 Joosu Kuivanen
Joosu Kuivanen Mikko Arvas†
Mikko Arvas†- VTT Technical Research Centre of Finland Ltd, Espoo, Finland
D-Glucuronic acid is a biomass component that occurs in plant cell wall polysaccharides and is catabolized by saprotrophic microorganisms including fungi. A pathway for D-glucuronic acid catabolism in fungal microorganisms is only partly known. In the filamentous fungus Aspergillus niger, the enzymes that are known to be part of the pathway are the NADPH requiring D-glucuronic acid reductase forming L-gulonate and the NADH requiring 2-keto-L-gulonate reductase that forms L-idonate. With the aid of RNA sequencing we identified two more enzymes of the pathway. The first is a NADPH requiring 2-keto-L-gulonate reductase that forms L-idonate, GluD. The second is a NAD+ requiring L-idonate 5-dehydrogenase forming 5-keto-gluconate, GluE. The genes coding for these two enzymes are clustered and share the same bidirectional promoter. The GluD is an enzyme with a strict requirement for NADP+/NADPH as cofactors. The kcat for 2-keto-L-gulonate and L-idonate is 21.4 and 1.1 s-1, and the Km 25.3 and 12.6 mM, respectively, when using the purified protein. In contrast, the GluE has a strict requirement for NAD+/NADH. The kcat for L-idonate and 5-keto-D-gluconate is 5.5 and 7.2 s-1, and the Km 30.9 and 8.4 mM, respectively. These values also refer to the purified protein. The gluD deletion resulted in accumulation of 2-keto-L-gulonate in the liquid cultivation while the gluE deletion resulted in reduced growth and cessation of the D-glucuronic acid catabolism.
Introduction
The genus Aspergillus is a large group of filamentous fungi containing species that are known to be versatile decomposers of biomass polymers (de Vries and Visser, 2001). Aspergillus niger – a member of the group of black aspergilli – is widely used in industrial biotechnology due to its useful characteristics such as capacity to produce organic acids and biomass hydrolysing enzymes in high yields. Several different sugars and sugar acids resulting from the extracellular biomass hydrolysis by a mixture of secreted enzymes are catabolized by the organism through metabolic pathways. Many of these pathways are known and characterized; however, some remain still unknown and may contain enzymes and biochemical reactions that are not described earlier. These reactions may serve as source of enzymes for biotechnological applications such as production of fuels and chemicals from biomass.
One such a biomass component with limited knowledge on its catabolism is D-glucuronic acid (D-glcUA). It occurs in the cell wall polysaccharides such as glucuronoxylan (Reis et al., 1994) in plants and ulvan (Lahaye and Robic, 2007) in algae. In nature, D-glcUA resulting from biomass hydrolysis is catabolised by saprotrophic microorganisms through different metabolic pathways. In bacteria, two different catabolic pathways for D-glcUA are known: an isomerase pathway (Ashwell, 1962) and an oxidative pathway (Dagley and Trudgill, 1965; Chang and Feingold, 1970). D-GlcUA and its close structural isomer D-galacturonic acid (D-galUA), a pectin constituent, are catabolized analogously via these pathways in bacteria. Some of the enzymes in these pathways have dual functions and are used for the catabolism of both compounds. In addition to the bacterial pathways, a different catabolic D-glcUA pathway is known in animal cells (Hankes et al., 1969). The animal pathway, also known as glucuronate-xylulose-pentose phosphate pathway or uronate cycle, contains two reduction, two oxidation and one decarboxylation reactions resulting in formation of D-xylulose, which, after phosphorylation to D-xylulose 5-phosphate, is a metabolite of pentose phosphate pathway (Figure 1A). In fungi, the catabolic pathway for D-galUA is well known including reduction, dehydration, an aldolase reaction and second reduction (Figure 1B) (Kuorelahti et al., 2005, 2006; Liepins et al., 2006; Hilditch et al., 2007). However, a fungal pathway for D-glcUA catabolism is only partly known.
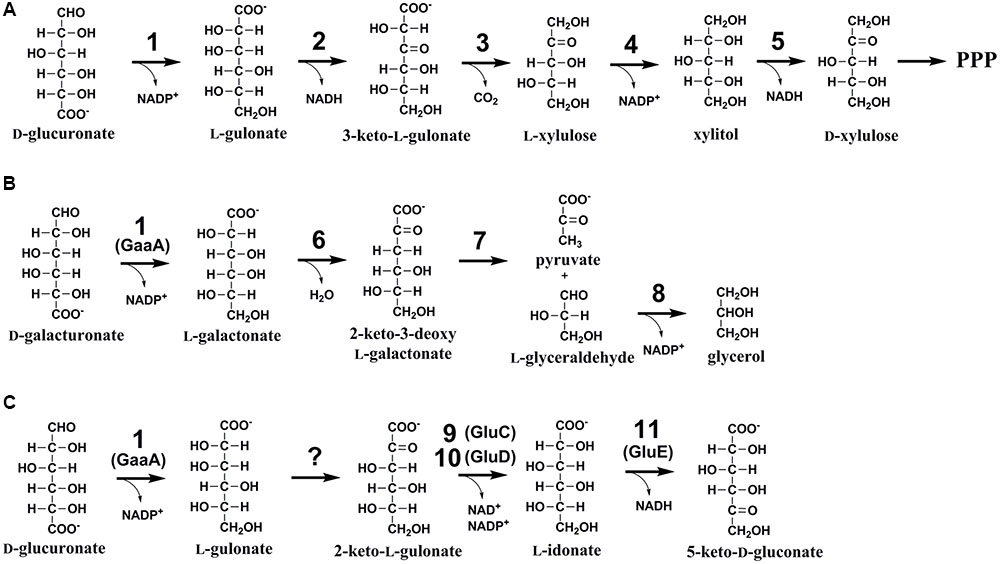
FIGURE 1. (A) The catabolic D-glucuronic acid pathway in animals, (B) the fungal D-galacturonic acid pathway, and (C) the first suggested reactions in the fungal D-glucuronic acid pathway. The enzymes are: (1) hexuronate reductase, (2) L-gulonate 3-dehydrogenase, (3) 3-keto-L-gulonate decarboxylase, (4) L-xylulose reductase, (5) xylitol dehydrogenase, (6) L-galactonate dehydratase, (7) 2-keto-3-deoxy-L-galactonate aldolase, (8) L-glyceraldehyde reductase, (9) NADH dependent 2-keto-L-gulonate reductase, GluC, (10) NADPH dependent 2-keto-L-gulonate reductase, GluD (in this study), and (11) L-idonate dehydrogenase, GluE (in this study).
The first enzyme for D-galUA catabolism in the filamentous fungus A. niger has most likely a dual function and is also the first enzyme in D-glcUA catabolism. The gaaA, encoding a hexuronate reductase is reducing D-galUA to L-galactonate and D-glcUA to L-gluconate (Martens-Uzunova and Schaap, 2008; Kuivanen et al., 2016). Transcription of gaaA was induced on both of these carbon sources and deletion of the gene reduced the catabolism of both carbon sources, however, did not block it completely (Kuivanen et al., 2016). In the following steps the pathways for D-galUA and D-glcUA differ, the L-galactonate dehydratase showed no activity with L-gulonate and an L-gulonate dehydratase activity was not found in A. niger (Motter et al., 2014). An enzyme that is essential for D-glcUA catabolism was identified to be a NADH dependent 2-keto-L-gulonate reductase, GluC (Kuivanen et al., 2016). Deletion of gluC gene resulted in reduced growth on D-glcUA plates and blocked the D-glcUA consumption in liquid cultivations. The L-gulonate is converted to 2-keto-L-gulonate by an unknown activity. For the further conversion of L-idonate, two enzyme activities have been described in the literature: the NAD+ and NADP+ dependent L-idonate 5-dehydrogenases (EC 1.1.1.366 and EC 1.1.1.264). The NAD+ dependent activity has been described in plants (Wen et al., 2010) in the pathway for L-ascorbic acid catabolism (DeBolt et al., 2006) and in bacteria as part of L-idonate catabolism (Bausch et al., 1998). The NADP+ dependent L-idonate 5-dehydrogenase activity was described for the first time already long time ago in the filamentous fungus Fusarium sp. (Takagi, 1962). However, there is no report on a fungal L-idonate 5-dehydrogenase gene or the biological function of such a gene.
In the present study, we identify a gene cluster encoding NADPH dependent, L-idonate forming, 2-keto-L-gulonate reductase and NAD+ dependent L-idonate 5-dehydrogenase which forms 5-keto-D-gluconate (Figure 1C). These genes are involved in the fungal D-glcUA catabolism and the reaction catalyzed by the latter enzyme is a direct continuation for the previously identified reaction by the action of GluC.
Materials and Methods
Strains
The A. niger strain ATCC 1015 (CBS 113.46) was used as a wild type. The A. niger mutant strain ΔpyrG (deleted orotidine-5′-phosphate decarboxylase) was described earlier (Mojzita et al., 2010). All the plasmids were produced in Escherichia coli TOP10 cells. The Saccharomyces cerevisiae strains ATCC 90845 and a modified CEN.PK2 (MATα, leu2-3/112, ura3-52, trp1-289, his3-Δ1, MAL2-8c, SUC2) were used in the homologous recombination for the plasmid construction and for the production of the purified GluD and GluE enzymes, respectively.
Media and Cultural Conditions
Luria Broth culture medium supplemented with 100 μg ml-1 of ampicillin and cultural conditions of 37°C and 250 rpm were used with E. coli. YPD medium (10 g yeast extract l-1, 20 g peptone l-1, and 20 g D-glucose l-1) was used for yeast pre-cultures. After the transformation of an expression plasmid in yeast, SCD-URA (uracil deficient synthetic complete media supplemented with 20 g D-glucose l-1) plates were used for uracil auxotrophic selection. SCD-URA medium was used in protein production. All the yeast cultivations were carried out at 30°C and the liquid cultivations at 250 rpm. A. niger spores were generated on potato-dextrose plates and ∼108 spores were inoculated to 50 ml of YP medium (10 g yeast extract l-1, 20 g peptone l-1) containing 30 g gelatin l-1 for pre-cultures. Mycelia were pre-grown in 250-ml Erlenmeyer flasks by incubating overnight at 28°C, 200 rpm and harvested by vacuum filtration, rinsed with sterile water and weighted. In A. niger transformations, SCD-URA plates supplemented with 1.2 M D-sorbitol and 20 g agar l-1 (pH 6.5) were used. A. nidulans defined minimal medium (Barratt et al., 1965) was used in the A. niger cultivations. The minimal medium used in the phenotypic characterization in liquid cultivations contained 20 g D-glcUA l-1 and the pH was adjusted to 3. These cultivations were inoculated with 20 g l-1 (wet) of pre-grown mycelia. The strain ΔgluD was cultivated in 24-well plates in 4 ml final volume and ΔgluE in shake flasks in 25 ml final volume. Agar plates used for phenotypic characterization contained uracil deficient SC-medium (synthetic complete), 15 g agar l-1 and 10 g D-glcUA l-1. Plates were inoculated with 1.5∗106 spores and incubated at 28°C for 3 days.
Transcriptional Analysis
The RNA sequencing for transcriptional analysis was carried out as described previously (Kuivanen et al., 2016). The protein ID numbers refer the numbers from the Join Genome Institute (JGI), MycoCosm, A. niger ATCC 1015 v.4.0 database, available at: http://genome.jgi.doe.gov/Aspni7/Aspni7.home.html.
Protein Production and Purification
The gene gluD was amplified by PCR (KAPA HiFi DNA polymerase, Kapa Biosystems, primers in Table 1) from A. niger cDNA extracted and generated from D-glcUA cultivated wild type strain. The resulting DNA fragment was digested with BamHI and NheI (both NEB) and ligated into a modified pYX212 plasmid (Verho et al., 2004) containing TPI1 promoter and URA3 selectable marker. The gluE gene was custom synthesized as a yeast codon optimized gene (GenScript, USA), released with EcoRI and BamHI (both NEB) and ligated into the modified pYX212 plasmid. For the histidine-tagged protein, gluE was amplified by PCR (primers in Table 1) and ligated in a similar manner to the modified pYX212 plasmid. A yeast strain was then transformed with the resulting plasmids using the lithium acetate method (Gietz and Schiestl, 2007). The procedure for protein production and purification was described previously (Kuivanen et al., 2016).

TABLE 1. Oligonucleotides used in the study.
Enzymatic Assays
The oxidoreductase activity of purified GluD and GluE proteins was assayed using Konelab 20XT Clinical Chemistry Analyzer (Thermo Scientific). The reaction mixture contained 50 mM Tris buffer, 400 μM NAD+ or NADH, a substrate in different concentrations and purified proteins in a final concentration of 3.6 mg l-1. The pH 8 was used with NAD+ and L-idonate and pH 7 with NADH and 2-keto-L-gulonate and 5-keto-D-gluconate. The reaction was started by addition of the purified protein and the formation/consumption of NADH was followed at 340 nm. The kinetic parameters were determined using the IC50 tool kit1. L-Idonate and 2-keto-L-gulonate were ordered as custom synthesized by Omicron Biochemicals Inc, USA while 5-keto-D-gluconate was ordered from Sigma-Aldrich.
Gene Deletions in A. niger
The deletion cassette for gluD contained homologous 5′ (∼450 bp) and 3′ flanks (∼650 bp) for targeted integration and the selectable marker pyrG. The 5′ and 3′ flanks were amplified by PCR with the primers as described in Table 1. The resulting PCR amplified fragments contained 40 bp compatible ends for homologous recombination with the A. niger pyrG and EcoRI and BamHI digested pRS426. The deletion cassette for gluE was constructed in a similar manner but contained homologous 5′ and 3′ flanks of 1.5 kb (primers in Table 1). All the fragments were joined using yeast homologous recombination as described earlier (Kuivanen et al., 2015). The resulting deletion cassette for gluD was produced by PCR amplification (primers in Table 1) from the resulting plasmid and the cassette for gluE deletion was produced by linearization of the plasmid with NotI (NEB). The gluD deletion cassette was transformed to A. nigerΔpyrG strain together with the CRISPR plasmid pFC-332 (Nodvig et al., 2015) and the in vitro synthesized sgRNA (CTCCTCCATCCTGACCTTGA) (GeneArtTM Precision Synthesis Kit). The gluE deletion cassette was transformed to A. nigerΔpyrG strain without the CRISPR plasmid. Mutants with successful integration of the cassette were selected for growth in the absence of uracil and, in the case of gluD deletion, in the presence of hygromycin (for pFC-332) and in the absence of uracil (for the deletion cassette containing pyrG). Resulting transformants were screened for the correct integration of the deletion cassette and for the deletion of gluD or gluE open reading frame using diagnostic PCR (Phire direct PCR kit, Thermo Scientific, primers in Table 1).
Chemical Analyses
Samples were removed from liquid cultivations at intervals and mycelium was separated from the supernatant by centrifugation or filtration. The concentration of D-glcUA and 2-keto-L-gulonate was determined by HPLC using a Fast Acid Analysis Column (100 mm × 7.8 mm, Bio-Rad Laboratories, Hercules, CA, USA) linked to an Aminex HPX-87H organic acid analysis column (300 mm × 7.8 mm, Bio-Rad Laboratories) with 5.0 mM H2SO4 as eluent and a flow rate of 0.5 ml min-1. The column was maintained at 55°C. Peaks were detected using a Waters 2487 dual wavelength UV (210 nm) detector. The retention times of the peaks resulting from the supernatant were compared with the retention times of standards.
Results
Clustered Genes Are Induced by D-Glucuronic Acid
RNA sequencing of the A. niger wild type strain ATCC 1015 cultivated in D-glcUA as sole carbon source revealed several putative genes with induced transcription (Figure 2). Figure 2 presents the induction of transcript levels between 0 and 4 hours (Y-axis) and the absolute transcript levels at 4 h (X-axis). We selected genes that were induced on D-glcUA (Figure 2, values on Y-axis clearly above 1), had absolute transcript levels around similar or higher than that of actin at 4 h (Figure 2, X-axis) and are predicted to code for a metabolic enzyme, such as oxidoreductases. The D-galUA/D-glcUA reductase gaaA and the 2-keto-L-gulonate reductase gluC were among the most induced genes as reported earlier (Kuivanen et al., 2016). In addition, two genes, with the protein identifiers 1114837 and 1099233 (JGI, MycoCosm, A. niger ATCC 1015 v.4.0 database), putatively encoding a D-isomer specific 2-hydroxy acid dehydrogenase and an alcohol dehydrogenase, respectively, were induced. These genes are clustered in the genome in opposite directions relative to each other and share a common promoter region of 455 bp (Figure 3A). The fold change in transcript levels after the shift to D-glcUA was exactly the same for these genes while 1114837 had slightly higher transcript abundancy (Figure 2).
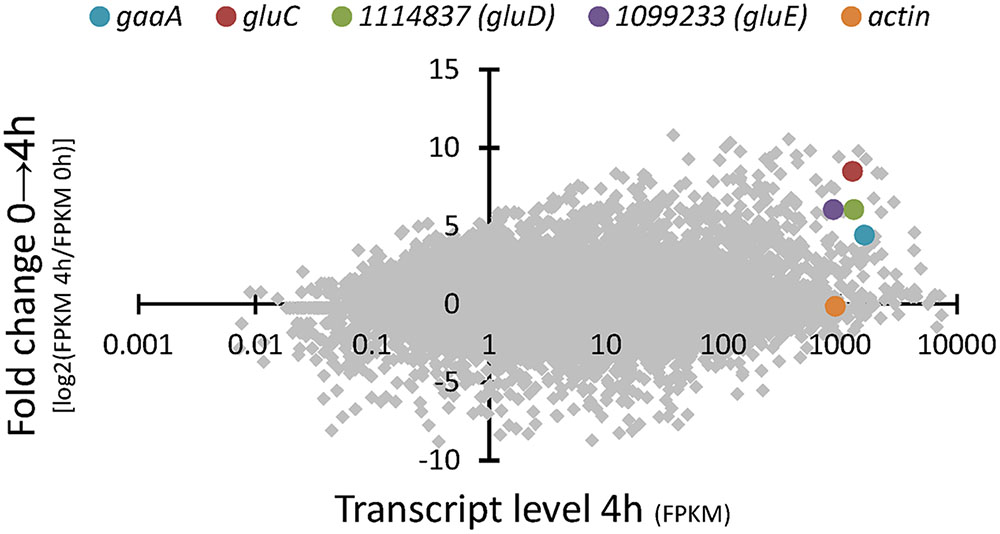
FIGURE 2. RNA sequencing of A. niger wild type strain – fold change in transcript abundancies 4 h after the shift to D-glucuronic acid (y-axis) and transcript level (x-axis) 4 h after the shift to D-glucuronic acid. The genes gaaA, gluC, actin and the genes with the protein IDs 1114837 (gluD) and 1099233 (gluE) are highlighted. Transcript levels are presented as fragments per kilobase of exon per million fragments mapped (FPKM). The protein ID numbers refer the numbers from the JGI MycoCosm A. niger ATCC 1015 v.4.0 database.
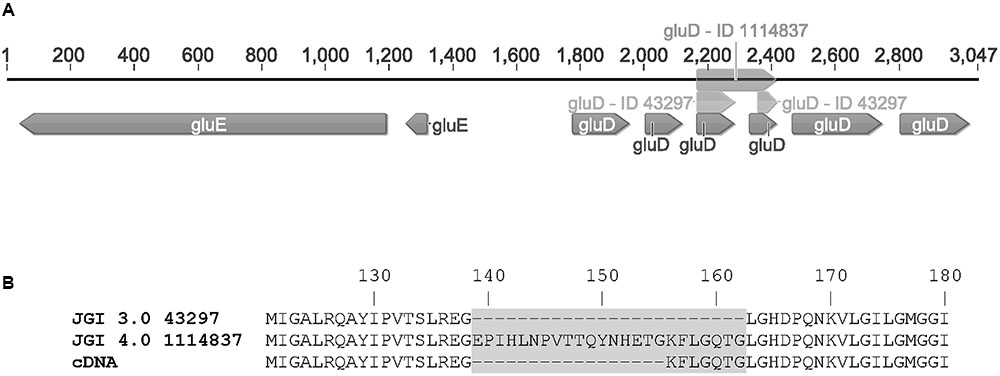
FIGURE 3. (A) The gene cluster of gluD (as predicted in JGI MycoCosm A. niger ATCC 1015 database v3.0 = ID 43297; v4.0 = ID 1114837 and sequenced from cDNA = gluD) and gluE (ID 1099233) and (B) the differences in protein sequences of GluD as predicted in the v3.0 (43297), v4.0 (1114837) and determined from cDNA.
The Clustered Genes gluD and gluE Code for 2-Keto-L-Gulonate Reductase and L-Idonate 5-Dehydrogenase
The open reading frames of the two genes were cloned in multicopy yeast expression vectors, expressed in yeast and the crude cell extracts were tested for activity with a small library of sugars and sugar acids. Both enzymes showed activity towards L-idonate. The 1114837 had activity with NADP+ as a cofactor whereas the 1099233 had activity when the cofactor was NAD+. In the case of 1114837 we noticed that the open reading frame that was custom synthetized according to the open reading frame as predicted in the DOE JGI A. niger ATCC 1015 v3.0 database (the protein ID in v3.0 is 43297), did not result in an active protein, however, when the gene was amplified from A. niger cDNA the resulting protein was active. In the current DOE JGI A. niger ATCC 1015 v4.0 database the exon prediction has been changed, however, both of the predictions (v3.0 and 4.0) are wrong. The sequence of the 1114837 amplified from cDNA differs from the predicted sequences: In the prediction v3.0, 21 nucleotides were predicted to be an intron and are missing in the open reading frame whereas in the prediction v4.0 the exons 3, 4 and the intron between them are combined. This is shown in the Figure 3A (v3.0 = 43297 and v4.0 = 1114837) and the differences in the resulting protein sequences are shown in Figure 3B. The correct gene sequence was deposited at GenBank with the accession number KX443112.
The enzyme 1114837 showed, besides the activity with NADP+ and L-idonate, also activity with 2-keto-L-gulonate and NADPH as cofactor. This suggests that the enzyme is a NADPH dependent 2-keto-L-gulonate reductase. We named the gene gluD. The gene 1099233 had activity with NAD+ and L-idonate but did not show activity with 2-keto-L-gulonate and NADH. It showed, however, activity with 5-keto-D-gluconate and NADH. We conclude that the enzyme is a NAD+ dependent L-idonate 5-dehydrogenase. We named the gene gluE.
For the more detailed characterization, histidine tagged GluD and GluE proteins were produced in yeast and the kinetic parameters of the purified proteins were investigated. Purified GluD showed NADPH/NADP+ dependent oxidoreductase activity toward 2-keto-L-gulonate and L-idonate with the kcat values of 21.4 and 1.1 s-1, respectively. The Km values for the substrates were 25.3 and 12.6 mM, respectively. Purified GluE protein had strictly NAD+/NADH dependent oxidoreductase activity towards L-idonate and 5-keto-D-gluconate with the kcat values of 5.5 and 7.2 s-1, respectively. The Km values for the substrates were 30.9 and 8.4 mM, respectively. Kinetic parameters of GluD and GluE are presented in Table 2 and Supplementary Figure 1.

TABLE 2. Kinetic parameters of the purified GluD and GluE proteins.
Deletion of gluD or gluE has an Effect on D-Glucuronic Acid Catabolism
We also deleted the genes gluD and gluE from A. niger and tested the resulting phenotypes. For the gluD gene deletion CRISPR technology was used to remove the native gene. This was implemented using the AMA-plasmid expressing Cas9 (Nodvig et al., 2015), an in vitro synthetized sgRNA and the deletion cassette with the selectable marker pyrG. GluE gene was deleted without CRISPR using only the deletion cassette containing pyrG marker. Both of the gene deletions were confirmed with diagnostic PCR and the mutant strains were tested for growth and ability to catabolize D-glcUA.
The mutant strain ΔgluD did not show reduced growth when cultivated on agar plate with D-glcUA as sole carbon source (Figure 4). However, in the liquid cultivation on D-glcUA, a phenotype was observed for ΔgluD: 2-keto-L-gulonate accumulated in the medium after D-glcUA was consumed (Figures 5A,C). This was not observed with the wild type strain (Figure 5B). In the case of the mutant strain ΔgluE, growth on D-glcUA plate was reduced (Figure 4). In addition, the consumption of D-glcUA in liquid cultivation was almost completely disrupted in ΔgluE (Table 3).

FIGURE 4. Growth of the A. niger strains wt, ΔgluD, and ΔgluE on agar plates with D-glucuronic acid or D-glucose as sole carbon source.
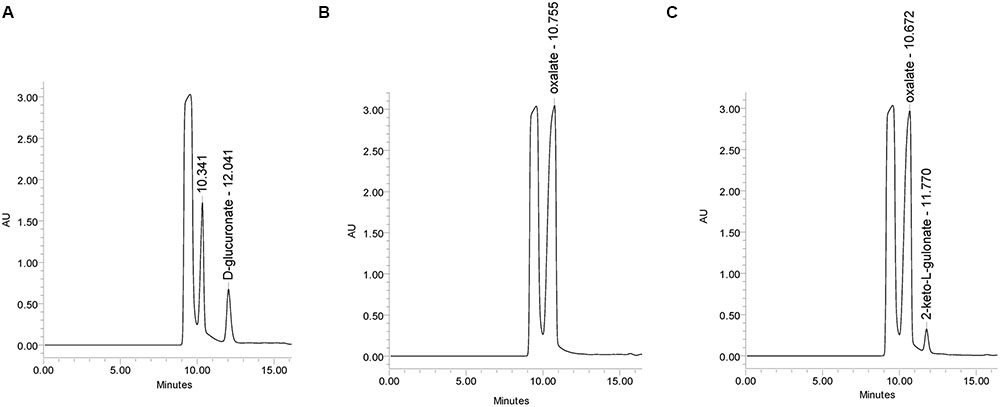
FIGURE 5. HPLC analysis of the A. niger growth medium (A) at 0 h, (B) at 72 h with the A. niger wild type strain, and (C) with ΔgluD.

TABLE 3. Concentration of D-glucuronic acid (D-glcUA) in submerged cultivations by the A. niger wild type strain (wt) and ΔgluE.
Discussion
D-GlcUA is a biomass component that is catabolised by many microorganisms including fungi. However, the catabolic pathway in fungi is only partly known. Recently, we identified the gene gluC that is essential for D-glcUA catabolism in the filamentous fungus A. niger (Kuivanen et al., 2016). The gene encoded an enzyme reducing 2-keto-L-gulonate to L-idonate using NAD+ as cofactor. We also showed that the gaaA gene encoding a D-galUA and D-glcUA reductase is induced on both substrates, D-galUA and D-glcUA. All this indicates that D-glcUA is first reduced to L-gulonate, then converted to 2-keto-L-gulonate by an unknown mechanism, and then reduced to L-idonate by the GluC. In the present study, we identified a gene cluster that is involved in D-glcUA catabolism in A. niger consisting of the genes gluD and gluE. In this cluster, gluD encodes a NADP+ dependent enzyme that, similar to GluC, catalyzes the reaction between 2-keto-L-gulonate and L-idonate. The other gene in the cluster, gluE, encodes a NADH dependent enzyme that catalyzes the reaction between L-idonate and 5-keto-D-gluconate. The latter reaction catalyzed by GluE seems to be the next step after the formation of L-idonate in the catabolic D-glcUA pathway in A. niger. This, still uncomplete pathway is summarized in the Figure 1C.
The D-glcUA pathway genes gluD and gluE are clustered in a similar manner as the D-galUA catabolic pathway genes gaaA and gaaC (Martens-Uzunova and Schaap, 2008) in the A. niger genome. An ortholog of gluD-gluE gene cluster is present in most of the sequenced aspergilli (AspGD)2. In fungi, genes of the same metabolic pathway are sometimes co-localized on chromosomes, i.e., they form chromosomal clusters (Wisecaver et al., 2014). What drives the formation of these clusters is debated. The need to ensure removal of toxic intermediates (McGary et al., 2013) has been proposed as the ultimate reason, but mere transcriptional co-regulation (Gordon et al., 2015) might have other benefits too. In this case, gluD and gluE share a common promoter and transcription of the genes is induced with a similar pattern on D-glcUA. Thus, transcriptional co-regulation is a possible explanation for the formation of gluD-gluE cluster. It is also suggested that soil-dwelling fungi may have obtained genes from bacteria for catabolism of unusual carbon sources through horizontal gene transfer (Wisecaver et al., 2014; Wisecaver and Rokas, 2015). In fact, it was suggested that fungal β-glucuronidase genes are derived from bacteria allowing fungi to hydrolyse glucuronides resulting in access to released monomeric D-glcUA (Wenzl et al., 2005). In bacteria, metabolic genes are often present in clusters such as in the case of catabolic L-idonate pathway in E. coli (Bausch et al., 2004). If such metabolic genes are acquired from bacteria via horizontal gene transfer, it may eventually lead to the formation of metabolic gene clusters in fungi as well.
In the previous study, deletion of gluC in A. niger disrupted the D-glcUA catabolism nearly completely (Kuivanen et al., 2016). Even though, GluC and GluD catalyze the same reaction and both genes are induced on D-glcUA, it seems that GluD cannot compensate the loss of GluC activity in the fungal D-glcUA pathway (deletion of gluC disrupted growth on D-glcUA; Kuivanen et al., 2016). This might be due to cofactor requirements: GluC requires NADH and GluD NADPH. In fact, it is surprising and unusual that two enzymes, in this case GluC and GluD, are present for the same reaction, but have different cofactor requirements. Since both reactions are reversible a possible interpretation is that the enzyme couple may act as an NAD(P)+ transhydrogenase adjusting the ratio of NAD+/NADH and NADP+/NADPH. Deletion of gluD did not result in reduced or no growth on D-glcUA as sole carbon source. However, it resulted in a phenotype of accumulating 2-keto-L-gulonate when cultivating on D-glcUA. This observation further supports the hypothesis that the fungal catabolic D-glcUA pathway proceeds through the oxidation of L-gulonate to 2-keto-L-gulonate. The oxidation of L-gulonate to 2-keto-L-gulonate is a biochemical reaction that is not described in the literature and the responsible enzyme in A. niger still remains unclear. In the case of 2-keto-L-gulonate reductase activity, an unspecific bacterial D-gluconate 2-dehydrogenase (EC 1.1.1.215) had been described that showed also activity for the reaction between L-idonate and 2-keto-L-gulonate (Yum et al., 1998). This bacterial enzyme used NADP+/NADPH as a cofactor similar to the GluD described in this study. However, we conclude that GluD is the first specific NADPH dependent 2-keto-L-gulonate reductase reported to date.
The protein product of the gene gluE, described in this study, catalyzed the reversible reaction from L-idonate to 5-keto-D-gluconate using NAD+/NADH as cofactor. A similar enzyme activity has been described in the filamentous fungus Fusarium sp. already in Takagi (1962), however, this enzyme activity was strictly NADP+/NADPH dependent. In plants, an NAD+/NADH enzyme (EC 1.1.1.366) oxidizing L-idonate to 5-keto-D-gluconate functions in the pathway converting L-ascorbic acid to L-tartaric acid (DeBolt et al., 2006). In addition, E. coli has an L-idonate 5-dehydrogenase, IdnD (EC 1.1.1.264), producing 5-keto-D-gluconate from L-idonate with NAD+ as cofactor in the catabolic L-idonate pathway (Bausch et al., 1998). GluE has only low sequence homology toward the other characterized L-idonate 5-dehydrogenases and it is the first reported fungal NAD+ dependent L-idonate 5-dehydrogenase. The gluE deletion in A. niger had also a phenotype – growth was reduced and D-glcUA consumption was ceased. This is a strong indication that the gene is part of the fungal catabolic D-glcUA pathway and the pathway passes through the oxidation of L-idonate to 5-keto-D-gluconate.
It is unclear how the fungal D-glcUA pathway continues after formation of 5-keto-D-gluconate. In plants, a NAD+ dependent L-idonate 5-dehydrogenase forming 5-keto-D-gluconate was described (Wen et al., 2010). This was suggested to be part of the pathway for L-ascorbic acid degradation (DeBolt et al., 2006). In this pathway, the resulting 5-keto-D-gluconate is split by an aldolase to L-threo-tetruronate and glycolaldehyde. The L-threo-tetruronate is then oxidized to L-tartaric acid. In this pathway, only the L-idonate 5-dehydrogenase gene had been identified. Another possibility would be a route similar to the L-idonate catabolism in bacteria. In E. coli, a NAD+ specific L-idonate 5-dehydrogenase is reducing the L-idonate to 5-keto-D-gluconate and the 5-keto-D-gluconate is subsequently reduced to D-gluconate (Bausch et al., 2004). D-Gluconate is then phosphorylated and the resulting 6-phosphogluconate enters the Entner–Doudoroff pathway. If 5-keto-D-gluconate is reduced to D-gluconate in the fungal D-glcUA pathway in A. niger, it would connect D-glcUA catabolism with the catabolism of D-glucose. A. niger oxidizes extracellular D-glucose to D-gluconate which is then taken up and catabolized further through the phosphorylation to D-gluconate-6-phosphate and subsequently via pentose phosphate pathway (Muller, 1985). It is also suggested that some strains of A. niger catabolize D-gluconate through the non-phosphorylative Entner–Doudoroff pathway including dehydratation of D-gluconate to 2-keto-3-deoxy-gluconate which is the split to D-glyceraldehyde and pyruvate by the action of an aldolase (Elzainy et al., 1973; Allam et al., 1975). However, the fate of 5-keto-D-gluconate in the fungal D-glcUA pathway still remains to be unraveled.
Author Contributions
JK and PR designed and JK carried out all the experimental work and analyzed the data. MA processed and analyzed the RNAseq data. JK and PR drafted the manuscript. PR designed the fundamental concept and participated in the coordination of the study. All the authors read and approved the final manuscript.
Funding
This work was supported by the Academy of Finland through the grant 271025 and the program ERA-Net LAC Energy 2016 (ELAC 2015/T03-0579 CPW Biorefinery).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
We thank the technical staff at VTT Industrial Biotechnology for their assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.00225/full#supplementary-material
FIGURE S1 | Oxidoreductase activity of purified GluD towards (A) 2-keto-L-gulonate and (B) L-idonate with NADPH and NADP+, respectively and oxidoreductase activity of purified GluE toward (C) L-idonate and (D) 5-keto-D-gluconate with NAD+ and NADH, respectively. Data represent means ± standard deviation from three biological repeats. If error bars not visible are smaller than the symbol.
Footnotes
- ^www.ic50.tk
- ^http://aspgd.broadinstitute.org/cgi-bin/asp2_v3/shared/show_protein_cluster.cgi?site=asp2_v9\&id=2050676
References
Allam, A. M., Hassan, M. M., and Elzainy, T. A. (1975). Formation and cleavage of 2-keto-3-deoxygluconate by 2-keto-3-deoxygluconate aldolase of Aspergillus niger. J. Bacteriol. 124, 1128–1131.
Ashwell, G. (1962). Enzymes of glucuronic and galacturonic acid metabolism in bacteria. Methods Enzymol. 5, 190–208.
Barratt, R., Johnson, G., and Ogata, W. (1965). Wild-type and mutant stocks of Aspergillus nidulans. Genetics 52, 233–246.
Bausch, C., Peekhaus, N., Utz, C., Blais, T., Murray, E., Lowary, T., et al. (1998). Sequence analysis of the GntII (Subsidiary) system for gluconate metabolism reveals a novel pathway for L-idonic acid catabolism in Escherichia coli. J. Bacteriol. 180, 3704–3710.
Bausch, C., Ramsey, M., and Conway, T. (2004). Transcriptional organization and regulation of the L-idonic acid pathway (GntII System) in Escherichia coli. J. Bacteriol. 186, 1388–1397.
Chang, Y. F., and Feingold, D. S. (1970). D-Glucaric acid and galactaric acid catabolism by Agrobacterium tumefaciens. J. Bacteriol. 102, 85–96.
Dagley, S., and Trudgill, P. W. (1965). The metabolism of galactarate, D-glucarate and various pentoses by species of Pseudomonas. Biochem. J. 95, 48–58.
de Vries, R. P., and Visser, J. (2001). Aspergillus enzymes involved in degradation of plant cell wall polysaccharides. Microbiol. Mol. Biol. Rev. 65, 497–522.
DeBolt, S., Cook, D. R., and Ford, C. M. (2006). L-Tartaric acid synthesis from vitamin C in higher plants. Proc. Natl. Acad. Sci. U.S.A. 103, 5608–5613.
Elzainy, T. A., Hassan, M. M., and Allam, A. M. (1973). New pathway for nonphosphorylated degradation of gluconate by Aspergillus niger. J. Bacteriol. 114, 457–459.
Gietz, R., and Schiestl, R. (2007). High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG methode. Nat. Protoc. 2, 31–34.
Gordon, S. P., Tseng, E., Salamov, A., Zhang, J., Meng, X., Zhao, Z., et al. (2015). Widespread polycistronic transcripts in fungi revealed by single-molecule mRNA sequencing. PLoS ONE 10:e0132628. doi: 10.1371/journal.pone.0132628
Hankes, L., Politzer, W., Touster, O., and Anderson, L. (1969). Myo-inositol catabolism in human pentosurics: the predominant role of the glucuronate-xylulose-pentose phosphate pathway. Ann. N. Y. Acad. Sci. 165, 564–576.
Hilditch, S., Berghäll, S., Kalkkinen, N., Penttilä, M., and Richard, P. (2007). The missing link in the fungal D-galacturonate pathway: identification of the L-threo-3-deoxy-hexulosonate aldolase. J. Biol. Chem. 282, 26195–26201.
Kuivanen, J., Penttilä, M., and Richard, P. (2015). Metabolic engineering of the fungal D-galacturonate pathway for L-ascorbic acid production. Microb. Cell Fact. 14, 1–9. doi: 10.1186/s12934-014-0184-2
Kuivanen, J., Sugai-Guérios, M. H., Arvas, M., and Richard, P. (2016). A novel pathway for fungal D-glucuronate catabolism contains an L-idonate forming 2-keto-L-gulonate reductase. Sci. Rep. 6:26329. doi: 10.1038/srep26329
Kuorelahti, S., Jouhten, P., Maaheimo, H., Penttilä, M., and Richard, P. (2006). L-Galactonate dehydratase is part of the fungal path for D-galacturonic acid catabolism. Mol. Microbiol. 61, 1060–1068.
Kuorelahti, S., Kalkkinen, N., Penttilä, M., Londesborough, J., and Richard, P. (2005). Identification in the mold Hypocrea jecorina of the first fungal D-galacturonic acid reductase. Biochemistry 44, 11234–11240.
Lahaye, M., and Robic, A. (2007). Structure and function properties of Ulvan, a polysaccharide from green seaweeds. Biomacromolecules 8, 1765–1774.
Liepins, J., Kuorelahti, S., Penttilä, M., and Richard, P. (2006). Enzymes for the NADPH-dependent reduction of dihydroxyacetone and D-glyceraldehyde and L-glyceraldehyde in the mould Hypocrea jecorina. FEBS J. 273, 4229–4235.
Martens-Uzunova, E. S., and Schaap, P. J. (2008). An evolutionary conserved D-galacturonic acid metabolic pathway operates across filamentous fungi capable of pectin degradation. Fungal Genet. Biol. 45, 1449–1457. doi: 10.1016/j.fgb.2008.08.002
McGary, K. L., Slot, J. C., and Rokas, A. (2013). Physical linkage of metabolic genes in fungi is an adaptation against the accumulation of toxic intermediate compounds. Proc. Natl. Acad. Sci. U.S.A. 110, 11481–11486. doi: 10.1073/pnas.1304461110
Mojzita, D., Wiebe, M., Hilditch, S., Boer, H., Penttila, M., and Richard, P. (2010). Metabolic engineering of fungal strains for conversion of D-galacturonate to meso-galactarate. Appl. Environ. Microbiol. 76, 169–175. doi: 10.1128/AEM.02273-09
Motter, F. A., Kuivanen, J., Keränen, H., Hilditch, S., Penttilä, M., and Richard, P. (2014). Categorisation of sugar acid dehydratases in Aspergillus niger. Fungal Genet. Biol. 64, 67–72. doi: 10.1016/j.fgb.2013.12.006
Muller, H. M. (1985). Utilization of gluconate by Aspergillus niger. I. Enzymes of phosphorylating and nonphosphorylating pathways. Zentralbl. Mikrobiol. 140, 475–484.
Nodvig, C. S., Nielsen, J. B., Kogle, M. E., and Mortensen, U. H. (2015). A CRISPR-Cas9 system for genetic engineering of filamentous fungi. PLoS ONE 10:e0133085. doi: 10.1371/journal.pone.0133085
Reis, D., Vian, B., and Roland, J.-C. (1994). Cellulose-glucuronoxylans and plant cell wall structure. Micron 25, 171–187.
Verho, R., Putkonen, M., Londesborough, J., Penttilä, M., and Richard, P. (2004). A Novel NADH-linked L-xylulose reductase in the L-arabinose catabolic pathway of yeast. J. Biol. Chem. 279, 14746–14751.
Wen, Y.-Q., Li, J.-M., Zhang, Z.-Z., Zhang, Y.-F., and Pan, Q.-H. (2010). Antibody preparation, gene expression and subcellular localization of L -idonate dehydrogenase in grape berry. Biosci. Biotechnol. Biochem. 74, 2413–2417.
Wenzl, P., Wong, L., Kwang-Won, K., and Jefferson, R. A. (2005). A functional screen identifies lateral transfer of beta-glucuronidase (gus) from bacteria to fungi. Mol. Biol. Evol. 22, 308–316.
Wisecaver, J. H., and Rokas, A. (2015). Fungal metabolic gene clusters-caravans traveling across genomes and environments. Front. Microbiol. 6:161. doi: 10.3389/fmicb.2015.00161
Wisecaver, J. H., Slot, J. C., and Rokas, A. (2014). The evolution of fungal metabolic pathways. PLoS Genet. 10:e1004816. doi: 10.1371/journal.pgen.1004816
Keywords: fungi, Aspergillus, metabolism, D-glucuronate, D-glucuronic acid, L-idonate, 2-keto-L-gulonate
Citation: Kuivanen J, Arvas M and Richard P (2017) Clustered Genes Encoding 2-Keto-l-Gulonate Reductase and l-Idonate 5-Dehydrogenase in the Novel Fungal d-Glucuronic Acid Pathway. Front. Microbiol. 8:225. doi: 10.3389/fmicb.2017.00225
Received: 09 December 2016; Accepted: 31 January 2017;
Published: 14 February 2017.
Edited by:
Michael Sauer, University of Natural Resources and Life Sciences, Vienna, AustriaReviewed by:
Sonia Cortassa, National Institutes of Health (NIH), USADavid Mousdale, Beocarta Ltd., UK
Copyright © 2017 Kuivanen, Arvas and Richard. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joosu Kuivanen, am9vc3Uua3VpdmFuZW5AdnR0LmZp
†Present address: Mikko Arvas, The Finnish Red Cross Blood Service, Helsinki, Finland