 Thais Louvain de Souza1,2
Thais Louvain de Souza1,2 Regina C. de Souza Campos Fernandes1,2
Regina C. de Souza Campos Fernandes1,2 Juliana Azevedo da Silva3Vladimir Gomes Alves Júnior1,2Adelia Gomes Coelho4Afonso C. Souza Faria5Nabia M. Moreira Salomão Simão6
Juliana Azevedo da Silva3Vladimir Gomes Alves Júnior1,2Adelia Gomes Coelho4Afonso C. Souza Faria5Nabia M. Moreira Salomão Simão6 João T. Souto Filho2Caroline Deswarte7,8Stéphanie Boisson-Dupuis7,8,9Dara Torgerson10Jean-Laurent Casanova7,8,9,11,12
João T. Souto Filho2Caroline Deswarte7,8Stéphanie Boisson-Dupuis7,8,9Dara Torgerson10Jean-Laurent Casanova7,8,9,11,12 Jacinta Bustamante7,8,9,13
Jacinta Bustamante7,8,9,13 Enrique Medina-Acosta1*
Enrique Medina-Acosta1*- 1Núcleo de Diagnóstico e Investigação Molecular, Laboratório de Biotecnologia, Universidade Estadual do Norte Fluminense, Campos dos Goytacazes, Brazil
- 2Faculdade de Medicina de Campos, Campos dos Goytacazes, Brazil
- 3Laboratório de Biologia do Reconhecer, Universidade Estadual do Norte Fluminense, Campos dos Goytacazes, Brazil
- 4Hospital Municipal Ferreira Machado, Campos dos Goytacazes, Brazil
- 5Pronto Atendimento UNIMED, Campos dos Goytacazes, Brazil
- 6Hospital Escola Álvaro Alvim, Campos dos Goytacazes, Brazil
- 7Laboratory of Human Genetics of Infectious Diseases, Institut National de la Santé et de la Recherche Médicale, Paris, France
- 8Laboratory of Human Genetics of Infectious Diseases: Mendelian Predisposition, Imagine Institute, Paris Descartes University, Paris, France
- 9St. Giles Laboratory of Human Genetics of Infectious Diseases, The Rockefeller University, New York, NY, USA
- 10Department of Medicine, University of California San Francisco, San Francisco, CA, USA
- 11Pediatric Hematology-Immunology Unit, Necker Hospital for Sick Children, Assistance Publique Hôpitaux de Paris, Paris, France
- 12Howard Hughes Medical Institute, The Rockefeller University, New York, NY, USA
- 13Study Center of Primary Immunodeficiencies, Assistance Publique Hôpitaux de Paris, Necker Hospital for Sick Children, Paris, France
Patients with Mendelian Susceptibility to Mycobacterial Diseases (MSMD) exhibit variable vulnerability to infections by mycobacteria and other intramacrophagic bacteria (e.g., Salmonella and Klebsiella) and fungi (e.g., Histoplasma, Candida, Paracoccidioides, Coccidioides, and Cryptococcus). The hallmark of MSMD is the inherited impaired production of interferon gamma (IFN-γ) or the lack of response to it. Mutations in the interleukin (IL)-12 receptor subunit beta 1 (IL12RB1) gene accounts for 38% of cases of MSMD. Most IL12RB1 pathogenic allele mutations, including ten known stop-gain variants, cause IL-12Rβ1 complete deficiency (immunodeficiency-30, IMD30) by knocking out receptor cell-surface expression. IL12RB1 loss-of-function genotypes impair both IL-12 and IL-23 responses. Here, we assess the health effects of a rare, novel IL12RB1 stop-gain homozygous genotype with paradoxical IL-12Rβ1 cell-surface expression. We appraise four MSMD children from three unrelated Brazilian kindreds by clinical consultation, medical records, and genetic and immunologic studies. The clinical spectrum narrowed down to Bacillus Calmette-Guerin (BCG) vaccine-related suppurative adenitis in all patients with one death, and recrudescence in two, histoplasmosis, and recurrence in one patient, extraintestinal salmonellosis in one child, and cutaneous vasculitis in another. In three patients, we established the homozygous Trp7Ter predicted loss-of-function inherited genotype and inferred it from the heterozygote parents of the fourth case. The Trp7Ter mutation maps to the predicted IL-12Rβ1 N-terminal signal peptide sequence. BCG- or phytohemagglutinin-blasts from the three patients have reduced cell-surface expression of IL-12Rβ1 with impaired production of IFN-γ and IL-17A. Screening of 227 unrelated healthy subjects from the same geographic region revealed one heterozygous genotype (allele frequency 0.0022) vs. one in over 841,883 public genome/exomes. We also show that the carriers bear European ancestry-informative alleles and share the extended CACCAGTCCGG IL12RB1 haplotype that occurs worldwide with a frequency of 8.4%. We conclude that the novel IL12RB1 N-terminal signal peptide stop-gain loss-of-function homozygous genotype confers IL-12Rβ1 deficiency with varying severity and early-onset age through diminished cell-surface expression of an impaired IL-12Rβ1 polypeptide. We firmly recommend attending to warning signs of IMD30 in children who are HIV-1 negative with a history of adverse effects to the BCG vaccine and presenting with recurrent Histoplasma spp. and extraintestinal Salmonella spp. infections.
Introduction
Here, we report four pediatric cases that exemplify the microbial and clinical spectra of infectious diseases associated with the impaired production of the Th1 signature cytokine interferon-gamma (IFN-γ) in subjects with Mendelian Susceptibility to Mycobacterial Diseases (MSMD; Zerbe and Holland, 2005; Kutukculer et al., 2006; de Beaucoudrey et al., 2010; Zahid et al., 2014). These comprised attenuated bacillus Calmette-Guerin (BCG) vaccine-related suppurative adenitis in all patients with one death, and recrudescence in two, histoplasmosis, and recurrence in one patient, extraintestinal salmonellosis in one child, and cutaneous vasculitis in another. Our subjects share a history of adverse events to the BCG vaccine in the first year of life. BCG vaccine adverse effects are a major warning sign of primary immunodeficiency disorder of the IFN-γ axis as the underlying cause (de Beaucoudrey et al., 2010; Carneiro-Sampaio et al., 2011; Costa-Carvalho et al., 2014; Abolhassani et al., 2015). The four children are from three apparently unrelated kindreds, living in 60-km2 proximity near the city of Campos dos Goytacazes, situated in the northern region of the State of Rio de Janeiro, Brazil. Moreover, the parents of Case 1 are first-degree relatives. Therefore, we based the framework of investigation on the hypothesis of a rare homozygous loss-of-function alteration in the interleukin (IL)-12 receptor subunit beta 1 (IL12RB1) gene occurring in a small population.
Kindred A
Case 1
White male, born in 1999, the only child of first-degree consanguineous parents. BCG-vaccinated during the first month of life. At age 3 months, he developed suppurative axillary adenitis, ipsilateral to the BCG vaccine site, and was treated with Isoniazid (10 mg/kg/day), with regression of the lymphadenopathy. At age 9 months, he was hospitalized due to fever, sweating, failure to thrive, severe anemia and hepatic and splenic enlargements. Findings included normal chest X-ray; cultures of blood and urine negative for bacteria; hemoglobin 6 g/dL; leukocytes 2,700 cells/mm3 (0% eosinophils, 1% bands, 2% monocytes, 47% neutrophils, and 50% lymphocytes); 40,000 platelets/mm3; normal AST (36 IU/L) and ALT (20 IU/L). A myelogram demonstrated erythroid and platelet hypoplasias and a blockage in granulocyte development. He was treated with Isoniazid (10 mg/kg/day) and with Ceftriaxone (100 mg/kg /day) and received two blood transfusions. Mother and child were both HIV-1 negative. After 7 days, he was transferred to the intensive treatment unit, with respiratory failure and died after massive pulmonary bleeding.
Kindred B
Case 2
White male, born in 1993, the first child of a non-consanguineous couple. BCG-vaccinated during the first month of life. At age 1 year, he presented with suppurative axillary adenopathy ipsilateral to the BCG vaccination site. He was treated with Isoniazid (10 mg/kg/day) for 6 months, with good clinical evolution. At age 13 years, he had cervical lymphadenopathy and fever. Histoplasmosis was confirmed by histopathology (Figure 1A) and was treated with ketoconazole (400 mg/day) with excellent response. At age 18 years, he had a relapse of histoplasmosis manifested by bulky cervical lymphadenopathy, hoarseness, dysphagia, and fever. Video laryngoscopy revealed bilateral granulomatous lesions of the aryepiglottic folds (Figure 1B). Immunological findings included hyper-IgE (17,300 and 17,937 IU/mL in two separate tests; Reference Value, RV: <87); IgG = 2,790 mg/dL (RV: 549–1584), IgA = 225 mg/dL (RV: 61–348), IgM = 106 mg/dL (RV: 23–259); CD4 = 350 cells/mm3 (41.5%), CD8 = 269 cells/mm3 (17.2%), CD4/CD8 = 1.30. Histoplasmosis recurrence was treated with Itraconazole (600 mg/day) for 2 years. He is currently healthy without prophylaxis.
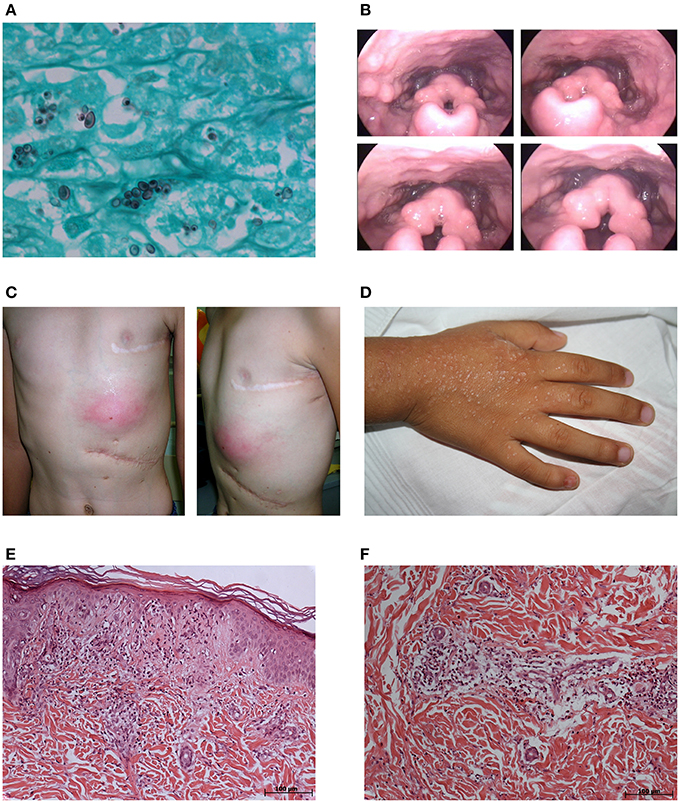
Figure 1. Clinicopathological findings in the affected children. Case 2: (A) Presence of many intracellular, small, narrow base, budding, and darkly pigmented yeast-like cells (1–5 mm diameter) in the lymph node (Grocott stain, original magnification × 1,000). (B) Screenshots of video laryngoscopy showing bilateral granulomatous lesions of aryepiglottic folds. Case 3: (C) Frontal and lateral photographs of the left infracostal large abscess. Case 4: (D) Photograph of the pruritic, maculopapular erythematous rash in the back of the right hand. (E) Hyperkeratosis and regional atrophy associated with basal vacuolization and focal necrosis in addition to perivascular and interstitial inflammatory mixed infiltrate with mild nuclear fragmentation in the erythematous maculopapular rash on the left hand (hematoxylin-eosin stain, × 100). (F) Perivascular and interstitial inflammatory mixed infiltrates with neutrophils in karyorrhexis, lymphocyte, and eosinophils in the erythematous lesions of the left limb (hematoxylin-eosin stain, × 100).
Case 3
White male, born in 2001, a full sibling of Case 2. BCG-vaccinated during the first month of life. At age 4 months, he had regional suppurative adenitis ipsilateral to the BCG vaccine site and was treated with Isoniazid (10 mg/kg/day) for 6 months with good clinical response. At age 5 years, when his older brother (Case 2) was diagnosed with histoplasmosis, he presented with multiple cervical lymphadenopathies and fever. He was treated with ketoconazole (400 mg/day). Treatment was interrupted 7 months later without recovery. At age 6 years, he was hospitalized with cervical lymphadenopathies and was treated with Amphotericin B without regression. At age 8 years, he was hospitalized with asthenia, anorexia, abdominal pain and cervical adenopathies, and evolved hepatic and splenic enlargements, respiratory distress, persistent fever, pericardial and pleural collections and splenic abscess, which demanded drainage and resection, respectively. Laboratory studies showed leukocytosis (26,900 cells/mm3; 14% bands, 80% segments, 5% lymphocytes) with normal platelet count (297,000/mm3), AST (38 U/L), ALT (25 IU/L), AP (342 IU/L), and altered albumin (2.30 g/dL), globulin (3.60 g/dL); total bilirubin (7.80 mg/dL); and direct bilirubin (6.60 mg/dL). The Wade, PAS and Giemsa stains of lymph nodes and spleen were negative. The PAS and Wade stains of the pericardial membrane were negative. The culture of bone marrow aspirate was positive for Salmonella choleraesuis. Besides antibacterial therapy (40 mg/kg/day of Vancomycin and 30 mg/kg/day of Meropenem), Rifampin (10 mg/kg/day), Isoniazid (10 mg/kg/day), and Pyrazinamide (20 mg/kg/day) were administered due to polyserositis and the unconfirmed possibility of household contacts with tuberculosis. Immunological evaluation revealed: hyper-IgE (902 and 2,848 IU/mL (RV at 87 IU/ml) in two separate evaluations as well as elevated values for IgG = 3,150 mg/dL (RV: 572–1,474), IgA = 943 mg/dL (RV: 34–305), IgM = 223 mg/dL (RV: 32–208), and T cell markers (CD4 = 955 cells/mm3 (17%), CD8 = 1,348 cells/mm3 (24%), CD4/CD8 = 0.7. Three months after suspension of the tuberculostatics, there was a recrudescence of cervical lymphadenopathy with fistulization despite the absence of fever and inflammatory signs. Cultures of ganglion aspirate were negative for bacteria, acid-fast bacilli, and fungi. Histopathology of cervical lymph nodes was negative for intracellular microbes. He received experimental treatment with Trimethoprim/Sulfamethoxazole for 5 months with complete recovery and prophylaxis with the same medication for 6 months. After the interruption of prophylaxis, he developed a left infracostal large abscess (Figure 1C), which was drained and found negative on culture. He was empirically prescribed with Ciprofloxacin (40 mg/kg/day) for Salmonella and Clarithromycin (15 mg/kg/day) for mycobacteria for 6 months. He is currently on prophylaxis with Trimethoprim/Sulfamethoxazole and is free of symptoms.
Kindred C
Case 4
White male, born in 2010, the only child of a non-consanguineous couple. BCG-vaccinated during the first month of life. At age 6 months, he developed axillary adenopathy ipsilateral to the BCG vaccine site and was treated with Isoniazid (10 mg/kg/day) for 6 months. Four months later, adenopathy recurred with fistulization and was successfully managed with Isoniazid (10 mg/kg/day) and Rifampin (10 mg/kg/day) for 6 months. Cultures from an adenopathy puncture sample were negative for bacteria, mycobacteria, and fungi. At age 5 years, presented with ankle arthritis, no fever, and non-pruritic erythematous lesions in the lower limbs and buttocks, which did not disappear with compression, and progressive flares for shorter periods during 4 months, which included one full body episode, with a clinical diagnosis of leukocytoclastic vasculitis. He also evolved a pruritic, maculopapular erythematous rash in the neck and the back of both hands (Figure 1D), with a clinical diagnosis of pityriasis. Laboratory studies showed leukocytosis (14,980 cells/mm3), ESR 25 mm/h (RV 0–20 mm/h), and LDH 730 IU/L (RV 200–480 IU/L). Immunological findings included elevated IgE (934 IU/ml [RV at 52 IU/ml]), IgG = 5,650 mg/dL (RV: 504–1,465), IgA = 340 mg/dL (RV: 27–195), and IgM = 537 mg/dL (RV: 24–210); CD4 = 1,280 cell/mm3 (23.26%); CD8 = 2,080 cells/mm3 (37.79%); and CD4/CD8 = 0.62. Normal renal function, with no proteinuria or hematuria. Positive for rheumatoid factor (512 IU/mL; RV <8 IU/mL). Negative for antinuclear antibodies. Histopathology of an erythematous maculopapular rash on the left hand revealed hyperkeratosis and regional atrophy associated with basal vacuolization and focal necrosis in addition to perivascular and interstitial inflammatory mixed infiltrates with mild nuclear fragmentation (Figure 1E) and, thus, inconsistent with the diagnosis of pityriasis. Histopathology of the erythematous lesions of the left limb showed inflammatory mixed perivascular, and interstitial infiltrates with neutrophils in karyorrhexis, lymphocytes, and eosinophils (Figure 1F), confirming the diagnosis of leukocytoclastic vasculitis. Positive Widal H agglutination test (flagella B reactive). Blood and stool cultures negative for bacteria. He was treated with Ciprofloxacin (40 mg/kg/day) for 2 weeks with regression of lesions and no flares. He is currently on Trimethoprim/Sulfamethoxazole prophylaxis.
We sequenced the exons of the IL12RB1 gene in Cases 2, 3, and 4 (no DNA sample was available from Case 1) and found all three to be homozygous for the autosomal recessive Trp7Ter predicted loss-of-function single nucleotide polymorphism (SNP) c.21G>A/p.Trp7Ter/TGG ⇒ TGA (Figure 2). We next sequenced the parents from the three unrelated kindreds and confirmed them all to be heterozygous for the stop-gain allele variant (Figure 2). Henceforth, we assume that Case 1 was homozygous for the stop-gain allele variant. We also genotyped a population subset of 227 unrelated individuals from the northern region of the State of Rio de Janeiro and established that the Trp7Ter predicted loss-of-function allele variant occurs at a frequency of 1 in 454 chromosomes (allele frequency of 0.0022). We note that the unrelated carrier in this subset lives in a rural area of the district of Travessão, 20.1 km from the city of Campos dos Goytacazes. Thus, we identified three unrelated couples being at high risk of recurrence in future pregnancies.
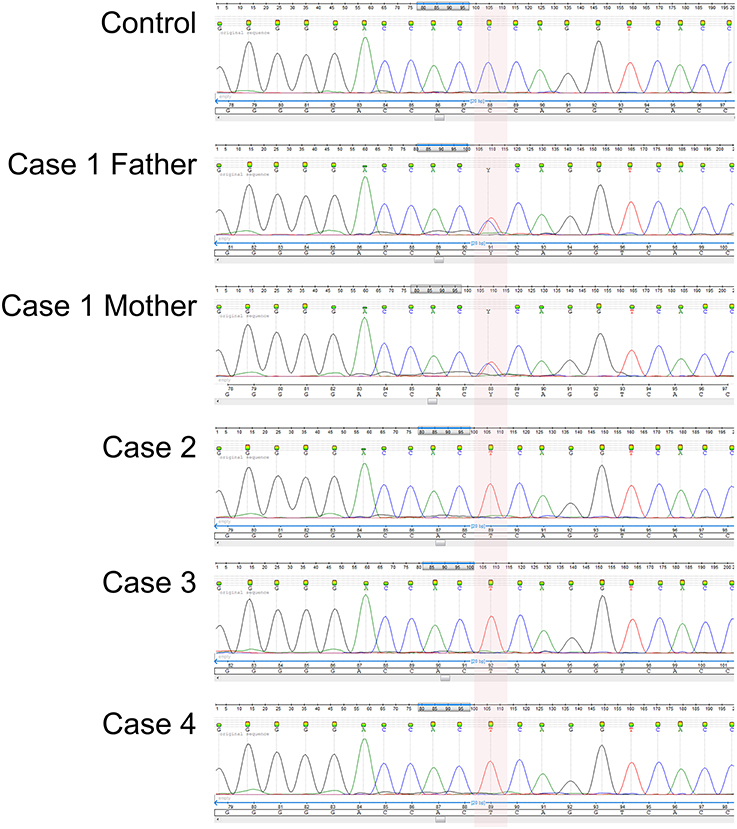
Figure 2. Electropherogram of the novel IL12RB1 predicted loss-of-function genotype confirmed by Sanger sequencing. The area shaded in pink indicates the homozygous Trp7Ter predicted loss-of-function genotype for the SNP variant rs150172855 (c.21G>A/p.Trp7Ter/TGG ⇒ TGA) identified in Cases 2, 3, and 4. As exemplified here with Case 1, the parents of all four cases were heterozygous carriers. The stop-gain mutation is located in exon 1 within the predicted coding region of the signal peptide. Shown are the sequencing traces for the forward strand of DNA.
The Trp7Ter stop-gain mutation is absent in 841,883 genotyped or sequenced genomes and whole exomes worldwide in public databases (NHLBI GO Exome Sequencing Project, 2012; Sulem et al., 2015; Chen et al., 2016; Lek et al., 2016; Narasimhan et al., 2016; Supplementary Data). However, a heterozygous carrier of the stop-gain mutation was identified in the Severe Asthma Research Program (SARP) at a frequency of 0.0005 (1 out of 2,056 chromosomes; Moore et al., 2007; Torgerson et al., 2012) and deposited into dbSNP under rs150172855. The subject is of self-reported European ancestry. To assess the genetic ancestry of this individual, we ran a Principal Component Analysis using genome-wide SNP genotypes with other subjects from the same cohort study (Torgerson et al., 2012). These included African American and European American population subsets as well as the HapMap phase 3 populations (International HapMap et al., 2010). Consistent with the participant's self-reported European ancestry, this individual clustered with the HapMap population with Northern and Western European ancestry (CEU) and other persons of self-reported European ancestry from the SARP (Supplementary Figure S1). We also determined that Cases 2, 3, and 4 share the CACCAGTCCGG (forward strand) extended homozygous genotype, which spans 27.3 kb across the IL12RB1 gene (Supplementary Figure S2, Supplementary Table S1, and Supplementary data). Moreover, the three probands are homozygous for seven out of eight tested informative SNP variant alleles of European ancestry (Supplementary Table S2, and Supplementary data).
We showed ex vivo that the stop-gain mutation at the signal peptide coding sequence, paradoxically, does not knock out cell-surface expression of the IL-12Rβ1 polypeptide (Figure 3). As observed in a non-carrier control subject (Figure 3A), and the two heterozygous carrier mothers (kindred B, Figure 3B and kindred C, Figure 3C), the IL-12Rβ1 protein antigen is present in the cell-surface of PHA-blasts from Case 2 (Figure 3D), Case 3 (Figure 3E), and Case 4 (Figure 3F). Importantly, the ex vivo and in vitro levels of the cell-surface expressed IL-12Rβ1 polypeptide are diminished in these children, and the receptor activity is affected because the production of IFN-γ in response to phytohemagglutinin (PHA; Figure 4A) and BCG lysate (Figure 4B) and of IL-17A in response to PHA (Figure 4C) and BCG (Figure 4D) stimuli is impaired vs. the control subjects. Worth of note, PHA-blasts from Case 2 produced >4,000 pg/mL of IFN-γ, which is close to the levels observed in the controls (Figure 4A), and contrasting to the situation in the younger affected brother (Case 3). There are reports of close to normal and even higher levels of IFN-γ production in PHA- or BCG-blasts of a few other patients with IMD30 (de Jong et al., 1998; Caragol et al., 2003; Lichtenauer-Kaligis et al., 2003; Moraes-Vasconcelos et al., 2005; Schepers et al., 2013; Ramirez-Alejo et al., 2014).
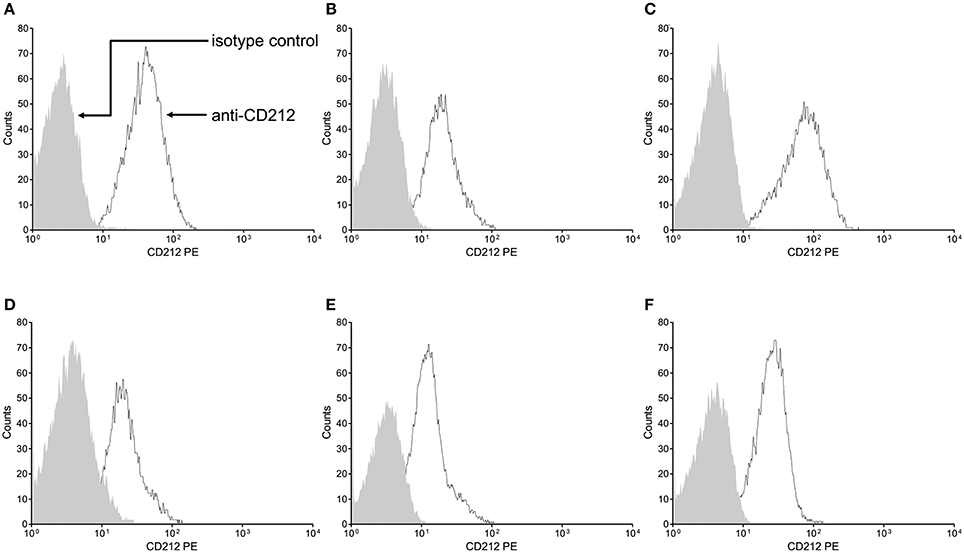
Figure 3. The Trp7Ter stop-gain mutation at the signal peptide sequence does not knock out cell-surface expression of the IL-12Rβ1 polypeptide. Cell-surface expression of IL-12Rβ1 protein antigen in peripheral blood mononuclear cells was ascertained by flow cytometry in a representative non-carrier control subject (A), and the heterozygous carrier mothers of case 2 (B) and case 4 (C) and the homozygous Trp7Ter predicted loss-of-function cases 2 (D), 3 (E), and 4 (F). The PHA-T-cell blasts were stained with phycoerythrin (PE)-labeled mouse 2.4E6 monoclonal antibody anti-Human CD212 (IL-12Rβ1), or PE-labeled matched control isotype. The arrows point to the matched control isotype and CD212 specific staining intensities depicted as the shaded and open areas under the curves, respectively. In all subjects, stimulated cells exhibited increased fluorescence intensity, reflecting the presence of the CD212 specific IL-12Rβ1 antigen at the cell surface, independently of the occurrence of the stop-gain mutation. For comparison, the expression mean percentage of positive cells in seven non-carrier controls was 88.2.
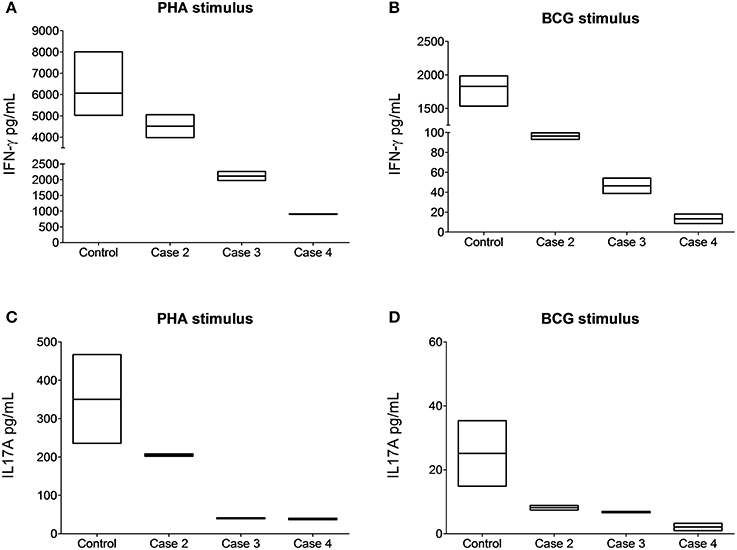
Figure 4. The cell-surface-expressed IL-12Rβ1 protein in the affected children is reduced and functionally impaired. Cytokine-specific Cytometric Bead Array analyses revealed reduced production of IFN-γ in response to PHA (A) and BCG lysate (B) and of IL-17A in response to PHA (C) and BCG lysate (D) stimuli in cultured peripheral blood monocular cells from the subjects homozygous for the IL12RB1 Trp7Ter predicted loss-of-function mutation vs. the control subjects. For comparison, in five control non-carriers, the IFN-γ mean expression values were 6,063 and 1,830 pg/mL upon stimulation with PHA or BCG lysate, respectively. For IL17-A cytokine, the mean expression values were 350.3 and 25.10 pg/mL under the same stimuli. Quantification assays were done on samples taken at least 4 months after remission (3 years for case 2, 4 months for case 3, and 2 years for case 4). Boxes, interquartile ranges; horizontal lines within boxes, median values.
IL-12Rβ1 is a type I transmembrane receptor chain; thus, it has an extracellular N-terminal domain topology. Interestingly, the Trp7Ter mutation maps to within the predicted 23-amino acid N-terminal signal peptide coding sequence. By imputation, the Trp7Ter stop-gain mutation creates either an exonic acceptor splice silencer (ESS) site, an acceptor splice site or a donor splice site [Supplementary Figure S3 with potentially deleterious effect(s) through RNA mis-splicing]. Whether the Trp7Ter stop-gain mutation is deleterious through exon 1 partial in-frame skipping in the IL12RB1 gene or there is transcriptional initiation from potentially alternative non-AUG start sites downstream of the annotated AUG (i.e., Val9, Leu16, or Leu17; Supplementary Figure S3) is still unclear.
Background
MSMD comprises 19 complete or partial primary immunodeficiencies of the IFN-γ-mediated immunity (IFN-γ axis). Mutations in seven autosomal genes (IFNGR1, IFNGR2, IL12B, IL12RB1, IRF8, ISG15, STAT1) and two X-linked genes (IKBKG and CYBB) caused MSMD (Bustamante et al., 2014). The hallmark of MSMD is the impaired production of IFN-γ or a lack of response to it. MSMD patients exhibit variable vulnerability to early and often life-threatening infections with intramacrophagic bacteria (Mycobacterium spp., Salmonella spp., and Klebsiella spp.) and fungi (Candida spp., Paracoccidioides spp., Coccidioides spp., Cryptococcus spp., and Histoplasma spp.; Moraes-Vasconcelos et al., 2005; de Beaucoudrey et al., 2010). Adverse events are an important warning sign of MSMD in the context of vaccination with BCG, BCG-itis (local or regional disease), or BCG-osis (disseminated disease; de Beaucoudrey et al., 2010; Costa-Carvalho et al., 2014; Abolhassani et al., 2015).
A recent database update compiled 77 pathogenic allele variants in the IL12RB1 gene on chromosome 19p13 in 211 patients (de Beaucoudrey et al., 2010; van de Vosse et al., 2013), which accounts for 38.2% of cases of MSMD. IL12RB1 loss-of-function impairs both IL-12 and IL-23 responses. Most pathogenic allele mutations in the IL12RB1 gene cause complete IL-12Rβ1 deficiency (immunodeficiency-30, IMD30) without cell-surface expression of the receptor. However, in homozygous carriers of the c.230T>C, c.632G>C, c.700+362_1619-944del, and p.(Pro233_Glu234insVGLVLIA) variants (Scheuerman et al., 2007; van de Vosse et al., 2013; Ramirez-Alejo et al., 2014), the cell-surface expression of the IL-12Rβ1 polypeptide is reduced (Lichtenauer-Kaligis et al., 2003; Fieschi et al., 2004). There are only ten reported stop-gain mutations linked to IMD30 with information on cell-surface expression (de Jong et al., 1998; de Beaucoudrey et al., 2010; Boisson-Dupuis et al., 2011; van de Vosse et al., 2013). Significantly, all ten stop-gain variants knockout cell-surface expression of the receptor.
Discussion
We describe the health effects of the IL12RB1 inactivation by the novel N-terminal signal peptide Trp7Ter stop-gain, predicted loss-of-function inherited homozygous genotype in three children from two apparently unrelated Brazilian kindreds and inferred the same genotype in a fourth case from a third geographically proximate kindred. All four probands have a history of BCG adverse events (suppurative adenitis in all four and recurrence in two patients) at an early age, three of them exhibited diminished cell-surface expression of the IL-12Rβ1 polypeptide and had impaired production of IFN-γ and IL-17A in response to in vitro stimuli. The genotype-phenotype correlations observed in these four children are a hallmark of MSMD (de Beaucoudrey et al., 2010). Besides the complications with the BCG vaccine, clinical features varied in breadth with different age of onset, and were associated mainly with Histoplasma spp. and Salmonella spp. infections. In all four cases, the health effects necessitated hospitalization with surgical interventions in two subjects. We believe that the cause of death of Case 1 was BCG-osis because the severe manifestations appeared shortly after the interruption of the intermittent use of Isoniazid and a lack of response to Ceftriaxone, which excluded a Salmonella infection.
Except for candidiasis, fungal diseases in IMD30 are rare. Reports in endemic countries are one case each by Histoplasma spp. in India (de Beaucoudrey et al., 2010), Paracoccidioides brasiliensis in Brazil (Moraes-Vasconcelos et al., 2005), Cryptococcus neoformans in Thailand (Jirapongsananuruk et al., 2012), and two cases by Coccidioides spp. in Palestine (Vinh et al., 2011). None of our four subjects had candidiasis. Case 2 had histoplasmosis and relapsed 5 years after the first episode. In the first episode, he was treated with ketoconazole. With Histoplasma spp. recrudescence, he was successfully managed with Itraconazole, following the updated guidelines (Wheat et al., 2007). Worth noting, in immunocompetent patients, histoplasmosis has been successfully managed using ketoconazole (Faiolla et al., 2013). Cases of disseminated histoplasmosis have been reported in individuals with IFN-γR1 deficiency (IMD27B; Zerbe and Holland, 2005), which indicates that the IFN-γ response is necessary for the control of histoplasmosis (Clemons et al., 2000). In Brazil, the number of reported cases of histoplasmosis is small, with male gender, HIV-1 infection, and rural environment being the main risks factors mentioned (Faiolla et al., 2013). All four our patients live in a rural environment, but only Case 2 had histoplasmosis.
Salmonella infections in patients with MSMD are mainly caused by non-typhoid serotypes and involve severe extraintestinal manifestations that include lymphadenopathy, bacteremia, and septicemia (MacLennan et al., 2004). Cases 3 and 4 presented with different clinical manifestations of Salmonella infections. Case 3 had sepsis by S. choleraesuis and was successfully managed with appropriate antibiotics and splenectomy. Recurrence of suppurative cervical adenopathies and the infracostal abscess in this patient were also probably related to Salmonella infection because the early onset of BCG disease has a protective role against reactivation of latent mycobacteria or a new infection (de Beaucoudrey et al., 2010) and the adequate management with Ciprofloxacin in the last episode.
Case 4 had recurrent cutaneous vasculitis. Leukocytoclastic vasculitis by Salmonella is a rare condition in MSMD patients. There are only three cases reported in IMD30 (Kutukculer et al., 2006; Sanal et al., 2006; Filiz et al., 2014). The last episode of cutaneous vasculitis in Case 4 was a full-body flare, which ruled out Henoch-Schönlein purpura. Because the child was positive for antibodies to Salmonella flagellar antigens and there was complete remission after treatment with Ciprofloxacin, we believe Salmonella infection caused the vasculitis in this subject. Other infectious diseases, use of medicines, connective tissue disorders, and malignancies were excluded.
Elevated IgE occurs in atopic dermatitis and allergic diseases (Woodfolk et al., 2015). Cases 2 and 3 evolved IgE >2,000 IU/mL but presented no symptoms of atopy, such as asthma or eczema, or mild allergy. Hyper-IgE syndrome (Job syndrome) was excluded in these children because of the clinical course. Elevated IgE occurred in some reported cases of IMD30, none of which were due to loss-of-function mutations (Altare et al., 1998; Caragol et al., 2003; Carvalho et al., 2003; Moraes-Vasconcelos et al., 2005; Luangwedchakarn et al., 2009), and in one case of Chronic Granulomatous Disease, which is a type of MSMD characterized by mycobacterial granuloma formation (Patiroglu et al., 2013). In the later case, IgA deficiency was also reported and no mutations in the STAT3 and DOCK8 genes, characteristic in hiper-IgE syndrome, were found. We believe the elevated IgE in Cases 2 and 3 reflects a state of chronic antigenic stimulation by the intracellular microbes. Thus, for children with no signs of atopy, we propose that elevated IgE is an important warning sign to investigate the narrow spectrum of MSMD. Measurement of IgE levels should be valuable also for patients seen in countries where BCG vaccination is not universal. Overall, the causative IL12RB1 mutations imply that Th1 signature cytokines have no major regulatory effect on Th2 cells in IMD30 patients (Döffinger et al., 1999). However, the Th1/Th2 polarization paradigm needs to be directly assessed in the IMD30 cases reported this far, by measuring IL-4, IL-5, IL-10, and IL-13 levels, and assessing the influence of the deficiency in the expression levels of the transcription factors T-bet, GATA-3 and RORγt.
We found that Cases 2, 3, and 4 were homozygous for the extended CACCAGTCCGG IL12RB1 haplotype. In the 1,000 Genomes Project database (Sudmant et al., 2015), this haplotype occurs at a rate of 8.4%, with 47.5, 18.2, and 11.6% of the haplotype counts, being distributed among individuals of European, American and African ancestry, respectively (Supplementary Table S3). Thus, the pathogenic Trp7Ter germline allele likely represents a founder mutation among individuals from the northern region of the State of Rio the Janeiro, Brazil, who carried an extended haplotype that is more common in European populations. These findings are significant for both estimating disease risk in the population and genetic counseling for the affected families.
Concluding Remarks
The novel Trp7Ter predicted loss-of-function homozygous IL12RB1 genotype was linked to the paradoxical cell-surface expression of an impaired IL-12Rβ1 polypeptide with health effects that varied in severity and early-onset age mainly regarding susceptibility to Mycobacteria spp., Histoplasma spp., and Salmonella spp. Our study expands the narrow spectrum of clinical manifestations observed in pediatric IMD30 by including histoplasmosis recurrence. We urge on health care professionals to evaluate IMD30 in HIV-1 negative children with a history of adverse effects to the BCG vaccine and presenting with recurrent Histoplasma spp. and extraintestinal Salmonella spp.
Ethics Statement
This study was carried out in accordance with the recommendations of the Brazilian National Ethics Committee CONEP with written informed consent from all subjects. All subjects gave written informed consent in accordance with the Declaration of Helsinki. The protocol was approved by the CONEP (national approval registry CAAE no. 35385714.0.0000.5244). The subjects were included from a pilot, observational, exploratory, qualitative study entitled Investigation of the molecular and genetic basis of the severe adverse events to the BCG vaccine in the context of the Chronic Granulomatous Disease and the Mendelian Susceptibility to Mycobacteria Disease. A major goal of the study was to determine the rates of adverse events to the BCG vaccine in children as warning signs of primary immunodeficiencies in otherwise healthy children. The study involved physical and history examinations of subjects, laboratory testing for primary immunodeficiencies, management of episodes of infectious diseases, and genetic testing and counseling. Sampling was by convenience or accessibility, in subjects who presented with or have a history of adverse events to the BCG vaccine. Peripheral blood samples from participating controls and the three Brazilian families were collected with written informed consent. For infants and children, a surrogate consent procedure was used, whereby the next of kin or a legally authorized representative approved in writing on behalf of the participants.
Author Contributions
TL, RF, JC, JB, and EM: conceived and designed experiments. TL: performed genotyping, sequencing, analyzed the data. TL and JA: performed flow cytometry studies. TL, VG, and EM: performed genotyping of extended haplotypes. RF, AG, AS, NM, and JS: recruited, attended patients. CD, SB, JC, and JB: performed genetic screening and sequencing. DT: performed principal component analysis. EM: carried out comprehensive computational analysis, contributed biological samples, reagents, materials, made figures, and wrote the manuscript.
Funding
This work was supported by grants from the Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (BR) (http://www.faperj.br/) [grant numbers E-26/110.775/2011, E-26/111.715/2012 and E26/010.001036/2015 to EM and E26/010.000892/2015 to RF], and from Conselho Nacional de Desenvolvimento Científico e Tecnológico (BR) (http://cnpq.br/) [grant number 301034/2012-5 to EM]. TL received a graduate fellowship (grant number 1121200877) from Universidade Estadual do Norte Fluminense Darcy Ribeiro (BR) (http://www.uenf.br/). VG received an undergraduate fellowship (grant number E-26/202.426/2015) from Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (BR). The agencies had no role in the study design, data collection, and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the three families for the willingness and trust to participate in this investigation. We are grateful to Doctors Laila Ayd de Oliveira de Lima, Rodrigo Aires de Morais, Andreya Moreira de Souza Soares Machado, Bianca Silveira Moreira, Felipe Eulalio Baldi Pessanha, Maira Terezinha Abel and Elizabeth Passebon Soares from the Municipal Health Units of the northern region of the State of Rio de Janeiro for their discussion of cases. We thank Dr. Deborah A. Meyers from Wake Forest School of Medicine for sharing genotype data from the Severe Asthma Research Program.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.00616/full#supplementary-material
References
Abolhassani, H., Rezaei, N., Mohammadinejad, P., Mirminachi, B., Hammarstrom, L., and Aghamohammadi, A. (2015). Important differences in the diagnostic spectrum of primary immunodeficiency in adults versus children. Expert Rev. Clin. Immunol. 11, 289–302. doi: 10.1586/1744666X.2015.990440
Altare, F., Durandy, A., Lammas, D., Emile, J. F., Lamhamedi, S., Le Deist, F., et al. (1998). Impairment of mycobacterial immunity in human interleukin-12 receptor deficiency. Science 280, 1432–1435.
Boisson-Dupuis, S., El Baghdadi, J., Parvaneh, N., Bousfiha, A., Bustamante, J., Feinberg, J., et al. (2011). IL-12Rbeta1 deficiency in two of fifty children with severe tuberculosis from Iran, Morocco, and Turkey. PLoS ONE 6:e18524. doi: 10.1371/journal.pone.0018524
Bustamante, J., Boisson-Dupuis, S., Abel, L., and Casanova, J. L. (2014). Mendelian susceptibility to mycobacterial disease: genetic, immunological, and clinical features of inborn errors of IFN-γ immunity. Semin. Immunol. 26, 454–470. doi: 10.1016/j.smim.2014.09.008
Caragol, I., Raspall, M., Fieschi, C., Feinberg, J., Larrosa, M. N., Hernández, M., et al. (2003). Clinical tuberculosis in 2 of 3 siblings with interleukin-12 receptor beta1 deficiency. Clin. Infect. Dis. 37, 302–306. doi: 10.1086/375587
Carneiro-Sampaio, M., Jacob, C. M., and Leone, C. R. (2011). A proposal of warning signs for primary immunodeficiencies in the first year of life. Pediatr. Allergy Immunol. 22, 345–346. doi: 10.1111/j.1399-3038.2010.01084.x
Carvalho, B. T., Iazzetti, A. V., Ferrarini, M. A., Campos, S. O., Iazzetti, M. A., and Carlesse, F. A. (2003). [Salmonella septicemia associated with interleukin 12 receptor β1 (IL-12β1) deficiency]. J. Pediatr. (Rio J) 79, 273–276. doi: 10.2223/JPED.1031
Chen, R., Shi, L., Hakenberg, J., Naughton, B., Sklar, P., Zhang, J., et al. (2016). Analysis of 589,306 genomes identifies individuals resilient to severe Mendelian childhood diseases. Nat. Biotechnol. 34, 531–538. doi: 10.1038/nbt.3514
Clemons, K. V., Darbonne, W. C., Curnutte, J. T., Sobel, R. A., and Stevens, D. A. (2000). Experimental histoplasmosis in mice treated with anti-murine interferon-γ antibody and in interferon-γ gene knockout mice. Microbes Infect. 2, 997–1001. doi: 10.1016/S1286-4579(00)01253-3
Costa-Carvalho, B. T., Grumach, A. S., Franco, J. L., Espinosa-Rosales, F. J., Leiva, L. E., King, A., et al. (2014). Attending to warning signs of primary immunodeficiency diseases across the range of clinical practice. J. Clin. Immunol. 34, 10–22. doi: 10.1007/s10875-013-9954-6
de Beaucoudrey, L., Samarina, A., Bustamante, J., Cobat, A., Boisson-Dupuis, S., Feinberg, J., et al. (2010). Revisiting human IL-12Rβ1 deficiency: a survey of 141 patients from 30 countries. Medicine (Baltimore) 89, 381–402. doi: 10.1097/MD.0b013e3181fdd832
de Jong, R., Altare, F., Haagen, I. A., Elferink, D. G., Boer, T., van Breda Vriesman, P. J., et al. (1998). Severe mycobacterial and Salmonella infections in interleukin-12 receptor-deficient patients. Science 280, 1435–1438.
Döffinger, R., Jouanguy, E., Altare, F., Wood, P., Shirakawa, T., Novelli, F., et al. (1999). Inheritable defects in interleukin-12- and interferon-γ-mediated immunity and the TH1/TH2 paradigm in man. Allergy 54, 409–412. doi: 10.1034/j.1398-9995.1999.00088.x
Faiolla, R. C., Coelho, M. C., Santana Rde, C., and Martinez, R. (2013). Histoplasmosis in immunocompetent individuals living in an endemic area in the Brazilian Southeast. Rev. Soc. Bras. Med. Trop. 46, 461–465. doi: 10.1590/0037-8682-0124-2013
Fieschi, C., Bosticardo, M., de Beaucoudrey, L., Boisson-Dupuis, S., Feinberg, J., Santos, O. F., et al. (2004). A novel form of complete IL-12/IL-23 receptor beta1 deficiency with cell surface-expressed nonfunctional receptors. Blood 104, 2095–2101. doi: 10.1182/blood-2004-02-0584
Filiz, S., Kocacik Uygun, D. F., Verhard, E. M., van Dissel, J. T., Uygun, V., Bassorgun, C., et al. (2014). Cutaneous leukocytoclastic vasculitis due to Salmonella enteritidis in a child with interleukin-12 receptor beta-1 deficiency. Pediatr. Dermatol. 31, 236–240. doi: 10.1111/j.1525-1470.2012.01856.x
International HapMap, C., Altshuler, D. M., Gibbs, R. A., Peltonen, L., Altshuler, D. M., Gibbs, R. A., et al. (2010). Integrating common and rare genetic variation in diverse human populations. Nature 467, 52–58. doi: 10.1038/nature09298
Jirapongsananuruk, O., Luangwedchakarn, V., Niemela, J. E., Pacharn, P., Visitsunthorn, N., Thepthai, C., et al. (2012). Cryptococcal osteomyelitis in a child with a novel compound mutation of the IL12RB1 gene. Asian Pac. J. Allergy Immunol. 30, 79–82.
Kutukculer, N., Genel, F., Aksu, G., Karapinar, B., Ozturk, C., Cavusoglu, C., et al. (2006). Cutaneous leukocytoclastic vasculitis in a child with interleukin-12 receptor beta-1 deficiency. J. Pediatr. 148, 407–409. doi: 10.1016/j.jpeds.2005.10.003
Lek, M., Karczewski, K. J., Minikel, E. V., Samocha, K. E., Banks, E., Fennell, T., et al. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291. doi: 10.1038/nature19057
Lichtenauer-Kaligis, E. G., de Boer, T., Verreck, F. A., van Voorden, S., Hoeve, M. A., van de Vosse, E., et al. (2003). Severe Mycobacterium bovis BCG infections in a large series of novel IL-12 receptor beta1 deficient patients and evidence for the existence of partial IL-12 receptor beta1 deficiency. Eur. J. Immunol. 33, 59–69. doi: 10.1002/immu.200390008
Luangwedchakarn, V., Jirapongsaranuruk, O., NiemeLa, J. E., Thepthai, C., Chokephaibulkit, K., Sukpanichnant, S., et al. (2009). A novel mutation of the IL12RB1 gene in a child with nocardiosis, recurrent salmonellosis and neurofibromatosis type I: first case report from Thailand. Asian Pac. J. Allergy Immunol. 27, 161–165.
MacLennan, C., Fieschi, C., Lammas, D. A., Picard, C., Dorman, S. E., Sanal, O., et al. (2004). Interleukin (IL)-12 and IL-23 are key cytokines for immunity against Salmonella in humans. J. Infect. Dis. 190, 1755–1757. doi: 10.1086/425021
Moore, W. C., Bleecker, E. R., Curran-Everett, D., Erzurum, S. C., Ameredes, B. T., Bacharier, L., et al. (2007). Characterization of the severe asthma phenotype by the National Heart, Lung, and Blood Institute's Severe Asthma Research Program. J. Allergy Clin. Immunol. 119, 405–413. doi: 10.1016/j.jaci.2006.11.639
Moraes-Vasconcelos, D. d., Grumach, A. S., Yamaguti, A., Andrade, M. E., Fieschi, C., de Beaucoudrey, L., et al. (2005). Paracoccidioides brasiliensis disseminated disease in a patient with inherited deficiency in the beta1 subunit of the interleukin (IL)-12/IL-23 receptor. Clin. Infect. Dis. 41, e31–37. doi: 10.1086/432119
Narasimhan, V. M., Hunt, K. A., Mason, D., Baker, C. L., Karczewski, K. J., Barnes, M. R., et al. (2016). Health and population effects of rare gene knockouts in adult humans with related parents. Science 352, 474–477. doi: 10.1126/science.aac8624
NHLBI GO Exome Sequencing Project (2012). Exome Variant Server. Seattle, WA. Available online at: http://evs.gs.washington.edu/EVS/ [Accessed April 29, 2016 2016].
Patiroglu, T., Gungor, H. E., Lazaroski, S., and Unal, E. (2013). Chronic granulomatous disease with markedly elevated IgE levels mimicking hyperimmunoglobulin E syndrome. Acta Microbiol. Immunol. Hung. 60, 155–162. doi: 10.1556/AMicr.60.2013.2.6
Ramirez-Alejo, N., Blancas-Galicia, L., Yamazaki-Nakashimada, M., García-Rodríguez, S. E., Rivas-Larrauri, F., Paolo-Cienfuegos, D. P., et al. (2014). Molecular analysis for patients with IL-12 receptor beta1 deficiency. Clin. Genet. 86, 161–166. doi: 10.1111/cge.12253
Sanal, O., Turul, T., De Boer, T., Van de Vosse, E., Yalcin, I., Tezcan, I., et al. (2006). Presentation of interleukin-12/-23 receptor β1 deficiency with various clinical symptoms of Salmonella infections. J. Clin. Immunol. 26, 1–6. doi: 10.1007/s10875-006-7830-3
Schepers, K., Schandené, L., Bustamante, J., Van Vooren, J. P., de Suremain, M., Casanova, J. L., et al. (2013). IL-12Rβ1 deficiency and disseminated Mycobacterium tilburgii disease. J. Clin. Immunol. 33, 1285–1288. doi: 10.1007/s10875-013-9941-y
Scheuerman, O., de Beaucoudrey, L., Hoffer, V., Feinberg, J., Casanova, J. L., and Garty, B. Z. (2007). Mycobacterial disease in a child with surface-expressed non-functional interleukin-12Rbeta1 chains. Isr. Med. Assoc. J. 9, 560–561.
Sudmant, P. H., Rausch, T., Gardner, E. J., Handsaker, R. E., Abyzov, A., Huddleston, J., et al. (2015). An integrated map of structural variation in 2,504 human genomes. Nature 526, 75–81. doi: 10.1038/nature15394
Sulem, P., Helgason, H., Oddson, A., Stefansson, H., Gudjonsson, S. A., Zink, F., et al. (2015). Identification of a large set of rare complete human knockouts. Nat. Genet. 47, 448–452. doi: 10.1038/ng.3243
Torgerson, D. G., Capurso, D., Ampleford, E. J., Li, X., Moore, W. C., Gignoux, C. R., et al. (2012). Genome-wide ancestry association testing identifies a common European variant on 6q14.1 as a risk factor for asthma in African American subjects. J. Allergy Clin. Immunol. 130, 622–629 e629. doi: 10.1016/j.jaci.2012.03.045
van de Vosse, E., Haverkamp, M. H., Ramirez-Alejo, N., Martinez-Gallo, M., Blancas-Galicia, L., Metin, A., et al. (2013). IL-12Rbeta1 deficiency: mutation update and description of the IL12RB1 variation database. Hum. Mutat. 34, 1329–1339. doi: 10.1002/humu.22380
Vinh, D. C., Schwartz, B., Hsu, A. P., Miranda, D. J., Valdez, P. A., Fink, D., et al. (2011). Interleukin-12 receptor beta1 deficiency predisposing to disseminated Coccidioidomycosis. Clin. Infect. Dis. 52, e99–e102. doi: 10.1093/cid/ciq215
Wheat, L. J., Freifeld, A. G., Kleiman, M. B., Baddley, J. W., McKinsey, D. S., Loyd, J. E., et al. (2007). Clinical practice guidelines for the management of patients with histoplasmosis: 2007 update by the Infectious Diseases Society of America. Clin. Infect. Dis. 45, 807–825. doi: 10.1086/521259
Woodfolk, J. A., Commins, S. P., Schuyler, A. J., Erwin, E. A., and Platts-Mills, T. A. (2015). Allergens, sources, particles, and molecules: Why do we make IgE responses? Allergol. Int. 64, 295–303. doi: 10.1016/j.alit.2015.06.001
Zahid, M. F., Ali, S. A., Jehan, F., Billo, A. G., Casanova, J. L., Bustamante, J., et al. (2014). Recurrent Salmonellosis in a Child with Complete IL-12Rβ1 Deficiency. J Immunodefic Disord. 3:1000109. doi: 10.4172/2324-853X.1000109
Keywords: IL-12Rβ1 deficiency, Mendelian susceptibility to mycobacterial diseases, impaired receptor, IFN-γ, signal peptide, founder stop-gain mutation
Citation: Louvain de Souza T, de Souza Campos Fernandes RC, Azevedo da Silva J, Gomes Alves Júnior V, Gomes Coelho A, Souza Faria AC, Moreira Salomão Simão NM, Souto Filho JT, Deswarte C, Boisson-Dupuis S, Torgerson D, Casanova J-L, Bustamante J and Medina-Acosta E (2017) Microbial Disease Spectrum Linked to a Novel IL-12Rβ1 N-Terminal Signal Peptide Stop-Gain Homozygous Mutation with Paradoxical Receptor Cell-Surface Expression. Front. Microbiol. 8:616. doi: 10.3389/fmicb.2017.00616
Received: 23 November 2016; Accepted: 27 March 2017;
Published: 13 April 2017.
Edited by:
Ying Zhang, Johns Hopkins University, USAReviewed by:
Miriam Wittmann, University of Leeds, UKLeopoldo Santos-Argumedo, Center for Advanced Research, The National Polytechnic Institute, Cinvestav-IPN, Mexico
Copyright © 2017 Louvain de Souza, de Souza Campos Fernandes, Azevedo da Silva, Gomes Alves Júnior, Gomes Coelho, Souza Faria, Moreira Salomão Simão, Souto Filho, Deswarte, Boisson-Dupuis, Torgerson, Casanova, Bustamante and Medina-Acosta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Enrique Medina-Acosta, cXVpcXVlQHVlbmYuYnI=