comparison with Genbank1 and Ribosomal Databases Project 2 (RDPII)2 using Basic Local Alignment Search Tool (BLAST) and Seqmatch programs, respectively. The sequence with the highest percentage was used for OTU affiliation as previously described by Ngo et al. (2015). Briefly, a sequence was affiliated to a species-level OTU when the percent of sequence similarity with the species type strain was above 99.0% (Drancourt et al., 2000). Below 99.0%, the sequence was affiliated to the genus of the reference sequence with the highest percentage. When several species reference sequences match equally, affiliation was done to the genus level or to a group of species if relevant. For example, sequence with 99.5% in similarity to both Aeromonas caviae and A. hydrophila was only assigned to the genus Aeromonas. The same rule was applied for the taxonomic level higher than the genus level if necessary. The 16S rRNA gene sequences from all cultured bacteria are available in GenBank (accession numbers KX449232–KX449311). Sequences from TTGE band are available by contacting corresponding author or by consulting the Supplementary Fasta File.
Diversity Index Calculation and Statistical Analysis
The microbiota diversity for each species of Anopheles was estimated by the calculation of crude diversity index (DI, number of different OTUs; Romano-Bertrand et al., 2012, 2014), Shannon–Wiener DI (H) and Simpson’s index (D; Gafan et al., 2005; Ledder et al., 2007; Romano-Bertrand et al., 2014).
The 100 Anopheles specimens were classified into the Dirus Complex or into the Funestus and Maculatus Groups according to their species identification. The bacterial diversity scores (DI, H, and D) for the different Anopheles species, complex and groups were then calculated.
Results
Anopheles Identification
Specific PCR assays identified eight Anopheles species among the 100 specimens collected in Binh Phuoc Province. Out of 100, 37 specimens belonged to the Dirus Complex including An. dirus (n = 33) and An. scanloni (n = 4), 43 to the Funestus Group with An. minimus (n = 34), An. jeyporiensis (n = 5), and An. pampanai (n = 4), and 20 to the Maculatus Group with An. sawadwongporni (n = 12), An. rampae (n = 6), and An. maculatus (n = 2; Figure 1A).
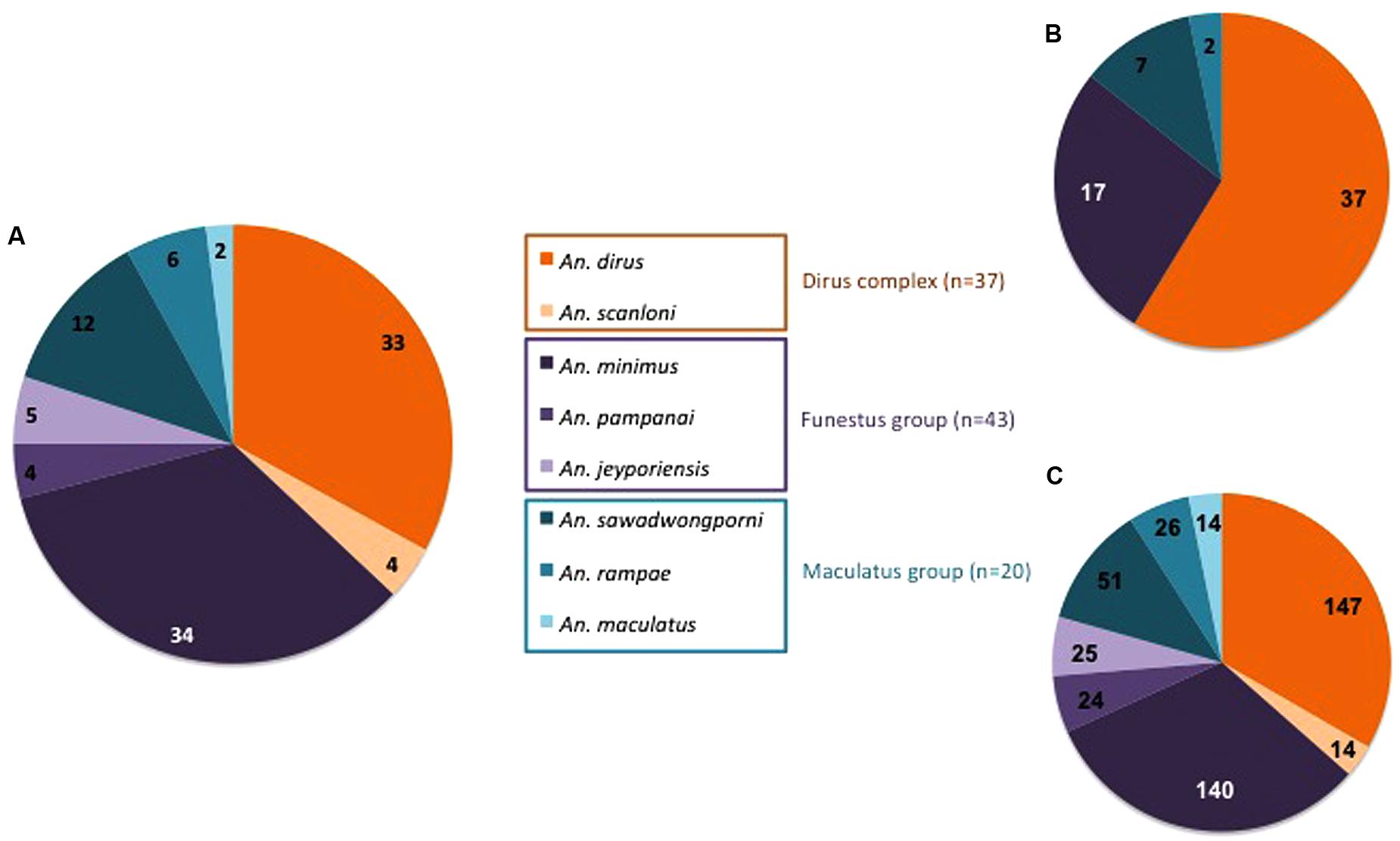
FIGURE 1. Distribution according to Anopheles species, complex and group of (A) mosquitoes specimens, (B) bacterial strains, and (C) TTGE bands.
Bacterial Diversity in the Abdomen of Anopheles Adults
The abdomens of 100 Anopheles were analyzed using both bacterial culture and PCR-TTGE methods. Only a few negative controls presented weak amplification bands in agarose gel after nested-PCR but these bands were never observed after TTGE migration, permitting to validate the profiles for mosquitoes’ samples. The regular positive controls highlighted a sensitivity of detection comprised between 101 and 102 bacterial inoculums throughout the TTGE process (data not shown). Bacteria were cultured or detected in 98% of the Anopheles specimens, the two negative specimens belonged to species An. minimus and An. scanloni. A total of 63 bacterial strains from 22 specimens positive by culture and 441 TTGE bands from 98 specimens positive by PCR were obtained. Distributions of strains and TTGE bands according to Anopheles species are shown in Figures 1B,C, respectively.
Both strains and 393 TTGE bands were further analyzed by 16S rRNA gene PCR and sequenced. The remaining 48 TTGE bands were identified by comparison with another co-migrating band on the same TTGE gel. Results were presented in Supplementary Tables. Strains and PCR-TTGE bands were affiliated to seven phyla, the most represented being Proteobacteria, Firmicutes, and Actinobacteria (Figure 2). Considering OTUs, 193 bacterial OTUs corresponding to 190 genera-level OTUs (including 85 OTUs for which identification was performed until species-level), one family-level and two phyla-level OTUs. The sequences obtained from 3 TTGE bands were not accurate enough to be affiliated to the genus level, probably due to mixed DNA amplified and sequenced from co-migrating bands; one of them was affiliated to the Phylum Acidobacteria and two others to the Family Planctomycetaceae (Phylum Planctomycetes). The OTU richness by phylum is summarized in Figure 3. Proteobacteria was the most diversified phylum with a richness corresponding to 57 genera-level OTUs, followed by Actinobacteria and Firmicutes with, respectively, 24 and 18 genera-level OTUs.
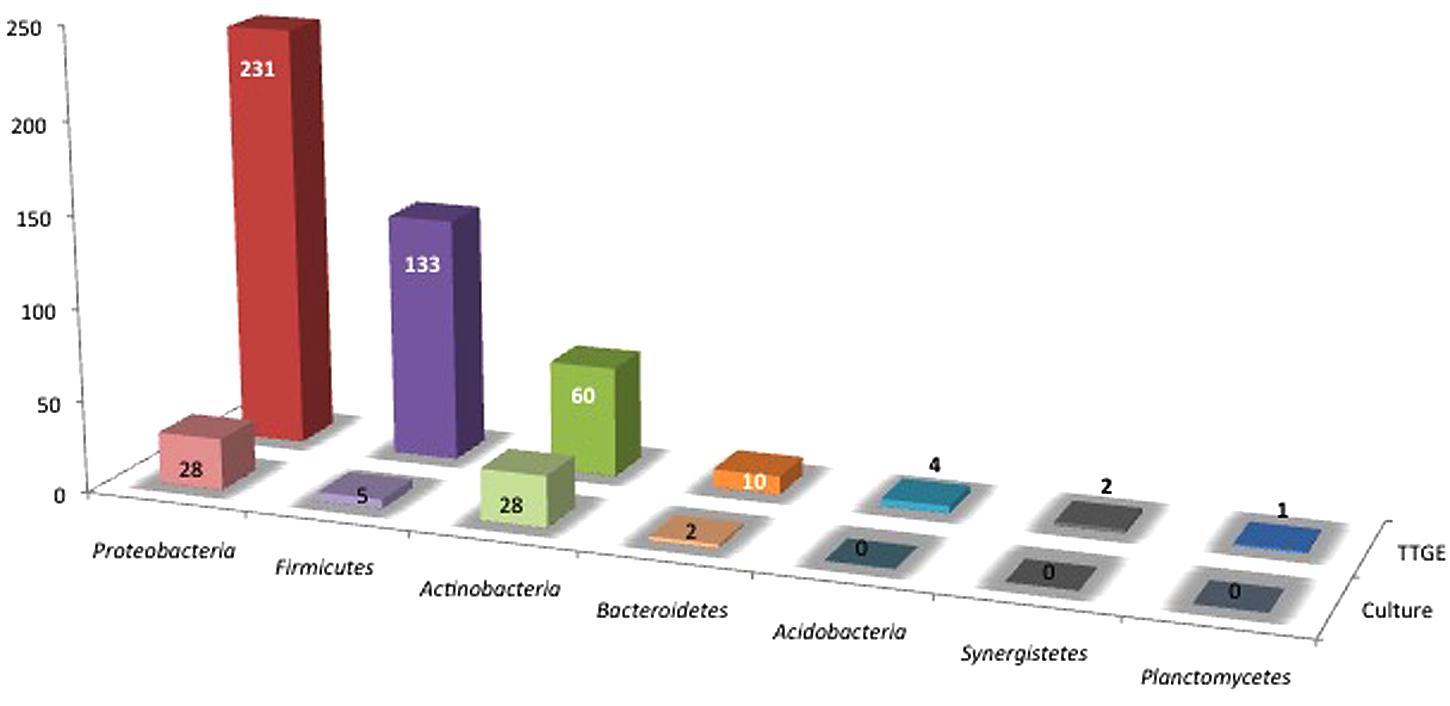
FIGURE 2. Distribution of TTGE bands and bacterial isolates according to phylum.

FIGURE 3. Repertory of genus-level OTUs according to phylum in adult Anopheles gut microbiota. ∗Cultivable genera; Genera only retrieved in culture are underlined; 1Genera identified in Anopheles abdomen for the first time; Genera previously found in Anopheles from Vietnam in bold face (Ngo et al., 2015); In parenthesis, the percent (50% and higher) of Anopheles species carrying genera frequently found in Binh Phuoc Anopheles microbiota.
Only 32 from the 190 genera-level OTUs were retrieved in culture, mainly from Actinobacteria with 18 cultivable genera (56%). Out of these 32 cultivable OTUs, 15 (7.9%) were not retrieved by the molecular approach (genera underlined in Figure 3 and Supplementary Tables).
In this study, molecular approach allowed the detection of more bacteria than culture, although culture improved the description of bacterial richness in the Anopheles abdomen microbiota and should be considered as complementary to PCR-based methods. The Actinobacteria richness was particularly widen thanks to culture.
Bacterial Richness and Diversity of the Abdomen Microbiota According to the Anopheles Species
Bacterial diversity according to Anopheles species was displayed in Figure 4A. No obvious link was observed between Anopheles complex or groups and microbiota diversity. However, the number of specimens in several Anopheles species is too low for robust conclusions, only tendencies can be drawn. Amongst the three major phyla, Firmicutes and Proteobacteria were present in all Anopheles species, while Actinobacteria were absent from An. jeyporiensis, An. scanloni, and An. rampae (Figure 4). An. minimus, considered as one of the primary vector of Plasmodium in Southeast Asia, including Vietnam, was the sole species colonized by members of the seven bacterial phyla. Two other malaria vectors well represented in the collection, An. dirus and An. sawadwongporni, were each colonized by five bacterial phyla (Figure 4A). Despite the low number of specimens (n = 6) in this species, An. rampae presented a particular diversity profile with the replacement of Actinobacteria by Bacteroidetes regarding other Anopheles species. Nevertheless, the phylum richness appeared more dependent to the number of specimens in a species of Anopheles than to the Anopheles complex and groups (Figures 4A,B). Indeed, if Anopheles complex or group were considered, the phyla distribution in percent was very similar among them (Figure 4B).
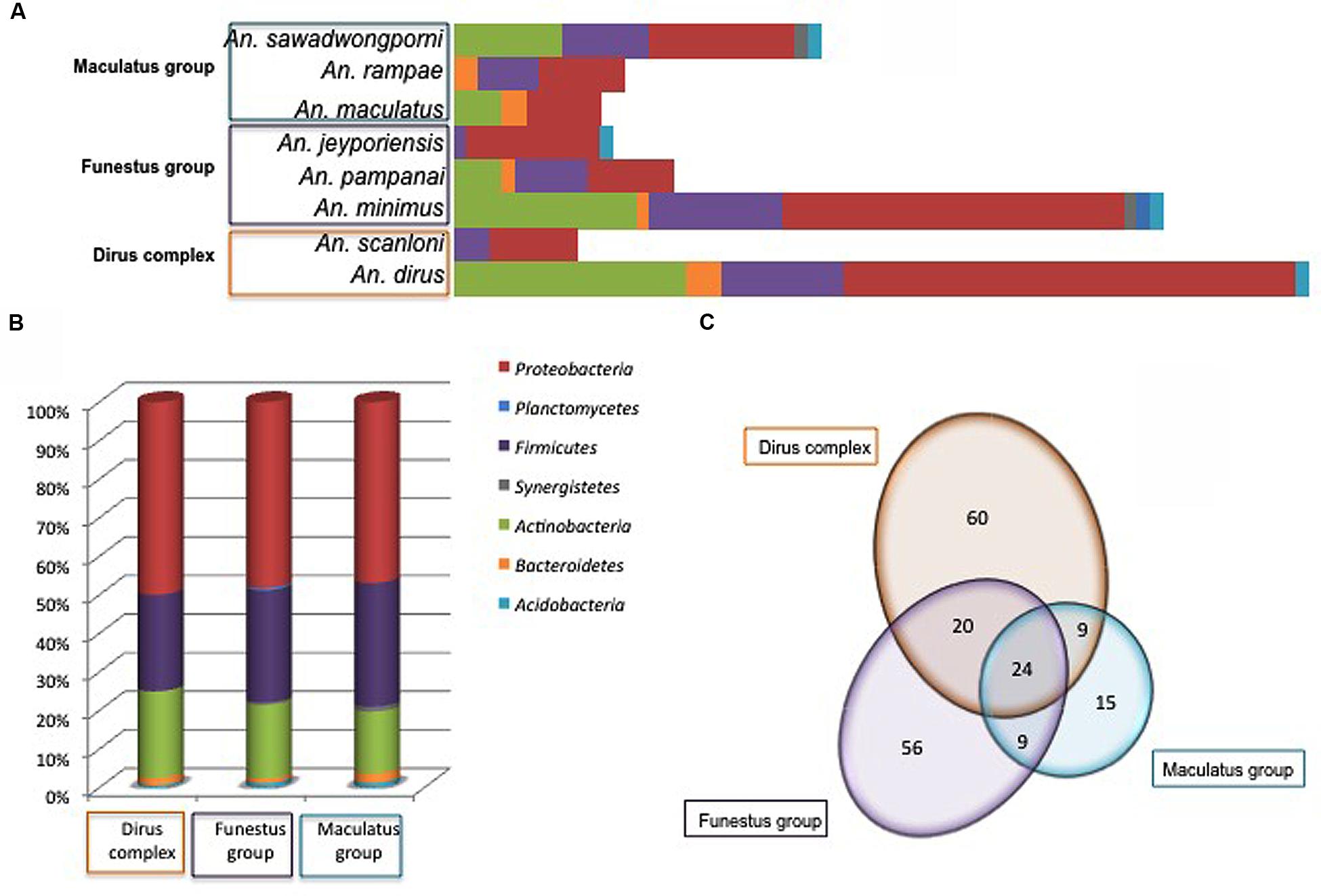
FIGURE 4. Number of different OTUs per phylum and Anopheles species (A), percent of OTUs in every phyla (B), and distribution of shared and specific OTUs (Venn diagram in C) according to Anopheies complex and groups. The phyla color code is given for (A) and (B).
However, at the OTU level the distribution varied according Anopheles complex and groups (Figure 4C). Among the 193 OTUs, 113, 109, and 57 OTUs were identified within the Dirus Complex, the Funestus and Maculatus Groups, respectively (Figure 4C). Out of them, 24 OTUs were shared by the three Anopheles taxa, 20 OTUs were shared by the Dirus Complex and the Funestus Group, and nine OTUs were commonly identified either between the Funestus and Maculatus Groups, or the Dirus Complex and Funestus Group (Figure 4C).
Anopheles abdomen microbiota diversity quantified by the mean index and scores was presented in Table 1. The average DI per specimen reached 4.843. An. maculatus (DI = 7.0) and An. jeyporiensis (DI = 6.0) microbiota were the most diverse, whereas An. minimus displayed lowest diversity (DI = 4.429) (Table 1). Considering both bacterial richness and equitability of the OTUs within Anopheles species, the average Shannon–Wiener score (H) for all species was 5.569. An. minimus had the higher H score (H = 6.213) indicating that the numbers of OTUs are evenly distributed between all specimens (Table 1). The Simpson index (D) takes into account both bacterial species richness and an evenness of abundance among the bacterial species present, and is inversely proportional to the diversity (DI). For all Anopheles species, except An. sawadwongporni (D = 0.308), the D-values were low ranging between 0 and 0.082, suggesting high diversities (Table 1). However, these results should be taken with caution because of the low number of specimens for An. scanloni (n = 3), An. jeyporiensis (n = 5), An. pampanai (n = 4), An. maculatus (n = 2), and An. rampae (n = 6).
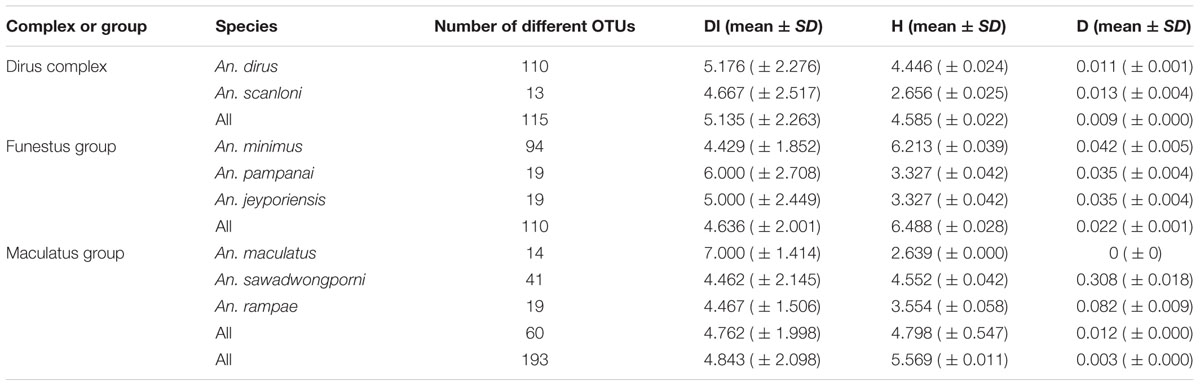
TABLE 1. Number of OTUs and diversity indexes for each species of Anopheles.
The Core Microbiota of Vietnam Anopheles Is Limited
Three Firmicutes genera, Staphylococcus, Bacillus, and Clostridium, were the most prevalent in the abdomen microbiota of Binh Phuoc Anopheles, respectively, identified in 29.6, 23.5, and 21.4% of the specimens. Besides, Propionibacterium, Peptostreptococcus, Acinetobacter, Burkholderia, Comamonas, Delftia, Lysobacter, and Sphingomonas were detected in more than 10% of the specimens, with frequencies ranging from 10.2 to 18.4%. The Family Enterobacteriaceae (Enterobacter, Klebsiella, Pantoea, and Proteus) was detected in 15.3% of the specimens. The other 90 genera were found at frequencies lower than 10%, including 81 genera detected in only 1–4 samples (<5%), certainly due to the important bacterial diversity in the mosquito microbiota (Supplementary Table 1).
The most represented genera in Binh Phuoc Anopheles microbiota were Staphylococcus and Sphingomonas (Supplementary Table 1 and Figure 3). Six other genera retrieved at lower frequencies were newly identified in our study: Agrococcus, Anoxybacillus, Peptostreptococcus, Lysobacter, Paracoccus, and Sphingopyxis (Supplementary Table 1 and Figure 3). Finally, a total of 21 genera were detected in more than 4 of 8 Anopheles species but these genera were present in <30% of the mosquito specimens confirming that the high bacterial diversity in Binh Phuoc Anopheles impaired the definition a core microbiota. Nevertheless, the presence of some frequent genera in Binh Phuoc Anopheles microbiota that were not yet described in Anopheles pan-microbiota, suggested the existence of specific microbiota according to the geographic region probably due to a strong influence of local environmental conditions.
Concerning the pan-microbiota of the whole genus Anopheles, meta-analysis of recent studies (Gendrin and Christophides, 2013; Manguin et al., 2013; Minard et al., 2013; Sharma et al., 2014; Villegas and Pimenta, 2014; Ngo et al., 2015) and bibliography databases survey for older publications, indicated that 51 bacterial genera (48.6%) were probably detected herein for the first time in Anopheles (1 in Figure 3 and Supplementary Table 1). These newly described genera could be slightly overestimated due to the meta-analysis approach and the comparison with non-fully exhaustive data but it clearly suggested that the overall richness of Anopheles microbiome is far from being fully explored. This is particularly true in Southeast Asia where very little work has been done on this topic (Gendrin and Christophides, 2013; Ngo et al., 2015).
In comparison with a previous study on the microbiota diversity in specimens from Dak Nong Province, Vietnam (Ngo et al., 2015), 17 genera (in bold in Figure 3) were common in Anopheles species from both provinces. Most of these genera were found at frequencies below 10% in the Anopheles specimens from Binh Phuoc Province, except four genera: Sphingomonas (13.3%), Acinetobacter (18.4%), Bacillus (23.5%), and Staphylococcus (29.6%). The genus Acinetobacter was detected in more than half Anopheles species in both Binh Phuoc and Dak Nong Anopheles microbiota. It could be considered as the sole genus forming the core microbiota of Vietnam Anopheles.
Characterization of the Abdomen Microbiota of Plasmodium Naturally Infected Anopheles Specimens
Two specimens were naturally infected by P. vivax, out of the 486 Anopheles specimens collected in Binh Phuoc Province (Ngo et al., 2014). They belonged to the species An. dirus and An. pampanai. The abdomen bacterial microbiota of these two specimens was described by PCR-TTGE only, because cultured isolates were not obtained from these two infected mosquitoes. The sequence analysis of seven TTGE bands showed the presence of six bacterial taxa: Acinetobacter sp., Geobacillus sp., Luteimonas sp., Methylobacterium sp., Rubellimicrobium sp., and Sphingomonas sp., among which five (83.3%) belonged to Proteobacteria and only one, Geobacillus sp., to Firmicutes. The genus Acinetobacter, Geobacillus, Methylobacter, and Sphingomonas belonged to the core50% microbiota and were also retrieved from other non-infected specimens of Anopheles, whereas Luteimonas was only retrieved from the infected An. pampanai specimen, and Rubellimicrobium from both the infected An. dirus and one specimen of An. rampae (Supplementary Table 1).
Discussion
The description of microbiota in vector arthropods presented increasing interest because of potential role in vector competence. Protective role of Anopheles bacterial microbiota against Plasmodium infections has been demonstrated because clearing midgut microbiota by antibiotic treatment resulted in enhanced Plasmodium infections rate (Beier et al., 1994; Favia et al., 2007; Dong et al., 2009; Rodrigues et al., 2010). Similarly but in semi-natural settings, antibiotics in ingested blood enhanced the susceptibility of An. gambiae to Plasmodium infection and increasing mosquito fitness through higher survival and fecundity. Moreover, the presence of antibiotics in the blood of malaria-infected people was identified as a risk for increased disease transmission (Gendrin et al., 2015). In addition, the experimental co-infections of bacteria with Plasmodium have showed the reduction of the number of developing oocysts in the mosquito midgut, both in laboratory and field conditions (Pumpuni et al., 1993; Lowenberger et al., 1999; Gonzalez-Ceron et al., 2003; Dong et al., 2009; Meister et al., 2009; Cirimotich et al., 2011). More recently, it was observed that expansion of midgut microbiota by negative regulation of An. gambiae immune response increased susceptibility to Plasmodium as observed before for microbiota depletion (Dennison et al., 2015). Finally, it’s probably the microbiota equilibrium that confers resistance to Plasmodium infection. Then, the development of new approaches in malaria control needs the description of the microbiota of wild Anopheles species and its variations (Rani et al., 2009; Boissière et al., 2012; Chavshin et al., 2012; Osei-Poku et al., 2012; Gendrin and Christophides, 2013; Manguin et al., 2013; Ngo et al., 2015).
As previously done (Ngo et al., 2015), we present herein a combined approach associating molecular detection and culture that improve the performance of each separate method. About one third of the bacterial genera detected in this study were obtained by culture, thus providing a valuable collection of isolates for further experiments such as experimental challenges (Ramirez et al., 2014) and in paratransgenesis projects (Chavshin et al., 2014). Due to the low resolution of TTGE fingerprints when the number of band is high, PCR-TTGE is only suitable for the description of microbiota with relative low richness as demonstrated for human neonatal microbiota (Jacquot et al., 2011) and surgical microbiota (Romano-Bertrand et al., 2014), but also for the description of mosquito microbiota (Manguin et al., 2013; Ngo et al., 2015). By this mean, we previously described most of the known repertory of bacterial taxa found in Asian Anopheles as shown by Villegas and Pimenta (2014). TTGE fingerprinting gave a good description of majority bacterial populations in mosquito microbiota, with a rather good sensitivity, as demonstrated by the extent of the taxonomic richness described herein compared to other studies even those which used Next Generation Sequencing (NGS; Villegas and Pimenta, 2014). Moreover, the use of PCR-TTGE in this study allowed a comparison with previous studies in Vietnam (Ngo et al., 2015), but also in Thailand (Manguin et al., 2013). NGS is now becoming a reference method for microbiota description and knowledge of mosquito-associated bacteria would likely benefit from studies by NGS. Because NGS provide huge amounts of data sometimes partially exploited, preliminary and comparative studies by TTGE or other community fingerprinting approaches allow asking scientific questions to be further in-depth explored by NGS.
The Anopheles collection of 100 wild specimens collected in Binh Phuoc Province was colonized by a great diversity of bacteria with, to our knowledge, 51 genera not yet described in anophelines. Thereby, this study widens the description of the Anopheles pan-microbiota. The huge diversity of Anopheles associated bacteria contrasts the low richness observed per specimen. This result is similar to that shown by Osei-Poku et al. (2012) in different mosquito genera, including Anopheles, in which bacterial richness per specimen is very low, nearly always dominated by a small number of taxa. This low richness could explain that only 78% of the specimens are positive in culture. Other reasons could be the trapping of some bacteria in the Anopheles tissues despite the preparative crushing step, the presence of viable but non-cultivable bacteria and the limited panel of culture medium used in the study. Culturomics approaches (Tandina et al., 2016) with the use of large panel of culture media and conditions could enhance the culture yield in next studies.
Within the bacteria detected in Anopheles specimens from Binh Phuoc, approximately half of the genera have previously been identified in the intra-abdominal microbiota of Anopheles populations from Thailand (Manguin et al., 2013), Vietnam (Manguin et al., 2013; Ngo et al., 2015), Cameroon (Boissière et al., 2012), Iran (Chavshin et al., 2012), India (Rani et al., 2009), or in reviews (Gendrin and Christophides, 2013; Minard et al., 2013; Hughes et al., 2014; Villegas and Pimenta, 2014).
Our results also showed that there is no link between bacterial diversity and host species or group of host species collected in Binh Phuoc Province. This result is in accordance with previous reports that rather showed the influence of the local environment of the sampling site, environment that gives a specific bacterial profile to mosquito specimens (Boissière et al., 2012; Manguin et al., 2013; Buck et al., 2016). The microbiota diversity of host species will also be significantly influenced by the origin of specimens like wild versus laboratory reared Anopheles mosquitoes (Chavshin et al., 2012). For instance, the selective pressure of laboratory conditions may limit bacteria acquisition at both larval and adult stages resulting in great reduction or nil cultivable bacteria in mosquito midgut (Riehle and Jacobs-Lorena, 2005; Favia et al., 2007; Gusmão et al., 2010). Studies on microbiota of An. gambiae larvae and pupae showed that only subset of bacteria from the aquatic habitat were able to inhabit the mosquito gut (Wang et al., 2011). The same influence of environment on Anopheles microbiota is likely to occur during the terrestrial life of adults. Considering the repertory of bacterial genera presented in Figure 3, the presence of soil- and plant-associated bacteria in Anopheles microbiota is probable. For instance, Agrococcus, Janibacter, Streptomyces, Bacillus (and related genera), and Lysobacter are classical soil inhabitants, whereas Asaia, Burkholderia, Comamonas, Delftia, and Sphingomonas and Methylobacterium are frequently associated with plants. In addition, several prevalent bacterial genera are associated with animal or human microbiota, mainly to cutaneous microbiota: Propionibacterium, Staphylococcus, Peptostreptococcus, Clostridium, and Acinetobacter. These results suggest that bacteria from diverse origins could colonize adult Anopheles.
Interestingly, the Anopheles species from Binh Phuoc Province contained members of the genus Enterobacter sp. recently isolated from wild An. gambiae from Zambia. Enterobacter sp. received lot of attention due its anti-Plasmodium effect through the production of reactive oxygen species that directly target Plasmodium parasites in the midgut of An. gambiae (Cirimotich et al., 2011). More recently, another bacteria, Chromobacterium Csp_P, has been described as responsible of vectors refractoriness to P. falciparum and dengue virus (Ramirez et al., 2014). Then, further studies on mosquito microbiota should provide new insights on pathogens transmission and might open new ways to prevent vector-borne diseases.
Recently published studies have described new bacterial species, first isolated from Anopheles mosquitoes, such as E. anophelis (Kampfer et al., 2011), Thorsellia anophelis (Kampfer et al., 2006a), and Janibacter anophelis (Kampfer et al., 2006b). The latter species was accurately identified herein after bacterial culture and the species J. terrae described in soil was also found in Binh Phuoc Anopheles. Bacterial identification to the species level or below, defining variants within a bacterial species (genovar, genomovar, biovar, etc.), is needed to determine whether strictly identical bacterial populations are shared between environment, host and vector, or if Anopheles-associated bacteria are specific.
As a tendency to be confirmed, we also described herein the microbiota of two Anopheles (An. dirus and An. pampanai) naturally infected by P. vivax. The genus Acinetobacter solely was common to the two infected specimens but it was also one of the most prevalent bacterial species forming Anopheles microbiota. Indeed, it was frequently identified in the microbiota of both Asian and African anopheline species (Cameroon, Iran, India, Kenya, Mali, and Vietnam; Straif et al., 1998; Rani et al., 2009; Chavshin et al., 2012). Nevertheless, there is no available report on the implication of this genus for the development of Plasmodium in Anopheles and the interaction between this bacterial genus and malarial parasites in Anopheles might be studied.
The huge diversity of the microbial world even within a single species urges to overpass genus or species level for microbiota descriptions. Indeed, it is likely that the informative taxonomic level for host and microbes interactions is the subspecies level. Such a precise characterization needs availability of bacterial strains, highlighting again the interest of culture in microbiota studies. Moreover, the phenotype of bacterial strains could be of great interest to understand mechanisms involved in bacteria/Plasmodium/Anopheles interactions. This study provides 51 strains available for further experiments. Moreover, to the niche level, functional metagenomic and metabolomic approaches should be undertaken to determine microbiota conditions that favor or not vector competence.
Author Contributions
CN made the bacterial analyses (bacterial cultures and molecular analyses), and results’ interpretation. SR-B helped for DNA sequencing, bacterial identification, and results’ interpretation. SM contributed to the study design, the specimens collection and the results’ interpretation. EJ-B supervised the study design, the bacterial analyses, and the results’ interpretation. All the authors contributed to the manuscript redaction.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was made possible through a PhD allowance provided to the CN by the Institute of Research for Development (IRD), France. This work was also supported by the association ADEREMPHA, Sauzet, France.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2016.02095/full#supplementary-material
Footnotes
References
Abe, T., Honda, S., Nakazawa, S., Tuong, T. D., Thieu, N. Q., Hung, L. X., et al. (2009). Risk factors for malaria infection among ethnic minorities in Binh Phuoc, Vietnam. Southeast Asian J. Trop. Med. Public Health 40, 18–29.
Beier, M. S., Pumpuni, C. B., Beier, J. C., and Davis, J. R. (1994). Effects of para-aminobenzoic acid, insulin, and gentamicin on Plasmodium falciparum development in anopheline mosquitoes (Diptera: Culicidae). J. Med. Entomol. 31, 561–565. doi: 10.1093/jmedent/31.4.561
Blumberg, B. J., Trop, S., Das, S., and Dimopoulos, G. (2013). Bacteria- and IMD pathway-independent immune defenses against Plasmodium falciparum in Anopheles gambiae. PLoS ONE 8:e72130. doi: 10.1371/journal.pone.0072130
Boissière, A., Tchioffo, M. T., Bachar, D., Abate, L., Marie, A., Nsango, S. E., et al. (2012). Midgut microbiota of the malaria mosquito vector Anopheles gambiae and interactions with Plasmodium falciparum infection. PLoS Pathog 8:e1002742. doi: 10.1371/journal.ppat.1002742
Briones, A. M., Shililu, J., Githure, J., Novak, R., and Raskin, L. (2008). Thorsellia anophelis is the dominant bacterium in a Kenyan population of adult Anopheles gambiae mosquitoes. ISME J. 2, 74–82. doi: 10.1038/ismej.2007.95
Buck, M., Nilsson, L. K., Brunius, C., Dabire, R. K., Hopkins, R., and Terenius, O. (2016). Bacterial associations reveal spatial population dynamics in Anopheles gambiae mosquitoes. Sci. Rep. 6, 22806. doi: 10.1038/srep22806
Chang, X. L., Xue, Y. Q., Zhang, A. D., Zhu, G. D., and Fang, Q. (2013). [Deltamethrin resistance, metabolic detoxification enzyme and kdr mutation in Anopheles sinensis in region along Huaihe River in Anhui Province]. Zhongguo Xue Xi Chong Bing Fang Zhi Za Zhi 25, 263–267.
Chavshin, A. R., Oshaghi, M. A., Vatandoost, H., Pourmand, M. R., Raeisi, A., Enayati, A. A., et al. (2012). Identification of bacterial microflora in the midgut of the larvae and adult of wild caught Anopheles stephensi: a step toward finding suitable paratransgenesis candidates. Acta Trop. 121, 129–134. doi: 10.1016/j.actatropica.2011.10.015
Chavshin, A. R., Oshaghi, M. A., Vatandoost, H., Pourmand, M. R., Raeisi, A., and Terenius, O. (2014). Isolation and identification of culturable bacteria from wild Anopheles culicifacies, a first step in a paratransgenesis approach. Parasit Vectors 7, 419. doi: 10.1186/1756-3305-7-419
Cirimotich, C. M., Dong, Y., Clayton, A. M., Sandiford, S. L., Souza-Neto, J. A., Mulenga, M., et al. (2011). Natural microbe-mediated refractoriness to Plasmodium infection in Anopheles gambiae. Science 332, 855–858. doi: 10.1126/science.1201618
Dabrowski, J. M., and Balderacchi, M. (2013). Development and field validation of an indicator to assess the relative mobility and risk of pesticides in the Lourens River catchment, South Africa. Chemosphere 93, 2433–2443. doi: 10.1016/j.chemosphere.2013.08.070
Dabrowski, J. M., Shadung, J. M., and Wepener, V. (2014). Prioritizing agricultural pesticides used in South Africa based on their environmental mobility and potential human health effects. Environ. Int. 62, 31–40. doi: 10.1016/j.envint.2013.10.001
Dennison, N. J., BenMarzouk-Hidalgo, O. J., and Dimopoulos, G. (2015). MicroRNA-regulation of Anopheles gambiae immunity to Plasmodium falciparum infection and midgut microbiota. Dev. Comp. Immunol. 49, 170–178. doi: 10.1016/j.dci.2014.10.016
Dennison, N. J., Jupatanakul, N., and Dimopoulos, G. (2014). The mosquito microbiota influences vector competence for human pathogens. Curr. Opin. Insect Sci. 3, 6–13. doi: 10.1016/j.cois.2014.07.004
Dong, Y., Aguilar, R., Xi, Z., Warr, E., Mongin, E., and Dimopoulos, G. (2006). Anopheles gambiae immune responses to human and rodent Plasmodium parasite species. PLoS Pathog 2:e52. doi: 10.1371/journal.ppat.0020052
Dong, Y., Das, S., Cirimotich, C., Souza-Neto, J. A., McLean, K. J., and Dimopoulos, G. (2011). Engineered anopheles immunity to Plasmodium infection. PLoS Pathog 7:e1002458. doi: 10.1371/journal.ppat.1002458
Dong, Y., Manfredini, F., and Dimopoulos, G. (2009). Implication of the mosquito midgut microbiota in the defense against malaria parasites. PLoS Pathog 5:e1000423. doi: 10.1371/journal.ppat.1000423
Drancourt, M., Bollet, C., Carlioz, A., Martelin, R., Gayral, J. P., and Raoult, D. (2000). 16S ribosomal DNA sequence analysis of a large collection of environmental and clinical unidentifiable bacterial isolates. J. Clin. Microbiol. 38, 3623–3630.
Favia, G., Ricci, I., Damiani, C., Raddadi, N., Crotti, E., Marzorati, M., et al. (2007). Bacteria of the genus Asaia stably associate with Anopheles stephensi, an Asian malarial mosquito vector. Proc. Natl. Acad. Sci. U.S.A. 104, 9047–9051. doi: 10.1073/pnas.0610451104
Gafan, G. P., Lucas, V. S., Roberts, G. J., Petrie, A., Wilson, M., and Spratt, D. A. (2005). Statistical analyses of complex denaturing gradient gel electrophoresis profiles. J. Clin. Microbiol. 43, 3971–3978. doi: 10.1128/JCM.43.8.3971-3978.2005
Garros, C., Harbach, R. E., and Manguin, S. (2005). Morphological assessment and molecular phylogenetics of the Funestus and Minimus groups of Anopheles (Cellia). J. Med. Entomol. 42, 522–536. doi: 10.1603/0022-25852005042[0522:MAAMPO]2.0.CO;2
Gendrin, M., and Christophides, G. K. (2013). “The Anopheles mosquitoes microbiota and their impact on pathogen transmission,” in Anopheles mosquitoes - New insights into malaria vectors, ed. S. Manguin (Rijeka: InTech Open Access), 525–548.
Gendrin, M., Rodgers, F. H., Yerbanga, R. S., Ouedraogo, J. B., Basanez, M. G., Cohuet, A., et al. (2015). Antibiotics in ingested human blood affect the mosquito microbiota and capacity to transmit malaria. Nat. Commun. 6, 5921. doi: 10.1038/ncomms6921
Gonzalez-Ceron, L., Santillan, F., Rodriguez, M. H., Mendez, D., and Hernandez-Avila, J. E. (2003). Bacteria in midguts of field-collected Anopheles albimanus block Plasmodium vivax sporogonic development. J. Med. Entomol. 40, 371–374. doi: 10.1603/0022-2585-40.3.371
Gusmão, D. S., Santos, A. V., Marini, D. C., Bacci, M. Jr., Berbert-Molina, M. A., and Lemos, F. J. (2010). Culture-dependent and culture-independent characterization of microorganisms associated with Aedes aegypti (Diptera: Culicidae) (L.) and dynamics of bacterial colonization in the midgut. Acta Trop 115, 275–281. doi: 10.1016/j.actatropica.2010.04.011
Hughes, G. L., Dodson, B. L., Johnson, R. M., Murdock, C. C., Tsujimoto, H., Suzuki, Y., et al. (2014). Native microbiome impedes vertical transmission of Wolbachia in Anopheles mosquitoes. Proc. Natl. Acad. Sci. U.S.A. 111, 12498–12503. doi: 10.1073/pnas.1408888111
Huong, N. M., Hewitt, S., Davis, T. M., Dao, L. D., Toan, T. Q., Kim, T. B., et al. (2001). Resistance of Plasmodium falciparum to antimalarial drugs in a highly endemic area of southern Viet Nam: a study in vivo and in vitro. Trans. R. Soc. Trop. Med. Hyg. 95, 325–329. doi: 10.1016/S0035-9203(01)90254-8
Jacquot, A., Neveu, D., Aujoulat, F., Mercier, G., Marchandin, H., Jumas-Bilak, E., et al. (2011). Dynamics and clinical evolution of bacterial gut microflora in extremely premature patients. J. Pediatr. 158, 390–396. doi: 10.1016/j.jpeds.2010.09.007
Kabula, B., Tungu, P., Malima, R., Rowland, M., Minja, J., Wililo, R., et al. (2013). Distribution and spread of pyrethroid and DDT resistance among the Anopheles gambiae complex in Tanzania. Med. Vet. Entomol. 28, 244–252. doi: 10.1111/mve.12036
Kampfer, P., Lindh, J. M., Terenius, O., Haghdoost, S., Falsen, E., Busse, H. J., et al. (2006a). Thorsellia anophelis gen. nov., sp. nov., a new member of the Gammaproteobacteria. Int. J. Syst. Evol. Microbiol. 56, 335–338. doi: 10.1099/ijs.0.63999-0
Kampfer, P., Terenius, O., Lindh, J. M., and Faye, I. (2006b). Janibacter anophelis sp. nov., isolated from the midgut of Anopheles arabiensis. Int. J. Syst. Evol. Microbiol. 56, 389–392. doi: 10.1099/ijs.0.63905-0
Kampfer, P., Matthews, H., Glaeser, S. P., Martin, K., Lodders, N., and Faye, I. (2011). Elizabethkingia anophelis sp. nov., isolated from the midgut of the mosquito Anopheles gambiae. Int. J. Syst. Evol. Microbiol. 61, 2670–2675. doi: 10.1099/ijs.0.026393-0
Ledder, R. G., Gilbert, P., Huws, S. A., Aarons, L., Ashley, M. P., Hull, P. S., et al. (2007). Molecular analysis of the subgingival microbiota in health and disease. Appl. Environ. Microbiol. 73, 516–523. doi: 10.1128/AEM.01419-06
Lowenberger, C. A., Kamal, S., Chiles, J., Paskewitz, S., Bulet, P., Hoffmann, J. A., et al. (1999). Mosquito-Plasmodium interactions in response to immune activation of the vector. Exp. Parasitol. 91, 59–69. doi: 10.1006/expr.1999.4350
Manguin, S., Ngo, C. T., Tainchum, K., Juntarajumnong, W., Chareonviriyaphap, T., Michon, A. L., et al. (2013). “Bacterial biodiversity in midguts of Anopheles mosquitoes, malaria vectors in Southeast Asia,” in Anopheles mosquitoes - New insights into malaria vectors, ed. S. Manguin (Rijeka: InTech Open Access), 549–576.
Meister, S., Agianian, B., Turlure, F., Relogio, A., Morlais, I., Kafatos, F. C., et al. (2009). Anopheles gambiae PGRPLC-mediated defense against bacteria modulates infections with malaria parasites. PLoS Pathog 5:e1000542. doi: 10.1371/journal.ppat.1000542
Minard, G., Mavingui, P., and Moro, C. V. (2013). Diversity and function of bacterial microbiota in the mosquito holobiont. Parasit Vectors 6, 146. doi: 10.1186/1756-3305-6-146
Ngo, C. T., Aujoulat, F., Veas, F., Jumas-Bilak, E., and Manguin, S. (2015). Bacterial diversity associated with wild caught Anopheles mosquitoes from Dak Nong Province, Vietnam using culture and DNA fingerprint. PLoS ONE 10:e0118634. doi: 10.1371/journal.pone.0118634
Ngo, C. T., Dubois, G., Sinou, V., Parzy, D., Le, H. Q., Harbach, R. E., et al. (2014). Diversity of Anopheles mosquitoes in Binh Phuoc and Dak Nong Provinces of Vietnam and their relation to disease. Parasit Vectors 7, 316. doi: 10.1186/1756-3305-7-316
Nguyen, M. H., Davis, T. M., Cox-Singh, J., Hewitt, S., Tran, Q. T., Tran, B. K., et al. (2003). Treatment of uncomplicated falciparum malaria in southern Vietnam: can chloroquine or sulfadoxine-pyrimethamine be reintroduced in combination with artesunate? Clin. Infect. Dis. 37, 1461–1466. doi: 10.1086/379323
Ogier, J. C., Son, O., Gruss, A., Tailliez, P., and Delacroix-Buchet, A. (2002). Identification of the bacterial microflora in dairy products by temporal temperature gradient gel electrophoresis. Appl. Environ. Microbiol. 68, 3691–3701. doi: 10.1128/AEM.68.8.3691-3701.2002
Osei-Poku, J., Mbogo, C. M., Palmer, W. J., and Jiggins, F. M. (2012). Deep sequencing reveals extensive variation in the gut microbiota of wild mosquitoes from Kenya. Mol. Ecol. 21, 5138–5150. doi: 10.1111/j.1365-294X.2012.05759.x
Pumpuni, C. B., Beier, M. S., Nataro, J. P., Guers, L. D., and Davis, J. R. (1993). Plasmodium falciparum: inhibition of sporogonic development in Anopheles stephensi by gram-negative bacteria. Exp. Parasitol. 77, 195–199. doi: 10.1006/expr.1993.1076
Ramirez, J. L., Short, S. M., Bahia, A. C., Saraiva, R. G., Dong, Y., Kang, S., et al. (2014). Chromobacterium Csp_P reduces malaria and dengue infection in vector mosquitoes and has entomopathogenic and in vitro anti-pathogen activities. PLoS Pathog 10:e1004398. doi: 10.1371/journal.ppat.1004398
Rani, A., Sharma, A., Rajagopal, R., Adak, T., and Bhatnagar, R. K. (2009). Bacterial diversity analysis of larvae and adult midgut microflora using culture-dependent and culture-independent methods in lab-reared and field-collected Anopheles stephensi-an Asian malarial vector. BMC Microbiol. 9:96. doi: 10.1186/1471-2180-9-96
Riehle, M. A., and Jacobs-Lorena, M. (2005). Using bacteria to express and display anti-parasite molecules in mosquitoes: current and future strategies. Insect Biochem. Mol. Biol. 35, 699–707. doi: 10.1016/j.ibmb.2005.02.008
Rodrigues, J., Brayner, F. A., Alves, L. C., Dixit, R., and Barillas-Mury, C. (2010). Hemocyte differentiation mediates innate immune memory in Anopheles gambiae mosquitoes. Science 329, 1353–1355. doi: 10.1126/science.1190689
Romano-Bertrand, S., Frapier, J. M., Calvet, B., Colson, P., Albat, B., Parer, S., et al. (2014). Dynamics of the surgical microbiota along the cardiothoracic surgery pathway. Front. Microbiol. 5:787. doi: 10.3389/fmicb.2014.00787
Romano-Bertrand, S., Parer, S., Lotthé, A., Colson, P., Albat, B., and Jumas-Bilak, E. (2012). “Temporal temperature gel electrophoresis to survey pathogenic bacterial communities: the case of surgical site infections,” in Gel Electrophoresis – Advanced Techniques, ed. S. Magdeldin (Rijeka: InTech Open Access), 500.
Roudiere, L., Jacquot, A., Marchandin, H., Aujoulat, F., Devine, R., Zorgniotti, I., et al. (2009). Optimized PCR-Temporal Temperature Gel Electrophoresis compared to cultivation to assess diversity of gut microbiota in neonates. J. Microbiol. Methods 79, 156–165. doi: 10.1016/j.mimet.2009.08.005
Sharma, P., Sharma, S., Maurya, R. K., Das De, T., Thomas, T., Lata, S., et al. (2014). Salivary glands harbor more diverse microbial communities than gut in Anopheles culicifacies. Parasit. Vectors 7:235. doi: 10.1186/1756-3305-7-235
Singleton, S. T., Lein, P. J., Farahat, F. M., Farahat, T., Bonner, M. R., Knaak, J. B., et al. (2013). Characterization of alpha-cypermethrin exposure in Egyptian agricultural workers. Int. J. Hyg. Environ. Health 217, 538–545. doi: 10.1016/j.ijheh.2013.10.003
Straif, S. C., Mbogo, C. N., Toure, A. M., Walker, E. D., Kaufman, M., Toure, Y. T., et al. (1998). Midgut bacteria in Anopheles gambiae and An. funestus (Diptera: Culicidae) from Kenya and Mali. J. Med. Entomol. 35, 222–226. doi: 10.1093/jmedent/35.3.222
Tandina, F., Almeras, L., Kone, A. K., Doumbo, O. K., Raoult, D., and Parola, P. (2016). Use of MALDI-TOF MS and culturomics to identify mosquitoes and their midgut microbiota. Parasit Vectors 9, 495. doi: 10.1186/s13071-016-1776-y
Tangena, J. A., Adiamoh, M., D’Alessandro, U., Jarju, L., Jawara, M., Jeffries, D., et al. (2013). Alternative Treatments for Indoor Residual Spraying for Malaria Control in a Village with Pyrethroid- and DDT-Resistant Vectors in The Gambia. PLoS ONE 8:e74351. doi: 10.1371/journal.pone.0074351
Thanh, N. V., Toan, T. Q., Cowman, A. F., Casey, G. J., Phuc, B. Q., Tien, N. T., et al. (2010). Monitoring for Plasmodium falciparum drug resistance to artemisinin and artesunate in Binh Phuoc Province, Vietnam: 1998-2009. Malar. J. 9, 181. doi: 10.1186/1475-2875-9-181
Tran, T. H., Nguyen, T. T., Nguyen, H. P., Boni, M. F., Ngo, V. T., Nguyen, T. N., et al. (2012). In vivo susceptibility of Plasmodium falciparum to artesunate in Binh Phuoc Province, Vietnam. Malar. J. 11, 355. doi: 10.1186/1475-2875-11-355
Verhaeghen, K., Van Bortel, W., Trung, H. D., Sochantha, T., and Coosemans, M. (2009). Absence of knockdown resistance suggests metabolic resistance in the main malaria vectors of the Mekong region. Malar. J. 8, 84. doi: 10.1186/1475-2875-8-84
Verhaeghen, K., Van Bortel, W., Trung, H. D., Sochantha, T., Keokenchanh, K., and Coosemans, M. (2010). Knockdown resistance in Anopheles vagus, An. sinensis, An. paraliae and An. peditaeniatus populations of the Mekong region. Parasit. Vectors 3, 59. doi: 10.1186/1756-3305-3-59
Villegas, L. M., and Pimenta, P. F. (2014). Metagenomics, paratransgenesis and the Anopheles microbiome: a portrait of the geographical distribution of the anopheline microbiota based on a meta-analysis of reported taxa. Mem. Inst. Oswaldo Cruz 109, 672–684. doi: 10.1590/0074-0276140194
Walton, C., Handley, J. M., Kuvangkadilok, C., Collins, F. H., Harbach, R. E., Baimai, V., et al. (1999). Identification of five species of the Anopheles dirus complex from Thailand, using allele-specific polymerase chain reaction. Med. Vet. Entomol. 13, 24–32. doi: 10.1046/j.1365-2915.1999.00142.x
Walton, C., Somboon, P., O’Loughlin, S. M., Zhang, S., Harbach, R. E., Linton, Y. M., et al. (2007). Genetic diversity and molecular identification of mosquito species in the Anopheles maculatus group using the ITS2 region of rDNA. Infect. Genet. Evol. 7, 93–102. doi: 10.1016/j.meegid.2006.05.001
Keywords: Anopheles mosquitoes, abdomen bacterial microbiota, 16S rRNA PCR – TTGE, malaria, Binh Phuoc, Vietnam
Citation: Ngo CT, Romano-Bertrand S, Manguin S and Jumas-Bilak E (2016) Diversity of the Bacterial Microbiota of Anopheles Mosquitoes from Binh Phuoc Province, Vietnam. Front. Microbiol. 7:2095. doi: 10.3389/fmicb.2016.02095
Received: 22 April 2016; Accepted: 12 December 2016;
Published: 23 December 2016.
Edited by:
Miguel Cacho Teixeira, University of Lisbon, PortugalReviewed by:
Guy Smagghe, Ghent University, BelgiumGeorge Christophides, Imperial College London, UK
Copyright © 2016 Ngo, Romano-Bertrand, Manguin and Jumas-Bilak. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sara Romano-Bertrand, c2FyYS5yb21hbm8tYmVydHJhbmRAdW1vbnRwZWxsaWVyLmZy