 Atmika Paudel1
Atmika Paudel1 Hiroshi Hamamoto1
Hiroshi Hamamoto1 Suresh Panthee1
Suresh Panthee1 Keiichi Kaneko2Shigeki Matsunaga2Motomu Kanai2Yutaka Suzuki3
Keiichi Kaneko2Shigeki Matsunaga2Motomu Kanai2Yutaka Suzuki3 Kazuhisa Sekimizu1,4*
Kazuhisa Sekimizu1,4*- 1Institute of Medical Mycology, Teikyo University, Hachioji, Tokyo, Japan
- 2Laboratory of Synthetic Organic Chemistry, Graduate School of Pharmaceutical Sciences, The University of Tokyo, Tokyo, Japan
- 3Department of Medical Genome Science, Graduate School of Frontier Sciences, The University of Tokyo, Kashiwa, Japan
- 4Genome Pharmaceuticals Institute Co., Ltd., Bunkyo, Tokyo, Japan
Synthetic compounds are a vital source of antimicrobial agents. To uncover therapeutically effective antimicrobial agents from a chemical library, we screened over 100,000 synthetic compounds for in vitro antimicrobial activity against methicillin-resistant Staphylococcus aureus and evaluated the in vivo therapeutic effectiveness of the hits in S. aureus-infected silkworms. Three antimicrobial agents exhibited therapeutic effects in the silkworm infection model. One of these, GPI0363, a novel spiro-heterocyclic compound, was bacteriostatic and inhibited RNA synthesis in S. aureus cells. GPI0363-resistant S. aureus strains harbored a point mutation in the gene encoding the primary sigma factor, SigA, of RNA polymerase, and this mutation was responsible for the resistance to GPI0363. We further revealed that GPI0363 could bind to SigA, inhibit promoter-specific transcription in vitro, and prolong the survival of mice infected with methicillin-resistant S. aureus. Thus, GPI0363 is an attractive candidate therapeutic agent against drug-resistant S. aureus infections.
Introduction
Infectious diseases are a major cause of disease and death around the world. The emergence of drug-resistant pathogens has drastically increased, resulting in a scarcity of effective antimicrobials. The worldwide spread of multidrug-resistant pathogens has greatly increased the economic and health burden (WHO, 2014). New antimicrobials with therapeutic effectiveness against drug-resistant pathogens are thus urgently needed. Staphylococcus aureus is a Gram-positive opportunistic pathogen that causes infectious diseases such as sepsis and pneumonia, and skin and soft tissue infections (Lowy, 1998). S. aureus strains resistant to clinically used antibiotics such as methicillin, linezolid, daptomycin, and vancomycin are a serious threat to global health (Jevons, 1961; Hiramatsu et al., 1997; Tsiodras et al., 2001; Mangili et al., 2005; Marty et al., 2006; Howden et al., 2010). The discovery and development of novel antibacterial agents effective against such drug-resistant staphylococci is therefore critical.
The screening of synthetic compounds for antimicrobial activities began with the discovery of arsphenamine by Paul Ehrlich in 1907 (Williams, 2009). The introduction of sulfanilamides, quinolones, and linezolid, among others, demonstrated that chemically synthesized molecules are promising for the treatment of infectious diseases caused by pathogenic bacteria (Emmerson and Jones, 2003; Shaw and Barbachyn, 2011; Aminov, in press). Of the 30 new antimicrobials launched since 2000, more than 50% are synthetic compounds, including two new classes of antimicrobial agents (Butler et al., 2017). Additionally, 14 synthetic antimicrobials are currently in phase II or phase III clinical trials (Butler et al., 2017). Therefore, synthetic compounds comprise a major and promising source of antimicrobial agents, and the screening of chemical libraries can reveal promising drug leads for the treatment of infectious diseases.
Apart from finding new compounds effective against drug-resistant pathogens, obtaining therapeutically effective agents for clinical application is crucial. This is a challenging task as not all agents with in vitro effectiveness are effective in vivo due to factors such as poor pharmacokinetic parameters and toxicity, which are difficult to predict based simply on the chemical structure. Individual assessment of the therapeutic activity of each newly discovered compound is therefore essential. The identification of therapeutically effective agents requires screening in animal models. The use of mammalian models for screening, however, is fraught with ethical issues in addition to high cost and the need for sophisticated facilities. The use of invertebrate models, on the other hand, avoids such issues, and is highly suitable for the early stages of screening and drug development.
In the present study, we utilized a silkworm infection model to screen a chemical library of synthetic compounds for the first time, and identified a spiro-heterocyclic molecule, GPI0363, as a novel anti-staphylococcal agent. We demonstrated that GPI0363 inhibited RNA synthesis in vivo in S. aureus, and found that the primary sigma factor, SigA, was involved in its anti-staphylococcal activity.
Materials and Methods
Chemical Library Screening
Primary Screening
The chemical library of the Drug Discovery Initiative at the University of Tokyo (http://www.ddi.u-tokyo.ac.jp/en/) was used. As of March 2016, this library comprises more than 230,000 “synthetic compounds regulating a biological function.” Chemical compounds obtained from the library were dissolved in dimethyl sulfoxide (DMSO) at a final concentration of 10 mM. Inhibition of MRSA growth was determined by broth microdilution assay (Clinical and Laboratory Standards Institute, 2012). Briefly, S. aureus MRSA4 was grown with aeration in 5 mL Tryptic Soy Broth (TSB; Becton Dickinson and Company, Franklin Lakes NJ, USA) in a shaker maintained at 37°C. The overnight culture was diluted with cation-adjusted Muller–Hinton Broth (MHB; Becton Dickinson and Company) to have ~5 × 105 colony forming units (CFU)/mL per well in a round bottom 96-well-plate. Compounds were added to each well to obtain a final concentration of 100 μM. Vancomycin was used as a positive control and DMSO as a vehicle control in each plate. The plates were incubated at 37°C for 20 h. Compounds that inhibited growth when vancomycin inhibited the growth and DMSO did not inhibit the growth were selected as hits.
Secondary Screening
Therapeutic activity in silkworms infected with methicillin-susceptible S. aureus (MSSA) was determined as previously described (Hamamoto et al., 2015). Briefly, hatched larvae from silkworm eggs (Hu•Yo × Tsukuba•Ne, Ehime Sanshu, Japan) were fed and grown at 27°C until the fourth molt stage. S. aureus MSSA1 was grown with aeration in 5 mL TSB at 37°C. The overnight culture was diluted with 0.9% NaCl to obtain ~6 × 108 CFU/mL. Fifty microliters of the culture was injected into the hemolymph of fifth instar 2nd-day larvae (n = 3). Compounds (10 mM) dissolved in DMSO were diluted with 0.9% NaCl to obtain a concentration of 2 mM, thus producing a final DMSO concentration of 20%. Silkworms were immediately injected with 50 μL of each compound separately and larvae were further incubated at 27°C without feeding. Control groups received S. aureus or 20% DMSO in 0.9% NaCl or S. aureus and vancomycin (10 μg/larva). Silkworm survival was judged on day 2 when all the silkworms injected with only bacteria died, those injected with vehicle survived, and those injected with bacteria and vancomycin survived. Compounds that cured at least 2 of 3 silkworms at day 2 post-injection were judged to be therapeutically effective. To determine the half-maximal effective dose (ED50), compounds dissolved in DMSO were diluted with 0.9% NaCl; 50 μL of which was injected into the hemolymph of the silkworm (n = 10) after injection of S. aureus. The final concentrations of the compounds were 200, 150, 100, 75, 50, 25, and 12.5 μg/larva. Survival was plotted against the concentration, and the concentration that allowed for 50% silkworm survival was calculated from the graph.
Antimicrobial Spectrum
Bacterial cultures were prepared in either Luria Bertani medium (tryptone 10g/L, yeast extract 5g/L, NaCl 10g/L, pH 7.0), TSB, or MHB. Cation-adjusted MHB was used for antimicrobial susceptibility tests. For streptococcus species, cation-adjusted MHB with 2.5% lysed horse blood (Nippon Biotest Laboratories Inc, Tokyo, Japan) was used. The minimum inhibitory concentration (MIC) was determined by broth microdilution assay (Clinical and Laboratory Standards Institute, 2012).
Bacteriostatic Activity
The bacteriostatic activity was tested according to the National Committee for Clinical Laboratory Standards guidelines (National Committee for Clinical Laboratory Standards, 1999). Briefly, overnight culture of S. aureus MSSA1 grown at 37°C in MHB was diluted 1,000 times in MHB and cultured for 2 h at 37°C. For daptomycin, MHB was supplemented with 50 mg/L Ca2+. GPI0363 (20 μg/mL) or daptomycin (5 μg/mL) was added to 1 mL of the culture and incubated for 24 h at 37°C. Culture aliquots were collected at the indicated time, diluted, spread on Luria Bertani agar plates, and incubated for 24 h at 37°C. Cell viability was determined by counting the CFU of bacteria per milliliter. The lower limit of detection was 104 CFU/mL. Data were analyzed using Prism 5 for Mac OS X, version 5.0d (GraphPad Software).
Incorporation of Radiolabeled N-acetyl-glucosamine, Uridine, Thymidine, and Methionine
The amount of incorporated radiolabeled precursors was measured as previously described (Maki et al., 2001; Paudel et al., 2012, 2013). S. aureus RN4220 was grown to exponential phase and used for the assay in the presence of 25 μci [3H] N-acetyl-glucosamine (American Radiolabeled Chemicals, St. Louis, MO, USA) or 2 μci/mL of either [3H] uridine (Moravek Biochemical, Brea, CA, USA), [3H] thymidine (Moravek Biochemical), or [35S] methionine (Institute of Isotopes, Budapest, Hungary). Vancomycin, rifampicin, norfloxacin, and chloramphenicol (100 μg/mL each) were used as inhibitors of peptidoglycan, RNA, DNA, and protein synthesis, respectively. Vancomycin (100 μg/mL) or ampicillin (100 μg/mL) and vehicle were used as controls. GPI0363 (125 μg/mL) was used for all the assays. Aliquots were collected at 5, 10, 15, 20, and 30 min. Radioactivity of the acid insoluble fraction was counted with a liquid scintillation counter (LS6000SE, Beckman Coulter, Carlsbad, CA, USA) and expressed as counts per minute (CPM). Data were analyzed using Prism 5 for Mac OS X, version 5.0d (GraphPad Software).
Isolation and Analysis of S. aureus Mutants Resistant to GPI0363
S. aureus RN4220 was treated with 0.2% ethyl methanesulfonate overnight, spread on Luria Bertani agar plates containing 12.5, 25, and 50 μg/mL GPI0363, and incubated overnight at 30°C. The isolated strains were further grown at two temperatures, 30°C and 43°C. Strains that were viable at 30°C and non-viable at 43°C were defined as temperature-sensitive strains. Temperature-sensitive GPI0363-resistant strains were selected for whole-genome sequencing. The genome was sequenced according to previous report (Panthee et al., 2017a) using the Illumina HiSeq2000 (Illumina, San Diego, CA, USA) and the data were analyzed using the CLC Genomic Workbench (CLCbio, Aarhus, Denmark) to determine the mutated genes. The sigA gene was amplified with Prime STAR Max DNA polymerase using the primers sigA_Fw and sigA_Rev (Table 1), and the mutation site was confirmed by sequencing using an ABI3130 sequencer.

Table 1. Primers used in this study.
Phage Transduction
Phage transduction using phage 80 α was performed as previously described (Novick, 1991). To 100 μL of overnight culture of the recipient bacteria, 200 μL of the donor phage was added, and the solution was mixed with 3 mL of top agar [50% 0.3GL (casamino acid 3g, yeast extract 3g, NaCl 5.9g, 60% sodium lactate syrup 3.3 mL, 25% glycerol 4 mL per liter) and 0.75% agar] and poured into a plate containing bottom agar (50% 0.3GL, 1.5% agar, and 37.5 μg/mL chloramphenicol) and middle agar (50% 0.3GL and 1.5% agar). The plates were grown at 30°C for 2–3 days. The transductants grown on the plates were isolated. The MIC value of GPI0363 against these isolates was determined and the mutation site of the sigA gene was identified by sequencing on an ABI3130 sequencer. Colony polymerase chain reaction (PCR) was performed using KOD fx Neo DNA polymerase and primers: sigA_Fw and sigA_Rev (Table 1). The primers used for sequencing were: sigA_Fw, sigA_Rev, sigA_seq_Fw, and sigA_seq_Rev (Table 1).
Preparation of DNA Template and S. aureus RNA Polymerase Holoenzyme
For template preparation, a 406-bp long portion of the fbaA gene including its promoter was amplified using Prime STAR max DNA polymerase with the primers fbaA_Rev and fbaA_Fw (Table 1). The obtained PCR product was electrophoresed in 1% agarose and purified by gel extraction (QIAquick Gel Extraction kit 250, Qiagen, Hilden, Germany). S. aureus RNA polymerase (RNAP) was partially purified as described previously (Deora and Misra, 1996) with slight modification. Briefly, S. aureus strain RN4220 or GPI0363-resistant cells were grown in nutrient broth containing 2% casein enzymatic hydrolysate and 1% yeast extract until OD600 was 1.0. The cells were then harvested by centrifugation at 8000 rpm for 10 min at 4°C and washed with buffer A [10 mM Tris-HCl (pH 7.9), 10 mM MgCl2, 1.0 M NaCl, 5 mM EDTA (pH 8.0), 0.2 mM dithiothreitol (DTT), and 5% glycerol], followed by grinding buffer (0.05 M Tris, 5% glycerol, 2 mM EDTA, 0.1 mM DTT, 1 mM 2-mercaptoethanol, 0.233 M NaCl, 130 μg/mL lysozyme, 23 μg/mL phenylmethylsulfonyl fluoride). Cells (~2g) were suspended in 10 mL of grinding buffer [0.05M Tris, 5% (v/v) glycerol, 2 mM EDTA, 0.1 mM DTT, 1 mM 2-mercaptoethanol, 0.233 M NaCl, 130 μg/mL lysozyme, and 23 μg/mL phenylmethylsulfonyl fluoride], treated with 1 mg lysostaphin and incubated for 30 min at room temperature. The cells were homogenized in a polytron homogenizer (Kinematica, Littau, Switzerland) and subjected to ultra-centrifugation at 80,000 rpm for 30 min at 4°C. Ammonium sulfate (35 g/100 mL) was added to the resulting supernatant and precipitate was collected by centrifugation at 8000 rpm for 30 min at 4°C, and washed with saturated ammonium sulfate solution. The precipitate was suspended in TGED buffer [10 mM Tris-HCl (pH 7.9), 5% glycerol, 0.1 mM EDTA, and 0.2 mM DTT], aliquoted, and stocked at −80°C until use.
In vitro Transcription Assay
The in vitro transcription assay was performed as previously described (Deora and Misra, 1996) with slight modification. The reaction mixture contained a final concentration of 40 mM Tris acetate (pH 7.9), 100 mM NaCl, 5 mM MgCl2, 0.2 mM DTT, 100 μg/mL bovine serum albumin (BSA), 0.25 mM each of ATP, CTP, GTP, 0.015 mM UTP, 10 μci of [α-32P] UTP (PerkinElmer, Waltham, MA, USA), 0.5 units of RNase inhibitor, and partially purified S. aureus RNAP. GPI0363 was prepared in DMSO for the assay. The total assay concentration of DMSO did not exceed 4%. GPI0363 or DMSO was added to the reaction mixture devoid of nucleoside triphosphates and template DNA, and incubated at room temperature for 10 min. The transcription reaction was started by adding nucleoside triphosphates and template DNA, and further incubated for 10 min at 35°C. The samples were placed on ice, and 100 μL of stop solution (0.4 M ammonium acetate, 20 mM EDTA, 0.3% SDS, 4 μg of tRNA) was added. Transcripts were extracted by phenol chloroform, electrophoresed on 7M urea 6% polyacrylamide gel, and visualized by autoradiography using Typhoon FLA 9000 (GE Healthcare, Tokyo, Japan).
Purification of His-Tagged Recombinant SigA
The sigA gene was amplified by PCR with Prime STAR max DNA polymerase using primers His_SigA_Fw and His-SigA_Rev from the genomic DNA of S. aureus RN4220 (Table 1). The PCR product was digested with BamHI and XhoI, and cloned in the pET28a vector (Novagen). The plasmids were introduced into Escherichia coli BL21(DE3)/pLysS, and cells were selected with 50 μg/mL kanamycin. Colonies were inoculated into Luria Bertani medium in the presence of 50 μg/mL kanamycin. After overnight incubation at 37°C, 5 mL of the culture was added to 500 mL of the fresh medium containing 50 μg/mL kanamycin and incubated with shaking at 37°C. After the OD600 reached 0.3, 1 mM of isopropyl β-D-1-thiogalactopyranoside was added, and the culture was further incubated for 5 h at 30°C. Cells were collected by centrifugation and frozen by liquid nitrogen. SigA was purified using the ProbondTM purification system according to manufacturer's protocol (Life Technologies, Carlsbad, CA, USA).
Binding of SigA with GPI0363
TALON® magnetic beads pre-charged with cobalt (Clontech Laboratories, Mountain View, CA, USA) were equilibrated with 50 mM Tris buffer, pH 7.5. The equilibrated beads were incubated with His-tagged recombinant SigA in the presence and absence of GPI0363 for 30 min in a rotary shaker. GPI0363 was pretreated with BSA (0.5 mg/mL) prior to incubation. The resulting beads were washed with the same buffer, separated, and eluted with 50% acetonitrile + 0.1% trifluoroacetic acid. The eluted fractions were analyzed by HPLC in TSKgel α-M size exclusion column (7.8 mm ID × 30 cm, 13 μm; TOSOH) with 50% acetonitrile + 0.1% trifluoroacetic acid at a flow rate of 0.5 mL/min.
Mouse Infection Model
All mouse protocols were approved by the Animal Use Committee at the Graduate School of Pharmaceutical Science at our previous affiliation at the University of Tokyo. MRSA USA300 was cultivated overnight in TSB at 37°C. The culture was centrifuged at 10,000 rpm for 1 min at 4°C and the pellet was suspended in phosphate-buffered saline (PBS). GPI0363 was dissolved in a mixture of 1:1:6 ethanol/cremophor EL/0.9% NaCl. Mice (ICR, female, 18–20 g, 4 weeks old, CLEA, Tokyo, Japan) were infected with the bacterial suspension of 1.0 × 109 CFU per mouse by intravenous injection, followed by intraperitoneal injection of 400 mg/kg GPI0363 (n = 7) 30 min after infection. Control groups were injected with bacteria only (n = 8) or vehicle only (n = 7). Survival was noted in a condition that all mice of the vehicle group survived. Data were analyzed using Prism 5 for Mac OS X, version 5.0d (GraphPad Software).
Results
Screening and Identification of In vivo Effective Anti-Staphylococcal Agent
We screened a synthetic chemical library derived from the Drug Discovery Initiative at the University of Tokyo for compounds that inhibit the growth of an MRSA strain. Of 103,873 synthetic compounds, 3383 (3.25%) compounds inhibited the growth of MRSA in vitro. Next, we used the silkworm infection model as a secondary screening tool, and identified three (0.003%) compounds with therapeutic activity in silkworms infected with methicillin-susceptible S. aureus (MSSA) (Table 2, Figure 1A). Given that current treatment options for MRSA are quite limited and antimicrobial agents that show activity against MRSA exert activity against MSSA, we used MRSA during in vitro screening. Further, to examine the therapeutic activity in silkworms, we used MSSA as this system is robust and well-established. We focused on a spiro-heterocyclic compound, GPI0363 (7-fluoro-N-propylspiro[5H-pyrrolo[1,2-a]quinoxaline-4,4′-piperidine]-1′-carboxamide) (Figure 1A), as this was the most potent of the three compounds with an ED50 value of 26 mg/kg (Figure 1B, Table 3). GPI0363 was effective against several Staphylococcus sp., including MRSA, S. pseudintermedius, and S. haemolyticus. It was not effective against other tested Gram-positive and Gram-negative bacteria (Table 4). We tested the killing effect of GPI0363 on exponentially growing S. aureus and found that treatment of S. aureus with GPI0363 did not significantly change the colony-forming capacities after 24 h, suggesting that the action of the compound is bacteriostatic (Figure 1C). We, further found that the rate of appearance of spontaneous resistance to GPI0363 was <10−7 (12.5 μg/mL).
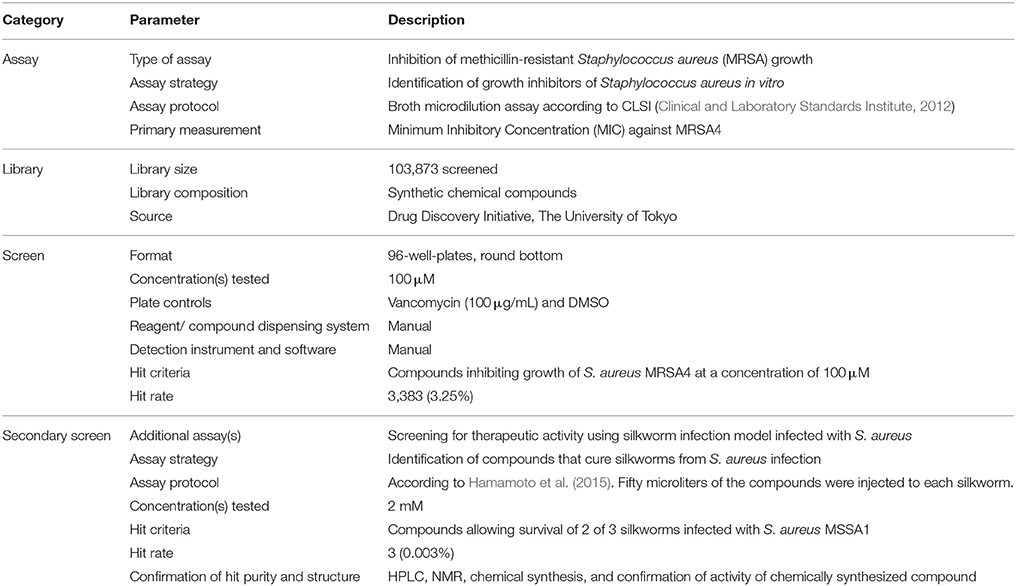
Table 2. Screening of chemical library for therapeutically effective anti-staphylococcal agent.
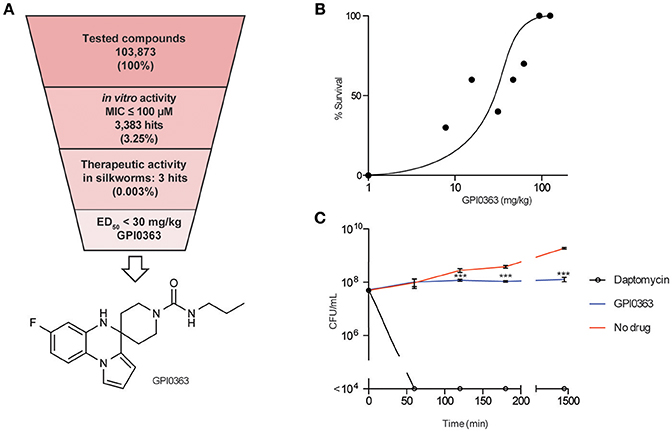
Figure 1. In vitro and in vivo activities of GPI0363. (A) Screening strategy and chemical structure of GPI0363. Compounds that displayed in vitro activity against S. aureus MRSA4 by broth microdilution assay were screened using the silkworm infection assay. Silkworms (n = 3) were injected with S. aureus MSSA1 suspension (3 × 107 CFU/larva) and immediately injected with 50 μL of 2 mM compounds. (B) In vivo activity of GPI0363 in the silkworm infection model. Silkworms were injected with S. aureus MSSA1 suspension (3 × 107 CFU/larva) and immediately injected into the hemolymph with GPI0363 at different doses (n = 10). (C) Bacteriostatic activity of GPI0363. Exponentially growing S. aureus MSSA1 was treated with 20 μg/mL GPI0363, 5 μg/mL daptomycin, or vehicle. Culture aliquots were collected at different intervals as shown, diluted, and spread on agar plates, and cell viability was determined by counting the CFU per mL. Data are shown as mean ± SD of three independent experiments. Data were analyzed by one-way ANOVA using Dunnett's Multiple Comparison test and significant differences compared with no drug are indicated by asterisks (***p ≤ 0.001).

Table 3. In vitro and in vivo activities of three candidate hits obtained from secondary screening.

Table 4. Antimicrobial spectrum of GPI0363.
GPI0363 Inhibits RNA Synthesis in S. aureus
To elucidate the mode of action of GPI0363, we determined its effect on macromolecule biosynthesis by measuring the incorporation of radiolabeled precursors into the acid-insoluble fractions in the presence of the compound. GPI0363 significantly inhibited the incorporation of [3H] uridine, a precursor of RNA synthesis, within 30 min in exponentially growing S. aureus (Figure 2A). We observed a slight inhibition of the incorporation of [3H] thymidine, a precursor of DNA synthesis, and no inhibition of the incorporation of [3H] N-acetyl-glucosamine and [35S] methionine, precursors of peptidoglycan and protein synthesis, respectively (Figures 2B–D). GPI0363 significantly inhibited RNA synthesis in S. aureus in a dose-dependent manner, and the half-maximal inhibitory concentration (IC50) value was 12 μg/mL (Figure 2E).
Figure 2. Effect of GPI0363 on macromolecule biosynthesis. The effect of GPI0363 on macromolecule biosynthesis was examined by measuring the incorporation of the corresponding radiolabeled precursors. Aliquots were collected at different intervals as indicated, and the radioactivity of acid-insoluble fractions was measured by a liquid scintillation counter and is shown as counts per minute (CPM). (A) Effect on RNA synthesis. Data are shown as mean ± SD of three independent experiments and analyzed by one-way ANOVA using Dunnett's Multiple Comparison test and significant differences compared with no drug are indicated by asterisks (*p ≤ 0.05, ***p ≤ 0.001) (B) Effect on DNA synthesis. (C) Effect on peptidoglycan synthesis. (D) Effect on protein synthesis. Data from a single experiment are shown in figures (B–D). (E) Dose response of RNA synthesis inhibition: Aliquots were collected after 30 min, and radioactivity of the acid insoluble fractions was measured. Data represents mean ± SD of triplicates. Data were analyzed by one-way ANOVA using Dunnett's Multiple Comparison test and significant differences compared with no treatment are indicated by asterisks (**p ≤ 0.01, ***p ≤ 0.001).
GPI0363-Resistant Strains Harbor a Mutation in the sigA Gene
To identify a cellular target of GPI0363, we treated S. aureus RN4220, a strain suitable for genetic manipulations (Monk and Foster, 2012), with a mutagen ethyl methanesulfonate and spread it on Luria Bertani agar plates containing 12.5, 25, or 50 μg/mL GPI0363. We observed no colonies on the plate containing 50 μg/mL GPI0363. From the plates containing 12.5 and 25 μg/mL GPI0363, a total of 150 colonies were isolated. Several studies have demonstrated that drug-resistant phenotypes showing temperature sensitivity harbor mutations in genes related to the action of the drug (Canepari et al., 1987; Hamamoto et al., 2015). Among the 150 colonies, we screened for temperature-sensitive phenotypes and checked their susceptibility to GPI0363. We identified two strains with a temperature-sensitive phenotype that displayed resistance to GPI0363 (MIC ≥ 16 μg/mL) (GPI0363R74 and GPI0363R108) (Table 5). We could not correlate the temperature-sensitive phenotype with resistance to GPI0363; therefore, we sequenced the whole genome of both the strains by a next generation sequencer. Along with other mutations, these strains commonly had a point mutation in the sigA gene (G601A) leading to an amino acid substitution, (D201N) (Table 6). Mutation in the same gene in the independent resistant strains led us to speculate the involvement of the sigA gene product in the anti-staphylococcal activity of GPI0363. To test whether the sigA gene is involved in resistance conferred by GPI0363, we performed genetic recombination with phage transduction experiments (Novick, 1991) using phage 80α. For the first phage transduction, wild-type sigA gene was introduced into the GPI0363-resistant mutant and transductants were isolated by chloramphenicol selection. The donor and recipient were phage 80α harboring wild-type sigA and a chloramphenicol-resistant (CmR) marker inserted into the SA1392 gene (Figure 3A), and GPI0363-resistant strain GPI0363R74, respectively. Next, we introduced the mutated sigA gene into the wild-type strain using phage 80α with mutant sigA (G601A) and CmR marker as the donor, and wild-type RN4220 strain as the recipient. The genotype of the transductants was confirmed by sequencing. In both the phage transduction experiments, we found that the transductants harboring the wild-type genotype had restored susceptibility to GPI0363 and strains with a mutant genotype showed resistance to GPI0363 (Figure 3B, Supplementary Tables 1, 2). Thus, we found that resistance to GPI0363 and the mutation in the sigA gene were co-transducible, suggesting that the single mutation in the sigA gene led to resistance to the compound in these strains. Further, we used BLAST to analyze the sequence of SigA of different bacteria used in our study. We found that the SigA in bacteria that were resistant to GPI0363 had <80% identity to that of staphylococci (Table 7). Hence, the specificity of GPI0363 toward staphylococci could be explained by the differences in SigA among different bacteria.

Table 5. MIC values of GPI0363 against wild-type and EMS treated selected strains.
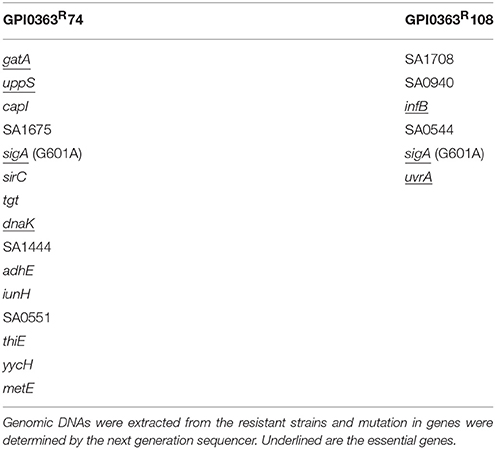
Table 6. GPI0363-resistant strains and the genes mutated.
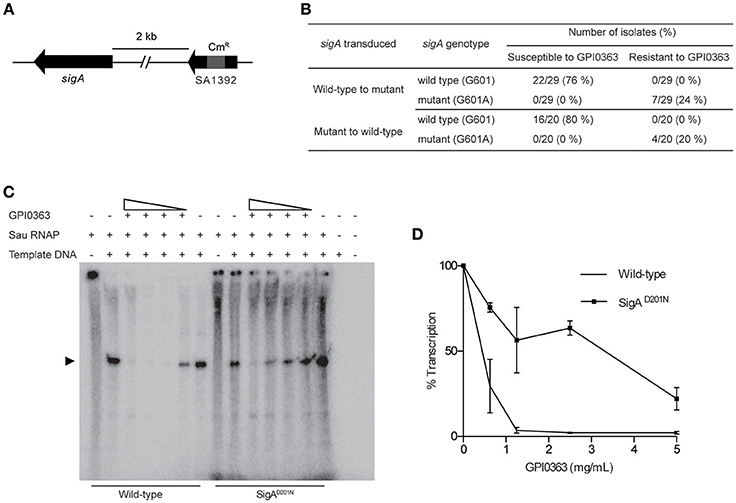
Figure 3. Involvement of SigA in transcription inhibition by GPI0363. (A) sigA gene and the resistance marker used for the phage transduction experiments. (B) Correlation between mutation in the sigA gene and resistance to GPI0363 by phage transduction analysis. Wild-type sigA gene and mutated sigA gene were transduced into the GPI0363-resistant strain and wild-type RN4220 strain, respectively, by phage transduction. The genotype of the sigA gene and the susceptibility of the strains toward GPI0363 were determined. (C) GPI0363 inhibits promoter-specific transcription in vitro. Sau RNAP from wild-type strain and SigAD201N mutant GPI0363-resistant strain were prepared as described in the Section Materials and Methods. Transcripts were extracted and electrophoresed on 7M urea and 6% polyacrylamide gel, and visualized by autoradiography. The promoter-specific transcript band is indicated by the arrowhead in the autoradiogram. (D)The intensities of the bands obtained were measured quantitatively using ImageJ software. The band intensity in the lanes containing the fraction and template was taken as 100%. Results are expressed as the average and range of the two independent experiments.

Table 7. Comparison of staphylococcal SigA with different bacteria.
GPI0363 Inhibits Promoter-Specific Transcription In vitro
The sigA gene is an essential gene encoding the primary sigma factor, SigA, of S. aureus. SigA binds to the RNAP core enzyme to form the holoenzyme and facilitates recognition of the promoter region, separation of DNA strands, and the initiation of transcription (Burgess et al., 1969; Gross et al., 1988; Helmann and dehaseth, 1999). We partially purified the S. aureus RNAP holoenzyme (hereafter referred to as Sau RNAP) from the wild-type strain and GPI0363-resistant strain (SigAD201N), and determined the effect of GPI0363 on their transcription ability using a DNA template harboring the fbaA gene promoter. GPI0363 inhibited the promoter-specific transcription by wild-type Sau RNAP while the SigAD201N mutant Sau RNAP was comparatively resistant to the compound (Figures 3C,D) with an IC50 value of 140 and 650 μg/mL for the wild-type and the SigAD201N mutant Sau RNAP, respectively.
We next examined whether GPI0363 directly binds to SigA. To establish the binding assay, we purified histidine-tagged recombinant SigA from the wild-type strain. Next, we used magnetic beads with an affinity for histidine and incubated them with His-tagged SigA, followed by incubation with GPI0363 pretreated with BSA. We separated the bound fraction, and then washed, eluted, and analyzed the eluted fractions using HPLC. The peak of GPI0363 appeared when mixed with SigA whereas the peak was not observed when SigA was omitted (Figure 4), suggesting the direct binding of GPI0363 to SigA.
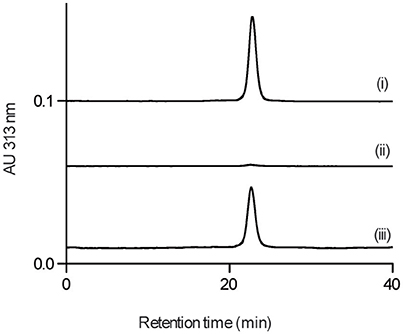
Figure 4. GPI0363 binds to SigA. Magnetic beads with (i) and without (ii) His-tagged SigA were incubated with GPI0363 pretreated with BSA; the bound fraction was washed, eluted, and analyzed by HPLC. The retention time of the peak appeared was confirmed to be GPI0363 by running reference GPI0363 (iii). Representative result of two independent experiments is shown.
Activity of GPI0363 in a Mouse Infection Model
To determine the effect of GPI0363 in infected mice, mice were injected intravenously with MRSA USA300 followed 30 min later by intraperitoneal administration of 400 mg/kg GPI0363 (n = 7). Administration of GPI0363 prolonged the survival of the mice compared to the control (Figure 5). The median survival time of the infection control group was 0.8 days while median survival of the GPI0363-treated group was 1.7 days. Thus, an antimicrobial compound identified using the silkworm model also exhibited activity in a mouse infection model.
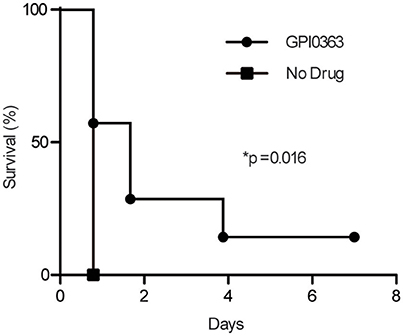
Figure 5. Effect of GPI0363 on a mouse model of infection. Mice were infected with S. aureus USA300 by intravenous injection, followed 30 min later by intraperitoneal injection of 400 mg/kg GPI0363. Data were analyzed using the Gehan-Breslow-Wilcoxon test (*p = 0.016) and log-rank test (*p = 0.016).
Discussion
In the current condition of the increased incidence of multi-drug resistant organisms due to the overuse of broad spectrum antimicrobials, the use of a narrow-spectrum antimicrobial agent is desired to treat specific infections as a tailor-made therapy. In addition to the increased effectiveness of treatment and the reduced incidence of multidrug resistance, these compounds will have fewer side effects on normal gut flora and the host immune system. Here we screened a chemical library of compounds using silkworms infected with S. aureus and identified three potential antimicrobial agents with therapeutic activity. One of these, GPI0363, was active against both MSSA and MRSA, and may be useful to treat severe staphylococcal infections.
Bacterial RNA polymerase comprises of five subunits, ααββ'ω, forming the core enzyme. A sigma factor binds to the core enzyme to form the holoenzyme and helps recognize the promoter region to initiate specific transcription (Burgess et al., 1969). The primary sigma factor, SigA, is unique to bacteria, essential for cell viability (Chaudhuri et al., 2009), and responsible for transcription of housekeeping genes during the exponential phase (Deora and Misra, 1996). S. aureus SigA differs from the eukaryotic transcription initiation factor TFIID (Horikoshi et al., 1989) and, because the transcription initiation mechanisms in prokaryotes and eukaryotes are entirely different, RNA polymerase is now a validated target for antibacterial agents (Ho et al., 2009). Only two classes of RNA polymerase inhibitors, rifampicin and lipiarmycin, are currently in clinical use (Sonenshein et al., 1974; Tupin et al., 2010). We found that GPI0363 inhibited transcription in S. aureus and that the staphylococcal SigA is involved in the mechanism of action of GPI0363. Based on this finding, we speculate that GPI0363 inhibits or alters the formation of the RNA polymerase holoenzyme or the binding to promoter region, or the steps following binding, but the exact mechanism of how GPI0363 inhibits transcription requires further investigation.
Our results further highlight the usefulness of the silkworm infection model as a screening system to eliminate compounds that are effective only in vitro. The use of invertebrate animal models for drug screening has many advantages over the use of mammalian models. Several invertebrate animal models and the benefits of silkworms for screening antimicrobial agents have been reviewed elsewhere (Panthee et al., 2017b). The silkworm infection model has been used to identify several novel therapeutically effective antibiotics, such as lysocin E (Hamamoto et al., 2015), nosokomycin (Uchida et al., 2010), and ASP2397 (Nakamura et al., 2017), from the culture broth of microorganisms. One limitation of the silkworm infection model is that the compounds are injected immediately after injection of the bacteria, which does not mimic real-life situations. In addition, antimicrobial compounds that are effective only in mammals might not be identified using the silkworm infection model. Here, GPI0363 protected both silkworm and mice from S. aureus-induced infection. These promising results highlight the potential of this drug as a novel therapy for infections caused by S. aureus. However, toxicity studies are necessary to support its application in to the clinic.
Conclusion
The discovery of therapeutically effective antimicrobial agents remains a challenge for drug-development. By using the silkworm infection model for screening, we were able to eliminate compounds that displayed only in vitro activity and identified a new anti-staphylococcal agent, GPI0363. GPI0363 inhibited the growth of S. aureus by inhibiting RNA synthesis. Our findings suggest the involvement of the primary sigma factor, SigA, in the anti-staphylococcal activity of GPI0363.
Author Contributions
AP, HH, and KS designed research; AP, SP, HH, and KS performed experiments; YS performed genetic analysis; MK, SM, and KK performed chemical synthesis and analysis. AP and HH wrote the manuscript; KS integrated the research and supervised the project. KS and MK critically revised the manuscript for important intellectual content. All the authors commented on and approved the manuscript. KS decided the final approval of the manuscript.
Funding
This work was supported by JSPS postdoctoral fellowship and TBRF postdoctoral fellowship to AP, MEXT KAKENHI (JP221S0002) and a Grant-in-Aid for challenging Exploratory Research (JP26670043) to HH; and Grant-in-Aid for scientific research (S) (JP15H05783) to KS.
Conflict of Interest Statement
KS is a consultant for Genome Pharmaceutical Institute Co., Ltd.
The other authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. H. Kojima (Drug Discovery Initiative, The University of Tokyo) for providing the chemical library, and A. Noguchi, K. Kyogoku, and Y. Matsuzawa (Genome Pharmaceutical Institute, Co., Ltd.) for technical assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.00712/full#supplementary-material
References
Aminov, R. (in press). History of antimicrobial drug discovery–Major classes health impact. Biochem. Pharmacol. doi: 10.1016/j.bcp.2016.10.001
Burgess, R. R., Travers, A. A., Dunn, J. J., and Bautz, E. K. F. (1969). Factor stimulating transcription by RNA polymerase. Nature 221, 43–46. doi: 10.1038/221043a0
Butler, M. S., Blaskovich, M. A., and Cooper, M. A. (2017). Antibiotics in the clinical pipeline at the end of 2015. J. Antibiot. 70, 3–24. doi: 10.1038/ja.2016.72
Canepari, P., Lleò, M. M., Fontana, R., and Satta, G. (1987). Streptococcus faecium mutants that are temperature sensitive for cell growth and show alterations in penicillin-binding proteins. J. Bacteriol. 169, 2432–2439. doi: 10.1128/jb.169.6.2432-2439.1987
Chaudhuri, R. R., Allen, A. G., Owen, P. J., Shalom, G., Stone, K., Harrison, M., et al. (2009). Comprehensive identification of essential Staphylococcus aureus genes using Transposon-Mediated Differential Hybridisation (TMDH). BMC Genomics 10:291. doi: 10.1186/1471-2164-10-291
Clinical Laboratory Standards Institute (2012). Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically; Approved Standard—9th Edn (CLSI document M07–A9). Wayne, PA: Clinical and Laboratory Standards Institute.
Deora, R., and Misra, T. K. (1996). Characterization of the primary factor of Staphylococcus aureus. J. Biol. Chem. 271, 21828–21834. doi: 10.1074/jbc.271.36.21828
Emmerson, A. M., and Jones, A. M. (2003). The quinolones: decades of development and use. J. Antimicrob. Chemother. 51, 13–20. doi: 10.1093/jac/dkg208
Gross, C., Chan, C., Dombroski, A., Gruber, T., Sharp, M., Tupy, J., et al. (1988). The functional and regulatory roles of sigma factors in transcription. Cold Spring Harb. Symp. Quant. Biol. 63, 141–156. doi: 10.1101/sqb.1998.63.141
Hamamoto, H., Urai, M., Ishii, K., Yasukawa, J., Paudel, A., Murai, M., et al. (2015). Lysocin E is a new antibiotic that targets menaquinone in the bacterial membrane. Nat. Chem. Biol. 11, 127–133. doi: 10.1038/nchembio.1710
Helmann, J. D., and dehaseth, P. L. (1999). Protein nucleic acid interactions during open complex formation investigated by systematic alteration of the protein and DNA binding partners. Biochemistry 38, 5959–5967. doi: 10.1021/bi990206g
Hiramatsu, K., Hanaki, H., Ino, T., Yabuta, K., Oguri, T., and Tenover, F. C. (1997). Methicillin-resistant Staphylococcus aureus clinical strain with reduced vancomycin susceptibility. J. Antimicrob. Chemother. 40, 135–136. doi: 10.1093/jac/40.1.135
Ho, M. X., Hudson, B. P., Das, K., Arnold, E., and Ebright, R. H. (2009). Structures of RNA polymerase-antibiotic complexes. Curr. Opin. Struct. Biol. 19, 715–723. doi: 10.1016/j.sbi.2009.10.010
Horikoshi, M., Wang, C. K., Fujii, H., Cromlish, J. A., Weil, P. A., and Roeder, R. G. (1989). Cloning and structure of a yeast gene encoding a general transcription initiation factor TFIID that binds to the TATA box. Nature 341, 299–303. doi: 10.1038/341299a0
Howden, B. P., Davies, J. K., Johnson, P. D. R., Stinear, T. P., and Grayson, M. L. (2010). Reduced vancomycin susceptibility in Staphylococcus aureus, including vancomycin-intermediate and heterogeneous vancomycin-intermediate strains: resistance mechanisms, laboratory detection, and clinical implications. Clin. Microbiol. Rev. 23, 99–139. doi: 10.1128/CMR.00042-09
Jevons, M. P. (1961). Celbenin - resistant Staphylococci. Br. Med. J. 1, 124–125. doi: 10.1136/bmj.1.5219.124-a
Lowy, F. D. (1998). Staphylococcus aureus infections. N.Engl. J. Med. 339, 520–532. doi: 10.1056/NEJM199808203390806
Maki, H., Miura, K., and Yamano, Y. (2001). Katanosin, B., and plusbacin A(3), inhibitors of peptidoglycan synthesis in methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 45, 1823–1827. doi: 10.1128/AAC.45.6.1823-1827.2001
Mangili, A., Bica, I., Snydman, D. R., and Hamer, D. H. (2005). Daptomycin-resistant, methicillin-resistant Staphylococcus aureus bacteremia. Clin. Infect. Dis. 40, 1058–1060. doi: 10.1086/428616
Marty, F. M., Yeh, W. W., Wennersten, C. B., Venkataraman, L., Albano, E., Alyea, E. P., et al. (2006). Emergence of a clinical daptomycin-resistant Staphylococcus aureus isolate during treatment of methicillin-resistant Staphylococcus aureus bacteremia and Osteomyelitis. J. Clin. Microb. 44, 595–597. doi: 10.1128/JCM.44.2.595-597.2006
Monk, I. R., and Foster, T. J. (2012). Genetic manipulation of Staphylococci breaking through the barrier. Front. Cell. Infect. Microbiol. 2:49. doi: 10.3389/fcimb.2012.00049
Nakamura, I., Kanasaki, R., Yoshikawa, K., Furukawa, S., Fujie, A., Hamamoto, H., et al. (2017). Discovery of a new antifungal agent ASP2397 using a silkworm model of Aspergillus fumigatus infection. J. Antibiot. 70, 41–44. doi: 10.1038/ja.2016.106
National Committee for Clinical Laboratory Standards (1999). Methods for Determining Bactericidal Activity of Antimicrobial Agents; Approved Guideline (NCCLS Document M26-A). Wayne, PA: National Committee for Clinical Laboratory Standards.
Novick, R. P. (1991). Genetic systems in Staphylococci. Methods Enzymol. 204, 587–636. doi: 10.1016/0076-6879(91)04029-N
Panthee, S., Hamamoto, H., Suzuki, Y., and Sekimizu, K. (2017a). In silico identification of lysocin biosynthetic gene cluster from Lysobacter sp. RH2180-5. J. Antibiot. 70, 204–207. doi: 10.1038/ja.2016.102
Panthee, S., Paudel, A., Hamamoto, H., and Sekimizu, K. (2017b). Advantages of the silkworm as an animal model for developing novel antimicrobial agents. Front. Microbiol. 8:373. doi: 10.3389/fmicb.2017.00373
Paudel, A., Hamamoto, H., Kobayashi, Y., Yokoshima, S., Fukuyama, T., and Sekimizu, K. (2012). Identification of novel deoxyribofuranosyl indole antimicrobial agents. J. Antibiot. 65, 53–57. doi: 10.1038/ja.2011.110
Paudel, A., Kaneko, K., Watanabe, A., Matsunaga, S., Kanai, M., Hamamoto, H., et al. (2013). Structure-activity relationship study of novel iminothiadiazolo-pyrimidinone antimicrobial agents. J. Antibiot. 66, 663–667. doi: 10.1038/ja.2013.69
Shaw, K. J., and Barbachyn, M. R. (2011). The oxazolidinones: past, present, and future. Ann. N.Y. Acad. Sci. 1241, 48–70. doi: 10.1111/j.1749-6632.2011.06330.x
Sonenshein, A. L., Cami, B., Brevet, J., and Cote, R. (1974). Isolation and characterization of rifampin-resistant and streptolydigin-resistant mutants of Bacillus subtilis with altered sporulation properties. J. Bacteriol. 120, 253–265.
Suzuki, M., Yamada, K., Nagao, M., Aoki, E., Matsumoto, M., Hirayama, T., et al. (2011). Antimicrobial ointments and methicillin-resistant Staphylococcus aureus USA300. Emerg. Infect. Dis. 17, 1917–1920. doi: 10.3201/eid1710.101365
Tsiodras, S., Gold, H. S., Sakoulas, G., Eliopoulos, G. M., Wennersten, C., Venkataraman, L., et al. (2001). Linezolid resistance in a clinical isolate of Staphylococcus aureus. Lancet 358, 207–208. doi: 10.1016/S0140-6736(01)05410-1
Tupin, A., Gualtieri, M., Leonetti, J. P., and Brodolin, K. (2010). The transcription inhibitor lipiarmycin blocks DNA fitting into the RNA polymerase catalytic site. EMBO J. 29, 2527–2537. doi: 10.1038/emboj.2010.135
Uchida, R., Iwatsuki, M., Kim, Y.-P., Ohte, S., Omura, S., and Tomoda, H. (2010). Nosokomycins, new antibiotics discovered in an in vivo-mimic infection model using silkworm larvae. I: fermentation, isolation and biological properties. J. Antibiot. 63, 151–155. doi: 10.1038/ja.2010.9
WHO (2014). Antimicrobial Resistance: Global Report on Surveillance. Geneva: World Health Organization.
Keywords: chemical library, antimicrobial agent, therapeutic activity, anti-staphylococcal, silkworm infection model, RNA polymerase, sigma factor
Citation: Paudel A, Hamamoto H, Panthee S, Kaneko K, Matsunaga S, Kanai M, Suzuki Y and Sekimizu K (2017) A Novel Spiro-Heterocyclic Compound Identified by the Silkworm Infection Model Inhibits Transcription in Staphylococcus aureus. Front. Microbiol. 8:712. doi: 10.3389/fmicb.2017.00712
Received: 06 December 2016; Accepted: 06 April 2017;
Published: 25 April 2017.
Edited by:
Yuji Morita, Aichi Gakuin University, JapanReviewed by:
Sheila Nathan, National University of Malaysia, MalaysiaAlexandra Gruss, INRA Centre Jouy-en-Josas, France
Elizabeth S. Fernandes, Universidade Ceuma, Brazil
Copyright © 2017 Paudel, Hamamoto, Panthee, Kaneko, Matsunaga, Kanai, Suzuki and Sekimizu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kazuhisa Sekimizu, c2VraW1penVAbWFpbi50ZWlreW8tdS5hYy5qcA==