 Stuart G. Dashper1†
Stuart G. Dashper1† Helen L. Mitchell1†
Helen L. Mitchell1† Christine A. Seers1†
Christine A. Seers1† Simon L. Gladman2Torsten Seemann2Dieter M. Bulach2
Simon L. Gladman2Torsten Seemann2Dieter M. Bulach2 P. Scott Chandry3Keith J. Cross1Steven M. Cleal1
P. Scott Chandry3Keith J. Cross1Steven M. Cleal1 Eric C. Reynolds1*
Eric C. Reynolds1*- 1Oral Health Cooperative Research Centre, Melbourne Dental School, Bio21 Institute, University of Melbourne, VIC, Australia
- 2Victorian Life Sciences Computation Initiative, Carlton, VIC, Australia
- 3CSIRO Food and Nutrition, Werribee, VIC, Australia
Porphyromonas gingivalis is a keystone pathogen of chronic periodontitis. The virulence of P. gingivalis is reported to be strain related and there are currently a number of strain typing schemes based on variation in capsular polysaccharide, the major and minor fimbriae and adhesin domains of Lys-gingipain (Kgp), amongst other surface proteins. P. gingivalis can exchange chromosomal DNA between strains by natural competence and conjugation. The aim of this study was to determine the genetic variability of P. gingivalis strains sourced from international locations over a 25-year period and to determine if variability in surface virulence factors has a phylogenetic basis. Whole genome sequencing was performed on 13 strains and comparison made to 10 previously sequenced strains. A single nucleotide polymorphism-based phylogenetic analysis demonstrated a shallow tri-lobed phylogeny. There was a high level of reticulation in the phylogenetic network, demonstrating extensive horizontal gene transfer between the strains. Two highly conserved variants of the catalytic domain of the major virulence factor the Kgp proteinase (KgpcatI and KgpcatII) were found. There were three variants of the fourth Kgp C-terminal cleaved adhesin domain. Specific variants of the cell surface proteins FimA, FimCDE, MfaI, RagAB, Tpr, and PrtT were also identified. The occurrence of all these variants in the P. gingivalis strains formed a mosaic that was not related to the SNP-based phylogeny. In conclusion P. gingivalis uses domain rearrangements and genetic exchange to generate diversity in specific surface virulence factors.
Introduction
Chronic periodontitis is an inflammatory disease associated with bacteria which results in destruction of the tooth's supporting tissues including the alveolar bone (Wiebe and Putnins, 2000). The disease is associated with specific bacteria in a polymicrobial biofilm, subgingival plaque, accreted to the surface of the root of the tooth. In the USA, 38% of the adult population 30 years and older and 65% of adults 65 years and older have either severe or moderate periodontitis (Eke et al., 2012). Epidemiological surveys have shown that clinical indicators of periodontitis are linked to an increased risk of cardiovascular diseases, certain cancers (orogastrointestinal tract and pancreas), rheumatoid arthritis, adverse pregnancy outcomes, and other systemic diseases related to the regular bacteraemia and chronic inflammation associated with the disease (Genco and Van Dyke, 2010; Lundberg et al., 2010; Linden et al., 2013; Madianos et al., 2013; Tonetti and Van Dyke, 2013).
P. gingivalis is a keystone pathogen of chronic periodontitis that dysregulates the host immune response to favor the proliferation of the polymicrobial biofilm thereby disrupting homeostasis with the host to cause dysbiosis and disease (Hajishengallis et al., 2012). The closely related Porphyromonas gulae is proposed to play a similar role to P. gingivalis in the development of periodontitis in dogs (Fournier et al., 2001; Lenzo et al., 2016).
The pathogenicity of P. gingivalis is attributed to a number of surface-associated virulence factors that include cysteine proteinases (gingipains), fimbriae, haem-binding proteins, and outer membrane transport proteins amongst others. In particular, the cysteine endoproteinases, the Arg-specific gingipains (RgpA and RgpB) and Lys-specific gingipain (Kgp) have multiple effects on both the innate and adaptive immune responses (Popadiak et al., 2007; Bostanci and Belibasakis, 2012). All three gingipain polyproteins contain a signal peptide of ~22 amino acids in length, an unusually long propeptide of over 200 amino acids in length, and a catalytic domain of ~360 amino acids. In addition RgpA and Kgp have repetitive haemagglutinin-adhesin (HA) domains in the polypeptide region C-terminal to the catalytic domains. Some of these have been alternatively described as C-terminal adhesin domains or cleaved adhesin domains (CADs) and some are DUF 2436 domains (O'Brien-Simpson et al., 2003; Li et al., 2010). The RgpA and Kgp precursor proteins are cleaved into multiple domains that remain non-covalently associated forming large outer membrane protein complexes (Bhogal et al., 1997). The Kgpcat domain structure consists of a central 10 stranded β-sheet surrounded by 10 α-helices forming an α-β sandwich domain. An immunoglobulin superfamily-like domain comprising six antiparallel β-strands, is situated on the opposite side from the substrate binding face of the α-β domain (de Diego et al., 2014; Gorman et al., 2015). RgpB lacks the adhesin domains and is located in a monomeric form on the outer membrane and has a structure similar to Kgpcat (Eichinger et al., 1999). P. gingivalis strains also contain the large polyprotein HagA which has multiple repeating CADs that associate with other cell surface proteins including the gingipains (Kozarov et al., 1998; Frazer et al., 2006).
Fimbriae facilitate adherence to both host cells and other bacterial members of the plaque microbiota. P. gingivalis has at least two types of fimbriae, including the major fimbriae encoded by fimA-E and the minor fimbriae encoded by mfa1-4. The major fimbriae are involved in colonization and cellular invasion and both types of fimbriae have proinflammatory capacity (Yilmaz et al., 2002; Bostanci and Belibasakis, 2012). The major fimbriae in P. gingivalis are comprised of the FimA fimbrillin structural protein and FimCDE accessory structural proteins that are involved in adhesion specificity. There are six reported FimA types, types I, Ib, II-V based on identified genome sequences with predicted molecular masses ranging from 41 to 49 kDa (Amano et al., 1999, 2004). The P. gingivalis minor fimbriae are encoded by mfa1-4 and two mfa1 genotypes encoding two distinct Mfa1 fibrillin monomers have been described (Nagano et al., 2015).
Virulence factors such as the fimbriae and gingipains are the basis for typing methods used to identify disease-associated strains (Yoshino et al., 2007). It is thought that only some strains may have the capacity to cause disease and differences between strains in the ability to cause localized or systemic infections have been demonstrated in animal models (Griffen et al., 1999).
Using multilocus sequence typing (MLST) it has been shown that up to eight sequence types of P. gingialis can be found in individual pockets at diseased sites in patients with chronic periodontitis (Enersen et al., 2008). Similarly, heteroduplex analysis of the ribosomal operon intergenic spacer region of P. gingivalis identified in subgingival plaque samples found 39% of these samples had multiple heteroduplex types (Igboin et al., 2009). At present very little is known about the genetic diversity and underlying population structure of P. gingivalis, the stability of strains within an individual over time or during disease development, or the transmission of P. gingivalis between individuals. P. gingivalis is naturally competent and able to exchange alleles between strains (Tribble et al., 2012; Kerr et al., 2014). Multilocus sequence typing indicates that there is frequent horizontal gene transfer and recombination (Frandsen et al., 2001; Koehler et al., 2003; Enersen et al., 2008; Enersen, 2011). To better understand the virulence and evolution of the bacterium a greater knowledge of its genetic diversity and genetic exchange is required. Genetic diversity, especially in genes encoding surface virulence factors can make a targeted immunotherapy more challenging.
Here we report the comparison of the genomes of 21 P. gingivalis strains sourced from around the world over a 25 year time period. We determined that there is extensive genomic recombination with overall limited sequence diversity. Notably, a subset of genes encoding specific surface virulence factors have multiple alleles that through recombination have produced a large variety of combinations.
Materials and Methods
Bacterial Strains
Thirteen P. gingivalis strains were sequenced in this study. These included 11 P. gingivalis isolates 11A, 7B TORR, 381, A74A1-28, 13–1, Afr-5B1, 3A1, 3_3, 15–9, ATCC 49417, and 84–3 that were a kind gift from R. Page, Washington University, Seattle, WA, USA. All clinical samples in this group were obtained by paper point sampling of subgingival plaque found in periodontal pockets at least 6mm deep in subjects that had at least four 6mm deep pockets, gingival bleeding on probing, no previous periodontal treatment, no antibiotic use in the past 6 months and good systemic health. These isolates were obtained from periodontitis patients residing in the United States, Sudan, Romania and Norway (Ali et al., 1994a,b, 1996, 1997; Slakeski et al., 2002). P. gingivalis W50 was a kind gift from P. Marsh (University of Leeds, UK) and has been in the laboratory stocks of the Melbourne Dental School for 24 years. P. gingivalis YH522 was obtained from H. Yoshimoto (Kanagawa Dental College, Japan) and has been in the laboratory stocks of the Melbourne Dental School for 18 years.
The publicly available closed genomic sequences of P. gingivalis strains W83 (Nelson et al., 2003), ATCC 33277 (Naito et al., 2008) and TDC60 (Watanabe et al., 2011); six draft genomes (FO185, FO566, FO568, FO569, FO570, W4087) available from the Forsyth Institute, Boston, USA; and a draft genome of a Chinese strain SJD2 (Liu et al., 2014) were included in the analyses. Background information for each strain and the relevant accession numbers are shown in Table 1.
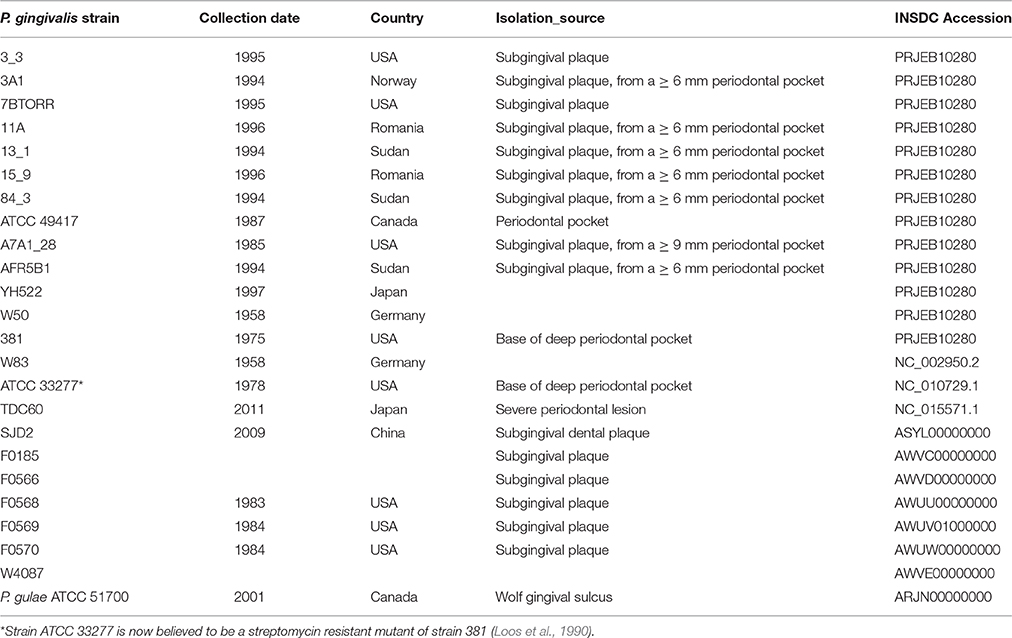
Table 1. P. gingivalis strains referred to in this study.
There were only 20 single nucleotide polymorphisms (SNPs) between the genome sequence of strain W50 from this study and the previously sequenced strain W83. These two isolates were collected at the same time from the same patient and presumably are the same strain. As a result, only W83 data are presented in these analyses to avoid bias.
Only a small number of SNPs were detected between strain (FDC) 381, sequenced in this study, and the genome sequence of ATCC 33277 (Naito et al., 2008) so only ATCC 33277 data is presented in this study. The recently published genome sequences of 12 P. gulae strains (Coil et al., 2015) were included in this study to provide a reference and phylogenetic outgroup. The Loup 1 P. gulae type strain (ATCC 51700) (Fournier et al., 2001) was obtained from the American Type Culture Collection.
Preparation of DNA and Sequencing
P. gingivalis strains and P. gulae were grown to stationary phase in Brain Heart Infusion medium supplemented with 0.5 g/L cysteine and 1 mg/L menadione. Cells were harvested by centrifugation and washed in 10 mM Tris-HCl pH 8.0. DNA was extracted using the Blood and Tissue Genomic DNA Isolation Kit (Qiagen) and concentrated to 100 ng/μL using ethanol precipitation then resuspended in 10 mM Tris-HCl.
Ilumina Nextera-based whole-genome sequencing producing 2 × 100 bp paired-end reads with an average insert size of ~500 bp was performed at The Australian Genome Research Facility (AGRF) using the Illumina HiSeq-2000 Platform with the CASAVA1.8.2 pipeline.
Assembly and Annotation of Genomes
Assembly and annotation of sequences was performed at the Victorian Life Sciences Computation Initiative—Life Science Computation Centre. Contigs were created by de novo assembly using Velvet v1.2.07 (Zerbino and Birney, 2008) in conjunction with VelvetOptimiser v2.2.5 (https://github.com/tseemann/VelvetOptimiser). The assembled sequences were annotated using Prokka v1.11 (Seemann, 2014).
Manual curation of the annotation to remove false overlapping genes and correct translation initiation start sites was performed using eCAMBER (Wozniak et al., 2014). Genes encoding hypothetical proteins < 61 aa in length, with no similar proteins in the NCBI databases and lacking any detectable conserved domains, were omitted (Salzberg et al., 1998). This conservative approach resulted in the exclusion of only three genes in the P. gingivalis W83 genome of 1822 genes. The genome of P. gulae ATCC 51700 was included as a more phylogenetically distant outgroup to improve structural annotation predictions. InterProScan was used from within Geneious v 8.1.3 (Biomatters Ltd.) to identify signal peptides and other protein domains signatures for genes predicted to encode gingipains. Type IX secretion system C-Terminal Domain (CTD) signal sequences were identified by alignment (Veith et al., 2013). Gene identifiers used throughout this manuscript refer to those from the first published genome of P. gingivalis W83 (Nelson et al., 2003) as commonly used in the literature. For the updated NC_002950.2 version of the W83 genome these “PGXXXX” identifiers are referred to under the old_locus_tag qualifier.
Phylogeny
The MLST gene data set used was the complete gene sequences from six of the seven genes (ftsQ, gpdxJ, mcmA, pepO, pga, recA) defined in the PubMLST P. gingivalis MLST Database http://pubmlst.org/pgingivalis/ (Jolley and Maiden, 2010). The hagB gene is a repetitive sequence and was not included in the phylogenetic analysis (as it is paralogous to hagC). Sequence manipulations were performed in Geneious v 8.1.3. Individual genes were codon aligned with MAFFT v 1.3.5 (Katoh and Standley, 2013) prior to testing several methods to generate phylogenetic trees. Inconsistent results in generating trees (and poor branch support) indicated that a bifurcating tree was unlikely to accurately represent the ancestry of these strains. Therefore, the MLST alignment was plotted as a NeighborNet network using SplitsTree4 (v 4.13.1) (Huson and Bryant, 2006). SplitsTree4 was used to calculate the δ score and Q-residual score as measures of “treelikeness” of the phylogenetic distance data (Holland et al., 2002). The Phi test implemented in Splits Tree4 was used to infer the likelihood of recombination in the DNA sequence alignment data.
Whole genome SNP phylogeny was computed for the P. gingivalis genomes with and without P. gulae using Parsnp v 1.0 (Treangen et al., 2014) with recombination detection enabled. Alignments without P. gulae were used to examine the phylogenetic relationship among the P. gingivalis strains. Phylogenetic trees were generated with FastTree (v 2.1.7) (Price et al., 2010) using the GTR evolutionary model and reliability tested with the Shimodaira-Hasegawa test for 2000 resampling steps. Consensus trees were calculated for branches with >75% support using a greedy clustering method. FASTA alignments were converted from the XML files generated by Parsnp using a custom script for analysis in SplitsTree4 as described above.
Selection Analysis
Selection analysis was conducted on codon aligned catalytic domains from proteins Kgp, RgpA, and RgpB. Positional references denote the location of the codon in the intact gene rather than the extracted catalytic domain sequence. The two variants of Kgp necessitated that selection analysis be conducted in each variant individually. Nucleotide diversity calculations (π) were performed with DNASP ver 5.10 (Librado and Rozas, 2009). In order to determine if codons in the catalytic domain were subjected to positive or negative selection, several methods within the HyPhy analysis suite were used (Pond et al., 2005). A conservative approach was taken for interpreting the results requiring that more than one method detected selection and that P-values or posterior probabilities were below 0.05 or 0.9, respectively. HyPhy analysis methods were implemented in the Datamonkey.org web portal (Pond and Frost, 2005). Methods tested included single likelihood ancestor counting method (SLAC; Kosakovsky Pond and Frost, 2005), fixed effects likelihood method (FEL, iFEL; Pond et al., 2006), and the fast unconstrained Bayesian approximation method (FUBAR; Murrell et al., 2013). All alignments were examined for signals indicative of recombination with GARD (Kosakovsky Pond et al., 2006) and the analysis adjusted accordingly.
Sequence Analysis
Average nucleotide identity (ANI) between strains was calculated as OrthoANI values calculated from the Orthologous Average Nucleotide Identity Tool, v 0.90 (Lee et al., 2016). Sequence alignments and manipulations were performed essentially as described above. The E-INS-I algorithm was used with a BLOSUM62 scoring matrix with a gap opening penalty of 1.03. Alignments of multiple gene regions were performed using the Geneious alignment tool using a global alignment with free end gaps, a 65% similarity cost matrix, a gap opening penalty of 12 and extension penalty of 3. To manually find and verify genes of interest across all 21 P. gingivalis genomes, a local database of genome nucleotide sequences was created within Geneious and the MegaBLAST search algorithm was used with default parameters to find and visualize hits.
Assembly of Gingipain and hagA Genes
The repeat regions within the hagA, rgpA, and kgp genes were unable to be resolved by using the Velvet assembler, resulting in contig breaks within each of these genes. The hagA, rgpA, and kgp genes and their surrounding regions in the three closed genomes were compared using global and local sequence alignments within Geneious. Extensive manual curation was performed to identify and annotate unique and repeat regions within and across these genes and identify any conservation of gene order of the surrounding genes. These unique regions were used to identify contigs that could be assigned to a specific gene. The connections extending out from these contigs were then visualized using Bandage v 0.8.0 (Wick et al., 2015) and connected contigs compared to the reference genomes using MegaBLAST and annotated. A plausible path for scaffolding contigs was manually determined based on the order of the specific regions found in the three closed genomes and the depth of coverage of each of the contigs.
Validation of Kgp Assemblies
Predicted kgp assemblies from the 11 P. gingivalis strains and single P. gulae strain sequenced in this study were validated using Sanger sequencing. Primers were designed using Primer3 v 2.3.4 (Untergasser et al., 2007). Strain specific primer sets were designed to amplify 500 bp regions and repeat regions co-ordinates were used to exclude target sites (Table 2). Primers kgp_F1 and kgp_Rii were used to produce a 5.2 kb amplicon for each strain, which was quantified and used as the template with each of the primers listed for capillary electrophoresis sequencing at The Melbourne Translational Genomics Platform.

Table 2. Primers used for validation of P. gingivalis and P. gulae kgp assemblies.
Pangenome Estimation and Visualisation
A visual overview of the relationship of each of these genome sequences to that of W83 was produced using BLAST Ring Image Generator, BRIG v 0.95 (Alikhan et al., 2011). Island Viewer v 3.0 was used to identify genomic islands in the P. gingivalis W83 genome (Dhillon et al., 2015).
Pangenome analysis was based on protein coding sequences only. Clustering of genes annotated as Coding DNA Sequences (CDSs) was performed using eCAMBER (Wozniak et al., 2014) to determine homologs. The parameter specifying the minimum percentage identity (PID) required for a match to be accepted was set at 50% and the default e-value score parameter was used.
Lys-Specific Proteolytic Activity
Whole cell Lys-specific proteolytic activity of P. gingivalis strains was determined using the chromogenic substrate N-(p-tosyl)-Gly-Pro-Lys 4-nitroanilide acetate salt (GPK-NA) as previously published (Toh et al., 2011).
Modeling of Kgp Adhesins
Modeling of the P. gingivalis K4 domain based on the known structure of the cleaved adhesin domain K3 (PDB: 3m1h) was performed using FUGUE (Shi et al., 2001) to generate a structural alignment, followed by ORCHESTRAR (Tripos International) to build a molecular model based on that alignment. Model quality was tested using the align_all algorithm from PyMOL (Schrodinger Inc.) to calculate the root-mean square deviation between structurally conserved atoms in the different CAD domains. Graphical representations of the CADs were generated using PyMOL.
Results
Phylogeny
The genomes of 13 P. gingivalis strains were sequenced and these data combined with a further 10 complete or draft genomes deposited in GenBank to provide an analysis data set (Table 1). The draft genome assemblies of the P. gingivalis strains ranged in size from 2,210,297 bp (W4087) to 2,424,225 bp (ATCC 49417) (Table 3). All P. gingivalis strains were closely related, with ANI values of orthologous fragment pairs between each of the 21 strains in the range 98.09–99.44%. In contrast pairwise comparison of each of the 12 published strains of P. gulae (Coil et al., 2015) with each of the P. gingivalis strains showed ANI values of between 91.88 and 92.77%, clearly indicating they are two distinct species, with values well below the 95–96% cutoff for species demarcation (Lee et al., 2016).
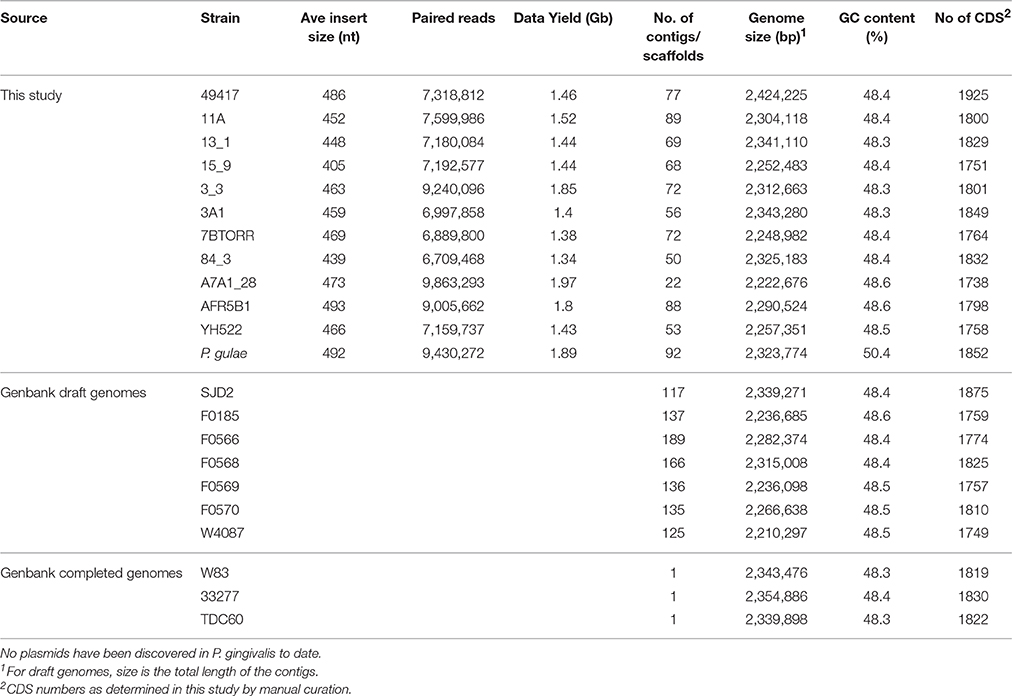
Table 3. P. gingivalis and P. gulae chromosomal contig assembly and annotation data.
An alignment derived from a concatenated set of MLST genes was examined using a NeighborNet network. The resultant network generated with uncorrected nucleotide distances was not tree-like displaying a high level of reticulation (δ score = 0.4196, Q-residual score = 0.02411; Figure S1). A Phi test for recombination suggested a high likelihood of recombination (p < 0.0001) among the selected MLST genes used for this analysis. These results are consistent with Koehler's previous MLST study that found a high rate of recombination (Koehler et al., 2003). The discriminatory power of the MLST method is entirely dependent on the selection of appropriate housekeeping genes. P. gingivalis genes including nah were excluded from this scheme by previous studies due to their limited variation (Enersen, 2011). The official MLST typing scheme for P. gingivalis (http://pubmlst.org/pgingivalis) still includes the hagB gene despite this being a paralog with two nearly identical copies in the W83 strain. This shows that typing systems can be inaccurate without genome wide information from a broad selection of strains before developing such systems.
Due to the limitations of the MLST method a SNP based approach was adopted using whole genome data. The P. gingivalis SNP alignment was subjected to approximate maximum likelihood analysis to yield an unrooted tree with strains separated into three broad clades (Figure 1). Resampling branch support was high for most branch points but three strains (13_1, 3A1 and TDC60) could not be clearly resolved resulting in a polytomy. A NeighborNet network derived from the whole genome SNP alignment yielded a more tree-like network than that MLST data set (δ score = 0.251, Q-residual score = 0.000533; Figure S2) confirming that a bifurcating tree adequately describes the phylogeny of these organisms.
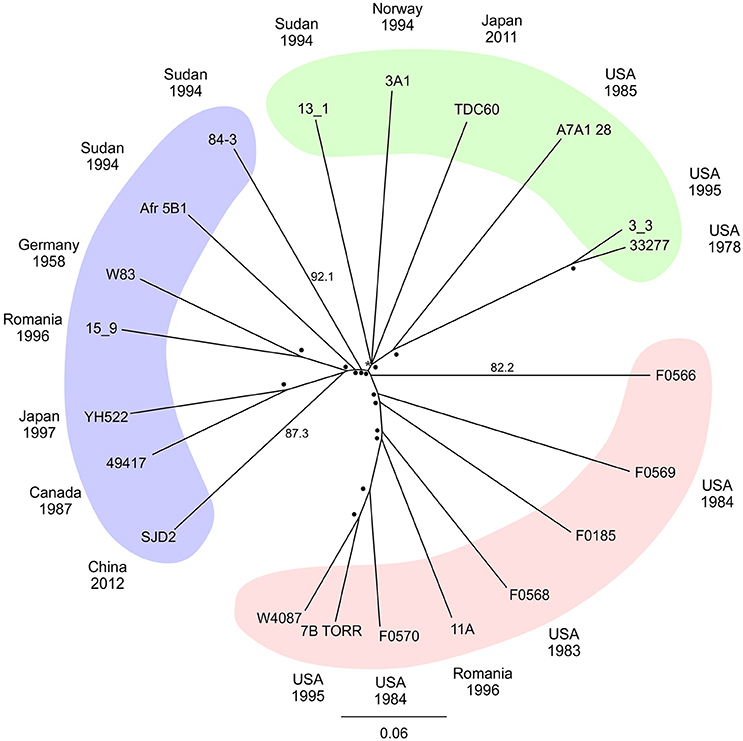
Figure 1. Radial phylogenetic tree. Unrooted approximate maximum likelihood consensus tree of whole genome P. gingivalis core SNP alignment. Plotted with percentage support values determined by Shimodiara-Hasegawa resampling test placed at the node or up the branch to improve readability, values of 99–100% are depicted by (•) with lower values written. Colored areas highlight major groupings of the taxa. The date and country of isolation of each strain where known are shown in the outer ring of the diagram.
Pangenome
The gene content of the P. gingivalis W83 strain was highly conserved in the 20 other strains as seen in the BRIG diagram (Figure 2). There are two major regions of sequence variability, a genomic island of ~70 kbp centered around 900 kbp on the chromosome and a homolog of the Bacteroides conjugative transposon CTnDot centered around 1550 bp. The predicted genome size of the ATCC 49417 strain was 70 kb larger than any of the other genomes (Table 3) and two relatively complete prophage genomes were detected in this strain, one of which was also present and highly conserved in the AFR5B1 strain (Figure S3). Although numerous genome rearrangements and different integration sites for insertion elements have been observed between the three strains with complete genomes, it is problematic to determine these sorts of changes with fragmented draft genomes, so our study has focused on a comparison of the predicted protein coding regions. A total of 3740 protein-coding gene clusters were found across the 21 P. gingivalis strains and 1488 high-frequency genes were present in 17 or more of the strains (Data Set S1). Of the remaining gene clusters, 345 were medium-frequency genes present in 6–16 of the P. gingivalis strains whilst 1907 were low-frequency genes present in five or less strains. The majority of gene clusters occurred only once per strain, but 145 contained paralogs, with two or more copies in at least one strain.
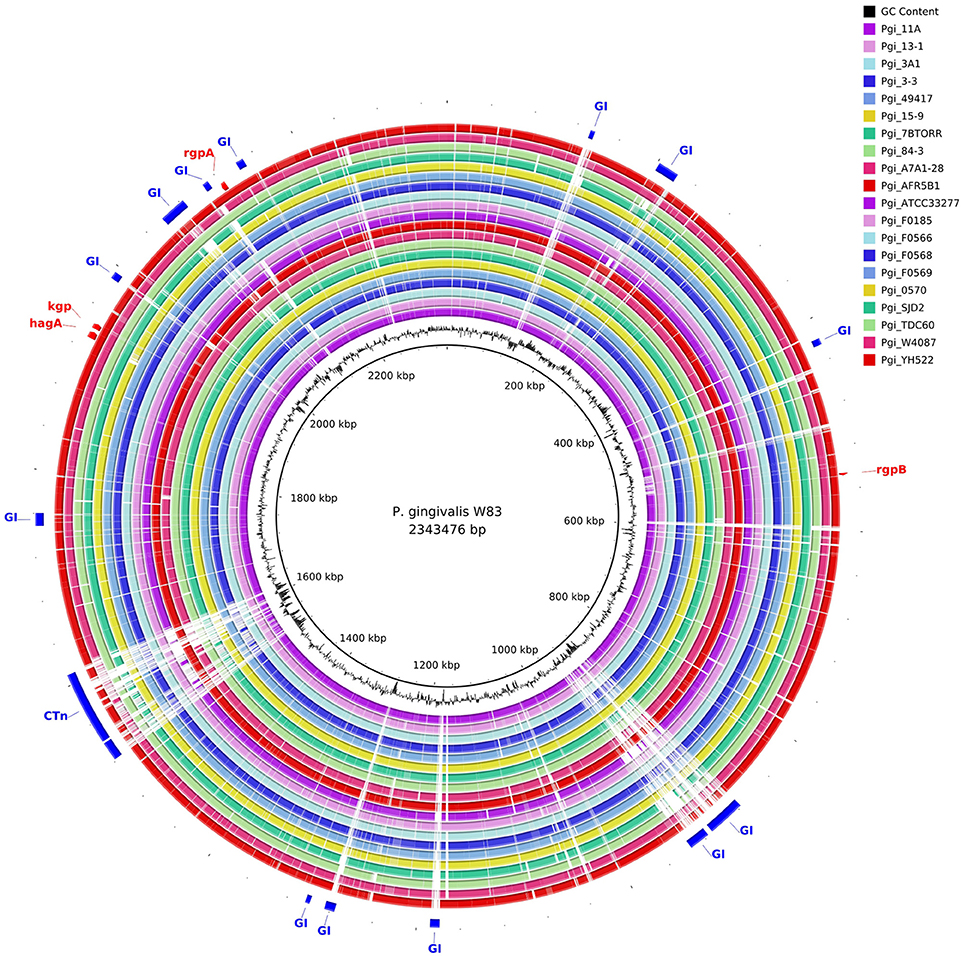
Figure 2. Comparison of P. gingivalis genomes by BLAST Ring Image Generator (BRIG). The contigs of 20 P. gingivalis genomes are compared with the complete nucleotide sequence of strain W83 as reference. There are two areas of high divergence. P. gingivalis W83 contains several predicted genomic islands, many of which are highly divergent between strains. The genomic island centered on 900 kbp is bounded by a series of repeats, integrases and a tRNA. It is likely that this genomic island has been built up by multiple insertion events given the large number of tandem integrases in this region. A highly variable region centered around 1550 kbp contains a putative conjugative transposon (CTn).
Gingipain Catalytic Domains
Gingipain Kgp catalytic domain crystal structure shows a caspase-like fold where catalysis occurs, followed by an immunoglobulin-like fold (de Diego et al., 2014; Gorman et al., 2015). The Kgp structure reveals a flat substrate binding site that would be amenable to interactions with a wide array of proteins. Cleavage site specificity is produced by the constraints of the S1 binding pocket into which the side chain of Lys residue is inserted. The RgpB catalytic domain has a structure similar to Kgpcat (Eichinger et al., 1999).
RgpA and RgpB catalytic domain sequences were highly conserved across all of the strains, with only 14 substitutions across the 346 amino acids of the RgpAcat domain in the context of overall nucleotide diversity of π = 0.0109 (Figure 3A). There were two positions in the RgpAcat domain that had specific conservative substitutions in approximately half of the strains, Ile302Val and Val433Ala. The remainder of substitutions occurred in a low number of strains and appeared to be random. The RgpA propeptide was highly conserved across all strains with only two substitutions in single strains relative to the W83 sequence. An analysis to detect signals of evolutionary selection using the 17 unique RgpAcat domains determined that RgpAcat domain had seven codon positions under negative selection and none under positive selection (Table S1). Similarly, the RgpBcat domain was highly conserved with a nucleotide diversity level of π = 0.01955 across 20 unique sequences. Selection analysis detected 10 codons under negative selection and none under positive selection (Table S1). There were significant differences between the RgpA and RgpB propeptides (43 substitutions and 3 indels across 199 amino acids) indicating that they have distinct binding sites or other constraints on sequence variation.
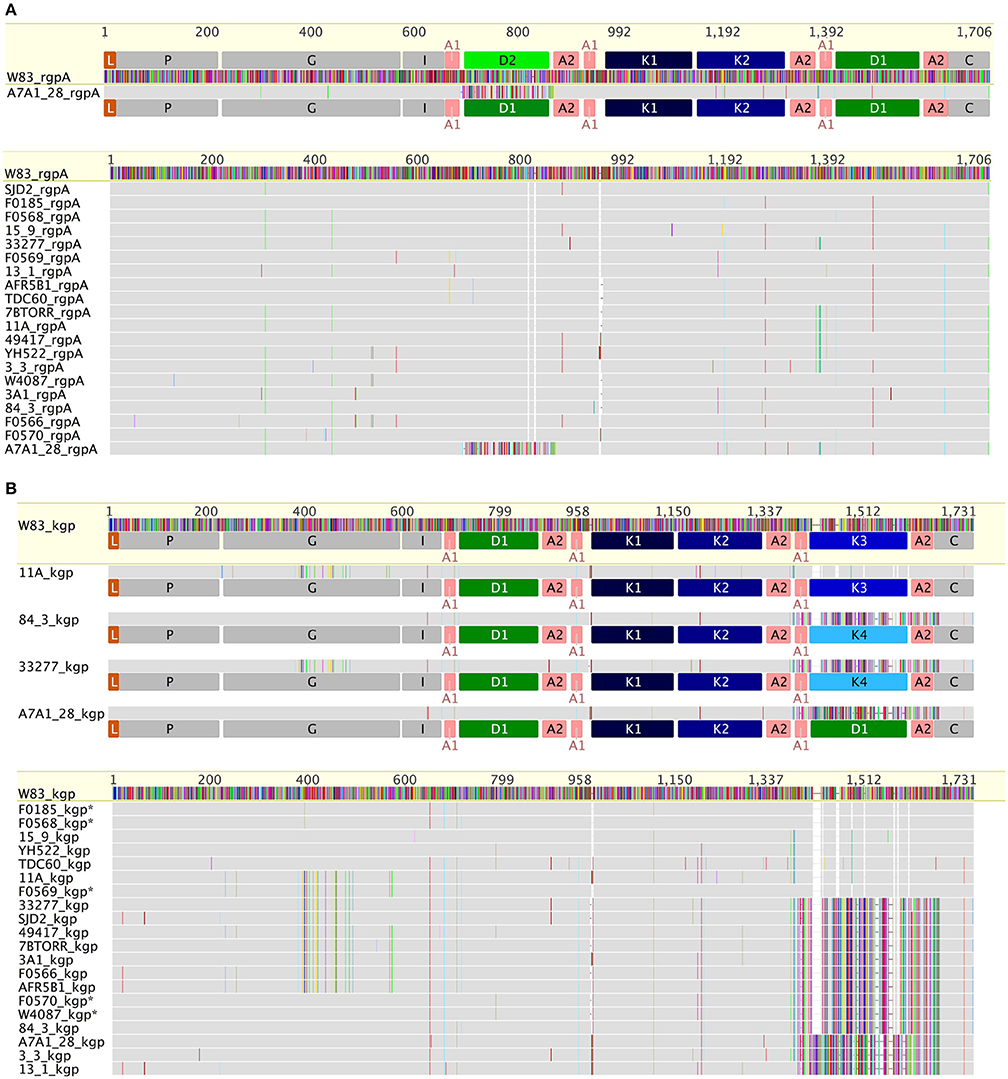
Figure 3. Comparison of RgpA (A) and Kgp (B) variability across the P. gingivalis strains. RgpA and Kgp are comprised of a leader peptide (L), a propeptide region (P), a catalytic domain (G) of around 360 amino acids, an immunoglobulin fold (I) followed by three or more of modular adhesin domains comprised of CADs (K1–K4) and DUF2436 domains (D1–D2) and a conserved C-terminal domain (C). Kgp separates into four groups based on specific substitutions in the catalytic domain combined with differences in the C-terminal CAD. The specific substitutions in the Kgp catalytic domains around the catalytic site that may impact catalysis, substrate binding or propeptide interactions are discussed in the text. In contrast to Kgp in RgpA there is a high level of conservation of the catalytic domain sequence and the CADs with only strain A7A1_28 showing a substituted D1 domain.
In constrast there were two distinct variants of the catalytic domain of Lys-gingipain (Kgpcat) with 21 specific amino acid substitutions in a 150 amino acid region that encompasses the Lys-gingipain catalytic site (Figure 3B). We refer to these variants as KgpcatI and KgpcatII. Mapping these substitutions to the known structure of Kgpcat (Gorman et al., 2015) showed that five of the variable residues occur in proximity to the active site pocket and 11 form an adjacent contiguous surface exposed patch (see Section Discussion). Examination of both Kgpcat variants for selection signals indicated that of the eight unique KgpcatI and five unique KgpcatII domain sequences, three and one codon were under negative selection respectively and no codons were detected to be under positive selection (Table S1). Determination of whole cell Lys-X proteolytic activity revealed that those strains that harbored KgpcatI had on average higher activity than those strains harboring KgpcatII (Figure 4).
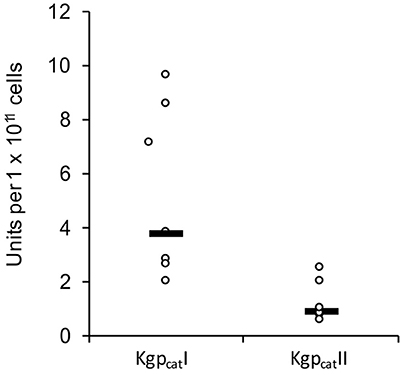
Figure 4. Whole culture Lys-specific proteolytic activity of 12 P. gingivalis strains determined using the chromogenic substrate N-(p-tosyl)-Gly-Pro-Lys 4-nitroanilide acetate salt (GPK-NA) as previously described (Toh et al., 2011).
Haemagglutinin-like Adhesins
Due to identical repeat sequences within and between the regions of the hagA, rgpA, and kgp genes encoding adhesins (CADs; IPR011628) these regions could not be fully resolved using automated assembly. Therefore, extensive manual assembly was performed with reference to the three closed genomes to identify contigs that could be assigned to a specific gene. The rgpA and kgp CAD encoding regions were assembled and the kgp assemblies for each strain were validated by Sanger sequencing (Table 2). Unfortunately, the highly repetitive nature of hagA and the length of the repeated units confounded assembly of hagA from draft genomes.
The CAD adhesins of Kgp of strain W83 are known to occur in the order K1-K2-K3. N-terminal to K1 is another domain module DUF2436 (conserved Pfam Domain of Unknown Function; IPR018832) (D1) giving the adhesin organization D1-K1-K2-K3. Defining a CAD or DUF2436-like domain subtype as having >90% amino acid identity indicates that in other P. gingivalis strains, following Kgpcat are also modules D1-K1-K2. However, the C-terminal domain varies, being an additional copy of D1 or a fourth CAD, K4 (Figure 3B). C-terminal to RgpAcat the CAD and DUF2436-like domains occur in the order D2-K1-K2-D1, with the exception of strain A7A1-28 which has D1-K1-K2-D1 (Figure 3A). This shows that there is combinatorial modularity in the adhesin regions of the gingipains. Similarly CAD and DUF2436 domain modularity is evident in P. gulae (Figures 3A,B).
Examination of HagA encoded in the completed W83 genome shows further DUF2436 and CAD domains, D3 and K5, respectively, which are followed by repeated K3-K2 pairs and an orphan C-terminal K3 (Figure 5). In contrast, strains TDC60 and ATCC 33277 do not have the orphan K3 domain and ATCC 33277 has an additional K3-K2 repeat.

Figure 5. Structure of the genes encoding known and putative gingipains and modular adhesins containing CADs. The gingipains are comprised of a leader peptide (L) that is cleaved during translocation across the inner membrane, an unusually long propeptide region (P; InterPro Accession number IPR012600) of ~200 amino acids and a catalytic domain (G; IPR001769) of around 360 amino acids that is followed by an immunoglobulin fold (I; IPR005536). RgpA and Kgp contain three or more modular adhesin domains comprised of CADs (K1–K3; IPR011628) and DUF2436 domains (D1-D2; IPR018832). There is a conserved C-terminal domain (C; IPR026444) of ~80 amino acids. HagA lacks a proteolytic domain and cognate propeptide. Interspersed between and flanking the immunoglobulin fold and modular adhesins are adhesin binding motifs (A1–A2).
Interestingly, the variants of the P. gingivalis Kgp CADs are not linked to the variants of Kgpcat (Table 4), which divides the Kgps into five discernible groups. These Kgp groupings are incongruent with the clades identified by genomic SNP-based analysis. Kgp phylogenetic analysis indicated a high level of recombination (Figure S4).
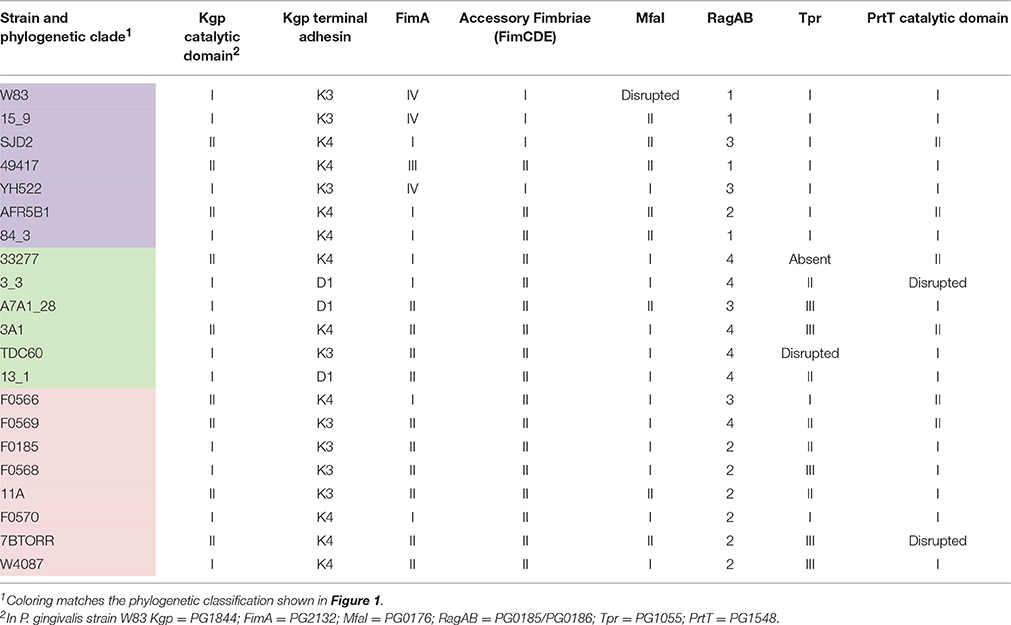
Table 4. Distribution of variable surface proteins across P. gingivalis strains.
PG0411, in a similar manner to HagA, is composed of adhesin domains and has an N-terminal Sec-dependent leader sequence and a type IX secretion system CTD signal sequence indicating it is exported to the cell surface and attached in the same manner as the other adhesin domain-containing proteins. Intriguingly in strain W83 the PG0410 gene is immediately upstream of PG0411 and encodes a gingipain proteinase that contains no adhesin domains but does have a type IX secretion system CTD sequence.
Fimbriae
Four distinct types of fimA were detected across the 21 strains in this study, corresponding to types I, II, III, and IV although only one strain, ATCC 49417, contained a type III fimA gene and no type V genes were detected (Figure S5; Table 4). A network analysis showed the very high level of divergence between fimA types and obvious signs of horizontal gene transfer (Figure S6).
In addition to the highly divergent fimA types, a DNA alignment of the fimA region for all 21 strains revealed that the genes encoding the fimbrial accessory proteins (fimCDE) have two distinct types (Figure S7). This divergent region, encompassing the last 370 bp of the 3′ end of the fimC gene as well as entire fimD and fimE genes, is likely to result from insertion of variant DNA at a second recombination site within the fimA gene cluster. The two distinct groups present (fimCDE type I and II) have < 43% aa identity for FimD and FimE, compared to the >96% nt identity across all strains for the 5′ region of the fimC gene. FimCDE type I was associated with fimA type IV however this linkage can be disrupted as observed in strain SJD2, a recent clinical isolate from China, which has fimCDE type I genes paired with fimA type I.
Both known alleles of the minor fimbriae fibrillin gene mfaI (Nagano et al., 2015) were found in the sequenced P. gingivalis strains with type II occurring in eight of the strains (Table 4). It is worth noting that both of the commonly used laboratory strains W83 and the ATCC 32277 type strain appear to be atypical for fimbriae production. The mfaI gene of W83 has been disrupted by an insertion element, which was detected in both W83 and W50 genomes. In addition, as previously published, FimA expression is greatly decreased in strain W50 because of a mutation in the ATP-binding site of the FimS histidine sensor kinase that regulates this operon. This mutation results in premature termination of the FimS protein and is present in both W83 and W50 but not in any other strain. ATCC 33277 has a non-sense mutation in fimB that results in unusually long major fimbriae (FimA) and an increased level of detachment and secretion of these long fimbriae (Nagano et al., 2013). This point mutation was not found in any of the other sequenced strains, including the 381 strain that was thought to be the provenance of the ATCC 33277 type strain (Loos et al., 1990).
Other Variable Surface Proteins
All four known variants of the TonB-linked integral outer membrane protein RagA and associated lipoprotein RagB were detected in this study. RagAB Type 2 and type 4 were the most prevalent, being found in seven and six strains, respectively (Table 4).
At least two P. gingivalis streptopain homologs have been reported, PrtT (Kuramitsu, 1998) and periodontain (PG1427; Nelson, 1999). PrtT (PG1548) is an 841 amino acid long CTD family protein with a predicted C10 cysteine peptidase catalytic domain of 195 amino acids that is preceded by a predicted propeptide of 185 amino acids in length. Two distinct catalytic domain alleles of PrtT were detected in the current study that differ by 11 specific amino acid substitutions in an 18 amino acid region of the catalytic domain. Type I was the most prevalent allele being found in 13 strains, whilst two strains had no functional alleles (Table 4; Figure S8).
Three distinct variants of the gene encoding the 503 aa outer-membrane bound Thiol Protease (Tpr; PG1055) were found in 20 of the 21 P. gingivalis strains although the gene was disrupted in TDC60 (Table 4; Figure S9). The gene was not found in strain 33277.
Discussion
P. gingivalis is a normal member of the oral microbiota and is widely considered to be a keystone pathogen in chronic periodontitis (Hajishengallis et al., 2012). Numerous attempts have been made to classify the bacterium into strains that are associated with disease (Griffen et al., 1999; Amano et al., 2004; Yoshino et al., 2007). The genomes of all 21 P. gingivalis strains examined in this study showed that global strains are closely related, with ANI values between strains of above 98%. The strains could be separated into three broad, shallow clades, and distribution of the strains between the clades was unrelated to the time and country of isolation (Figure 1).
P. gingivalis had a pangenome of 3740 gene clusters. Core genes are often defined as those that are present in all strains of a species. However, genes may not be detected in some strains due to errors in sequencing, assembly, gene calling, or through mutations arising from multiple passaging within the laboratory. Rather than defining core and variable/flexible genes, Cordero and Polz (2014) suggested categorizing gene occurrence as high, medium or low frequency, as these divisions are more accurate and can be linked to different evolutionary processes. Core metabolic and housekeeping functions are typically encoded by high frequency genes that are maintained through both vertical inheritance and horizontal recombination. The 1488 high-frequency genes detected in 17 or more P. gingivalis strains comprise 83% of the average genome size of 1803 predicted CDSs. This is comparable to the 1476 genes recently estimated by comparative genomic hybridization microarray analysis of seven P. gingivalis strains being representative of the seven capsular serotypes (Brunner et al., 2010). Generally the number of genes in the core genome slowly decreases as more genomes are added (Collins and Higgs, 2012). Low frequency genes are gained and lost at high rates from individual genomes and are often found as singletons. Negative frequency-dependent selection arising from interactions with the immune system or predation by bacteriophage can act to drive and stabilize diversity within a population and many low-frequency genes are thought to be under this type of selection (Cordero and Polz, 2014). For example, the PG0410 gingipain gene was detected in only two P. gingivalis strains (W83 and F0185), however an ortholog of this gene with 99.5% nucleotide identity was found in three of the 12 P. gulae strains in a syntenous location, suggesting the gene may be acquired through homologous recombination. The maintenance of this gene at low frequency in both species is indicative that negative frequency-dependent selection is occurring.
The surface of P. gingivalis is heavily decorated with ~32 proteins that are exported and attached by the Type IX secretion system (Sato et al., 2010; Veith et al., 2013) as well as a range of other proteins including fimbrial proteins, lipoproteins, and integral membrane proteins. A subset of these proteins comprises the major virulence factors of the bacterium. Antigenic variation is a strategy commonly employed by pathogens to avoid the human immune system. The ability to quickly alter antigens provides an obvious advantage to the pathogen.
Three gingipains, RgpA, Kgp, and RgpB, comprise the major P. gingivalis cell-surface associated proteinases. They are capable of degrading a variety of host proteins and dysregulating host defenses and are critical for bacterial colonization and the establishment of dysbiosis and disease (O'Brien-Simpson et al., 2001, 2005, 2009; Hajishengallis et al., 2012). The catalytic domains of the RgpA, Kgp, and RgpB gingipains that belong to the distinct cysteine proteinase clan C25 (Barrett and Rawlings, 2001), are found in all strains of P. gingivalis examined to date (Bhogal et al., 1997; Slakeski et al., 1998; Curtis et al., 1999a; Potempa and Nguyen, 2007). The rgpA, kgp, and rgpB gene products all contain a signal peptide of ~22 amino acids in length, an unusually long propeptide of over 200 amino acids in length, and a catalytic domain of ~480 amino acids. The RgpA and RgpB catalytic domains were nearly identical to each other and there was little variation across all P. gingivalis strains, whereas two distinct variants of the Kgp catalytic domain (KgpcatI and KgpcatII) were observed (Figure 3). These variants were not associated with phylogenetic clustering of the strains. KgpcatI was present in 12 P. gingivalis strains including W50/W83 and TDC60 and KgpcatII was present in nine strains including ATCC 33277 and ATCC 49417 (Table 4). The cell associated enzymatic activity of the KgpcatII variant appeared to be lower than that of the KgpcatI variant. Examination of the known crystal structure of the P. gingivalis W50 Kgpcat domain, that consists of a central ten stranded β-sheet surrounded by ten α-helices forming an α-β sandwich (Gorman et al., 2015), revealed that five of these variable residues have at least one atom within 10 Angstrom of the catalytic site which may directly affect catalytic activity (Figure 6A). In addition, 11 of these residues (Tyr389Ser, Ser390Tyr, Ser393Pro, Gln394Lys, Val395Ile, Pro398Gln, Gly403Arg, Met404Val, Glu411Asp, Asn419Ser, and Leu421Pro) form a contiguous, surface exposed patch adjacent to the catalytic site that could alter protein:protein interactions with either another P. gingivalis surface protein (adhesin) or an exogenous protein (substrate; Figure 6A).
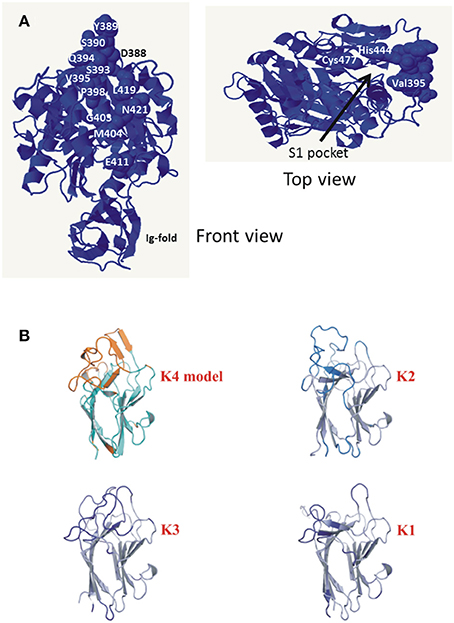
Figure 6. (A) Amino acid residues of KgpcatI that vary in KgpcatII. Front view shows the variable residues clustered on one face of the gingipain catalytic domain. The top view is a rotation of the front view ~90° to the right and ~90° down and shows the catalytic Cys477 and His444 relative to variable residue Val395. Structure rendered is 4TKX (Gorman et al., 2015). Modeling of the specific amino acid residue changes in KgpcatII relative to KgpcatI based on the known structure of P. gingivalis W50 KgpcatI showing the surface-exposed nature of the residues (Gorman et al., 2015). (B) Comparison of the modeled K4 domain against the known structures of the cleaved adhesin domains of P. gingivalis Kgp.
Closer examination of the catalytic domains for evolutionary selection detected negative selection acting on a number of amino acids in these regions. RgpA and RgpB had the greatest number of codons suggested to be under negative selection (8 and 10, respectively) while both Kgp variants had only three codons under negative selection. Interestingly, none of the amino acids involved in the surface exposed patch described above was demonstrated to be subject to positive selection as might be expected for such a region. The combination of a conservative analytic approach and the limited amount of nucleotide sequence diversity likely explains the low number of codons demonstrating signals for selection. Detection of only negative selection could be attributed to the fact that only the catalytic domain, which is likely to be under significant structural constraints, was examined.
The genes rgpA and kgp also encode four repetitive haemagglutinin-adhesin domains known as CADs that are located in the polypeptide region C-terminal to the catalytic domains (Figures 3, 5; O'Brien-Simpson et al., 2003; Li et al., 2010). Only the C-terminal CAD showed inter-strain variability with one of three variants present (Figure 3B). The crystal structures of three of the P. gingivalis W50/W83 Kgp CADs (K1, K2, K3) have recently been reported. The three CAD domains all have a conserved β-sheet core with structural differences limited to a series of solvent exposed loops (Li et al., 2010, 2011). The known crystal structure of P. gingivalis K3 (Li et al., 2011) was used to model the variant that we refer to as K4 and determine the location of the variable residues between K3 and K4. The differences between the K3 and K4 domains were restricted to the exposed surface loops—these are predicted to be structural differences as the sequences have inserts relative to each other (Figure 6B). Our modeling is consistent with these variable regions being the areas that interact with other molecules. The differences in exposed surface loops between CADs are proposed to affect the interaction of these CADs with other proteins and may potentially alter the host immune response to the bacterium or specificity of binding to host proteins.
RgpA, Kgp, and HagA also contain DUF2436 (conserved Pfam Domain of Unknown Function) domains that are similar in length (~160 aa) to the CAD domains and are similarly thought to be adhesins. These CAD and DUF2436 domains were found in five P. gingivalis proteins (Figure 5) and nine distinct types of CAD domains (K1–K9) and three types of DUF domains (D1–D3), based on amino acid sequence, were encoded in the P. gingivalis pangenome (Figure S10). Interspersed between and flanking CAD and DUF domains are adhesin binding motifs (ABM) ABM1 and ABM2 that may function in binding to host proteins and in assembly of the processed proteinase and adhesin domains into complexes at the cell surface (Slakeski et al., 1998; O'Brien-Simpson et al., 2003, 2005). Homologs of RgpA, RgpB, and Kgp are encoded in the P. gulae genome although there were significant differences with the P. gingivalis sequence (Lenzo et al., 2016; Figure 3).
The P. gingivalis major fimbrillin protein FimA, polymerises to produce long structures of up to 6 μm that protrude from the cell (61). FimA has been classified into five types (I–V) based on identified N-terminal amino acid sequences, nucleotide sequences and serological specificity (Lee et al., 1991; Fujiwara et al., 1993; Nakagawa et al., 2000). The fimA gene occurs within a seven gene cluster of fimX, pgmA, fimA-fimE (Frandsen et al., 2001). FimB modulates fimbrial length and FimC-E are accessory fimbrial proteins (Yoshimura, 1993; Watanabe et al., 1996; Nishiyama et al., 2007; Nagano et al., 2010). Based on a PCR typing system, type II has often been associated with disease and with increased virulence in laboratory studies (Amano et al., 1999; Fabrizi et al., 2013; Feng et al., 2014). However, studies using these PCR based typing systems have also shown that type II is the most widely distributed type among both healthy subjects as well as those with periodontitis (Moon et al., 2013). Four highly divergent types of fimA, corresponding to the known types I–IV were detected across the 21 strains in this study (Figure S4; Table 4). Two distinct forms of the FimC-E fimbrial accessory proteins that are thought to be localized to the tips of the fimbrial filaments and involved in host adhesion were also identified. Mutant strains devoid of accessory proteins have been shown to be less virulent in a mouse periodontitis model (Pierce et al., 2009). Kerr et al. (2014) demonstrated fimA transfer between P. gingivalis strains and also found transfer of the fimCDE region in some cases. The two known alleles of the minor fimbriae fimbrillin gene mfaI (Nagano et al., 2015) were found in the sequenced P. gingivalis strains. The Mfa minor fimbriae are important in interactions and biofilm formation with other oral bacteria (Park et al., 2005).
Several other surface proteins with variable alleles were detected that may also play a role in antigenic variation through gene conversion. RagAB are a TonB-linked integral outer membrane protein and associated lipoprotein that are reported to be involved in solute transport across the P. gingivalis outer membrane (Hanley et al., 1999; Goulas et al., 2016). They have been shown to form oligomeric complexes (Glew et al., 2014) and were originally described due to their immunogenicity in periodontal patients (Curtis et al., 1991, 1999b). Four distinct sequence types of RagAB have been described and linked to the virulence of the bacterium (Hall et al., 2005). These four sequence types were confirmed in this study with type 2 and type 4 being the most prevalent. Three distinct variants of the gene encoding the outer-membrane bound thiol protease Tpr were found in this study. Tpr is capable of degrading both complement proteins and the LL-37 antimicrobial peptide and may play a role in disruption of the immune system (Bourgeau et al., 1992; Staniec et al., 2015). Two distinct catalytic domain variants of the P. gingivalis streptopain homolog PrtT were also detected in this study.
In contrast to the variable surface virulence factors described above, Hbp35 (PG0616), LptO (PG0027), CPG70 (PG0232), integral outer membrane protein PG1823, peptidyl arginine deiminase (PPAD, PG1424) and P59 (PG2102) that are amongst the most abundant P. gingivalis surface proteins showed little variation between strains.
A comparison of the occurrence of all the variable surface proteins in the P. gingivalis strains formed a mosaic that was not related to the SNP based phylogeny (Table 4). In conjunction with the other surface proteins discussed above and assuming no genetic linkage of genes, the variability in these eight protein domains alone is enough to generate nearly 7000 different combinations. None of the 21 P. gingivalis strains in this study had the exact same combination of variants of these proteins. These variable surface proteins all had multiple variants present within P. gingivalis suggesting that the alleles encoding variants may be under negative frequency-dependent selection resulting in high rates of gene turnover, thus maintaining the diversity of these genes within the population. P. gingivalis is naturally competent, is not reported to contain plasmids, and is capable of gene conversion through homologous recombination, as well as possessing a suite of other mobile elements including a conjugative transposon system and phage that may aid in horizontal gene transfer.
The co-location of a number of P. gingivalis strains in the oral cavity provides a larger gene pool for P. gingivalis to draw on, and allelic exchange and recombination enables the bacterium to produce a wide range of phenotypes some of which may be more suited to the prevailing environmental conditions. This may permit P. gingivalis to increase in abundance by avoiding the host response or more efficiently obtaining nutrients. Rather than the presence of disease-associated strains or types, it may be the ability to achieve antigenic and/or functional variation through gene conversion that allows P. gingivalis to dysregulate host defenses and tip the homeostatic balance that exists within the host to trigger dysbiosis.
In conclusion, P. gingivalis uses specific domain rearrangements and allelic exchange through horizontal gene transfer to generate diversity in specific surface virulence factors. This diversity of specific genes and recombination means that typing schemes only provide limited useful information unless the complete diversity of P. gingivalis genes within an individual oral cavity or indeed periodontal pocket is captured. It is likely that the specific combination of genes in the genome of the dominant P. gingivalis strain may vary with the disease state and host response. Obtaining P. gingivalis genome sequences from strains isolated from both healthy and diseased individuals/sites may elucidate the full variety and number of strains that exist within the oral cavity.
Author Contributions
Conceived and designed the experiments: SD, HM, CS, TS, DB, and ER. Performed the experiments: HM, CS, SG, DB, PC, KC, and SC. Analyzed the data: SD, HM, CS, SG, TS, DB, PC, KC, and ER. Contributed to reagents/materials/analysis tools: SG, TS, DB, PC, KC, and ER. Wrote the paper: SD, HM, and CS. Collected the samples: HM, CS, and SC. Critically revised the manuscript: SD, HM, CS, SG, TS, DB, PC, KC, SC, and ER.
Funding
This work was supported by the Australian Government, Department of Industry, Innovation and Science and the Australian National Health and Medical Research Council Project 1081252.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.00048/full#supplementary-material
References
Ali, R. W., Bakken, V., Nilsen, R., and Skaug, N. (1994a). Comparative detection frequency of 6 putative periodontal pathogens in Sudanese and Norwegian adult periodontitis patients. J. Periodontol. 65, 1046–1052. doi: 10.1902/jop.1994.65.11.1046
Ali, R. W., Johannessen, A. C., Dahlén, G., Socransky, S. S., and Skaug, N. (1997). Comparison of the subgingival microbiota of periodontally healthy and diseased adults in northern Cameroon. J. Clin. Periodontol. 24, 830–835. doi: 10.1111/j.1600-051X.1997.tb01197.x
Ali, R. W., Skaug, N., Nilsen, R., and Bakken, V. (1994b). Microbial associations of 4 putative periodontal pathogens in Sudanese adult periodontitis patients determined by DNA probe analysis. J. Periodontol. 65, 1053–1057. doi: 10.1902/jop.1994.65.11.1053
Ali, R. W., Velcescu, C., Jivanescu, M. C., Lofthus, B., and Skaug, N. (1996). Prevalence of 6 putative periodontal pathogens in subgingival plaque samples from Romanian adult periodontitis patients. J. Clin. Periodontol. 23, 133–139. doi: 10.1111/j.1600-051X.1996.tb00546.x
Alikhan, N. F., Petty, N. K., Ben Zakour, N. L., and Beatson, S. A. (2011). BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12:402. doi: 10.1186/1471-2164-12-402
Amano, A., Nakagawa, I., Kataoka, K., Morisaki, I., and Hamada, S. (1999). Distribution of Porphyromonas gingivalis strains with fimA genotypes in periodontitis patients. J. Clin. Microbiol. 37, 1426–1430.
Amano, A., Nakagawa, I., Okahashi, N., and Hamada, N. (2004). Variations of Porphyromonas gingivalis fimbriae in relation to microbial pathogenesis. J. Periodont. Res. 39, 136–142. doi: 10.1111/j.1600-0765.2004.00719.x
Barrett, A. J., and Rawlings, N. D. (2001). Evolutionary lines of cysteine peptidases. Biol. Chem. 382, 727–733. doi: 10.1515/bchm.2001.382.5.727
Bhogal, P. S., Slakeski, N., and Reynolds, E. C. (1997). A cell-associated protein complex of Porphyromonas gingivalis W50 composed of Arg- and Lys-specific cysteine proteinases and adhesins. Microbiology143, 2485–2495. doi: 10.1099/00221287-143-7-2485
Bostanci, N., and Belibasakis, G. N. (2012). Porphyromonas gingivalis: an invasive and evasive opportunistic oral pathogen. FEMS Microbiol. Lett. 333, 1–9. doi: 10.1111/j.1574-6968.2012.02579.x
Bourgeau, G., Lapointe, H., Peloquin, P., and Mayrand, D. (1992). Cloning, expression, and sequencing of a protease gene (tpr) from Porphyromonas gingivalis W83 in Escherichia coli. Infect. Immun. 60, 3186–3192.
Brunner, J., Wittink, F. R., Jonker, M. J., De Jong, M., Breit, T. M., Laine, M. L., et al. (2010). The core genome of the anaerobic oral pathogenic bacterium Porphyromonas gingivalis. BMC Microbiol. 10:252. doi: 10.1186/1471-2180-10-252
Coil, D. A., Alexiev, A., Wallis, C., O'flynn, C., Deusch, O., Davis, I., et al. (2015). Draft genome sequences of 26 Porphyromonas strains isolated from the canine oral microbiome. Genome Announc. 3:e00187-15. doi: 10.1128/genomeA.00187-15
Collins, R. E., and Higgs, P. G. (2012). Testing the infinitely many genes model for the evolution of the bacterial core genome and pangenome. Mol. Biol. Evol. 29, 3413–3425. doi: 10.1093/molbev/mss163
Cordero, O. X., and Polz, M. F. (2014). Explaining microbial genomic diversity in light of evolutionary ecology. Nat. Rev. Microbiol. 12, 263–273. doi: 10.1038/nrmicro3218
Curtis, M. A., Hanley, S. A., and Aduse-Opoku, J. (1999b). The rag locus of Porphyromonas gingivalis: a novel pathogenicity island. J. Periodont. Res. 34, 400–405. doi: 10.1111/j.1600-0765.1999.tb02273.x
Curtis, M. A., Slaney, J. M., Carman, R. J., and Johnson, N. W. (1991). Identification of the major surface protein antigens of Porphyromonas gingivalis using IgG antibody reactivity of periodontal case-control serum. Oral Microbiol. Immunol. 6, 321–326. doi: 10.1111/j.1399-302X.1991.tb00502.x
Curtis, M. A., Kuramitsu, H. K., Lantz, M., Macrina, F.L., Nakayama, K., Potempa, J., et al. (1999a). Molecular genetics and nomenclature of proteases of Porphyromonas gingivalis. J. Periodont. Res. 34, 464–472. doi: 10.1111/j.1600-0765.1999.tb02282.x
de Diego, I., Veillard, F., Sztukowska, M. N., Guevara, T., Potempa, B., Pomowski, A., et al. (2014). Structure and mechanism of cysteine peptidase gingipain K (Kgp), a major virulence factor of Porphyromonas gingivalis in periodontitis. J. Biol. Chem. 289, 32291–32302. doi: 10.1074/jbc.M114.602052
Dhillon, B. K., Laird, M. R., Shay, J. A., Winsor, G. L., Lo, R., Nizam, F., et al. (2015). IslandViewer 3: more flexible, interactive genomic island discovery, visualization and analysis. Nucleic Acids Res. 43, W104–W108. doi: 10.1093/nar/gkv401
Eichinger, A., Beisel, H. G., Jacob, U., Huber, R., Medrano, F. J., Banbula, A., et al. (1999). Crystal structure of gingipain R: an Arg-specific bacterial cysteine proteinase with a caspase-like fold. EMBO J. 18, 5453–5462. doi: 10.1093/emboj/18.20.5453
Eke, P. I., Dye, B. A., Wei, L., Thornton-Evans, G. O., and Genco, R. J. (2012). Prevalence of periodontitis in adults in the United States: 2009 and 2010. J. Dent. Res. 91, 914–920. doi: 10.1177/0022034512457373
Enersen, M. (2011). Porphyromonas gingivalis: a clonal pathogen? Diversities in housekeeping genes and the major fimbriae gene. J. Oral Microbiol. 3:8487. doi: 10.3402/jom.v3i0.8487
Enersen, M., Olsen, I., and Caugant, D. A. (2008). Genetic diversity of Porphyromonas gingivalis isolates recovered from single “refractory” periodontitis sites. Appl. Environ. Microbiol. 74, 5817–5821. doi: 10.1128/AEM.00225-08
Fabrizi, S., León, R., Blanc, V., Herrera, D., and Sanz, M. (2013). Variability of the fimA gene in Porphyromonas gingivalis isolated from periodontitis and non-periodontitis patients. Med. Oral Patol. Oral Cir. Bucal 18, e100–e105. doi: 10.4317/medoral.18042
Feng, X., Zhang, L., Xu, L., Meng, H., Lu, R., Chen, Z., et al. (2014). Detection of eight periodontal microorganisms and distribution of Porphyromonas gingivalis fimA genotypes in Chinese patients with aggressive periodontitis. J. Periodontol. 85, 150–159. doi: 10.1902/jop.2013.120677
Fournier, D., Mouton, C., Lapierre, P., Kato, T., Okuda, K., and Ménard, C. (2001). Porphyromonas gulae sp. nov., an anaerobic, gram-negative coccobacillus from the gingival sulcus of various animal hosts. Int. J. Syst. Evol. Microbiol. 51, 1179–1189. doi: 10.1099/00207713-51-3-1179
Frandsen, E. V., Poulsen, K., Curtis, M. A., and Kilian, M. (2001). Evidence of recombination in Porphyromonas gingivalis and random distribution of putative virulence markers. Infect. Immun. 69, 4479–4485. doi: 10.1128/IAI.69.7.4479-4485.2001
Frazer, L. T., O'Brien-Simpson, N. M., Slakeski, N., Walsh, K. A., Veith, P. D., Chen, C. G., et al. (2006). Vaccination with recombinant adhesins from the RgpA-Kgp proteinase-adhesin complex protects against Porphyromonas gingivalis infection. Vaccine 24, 6542–6554. doi: 10.1016/j.vaccine.2006.06.013
Fujiwara, T., Morishima, S., Takahashi, I., and Hamada, S. (1993). Molecular cloning and sequencing of the fimbrilin gene of Porphyromonas gingivalis strains and characterization of recombinant proteins. Biochem. Biophys. Res. Commun. 197, 241–247. doi: 10.1006/bbrc.1993.2467
Genco, R. J., and Van Dyke, T. E. (2010). Prevention: reducing the risk of CVD in patients with periodontitis. Nat. Rev. Cardiol. 7, 479–480. doi: 10.1038/nrcardio.2010.120
Glew, M. D., Veith, P. D., Chen, D., Seers, C. A., Chen, Y. Y., and Reynolds, E. C. (2014). Blue native-PAGE analysis of membrane protein complexes in Porphyromonas gingivalis. J. Proteomics 110, 72–92. doi: 10.1016/j.jprot.2014.07.033
Gorman, M. A., Seers, C. A., Michell, B. J., Feil, S. C., Huq, N. L., Cross, K. J., et al. (2015). Structure of the lysine specific protease Kgp from Porphyromonas gingivalis, a target for improved oral health. Protein Sci. 24, 162–166. doi: 10.1002/pro.2589
Goulas, T., Garcia-Ferrer, I., Hutcherson, J. A., Potempa, B. A., Potempa, J., Scott, D. A., et al. (2016). Structure of RagB, a major immunodominant outer-membrane surface receptor antigen of Porphyromonas gingivalis. Mol. Oral Microbiol. 31, 472–485. doi: 10.1111/omi.12140
Griffen, A. L., Lyons, S. R., Becker, M. R., Moeschberger, M. L., and Leys, E. J. (1999). Porphyromonas gingivalis strain variability and periodontitis. J. Clin. Microbiol. 37, 4028–4033.
Hajishengallis, G., Darveau, R. P., and Curtis, M. A. (2012). The keystone-pathogen hypothesis. Nat. Rev. Microbiol. 10, 717–725. doi: 10.1038/nrmicro2873
Hall, L. M., Fawell, S. C., Shi, X., Faray-Kele, M. C., Aduse-Opoku, J., Whiley, R. A., et al. (2005). Sequence diversity and antigenic variation at the rag locus of Porphyromonas gingivalis. Infect. Immun. 73, 4253–4262. doi: 10.1128/IAI.73.7.4253-4262.2005
Hanley, S. A., Aduse-Opoku, J., and Curtis, M. A. (1999). A 55-kilodalton immunodominant antigen of Porphyromonas gingivalis W50 has arisen via horizontal gene transfer. Infect. Immun. 67, 1157–1171.
Holland, B. R., Huber, K. T., Dress, A., and Moulton, V. (2002). Delta plots: a tool for analyzing phylogenetic distance data. Mol. Biol. Evol. 19, 2051–2059. doi: 10.1093/oxfordjournals.molbev.a004030
Huson, D. H., and Bryant, D. (2006). Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23, 254–267. doi: 10.1093/molbev/msj030
Igboin, C. O., Griffen, A. L., and Leys, E. J. (2009). Porphyromonas gingivalis strain diversity. J. Clin. Microbiol. 47, 3073–3081. doi: 10.1128/JCM.00569-09
Jolley, K. A., and Maiden, M. C. J. (2010). BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics 11:595. doi: 10.1186/1471-2105-11-595
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kerr, J. E., Abramian, J. R., Dao, D. H., Rigney, T. W., Fritz, J., Pham, T., et al. (2014). Genetic exchange of fimbrial alleles exemplifies the adaptive virulence strategy of Porphyromonas gingivalis. PLoS ONE 9:e91696. doi: 10.1371/journal.pone.0091696
Koehler, A., Karch, H., Beikler, T., Flemmig, T. F., Suerbaum, S., and Schmidt, H. (2003). Multilocus sequence analysis of Porphyromonas gingivalis indicates frequent recombination. Microbiology 149, 2407–2415. doi: 10.1099/mic.0.26267-0
Kosakovsky Pond, S. L., and Frost, S. D. (2005). Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 22, 1208–1222. doi: 10.1093/molbev/msi105
Kosakovsky Pond, S. L., Posada, D., Gravenor, M. B., Woelk, C. H., and Frost, S. D. (2006). Automated phylogenetic detection of recombination using a genetic algorithm. Mol. Biol. Evol. 23, 1891–1901. doi: 10.1093/molbev/msl051
Kozarov, E., Whitlock, J., Dong, H., Carrasco, E., and Progulske-Fox, A. (1998). The number of direct repeats in hagA is variable among Porphyromonas gingivalis strains. Infect. Immun. 66, 4721–4725.
Kuramitsu, H. K. (1998). Proteases of Porphyromonas gingivalis: what don't they do? Oral Microbiol. Immunol. 13, 263–270. doi: 10.1111/j.1399-302X.1998.tb00706.x
Lee, I., Kim, Y. O., Park, S.-C., and Chun, J. (2016). OrthoANI: an improved algorithm and software for calculating average nucleotide identity. Int. J. Syst. Evol. Microbiol. 66, 1100–1103. doi: 10.1099/ijsem.0.000760
Lee, J. Y., Sojar, H. T., Bedi, G. S., and Genco, R. J. (1991). Porphyromonas (Bacteroides) gingivalis fimbrillin: size, amino-terminal sequence, and antigenic heterogeneity. Infect. Immun. 59, 383–389.
Lenzo, J. C., O'Brien-Simpson, N. M., Orth, R. K., Mitchell, H. L., Dashper, S. G., and Reynolds, E. C. (2016). Porphyromonas gulae has virulence and immunological characteristics similar to those of the human periodontal pathogen Porphyromonas gingivalis. Infect. Immun. 84, 2575–2585. doi: 10.1128/IAI.01500-15
Li, N., Yun, P., Jeffries, C. M., Langley, D., Gamsjaeger, R., Church, W. B., et al. (2011). The modular structure of haemagglutinin/adhesin regions in gingipains of Porphyromonas gingivalis. Mol. Microbiol. 81, 1358–1373. doi: 10.1111/j.1365-2958.2011.07768.x
Li, N., Yun, P., Nadkarni, M. A., Ghadikolaee, N. B., Nguyen, K.-A., Lee, M., et al. (2010). Structure determination and analysis of a haemolytic gingipain adhesin domain from Porphyromonas gingivalis. Mol. Microbiol. 76, 861–873. doi: 10.1111/j.1365-2958.2010.07123.x
Librado, P., and Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. doi: 10.1093/bioinformatics/btp187
Linden, G. J., Lyons, A., and Scannapieco, F. A. (2013). Periodontal systemic associations: review of the evidence. J. Clin. Periodontol. 40, 8–19. doi: 10.1111/jcpe.12064
Liu, D., Zhou, Y., Naito, M., Yumoto, H., Li, Q., Miyake, Y., et al. (2014). Draft genome sequence of Porphyromonas gingivalis strain SJD2, isolated from the periodontal pocket of a patient with periodontitis in China. Genome Announc. 2:e01091-13. doi: 10.1128/genomeA.01091-13
Loos, B. G., Mayrand, D., Genco, R. J., and Dickinson, D. P. (1990). Genetic heterogeneity of Porphyromonas (Bacteroides) gingivalis by genomic DNA fingerprinting. J. Dent. Res. 69, 1488–1493. doi: 10.1177/00220345900690080801
Lundberg, K., Wegner, N., Yucel-Lindberg, T., and Venables, P. J. (2010). Periodontitis in RA-the citrullinated enolase connection. Nat. Rev. Rheumatol. 6, 727–730. doi: 10.1038/nrrheum.2010.139
Madianos, P. N., Bobetsis, Y. A., and Offenbacher, S. (2013). Adverse pregnancy outcomes (APOs) and periodontal disease: pathogenic mechanisms. J. Periodontol. 84, S170–S180. doi: 10.1902/jop.2013.1340015
Moon, J. H., Herr, Y., Lee, H. W., Shin, S. I., Kim, C., Amano, A., et al. (2013). Genotype analysis of Porphyromonas gingivalis fimA in Korean adults using new primers. J. Med. Microbiol. 62, 1290–1294. doi: 10.1099/jmm.0.054247-0
Murrell, B., Moola, S., Mabona, A., Weighill, T., Sheward, D., Kosakovsky Pond, S. L., et al. (2013). FUBAR: a fast, unconstrained bayesian approximation for inferring selection. Mol. Biol. Evol. 30, 1196–1205. doi: 10.1093/molbev/mst030
Nagano, K., Abiko, Y., Yoshida, Y., and Yoshimura, F. (2013). Genetic and antigenic analyses of Porphyromonas gingivalis FimA fimbriae. Mol. Oral Microbiol. 28, 392–403. doi: 10.1111/omi.12032
Nagano, K., Hasegawa, Y., Murakami, Y., Nishiyama, S., and Yoshimura, F. (2010). FimB regulates FimA fimbriation in Porphyromonas gingivalis. J. Dent. Res. 89, 903–908. doi: 10.1177/0022034510370089
Nagano, K., Hasegawa, Y., Yoshida, Y., and Yoshimura, F. (2015). A major fimbrilin variant of Mfa1 fimbriae in Porphyromonas gingivalis. J. Dent. Res. 94, 1143–1148. doi: 10.1177/0022034515588275
Naito, M., Hirakawa, H., Yamashita, A., Ohara, N., Shoji, M., Yukitake, H., et al. (2008). Determination of the genome sequence of Porphyromonas gingivalis strain ATCC 33277 and genomic comparison with strain W83 revealed extensive genome rearrangements in P. gingivalis. DNA Res. 15, 215–225. doi: 10.1093/dnares/dsn013
Nakagawa, I., Amano, A., Kimura, R. K., Nakamura, T., Kawabata, S., and Hamada, S. (2000). Distribution and molecular characterization of Porphyromonas gingivalis carrying a new type of fimA gene. J. Clin. Microbiol. 38, 1909–1914.
Nelson, D. (1999). Purification and characterization of a novel cysteine proteinase (periodontain) from Porphyromonas gingivalis. Evidence for a role in the inactivation of human α 1-proteinase inhibitor. J. Biol. Chem. 274, 12245–12251. doi: 10.1074/jbc.274.18.12245
Nelson, K. E., Fleischmann, R. D., Deboy, R. T., Paulsen, I. T., Fouts, D. E., Eisen, J. A., et al. (2003). Complete genome sequence of the oral pathogenic bacterium Porphyromonas gingivalis Strain W83. J. Bacteriol. 185, 5591–5601. doi: 10.1128/JB.185.18.5591-5601.2003
Nishiyama, S., Murakami, Y., Nagata, H., Shizukuishi, S., Kawagishi, I., and Yoshimura, F. (2007). Involvement of minor components associated with the FimA fimbriae of Porphyromonas gingivalis in adhesive functions. Microbiology 153, 1916–1925. doi: 10.1099/mic.0.2006/005561-0
O'Brien-Simpson, N. M., Paolini, R. A., Hoffmann, B., Slakeski, N., Dashper, S. G., and Reynolds, E. C. (2001). Role of RgpA, RgpB, and Kgp proteinases in virulence of Porphyromonas gingivalis W50 in a murine lesion model. Infect. Immun. 69, 7527–7534. doi: 10.1128/IAI.69.12.7527-7534.2001
O'Brien-Simpson, N. M., Pathirana, R. D., Paolini, R. A., Chen, Y.-Y., Veith, P. D., Tam, V., et al. (2005). An immune response directed to proteinase and adhesin functional epitopes protects against Porphyromonas gingivalis-induced periodontal bone loss. J. Immunol. 175, 3980–3989. doi: 10.4049/jimmunol.175.6.3980
O'Brien-Simpson, N. M., Pathirana, R. D., Walker, G. D., and Reynolds, E. C. (2009). Porphyromonas gingivalis RgpA-Kgp proteinase-adhesin complexes penetrate gingival tissue and induce proinflammatory cytokines or apoptosis in a concentration-dependent manner. Infect. Immun. 77, 1246–1261. doi: 10.1128/IAI.01038-08
O'Brien-Simpson, N. M., Veith, P. D., Dashper, S. G., and Reynolds, E. C. (2003). Porphyromonas gingivalis gingipains: the molecular teeth of a microbial vampire. Curr. Protein Pept. Sci. 4, 409–426. doi: 10.2174/1389203033487009
Park, Y., Simionato, M. R., Sekiya, K., Murakami, Y., James, D., Chen, W., et al. (2005). Short fimbriae of Porphyromonas gingivalis and their role in coadhesion with Streptococcus gordonii. Infect. Immun. 73, 3983–3989. doi: 10.1128/IAI.73.7.3983-3989.2005
Pierce, D. L., Nishiyama, S.-I., Liang, S., Wang, M., Triantafilou, M., Triantafilou, K., et al. (2009). Host adhesive activities and virulence of novel fimbrial proteins of Porphyromonas gingivalis. Infect. Immun. 77, 3294–3301. doi: 10.1128/IAI.00262-09
Pond, S. L., and Frost, S. D. (2005). Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 21, 2531–2533. doi: 10.1093/bioinformatics/bti320
Pond, S. L., Frost, S. D., and Muse, S. V. (2005). HyPhy: hypothesis testing using phylogenies. Bioinformatics 21, 676–679. doi: 10.1093/bioinformatics/bti079
Pond, S. L., Frost, S. D., Grossman, Z., Gravenor, M. B., Richman, D. D., and Brown, A. J. (2006). Adaptation to different human populations by HIV-1 revealed by codon-based analyses. PLoS Comput. Biol. 2:e62. doi: 10.1371/journal.pcbi.0020062
Popadiak, K., Potempa, J., Riesbeck, K., and Blom, A. M. (2007). Biphasic effect of gingipains from Porphyromonas gingivalis on the human complement system. J. Immunol. 178, 7242–7250. doi: 10.4049/jimmunol.178.11.7242
Potempa, J., and Nguyen, K. A. (2007). Purification and characterization of gingipains. Curr. Protoc. Protein Sci. Chapter 21: Unit 21.20. doi: 10.1002/0471140864.ps2120s49
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE 5:e9490. doi: 10.1371/journal.pone.0009490
Salzberg, S. L., Delcher, A. L., Kasif, S., and White, O. (1998). Microbial gene identification using interpolated Markov models. Nucleic Acids Res. 26, 544–548. doi: 10.1093/nar/26.2.544
Sato, K., Naito, M., Yukitake, H., Hirakawa, H., Shoji, M., McBride, M. J., et al. (2010). A protein secretion system linked to bacteroidete gliding motility and pathogenesis. Proc. Natl. Acad. Sci. U.S.A. 107, 276–281. doi: 10.1073/pnas.0912010107
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Shi, J., Blundell, T. L., and Mizuguchi, K. (2001). FUGUE: sequence-structure homology recognition using environment-specific substitution tables and structure-dependent gap penalties. J. Mol. Biol. 310, 243–257. doi: 10.1006/jmbi.2001.4762
Slakeski, N., Bhogal, P. S., O'brien-Simpson, N. M., and Reynolds, E. C. (1998). Characterization of a second cell-associated Arg-specific cysteine proteinase of Porphyromonas gingivalis and identification of an adhesin-binding motif involved in association of the PrtR and PrtK proteinases and adhesins into large complexes. Microbiology 144, 1583–1592. doi: 10.1099/00221287-144-6-1583
Slakeski, N., Margetts, M., Moore, C., Czajkowski, L., Barr, I. G., and Reynolds, E. C. (2002). Characterization and expression of a novel Porphyromonas gingivalis outer membrane protein, Omp28. Oral Microbiol. Immunol. 17, 150–156. doi: 10.1034/j.1399-302X.2002.170303.x
Staniec, D., Ksiazek, M., Thogersen, I. B., Enghild, J. J., Sroka, A., Bryzek, D., et al. (2015). Calcium regulates the activity and structural stability of Tpr, a bacterial calpain-like peptidase. J. Biol. Chem. 290, 27248–27260. doi: 10.1074/jbc.M115.648782
Toh, E. C., Dashper, S. G., Huq, N. L., Attard, T. J., O'brien-Simpson, N. M., Chen, Y. Y., et al. (2011). Porphyromonas gingivalis cysteine proteinase inhibition by kappa-casein peptides. Antimicrob. Agents Chemother. 55, 1155–1161. doi: 10.1128/AAC.00466-10
Tonetti, M. S., and Van Dyke, T. E. (2013). Periodontitis and atherosclerotic cardiovascular disease: consensus report of the Joint EFP/AAP workshop on periodontitis and systemic diseases. J. Clin. Periodontol. 40, 24–29. doi: 10.1111/jcpe.12089
Treangen, T. J., Ondov, B. D., Koren, S., and Phillippy, A. M. (2014). The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 15:524. doi: 10.1186/s13059-014-0524-x
Tribble, G. D., Rigney, T. W., Dao, D. H., Wong, C. T., Kerr, J. E., Taylor, B. E., et al. (2012). Natural competence is a major mechanism for horizontal DNA transfer in the oral pathogen Porphyromonas gingivalis. MBio 3:e00231-11. doi: 10.1128/mBio.00231-11
Untergasser, A., Nijveen, H., Rao, X., Bisseling, T., Geurts, R., and Leunissen, J. A. (2007). Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res. 35, W71–W74. doi: 10.1093/nar/gkm306
Veith, P. D., Nor Muhammad, N. A., Dashper, S. G., Likić, V. A., Gorasia, D. G., Chen, D., et al. (2013). Protein substrates of a novel secretion system are numerous in the Bacteroidetes phylum and have in common a cleavable C-terminal secretion signal, extensive post-translational modification, and cell-surface attachment. J. Proteome Res. 12, 4449–4461. doi: 10.1021/pr400487b
Watanabe, K., Onoe, T., Ozeki, M., Shimizu, Y., Sakayori, T., Nakamura, H., et al. (1996). Sequence and product analyses of the four genes downstream from the fimbrilin gene (fimA) of the oral anaerobe Porphyromonas gingivalis. Microbiol. Immunol. 40, 725–734. doi: 10.1111/j.1348-0421.1996.tb01133.x
Watanabe, T., Maruyama, F., Nozawa, T., Aoki, A., Okano, S., Shibata, Y., et al. (2011). Complete genome sequence of the bacterium Porphyromonas gingivalis TDC60, which causes periodontal disease. J. Bacteriol. 193, 4259–4260. doi: 10.1128/JB.05269-11
Wick, R. R., Schultz, M. B., Zobel, J., and Holt, K. E. (2015). Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 31, 3350–3352. doi: 10.1093/bioinformatics/btv383
Wiebe, C. B., and Putnins, E. E. (2000). The periodontal disease classification system of the American Academy of Periodontology-an update. J. Can. Dent. Assoc. 66, 594–597.
Wozniak, M., Wong, L., and Tiuryn, J. (2014). eCAMBer: efficient support for large-scale comparative analysis of multiple bacterial strains. BMC Bioinformatics 15:65. doi: 10.1186/1471-2105-15-65
Yilmaz, Ö., Watanabe, K., and Lamont, R. J. (2002). Involvement of integrins in fimbriae-mediated binding and invasion by Porphyromonas gingivalis. Cell. Microbiol. 4, 305–314. doi: 10.1046/j.1462-5822.2002.00192.x
Yoshimura, F. (1993). “Molecular biology of P. gingivalis fimbriae,” in Biology of the Species Porphyromonas gingivalis, eds H. N. Shah, D. Mayrand, and R. J. Genco. (Boca Raton, FL: CRC Press), 321–338.
Yoshino, T., Laine, M. L., Van Winkelhoff, A. J., and Dahlen, G. (2007). Genotype variation and capsular serotypes of Porphyromonas gingivalis from chronic periodontitis and periodontal abscesses. FEMS Microbiol. Lett. 270, 75–81. doi: 10.1111/j.1574-6968.2007.00651.x
Keywords: periodontal pathogen, Porphyromonas gingivalis, genetic diversity, specific domain rearrangement, surface virulence factors
Citation: Dashper SG, Mitchell HL, Seers CA, Gladman SL, Seemann T, Bulach DM, Chandry PS, Cross KJ, Cleal SM and Reynolds EC (2017) Porphyromonas gingivalis Uses Specific Domain Rearrangements and Allelic Exchange to Generate Diversity in Surface Virulence Factors. Front. Microbiol. 8:48. doi: 10.3389/fmicb.2017.00048
Received: 28 August 2016; Accepted: 06 January 2017;
Published: 26 January 2017.
Edited by:
John R. Battista, Louisiana State University, USAReviewed by:
Zhuofei Xu, Huazhong Agricultural University, ChinaSoo Rin Kim, Kyungpook National University, South Korea
Copyright © 2017 Dashper, Mitchell, Seers, Gladman, Seemann, Bulach, Chandry, Cross, Cleal and Reynolds. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eric C. Reynolds, ZS5yZXlub2xkc0B1bmltZWxiLmVkdS5hdQ==
†These authors have contributed equally to this work.