 Yong Wang1†
Yong Wang1† Xiang Wu
Xiang Wu- 1School of Basic Medical Sciences, Xiangya School of Medicine, Central South University, Changsha, China
- 2Department of Cardiovascular Diseases, Xiangya Hospital, Central South University, Changsha, China
- 3Department of Microbiology and Immunology, University of Texas Health Science Center at San Antonio, San Antonio, TX, USA
Chlamydiae are very important pathogens which could cause several types of diseases in human, but little is known about its pathogenic mechanism. In order to elucidate host inflammatory response and the signal pathway induced by Chlamydial lipoproteins, the predicted lipoproteins of Chlamydia trachomatis were tested for their ability to induce the release of proinflammatory cytokines by mouse macrophages or human TLR (Toll-Like Receptor) expressing cell lines. The results showed that recombinant proteins of C. trachomatis D381, D541, D067, and D775 displayed a strong ability to induce the release of IL-8 in TLR expressing cell line. The signal pathways involved TLR1/2 and TLR2/CD14 but not TLR4. Moreover, except D067, the proinflammatory cytokine induction by D381, D541, and D775 required the thioacylation site (cysteine) for lipid modification and the induction was through MyD88-mediated pathway. Our data supported that lipoproteins played a vital role in pathogenesis of C. trachomatis-induced inflammatory responses via TLR pathway. It was the first study to characterize other chlamydial lipoproteins after identifying the role of MIP (D541) on pathogenesis of Chlamydial diseases.
Introduction
Chlamydiae are obligate intracellular bacteria which can infect both human and animals, causing a wide range of diseases (Hahn et al., 1991; Centers for Disease Control and Prevention, 1997; Mertz et al., 1998; Kalayoglu et al., 2002; Corsaro and Greub, 2006). Among the species of Chlamydiae, Chlamydia trachomatis infects human ocular (serovars A–C) and urogenital/colorectal (serovars D–K and L1–L3) epithelial tissues, causing trachoma (Wright et al., 2008), and sexually transmitted diseases (Sherman et al., 1990; Peterman et al., 2006), respectively. Although, it is known that chlamydia can induce serious damage and proinflammatory immune response after they invade into human epithelia cells (Patton and Kuo, 1989; Kiviat et al., 1990), few details were known about which chlamydial components led to inflammatory diseases. It is unclear about the relationship between the inflammation caused by C. trachomatis and the innate immunity of the host yet.
As major components of the bacterial cell wall, lipoproteins played an important role in pathogenesis of many bacterial diseases. However, there was no enough evidence supporting the presence of chlamydial lipoproteins participated in the pathogenic and host response process. One of the lipoproteins characterized in C. trachomatis is macrophage infectivity potentiator (MIP), similar to the murein lipoprotein (LppA) from Escherichia coli (Bas et al., 2010). Dr. Syvette Bas's group further verified that MIP lipoprotein induced inflammatory response in the pathogenesis of C. trachomatis through TLR2/TLR1/TLR6 and CD14 (Bas et al., 2008). Besides MIP and LPS, chlamydial Hsp60 (cHsp60) was another reported molecule responsible for innate immunity via TLR (Toll-Like Receptor) pathway, and cHsp60 acts as the intrinsic ligands of TLRs that evoked the essential signaling pathway of immune responses. Hsp60 activated mononuclear macrophage through TLR4 signal pathway (Ohashi et al., 2000), and Hsp60-mediated B cell initial activation was through TLR4-MyD88 signaling pathway (Flohé et al., 2003; Cohen-Sfady et al., 2005). Bulut found that cHsp60 induced NF-κB activation via TLR4-MD2 and MyD88 pathway as endotoxin and the effect of cHsp60 could be blocked by anti-TLR4 antibody rather than anti-TLR2 antibody (Bulut et al., 2002). Thus, in this study, Hsp60 and LPS were chosen as controls for identifying the unknown chlamydial components involved in TLR4 pathway.
The aim of our study was to illuminate the role of chlamydial lipoproteins in host inflammatory responses of chlamydial diseases. To achieve these aims, we tested the ability of the predicted chlamydial lipoproteins to induce the production of proinflammatory cytokines. As TLRs and CD14 are commonly involved in bacterial lipoprotein recognition (Janeway and Medzhitov, 2002), our experiments were conducted in the cells expressing TLR1/2, TLR2, TLR2/6, and TLR2/CD14 but not TLR4. These receptors are expressed on cell surface of HEK-293 cells which are stably transfected with the empty plasmid (293-Null), human TLR1/2/TLR2/TLR2/6, or TLR2/CD14 genes.
Materials and Methods
Lipoprotein Prediction
The lipoproteins of C. trachomatis were predicted using DOLOP (A database of bacterial lipo-proteins). The result can be find at http://www.mrc-lmb.cam.ac.uk/genomes/dolop/predicted/ct.shtml.
Bacteria and Proteins
All chlamydial organisms were either purchased from ATCC (Manassas, VA) or acquired from Dr. Harlan Caldwell at the Rocky Mountain Laboratory, NIAID/NIH and Dr. Ted Kou at the University of Washington (Seattle, WA). The chlamydial organisms were propagated, purified, aliquoted, and stored as previously described (Zhong et al., 2001). All chlamydial organisms were routinely checked for mycoplasma contamination.
Chlamydial Gene Cloning and Fusion Protein Expression
The ORFs of D381, D541, D067, and D775 from C. trachomatis serovar D were cloned into pGEX-6P-2 vectors (Amersham Pharmacia Biotech, Inc., Piscataway, NJ). According the prediction site of the lipobox, the target amino acid mutagenesis primers were designed and constructed into pGEX-6P-2 vector according the protocol of quick change R II site directed mutagenesis Kit. The following primers were used for cloning the ORFs:
CT567 -P1# 5′-CGCGGATCC-ATGTTTAGAAAGATAAAAAAGAA-3′
CT 567-P2# 5′-CCGGAGCTC-TCTACCCACAGCAAAAATATAG-3′
CT 067-P1# 5′-CGCGGATCC-ATGTCTTTTTTTCATACTAGAA-3′
CT 067-P2# 5′-CCGGAGCTC-TTCAAGAACAGTCCCTCCCAAT-3′
CT 381-P1# 5′-CGCGGATCC-ATGTGCATAAAAAGAAAAAAAACATGGAT-3′
CT 381-P2# 5′-CCGGAGCTC-GTTGTTCAAACCCCACTTCTC-3′
CT 775-P1# 5′-CGCGGATCC-ATGAAGATAGGGTTTTGGCGTA-3′
CT 775-P2# 5′-CCGGAGCTC-GGGGACCTCTAAAGGAG-3′
CT 567-P1#(C20A-site mutation) 5′-CTTTCAGAACTTCTCA
TCGCTGCCGTGCTTATC
AGTTTGCTGCTC-3′
CT 567-P2#(C20A-site mutation) 5′-GAGCAGCAAACTGAT
AAGCACGGCAGCGATGA
GAAGTTCTGAAAG-3′
CT 067-P1#(C22A-site mutation) 5′-GGACTCTTGTGTTTAG
CAGGCGCTTTCTTAATG
AACAGCTGTTCC-3′
CT 067-P2#(C22A-site mutation) 5′-GGAACAGCTGTTCATT
AAGAAAGCGCCTGCTAA
ACACAAGAGTCC-3′
CT 381-P1#(C24A-site mutation) 5′-TGTAGTTTTTGTTTGA
CGGGTGCTTTAAAAGAA
GGGGGAGACTCC-3′
CT 381-P2#(C24A-site mutation) 5′-GGAGTCTCCCCCTTCT
TTTAAAGCACCCGTCAA
ACAAAAACTACA-3′
CT 775-P1#(C20A-site mutation) 5′-TGGCGTAGACTGTATG
AGGTGTGTTATACTTCTC
TTATAGGGGCC-3′
CT 775-P2#(C20A-site mutation) 5′-GGCCCCTATAAGAGA
AGTATAACACACCTCAT
ACAGTCTACGCCA-3′
CT 775-P1#(C13A-site mutation) 5′-TGGCGTAGACTGTATG
AGGTGGCTTATACTTCT
CTTATAGGGTGC-3′
CT 775-P2#(C13A-site mutation) 5′-GCACCCTATAAGAGA
AGTATAAGCCACCTCAT
ACAGTCTACGCCA-3′
CT 775-P1#(C13A20A-site mutation) 5′-TGGCGTAGACTGT
ATGAGGTGGCTTATA
CTTCTCTTATAGGGGCC-3′
CT 775-P2#(C13A20A-site mutation) 5′-GGCCCCTATAAGA
GAAGTATAAGCCACC
TCATACAGTCCGCCA-3′
The ORF was expressed as a fusion protein with glutathione- S- transferase (GST) fused to the N-terminuses as previously described (Wu et al., 2011). Expression of the fusion protein was induced with isopropyl-beta-D-thiogalactoside (IPTG; Invitrogen, Waltham, MA) and the fusion proteins were extracted by lysing the bacteria via sonication in a Triton-X100 lysis buffer (1% TritonX-100, 1 mM PMSF, 75 units/ml Aprotinin, 20 μM Leupeptin, and 1.6 μM Pepstatin; Sigma-Aldrich, St. Louis, MO). After removing cell debris by high-speed centrifugation, the fusion protein was purified using glutathione-conjugated agarose beads (Pfizer Inc., New York, NY).
Cell Culture
TLR4−/−, myD88−/−, Tirap/Mal,−/− Trif−/−, and female Balb/c mice at the age of 3–4 weeks old were purchased from Charles River Laboratories, Inc. (Wilmington, MA). The isolated monocytes were seeded in 96-well plates (100 cells/μl) in DMEM supplemented with GlutaMAX-I (2 mM), streptomycin (100 μg/ml), penicillin (100 U/ml), and heat inactivated 10% (v/v) FBS (Invitrogen, Waltham, MA). After the cells were stimulated for 24 h at 37°C, the supernatants were collected by centrifugation (400 × g, 4°C, 10 min). The LPS (E. coli serotype O55:B5) supplemented in cell culture and triacylated control lipopeptide, racemic Pam3, were purchased from Sigma-Aldrich (St. Louis, MO). Affinity chromatography on polymyxin B agarose (Sigma-Aldrich, St. Louis, MO) was used to remove LPS from recombinant proteins.
Cell Stimulation Experiment
Transfected HEK-293 cell lines stably expressing human TLR1/2, TLR2, TLR2/6, or TLR2/CD14 genes were gifts from Dr. Yan Xiang (University of Texas, Health Science Center at San Antonio). The cells carrying an empty plasmid, TLR1/2 plasmids, or TLR2/6 plasmids were cultured in high glucose DMEM (Dulbecco's Modified Eagle's Medium; Invitrogen, Gaithersburg, MD) with 10% FBS, streptomycin (100 μg/ml), penicillin (100 U/ml), and blasticidin S (10 mg/ml; InvivoGen, San Diego, CA). The cells carrying TLR2/CD14 plasmids were cultured in in high glucose DMEM supplemented with 50 mg/ml Hygromycin B Gold (InvivoGen, San Diego, CA). After 24 h culture in a 96-well microplate with an initial concentration of 500 cells/μl, the cells were co-cultured with E. coli LPS (1 μg/ml) or lipoproteins (1 to 0.001 μg/ml) for another 24 h. The lipoproteins were diluted in D10 medium (with 20 μg/ml gentamycin and with or without polymycin B 50 μg/ml) and incubated at 37°C for 1 h. The culture supernatants were collected for the ELISA analysis of IL-8 or mMIP-2 levels (DuoSet ELISA; R&D Systems, Inc. Minneapolis, MN). Results were obtained from cultures performed in triplicates.
Proteinase and Lipase Treatments of Lipoproteins
Proteinase K sensitivity was tested by incubating D775 or D541 with 100 μg/ml proteinase K (Promega) in 100 mM Tris-HCl (pH 8.0) at 37°C for 2 h, followed by addition of 200 mM PMSF (phenylmethylsulfonyl fluoride). Lipoproteins D775 and D541 in a buffer with 50 mM HEPES (pH 7.5) and 10 mM CaCl2 were incubated with1000 U/ml pig pancreas lipase (type VI-S lipase; Sigma-Aldrich) at 37°C for 16 h, followed by heating at 100°C before beginning the assay (Bas et al., 2008). The lipoproteins were also incubated in enzyme free buffer as controls. TLR4−/− macrophage were incubated with lipoprotein with lipase, lipoprotein with protease, lipase alone, or protease alone. Cell-free supernatants were harvested after 4 h incubation for cytokine measurement.
Lipoprotein Structure Analysis
The lipoprotein structures (D067, D381, D541, and D775) were initially built by homologous modeling using SWISS-MODEL (swissmodel.expasy.org). The initial models were then solvated in a water box and run 10,000 steps of energy minimization and 10 ns molecular dynamics using NAMD (NAnoscale Molecular Dynamics) v 2.9. The modeled lipoprotein-TLR complex structures were built by manually aligning the lipobox cysteine to the Pam3-cysteine based on the crystal structure of TLR2/TLR6- Pam2CSK4 complex (PDB code: 3A79).
Results
Fourteen Lipoproteins of Chlamydia trachomatis were Predicted
Fourteen chlamydial lipoproteins were predicted from its genome. They are D067, D175, D253, D381, D386, D444, D541, D548, D567, D600, D734, D770, D775, and D780. The possible function and putative lipobox were also predicted (Table 1). Our previous animal experiments data suggested that CT381 (ABC transporter, solute binding pr; R-binding periplasmic pr 2), CT567 (Hypothetical pr), CT541 (MIP-like pr), CT067 (ABC transporter, solute binding pr; adhesin), CT775 (possible sn-Glycerol-3-P Acyltransferase) were related to the inflammatory reaction. The modeled structures of D067, D381, D541, and D775 were displayed in Supplementary Figure 1. Five proteins were chosen for expression and mutation to explore their roles in host innate immunity.
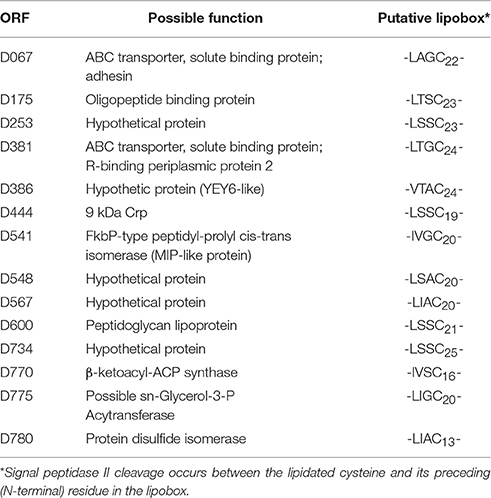
Table 1. Fourteen predicted Chlamydia trachomatis (serovar D) lipoproteins using DOLOP program.
TLR1/2 and CD14 Rather than TLR4 were Involved in Stimulation of Inflammatory Cytokine Production by C. trachomatis Lipoproteins
The mouse macrophage were stimulated using C. trachomatis recombinant lipoproteins, including D541, D541 mutant (D541C20A), D381, D381 mutant (D381C24A), D067, D067 mutant (D067C22A), D775, D775 mutants (D775C13A, D775C20A, and D775C13AC20A) as well as Hsp60, E. coli LPS, Pam3, and GST. Compared to GST and blank control groups, very high levels of mMIP-2 secretion were detected in treatment groups, especially in Hsp60 and D381 treatment groups (Figure 1A). The stimulation effect of LPS decreased by the addition of polymyxin B (PMB), as shown in Figure 1A. The results demonstrated that 50 μg/ml polymycin B could prevent E. coli LPS contamination. These data also indicated that the predicted lipoproteins induced proinflammatory cytokines in mouse macrophages.
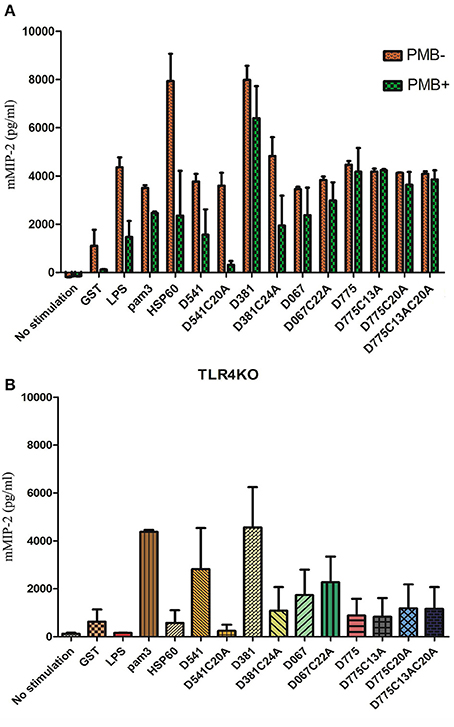
Figure 1. Cytokine profile of mouse macrophage after Chlamydia trachomatis lipoprotein stimulation. (A) Cytokine productions by mouse macrophages in response to lipoproteins of Chlamydia trachomatis. Mouse macrophages (105 cells/ml per well) were stimulated by recombinant C. trachomatis lipoproteins (D381, D541, D067, D775), lipobox mutated proteins (D381C24A, D541C20A, D067C22A, D775C13A, D775C20A, and D775C13AC20A), Hsp60, E. coli LPS, pam3, and GST. The stimulators were incubated with or without polymycin B (50 μg/ml), and supernatants were collected and the cytokine levels were analyzed by sandwich ELISA. (B) Cytokine productions by TLR4−/− mouse macrophages in response to lipoproteins of Chlamydia trachomatis. Macrophages (105 cells/ml per well) were stimulated by Hsp60, D541, D541 mutant (D541C20A), D381, D381 mutant (D381C24A), D067, D067 mutant (D067C22A), D775, D775 mutants (D775C13A, D775C20A, and D775C13AC20A), E. coli LPS, Pam3, and GST. After 24 h culture, supernatants were collected to analyze mMIP-2 levels by ELISA. D381, D541, D067, and D775 could elicit TLR4−/− mouse macrophages to release proinflammatory cytokine, mouse Macrophage Inflammatory Protein-2 (mMIP-2), which was not through TLR4 signal pathway. Except D067, the other 3 proteins required lipobox cysteine for inflammatory cytokine stimulation.
When the assay were repeated using TLR4−/− mouse macrophage, very few mMIP-2 was detected in LPS and Hsp60 treatment groups while high levels of mMIP-2 were detected in Pam3, D541, D381, D067, and D775 groups (Figure 1B). These result was consistent with previously reported data. However, in the groups of lipobox mutated proteins (D381C24A, D541C20A, D775C13A, D775C20A, and D775C13AC20A), proinflammatory cytokine levels were much lower than the wild type protein groups. It suggested that the stimulation by D541, D381, and D775 required the lipid modification on lipobox cysteine, which was consistent with the lipoprotein prediction results. Therefore, these data indicated that D541, D381, D067, D775 could induce proinflammatory cytokines in macrophages, which might not be only through TLR4 pathway. The stimulation by D541 and D381 required lipid modification. In the presence of lipoproteins, the secretion level of proinflammatory cytokines was correlated with the stimulator concentrations (Figure 1B).
Cytokine productions were measured for the HEK-293 cells expressing human TLR1/2, TLR2, TLR2/6, or TLR2/CD14 in response to C. trachomatis D541, D541 mutant, D381, D381 mutant, D067, D067 mutant, D775, and D775 mutants, as well as Hsp60, E. coli LPS, Pam3, and GST. As showed in Figure 2B, high levels of IL-8 were stimulated in TLR1/2 expressing cell line. However, much lower IL-8 levels were detected in their lipobox cysteine mutated proteins compared with that stimulated by wild-type proteins (Figure 2B), suggesting that TLR1/2 signal pathway was involved in Pam3, D541, D381, and D775 stimulations and the stimulation required the lipobox cysteine. There were very low cytokine levels detected in stimulation assays of TLR2 and TLR2/6 expressing cells (Figures 2A,B). In addition, TLR2/CD14 could be stimulated by D381 and Pam3. The results indicated that most of the C. trachomatis lipoproteins stimulated proinflammatory cytokines through the TLR1/2 pathway and D381 could also use TLR2/CD14 pathway.
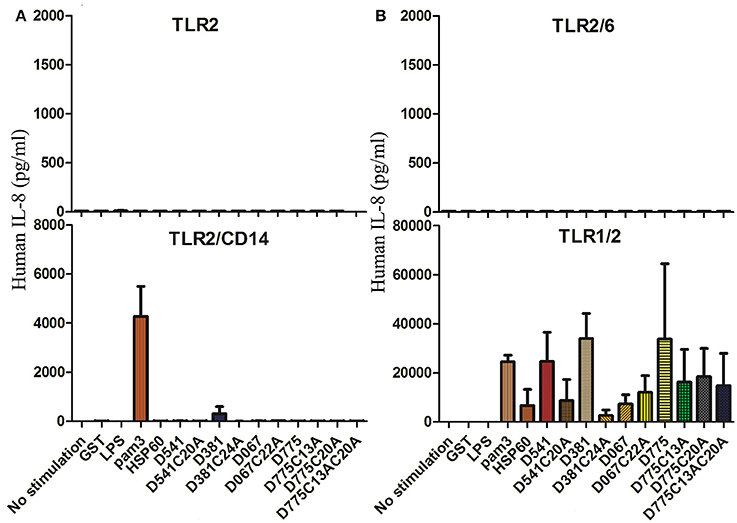
Figure 2. Toll-Like receptors required for the recognition of chlamydial inflammatory lipoproteins. (A) IL-8 productions of stimulated TLR2 and TLR2/CD14 expressing cells. (B) IL-8 productions of stimulated TLR2/6 and TLR1/2 expressing cells. Cytokine productions of HEK-293 cell lines transfected with human TLR1/2, TLR2, TLR2/6, or TLR2/CD14 genes (105 cells/ml per well) were tested, respectively, after stimulation of Chlamydia trachomatis lipoproteins, D541, D541 mutant, D381, D381 mutant, D067, D067 mutant, D775, D775 mutants, Hsp60, E. coli LPS, pam3, and GST. The stimulators were diluted in D10 medium with 20 μg/ml gentamycin. After 24 h of culture, supernatants were collected and human IL-8 were analyzed by ELISA.
The Induction of Proinflammatory Cytokines by D775 Required Lipid Modification
To explore whether the lipid or protein part contributes to the proinflammatory activity of recombinant lipoproteins, D775 and D541 were first treated with proteinase K. Results showed that proinflammatory activity of D775 and D541 was maintained after proteinase digestion (Figure 3), which was similar to the result of a macrophage-activating lipopeptide from Mycoplasma fermentans (Mühlradt et al., 1996). However, lipase treatment of Chlamydial lipoprotein D775 led to dramatic decrease of mMIP-2 release after cell stimulation. These results confirmed that D775 required lipid modification to stimulate proinflammatory cytokine production. But our data indicated that D541 did not significantly influenced by lipase which was inconsistent with a previous report (Bas et al., 2008).
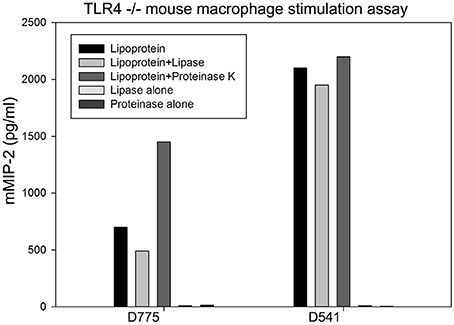
Figure 3. Production of proinflammatory cytokines induced by D775 required lipid modification while D541 did not. TLR4−/− mouse macrophage were incubated with lipoproteins treated with lipase, lipoproteins treated with proteinase, lipase alone, or proteinase alone. Lipoprotein+lipase groups: D775 and D541 were incubated with1000 U/ml pig pancreas lipase at 37°C for 16 h, followed by heating at 100°C before beginning cytokine stimulation assay. Lipoprotein+ proteinase groups: D775 or D541 with 100 μg/ml proteinase K at 37°C for 2 h, followed by addition of 200 mM PMSF. Lipoprotein groups: D775 and D541 were incubated in enzyme free buffer as controls. Cell-free supernatants were harvested after 4 h incubation for cytokine mMIP-2 measurement by ELISA. It confirmed that lipid modification of D775 was essential for its stimulation of proinflammatory cytokine production, but D541 stimulation was not greatly affected by lipase.
Production of Proinflammatory Cytokines Induced by D775 and D541 was through MyD88-Dependent Pathway
To determine whether the ability of proinflammatory cytokine production induced by D541 and D775 was in a MyD88-dependent manner, cytokine productions in MyD88 KO, Tirap/Mal KO, and Trif KO macrophages were stimulated by C. trachomatis recombinant lipoproteins (D541 and D775), LPS, and Pam3. When MyD88 were knocked out, mMIP-2 production decreased to an extreme low level in the stimulation assay of LPS, Pam3, D541, and D775 (Figure 4). It showed that the proinflammatory cytokines induced by D775 and D541 were through MyD88-dependent pathway. When Tirap/Mal were knocked out, mMIP-2 release remained high level after stimulation by Pam3 and D775 while the stimulation of D541 relied on both MyD88 and Tirap/Mal. The knock-out of Trif substantially decreased the stimulation effects of D541.
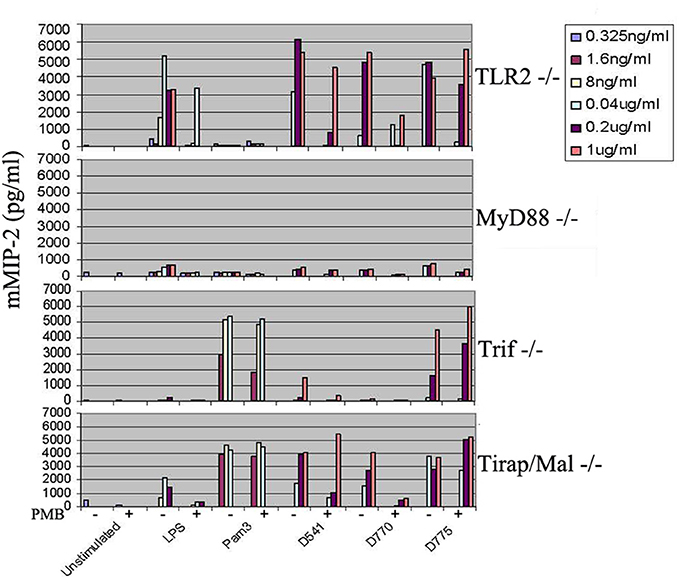
Figure 4. Cytokine productions by myD88−/−, Tirap/Mal−/−, and Trif−/−mouse macrophages in response to Chlamydia trachomatis lipoproteins. The mouse macrophages (105 cells/ml per well) were stimulated by recombinant C. trachomatis lipoproteins D541 and D775 as well as E. coli LPS and Pam3, and then mMIP-2 level was determined by ELISA. The “+” symbol represents the addition of polymycin B (50 μg/ml) while the “−” symbol in the figure indicates that no polymycin B is supplemented. After 24 h culture, supernatants were collected and the mMIP-2 were analyzed. Results were obtained from 6 different dilution concentrations of stimulators and cell cultures performed in triplicates. The results suggested the proinflammatory cytokine production induced by D775 and D541 relied on MyD88 while D541 stimulation depended on both MyD88 and Tirap/Mal.
Discussion
Chlamydia infection can cause high levels of inflammatory cytokines in the culture supernatants or at the site of infection. However, it is unclear how chlamydial organisms activate inflammatory cytokine genes and what chlamydial components are responsible for or participate in the host cytokine gene activation. Therefore, looking for chlamydial lipoproteins that activate the host cells cytokine secretion has become an interesting topic in chlamydia research field. To date, the only study of chlamydial lipoprotein was the D541 (MIP) which induces cytokines release in a TLR4-independent fashion (Bas et al., 2008).
TLRs are a class of receptors that play a key role in the innate immune system and initiating the inflammatory reactions (Lien et al., 1999; Akira and Takeda, 2004; Kufer and Sansonetti, 2007). They are single, membrane-spanning, non-catalytic receptors usually expressed in sentinel cells such as macrophages and dendritic cells and recognize structurally conserved molecules derived from microbes. It has been verified that different TLRs recognized PAMPs by matching the steric configuration of the ligands in TLR knock out mouse. TLR4 recognizes LPS while TLR2 recognizes bacterial lipoprotein and glycolipid. TLR2/TLR1 heterodimers bind triacylated lipopeptides, whereas TLR2/TLR6 heterodimers recognize diacylated lipopeptides (Takeuchi et al., 2001; Takeda et al., 2002; Akira, 2003).
Our study demonstrated that among the 14 predicted lipoproteins of C. trachomatis, D381, D541, D067, and D775 have displayed greater ability of proinflammatory cytokine induction. Their proinflammatory activity could not be attributed to E. coli LPS contamination since the induction was insensitivity to polymyxin B. The induction may involve TLR1/2 and TLR2/CD14, but not TLR4 pathway. These results are consistent with previous reports of the predominant role of TLR2 in C. trachomatis inflammatory reaction (Darville et al., 2003). They demonstrated that TLR2 was directly involved in the disease development since there was no distinct pathologic change in oviduct after C. trachomatis infection in TLR2-deficient mice (Darville et al., 2003; He et al., 2011). On the other hand, CD14 could enhance TLR2-mediated responses to many PAMPs including peptidoglycans and lipopeptides. Therefore, it is postulated that CD14 participated in transferring signal from these ligands to TLR2 (Sellati et al., 1998; Wetzler, 2003; Akira and Takeda, 2004; Vasselon et al., 2004), which was the first step of the activation and transduction of immune response signals (Tsan and Gao, 2009; Casanova et al., 2011).
In TLR signal system, there are 4 adaptor proteins including Mal (MyD88 adaptor- like protein), TRIF (Toll-interleukin-1 receptor domain-containing adaptor inducing interferon-γ), TRAM (Trif-related adaptor molecule), and SARM (Sterile and HEAT-Armadillo motifs) which participated in TLR-associated signaling cascades. It has been proved that the interaction among TRAM and TRIF, Mal, TLR4 could affect the function of non-MyD88-dependent TLR4 signal pathway (Fitzgerald et al., 2003). In our study, results showed that the proinflammatory cytokines induced by D775 and D541 were through MyD88-dependent pathway. Most of our results on D541 were consistent with the results from Dr. Sylvette Bas's group (Bas et al., 2008). Furthermore, we found that the induction by D541 and Pam3 was partially relied on MyD88 and Tirap/Mal.
Except D067, the stimulatory activities of D381, D541, and D775 required lipid modification. Their effects on cytokine release were greatly decreased when the lipobox cysteines were mutated. In addition, cytokine induction activity of D775 was greatly reduced by lipase treatment, but proteinase K treatments did not affected its activity. It was strange that the stimulation effect of D541 was not significantly decreased by lipase treatment. This was contradicted with Sylvette Bas's results. The detail mechanism required further studies.
To gain the insight of structural characteristics of C. trachomatis lipoproteins, the homologous protein structures of D067, D381, D541, and D775 were predicted by homologous sequence searching (Supplementary Figure 1). The homolog of D541 is a peptidyl-prolyl cis-trans isomerase which could serve as the ligand of TLR. For example, a peptidyl prolyl cis-trans isomerase, HP0175, secreted by Helicobacter pylori is able to TLR4-dependently induce apoptosis, and could also be specifically pulled down by TLR4 (Basak et al., 2005).
In conclusion, the predicted Chlamydial lipoproteins D381, D541, D067, and D775 are very important factors in host inflammatory responses to the diseases caused by C. trachomatis via TLR pathways. This is the first exploring study on the effect of Chlamydial lipoproteins on pathogenesis of Chlamydial diseases after the report of MIP (D541). This study may provided the theoretical basis for drug development, molecular modification of vaccine and the prevention and cure of the diseases caused by C. trachomatis.
Author Contributions
YW and QL finished parts of the experiment and did the statistics. DC, JG, and LM contributed to experimental technique supports. GZ and HS provided much scientific advice for this study. XW finished parts of the experiments and wrote the paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This project was supported by National Science foundation of China (No. 81371834, No. 81171597, and No. 81501791) Natural Science Foundation of Hunan Province (2015JJ2179).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.00078/full#supplementary-material
Supplementary Figure 1. The modeled protein structures of chlamydial inflammatory lipoproteins. The predicted 3-dimensional structures of Chlamydia trachomatis lipoproteins are listed in different panels. (A) The structure model of D541. (B) The structure model of D067-Toll like receptor complex. (C) The structure model of D381. (D) The structure model of D775-Toll like receptor complex.
References
Akira, S. (2003). Mammalian Toll-like receptors. Curr. Opin. Immunol. 15, 5–11. doi: 10.1016/S0952-7915(02)00013-4
Akira, S., and Takeda, K. (2004). Toll-like receptor signalling. Nat. Rev. Immunol. 4, 499–511. doi: 10.1038/nri1391
Bas, S., James, R. W., and Gabay, C. (2010). Serum lipoproteins attenuate macrophage activation and Toll-Like Receptor stimulation by bacterial lipoproteins. BMC Immunol. 11:46. doi: 10.1186/1471-2172-11-46
Bas, S., Neff, L., Vuillet, M., Spenato, U., Seya, T., Matsumoto, M., et al. (2008). The proinflammatory cytokine response to Chlamydia trachomatis elementary bodies in human macrophages is partly mediated by a lipoprotein, the macrophage infectivity potentiator, through TLR2/TLR1/TLR6 and CD14. J. Immunol. 180, 1158–1168. doi: 10.4049/jimmunol.180.2.1158
Basak, C., Pathak, S. K., Bhattacharyya, A., Pathak, S., Basu, J., and Kundu, M. (2005). The secreted peptidyl prolyl cis,trans-isomerase HP0175 of Helicobacter pylori induces apoptosis of gastric epithelial cells in a TLR4- and apoptosis signal-regulating kinase 1-dependent manner. J. Immunol. 174, 5672–5680. doi: 10.4049/jimmunol.174.9.5672
Bulut, Y., Faure, E., Thomas, L., Karahashi, H., Michelsen, K. S., Equils, O., et al. (2002). Chlamydial heat shock protein 60 activates macrophages and endothelial cells through Toll-like receptor 4 and MD2 in a MyD88-dependent pathway. J. Immunol. 168, 1435–1440. doi: 10.4049/jimmunol.168.3.1435
Casanova, J. L., Abel, L., and Quintana-Murci, L. (2011). Human TLRs and IL-1Rs in host defense: natural insights from evolutionary, epidemiological, and clinical genetics. Annu. Rev. Immunol. 29, 447–491. doi: 10.1146/annurev-immunol-030409-101335
Centers for Disease Control Prevention (1997). Chlamydia trachomatis genital infections–United States, 1995. MMWR Morb. Mortal. Wkly. Rep. 46, 193–198.
Cohen-Sfady, M., Nussbaum, G., Pevsner-Fischer, M., Mor, F., Carmi, P., Zanin-Zhorov, A., et al. (2005). Heat shock protein 60 activates B cells via the TLR4-MyD88 pathway. J. Immunol. 175, 3594–3602. doi: 10.4049/jimmunol.175.6.3594
Corsaro, D., and Greub, G. (2006). Pathogenic potential of novel Chlamydiae and diagnostic approaches to infections due to these obligate intracellular bacteria. Clin. Microbiol. Rev. 19, 283–297. doi: 10.1128/CMR.19.2.283-297.2006
Darville, T., O'neill, J. M., Andrews, C. W. Jr., Nagarajan, U. M., Stahl, L., and Ojcius, D. M. (2003). Toll-like receptor-2, but not Toll-like receptor-4, is essential for development of oviduct pathology in chlamydial genital tract infection. J. Immunol. 171, 6187–6197. doi: 10.4049/jimmunol.171.11.6187
Fitzgerald, K. A., Rowe, D. C., Barnes, B. J., Caffrey, D. R., Visintin, A., Latz, E., et al. (2003). LPS-TLR4 signaling to IRF-3/7 and NF-kappaB involves the toll adapters TRAM and TRIF. J. Exp. Med. 198, 1043–1055. doi: 10.1084/jem.20031023
Flohé, S. B., Brüggemann, J., Lendemans, S., Nikulina, M., Meierhoff, G., Flohé, S., et al. (2003). Human heat shock protein 60 induces maturation of dendritic cells versus a Th1-promoting phenotype. J. Immunol. 170, 2340–2348. doi: 10.4049/jimmunol.170.5.2340
Hahn, D. L., Dodge, R. W., and Golubjatnikov, R. (1991). Association of Chlamydia pneumoniae (strain TWAR) infection with wheezing, asthmatic bronchitis, and adult-onset asthma. JAMA 266, 225–230. doi: 10.1001/jama.1991.03470020051031
He, X., Nair, A., Mekasha, S., Alroy, J., O'connell, C. M., and Ingalls, R. R. (2011). Enhanced virulence of Chlamydia muridarum respiratory infections in the absence of TLR2 activation. PLoS ONE 6:e20846. doi: 10.1371/journal.pone.0020846
Janeway, C. A. Jr., and Medzhitov, R. (2002). Innate immune recognition. Annu. Rev. Immunol. 20, 197–216. doi: 10.1146/annurev.immunol.20.083001.084359
Kalayoglu, M. V., Libby, P., and Byrne, G. I. (2002). Chlamydia pneumoniae as an emerging risk factor in cardiovascular disease. JAMA 288, 2724–2731. doi: 10.1001/jama.288.21.2724
Kiviat, N. B., Paavonen, J. A., Wølner-Hanssen, P., Critchlow, C. W., Stamm, W. E., Douglas, J., et al. (1990). Histopathology of endocervical infection caused by Chlamydia trachomatis, herpes simplex virus, Trichomonas vaginalis, and Neisseria gonorrhoeae. Hum. Pathol. 21, 831–837. doi: 10.1016/0046-8177(90)90052-7
Kufer, T. A., and Sansonetti, P. J. (2007). Sensing of bacteria: NOD a lonely job. Curr. Opin. Microbiol. 10, 62–69. doi: 10.1016/j.mib.2006.11.003
Lien, E., Sellati, T. J., Yoshimura, A., Flo, T. H., Rawadi, G., Finberg, R. W., et al. (1999). Toll-like receptor 2 functions as a pattern recognition receptor for diverse bacterial products. J. Biol. Chem. 274, 33419–33425. doi: 10.1074/jbc.274.47.33419
Mertz, K. J., McQuillan, G. M., Levine, W. C., Candal, D. H., Bullard, J. C., Johnson, R. E., et al. (1998). A pilot study of the prevalence of chlamydial infection in a national household survey. Sex. Transm. Dis. 25, 225–228. doi: 10.1097/00007435-199805000-00001
Mühlradt, P. F., Meyer, H., and Jansen, R. (1996). Identification of S-(2,3-dihydroxypropyl)cystein in a macrophage-activating lipopeptide from Mycoplasma fermentans. Biochemistry 35, 7781–7786. doi: 10.1021/bi9602831
Ohashi, K., Burkart, V., Flohé, S., and Kolb, H. (2000). Cutting edge: heat shock protein 60 is a putative endogenous ligand of the toll-like receptor-4 complex. J. Immunol. 164, 558–561. doi: 10.4049/jimmunol.164.2.558
Patton, D. L., and Kuo, C. C. (1989). Histopathology of Chlamydia trachomatis salpingitis after primary and repeated reinfections in the monkey subcutaneous pocket model. J. Reprod. Fertil. 85, 647–656. doi: 10.1530/jrf.0.0850647
Peterman, T. A., Tian, L. H., Metcalf, C. A., Satterwhite, C. L., Malotte, C. K., Deaugustine, N., et al. (2006). High incidence of new sexually transmitted infections in the year following a sexually transmitted infection: a case for rescreening. Ann. Intern. Med. 145, 564–572. doi: 10.7326/0003-4819-145-8-200610170-00005
Sellati, T. J., Bouis, D. A., Kitchens, R. L., Darveau, R. P., Pugin, J., Ulevitch, R. J., et al. (1998). Treponema pallidum and Borrelia burgdorferi lipoproteins and synthetic lipopeptides activate monocytic cells via a CD14-dependent pathway distinct from that used by lipopolysaccharide. J. Immunol. 160, 5455–5464.
Sherman, K. J., Daling, J. R., Stergachis, A., Weiss, N. S., Foy, H. M., Wang, S. P., et al. (1990). Sexually transmitted diseases and tubal pregnancy. Sex. Transm. Dis. 17, 115–121. doi: 10.1097/00007435-199007000-00001
Takeda, K., Takeuchi, O., and Akira, S. (2002). Recognition of lipopeptides by Toll-like receptors. J. Endotoxin Res. 8, 459–463. doi: 10.1179/096805102125001073
Takeuchi, O., Kawai, T., Mühlradt, P. F., Morr, M., Radolf, J. D., Zychlinsky, A., et al. (2001). Discrimination of bacterial lipoproteins by Toll-like receptor 6. Int. Immunol. 13, 933–940. doi: 10.1093/intimm/13.7.933
Tsan, M. F., and Gao, B. (2009). Heat shock proteins and immune system. J. Leukoc. Biol. 85, 905–910. doi: 10.1189/jlb.0109005
Vasselon, T., Detmers, P. A., Charron, D., and Haziot, A. (2004). TLR2 recognizes a bacterial lipopeptide through direct binding. J. Immunol. 173, 7401–7405. doi: 10.4049/jimmunol.173.12.7401
Wetzler, L. M. (2003). The role of Toll-like receptor 2 in microbial disease and immunity. Vaccine 21(Suppl. 2), S55–S60. doi: 10.1016/s0264-410x(03)00201-9
Wright, H. R., Turner, A., and Taylor, H. R. (2008). Trachoma. Lancet 371, 1945–1954. doi: 10.1016/S0140-6736(08)60836-3
Wu, X., Lei, L., Gong, S., Chen, D., Flores, R., and Zhong, G. (2011). The chlamydial periplasmic stress response serine protease cHtrA is secreted into host cell cytosol. BMC Microbiol. 11:87. doi: 10.1186/1471-2180-11-87
Keywords: Chlamydial trachoma, lipoproteins, immune response, Toll-Like Receptor, cytokine
Citation: Wang Y, Liu Q, Chen D, Guan J, Ma L, Zhong G, Shu H and Wu X (2017) Chlamydial Lipoproteins Stimulate Toll-Like Receptors 1/2 Mediated Inflammatory Responses through MyD88-Dependent Pathway. Front. Microbiol. 8:78. doi: 10.3389/fmicb.2017.00078
Received: 12 July 2016; Accepted: 11 January 2017;
Published: 26 January 2017.
Edited by:
Sunil Kumar Lal, Monash University, AustraliaReviewed by:
Paras Jain, Albert Einstein College of Medicine, USADebora Decote-Ricardo, Federal Rural University of Rio de Janeiro, Brazil
Carlo Riccardi, University of Perugia, Italy
Copyright © 2017 Wang, Liu, Chen, Guan, Ma, Zhong, Shu and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiang Wu, d3hzcHJpbmdAaG90bWFpbC5jb20=
†These authors have contributed equally to this work.
‡These authors have contributed equally to this work.