 Shuting Liu
Shuting Liu Boris Wawrik
Boris Wawrik Zhanfei Liu
Zhanfei Liu- 1Marine Science Institute, The University of Texas at Austin, Port Aransas, TX, USA
- 2Department of Microbiology and Plant Biology, The University of Oklahoma, Norman, OK, USA
Proteins and peptides are key components of the labile dissolved organic matter pool in marine environments. Knowing which types of bacteria metabolize peptides can inform the factors that govern peptide decomposition and further carbon and nitrogen remineralization in marine environments. A 13C-labeled tetrapeptide, alanine-valine-phenylalanine-alanine (AVFA), was added to both surface (normoxic) and bottom (hypoxic) seawater from a coastal station in the northern Gulf of Mexico for a 2-day incubation experiment, and bacteria that incorporated the peptide were identified using DNA stable isotope probing (SIP). The decomposition rate of AVFA in the bottom hypoxic seawater (0.018–0.035 μM h-1) was twice as fast as that in the surface normoxic seawater (0.011–0.017 μM h-1). SIP experiments indicated that incorporation of 13C was highest among the Flavobacteria, Sphingobacteria, Alphaproteobacteria, Acidimicrobiia, Verrucomicrobiae, Cyanobacteria, and Actinobacteria in surface waters. In contrast, highest 13C-enrichment was mainly observed in several Alphaproteobacteria (Thalassococcus, Rhodobacteraceae, Ruegeria) and Gammaproteobacteria genera (Colwellia, Balneatrix, Thalassomonas) in the bottom water. These data suggest that a more diverse group of both oligotrophic and copiotrophic bacteria may be involved in metabolizing labile organic matter such as peptides in normoxic coastal waters, and several copiotrophic genera belonging to Alphaproteobacteria and Gammaproteobacteria and known to be widely distributed may contribute to faster peptide decomposition in the hypoxic waters.
Introduction
Proteins and peptides are key components of labile dissolved organic matter (DOM) that supports bacterial growth (Azam, 1998). Small peptides (ca. <600 Da) are key immediate products of microbial protein decomposition owing to the size constraints of bacterial cell membrane transport systems, i.e., porins (Weiss et al., 1991). After proteins are degraded to small peptides, these small peptides can be either taken up directly by bacteria, or hydrolyzed further to individual amino acids via extracellular enzymes with subsequent uptake. The interaction between peptide decomposition and bacteria plays an important role in the cycling of carbon and nitrogen, regeneration of nutrients, and preservation of refractory dissolved organic nitrogen (DON) in marine environments (Aluwihare et al., 2005; Nagata, 2008).
Our previous studies have demonstrated that small peptides decompose more quickly in bottom hypoxic than surface normoxic (normal oxygen-saturated) waters in the northern Gulf of Mexico (nGOM), and that the growth of certain bacterial genera such as Vibrio, Marinobacterium, Neptuniibacter, Pseudoalteromonas, Thalassomonas, Amphritea, Roseobacter and Ruegeria, appears to respond to peptide addition (Liu et al., 2013; Liu and Liu, 2016). These results suggest that some bacterial groups may be more effective at metabolizing peptide-derived organic matter in hypoxic seawater, but direct evidence linking specific bacterial lineages to peptide decomposition has not been reported. Knowing which types of bacteria metabolize peptides may be useful in the assessment of factors that control hypoxia formation, as decomposition of labile organic matter leads to consumption of dissolved oxygen (DO) (Wright et al., 2012; Liu et al., 2013). As succession of microbial communities often occurs along with development of oxygen minimum zones (Crump et al., 2007; Zaikova et al., 2010; Parsons et al., 2015), studying the response of microbial communities to labile organic matter at different DO levels can also provide clues about linkages between microbial niche specialization and their resource utilization (Nelson and Wear, 2014).
Previous studies have demonstrated that some bacterial groups can outcompete others during the utilization of labile DOM (Eilers et al., 2000; Teske et al., 2011; Liu et al., 2015). For instance, the bacterial community shifted to Alphaproteobacteria and Betaproteobacteria dominated phylotypes in mesocosm tanks with diatom blooms that produced labile proteins, peptides and polysaccharides exudates (Murray et al., 2007). After bovine serum albumin (BSA) amendment, Gammaproteobacteria became the dominant bacterial class in the Chesapeake Bay water, while Bacteroidetes became dominant in the lower Delaware Bay water (Harvey et al., 2006). However, phylogeny-based incubation studies provide only indirect evidence of the role that different bacterial groups play in labile DOM mineralization, and only a few studies to date have linked specific bacteria groups with labile DOM decomposition directly using radioisotope-labeled substrate and microautoradiography combined with fluorescent in situ hybridization (MAR-FISH) technique (Tabor and Neihof, 1982; Ouverney and Fuhrman, 1999). For example, Cottrell and Kirchman (2000) identified that Bacteroidetes and Gammaproteobacteria actively utilized 3H-labeled protein in two estuarine waters. While powerful, hybridization techniques such as MAR-FISH are a targeted approach and require the design of unique probes to detect individual phylogenetic groups. These techniques often do not allow the identification of active bacteria beyond limited taxonomic depth due to probe hybridization constraints. In contrast, DNA-stable isotope probing (SIP) techniques provide an opportunity to interrogate activity in situ and to identify bacteria at several phylogenetic levels without a priori selection of specific phylotypes. SIP techniques were initially applied to identify bacteria that can degrade one-carbon (C1) compounds or specific pollutants in many environmental studies, such as discovering novel bacteria that degrade methanol, toluene, or alkanes in soils, sediments or marine seeps (Radajewski et al., 2000; Neufeld et al., 2007b; Luo et al., 2009; Redmond et al., 2010; Kleindienst et al., 2014). More recently, the application of DNA-SIP has been extended to marine environments. Examples include studies of urea uptake by marine pelagic bacteria and archaea in Arctic water, comparing bacteria incorporating glucose and cyanobacteria exudates in the Sargasso Sea, and exploring acetate-utilizing bacteria at the oxic–anoxic interface in the Baltic Sea (Gihring et al., 2009; Wawrik et al., 2009; Nelson and Carlson, 2012; Wawrik et al., 2012a; Berg et al., 2013; Connelly et al., 2014), demonstrating the utility of SIP to understand the marine C and N cycles.
The objective of this study was to gain insight into the identities of bacteria that utilize peptides in seawater. In particular, nGOM normoxic and hypoxic waters were targeted because they are characterized by contrasting biogeochemical processes and are known to harbor different bacterial communities that differentially respond to peptide addition. The13C-labeled tetrapeptide alanine-valine-phenylalanine-alanine (AVFA) was used as a model compound and incubated in both surface normoxic and bottom hypoxic seawater in the nGOM. The AVFA sequence is derived from the protein sequence of the large subunit of ribulose-1,5-biphosphate carboxylase/oxygenase (RuBisCO) that is ubiquitous in photosynthesis and has been used to investigate peptide hydrolysis (Liu et al., 2010, 2015; Orellana and Hansell, 2012; Liu and Liu, 2014, 2015). Although individual peptides are often undetectable in natural seawater due to their rapid turnover, they play an important role in supporting bacterial growth as intermediates released from sloppy-feeding or lysis of cells (Nagata, 2008; Sipler and Bronk, 2015).
Materials and Methods
Seawater Sampling
Surface (2 m) and bottom (16 m) seawater were collected at Sta. C6 (28°52′ N, 90°30′ W) in the nGOM during a May 2013 cruise on the R/V Pelican. This station, with a depth of 18 m and ca. 20 km offshore, is heavily influenced by Mississippi River discharge and often subjected to hypoxia during summer (Rabalais et al., 2001). Seawater was sampled using 10 L Niskin bottles mounted on a conductivity-temperature-depth (CTD) rosette (Seabird 911). Temperature, salinity, DO and chlorophyll a of seawater were monitored through the CTD device (Table 1). Seawater was filtered immediately onboard through a 0.2 μm Nylon filter (diameter 47 mm, Whatman) and preserved under -20°C for the analysis of dissolved organic carbon (DOC), total dissolved nitrogen (TDN), total dissolved amino acids (TDAAs), dissolved combined amino acids (DCAAs), dissolved free amino acids (DFAAs) and nutrients.
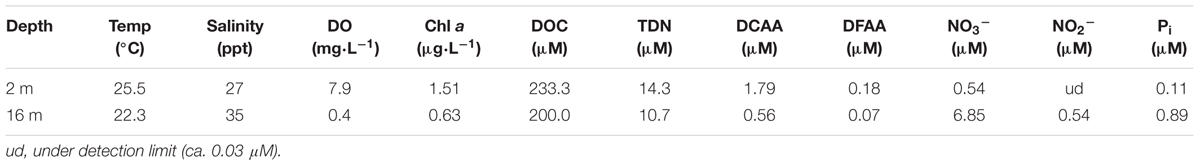
TABLE 1. Chemical parameters of initial surface (2 m) and bottom (16 m) seawater at Sta. C6.
Peptide Incubation
Peptides 12C-AVFA and 13C-AVFA were custom-synthesized (C.S Bio, Menlo Park, CA, USA), and had a >95% compound purity (Liu et al., 2013). In 13C-AVFA, 17 (all three carbons in A, all five carbons in V and six carbons of the aromatic ring in F) out of total 20 carbon atoms were labeled isotopically. As DNA of specific bacteria groups might only be partially labeled (<100%) with 13C, bacteria groups incorporating 13C can be spread through all SIP fractions with different density (see related method below). The unlabeled 12C-AVFA incubation was thus included as a reference for comparison with 13C-AVFA incubation to confirm isotopic enrichment in DNA. The unlabeled SIP gradient should contain less DNA in the more dense fractions than that in the samples incubated with 13C substrates (Neufeld et al., 2007a). AVFA was incubated onboard in the surface normoxic and bottom hypoxic seawater. Briefly, either 12C-AVFA or 13C-AVFA was respectively amended in a series of 125 mL amber round bottles filled with 120 mL seawater at final concentrations of 0.25–0.47 μM. Duplicate incubations were conducted in the dark for 48 h at 24°C, close to the ambient seawater temperature (Table 1). At different time points (0, 8, 13, 24, and 48 h), 1 mL aliquots of unfiltered water were collected and fixed with formaldehyde at a final concentration of 3% and stored at 4°C for bacterial abundance analysis. The remaining 119 mL were filtered through the 0.2 μm Nylon filter and preserved at -20°C for the analysis of peptides, amino acids, ammonium, and orthophosphate (Pi). The filters were preserved in 750 μL 1× STE (10 mM Tris-HCl [pH 8.0], 0.1 M NaCl, 1 mM EDTA [pH 8.0]) buffer at -20°C for DNA extraction and sequencing. DO was not monitored throughout the incubation, but the parallel incubation experiment showed that it remained relatively constant throughout the 72 h (Liu and Liu, 2016). Two kinds of controls were included for the incubation experiment: a seawater control without peptide amendment and a killed control with 0.48–0.58 μM 12C-AVFA and 180 μM HgCl2 to inhibit bacterial activity (Lee et al., 1992). The incubation and aliquot sampling for the controls followed the same procedures as described above, but only AVFA was analyzed in the killed control.
Chemical Analyses
DOC and TDN of the filtered initial seawater were analyzed using a Shimadzu total organic carbon (TOC-V) analyzer coupled with a TNM-1 TDN analyzer with <6% error between duplicates (Table 1). DFAA were calculated as sum of individual amino acids analyzed in high performance liquid chromatography (HPLC, Shimadzu Prominence) equipped with a fluorescence detector after pre-column o-phthaldialdehyde (OPA) derivatization (Lindroth and Mopper, 1979; Lee et al., 2000). TDAA were analyzed in the same way as DFAA but after hydrolysis in 6 N HCl under nitrogen at 110°C for 20 h (Kuznetsova and Lee, 2002). DCAA were calculated as TDAA subtracting DFAA. Nitrate, nitrite and orthophosphate (Pi) were measured following established protocols (Strickland and Parsons, 1968; Jones, 1984).
Alanine-valine-phenylalanine-alanine was analyzed in an HPLC-mass spectrometry (HPLC-MS) system (Shimadzu Prominence) following the method in Liu and Liu (2014). In brief, the mobile phase A was 10 mM ammonium acetate and mobile phase B was methanol. Samples were eluted through a C18 column (Alltima 5 μm, 150 mm × 4.6 mm) and a six-way valve was programmed to direct the sea salt peak to waste before introducing the AVFA peak to the MS detector that is equipped with an electrospray ionization (ESI) source and a quadrupole mass analyzer. 12C-AVFA and 13C-AVFA were quantified in positive ion mode under selective ion monitoring (SIM) at m/z = 407 and 424, respectively.
Alanine-valine-phenylalanine-alanine hydrolysis products including peptide fragments (AV, VF, FA, VFA) and amino acids (A, V, F) were analyzed by HPLC after pre-column OPA derivatization (Liu et al., 2013). Standard deviations of amino acid analysis among replicates were 10–20%. Ammonium, a main metabolite of AVFA, was analyzed using HPLC with post-column OPA derivatization (Gardner and St. John, 1991).
Bacterial Abundance Analysis
Bacterial cells in the formaldehyde-preserved samples were stained with SYBR Green II (Molecular Probes, 1:100 v/v) and enumerated in a flow cytometer (BD Accuri C6) under blue laser excitation at 488 nm (Marie et al., 1997; Liu et al., 2013). Bacterial cells were counted in a fixed volume mode with a flow rate below 300 events per second and cell counts were determined in a dot plot of side scatter (SSC-H) vs. green fluorescence signal (FL1-H) on a logarithmic scale.
DNA Extraction and Ultracentrifugation in CsCl Gradients
DNA was extracted from filtered cells using MoBio PowerSoil® DNA isolation kits (MoBio Laboratories, Carlsbad, CA, USA). Leftover STE buffer was spun in a centrifuge and the supernatant was discarded. The pellet was re-dissolved in 100 μL lysis solution from MoBio Powerbead tube and then combined with filter in the tube for DNA extraction. A subsample (ca. 10 μL) at each incubation time point was saved for microbial community structure analysis (unamended control 0 h, one duplicate of 12C-AVFA 2 m 0 h, and one duplicate of 12C-AVFA 16 m 0 h samples were lost during DNA extraction), and the remainder (ca. 80 μL) was for the ultracentrifugation in CsCl gradients. Duplicate DNA samples (each account for ca. 89% (80 μL out of 90 μL) of extracted DNA) from all three time points (13, 24, and 48 h) for surface and bottom seawater incubations respectively were pooled (i.e., six samples were pooled) to obtain sufficient DNA for ultracentrifugation and fractionation. The pooled DNA was precipitated using isopropanol, and the DNA pellet was then re-suspended in 50 μL TE buffer (50 mM Tris-HCl, 15 mM EDTA [pH 8.0]) as previously described (Wawrik et al., 2009). Four pooled DNA samples (12C-AVFA surface, 13C-AVFA surface, 12C-AVFA bottom, 13C-AVFA bottom) were prepared for ultracentrifugation. Note that ultracentrifugation and following fractionation were not performed on killed control samples and unamended samples. CsCl gradient ultracentrifugation and fractionation followed protocols as described previously (Buckley et al., 2007; Luo et al., 2009; Wawrik et al., 2012a). In brief, 61–170 ng DNA, quantified through a Qubit® 2.0 Fluorometer (Life Technologies), were mixed with 0.26 mL TE buffer and 4.45 mL of 1.295 g mL-1 CsCl in gradient buffer A (15 mM Tris-HCl [pH 8.0], 15 mM KCl, 15 mM EDTA [pH 8.0], 2 mg mL-1 ethidium bromide) in 4.7 mL polyallomer Optiseal tubes (Beckman). The tubes were centrifuged in a Beckman rotor VTi 65.2 at ca. 140,000 × g for 48 h. After ultracentrifugation, 30 150 μL fractions were collected from each tube in a Beckman fraction recovery system by replacing samples with mineral oil on top of the tubes at a constant rate using a peristaltic pump. The density of each fraction was calculated based on the refractive index that was measured in a Reichert AR200 refractometer (Wawrik et al., 2009). DNA was purified from each CsCl fraction by isopropanol precipitation and dissolved in 50 μL sterile nuclease-free water.
Quantitative PCR (qPCR) of 16S rRNA Gene
The purified DNA from each SIP fraction was used to determine 16S rRNA gene copy numbers of bacteria via quantitative polymerase chain reaction (qPCR). Primers were 27F (5′-AGA GTT TGA TCM TGG CTC AG-3′) and 519R (5′-GWA TTA CCG CGG CKG CTG-3′) (Nakatsu and Marsh, 2007). Every 30 μL reaction mix for qPCR included 13.9 μL 2X Power SYBR Green PCR master mix (Applied Biosystems), 13.9 μL nuclease-free water, 200 nM (final concentration) of each primer, and 2 μL DNA template. qPCR was conducted in a real-time PCR system (Applied Biosystems, ABI 7300) followed the program: 2 min at 50°C, 8 min at 95°C, 40 cycles of 30 s at 95°C, 1 min at 55°C and 1 min at 72°C. Genomic DNA of Roseobacter denitrificans Och 114 (DSMZ 7001) was used as the standard DNA for bacteria (standard concentrations ranging from 10-4 to 10 ng μL-1). The qPCR detection limit for bacteria was ca. 0.002 ng (corresponding gene copy number of 442) and cycle threshold was ca. 31. The qPCR data was then normalized to the highest quantities of 16S rRNA gene copy numbers observed among all fractions in that gradient, in order to account for the differential abundances and distributions of their DNA in gradients (Wawrik et al., 2009; Connelly et al., 2014). These normalized numbers were named as ratios of quantities in which the highest normalized frequency measured equaled 1.
16S rRNA Gene PCR, Barcoding, and Illumina Sequencing
DNA from each incubation time point and each fraction collected from SIP gradients was amplified by PCR using Phusion high-fidelity DNA polymerase (Thermo Scientific) and barcoded for Illumina sequencing. PCR reactions utilized the universal forward primer 519F containing a 5′ M13 tag (5′-GTA AAA CGA CGG CCA GCA CMG CCG C-3′) and the reverse primer Bac-785R (5′-TAC NVG GGT ATC TAA TCC-3′) as previously described (Wawrik et al., 2012b; Klindworth et al., 2013). PCR started with 94°C for 2 min, followed by 28–32 cycles of 95°C for 30 s, 52.8°C for 30 s, and 72°C for 30 s, then 72°C for 5 min. The number of PCR cycles was optimized based on qPCR results and agarose gel check of PCR products to make sure enough PCR products were obtained but not reaching PCR plateau. PCR products were purified by QIAquick PCR purification kit (Qiagen) and barcoded using a unique M13-contianing primer for each sample that contained a 12 bp barcode for bioinformatical parsing of data (Wawrik et al., 2012b). Barcode tagging was checked by gel electrophoresis, amplifications were mixed at equimolar ratios, and sent to the Oklahoma Medical Research Foundation for MiSeq PE250 Illumina sequencing. Sequence reads were trimmed to a quality score of Q30 and adapter and primer sequences were trimmed from raw Illumina sequences. Overlapping forward and reverse reads were stitched and all non-overlapping sequence reads were discarded. Processed sequences were clustered into OTUs using UCLUST, checked for chimeras using USEARCH and classified into taxonomy through the QIIME pipeline (Caporaso et al., 2010b). A randomly chosen set of representative sequences from each OTU was aligned to the SILVA small-subunit rRNA reference alignment1 using the PyNAST algorithm (Caporaso et al., 2010a). Sequences were assigned to the genus level at the 95% identity as a compromise between resolution and conservative interpretation due to the short reads (250 bp) used here (Connelly et al., 2014). Sequences were deposited in National Center for Biotechnology Information (NCBI) GenBank under BioProject accession number PRJNA297372.
Bacterial community structures (relative abundance of genera presented in percentage) of the initial samples were assumed to be the same among unamended control, 12C-AVFA and 13C-AVFA samples. As unamended control and one duplicate of the 12C-AVFA 0 h samples were lost, initial bacterial community data of 13C-AVFA samples was more reliable to represent the initial bacterial community structure of all. To be consistent with the pooled SIP samples, results of the bacterial community structures from the three time points (13, 24, and 48 h) were also pooled and compared with others by non-metric multidimensional scaling (NMDS) using Matlab®. Analysis of similarity (ANOSIM) was applied to compare the bacterial community structures between surface and bottom seawater and between unamended control and AVFA treatment time-point samples using vegan package in R (Oksanen et al., 2016).
Calculating percentage enrichment of each bacterial taxa in the 13C-AVFA samples relative to the 12C-AVFA SIP samples followed the protocol of Bell et al. (2011). In brief, 16S rRNA gene copy numbers for each SIP fraction were quantified through qPCR. Then the proportion of each bacterial taxonomic group sequences in a given density range was multiplied by the 16S rRNA gene copy number in that same density range, and the derived copy number of each bacterial taxonomic group was normalized to total gene copy number within that density range to correct for the slight difference in total DNA between the 12C-AVFA and 13C-AVFA samples and this normalized gene copy number was referred as relative gene copy number. Percentage enrichment of a certain bacterial taxonomic group was defined as dividing the difference of the relative copy number summed in the heavy density fractions between the 13C-AVFA samples and the 12C-AVFA samples by the relative copy number in the 12C-AVFA samples within the same density range. The percentage enrichment was used as an indicator of the relative amount of 13C enrichment among bacterial groups. A higher percentage enrichment in one bacterial taxa relative to other taxa indicates increased cell replication responding to amended 13C-AVFA. For example, 100% enrichment of a given taxonomic group in the same density range indicates that its 16S rRNA gene copy number in the 13C treatment is twice as abundant as that in the 12C treatment. To estimate the error/noise level of the percentage enrichment, 95% confidence interval was calculated from all positive percentage enrichment values among bacterial taxa for both surface and bottom samples.
Results
Peptide Decomposition
The 12C- and 13C-AVFA decomposition patterns were nearly identical during the 48 h incubation, as expected (Figures 1A,B). The AVFA concentrations decreased linearly with time in both the surface (2 m) and bottom (16 m) seawater, but the decomposition rate in the bottom seawater (0.018–0.035 μM h-1) was twice as high as in the surface seawater (0.011–0.017 μM h-1). The decomposition rate of AVFA in the bottom seawater was significantly higher than that in the surface seawater (t-test, p < 0.03). AVFA was completely degraded within 24–48 h in the surface water and within 13–24 h in the bottom water. In contrast, AVFA concentration in the killed control remained nearly unchanged during the 48 h incubation (Figure 1C), indicating that the peptide disappearance in the seawater was due to microbial activity.
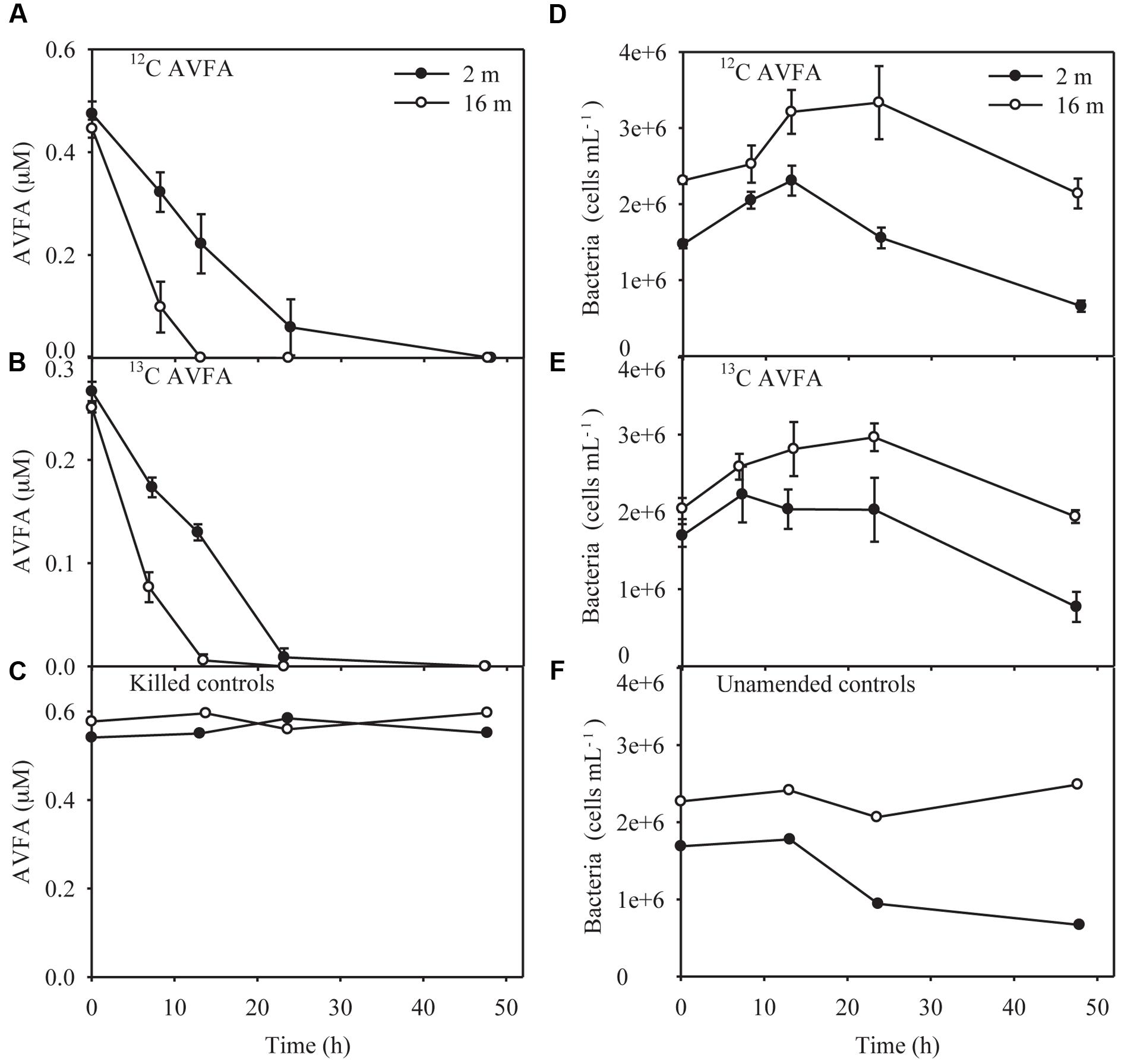
FIGURE 1. Alanine-valine-phenylalanine-alanine (AVFA) concentrations (A–C) and bacterial abundance counted in flow cytometer (D–F) with incubation time in the surface 2 m and bottom 16 m seawater of 12C-AVFA, 13C-AVFA, and control samples. Note controls samples in (C) are killed controls, and in (F) are unamended (no-AVFA) controls. Data points were presented as average ± absolute error of duplicate samples except control samples.
AV, FA, VF, and VFA produced during hydrolysis of AVFA (Liu et al., 2013) remained at low levels <0.012 μM throughout the incubation, but more amino acids and peptide fragments were produced in the surface than in the bottom incubation (Figure 2). Concentrations of amino acid F were significantly higher in the surface than in the bottom seawater (t-test, p = 0.04). Concentrations of free amino acids (A, V, and F) were 2–30 times greater than those of peptide fragments. As individual amino acids have been shown in similar incubations to be taken up at different rates (Liu et al., 2013), particularly amino acid A was taken up much faster than V and F, production of amino acids might not follow the stoichiometry of added peptides. F was the dominant amino acid, reaching up to 0.17 μM in the surface at 24 h and 0.082–0.11 μM in the bottom at 8 h, and then decreased to the background level at the end of the incubation. V and A followed a similar pattern to F, but with smaller changes, indicating they were taken up faster than F, consistent with a previous study at this station (Liu et al., 2013). Compared to the AVFA treatment, concentrations of amino acids in the control without peptide amendment remained relatively low (<0.011 μM) and constant throughout the incubation.
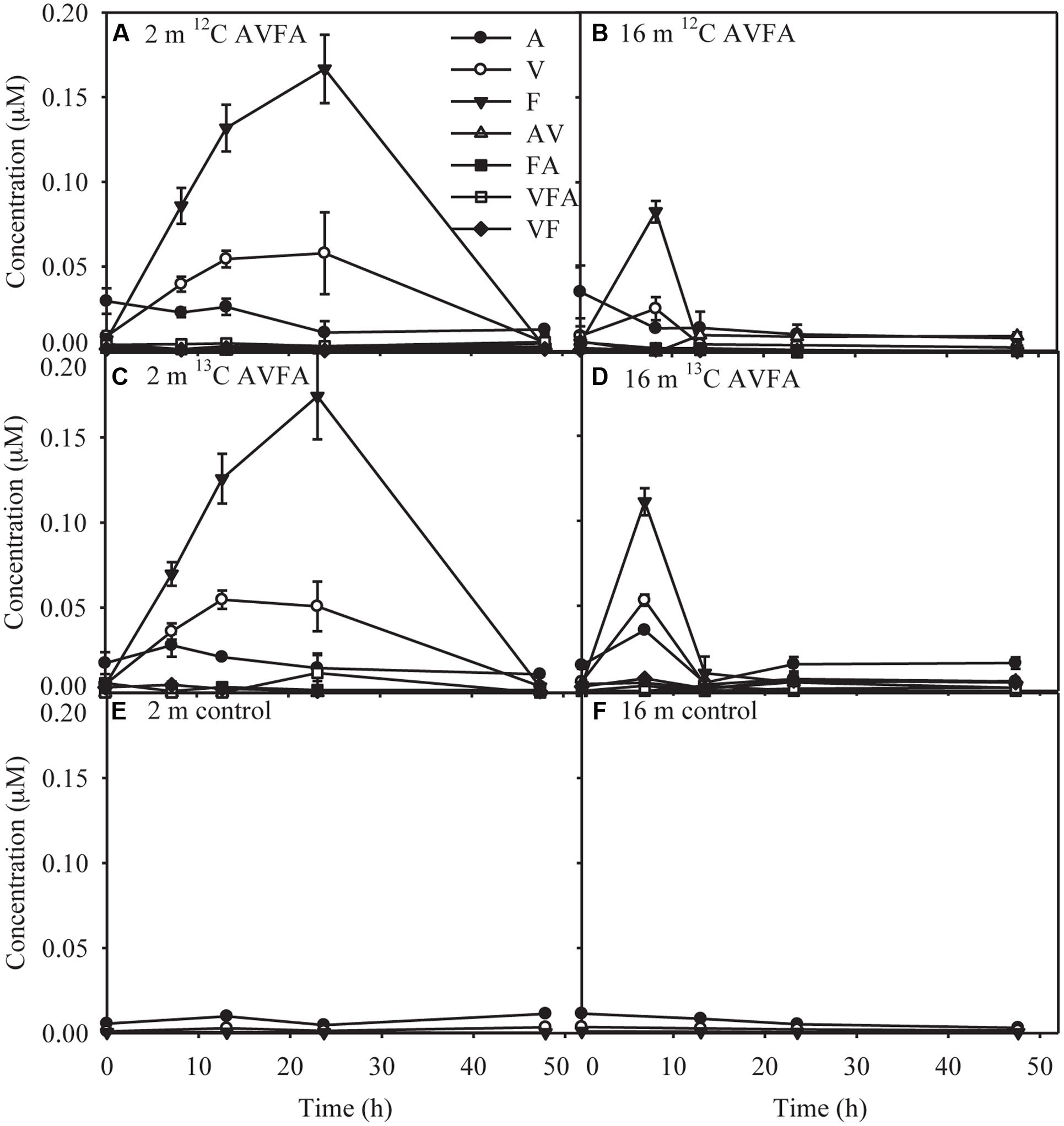
FIGURE 2. Concentrations of produced amino acids and peptide fragments with incubation time in the (A) surface 2 m seawater of 12C-AVFA incubation, (B) bottom 16 m seawater of 12C-AVFA incubation, (C) surface 2 m seawater of 13C-AVFA incubation, (D) bottom 16 m seawater of 13C-AVFA incubation samples, and amino acid concentrations with time in the (E) surface 2 m seawater of no-AVFA control and (F) bottom 16 m seawater of no-AVFA control samples. Data points were presented as average ± absolute error of duplicate samples except control samples.
Ammonium is a key metabolite of peptides (Liu et al., 2013). During the first 24 h in the surface seawater incubation, ammonium concentrations increased by 0.66–1.45 μM in the 12C- and 13C-AVFA samples (Supplementary Figures S1A–C). In contrast, ammonium concentrations in the bottom seawater changed little before AVFA was completely degraded (0–13 h). However, the difference of produced ammonium between surface and bottom seawater was not significant (t-test, p = 0.38). After 13 h, ammonium concentrations kept increasing to 2.6–2.9 μM in the surface seawater and remained constant at about 2.8 μM or increased by 1.5 μM to reach 4.1 μM in the bottom seawater. In the control without AVFA, ammonium concentrations increased by 0.97 μM in the surface seawater and decreased by 0.63 μM in the bottom seawater during the 48 h incubation.
Pi is an essential element for bacterial growth (Elser et al., 2000; Karl, 2014; Liu and Liu, 2016). However, Pi concentrations remained relatively constant throughout the 48-h incubation in both the peptide and control treatments (Supplementary Figures S1D–F). Pi concentrations in the bottom water (1.1–1.5 μM) were more than one order of magnitude higher than those in the surface water (0.02–0.09 μM).
Bacterial Abundance and Community Structure
In the surface 12C- and 13C-AVFA incubations, bacterial abundance increased by 31–57% within the initial 8–13 h, and then decreased afterward, while in the bottom, bacterial abundance increased by 44–45% during the initial 24 h and then decreased afterward (Figures 1D,E). Bacterial abundances in the control either decreased over time in the surface seawater or remained nearly constant in the bottom seawater (Figure 1F). Ambient surface water bacterial communities were dominated by Synechococcus (15–49%), whereas bottom samples were more evenly populated by Rhodobacteraceae (11–13%), Acidimicrobiaceae OCS155 marine group (3–8%), Saprospiraceae (5–7%), Planctomycetaceae (2–7%), SAR11 clade Surface 1 (3–6%), and Acidimicrobiales TM214 (3–5%) (Supplementary Figures S2A,B). Different initial surface community structures between 12C-AVFA and 13C-AVFA samples might be related to the loss of one duplicate 12C-AVFA DNA sample or contamination of Synechococcus in one sample. Thus, initial surface community structure of the 13C-AVFA duplicate samples might be more reliable than that of the 12C-AVFA samples. Throughout the incubation, the relative abundance of Rhodobacteraceae, Thalassococcus, and Ruegeria increased by 6–14, 7–13, and 1–3%, respectively, in both surface and bottom seawater, while other bacterial genera developed differently in the surface and bottom seawater incubations. For instance, OTUs classified within the Roseovarius clade increased by 7% only in the surface seawater, whereas Colwellia increased by 3% only in the bottom seawater. The surface and bottom bacterial community structures were well-separated in the NMDS plot (Supplementary Figure S2C); ANOSIM showed significant difference between the surface and bottom bacterial community structures (p = 0.002), further suggesting that bacterial community structures developed differently between the two water layers. To further evaluate the bacterial community development in unamended controls vs. AVFA treatments, NMDS was also plotted separately on surface and bottom bacterial communities (Figure 3). At the later incubation time points, unamended treatments and AVFA treatments formed separate clusters especially at NMDS1 axis, but this separation was not significant as shown in ANOSIM analysis (p > 0.05), indicating peptide amendement did not significantly change bacterial community structures during the 48-h incubation as compared to controls.
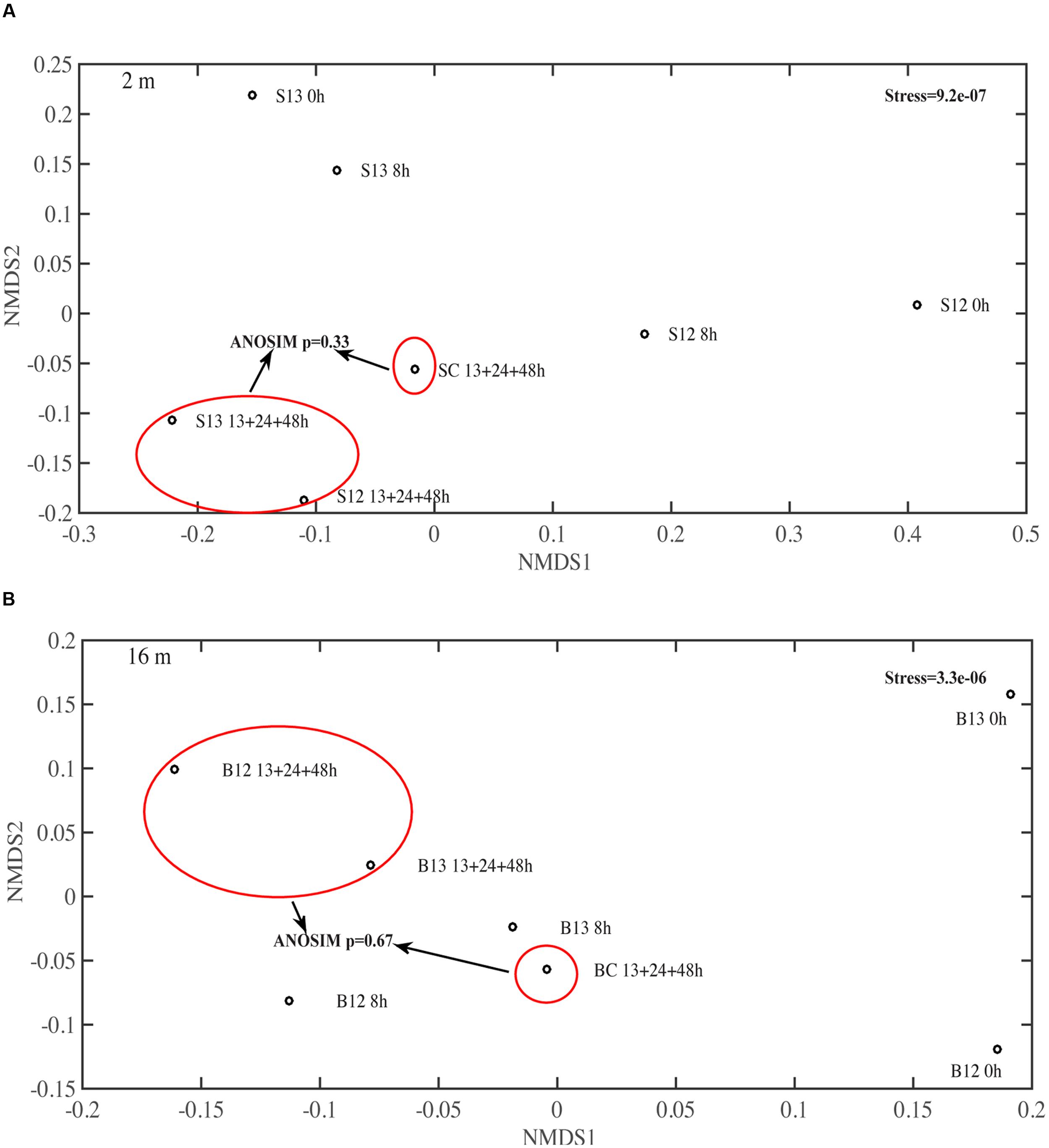
FIGURE 3. Non-metric multidimensional scaling (NMDS) on the bacterial community structure at genera level in (A) surface 2 m and (B) bottom 16 m time-point samples. 13, 24, and 48 h samples were pooled to be consistent with SIP data. S12, surface 2 m 12C-AVFA samples; S13, surface 2 m 13C-AVFA samples; SC, surface 2 m no-AVFA control samples; B12, bottom 16 m 12C-AVFA samples; B13, bottom 16 m 13C-AVFA samples; BC, bottom 16 m no-AVFA control samples. Stress was 9.2e-07 at 2 m and 3.3e-06 at 16 m, indicating excellent fitting of solution to recreate the dissimilarity (stress < 0.02). Bacterial composition formed separate clusters between unamended control and AVFA incubations at the later time points at both 2 and 16 m as shown in red circles, but ANOSIM showed this separation was not significant (p > 0.05).
Identifying Bacteria that Incorporated Peptides through DNA-SIP
Quantitative polymerase chain reaction of SIP fractions indicates that the density distribution of the 16S genes in bulk DNA of the 13C-AVFA samples shifted to heavier densities as compared to the 12C-AVFA samples in both the surface and bottom incubations (Figures 4A,B). The 16S PCR products from respective fractions were bar-coded and sequenced using Illumina Miseq to generate sequence libraries. A positive percentage enrichment is an indicator of the bacterial potential in incorporating 13C (Bell et al., 2011). Estimated error/noise of percentage enrichment was 46 and 65% at the class and genus level, respectively, as calculated from the 95% confidence interval. From this error/noise estimate, percentage enrichment above 84% at the class level and above 168% at the genus level was above the 95% confidence interval. 13C uptake, as suggested by SIP, was more evenly distributed among the bacterial classes in the surface seawater than in the bottom seawater (Figures 4C,D). Flavobacteria, Sphingobacteria, Alphaproteobacteria, Acidimicrobiia, Verrucomicrobiae, Cyanobacteria, and Actinobacteria showed highest enrichment ranging from 96 to 275% in the surface water, whereas Alphaproteobacteria and Gammaproteobacteria showed highest enrichment ranging from 175 to 279% in the bottom water.
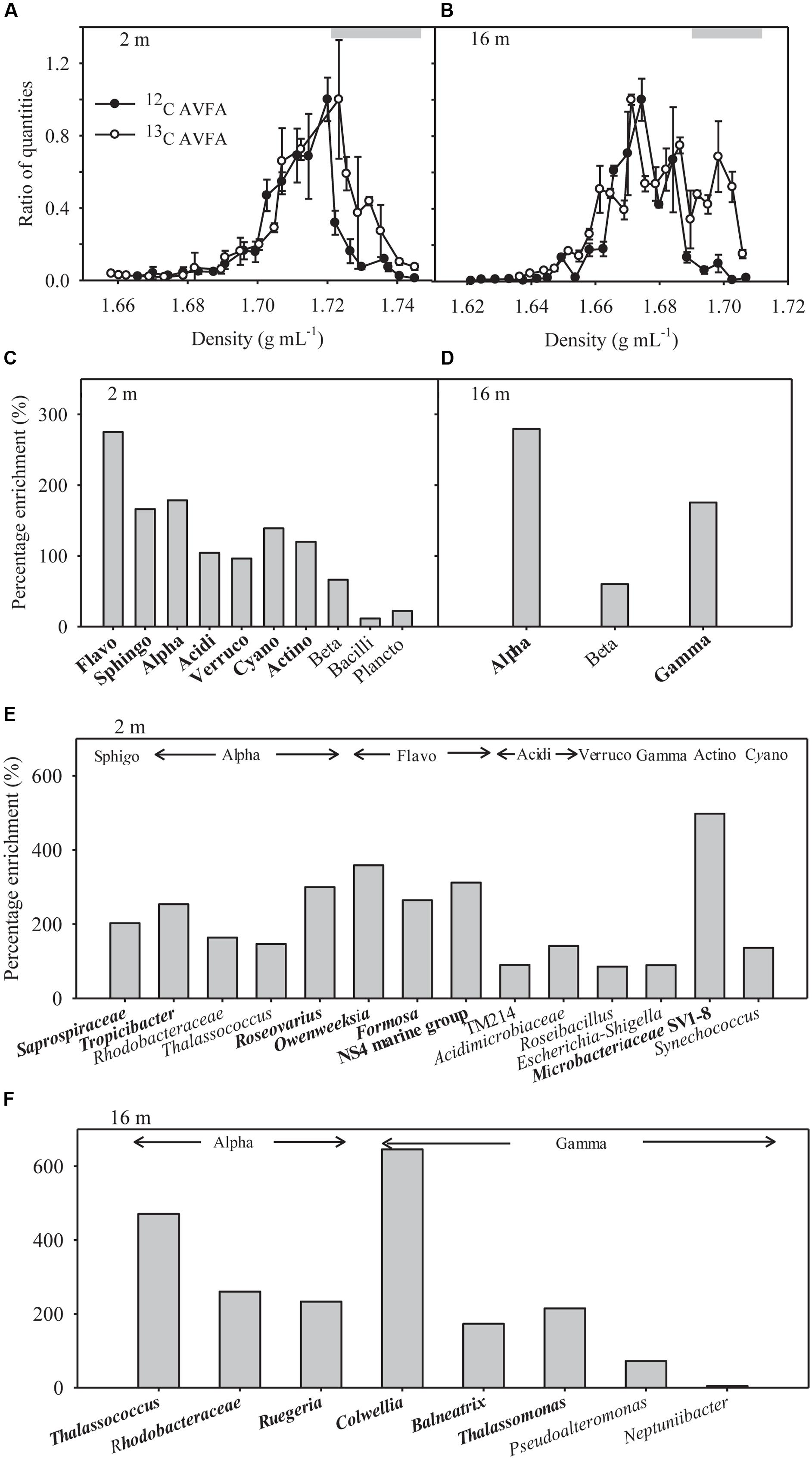
FIGURE 4. (A,B) Quantitative polymerase chain reaction (qPCR) analysis results shown as relative quantities vs. density of SIP gradient fractions for bacterial 16S rRNA gene copies in the surface 2 m and bottom 16 m samples. The ratio of quantities was calculated as 16S rRNA gene copy numbers in specific fraction normalized to the highest quantities of 16S rRNA gene copy numbers observed among all fractions in that sample, and 1 equals the highest value observed. Data points were presented as average ± standard deviation of three replicate qPCR measurements. Gray bars indicate heavy density ranges used for percentage enrichment calculations in (C–F). (C,D) Percentage enrichment of major bacterial classes in the heavy density range in the 13C-AVFA SIP fractions compared to the 12C-AVFA SIP fractions. The 13, 24, and 48 h DNA samples were pooled together for SIP results. Bacterial class chosen were at least 0.1% abundance of the community. Flavo, Flavobacteria; Sphingo, Sphingobacteria; Alpha, Alphaproteobacteria; Acidi, Acidimicrobiia; Verruco, Verrucomicrobiae; Cyano, Cyanobacteria subsection I; Actino, Actinobacteria; Beta, Betaproteobacteria; Plancto, Planctomycetacia; Gamma, Gammaproteobacteria. Class with percentage enrichment >84% at 95% confidence interval was in bold. (E,F) Percentage enrichment of major bacterial genera within each class (listed above the bars) in the heavy density range of the 13C AVFA sample SIP fractions compared to the 12C AVFA sample SIP fractions in the surface 2 m and bottom 16 m seawater. Bacterial genera chosen were at least 0.1% abundance of the community. Bacterial class abbreviation was same as before. Genus with percentage enrichment >168% at 95% confidence interval was in bold.
Communities taking up 13C in the surface and bottom seawater also differed at the level of dominant genera (>0.1% of the total bacterial community) (Figures 4E,F). In the surface seawater, Saprospiraceae, Tropicibacter, Roseovarius, Owenweeksia, Formosa, Flavobacteria NS4 marine group, and Microbacteriaceae SV1-8 dominated the 13C uptake. In the bottom samples, major 13C enriched groups included Thalassococcus, Rhodobacteraceae, Ruegeria, Colwellia, Balneatrix, and Thalassomonas. The extent of taxonomic enrichment in the heavier fractions varied widely among different bacterial genera, ranging from 86 to 498% in the surface incubation and from 4 to 646% in the bottom incubation. Within the same class, the enrichment of Roseovarius and Thalassococcus was almost twice as high as that of other genera in Alphaproteobacteria, and the enrichment of Colwellia was more than three times higher than that of other genera in Gammaproteobacteria.
Discussion
Faster AVFA Decomposition in the Hypoxic than in the Normoxic Seawater
Peptide decomposition in the bottom incubation was twice as fast as that in the surface incubation (Figures 1A,B). Normalized to initial bacterial abundance, cell-specific rate of peptide decomposition in the bottom incubation (9.0 × 10-9–1.5 × 10-8 nM h-1) was 1.3–1.4 times as high as that in the surface incubation (6.4 × 10-9–1.2 × 10-8 nM h-1). Also, AVFA decomposition produced less hydrolyzed fragments, including amino acids and peptides, in the bottom than surface incubations (Figure 2), indicating direct uptake of AVFA or tightly coupled hydrolysis-uptake in the bottom water but extracellular hydrolysis in the surface water. In contrast to previous studies, which used relatively high concentrations of added peptides (5–10 μM) (Liu et al., 2013; Liu and Liu, 2016), the much lower concentrations of AVFA (0.25–0.47 μM) added here accounted for only 14–84% of ambient DCAA that consist of all hydrolyzable proteins and peptides in seawater (Table 1). As individual peptides may exist only at trace levels in ambient seawater, adding low concentration of peptides may simulate natural processes better. However, the low concentration amendments conducted here resulted in uptake patterns generally consistent with previous studies (Liu et al., 2013; Liu and Liu, 2016). Peptide decomposition all followed zero-order reaction with a linear decrease of concentration with time, indicating that the peptide decomposition in this study and previous studies was limited by the availability of enzymes produced by bacteria and the added peptide concentrations were probably all above a threshold of enzyme capacity in the ambient seawater. Faster peptide decomposition and less fragments produced in the bottom than in the surface incubations at both high and low added concentrations suggest that added peptides within this concentration range (0.25–10 μM) may trigger similar peptide decomposition mechanisms and bacterial response.
The peptide decomposition mechanism can be interrogated through a mass balance of the fate of added nitrogen, which may include: (1) extracellular hydrolysis to produce peptide fragments and amino acids, (2) remineralization to ammonium, and (3) incorporation into bacterial biomass. The percentage of extracellular hydrolysis can be estimated using the amino acid F and peptide fragments containing F, as bacterial uptake of F is limited within 24 h (Liu et al., 2013). The degree of remineralization can be estimated via changes in ammonium concentrations in peptide treatments compared to controls assuming nitrification is negligible during the 24 h (Liu et al., 2013). To calculate the incorporation percentage to microbial biomass, we assume a carbon conversion value of 20 fg C per bacterial cell and a C/N ratio of 4 for bacteria (Lee and Fuhrman, 1987). Based on these parameters, extracellular hydrolysis (40–56%) dominated the decomposition of AVFA in the surface water, whereas biomass production (4–20%) dominated in the bottom water throughout the incubation, leaving a major fraction (29–81%) of the AVFA nitrogen uncounted for in both layers, possibly in other forms of DON (Figure 5), which might be semi-labile and refractory DON formed via microbial production or transformation processes (Jiao et al., 2010; Benner and Amon, 2015; Walker et al., 2016). For example, at 24 h, ca. 40% of decreased AVFA in the surface seawater was hydrolyzed to peptide fragments and amino acids, ca. 6% was converted to ammonium, 2–11% to bacterial biomass (8.1 × 104–3.3 × 105 cells per mL), and about 50% to other DON. In contrast, in the bottom seawater, less than 5% was hydrolyzed to peptide fragments and amino acids, hardly any ammonium was produced, and 18–28% was incorporated into bacterial biomass (7.7 × 105–9.0 × 105 cells per mL) at 13 h when AVFA disappeared, resulting in about 70–80% of AVFA nitrogen as other DON. This contrasting pattern suggests that the fast disappearance of AVFA in the bottom water incubation may relate to the higher percentage of peptide incorporation into bacterial biomass, i.e., bacterial growth. The efficiency of AVFA decomposition may depend on the fraction of nitrogen allocated to those fast-growing bacteria.
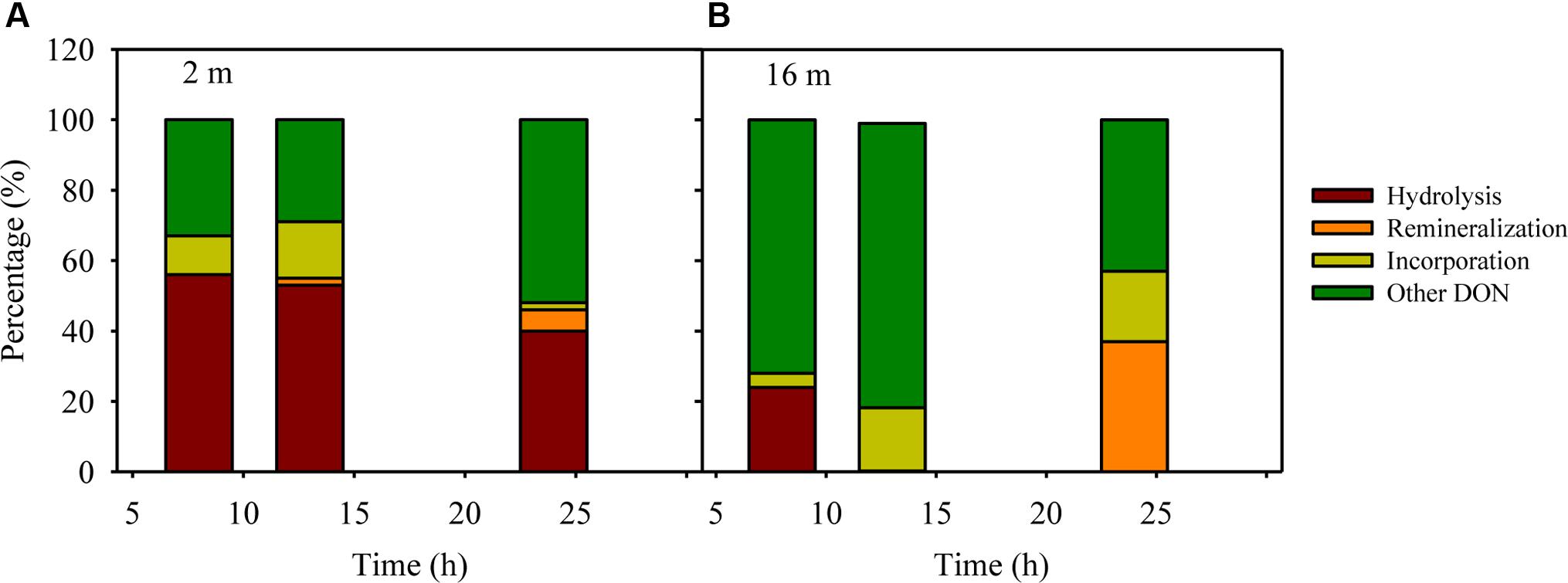
FIGURE 5. An example of mass balance (including percentages of decreased peptide due to hydrolysis, remineralization to ammonium, incorporation into bacterial biomass and other unaccounted transformation to DON besides AVFA and amino acids) throughout the 12C-AVFA decomposition in the (A) surface (2 m) and (B) bottom (16 m) seawaters.
Uptake of Peptide in the Normoxic vs. Hypoxic Seawater
In the surface seawater incubation, the incorporation of 13C was greatest for Flavobacteria, Sphingobacteria, Alphaproteobacteria, Cyanobacteria, Acidimicrobiia, Verrucomicrobiae, and Actinobacteria (Figure 4C), indicating that both oligotrophic (such as Cyanobacteria) and copiotrophic bacteria were involved in peptide decomposition in the surface seawater. Copiotrophic, perhaps r-selected, bacteria use labile organic matter in nutrient-enriched environments. This is in contrast to more K-selected oligotrophic species that maintain efficient metabolism by growing more slowly on complex refractory substrates (Fierer et al., 2007; Wang et al., 2015). At the genus level, Saprospiraceae (Sphingobacteria), Tropicibacter (Alphaproteobacteria), Roseovarius (Alphaproteobacteria), Owenweeksia (Flavobacteria), Formosa (Flavobacteria), Flavobacteria NS4 marine group (Flavobacteria), and Microbacteriaceae SV1-8 (Actinobacteria) took up the most 13C in the surface seawater (Figure 4E). Sphingobacteria showed a responsive role during peptone incubation in the seawater (Simon et al., 2012). The Roseobacter clade is often associated with plankton aggregates (Moran et al., 2007; Teeling et al., 2012; Yau et al., 2013), so Tropicibacter and Roseovarius belonging to the Roseobacter clade may be opportunistic in nutrient exploitation. Therefore, the observation that these populations can utilize the added peptide is expected. Flavobacteria are often effective in degrading high-molecular-weight DOM including proteins (Cottrell and Kirchman, 2000). Some Actinobacteria can produce a wide range of bioactive metabolites including extracellular peptidases that are sometimes involved in pathogenic processes (Ventura et al., 2007; Chen et al., 2011), suggesting their potential in peptide utilization. Consistent with our results, Orsi et al. (2016) also found that diverse bacterial taxa, such as Flavobacteria, Verrucomicrobia, Gammaproteobacteria, Alphaproteobacteria, Actinobacteria, and Planctomycetes, utilized added dissolved proteins in coastal California waters. The phylogenetic widespread of bacterial classes incorporating peptides in this study agrees with ecological theory and previous studies indicating that heterogeneity of the coastal oceans favors generalist bacteria in DOC utilization (Mou et al., 2008). Alternatively, this diverse bacterial pattern may result from the significant production of individual amino acids from extracellular hydrolysis (Figures 2A,C). Since uptake of amino acids is generally constitutive among marine bacterial taxa (Payne and Gilvarg, 1971; Poretsky et al., 2010) and uptake of amino acids is also part of peptide metabolizing process, bacterial groups possessing the ability to take up amino acids A, V, and F should be widespread, thus increasing the range of bacteria taxa showing positive percentage enrichment in the surface seawater.
In contrast to the surface seawater incubation, bacteria incorporating 13C in bottom waters were associated with fewer taxonomic groups, primarily belonging to the Alphaproteobacteria and Gammaproteobacteria (Figure 4D). The bacteria that metabolized the peptide differed between the surface normoxic and bottom hypoxic seawater. This depth-differential response was strikingly consistent with the study of Nelson and Carlson (2012), showing that while both oligotrophic and copiotrophic bacteria incorporated amended DOC sources such as Synechococcus exudate, Synechococcus lysate, and gluconic acid in the euphotic seawater, no oligotrophic bacteria showed evidence of incorporation of amended DOC sources in the mesopelagic seawater. This concomitant pattern in different seawater systems implies that different water parameters between depths are likely to be the driving force in bacterial response to DOM sources. The dominant percentage enrichment of Alphaproteobacteria and Gammaproteobacteria in the bottom seawater suggested they can outcompete other bacteria in incorporating AVFA, thus leading to the faster decomposition of the peptide in the bottom seawater. This result is consistent with previous studies in DOM utilization, which quantified this process either directly through tracing radioisotope incorporation by bacteria or indirectly via analyzing changes of bacterial community structure (Gihring et al., 2009; McCarren et al., 2010; Carney et al., 2015). For example, the percentage of Gammaproteobacteria consuming proteins was higher than their abundance percentage among all bacterial phylogenetic groups in estuarine and coastal environments, indicating they are efficient at metabolizing proteins (Cottrell and Kirchman, 2000). Alphaproteobacteria or Gammaproteobacteria can dominate the bacterial community during DOM incubation in certain marine environments, indicating they can outcompete other bacteria in using DOM substrates (Harvey et al., 2006).
At the genus level, the highest percentage enrichment in the bottom seawater occurred to the Thalassococcus (Alphaproteobacteria), Rhodobacteraceae (Alphaproteobacteria), Ruegeria (Alphaproteobacteria), Colwellia (Gammaproteo-bacteria), Balneatrix (Gammaproteobacteria), and Thalassomonas (Gammaproteobacteria) (Figure 4F). Thalassococcus has been shown to be capable of utilizing phthalate (Iwaki et al., 2012), but its ability to metabolize peptides, as suggested here, has not yet been explored. Previous studies have shown that Rhodobacterales are often one of the dominant groups in coastal seawaters, accounting for as high as 75% of the Alphaproteobacteria (Dong et al., 2014). Their abundance is thought to be related to DOC concentrations in nutrient-enriched habitats and they are frequently involved in taking up labile organic molecules, such as peptides and amino acids, as detected by metaproteomics (Dong et al., 2014; Fodelianakis et al., 2014). Our previous study also showed that populations of Ruegeria, Thalassomonas, Pseudoalteromonas, and Neptuniibacter grew rapidly when AVFA was amended to the same Sta. C6 bottom water (Liu et al., 2013). These genera contain many copiotrophs. Copiotrophs, such as Ruegeria, Vibrio, Alteromonas, and Colwellia, grow rapidly when substrates are available, but also maintain growth potential under starvation conditions. This is akin to a “feast or famine” strategy that allows adaptation to rapidly changing environments (Eilers et al., 2000; Christie-Oleza et al., 2012). Their high capability to assimilate peptide is thus consistent with their ecology strategy. The growth of Pseudoalteromonadaceae and Colwellia increased when peptone was incubated in the Southern Ocean seawater (Simon et al., 2012). Consistently, copiotrophic bacteria, such as Vibrio, Roseobacter, Pseudoalteromonas, Photobacterium, Marinomonas, Marinobacter, and Alteromonas, dominated the incorporation of DOC sources from Synechococcus exudate or lysate in seawater culture incubations (Nelson and Carlson, 2012). Particle-attached Colwellia and Pseudoalteromonas also showed high incorporation of proteins in marine microcosms (Mayali et al., 2015). DOC-related transporter genes, such as amino acids, oligopeptides, carbohydrates, carboxylic acids, polyamines, and lipids transporters, in coastal seawater were associated with Rhodobacterales (primarily Roseobacter), Rickettsiales, Flavobacteriales, and five orders of Gammaproteobacteria, including Alteromonadales, Oceanospirallales, Pseudomonadales, Vibrionales, and an uncharacterized taxon related to sulfur-oxidizing symbionts (Poretsky et al., 2010). Most of these bacteria also assimilated the peptide used in our study.
Changes in bacterial community structure that developed through incubations were not significantly different among the peptide treatment and control samples (Figure 3, ANOSIM p > 0.05, Supplementary Figure S2). These data suggest that peptide addition at relatively low concentrations had a minimal effect on the overall community structure. This also highlights that bacterial community structure cannot necessarily be used to infer roles of individual bacterial populations in incubation experiments. In contrast, the SIP technique directly links bacterial taxa with a metabolic function such as peptide decomposition. A direct comparison between the relative change in bacterial community structure and AVFA utilizing taxa, as determined via SIP, points to the interpretation that abundant bacterial taxa are not necessarily the most active ones (Table 2). For instance, Saprospricaea, Escherichia–Shigella, Balneatrix, and Thalassomonas accounted for <2% of communities and changed only <1% throughout the incubation while their abundance enrichment in heavy SIP fractions was 90–215%. Microbacteriaceae remained below 5% and their abundance did not increase with time during incubations, while frequencies increase by nearly 500% in heavy SIP fractions from surface seawater incubations. Similar observations have been reported elsewhere (Zemb et al., 2012), and indicate that some bacteria can be highly enriched in 13C, but they may represent only a small proportion of the overall community. These rare bacteria may have long generation time with 10s of hour or more (Brock, 1971). During our short 48 h incubation, certain bacteria might be at lag phase of growth, which changed little in the community structure. For example, if these rare bacteria only doubled once during 48 h, their increase from ca. <1% to ca. <2% would not contribute much the overall community structure. Alternatively, these rare bacteria might have utilized the assimilated peptides mostly for respiration instead of for biomass building, leading to the mismatch between abundance and SIP incorporation. These data showed the potential role of some rare and uncultivable bacteria in peptide utilization, which is often overlooked based on bacterial community structure analysis. This uncoupling between microbial abundance and activity is also consistent with other studies showing uncoupled pattern between rDNA and rRNA for some bacterial populations (Campbell and Kirchman, 2013; Caruso et al., 2013; Hunt et al., 2013), reflecting bacterial activity rates are not necessarily correlated to their abundance and these two parameters may be controlled by different factors.

TABLE 2. Comparison between the relative percentage change of bacteria genera (average percentage at 13+24+48 h relative to percentage at 0 h in quasi-replicates (n = 4) of 12C-AVFA and 13C-AVFA treatments) in the bacterial community structure and the percentage enrichment of bacteria genera showing positive enrichment in the SIP heavy fractions.
Factors Leading to the Development of Different Bacterial Communities
It is intriguing that bacterial communities that incorporated the added peptide differed in surface and bottom incubations. The two layers differed in chemical and biological parameters (Table 1, Figures 1D–F, 3, and Supplementary Figure S2), such as DO, DOC, and initial bacterial community structure, which probably contributed to the development of different bacterial communities, but the role of these factors seems to be limited (Liu et al., 2013; Liu and Liu, 2016). Other than these parameters, high levels of Pi (>0.4 μM) in the bottom seawater may stimulate the growth of fast-growing bacteria with high RNA content (Liu and Liu, 2016), such as Alphaproteobacteria and Gammaproteobacteria, consistent with the Growth Rate Hypothesis (Elser et al., 2000; Makino et al., 2003). The fast-growing bacteria may lead to faster peptide decomposition observed in the bottom than in the surface seawater. Assuming 0.2 pg dry mass bacterial cells and a P content of 1.3% (Sterner and Elser, 2002), the bacterial abundance increase observed here would have required 0.01–0.08 μM Pi. These small values are close to the standard deviation (ca. 0.02 μM) of Pi measurement, which may explain why no obvious decrease of Pi was observed during our incubations (Supplementary Figures S1D,E). On the other hand, these results further suggest that the level of Pi, rather than its absence, is the key factor limiting the development of fast-growing bacteria, supporting our previous hypothesis (Liu and Liu, 2016). The unique development of certain alphaproteobacterial and gammaproteobacterial genera may also explain the much lower production of AVFA fragments during the bottom water incubation compared to the surface water incubation (Figures 2B,D). Either these bacteria directly took up the peptide, or the hydrolysis and subsequent uptake of the fragments were tightly coupled (Fuhrman, 1987; Kuznetsova and Lee, 2002; Liu et al., 2013). These two processes cannot be differentiated with these data, but regardless, both pathways differ from that of the surface incubation, where hydrolysis and uptake seem uncoupled.
Factors to be Considered for the DNA-SIP Approach
A successful DNA-SIP experiment depends on the amount of isotopically labeled substrate being assimilated and the length of the incubation time (Radajewski et al., 2003; Neufeld et al., 2007a). The substrate concentration must be high enough to ensure sufficient isotopic labeling of nucleic acids relative to unlabeled background substrates that are relatively abundant. However, if the substrate concentrations are too high, the incubation may deviate from the in situ situation. In our incubations, we added relatively low concentrations (0.25–0.47 μM) of AVFA to minimize disturbance to the natural substrates. qPCR results support the notion that sufficient isotope was incorporated into bacterial DNA. Successful uptake of peptide by bacteria is also indicated through the increased bacterial abundance in peptide treatments compared to control. Longer incubation time often results in greater isotope incorporation, but may also lead to cross-feeding, such as bacterial assimilation of labeled byproducts, intermediates or dead cells, produced from substrate metabolism (Neufeld et al., 2007a,c; Wang et al., 2015). To reduce cross-feeding, we applied relatively short incubation time (48 h) that was nonetheless sufficient to allow complete peptide loss.
A potential limitation of the DNA-SIP approach is that the buoyant density of DNA varies with G+C content. As G+C content may vary among different bacteria, this may result in a loss of power to identify bacteria that have incorporated the labeled substrate based on density shift (Buckley et al., 2007). However, it is more problematic for 15N than for 13C substrates given the greater buoyant density differential for nucleic acids labeled with 13C. The density shift in our results was >0.01 g mL-1, equating to ca. 28% of 13C incorporation, which is more than the minimum percentage (20%) that is typically required for separating 13C and unlabeled organisms (Uhlik et al., 2009). Note that the overall buoyant density differed somewhat between the surface and bottom DNA fractions (Figures 4A,B). It is unclear why this difference was observed, but may be related to the different bacterial community composition in the surface and bottom incubations, as %G+C contents of DNA vary among different bacterial taxa and higher %G+C leads to heavier density (Buckley et al., 2007; Holben, 2011). However, it is presumed that this density difference will not affect our ability to identify bacteria incorporating 13C, because the taxonomic percentage enrichment was derived relative to the corresponding 12C-AVFA incubations within the surface or bottom samples. We note that DNA from three incubation time points (13, 24, 48 h) was pooled. This approach results in ‘smearing’ of the signal by spreading the DNA of active bacterial taxa across the gradient density range to a greater degree than for a single time point. However, this smearing should not be problematic with respect to the objectives of this study, because major bacterial taxa in the bacterial community structure were similar at all these three time points and the chemistry data showed a continuous pattern among these three time points. AVFA were completely degraded during 24–48 h in the surface seawater and during 13–24 h in the bottom seawater. Bacterial cell replication and DNA synthesis may have time lag after peptide incorporation due to their 9–12 h generation time (Eilers et al., 2000). Bacterial abundance was still increasing after 13 h in the bottom seawater (Figures 1D,E), indicating bacteria were still utilizing peptides for their growth shortly after peptide was completely degraded. To be consistent between two depths and make sure enough 13C signal is obtained, pooling the last three time points seems an appropriate choice. As surface and bottom incubations were treated in the same way for SIP samples, the comparison between two waters still holds with pooled samples. While the exact degree of isotopic labeling or peptide incorporation may therefore not be attainable from our experiments, the high degree of enrichment observed for some bacteria (Figures 4C–F) supports the notion of active 13C incorporation.
Conclusion
Work presented here builds on prior observations with respect to the inferred role of bacteria in peptide decomposition (Liu et al., 2013; Liu and Liu, 2016). Here, we directly linked specific bacterial taxa with peptide decomposition in surface and bottom waters in the hypoxic region of northern Gulf of Mexico. Major conclusions and implications from this study are as follows:
(1) Bacterial groups metabolizing peptide appear to differ between the surface normoxic and bottom hypoxic seawater. A more diverse group of bacteria including both oligotrophs and copiotrophs might be involved in peptide decomposition in the surface normoxic seawater, while peptide substrates appear to favor several copiotrophic marine bacterial lineages in the bottom hypoxic seawater. With a combination of detailed chemical and biological data in an isolation-independent way, this study expands our understanding of linkage between peptide decomposition and bacterial communities, especially with low concentrations of peptide amendment, and sheds new light on microbial behavior as single cells, populations and communities in microbial ecology.
(2) Peptide decomposition efficiency, pathway and fate differed between the surface normoxic and bottom hypoxic seawater, which might be related to the energy and element allocation to different bacterial taxa under distinct marine environments. By combining these chemical analyses with SIP results, the role of bacteria in contributing to this difference can thus be inferred. This serves as the first step to explore marine C and N cycle efficiency and mechanisms in various marine environments.
(3) This study further implies that some bacteria taxa can rapidly metabolize peptide in the context of high Pi concentration in the hypoxic seawater. It provides insights into the interactions among bacteria, labile DOM, nutrient, and DO in seawater, which stimulates more ecological hypotheses about diverse microbial groups and their functions in marine environments. As hypoxia may be intensified in the future scenario, investigating bacterial decomposition of labile DOM under different nutrient conditions is necessary to pinpoint the factors controlling hypoxia formation.
Author Contributions
All authors listed have made substantial, direct and intellectual contributions to the work and approved its final version for publication.
Funding
This work is funded by the Chemical and Biological Oceanography Programs of the National Science Foundation (OCE-1129659 & OCE-1634630).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the help from the crew of R/V Pelican. We appreciate J. Liu for his help with the incubation experiment, Dr. C. Shank for analyzing DOC samples and Dr. T. Villareal for his help with bacterial abundance analysis. We thank comments from Dr. W. Gardner, Dr. D. Erdner, Dr. J. McClelland and Dr. D. Kirchman. We are grateful for the DNA sequencing by the Oklahoma Medical Research Foundation.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.00353/full#supplementary-material
Footnotes
References
Aluwihare, L. I., Repeta, D. J., Pantoja, S., and Johnson, C. G. (2005). Two chemically distinct pools of organic nitrogen accumulate in the ocean. Science 308, 1007–1010. doi: 10.1126/science.1108925
Azam, F. (1998). Microbial control of oceanic carbon flux: the plot thickens. Science 280, 694–696. doi: 10.1126/science.280.5364.694
Bell, T. H., Yergeau, E., Martineau, C., Juck, D., Whyte, L. G., and Greer, C. W. (2011). Identification of nitrogen-incorporating bacteria in petroleum-contaminated Arctic soils by using [15N]DNA-based stable isotope probing and pyrosequencing. Appl. Environ. Microbiol. 77, 4163–4171. doi: 10.1128/AEM.00172-11
Benner, R., and Amon, R. M. W. (2015). The size-reactivity continuum of major bioelements in the ocean. Ann. Rev. Mar. Sci. 7, 185–205. doi: 10.1146/annurev-marine-010213-135126
Berg, C., Beckmann, S., Jost, G., Labrenz, M., and Jurgens, K. (2013). Acetate-utilizing bacteria at an oxic-anoxic interface in the Baltic Sea. FEMS Microbiol. Ecol. 85, 251–261. doi: 10.1111/1574-6941.12114
Buckley, D. H., Huangyutitham, V., Hsu, S. F., and Nelson, T. A. (2007). Stable isotope probing with 15N achieved by disentangling the effects of genome G+C content and isotope enrichment on DNA density. Appl. Environ. Microbiol. 73, 3189–3195. doi: 10.1128/AEM.02609-06
Campbell, B. J., and Kirchman, D. L. (2013). Bacterial diversity, community structure and potential growth rates along an estuarine salinity gradient. ISME J. 7, 210–220. doi: 10.1038/ismej.2012.93
Caporaso, J. G., Bittinger, K., Bushman, F. D., DeSantis, T. Z., Andersen, G. L., and Knight, R. (2010a). PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26, 266–267. doi: 10.1093/bioinformatics/btp636
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010b). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/NMETH.F.303
Carney, R. L., Mitrovic, S. M., Jeffries, T., Westhorpe, D., Curlevski, N., and Seymour, J. R. (2015). River bacterioplankton community responses to a high inflow event. Aquat. Microb. Ecol. 75, 187–205. doi: 10.3354/ame01758
Caruso, G., Azzaro, F., Azzaro, M., Decembrini, F., Ferla, R. L., Maimone, G., et al. (2013). Environmental variability in a transitional Mediterranean system (OliverieTindari, Italy): focusing on the response of microbial activities and prokaryotic abundance. Estuar. Coast. Shelf Sci. 135, 158–170. doi: 10.1016/j.ecss.2013.10.002
Chen, Y. Q., Ntai, I., and Kelleher, N. L. (2011). A proteomic survey of nonribosomal peptide and polyketide biosynthesis in Actinobacteria. J. Proteome Res. 11, 85–94. doi: 10.1021/pr2009115
Christie-Oleza, J. A., Fernandez, B., Nogales, B., Bosch, R., and Armengaud, J. (2012). Proteomic insights into the lifestyle of an environmentally relevant marine bacterium. ISME J. 6, 124–135. doi: 10.1038/ismej.2011.86
Connelly, T. L., Baer, S. E., Cooper, J. T., Bronk, D. A., and Wawrik, B. (2014). Urea uptake and carbon fixation by marine pelagic bacteria and archaea during the Arctic summer and winter seasons. Appl. Environ. Microbiol 80, 6013–6022. doi: 10.1128/AEM.01431-14
Cottrell, M. T., and Kirchman, D. L. (2000). Natural assemblages of marine proteobacteria and members of the Cytophaga-Flavobacter cluster consuming low- and high-molecular-weight dissolved organic matter. Appl. Environ. Microbiol. 66, 1692–1697. doi: 10.1128/AEM.66.4.1692-1697.2000
Crump, B. C., Peranteau, C., Beckingham, B., and Cornwell, J. C. (2007). Respiratory succession and community succession of bacterioplankton in seasonally anoxic estuarine waters. Appl. Environ. Microbiol. 73, 6802–6810. doi: 10.1128/AEM.00648-07
Dong, H. P., Hong, Y. G., Lu, S. H., and Xie, L. Y. (2014). Metaproteomics reveals the major microbial players and their biogeochemical functions in a productive coastal system in the northern South China Sea. Environ. Microbiol. Rep. 6, 683–695. doi: 10.1111/1758-2229.12188
Eilers, H., Pernthaler, J., and Amann, R. (2000). Succession of pelagic marine bacteria during enrichment: a close look at cultivation-induced shifts. Appl. Environ. Microbiol. 66, 4634–4640. doi: 10.1128/AEM.66.11.4634-4640.2000
Elser, J. J., Sterner, R. W., Gorokhova, E., Fagan, W. F., Markow, T. A., Cotner, J. B., et al. (2000). Biological stoichiometry from genes to ecosystems. Ecol. Lett. 3, 540–550. doi: 10.1111/j.1461-0248.2000.00185.x
Fierer, N., Bradford, M. A., and Jackson, R. B. (2007). Toward an ecological classification of soil bacteria. Ecology 88, 1354–1364. doi: 10.1890/05-1839
Fodelianakis, S., Papageorgiou, N., Pitta, P., Kasapidis, P., Karakassis, I., and Ladoukakis, E. D. (2014). The pattern of change in the abundances of specific bacterioplankton groups is consistent across different nutrient-enriched habitats in Crete. Appl. Environ. Microbiol. 80, 3784–3792. doi: 10.1128/AEM.00088-14
Fuhrman, J. A. (1987). Close coupling between release and uptake of dissolved free amino-acids in seawater studied by an isotope-dilution approach. Mar. Ecol. Prog. Ser. 37, 45–52. doi: 10.3354/meps037045
Gardner, W. S., and St. John, P. A. (1991). High-performance liquid chromatographic method to determine ammonium ion and primary amines in seawater. Anal. Chem. 63, 537–540. doi: 10.1021/ac00005a032
Gihring, T. M., Humphrys, M., Mills, H. J., Huettel, M., and Kostka, J. E. (2009). Identification of phytodetritus-degrading microbial communities in sublittoral Gulf of Mexico sands. Limnol. Oceanogr. 54, 1073–1083. doi: 10.4319/lo.2009.54.4.1073
Harvey, H. R., Dyda, R. Y., and Kirchman, D. L. (2006). Impact of DOM composition on bacterial lipids and community structure in estuaries. Aquat. Microb. Ecol. 42, 105–117. doi: 10.3354/ame042105
Holben, W. E. (2011). “GC fractionation allows comparative total microbial community analysis, enhances diversity assessment, and facilitates detection of minority populations of bacteria,” in Handbook of Molecular Micorbial Ecology: Metagenomics and Complementary Approaches, Vol. I, ed. F. J. de Bruijn (Hoboken, NJ: Wiley-Blackwell), 183–196.
Hunt, D. E., Lin, Y., Church, M. J., Karl, D. M., Tringe, S. G., Izzo, L. K., et al. (2013). Relationship between abundance and specific activity of bacterioplankton in open ocean surface waters. Appl. Environ. Microbiol. 79, 177–184. doi: 10.1128/AEM.02155-12
Iwaki, H., Nishimura, A., and Hasegawa, Y. (2012). Isolation and characterization of marine bacteria capable of utilizing phthalate. World J. Microbiol. Biot. 28, 1321–1325. doi: 10.1007/s11274-011-0925-x
Jiao, N., Herndl, G. J., Hansell, D. A., Benner, R., Kattner, G., Wilhelm, S. W., et al. (2010). Microbial production of recalcitrant dissolved organic matter: long-term carbon storage in the global ocean. Nat. Rev. Microbiol. 8, 593–599. doi: 10.1038/nrmicro2386
Jones, M. N. (1984). Nitrate reduction by shaking with Cadmium – alternative to Cadmium columns. Water Res. 18, 643–646. doi: 10.1016/0043-1354(84)90215-X
Karl, D. M. (2014). Microbially mediated transformations of phosphorus in the sea: new views of an old cycle. Ann. Rev. Mar. Sci. 6, 279–337. doi: 10.1146/annurev-marine-010213-135046
Kleindienst, S., Herbst, F. A., Stagars, M., von Netzer, F., von Bergen, M., Seifert, J., et al. (2014). Diverse sulfate-reducing bacteria of the Desulfosarcina/Desulfococcus clade are the key alkane degraders at marine seeps. ISME J. 8, 2029–2044. doi: 10.1038/ismej.2014.51
Klindworth, A., Pruesse, E., Schweer, T., Peplies, J., Quast, C., Horn, M., et al. (2013). Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41, e1. doi: 10.1093/nar/gks808
Kuznetsova, M., and Lee, C. (2002). Dissolved free and combined amino acids in nearshore seawater, sea surface microlayers and foams: influence of extracellular hydrolysis. Aquat. Sci. 64, 252–268. doi: 10.1007/s00027-002-8070-0
Lee, C., Hedges, J. I., Wakeham, S. G., and Zhu, N. (1992). Effectiveness of various treatments in retarding microbial activity in sediment trap material and their effects on the collection of swimmers. Limnol. Oceanogr. 37, 117–130. doi: 10.4319/lo.1992.37.1.0117
Lee, C., Wakeham, S. G., and Hedges, J. I. (2000). Composition and flux of particulate amino acids and chloropigments in equatorial Pacific seawater and sediments. Deep Sea Res. I 47, 1535–1568. doi: 10.1016/S0967-0637(99)00116-8
Lee, S., and Fuhrman, J. A. (1987). Relationships between biovolume and biomass of naturally derived marine bacterioplankton. Appl. Environ. Microbiol. 53, 1298–1303.
Lindroth, P., and Mopper, K. (1979). High-performance liquid-chromatographic determination of subpicomole amounts of amino-acids by precolumn fluorescence derivatization with o-phthaldialdehyde. Anal. Chem. 51, 1667–1674. doi: 10.1021/ac50047a019
Liu, S., and Liu, Z. (2014). A new method to measure small peptides amended in seawater using high performance liquid chromatography coupled with mass spectrometry. Mar. Chem. 164, 16–24. doi: 10.1016/j.marchem.2014.05.006
Liu, S., and Liu, Z. (2015). Comparing extracellular enzymatic hydrolysis between plain peptides and their corresponding analogs in the northern Gulf of Mexico Mississippi River plume. Mar. Chem. 177, 398–407. doi: 10.1016/j.marchem.2015.06.021
Liu, S., Riesen, A., and Liu, Z. (2015). Differentiating the role of different-sized microorganisms in peptide decomposition during incubations using size-fractioned coastal seawater. J. Exp. Mar. Biol. Ecol. 472, 97–106. doi: 10.1016/j.jembe.2015.07.004
Liu, Z., Kobiela, M. E., McKee, G. A., Tang, T. T., Lee, C., Mulholland, M. R., et al. (2010). The effect of chemical structure on the hydrolysis of tetrapeptides along a river-to-ocean transect: AVFA and SWGA. Mar. Chem. 119, 108–120. doi: 10.1016/j.marchem.2010.01.005
Liu, Z., and Liu, S. (2016). High phosphate concentrations accelerate bacterial peptide degradation in hypoxic bottom waters of the northern Gulf of Mexico. Environ. Sci. Technol. 50, 676–684. doi: 10.1021/ac50047a019
Liu, Z., Liu, S., Liu, J., and Gardner, W. S. (2013). Differences in peptide decomposition rates and pathways in hypoxic and oxic coastal environments. Mar. Chem. 157, 67–77. doi: 10.1016/j.marchem.2013.08.003
Luo, C. L., Xie, S. G., Sun, W. M., Li, X. D., and Cupples, A. M. (2009). Identification of a novel toluene-degrading bacterium from the candidate phylum TM7, as determined by DNA stable isotope probing. Appl. Environ. Microbiol. 75, 4644–4647. doi: 10.1128/AEM.00283-09
Makino, W., Cotner, J. B., Sterner, R. W., and Elser, J. J. (2003). Are bacteria more like plants or animals? Growth rate and resource dependence of bacterial C : N : P stoichiometry. Funct. Ecol. 17, 121–130. doi: 10.1046/j.1365-2435.2003.00712.x
Marie, D., Partensky, F., Jacquet, S., and Vaulot, D. (1997). Enumeration and cell cycle analysis of natural populations of marine picoplankton by flow cytometry using the nucleic acid stain SYBR Green I. Appl. Environ. Microbiol. 63, 186–193.
Mayali, X., Stewart, B., Mabery, S., and Weber, P. K. (2015). Temporal succession in carbon incorporation from macromolecules by particle-attached bacteria in marine microcosms. Environ. Microbiol. Rep. 8, 68–75. doi: 10.1111/1758-2229.12352
McCarren, J., Becker, J. W., Repeta, D. J., Shi, Y., Young, C. R., Malmstrom, R. R., et al. (2010). Microbial community transcriptomes reveal microbes and metabolic pathways associated with dissolved organic matter turnover in the sea. Proc. Natl. Acad. Sci. U.S.A. 107, 16420–16427. doi: 10.1073/pnas.1010732107
Moran, M. A., Belas, R., Schell, M. A., Gonzalez, J. M., Sun, F., Sun, S., et al. (2007). Ecological genomics of marine roseobacters. Appl. Environ. Microbiol. 73, 4559–4569. doi: 10.1128/AEM.02580-06
Mou, X., Sun, S., Edwards, R. A., Hodson, R. E., and Moran, M. A. (2008). Bacterial carbon processing by generalist species in the coastal ocean. Nature 451, 708–712. doi: 10.1038/nature06513
Murray, A. E., Arnosti, C., De La Rocha, C. L., Grossart, H. P., and Passow, U. (2007). Microbial dynamics in autotrophic and heterotrophic seawater mesocosms. II. Bacterioplankton community structure and hydrolytic enzyme activities. Aquat. Microb. Ecol. 49, 123–141. doi: 10.3354/ame01139
Nagata, T. (2008). “Organic matter-bacteria interactions in seawater,” in Microbial Ecology of the Oceans, ed. D. L. Kirchman (New York, NY: Wiley-Blackwell), 207–242.
Nakatsu, C. H., and Marsh, T. L. (2007). “Analysis of microbial communities with denaturing gradient gel eletrophoresis and terminal restriction fragment length polymorphism,” in Methods for General and Molecular Microbiology, eds C. A. Reddy, T. J. Beveridge, J. A. Breznak, G. A. Marzluf, T. M. Schmidt, and L. R. Snyder (Wahsington, DC: ASM Press), 909–923.
Nelson, C. E., and Carlson, C. A. (2012). Tracking differential incorporation of dissolved organic carbon types among diverse lineages of Sargasso Sea bacterioplankton. Environ. Microbiol. 14, 1500–1516. doi: 10.1111/j.1462-2920.2012.02738.x
Nelson, C. E., and Wear, E. K. (2014). Microbial diversity and the lability of dissolved organic carbon. Proc. Natl. Acad. Sci. U.S.A. 111, 7166–7167. doi: 10.1073/pnas.1405751111
Neufeld, J. D., Dumont, M. G., Vohra, J., and Murrell, J. C. (2007a). Methodological considerations for the use of stable isotope probing in microbial ecology. Microb. Ecol. 53, 435–442. doi: 10.1007/s00248-006-9125-x
Neufeld, J. D., Schafer, H., Cox, M. J., Boden, R., McDonald, I. R., and Murrell, J. C. (2007b). Stable-isotope probing implicates Methylophaga spp and novel Gammaproteobacteria in marine methanol and methylamine metabolism. ISME J. 1, 480–491. doi: 10.1038/ismej.2007.65
Neufeld, J. D., Wagner, M., and Murrell, J. C. (2007c). Who eats what, where and when? Isotope-labelling experiments are coming of age. ISME J. 1, 103–110. doi: 10.1038/ismej.2007.30
Oksanen, J., Blanchet, F. G., Kindt, R., Legendre, P., Minchin, P. R., O’Hara, R. B., et al. (2016). Package ’Vegan’. Available at: http://cran.r-project.org
Orellana, M. V., and Hansell, D. A. (2012). Ribulose-1,5-bisphosphate carboxylase/oxygenase (RuBisCO): a long-lived protein in the deep ocean. Limnol. Oceanogr. 57, 826–834. doi: 10.4319/lo.2012.57.3.0826
Orsi, W. D., Smith, J. M., Liu, S., Liu, Z., Sakamoto, C. M., Wilken, S., et al. (2016). Diverse, uncultivated bacteria and archaea underlying the cycling of dissolved protein in the ocean. ISME J. 10, 2158–2173. doi: 10.1038/ismej.2016.20
Ouverney, C. C., and Fuhrman, J. A. (1999). Combined Microautoradiography-16S rRNA probe technique for determination of radioisotope uptake by specific microbial cell types in situ. Appl. Environ. Microbiol. 65, 3264–3264.
Parsons, R. J., Nelson, C. E., Carlson, C. A., Denman, C. C., Andersson, A. J., Kledzik, A. L., et al. (2015). Microbial bacterioplankton community turnover within seasonally hypoxic waters of a subtropical sound: Devil’s Hole, Bermuda. Environ. Microbiol. 17, 3481–3499. doi: 10.1111/1462-2920.12445
Payne, J. W., and Gilvarg, C. (1971). Peptide transport. Adv. Enzymol. Relat. Areas Mol. Biol. 350, 187–244. doi: 10.1002/9780470122808.ch5
Poretsky, R. S., Sun, S. L., Mou, X. Z., and Moran, M. A. (2010). Transporter genes expressed by coastal bacterioplankton in response to dissolved organic carbon. Environ. Microbiol. 12, 616–627. doi: 10.1111/j.1462-2920.2009.02102.x
Rabalais, N. N., Turner, R. E., and Wiseman, W. J. (2001). Hypoxia in the gulf of Mexico. J. Environ. Qual. 30, 320–329. doi: 10.2134/jeq2001.302320x
Radajewski, S., Ineson, P., Parekh, N. R., and Murrell, J. C. (2000). Stable-isotope probing as a tool in microbial ecology. Nature 403, 646–649. doi: 10.1038/35001054
Radajewski, S., McDonald, I. R., and Murrell, J. C. (2003). Stable-isotope probing of nucleic acids: a window to the function of uncultured microorganisms. Curr. Opin. Biotechnol. 14, 296–302. doi: 10.1016/S0958-1669(03)00064-8
Redmond, M. C., Valentine, D. L., and Sessions, A. L. (2010). Identification of novel methane-, ethane-, and propane-oxidizing bacteria at marine hydrocarbon seeps by stable isotope probing. Appl. Environ. Microbiol. 76, 6412–6422. doi: 10.1128/AEM.00271-10
Simon, M., Billerbeck, S., Kessler, D., Selje, N., and Schlingloff, A. (2012). Bacterioplankton communities in the Southern Ocean: composition and growth response to various substrate regimes. Aquat. Microb. Ecol. 68, 13–28. doi: 10.3354/ame01597
Sipler, R. E., and Bronk, D. A. (2015). “Dynamics of dissolved organic nitrogen,” in Biogeochemistry of Marine Dissolved Organic Matter, eds D. A. Hansell and C. A. Carlson (San Diego, CA: Academic Press), 127–232.
Sterner, R. W., and Elser, J. J. (2002). Ecological Stoichiometry: The biology of Elements from Molecules to the Biosphere. Princeton, NJ: Princeton University Press.
Strickland, J. D. H., and Parsons, T. R. (1968). A Practical Handbook of Seawater Analysis. Ottawa, ON: Queen’s Printer.
Tabor, P. S., and Neihof, R. A. (1982). Improved microautoradiographic method to determine individual microorganisms active in substrate uptake in natural waters. Appl. Environ. Microbiol. 44, 945–953.
Teeling, H., Fuchs, B. M., Becher, D., Klockow, C., Gardebrecht, A., Bennke, C. M., et al. (2012). Substrate-controlled succession of marine bacterioplankton populations induced by a phytoplankton bloom. Science 336, 608–611. doi: 10.1126/science.1218344
Teske, A., Durbin, A., Ziervogel, K., Cox, C., and Arnosti, C. (2011). Microbial community composition and function in permanently cold seawater and sediments from an Arctic fjord of Svalbard. Appl. Environ. Microbiol. 77, 2008–2018. doi: 10.1128/AEM.01507-10
Uhlik, O., Jecna, K., Leigh, M. B., Mackova, M., and Macek, T. (2009). DNA-based stable isotope probing: a link between community structure and function. Sci. Total Environ. 407, 3611–3619. doi: 10.1016/j.scitotenv.2008.05.012
Ventura, M., Canchaya, C., Tauch, A., Chandra, G., Fitzgerald, G. F., Chater, K. F., et al. (2007). Genomics of Actinobacteria: tracing the evolutionary history of an ancient phylum. Microbiol. Mol. Biol. Rev. 71, 495–548. doi: 10.1128/MMBR.00005-07
Walker, B. D., Beaupré, S. R., Guilderson, T. P., McCarthy, M. D., and Druffel, E. R. M. (2016). Pacific carbon cycling constrained by organic matter size, age and composition relationships. Nat. Geosci. 9, 888–891. doi: 10.1038/ngeo2830
Wang, X., Sharp, C. E., Jones, G. M., Grasby, S. E., Brady, A. L., and Dunfield, P. F. (2015). Stable-isotope probing identifies uncultured Planctomycetes as primary degraders of a complex heteropolysaccharide in soil. Appl. Environ. Microbiol. 81, 4607–4615. doi: 10.1128/AEM.00055-15
Wawrik, B., Boling, W. B., Van Nostrand, J. D., Xie, J. P., Zhou, J. Z., and Bronk, D. A. (2012a). Assimilatory nitrate utilization by bacteria on the West Florida Shelf as determined by stable isotope probing and functional microarray analysis. FEMS Microbiol. Ecol. 79, 400–411. doi: 10.1111/j.1574-6941.2011.01226.x
Wawrik, B., Callaghan, A. V., and Bronk, D. A. (2009). Use of inorganic and organic nitrogen by Synechococcus spp. and diatoms on the West Florida Shelf as measured using stable isotope probing. Appl. Environ. Microbiol. 75, 6662–6670. doi: 10.1128/AEM.01002-09
Wawrik, B., Mendivelso, M., Parisi, V. A., Suflita, J. M., Davidova, I. A., Marks, C. R., et al. (2012b). Field and laboratory studies on the bioconversion of coal to methane in the San Juan Basin. FEMS Microbiol. Ecol. 81, 26–42. doi: 10.1111/j.1574-6941.2011.01272.x
Weiss, M. S., Abele, U., Weckesser, J., Welte, W., Schiltz, E., and Schulz, G. E. (1991). Molecular architecture and electrostatic properties of a bacterial porin. Science 254, 1627–1630. doi: 10.1126/science.1721242
Wright, J. J., Konwar, K. M., and Hallam, S. J. (2012). Microbial ecology of expanding oxygen minimum zones. Nat. Rev. Microbiol. 10, 381–394. doi: 10.1038/nrmicro2778
Yau, S., Lauro, F. M., Williams, T. J., DeMaere, M. Z., Brown, M. V., Rich, J., et al. (2013). Metagenomic insights into strategies of carbon conservation and unusual sulfur biogeochemistry in a hypersaline Antarctic lake. ISME J. 7, 1944–1961. doi: 10.1038/ismej.2013.69
Zaikova, E., Walsh, D. A., Stilwell, C. P., Mohn, W. W., Tortell, P. D., and Hallam, S. J. (2010). Microbial community dynamics in a seasonally anoxic fjord: Sannich Inlet, British Columbia. Environ. Microbiol. 12, 172–191. doi: 10.1111/j.1462-2920.2009.02058.x
Keywords: peptide, bacteria, DNA stable isotope probing, nitrogen, hypoxia, Gulf of Mexico
Citation: Liu S, Wawrik B and Liu Z (2017) Different Bacterial Communities Involved in Peptide Decomposition between Normoxic and Hypoxic Coastal Waters. Front. Microbiol. 8:353. doi: 10.3389/fmicb.2017.00353
Received: 16 November 2016; Accepted: 20 February 2017;
Published: 07 March 2017.
Edited by:
Andrew Decker Steen, University of Tennessee, USAReviewed by:
Xavier Mayali, Lawrence Livermore National Laboratory, USACraig E. Nelson, University of Hawaii at Manoa, USA
Nagissa Mahmoudi, Harvard University, USA
Copyright © 2017 Liu, Wawrik and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhanfei Liu, emhhbmZlaS5saXVAdXRleGFzLmVkdQ==