 María Dolores Rodríguez-Torres1África Islas-Robles1
María Dolores Rodríguez-Torres1África Islas-Robles1 Zulema Gómez-Lunar1
Zulema Gómez-Lunar1 Luis Delaye1
Luis Delaye1 Ismael Hernández-González1
Ismael Hernández-González1 Valeria Souza2
Valeria Souza2 Michael Travisano3
Michael Travisano3 Gabriela Olmedo-Álvarez1*
Gabriela Olmedo-Álvarez1*- 1Laboratorio de Biología Molecular y Ecología Microbiana, Departamento de Ingeniería Genética, Unidad Irapuato, Centro de Investigación y de Estudios Avanzados del Instituto Politécnico Nacional, Irapuato, México
- 2Laboratorio de Evolución Molecular y Experimental, Departamento de Ecología Evolutiva, Instituto de Ecología Universidad Nacional Autónoma de México, Mexico City, México
- 3Department of Ecology, Evolution and Behavior, University of Minnesota, Saint Paul, MN, USA
Understanding the relationship between phylogeny and predicted traits is important to uncover the dimension of the predictive power of a microbial composition approach. Numerous works have addressed the taxonomic composition of bacteria in communities, but little is known about trait heterogeneity in closely related bacteria that co-occur in communities. We evaluated a sample of 467 isolates from the Churince water system of the Cuatro Cienegas Basin (CCB), enriched for Bacillus spp. The 16S rRNA gene revealed a random distribution of taxonomic groups within this genus among 11 sampling sites. A subsample of 141 Bacillus spp. isolates from sediment, with seven well-represented species was chosen to evaluate the heterogeneity and the phylogenetic signal of phenotypic traits that are known to diverge within small clades, such as substrate utilization, and traits that are conserved deep in the lineage, such as prototrophy, swarming and biofilm formation. We were especially interested in evaluating social traits, such as swarming and biofilm formation, for which cooperation is needed to accomplish a multicellular behavior and for which there is little information from natural communities. The phylogenetic distribution of traits, evaluated by the Purvis and Fritz’s D statistics approached a Brownian model of evolution. Analysis of the phylogenetic relatedness of the clusters of members sharing the trait using consenTRAIT algorithm, revealed more clustering and deeper phylogenetic signal for prototrophy, biofilm and swimming compared to the data obtained for substrate utilization. The explanation to the observed Brownian evolution of social traits could be either loss due to complete dispensability or to compensated trait loss due to the availability of public goods. Since many of the evaluated traits can be considered to be collective action traits, such as swarming, motility and biofilm formation, the observed microdiversity within taxonomic groups might be explained by distributed functions in structured communities.
Introduction
Communities are assemblages of different species in which organisms co-exist and interact within a given environment. Molecular strategies have been instrumental in uncovering the great taxonomic diversity of microbial communities. Today, however, one of the fundamental pursuits in microbial ecology is to understand what the taxonomic classification of an organism means at the functional level. Thus, simply knowing “who is there” is no longer the most relevant question. The relationship between phylogeny and predicted functional traits revealed great heterogeneity that limits the predictive power of a microbial composition approach. Most previous studies showed trait consistency to the phylum level (Philippot et al., 2010; Goldfarb et al., 2011; Koeppel and Wu, 2012). However, some ecological traits may be species- or strain-specific since microbial genomes are highly dynamic and can change rapidly through loss or acquisition of genes from distant lineages via horizontal gene transfer (HGT, Boon et al., 2014). Also, the gene content of strains within a given species may differ by up to 30–35% (Konstantinidis and Tiedje, 2005). Bacteria species are therefore considered to be a mosaic of transferred genes since their great genetic diversity is obtained from distantly related organisms (Ochman et al., 2000). Differences in lifestyle correlate with variations in genes that are required for interactions with specific environments (Ochman et al., 2000; Polz et al., 2013). Capabilities for nutrient uptake, such as the capability to grow on different carbon sources, have been shown to be easily transferred or to have evolved rapidly from one function to another, probably because few genes are involved (Martiny et al., 2013). Therefore, these traits have been observed to be taxonomically dispersed, and move at shallow depths in phylogenies (Martiny et al., 2013). In contrast, traits associated with complex functions, such as photosynthesis and methanogenesis, are found only in a few deep clades (Martiny et al., 2013).
Community ecology investigates the complex interactions between organisms and the ecological and evolutionary consequences of sharing in a community. In the context of community, genes and functions can be lost when they are no longer needed in the habitat or when their function can be performed by community members such that the production of public goods is sufficient to support community stability (Visser et al., 2010; McInerney et al., 2011; Morris et al., 2012; Boon et al., 2014). Another fundamental aspect of microbial communities is social interactions among cells. At an individual level, many phenotypes affect their neighbor’s environment and thus influence their growth and reproduction. Important ecological processes, in fact, rely on a range of social traits such as biofilms, swarming, and quorum sensing, which are conserved in deep clades (Daniels et al., 2004). Such traits are considered to be collective action traits as they act at group-level (Stoodley et al., 2002; Daniels et al., 2004). Biofilm formation is dependent on the expression of costly exopolymers while swarming requires the production of biosurfactants and extracellular DNA (Velicer and Yu, 2003; Daniels et al., 2004; Spoering and Gilmore, 2006). These secreted substances can be considered as public goods. Swimming, swarming, and biofilm formation are traits that are also deeply rooted among the Firmicutes (Stoodley et al., 2002; Daniels et al., 2004). These traits are typically observed and have been characterized, in the extensively studied soil and pathogenic B. subtilis and the B. cereus groups. B. subtilis displays multicellular and social features, such as swarming and biofilm formation (González-Pastor et al., 2003). These traits can be assumed to be a fixed trait of Bacillus spp. and are rarely evaluated in wild isolates.
Swimming motility depends on flagella of individual cells (unorganized movement) that move independently upon perceiving chemical signals (Calvio et al., 2005). On the other hand, swarming is a multicellular behavior that allows rapid movement of differentiated bacterial cells (Kearns, 2010), and depends not only on the synthesis of flagella but also on the secretion of surfactants (Cosmina et al., 1993; Kearns and Losick, 2003). Both swarming ability and biofilm formation provide benefits at a group level since they depend on the coordinated expression of genes and signals (Stoodley et al., 2002; Daniels et al., 2004).
Many studies have focused on the evolution of social traits (Crespi, 2001; Foster et al., 2006; West et al., 2006; Diggle et al., 2007; de Vargas et al., 2011). A study by Whitham et al. (2006) provides a framework to understand how genetically based traits can have an influence at the species level and up to the ecosystem level; this hypothesis is supported by studies of multi-level selection and the fitness consequences of indirect genetic effects. Most efforts to address trait evolution were made in laboratory systems and thus there is very little information regarding trait heterogeneity in bacteria from natural communities, and, in particular, traits that are involved in social interaction.
The Cuatro Cienegas Basin (CCB), geographically isolated by high mountains, is located in the Chihuahuan desert. The CCB harbors a large number of springs among its water systems, and the area features high biological richness, numerous endemisms, and an enormous microbial diversity (Souza et al., 2012). We have systematically studied the diversity of bacteria in this region, particularly in the Churince water system. A genus we have extensively studied is the Bacillus, easily isolated due to their thermo-resistance property. The Bacillus spp. are abundant in this system and possess traits that correlate with an oligotrophic, phosphorus-limited environment (Tapia-Torres et al., 2016). HGT appears to be involved in the acquisition by Bacillus coahuilensis of genes from cyanobacteria that are involved in phosphorous utilization and adaptation to high-light environments (Alcaraz et al., 2008). We previously showed that Bacillus spp. from sediment communities are not assembled randomly and that biotic interaction among Bacillus members (specifically antagonism) have played a role in shaping community structure in this system (Pérez-Gutiérrez et al., 2013). The Bacillus spp. collection from the different sampling sites in this water system has therefore become an excellent model to understand bacterial interactions in natural communities.
In this work, we evaluated phenotypic heterogeneity and phylogenetic signal of phenotypic traits that are known to diverge within small clades and traits that are conserved deep in the lineage. Our results suggest that despite co-occurring in the same communities, different Bacillus species have different ecological strategies. The explanation to the observed evolution of these traits could be either loss due to complete dispensability of the trait, or compensated trait loss due to the availability of public goods. The observed microdiversity within taxonomic groups could be explained by distributed functions in structured communities.
Materials and Methods
Study Site
The Churince water system is located on the west side of CCB, and is formed by a spring, an intermediate lagoon, and a desiccation lagoon. The water and sediment samples described in this work were obtained from the intermediate lagoon. Physical and chemical measurements revealed homogeneous conditions along the sampling points, including pH (7.8–8), salinity (∼1.7–2.1 ppt), and daily temperature fluctuation (from 20 to 35°C; Pérez-Gutiérrez et al., 2013).
Bacterial Sampling and Isolation
The isolates in the collection were obtained in October 2007 from water column and sediment samples of the Intermediate Lagoon. Eleven sites, each about 30 m apart within the Churince water system, were chosen for small scale community sampling. At any point water samples were taken carefully as to not disturb the sediment (Column samples). Then approximately 50 μL of superficial (first millimeters TOP sample) sediment were obtained, and then another approximately 50 μL sample 2 cm below this (DEEP sample) (Figure 1). The samples were taken to an improvised in field laboratory, and a few hours later PBS buffer was added to the sample, and it was heat treated to select for thermo-resistant bacteria and plated out on Marine Medium (MM; Cerritos et al., 2008), and incubated at 37°C for 2 days. Different colony morphotypes were selected by size, shape, and color. Isolates were purified by subculture on the same medium to ensure that the culture was axenic; all isolates were stored at -80°C in MM with 15% (w/v) glycerol. In this work we show results for 467 thermo-resistant isolates that were obtained from eleven sampling points (Figure 1).
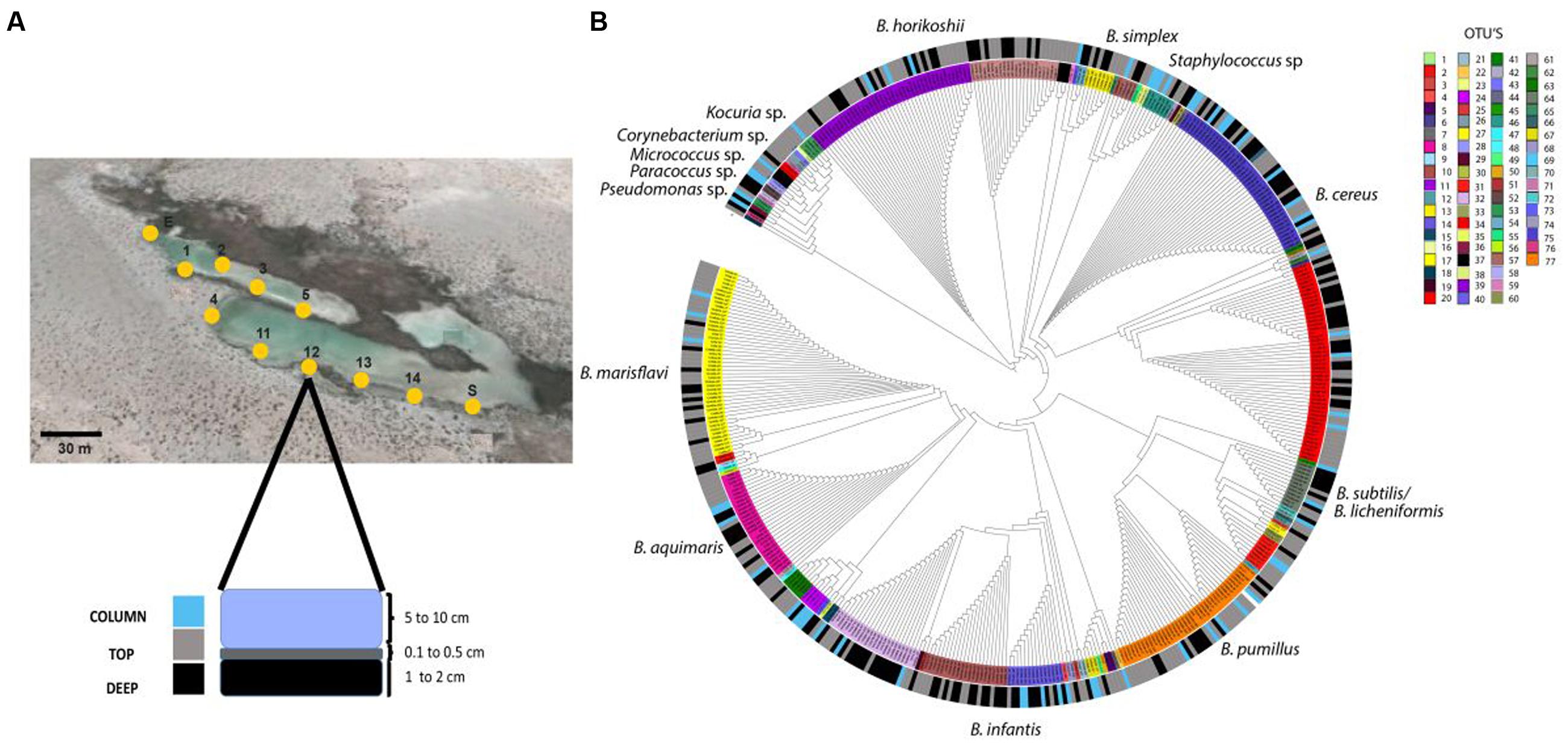
FIGURE 1. Spatial distribution of Bacillus isolated from the Churince System. (A) Map of sampling location in the Churince water system and method for sampling water (COLUMN), superficial sediment (TOP) and subsuperficial sediment (DEEP); (B) Phylogenetic reconstruction of thermo-resistant isolates sequences and their association habitat. The inner ring shows the OTU’s from the different sites (27 for COLUMN, 53 for TOP and 32 for DEEP). The outer ring shows the habitat (blue, water column; gray, TOP sediment; black, DEEP sediment). Map obtained from Google Maps (2013).
DNA Isolation, Molecular Markers and Phylogenetic Reconstruction
DNA from isolates was purified using the QuickGene DNA tissue Kit S (Fujifilm Corporation, Minato-Ku, Japan) following the manufacturer’s instructions. We amplified the 16S rRNA gene by PCR using previously published primer sequences 27F and 1492R (Lane, 1991). This included regions V1 to V3 (∼275 bp of the 5′ end region), considered to be the most informative for the Bacillus spp. (Goto et al., 2000). A 700 bp segment of the gltX gene (glutamyl-tRNA synthetase) was amplified using the primers gltX-for (5′ CGYGGBGADGAYCAYATYT 3′) and gltX-rev (5′ CRATTTCMGCDCCRWARCT 3′) and PCR-amplified by 30 cycles of 94°C for 30 s, 47°C for 1 min, 72°C for 2 min, and then a final elongation step at 72°C for 8 min with a thermocycler Palm-Cycler (Corbett Research). The PCR products were sequenced using the Sanger method (Sanger et al., 1977) at Cinvestav-Langebio (Irapuato, México). The quality of the 16S rRNA gene sequences was assessed using Phred (Ewing et al., 1998), and sequences of at least 500 bp were used for the analysis. The gltX gene sequences were assembled with CAP 3 (Huang and Madan, 1999). Gene accession numbers and lists of all 467 strains can be found in Supplementary Table S1. Maximum likelihood (Guindon et al., 2010) was used to reconstruct a phylogenetic history of all thermo-resistant bacteria through the 16S rRNA gene. A second phylogenetic reconstruction was done using the MrBayes program with sequences from a subsample of 138 Bacillus spp. for which phenotypic traits were obtained. Sequences were aligned with Muscle Software (Edgar, 2004). 16S rRNA Maximum-Likelihood tree was reconstructed with MEGA6 Software (Tamura et al., 2013). Following Bayesian Information Criteria (BIC) the Tamura 3-parameter + Gamma model was used to estimate distances between sequences on the tree generated with 100 bootstrap replicates. The 16S rRNA Bayesian tree was reconstructed with MrBayes (Ronquist et al., 2012). We used 5,000,000 Markov chain Monte Carlo generations and the GTR + G model of evolution. Average standard deviation of split frequencies dropped below 0.01. The gltX sequences of these 138 Bacillus spp. were also obtained to increase the resolution of the particular groups. Data from the group had shown that this gene allows more resolution than gyrA and rpoD. Phylogenetic reconstruction was done for these sequences using MrBayes and the sequences were grouped with CLANS (Huelsenbeck and Ronquist, 2001; Frickey and Lupas, 2004).
Phylogenetic Community Analysis
A total of 467 16S rRNA gene sequences were analyzed (TOP 240, DEEP 164 and COLUMN 63) using Mothur to assign to operational taxonomic units (OTUs) having 97% similarity thresholds (Schloss et al., 2009). The ribosomal database project (RDP) classifier was used for taxonomic assignment, with a minimum confidence of 0.8 to record an assignment (Cole et al., 2007). To identify ecologically distinct populations we examined the phylogenetic data for the group using AdaptML software, since it classifies a phylogeny based on genetic and ecological data and identifies populations as groups of related strains with distinct environmental distributions (Hunt et al., 2008). Tree drawings were generated with iTOL (Letunic and Bork, 2011). The net relatedness index (NRI) measures the degree of phylogenetic clustering of taxa across a phylogenetic tree in a given sample relative to the regional pool of taxa and was calculated with the Picante package (Webb, 2000; Kembel et al., 2010).
Phenotypic Assay of a 141 Bacillus spp. Isolate Subsample
To evaluate some phenotypic traits of the isolates, including carbon source utilization, growth kinetics, auxotrophies, motility assays, and sporulation frequency, we used a modified LPDM medium (Muller et al., 1997) that contained: Tris (pH 8.0) [50 mM], NH4NO3 [3.3 mM], K2HPO4 [0.065 mM], MgSO4 [3.5 mM], sodium citrate dehydrate [6.8 mM], MnCl2 [1 mM], ZnCl2 [0.01 mM], NaCl [0.17 M], FeCl3 [49.9 μM], CaCl2 [3.60 mM], KCl [0.67 mM], glucose [100 mM], vitamin B complex [100 μL/L], biotin [0.1 mg/mL], nicotinic acid [0.1 mg/mL], peptone [0.05%] and 20 amino acids (Harwood and Cutting, 1990).
To determine preferences for carbon source utilization we used five carbon sources: xylose, raffinose, sorbitol, trehalose, and glucose, all at a concentration of 100 mM in the LPDM medium. The assays were performed using microtiter plates (96 wells) and read on a BioTek μQuant Microplate Spectrophotometer. To estimate the maximum growth rate (μmax) and saturation density, growth kinetics were monitored under two concentrations of glucose (5 and 50 mM). The plates were incubated at 30°C, with constant vibration (100 Hz) and 90% humidity. Growth was monitored every 30 min for 24 h at OD600 using a microtiter plate reader (Tecan Infinite® M1000) assisted by a TECAN EVO robot (Tecan Infinite® M1000).
To evaluate whether the different strains could grow in the absence of a given amino acid, peptone or yeast extract, cultures were first grown in liquid MM, and then passed twice to LPDM lacking amino acids, yeast extract or peptone. Strains that grew under these conditions were considered to be prototrophs. Those strains that required addition of amino acids, peptone or yeast extract were further evaluated by transferring them to LPDM that contained either all 20 amino acids, and/or peptone or yeast extract. Those strains that could grow without peptone and yeast extract were then evaluated in LPDM medium containing all but one of the 20 amino acids. Growth was monitored at 24 h (Supplementary Table S2).
Motility assays were performed according to the protocol described in Hamze et al. (2009) for swarming, and Kearns and Losick (2003) for swimming. Briefly, Petri dishes were prepared with either 0.6% (swarming) or 0.3% (swimming) MM agar and allowed to dry at room temperature. Each Petri dish was inoculated at the center and the culture was allowed to grow for 24 h at 37°C. The ability to form biofilms was determined in a microtiter plate (24 wells) in MM liquid after static incubation for 48 h at 37°C.
Phylogenetic Signals of Bacillus spp. Traits
We compared the phylogenetic signal for the different phenotypic traits under evaluation. We determined the K value (Blomberg et al., 2003), a parameter that describes whether a pattern that arises when closely related taxa are more ecologically similar to each other than to distantly related taxa, and used this value to describe a tendency (pattern) for evolutionarily related organisms to resemble each other (K value is used for continuous traits, such as growth). K = 1 indicates a Brownian expectation, in which trait changes along each branch are random, and K > 1 indicates traits that are more conserved than the Brownian expectation. K = 0 indicates absence of a phylogenetic signal, and values between 0 and 1 indicate a significant phylogenetic signal but less conserved than under a Brownian motion model of evolution. We also carried out a Fritz and Purvis D-test applied to traits with discrete values. A D < 0 suggests a highly clustered trait, D ∼ 0 indicates a Brownian motion mode of evolution (BM), D = 0 suggests a random mode of evolution and D > 1 suggests phylogenetic overdispersion (Fritz and Purvis, 2010). D statistics can also be expressed in the same terms as the K statistics: -D + 1 = 0 value does not show a significant signal, -D + 1 > 0 is more conserved than expected by chance, 0 < -D + 1 < 1 indicates that is less conserved than expected under Brownian Motion model, -D + 1 = 1 is as conserved as expected under Brownian Motion model and – D + 1 > 1 suggest that is more conserved than expected under Brownian Motion model (Goberna and Verdú, 2016). Prandom and PBrownian convey the probability that the presence of a trait is the result of either a random or a Brownian motion mode of evolution, respectively. These tests help to identify the phylogenetic conservatism. The K and D values were calculated using the software packages Picante and Caper, respectively (Kembel et al., 2010; Fritz and Purvis, 2010), and both analyses were conducted in the R software environment (R Development Core Team, 2009). We used the consenTRAIT algorithm, that allows determining the cluster size where most organisms share a trait or, in other terms, the phylogenetic depth at which traits are conserved (Martiny et al., 2013).
The statistical analysis one-factor ANOVA was used to analyze the most numerous groups of Bacillus such as B. subtilis/licheniformis. B. pumillus. B. cereus, group 2, and the aquatic members. The traits subjected to this analysis were: carbon source utilization, maximum growth rate, and saturation density. The statistical analysis was carried out in the R programming environment (R Development Core Team, 2009).
Results
Phylogenetic Relatedness Analysis Revealed More Clustering from Thermo-Resistant Isolates in Sediment Communities Than in Water Column
We first characterized the diversity of culturable thermo-resistant bacteria from 11 sites within the Churince intermediate lagoon at CCB. We recovered 467 thermo-resistant isolates and analyzed partial 16S rRNA sequences that resulted in 77 OTUs (using Mothur with 97% similarity thresholds; Figure 1; Schloss et al., 2009). Among the 467 isolates, 428 (91.6%) were classified as Bacillus spp., 26 were classified as Koccuria spp., and 16 as Staphylococcus spp. (Figure 1). Sequences were assigned to a habitat category: COLUMN (water column, 63 members), TOP (surface sediment, 240 members), and DEEP (sub-surface sediment, 164 members). In order to know the phylogenetic structure among the different environments sampled, we determined if there was phylogenetic clustering among the 467 isolates using NRI, an index of community phylogenetic relatedness. We obtained positive NRI values for the sediment samples (TOP with 0.58 and DEEP with 1.83) indicative of phylogenetic clustering. For COLUMN sequences, however, the NRI values were negative (-2.28), suggesting a dispersed phylogenetic pattern of the thermo-resistant isolates from water column in comparison with the sediment sample. OTUs present in the COLUMN sample were more genetically dispersed, while those from sediment are more clustered, particularly those from the DEEP sample. We examined whether there was spatial resource partitioning among thermo-resistant isolates from COLUMN, TOP and DEEP samples. To do this, we used AdaptML which uses genetic and ecological similarity and classifies populations forming a common habitat into “projected habitats.” The populations were not classified and instead the analysis identified a single projected habitat, suggesting that there is no niche differentiation (data not shown).
Preferences for Carbohydrate Utilization among the Bacillus spp. Lineages
Clustering based on 16S rRNA gene revealed that several culturable Bacillus spp. lineages co-occur in the Churince system. To determine the phenotypic coherence of the different taxonomic lineages, we carried out a phenotypic trait analysis in a subsample of 141 isolates from 5 sampling sites. All strains in the subsample belonged to the Bacillus genus and represented the dominant taxa. The chosen isolates were distributed in 13 clades (according to CLANS analysis, Frickey and Lupas, 2004), of which 6 included 80% of all members. The selected isolates were phylogenetically closer to B. subtilis/B. licheniformis. B. pumillus. B. marisflavi. B. aquimaris. B. horikoshii, and B. cereus. According to a previous classification of Bacillus species based on evolutionary and functional relationships, we classified the isolates into three different groups: (1) soil members (B. subtilis/B. licheniformis, and B. pumillus), (2) aquatic members (B. marisflavi, B. aquimaris, and B. horikoshii), and (3) B. cereus-related species (Alcaraz et al., 2010).
The Bacillus spp. exhibited differences in the preference for different carbon sources. We addressed whether this was an overdispersed trait or if there was a phylogenetic signal associated to the phenotype. We chose the Purvis and Fritz’s D value that permits evaluating discrete traits and the significance of their genetic grouping. All isolates could grow on glucose and all clades had some members capable of using xylose, raffinose, sorbitol, and trehalose for growth (Figure 2). The soil member group exhibited the broadest ability to use different carbon sources, followed by the aquatic group, and finally the B. cereus group. All Bacillus spp. were able to consume glucose, so this is a highly conserved trait. On the other hand, the Fritz and Purvis’ D value for trehalose, raffinose and sorbitol show a significant phylogenetic signal (D = 0.77, 0.76 and 0.81, respectively) and differs significantly from the random and Brownian motion model expectations (Prandom < 0.05 and PBrownian < 0.05). Finally, xylose does not show a significant phylogenetic signal (D = 0.85), since it does not differ significantly from the random model expectations.
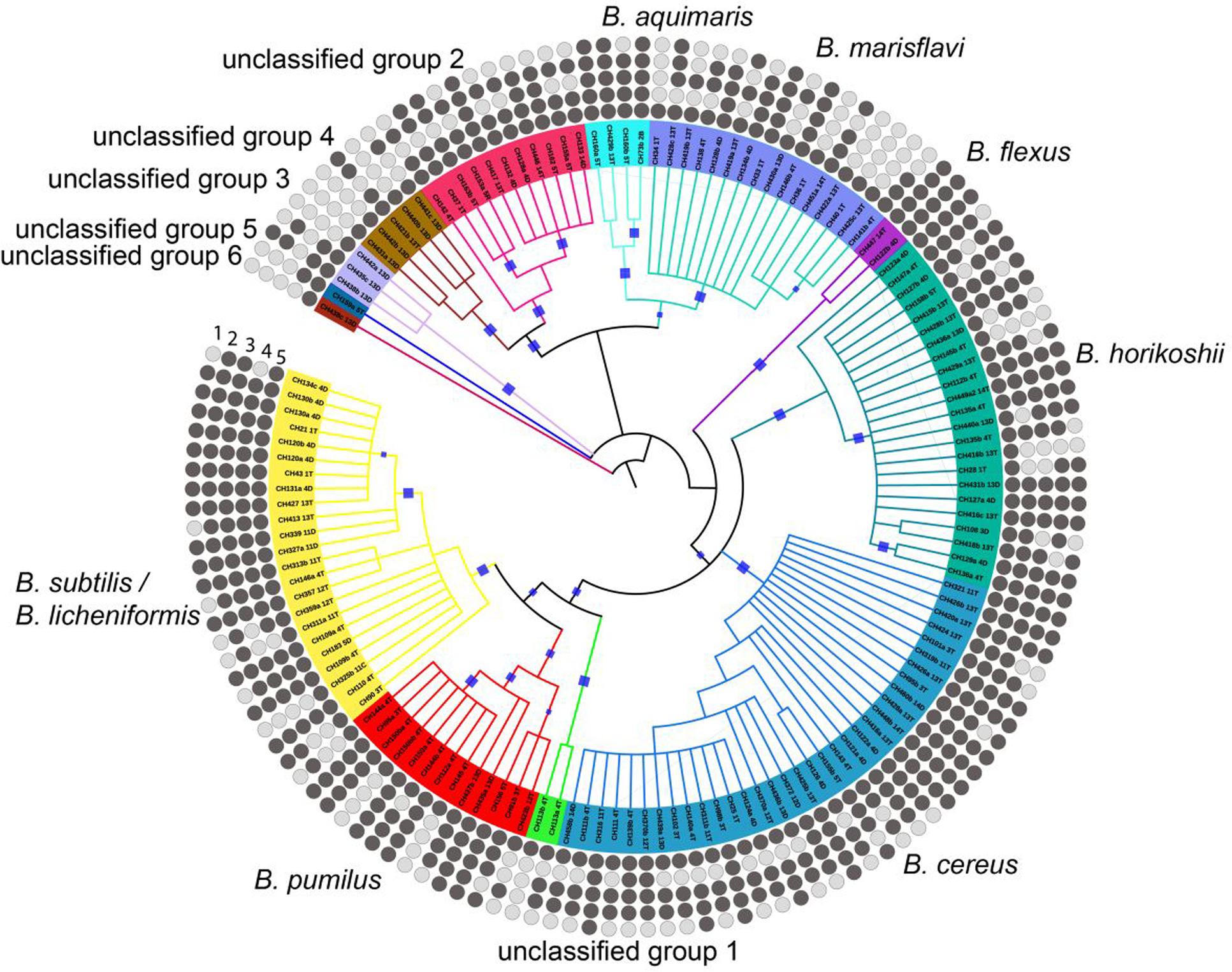
FIGURE 2. Variability in the utilization of different carbon sources by the different Bacillus taxonomic groups. Rings from the outside to the inside are designated by numbers for the carbon sources tested: (1) sorbitol, (2) raffinose, (3) trehalose, (4) xylose, (5) glucose. Dark and light circles indicate utilization and no utilization, respectively, of carbon sources. The phylogenetic reconstruction was made using 16S rRNA gene alignment and was based on the maximum likelihood method and the bootstrap values in the range from 0.8 to 1 were marked with blue squares.
Since the Churince water system is an oligotrophic environment, we also evaluated the ability of Bacillus spp. to respond to changes in nutrient availability as well as the saturation density (maximum growth density) the community members could reach. Isolates from mainly the B. subtilis/B. licheniformis group responded to increased glucose concentrations, but a significant growth increase was also observed in strains that were scattered throughout the phylogeny (Supplementary Figures S1A,B). All groups exhibited higher saturation density at higher glucose concentrations, but there were no statistically significant differences in saturation density or maximal growth rate in either of the two glucose concentrations across the taxonomic groups (ANOVA analysis did not give a significant value for Vmax or saturation density at 5 and 50 mM glucose; Supplementary Figure S1A). For this data, the Blomberg K value from Vmax was 1.27 at 5 mM glucose, and 3.75 at 50 mM glucose, which indicates that the observed value is more conserved than that expected in a Brownian model. The Blomberg K values for saturation density were 0.83 and 0.56 at 5 mM and 50 mM glucose, respectively, suggesting that the traits are conserved (all P-values observed were significant Vs. random variance of 0.001; Supplementary Table S3B). These data suggest that Vmax growth and saturation density values are conserved traits for the different taxonomic groups.
Most Bacillus spp. Isolates Are Auxotrophs, Suggesting a Strong Dependence on the Community
Co-occurrence of bacteria in a community could allow metabolic sharing or simply provide ample opportunity for scavenging and, either way, there would be no need for all members to be self-sufficient. We, therefore, evaluated metabolic self-sufficiency by analyzing auxotrophy for the different amino acids among members of the Bacillus spp. taxa from the sediment communities. Close to 60% of the evaluated Bacillus spp. isolates had an auxotrophy. Some members exhibited a strong dependency on peptone or yeast extract for growth even in the presence of all 20 amino acids, and this too was scored as an auxotrophic trait (44 strains required these supplements). These strains may obtain their amino acids in the form of peptides. For example, growth of aquatic Bacillus spp. (B. horikoshii. B. aquimaris, and B. marisflavi) exhibits extensive dependence on added amino acids or peptone (Figure 3). The same is true for B. cereus, for which prototrophy was rarely observed. The analysis of phylogenetic signal for this trait was consistent when measured with K and D statistics. It is compatible with a Brownian model of evolution using Bloomberg’s analysis (K of 1.28 with a PICrandom and a PBrownian of 0.0009) implying a non-random phylogenetic signal. The D statistic was 0.27 with a Prandom = 0 and PBrownian = 0.12, a pattern that differs significantly from the random model, but the Brownian Motion model of evolution can not be rejected (Orme, 2013). Analysis with calculated a depth for 16S of τD 0.0095 16S rRNA distance, deeper than those computed for individual sugars (τD range from 0.001 to 0.003 16S rRNA distance), suggesting deeper lineage conservation (Supplementary Table S3C).
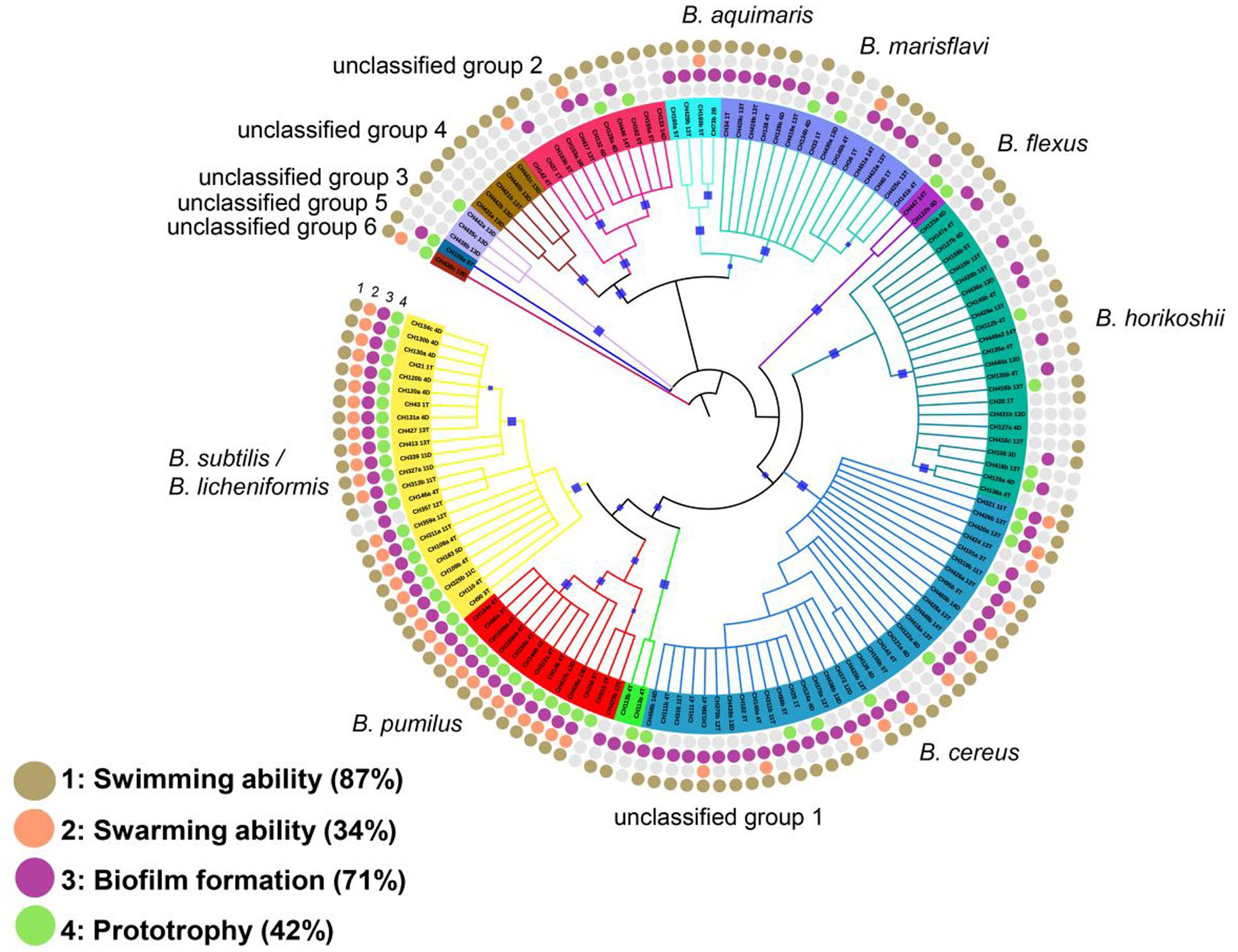
FIGURE 3. Traits of the isolates in the different taxonomic groups of Bacillus spp. Rings from the outside to the inside indicate the traits tested: (1) Swimming, (2) Swarming, (3) Biofilm and (4) Prototrophy. Filled circles indicate positive results for the described trait. Open circles indicate negative results for the evaluated trait. Bootstrap values in the range from 0.8 to 1 were marked with blue squares.
Microdiversity in Social Traits of the Bacillus spp. in Sediment Communities and a Phylogenetic Signal Compatible With a Brownian Model of Evolution
We determined the frequency of swimming and swarming motility and the ability to form biofilm in this Bacillus spp. collection. Our results showed microdiversity for these traits, but more so in certain taxonomic groups. Although swimming seems to be a conserved trait among all clades, some members in the aquatic group seem to have lost this feature. For example, 9 out of 23 B. horikoshii strains could not swim (Figure 3). Notably, swarming was exhibited by most members of the B. subtilis/licheniformis and B. pumilus clade, but 75% of B. cereus members scored negatively for this feature (22 of a total of 34). Swarming was absent in all members of the B. horikoshii clade and was seen in only one member of the B. marisflavi and B. aquimaris clade (Figure 3).
For biofilm formation, heterogeneity was most noticeable among the aquatic Bacillus spp., but this trait was almost always present in members of the B. subtilis/licheniformis-pumilus or B. cereus groups (Figures 3 and 4). Moreover, almost all soil group members could form biofilms (only 1 of 36 members of the B. subtilis/licheniformis. B. pumilus group did not, and 3 out of 34 of the B. cereus members did not display this feature) but less than half of the B. horikoshii clade formed biofilms.
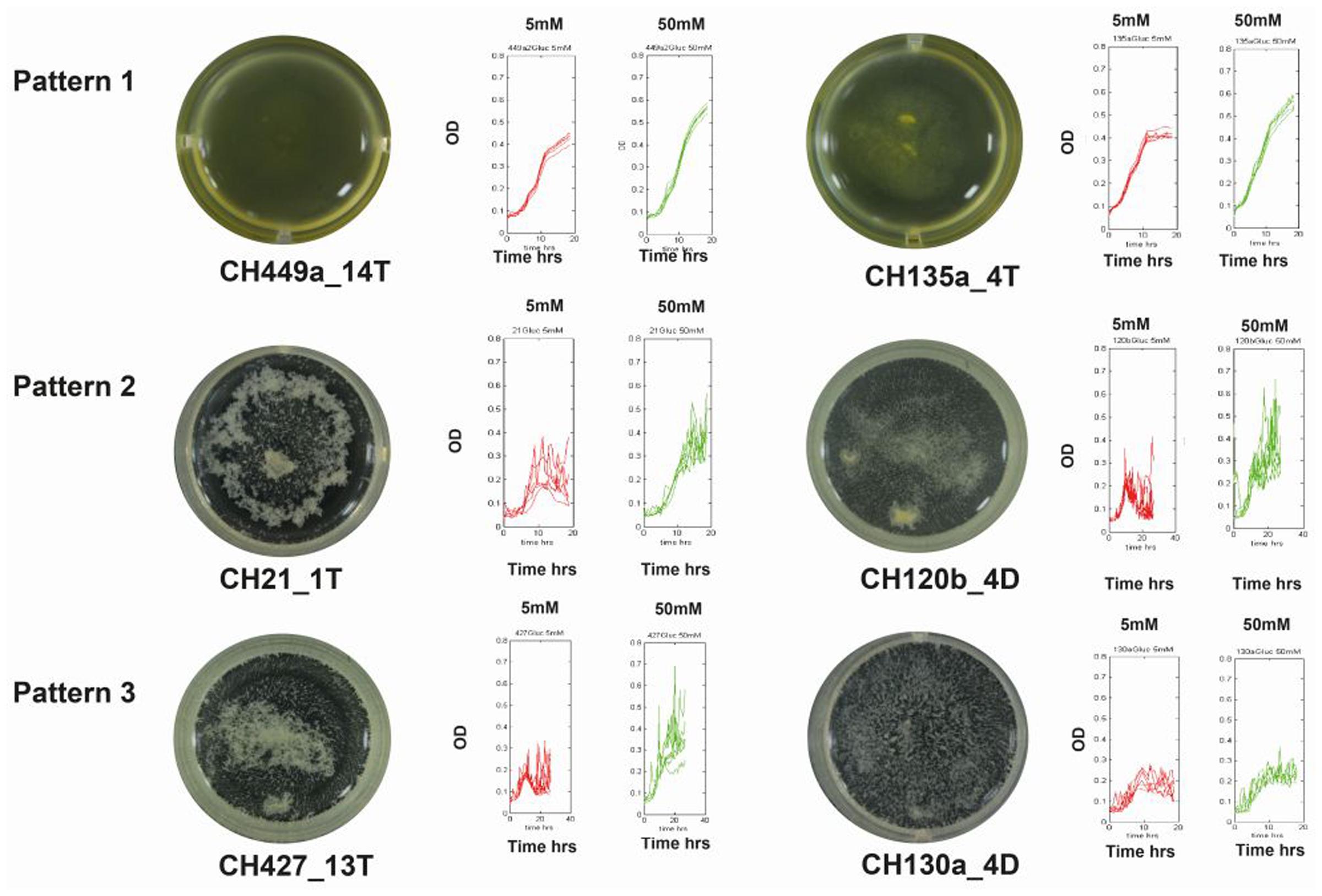
FIGURE 4. Relationship between growth patterns and biofilm production. Graphic representation of the three typical growth curve patterns at two glucose concentrations. The red and green curves describe growth with 5 and 50 mM glucose, respectively. Photographs at the ends of the figure depict strains at the end of the growth curve for each pattern.
Growth kinetics analyses for the different isolates revealed three different but consistent patterns for the assayed Bacillus strains: Pattern 1, a characteristic smooth growth curve was present throughout the growth period (47% of the isolates); Pattern 2, a smooth logarithmic growth phase was followed by aggregation at the end of growth period (resulting in fluctuating readings during the stationary phase in 50% of the isolates); and Pattern 3, readings fluctuated throughout growth (this pattern occurred for 3% of the evaluated strains) (Figure 4). These patterns indicate that there were differences in the growth strategies of the different strains, wherein some could display a planktonic behavior (Pattern 1) with no aggregation at the end of growth (although our evaluation for biofilm formation showed that many strains had this ability) and some had aggregation that was triggered immediately at the end of the exponential growth phase (Pattern 2).
Analysis of phylogenetic signal showed that the distribution of biofilm-forming ability across the taxonomic groups of Bacillus spp. from these sediment communities exhibited only a moderate level of phylogenetic clustering (D = 0.51), and differs from a random and Brownian model (PBrownian 0.02 and PRandom 0). K is 0.8 and is significantly different from random (Supplementary Tables S3A,B). We conclude that for this trait there is a departure from a random model and a Brownian model of evolution. Regarding swimming, the D value obtained suggested that this phylogenetic exhibited a random mode of evolution (D = 0.90; PRandom = 0.19, PBrownian = 0). A Blomberg analysis, in contrast, resulted in a K = 0.78 with a P of 0.0009 for a non-random mode of evolution. For swarming and prototrophy the D values were 0.21 and 0.28, respectively. The statistic indicates that both swarming and prototrophy traits differed from a random pattern of evolution and resembled a Brownian motion (both have a PRandom = 0; swarming PBrownian = 0.181, prototrophy PBrownian = 0.106).
We also computed the phylogenetic depth at which the observed traits were conserved using consenTRAIT (Martiny et al., 2013). It calculates the mean depth of clades containing above 90% of members sharing a trait. Cluster size is therefore an important data to take into account for this analysis. For all traits evaluated the 16S rRNA distance was τD = 0.00073 to 0.0095 16S rRNA distance. As a reference, the deepest value, τD 0.07416S rRNA distance, would be that for glucose (used by all the strains in the study). Lower distances (τD) were obtained for substrate utilization than for social traits (except for swarming and xylose). The largest size clusters computed were for prototrophy, swimming, and biofilm formation (size 38), compared to swarming (size 23), but the mean cluster size was higher for the social traits (biofilm, swimming, prototrophy). Social traits also had more depth signal (for biofilm, swimming, prototrophy τD = 0.005, 0.0056, 0.0095, 16S rRNA distance respectively) compared to the genetic distance for traits involved in substrate utilization (i.e., trehalose τD = 0.0011 and raffinose τD = 0.0015 16S rRNA distance) (Supplementary Table S3C).
Overall, the evolutionary models that describe the distribution of social traits in the phylogeny (except for swimming) suggest a Brownian model of evolution and a deeper phylogenetic signal than that observed for substrate utilization.
Colony Morphology and Pigmentation Are Highly Conserved, Clade-Specific Traits
Colony morphology, size, and pigmentation were observed to be very conserved and are distinctive features of some clades. The colony appearance in our sample is a good proxy for the phylogenetic membership of a strain. Although colony color may exhibit differences in intensity and can change with growth stage and medium, color is generally consistent for members of the same clade. Colony morphology and color probably involve a large number of genes in cellular processes. These genes are differentially and spatially expressed to form a highly organized and structured community. The fact that morphology conservation occurs suggests that it is not neutral but instead arises from pressure to maintain a given developmental pattern that may be relevant for survival. However, this would have to be further studied.
Finally, the 16S rRNA gene phylogeny was coherent with respect to the phenotypes of the clade members. We added one more gene, gltX, to increase phylogeny resolution. As expected, this gene maintained the deeply rooted clades in the same order as the 16S, although some of the tips of the tree changed position within their own basal clade. The so-called soil Bacillus (B. subtilis/licheniformis and B. pumilus) as well as B. cereus members remained as a group. Meanwhile several of the aquatic groups changed position at the tips of the tree (Figure 5) suggesting that gene transfer is more common in the aquatic clades than in soil clades’ members.
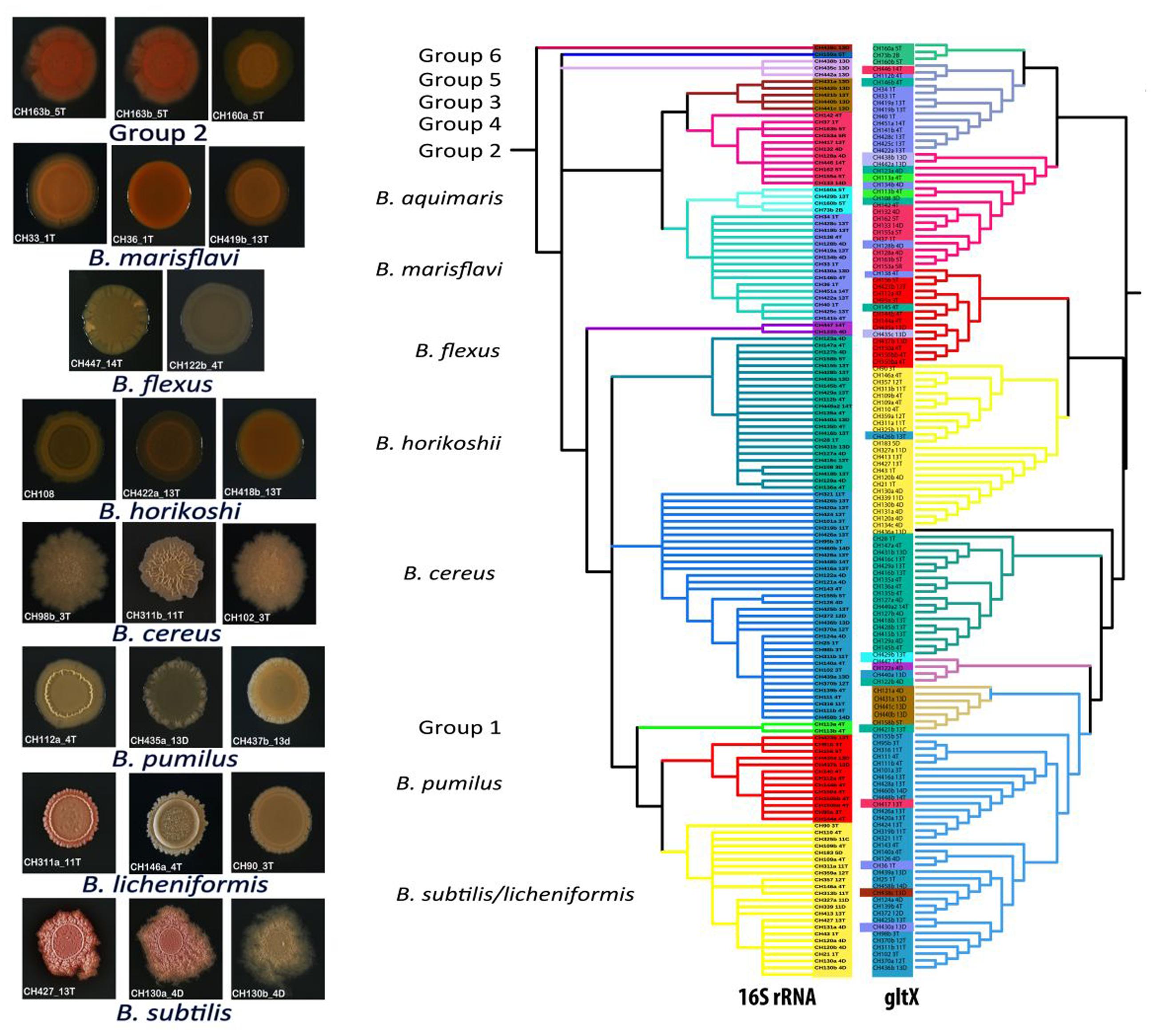
FIGURE 5. Distinct colony morphology of members of different Bacillus taxonomic groups and comparison between 16S rRNA gene and gltX gene phylogenies. The colored rectangles indicate strains that have moved within the phylogenetic group in the tree of 16S rRNA genes with respect to the gltX gene. On the left side of the figure, photographs of colonies of strains belonging to the same taxonomic clade.
Discussion
Herein, we addressed individual phenotypic trait heterogeneity in members of Bacillus spp. co-occurring in sediment communities in an extremely oligotrophic environment and compared the phylogenetic signal associated with substrate utilization and social phenotypic traits.
The Bacillus spp. collection in this study provided a strong model to explore trait variability because, as we have shown previously, its assemblage in the sediment community depends more on ecological interactions than on abiotic factors (Pérez-Gutiérrez et al., 2013). Our data showed ecologically phylogenetic clustering of the sediment samples, compared to those of the water column. Sediment and water may be acting as habitat filters (Webb et al., 2002) explaining the clustered structure of the soil taxons. A similar observation was made by Rebollar et al. (2012) in a study of Exiguobacterium genus sampled from different ponds at CCB that found ecologically differentiated clusters associated with either sediment or water environments. These results are also consistent with those from Burke et al. (2011) showing that bacterial communities associated with Ulva australis, an alga found in tidal pools, differed from those found in sea water.
Unpublished data from our group has shown that upon plating, two orders of magnitude more colonies belonging to Bacillus spp. are consistently recovered from sediment than from water column. The fact that sediment seems to be the preferred habitat for the Bacillus from the lagoon under study could imply a preference for spatially structured communities with a predictable neighborhood. This is consistent with the idea that bacteria in nature predominate as adherent populations (Costerton et al., 1995.). This arrangement would not be unexpected since biofilms are thought to represent the dominant state of bacteria in nature. Biofilms are extremely complex structures and, in the particular case of B. subtilis, up to five cell types have been shown to contribute to the spatial organization and function of biofilms: motility, sporulation, and production of diverse matrix molecules, surfactants and proteases (Vlamakis et al., 2013).
Despite the apparent preference for sediment of the different Bacillus taxa evaluated and that all taxa have members with lineage-derived social traits, we observed heterogeneity in the social trait distribution at group (species) level. What does this microdiversity mean for the ecological strategies of the different Bacillus taxonomic groups in the community? Our evaluation of individual isolates, representing seven species within the Bacillus genus, revealed that for the different clades there were differences in the number of isolates scoring negatively for social traits, biofilm, swimming, and swarming, and suggests that within the same genus, different species co-occurring in the same environment (sediment communities in an oligotrophic water system) have different ecological strategies. Members of the B. cereus group exhibited scattered swarming capabilities and dependency on the community to obtain amino acids; in this clade, biofilm formation is a trait that is conserved in almost every member, thus stressing the importance of this strategy for the group. On the other hand, well-known members of the B. subtilis/pumilus and licheniformis group rarely score negatively for a social trait. This robustness could allow members of this group to move between communities and in different environments. In contrast, some members of the more endemic and local B. horikoshii clade do not exhibit swimming, biofilm formation, and prototrophy, and swarming is completely absent. Thus, this group would have to rely on members of the community for nutrition and perhaps for movement as well.
As reported before, most functional traits are phylogenetically conserved in bacteria (Martiny et al., 2013; Goberna and Verdú, 2016). For our data, the Fritz and Purvis distance analysis, when applied to social traits (biofilm and swarming) calculated a D probability closer to a Brownian phylogenetic evolutionary model. Similar to what Martiny et al. (2013) reported, all D values for substrate utilization were between 0 and 1, exhibiting dispersion and falling between a random and a Brownian model of evolution. A Bloomberg analysis to detect phylogenetic signal resulted in K value > 1 only for some traits related to substrate utilization (sorbitol, and trehalose) suggesting that those traits were clustered, that is, with closely related species being more similar than expected by chance. However, for other substrates (xylose and raffinose) and for the social traits (swimming, swarming and biofilm), K was below 1, though still more similar to a Brownian model than to a random model. With both measures phylogenetic signal was significantly different from random for the two most relevant social traits, swarming and biofilm, suggesting that these exhibited microdiversity but were not phylogenetically dispersed. The data obtained with consenTRAIT resulted in lower distances (τD) for substrate utilization than for social traits (except for swarming and xylose), and the clusters’ mean was higher for social traits, suggesting that despite exhibiting microdiversity there is a stronger phylogenetic signal for these social traits than for substrate utilization. From what it is known about the Biology of the Bacillus genus, the evaluated traits can be traced back in the lineage to phylum level (Firmicutes) but are also present in several phyla (Kearns, 2010). It is intriguing why these traits exhibit a high intraspecific variation.
Differentiation in the ability to use various carbon substrates was expected and explains why numerous bacteria can co-exist in a community. Substrate utilization depends on only on few genes and is thus easily transferred (Martiny et al., 2013). Similarly, we have consistently observed microdiversity in traits for phosphorus scavenging in a large sample of Bacillus spp. from the CCB (Tapia-Torres et al., 2016). But, why is the phylogenetic signal of complex traits, that requires numerous genes, similar to that observed for simple traits (few genes)? An elementary explanation could be that expression of these complex traits finally depends on simple traits: sugars, such as exopolysacharides, in the case of biofilm, surfactin, for swarming, etc. If simple genes are behind the control of social (complex) traits, the signal will resemble that observed for the distribution of genes encoding enzymes involved in substrate utilization.
There are at least two explanations for what seems to be the intra-specific loss of traits: (i) real loss due to complete dispensability of the trait, and (ii) compensated loss due to the availability of public goods. For the first explanation, some traits would be lost by genetic drift and mutation because natural selection does not maintain them. For example, under the water-abundant conditions of sediment, movement by swimming might allow sufficient mobility for most cells and thus swarming might be dispensable. However, this situation can also be explained by compensated trait loss, as the addition of surfactants has been reported to complement bacteria that are defective in swarming (Kearns and Losick, 2003). Compensated trait loss (Visser et al., 2010) is promoted by extensive sharing in the community, as that occurring extensively in biofilms. Ellers et al. (2012) provide a useful framework to distinguish between the different terms that describe the evolutionary dynamics of interacting organisms. The “black queen hypothesis” (Morris et al., 2012) suggests that gene loss is adaptive in oligotrophic environments since essential functions are shared. Although we observed loss of different phenotypes, fitness analysis with non-isogenic strains complicates testing of this hypothesis. Also, we did not observe increased growth rates that were associated with the loss of phenotypic traits, which would have suggested a trade-off. Compensated trait loss through species interactions has been observed in the loss of lipogenic capacity of the parasitic fungus Malassezia globosa that feeds on lipids found on the skin of the host (Xu et al., 2007). Another example is loss of arginine biosynthesis in two leaf-cutter ant genera (Nygaard et al., 2011; Suen et al., 2011). The extensive auxotrophy observed is consistent with the notion that compensated trait loss occurs in communities where availability of resources permits the individual loss of traits as long as the community can maintain the given function. Some traits for the Bacillus spp. may not have been lost but their expression may instead depend either on specific interactions or triggered by metabolites produced by the community. All clades had members that scored positive for the evaluated trait, suggesting that members that scored negatively for the phenotype have the potential to manifest the function under different conditions or upon interaction with members of the community. There is evidence of the extensive signaling that controls biofilm formation (Vlamakis et al., 2013) and many reviews have discussed the principles of social evolution in microorganisms (Crespi, 2001; Foster et al., 2006; West et al., 2006; Diggle et al., 2007; de Vargas et al., 2011), some specifically focusing on biofilm (Nadell et al., 2008; Mitri et al., 2011), which has been argued to constitute an example of division of labor (van Gestel et al., 2015).
Sequencing of the genomes of the Bacillus spp. in this study will allow us to explore the nature of the observed phenotypic microdiversity at the genetic level, but the challenge remains of evaluating the phenotypic traits of individual bacteria in their communities and understanding how intra-species heterogeneity evolves.
Conclusion
The different lineages of the Bacillus genus that co-exist in sediment communities of the Churince system are an excellent model for an in-depth study of intra-genus and intra-species trait heterogeneity. We found microdiversity in social traits suggestive of different ecological strategies of the taxonomic groups. The phylogenetic signal suggests that substrate utilization and social traits conform to a Brownian model of evolution that could be explained by distributed functions in structured communities.
Author Contributions
MR-T contributed to the conception and design of the study, data acquisition and analysis, interpretation of the results, and preparation of the manuscript; AI-R and IH-G contributed to data acquisition and analysis; ZG-L and LD contributed to data acquisition and analysis, and to interpretation of the results; GO-A participated in the conception and design of the study, and to manuscript preparation; MT contributed to the design, discussion and critical revision of the manuscript, VS participated in obtaining sampling permits, discussion of the results and in a critical revision of the manuscript.
Funding
Investigations were supported by grants from a CONACyT scholarship (to MR-T) and the project was funded by a CINVESTAV multidisciplinary project award (to GO-A), Conacyt grant 220536 (CB-2013-01, to GO-A) and by Alianza WWF-FCS (to VS). MT is supported by the John Templeton Foundation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Julio Cruz-Medina, Susana de la Torre, Mayra E. Covaxin, and Adrian Jinich for excellent laboratory collaboration, José Antonio Cisneros for photographs, and J. Eugenio Reynoso for specialized technical assistance. The authors acknowledge reviewer for careful reading and expert advice on results interpretation.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.00029/full#supplementary-material
FIGURE S1 | Growth and saturation density of the different Bacillus spp. analyzed by taxonomic group. (A) Maximal growth rate was evaluated for each strain at two different glucose concentrations, 5 and 50 mM. (B) Saturation density was recorded for the different strains at 5 and 50 mM glucose. Mainly isolates from the B. subtilis/B. licheniformis group responded to increased glucose concentrations, but a significant growth increase was also observed in strains that were scattered throughout the phylogeny. Anova analysis did not give a significant value for Vmax or saturation density across taxonomic groups.
References
Alcaraz, L. D., Moreno-Hagelsieb, G., Eguiarte, L. E., Souza, V., Herrera-Estrella, L., and Olmedo, G. (2010). Understanding the evolutionary relationships and major traits of Bacillus through comparative genomics. BMC Genomics 11:332. doi: 10.1186/1471-2164-11-332
Alcaraz, L. D., Olmedo, G., Bonilla, G., Cerritos, R., Hernandez, G., Cruz, A., et al. (2008). The genome of Bacillus coahuilensis reveals adaptations essential for survival in the relic of an ancient marine environment. Proc. Natl. Acad. Sci. U.S.A. 105, 5803–5808. doi: 10.1073/pnas.0800981105
Blomberg, S. P., Garland, T., and Ives, A. R. (2003). Testing for phylogenetic signal in comparative data: behavioral traits are more labile. Evolution 57, 717–745. doi: 10.1111/j.0014-3820.2003.tb00285.x
Boon, E., Meehan, C. J., Whidden, C., Wong, D. H., Langille, M. G., and Beiko, R. G. (2014). Interactions in the microbiome: communities of organisms and communities of genes. FEMS Microbiol. Rev. 38, 90–118. doi: 10.1111/1574-6976.12035
Burke, C., Thomas, T., Lewis, M., Steinberg, P., and Kjelleberg, S. (2011). Composition, uniqueness and variability of the epiphytic bacterial community of the green alga Ulva australis. ISME J. 5, 590–600. doi: 10.1038/ismej.2010.164
Calvio, C., Celandroni, F., Ghelardi, E., Amati, G., Salvetti, S., Ceciliani, F., et al. (2005). Swarming differentiation and swimming motility in Bacillus subtilis are controlled by swrA, a newly identified dicistronic operon. J. Bacteriol. 187, 5356–5366. doi: 10.1128/JB.187.15.5356-5366.2005
Cerritos, R., Vinuesa, P., Eguiarte, L. E., Herrera-Estrella, L., Alcaraz-Peraza, L. D., Arvizu-Gómez, J. L., et al. (2008). Bacillus coahuilensis sp. nov., a moderately halophilic species from a desiccation lagoon in the Cuatro Cienegas Valley in Coahuila. Mexico. Int. J. Syst. Evol. Microbiol. 58, 919–923. doi: 10.1099/ijs.0.64959-0
Cole, J. R., Chai, B., Farris, R. J., Wang, Q., Kulam-Syed-Mohideen, A. S., McGarrell, D. M., et al. (2007). The ribosomal database project (RDP-II): introducing myRDP space and quality controlled public data. Nucleic. Acids Res. 35, D169–D172. doi: 10.1093/nar/gkl889
Cosmina, P., Rodriguez, F., Ferra, F., Grandi, G., Perego, M., Venema, G., et al. (1993). Sequence and analysis of the genetic locus responsible for surfactin synthesis in Bacillus subtilis. Mol. Microbiol. 8, 821–831. doi: 10.1111/j.1365-2958.1993.tb01629.x
Costerton, J. W., Lewandowski, Z., Caldwell, D. E., Korber, D. R., and Lappin-Scott, H. M. (1995). Microbial biofilms. Annu. Rev. Microbiol. 49, 711–745. doi: 10.1146/annurev.mi.49.100195.003431
Crespi, B. J. (2001). The evolution of social behavior in microorganisms. Trends Ecol. Evol. 16, 178–183. doi: 10.1016/S0169-5347(01)02115-2
Daniels, R., Vanderleyden, J., and Michiels, J. (2004). Quorum sensing and swarming migration in bacteria. FEMS Microbiol. Rev. 28, 261–289. doi: 10.1016/j.femsre.2003.09.004
de Vargas, L. R., Boyle, K. E., and Xaviera, J. B. (2011). Multilevel selection analysis of a microbial social trait. Mol. Syst. Biol. 9:684. doi: 10.1038/msb.2013.42
Diggle, S. P., Gardner, A., West, S. A., and Griffin, A. S. (2007). Evolutionary theory of bacterial quorum sensing: when is a signal not a signal? Philos. Trans. R. Soc. Lond. B Biol. Sci. 362, 1241–1249. doi: 10.1098/rstb.2007.2049
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Ellers, J., Kiers, E. T., Currie, C. R., McDonald, B. R., and Visser, B. (2012). Ecological interactions drive evolutionary loss of traits. Ecol. Lett. 15, 1071–1082. doi: 10.1111/j.1461-0248.2012.01830.x
Ewing, B., Hillier, L., Wendl, M., and Green, P. (1998). Basecalling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 8, 175–185. doi: 10.1101/gr.8.3.175
Foster, K. R., Wenseleers, T., and Ratnieks, F. L. W. (2006). Kin selection is the key to altruism. Trends Ecol. Evol. 21, 57–60. doi: 10.1016/j.tree.2005.11.020
Frickey, T., and Lupas, A. (2004). CLANS: a Java application for visualizing protein families based on pairwise similarity. Bioinformatics 20, 3702–3704. doi: 10.1093/bioinformatics/bth444
Fritz, S. A., and Purvis, A. (2010). Selectivity in mammalian extinction risk and threat types: a new measure of phylogenetic signal strength in binary traits. Conserv. Biol. 24, 1042–1051. doi: 10.1111/j.1523-1739.2010.01455.x
Goberna, M., and Verdú, M. (2016). Predicting microbial traits with phylogenies. ISME J. 10, 959–967. doi: 10.1038/ismej.2015.171
Goldfarb, K. C., Karaoz, U., Hanson, C. A., Santee, C. A., Bradford, M. A., Treseder, K. K., et al. (2011). Differential growth responses of soil bacterial taxa to carbon substrates of varying chemical recalcitrance. Front. Microbiol. 2:94. doi: 10.3389/fmicb.2011.00094
Google Maps (2013). Map of Churince. Available at: https://www.google.com.mx/maps/place/Churince/@26.8402503,-102.1348029,261m/data=!3m1!1e3!4m5!3m4!1s0x868c089c2f799535:0x20df7465fded2ec0!8m2!3d26.8402503!4d-102.1341687 (accessed October 10, 2013).
González-Pastor, J. E., Hobbs, E. C., and Losick, R. (2003). Cannibalism by sporulating bacteria. Science 301, 510–513. doi: 10.1126/science.1086462
Goto, K., Omura, T., Hara, Y., and Sadaie, Y. (2000). Application of the partial 16S rDNA sequence as an index for rapid identification of species in the genus Bacillus. J. Gen. Appl. Microbiol. 46, 1–8. doi: 10.2323/jgam.46.1
Guindon, S., Dufayard, J. F., Lefort, V., Anisimova, M., Hordijk, W., and Gascuel, O. (2010). New Algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Hamze, K., Julkowska, D., Autret, S., Hinc, K., Nagorska, K., Sekowska, A., et al. (2009). Identification of genes required for different stages of dendritic swarming in Bacillus subtilis, with a novel role for phrC. Microbiology 155, 398–412. doi: 10.1099/mic.0.021477-0
Harwood, C. R., and Cutting, S. M. (1990). Molecular Biological Methods for Bacillus, 1st Edn. Chichester: John Wylie and Sons.
Huang, X., and Madan, A. (1999). CAP3: a DNA sequence assembly program. Genome Res. 9, 868–877. doi: 10.1101/gr.9.9.868
Huelsenbeck, J. P., and Ronquist, F. (2001). MRBAYES: bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755. doi: 10.1093/bioinformatics/17.8.754
Hunt, D. E., David, L. A., Gevers, D., Preheim, S. P., Alm, E. J., and Polz, M. F. (2008). Resource partitioning and sympatric differentiation among closely related bacterioplankton. Science 320, 1081–1085. doi: 10.1126/science.1157890
Kearns, D. B. (2010). A field guide to bacterial swarming motility. Nat. Rev. Microbiol. 8, 634–644. doi: 10.1038/nrmicro2405
Kearns, D. B., and Losick, R. (2003). Swarming motility in undomesticated Bacillus subtilis. Mol. Microbiol. 49, 581–590. doi: 10.1046/j.1365-2958.2003.03584.x
Kembel, S. W., Cowan, P. D., Helmus, M. R., Cornwell, W. K., Morlon, H., Ackerly, D. D., et al. (2010). Picante: R tools for integrating phylogenies and ecology. Bioinformatics 26, 1463–1464. doi: 10.1093/bioinformatics/btq166
Koeppel, A. F., and Wu, M. (2012). Lineage-dependent ecological coherence in bacteria. FEMS Microbiol. Ecol. 81, 574–582. doi: 10.1111/j.1574-6941.2012.01387.x
Konstantinidis, K. T., and Tiedje, J. M. (2005). Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. U.S.A. 102, 2567–2572. doi: 10.1038/ismej.2008.62
Lane, D. J. (1991). “16S/23S rRNA sequencing,” in Nucleic Acid Techniques in Bacterial Systematics, eds E. Stackebrandt and M. Goodfellow (New York, NY: Wiley), 115–175.
Letunic, I., and Bork, P. (2011). Interactive tree of life v2: online annotation and display of phylogenetic trees made easy. Nucleic Acids Res. 39, W475–W478. doi: 10.1093/nar/gkr201
Martiny, A. C., Treseder, K., and Pusch, G. (2013). Phylogenetic conservatism of functional traits in microorganisms. ISME J. 7, 830–838. doi: 10.1038/ismej.2012.160
McInerney, J. O., Pisani, D., Bapteste, E., and O’Connell, M. J. (2011). The public goods hypothesis for the evolution of life on Earth. Biol. Direct 6:41. doi: 10.1186/1745-6150-6-41
Mitri, S., Xavier, J. B., and Foster, K. R. (2011). Social evolution in multispecies biofilms. Proc. Natl. Acad. Sci. U.S.A. 108, 10839–10846. doi: 10.1073/pnas.1100292108
Morris, J. J., Lenski, R. E., and Zinser, E. R. (2012). The black queen hypothesis: evolution of dependencies through adaptative gene loss. MBio 3, 1–7. doi: 10.1128/mBio.00036-12
Muller, J. P., An, Z., Merad, T., Hancock, I. C., and Harwood, C. R. (1997). Influence of Bacillus subtilis phoR on cell wall anionic polymers. Microbiology 143, 947–956. doi: 10.1099/00221287-143-3-947
Nadell, C. D., Xavier, J. B., Levin, S. A., and Foster, K. R. (2008). The evolution of quorum sensing in bacterial biofilms. PLoS Biol. 6:e14. doi: 10.1371/journal.pbio.0060014
Nygaard, S., Zhang, G., Schiøtt, M., Li, C., Wurm, Y., Hu, H., et al. (2011). The genome of the leaf-cutting ant Acromyrmex echinatior suggests key adaptations to advanced social life and fungus farming. Genome Res. 21, 1339–1348. doi: 10.1101/gr.121392.111
Ochman, H., Lawrence, J. G., and Groisman, E. A. (2000). Lateral gene transfer and the nature of bacterial innovation. Nature 405, 299–304. doi: 10.1038/35012500
Orme, D. (2013). The Caper Package: Comparative Analysis of Phylogenetics and Evolution in R. Reference Manual. Available at: https://cran.csiro.au/web/packages/caper/vignettes/caper.pdf
Pérez-Gutiérrez, R. A., López-Ramírez, V., Islas, A., Alcaraz, L. D., Hernández-González, I., Luna-Olivera, B. C., et al. (2013). Antagonism influences assembly of a Bacillus guild in a local community and is depicted as a food-chain network. ISME J. 7, 487–497. doi: 10.1038/ismej.2012.119
Philippot, L., Andersson, S. G., Battin, T. J., Prosser, J. I., Schimel, J. P., Whitman, W. B., et al. (2010). The ecological coherence of high bacterial taxonomic ranks. Nat. Rev. Microbiol. 8, 523–529. doi: 10.1038/nrmicro2367
Polz, M. F., Alm, E. J., and Hanage, W. P. (2013). Horizontal gene transfer and the evolution of bacterial and archaeal population structure. Trends Genet. 29, 170–175. doi: 10.1016/j.tig.2012.12.006
R Development Core Team (2009). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Rebollar, A. E., Avitia, M., Eguiarte, L. E., González-González, A., Mora, L., Bonilla-Rosso, G., et al. (2012). Water–sediment niche differentiation in ancient marine lineages of Exiguobacterium endemic to the Cuatro Cienegas Basin. Environ. Microbiol. 14, 2323–2333. doi: 10.1111/j.1462-2920.2012.02784.x
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Sanger, F., Nicklen, S., and Coulson, A. R. (1977). DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. U.S.A. 74, 5463–5467. doi: 10.1073/pnas.74.12.5463
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Souza, V., Siefert, J. L., Escalante, A. E., Elser, J. J., and Eguiarte, L. E. (2012). The Cuatro Ciénegas Basin in Coahuila, Mexico: an astrobiological precambrian park. Astrobiology 12, 641–647. doi: 10.1089/ast.2011.0675
Spoering, A. L., and Gilmore, M. S. (2006). Quorum sensing and DNA release in bacterial biofilms. Curr. Opin. Microbiol. 9, 133–137. doi: 10.1016/j.mib.2006.02.004
Stoodley, P., Sauer, K., Davies, D. G., and Costerton, J. W. (2002). Biofilms as complex differentiated communities. Annu. Rev. Microbiol. 56, 187–209. doi: 10.1146/annurev.micro.56.012302.160705
Suen, G., Teiling, C., Li, L., Holt, C., Abouheif, E., Bornberg-Bauer, E., et al. (2011). The genome sequence of the leaf-cutter ant Atta cephalotes reveals insights into its obligate symbiotic lifestyle. PLOS Genetics 7:e1002007. doi: 10.1371/journal.pgen.1002007
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Tapia-Torres, Y., Rodríguez-Torres, M. D., Elser, J. J., Islas, A., Souza, V., García-Oliva, F., et al. (2016). How to live with phosphorus scarcity in soil and sediment: lessons from bacteria. Appl. Environ. Microbiol. 82, 4652–4662. doi: 10.1128/AEM.00160-16
van Gestel, J., Vlamakis, H., and Kolter, R. (2015). From cell differentiation to cell collectives: Bacillus subtilis uses division of labor to migrate. PLoS Biol. 13:e1002141. doi: 10.1371/journal.pbio.1002141
Velicer, G. J., and Yu, Y. T. N. (2003). Evolution of novel cooperative swarming in the bacterium Myxococcus xanthus. Nature 425, 75–78. doi: 10.1038/nature01908
Visser, B., Le Lann, C., den Blanken, F. J., Harvey, J. A., van Alphen, J. J. M., and Ellers, J. (2010). Loss of lipid synthesis as an evolutionary consequence of a parasitic lifestyle. Proc. Natl. Acad. Sci. U.S.A. 107, 8677–8682. doi: 10.1073/pnas.1001744107
Vlamakis, H., Chai, Y., Beauregard, P., Losick, R., and Kolter, R. (2013). Sticking together: building a biofilm the Bacillus subtilis way. Nat. Rev. Microbiol. 11, 157–168. doi: 10.1038/nrmicro2960
Webb, C. O. (2000). Exploring the phylogenetic structure of ecological communities: an example for rain forest trees. Am. Nat. 156, 145–155. doi: 10.1086/303378
Webb, C. O., Ackerly, D. D., McPeek, M. A., and Donoghue, M. J. (2002). Phylogenies and community ecology. Annu. Rev. Ecol. Syst. 33, 475–505. doi: 10.1146/annurev.ecolsys.33.010802.150448
West, S. A., Griffin, A. S., Gardner, A., and Diggle, S. P. (2006). Social evolution theory for microorganisms. Nat. Rev. Microbiol. 4, 597–607. doi: 10.1038/nrmicro1461
Whitham, T. G., Bailey, J. K., Schweitzer, J. A., Shuster, S. M., Bangert, R. K., LeRoy, C. J., et al. (2006). A framework for community and ecosystem genetics: from genes to ecosystems. Nat. Rev. Genet. 7, 510–523. doi: 10.1038/nrg1877
Xu, J., Saunders, C. W., Hu, P., Grant, R. A., Boekhout, T., Kuramae, E. E., et al. (2007). Dandruff-associated Malassezia genomes reveal convergent and divergent virulence traits shared with plant and human fungal pathogens. Proc. Natl. Acad. Sci. U.S.A. 104, 18730–18735. doi: 10.1073/pnas.0706756104
Keywords: microbial communities, phylogenetic signal, phenotypic trait loss, collective action, Bacillus spp.
Citation: Rodríguez-Torres MD, Islas-Robles Á, Gómez-Lunar Z, Delaye L, Hernández-González I, Souza V, Travisano M and Olmedo-Álvarez G (2017) Phenotypic Microdiversity and Phylogenetic Signal Analysis of Traits Related to Social Interaction in Bacillus spp. from Sediment Communities. Front. Microbiol. 8:29. doi: 10.3389/fmicb.2017.00029
Received: 02 May 2016; Accepted: 05 January 2017;
Published: 30 January 2017.
Edited by:
Ludmila Chistoserdova, University of Washington, USAReviewed by:
Tim Cooper, University of Houston, USAMarta Goberna, Consejo Superior de Investigaciones Científicas, Spain
Copyright © 2017 Rodríguez-Torres, Islas-Robles, Gómez-Lunar, Delaye, Hernández-González, Souza, Travisano and Olmedo-Álvarez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gabriela Olmedo-Álvarez, Z29sbWVkb0BpcmEuY2ludmVzdGF2Lm14