 Tyler J. Carrier
Tyler J. Carrier Adam M. Reitzel
Adam M. Reitzel- Department of Biological Sciences, University of North Charlotte at Charlotte, Charlotte, NC, USA
Our understanding of the diverse interactions between hosts and microbes has grown profoundly over the past two decades and, as a product, has revolutionized our knowledge of the life sciences. Through primarily laboratory experiments, the current framework for holobionts and their respective hologenomes aims to decipher the underpinnings and implications of symbioses between host and microbiome. However, the laboratory setting restricts the full spectrum of host-associated symbionts as compared to those found in nature; thus, limiting the potential for a holistic interpretation of the functional roles the microbiome plays in host biology. When holobionts are studied in nature, associated microbial communities vary considerably between conditions, resulting in more microbial associates as part of the “hologenome” across environments than in either environment alone. We review and synthesize empirical evidence suggesting that hosts may associate with a larger microbial network that, in part, corresponds to experiencing diverse environmental conditions. To conceptualize the interactions between host and microbiome in an ecological context, we suggest the “host-associated microbial repertoire,” which is the sum of microbial species a host may associate with over the course of its life-history under all encountered environmental circumstances. Furthermore, using examples from both terrestrial and marine ecosystems, we discuss how this concept may be used as a framework to compare the ability of the holobiont to acclimate and adapt to environmental variation, and propose three “signatures” of the concept.
Nature of the Holobiont
Partnerships between host and microbes within an environmental setting exemplify a network of biotic relationships that are common across the tree of life (Rosenberg and Zilber-Rosenberg, 2013; Bosch and Miller, 2016; Hurst, 2016). The phenomena underlying these relationships complement more than a century of biological research that has focused on the evolution and ecology of individual species. To conceptualize the functional importance between host and microbiota, Zilber-Rosenberg and Rosenberg (2008) proposed the hologenome theory of evolution, stating that animals and plants, along with their microbiome serve as a unit of selection. This paradigm shift has led to advancements in our understanding of the spectrum of organismal symbioses in the life sciences (Bordenstein and Theis, 2015; Theis et al., 2016), with particular attention to developmental (McFall-Ngai and Ruby, 2000; McFall-Ngai, 2002), evolutionary (Brucker and Bordenstein, 2012, 2013a,b), and genetic modifications (Husnik et al., 2013) to the host.
The hologenome theory emphasizes the role microbes play in animal and plant evolution as integrated units of biological organization that intertwine Darwinian and Lamarckian principles (Zilber-Rosenberg and Rosenberg, 2008; Rosenberg et al., 2009; Bordenstein and Theis, 2015). The hologenome theory provides functional explanations for the role of the microbiome in a Darwinian framework as it relates to speciation (Brucker and Bordenstein, 2012, 2013b) and potentially host fitness (e.g., Callens et al., 2016). A Lamarckian framework (Rosenberg et al., 2009), on the other hand, is complementary to this, as it details the mechanisms whereby microbes are acquired or lost during an organism's lifetime, and that the acquisition of a novel species or strain of microorganism may be integrated into the hologenome. Therefore, hologenomes (and as a direct extension, the holobiont) integrate principles from multiple disciplines (Rosenberg et al., 2009) spanning the diverse fields of evolutionary genetics (Brucker and Bordenstein, 2012, 2013b) and evolutionary ecology (Macke et al., 2016; Theis et al., 2016).
One major challenge the field currently faces is melding insights from the evolutionary, genetic, and molecular underpinnings of host-microbe partnerships with the ecological conditions in which they formed and evolved. Recent work has begun addressing these disciplines as an integrative discipline (Gilbert et al., 2015; Theis et al., 2016); however, they remain largely as separate conceptual entities. Since the hologenome concept was proposed nearly a decade ago, other multi-disciplinary fields, such as evolutionary developmental biology (Moczek et al., 2015), have recognized and, in a conceptual as well as empirical manner, addressed this challenge in two successive steps. The first of these emphasizes the value of applying hypotheses and/or mechanisms derived from laboratory studies to complementary experiments in nature (Gilbert, 2016), while the second tests evolutionary principles across different environments. Based on our current interpretation of animal-microbe partnerships, we provide an initial assessment of these two steps, whereby using published data we quantify the degree that host-associated microbiomes differ between laboratory- and field-based studies (Table 1), as well as when the hologenome faces an environmental stress (Table 2).
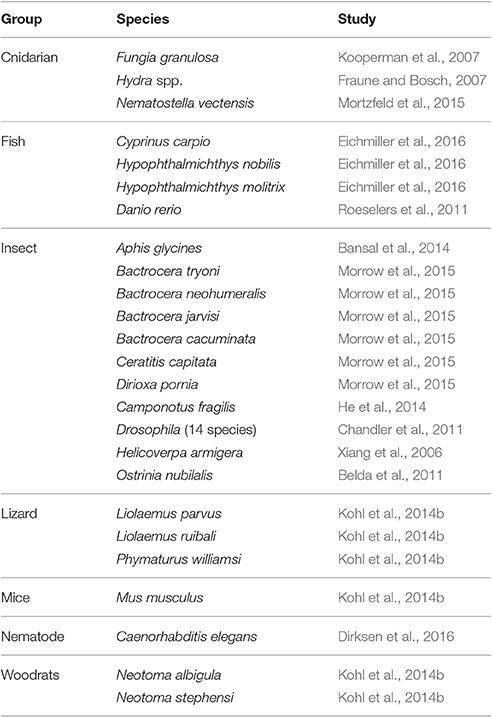
Table 1. Representative list of studies in different animals comparing host-associated microbiota in the laboratory and field.
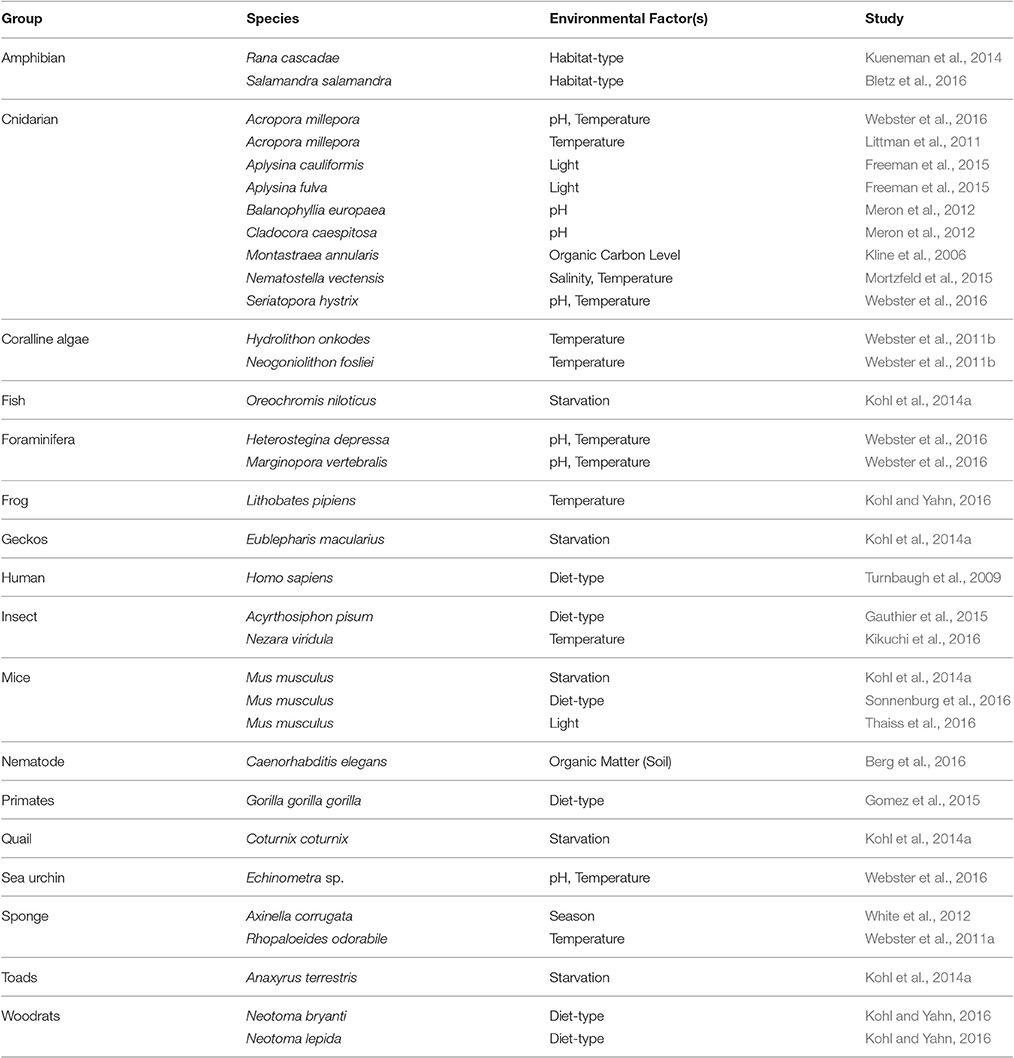
Table 2. Representative list of studies comparing host-associated microbiota between environments for different animals (please note that social environment/interactions were not addressed but may contribute to hologenomic composition, e.g., see Tung et al., 2015).
Toward Nature's Laboratory
Traditional animal models (e.g., Hydra: Fraune and Bosch, 2007; Drosophila: Shin et al., 2011; Mus: Sonnenburg et al., 2016) are powerful systems for laboratory experiments, particularly when dissecting the molecular mechanisms of organism-level processes under controlled settings. Substantial research with these species has led to fundamental discoveries in the formation, regulation, and diversity of microbial symbioses (e.g., Ley et al., 2005; Turnbaugh et al., 2006, 2009). However, these species, or any other for that matter, studied in the laboratory only partially represent the full spectrum of associations they may have with microbes in the natural setting where they have evolved (Chandler et al., 2011). In addition, species reared in the laboratory for many generations may have undergone artificial selection, intended or not, wherein phenotypic and genotypic traits having been selected for in response to abiotic and biotic environmental pressures may have been modified or lost (Kiers et al., 2007). As others have acknowledged before (e.g., Chandler et al., 2011; Har et al., 2015; Dirksen et al., 2016; Table 1) and as we do here, to what extent does the microbiome of laboratory animals reflect that of wild counterparts?
Marked declines in diversity and shifts in composition, and likely function, of the microbiome follow the onset of captivity, domestication, or other anthropogenically modified environments. Decreased microbial diversity and shifts in the species present have been reported for a number of the taxa, including but not limited to insects (Figure 1A), cnidarians (Figure 1B), lizards, fish, woodrats, and other mammals (Ley et al., 2008; Fraune et al., 2010; Wang et al., 2014; Kohl et al., 2014b, 2017; Mortzfeld et al., 2015; Clayton et al., 2016; Table 1). Therefore, defining the full diversity and functional traits of the microbiome with respect to the host would benefit from considering individuals in their native ecological niches to quantify if and how these differences shape the hologenome.
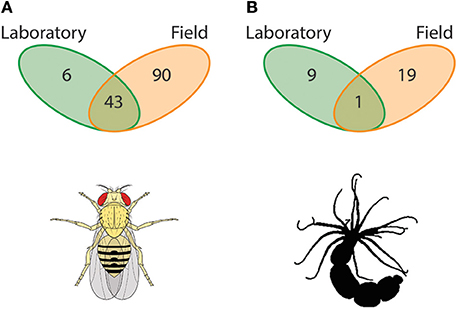
Figure 1. Host-associated microbial symbionts in laboratory vs. field. An overwhelming majority of host-associated microbiome studies have been conducted in the laboratory. This artificial and highly regulated setting inhibits the host from maintaining symbioses with the diversity of microbes that they do in the field, a phenomenon found broadly across the animal kingdom. Here, we present data from Chandler et al. (2011) and Har et al. (2015) that compared the microbiome of laboratory-entrained and wild caught Drosophilids and starlet sea anemone Nematostella vectensis. (A) For the Drosophila spp. system, there are a total of 139 associated OTUs in these settings with 90 specific to wild individuals, six unique to laboratory strains, and 43 shared between environments (Chandler et al., 2011). (B) For N. vectensis, on the other hand, there are a total of 29 associated OTUs with 19 specific to wild individuals, nine unique to laboratory strains, and one shared between environments (Har et al., 2015). These data imply that laboratory-based microbiome studies do not represent the symbiotic microbial community of the host and have large quantitative differences in associated OTUs, and thus potentially its ability to withstand, for example, environmental stressors.
Recent studies using traditional model animals (e.g., Drosophila: Chandler et al., 2011; Caenorhabditis elegans: Berg et al., 2016; Dirksen et al., 2016; Samuel et al., 2016) have addressed the limitations of the laboratory and have begun performing complementary studies with wild populations. Drosophila, for example, has shown to be a tractable system for characterizing the role of the host genome in mutualistic partnerships with microbes. Using 14 Drosophila species spanning a broad geographical distribution and in direct comparison with common laboratory strains, Chandler et al. (2011) showed that the assembly of the microbiome is primarily bound by host diet and physiology, and the degree (i.e., diversity) of this assembly is a function of the environment (laboratory vs. wild) in which the individual resides. As such, laboratory-reared Drosophilids associate with a microbiome much lower in richness and diversity than wild conspecifics. Of the 139 Operational Taxonomic Units (OTUs) found to associate with Drosophila spp. in these settings, 90 (64.7%) were specific to wild individuals, six (4.4%) were unique to laboratory strains, and 43 (30.9%) were shared between environments (Chandler et al., 2011; Figure 1A).
Aquatic organisms, both marine and freshwater, inhabit fluids that are particularly rich in environmental microbiota. Similar to traditional terrestrial models (e.g., Drosophila and Caenorhabditis elegans), aquatic animals are amendable to both field and laboratory experiments. A rising aquatic model species in the fields of evolutionary ecology and genomics is the estuarine cnidarian Nematostella vectensis (Darling et al., 2005; Tarrant et al., 2015). Recent studies using laboratory populations have shown that the microbial diversity associated with N. vectensis individuals is influenced significantly by developmental stage, temperature, and salinity, as well as the geographic location from which individuals were collected, despite ten years of laboratory culture (Mortzfeld et al., 2015). Like with the Drosophilids, N. vectensis collected from their natural habitat and subsequently cultured in the laboratory lose a significant portion of their microbial diversity and, in turn, associate with species of microbes not identified in the field (Figure 1B) (see, Figure 3 in Har et al., 2015). Comparing field and laboratory cultured individuals from the same geographical location showed that of the total 29 OTUs found to associate with N. vectensis, 19 (66.5%) were specific to wild individuals, nine (31.0%) were unique to laboratory strains, and one (3.4%) was shared between environments (Har et al., 2015).
Like the examples above, Caenorhabditis elegans taken directly from native habitats associates with a rich community of microbial symbionts (Dirksen et al., 2016), which has significant impacts on individual physiology and life-history (Cabreiro and Gems, 2013). Similarly, gut microbiota of wild mice (Mus musculus) are rich in diversity (Weldon et al., 2015) and dominated by Bacteroides and Robinsoniella enterotypes, a term coined by Arumugam et al. (2011) in reference to clusters of microbiota communities. Following the onset of captivity, mice were found to only maintain an association with the Robinsoniella-dominated enterotype (Wang et al., 2014). In another rodent species, Kohl and Dearing (2014) showed that individual desert woodrats (Neotoma lepida) transferred to the laboratory share 64% of their gut microbiota with individuals found in the wild. Therefore, although model organisms have historically served as exquisite laboratory-based systems for studying animal genomics and development, this approach remains more limited to study the role of the microbiome on host ecology and evolution.
In nature, holobionts face a diversity of biotic and abiotic stressors that may challenge the host to associate with a microbial community that performs an appropriate physiological response. What this implies is that when facing an ecological “task” a holobiont maximizes fitness through changes in the associated microbiota, as derived from a larger network of microbial partners, to form a complementary metabolic and physiologic profile. As stated in the hologenome theory (see, Zilber-Rosenberg and Rosenberg, 2008; Rosenberg and Zilber-Rosenberg, 2013, 2016; Bordenstein and Theis, 2015), and as presented as a conceptual extension here, a network of microbial partners would enable the holobiont, with its respective hologenome, to acclimate to short-term changes in the local environment as well as serve as a fitness landscape for adaptation (e.g., Soen, 2014). We next compare the composition of host-associated microbiota across the host's natural environments and describe implications of an associated microbial repertoire.
Host-Associated Microbial Repertoire
The environment is a selective filter where variation in microbial communities is sorted, resulting in the opportunity for evolutionary innovation, whether the origin of eukaryotic cells (Margulis, 1970) or the gut microbiota of invertebrates and vertebrates (Alberdi et al., 2016; Shapira, 2016). The microbial community in association with a eukaryotic host is generally considered to be a mixture of resident and transient species that vary over space and time. Regardless of the host-microbe interaction, the associated microbial community represents a small subset of the total species present in the surrounding environment (Ley R. E. et al., 2006; Lynch and Neufeld, 2015). The genomes of these microbes, which are found in the environment, a fraction of which associate with a particular host, constitute the environmental metagenome (Lynch and Neufeld, 2015). The combination of obligate and facultative microbial symbionts represents a complex set of potential interactions between host and each individual microbe as well as amongst members of the microbial community. This set of interactions is vast and discerning the type of association (i.e., mutualistic, pathogenic, neutral; Mushegian and Ebert, 2015) is an immense but important goal for determining the relative role of a microbe to the holobiont. These interactions should, in part, be dependent on the specific environment experienced by the holobiont (Steinert et al., 2000) or more indirectly due to the host's condition, which may also depend on the environment.
Recent investigations have begun resolving questions focused on how and why microbial communities shift under abiotic and/or biotic environmental stressors (e.g., Casey et al., 2015; Lokmer and Wegner, 2015; Webster and Thomas, 2016). These associations can be broadly viewed as the product of a host genome by microbial metagenome by environment interaction, or GH × GM × E (Bordenstein and Theis, 2015). Further determining the functional role of the microbiome in the face of environmental stressors would require manipulating E and measuring GM in the context of GH. This would mean that, in some or all cases, GH + GM1 in E1 differs to some degree from GH + GM2 in E2. The sum of unique microbial associates (GMtotal) in E1+2 therefore exceeds that of GM1 and/or GM2. GMtotal thus represents the microbial associates as part of the “hologenome” that exceeds either individual environment.
For example, when exposed to stressful temperatures, larvae of the Great Barrier Reef sponge Rhopaloeides odorabile lose partnerships with microbiota formed under ambient temperature (in particular the Nitrospira, Chloroflexi and a Roseobacter lineage) while forming partnerships with other microbiota (e.g., γ-proteobacteria) not previously part of the hologenome (Webster et al., 2011a). Under these two temperature regimes, Rhopaloeides odorabile larvae associated with 56 unique OTUs; 27 OTUs (48.2%) being specific to ambient temperature, 19 OTUs (33.9%) when faced with temperature stress, and 10 OTUs (17.9%) being shared between conditions (Webster et al., 2011a; Figure 2A). On the other hand, individual aphids (Acyrthosiphon pisum) specialized to the pea Pisum sativum as opposed to the red clover Trifolium pratense have five and two unique facultative associates, respectively, with three associates being shared between these diets (Figure 2B; Gauthier et al., 2015). Therefore, in these as well as other instances that span much of the animal kingdom (e.g., see Webster et al., 2011a; Gilbert et al., 2015; Har et al., 2015; Mortzfeld et al., 2015; Alberdi et al., 2016; Gilbert, 2016; Kohl and Yahn, 2016; Shapira, 2016; Webster et al., 2016; Webster and Thomas, 2016, as well as references therein; Figure 2), the collection of unique microbes as part of the hologenome in just two environmental conditions exceed that of either hologenome alone, such that by considering two environments in these focal cases (Webster et al., 2011a; Gauthier et al., 2015), the microbe associated with one host can more than double.
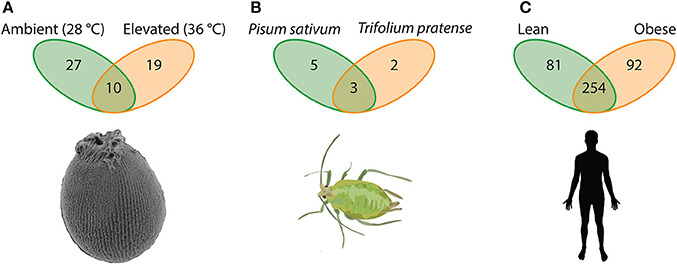
Figure 2. Elements of a host-associated microbial repertoire. Hosts in their natural setting experience a complex environment whereby abiotic and biotic factors vary spatially and temporally, and to cope with these factors the host must associated with appropriate microbial symbionts. (A) Larvae of the Great Barrier Reef sponge Rhopaloeides odorabile are exceptionally vulnerable to ocean warming. One mechanism R. odorabile larvae (and likely others) use to cope with temperature stress is by altering their microbial community (Webster et al., 2011a). (B) Aphids have adapted to effectively utilize diverse saps from plants. Some species of aphids, such as Acyrthosiphon pisum, exploit nutrients from multiple sap-types and, as a product, require unique microbial communities to process each sap-type (Gauthier et al., 2015). (C) Gut microbiota of humans provides an important metabolic complement to digest complex diet-derived biomolecules. An imbalance in the abundance and composition of these members results in a differential ability to utilize energy (Turnbaugh et al., 2006, 2008, 2009). Collectively in these three examples the total OTUs between “environments” exceeds that of an individual environment; thus, suggesting that these taxa (and likely others) have a microbial repertoire that they may associate with in a given environmental setting.
The properties of the environment can continually change, such that there are a nearly infinite number of unique environments, some combination of which must be taken into consideration when describing the evolutionary and ecological history of holobionts and their hologenomes. It is instructive to consider this interaction between the hologenome and the environment as GH × GM × E∞. This raises the question: what is the maximum number of microbial species a host may associate with its hologenome over the course of its life cycle in the presence and absence of all natural and anthropogenic biotic and abiotic factors? The composition of GM changes with respect to E due to the acquisition and loss of “transient” microbial symbionts while GH is nearly constant per generation but dynamic over evolutionary time. For simplicity, if we first consider GH as a constant for a given host and that the composition of GM differs with respect to E and is subsequently integrated across this continuum, then GH plus the sum of GM, or GH+M, should represent the host genome plus the maximum number of microbial species a host may associate with over the course of its life-history under all encountered environmental circumstances. As such, we define this as the “host-associated microbial repertoire” (H):
Furthermore, for a given species, GH is variable (e.g., Wegner et al., 2013; Mortzfeld et al., 2015; Chong and Moran, 2016) between individuals within a population and dynamic over evolutionary time, implying that the magnitude and composition of H may vary with respect to the interaction between GM and E. Thus, each GH would have a specific capacity to establish partnerships with a set number of microbial species due to the host genome and the environment (see, Sonnenburg et al., 2016).
The environmental factors contributing to the structure and composition of the hologenome is also influenced by time (t). In the context of the host-associated microbial repertoire, t can be represented in two primary ways: (i) absolute time of the host genome, microbial metagenome, and environment (“ecological time”) or (ii) accumulative time for co-evolution of the holobiont (“evolutionary time”). For a holobiont, absolute time is the duration of a specific cycle at each level of the GH × GM × E interaction, whereas the generation time for a bacterium is typically minutes to days, while that of the host could be decades. Environmental cycles, on the other hand, can encompass all of these time scales: the North Atlantic Oscillation, one of the most prominent and recurrent patterns of atmospheric variability, differs on a decadal scale (Hurrell et al., 2003), but temperature, oxygen, pH, and other abiotic factors may vary daily. Accumulative time, however, more explicitly integrates across timescales when describing chronologically distinct events over the course of development or selection of advantageous characteristics. Thus, this suggests that the repertoire of microbial species a host genome can associate with should plateau (i.e., reaching H) over evolutionary time based on environment-specific physiological requirements (Figure 3) (e.g., Reveillaud et al., 2014). We may now incorporate t into Equation 1 and, in doing so, create an equation that is conceptually analogous:
Here, instead of integrating over E specifically, we integrate over t; thus, demonstrating that both t and E are important components driving ecological and evolutionary change of holobionts and hologenomes.
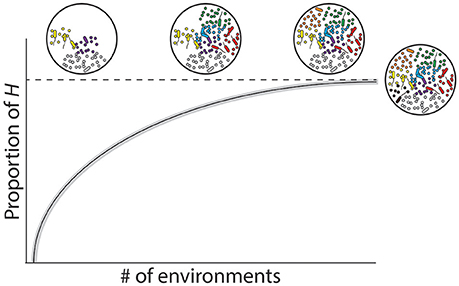
Figure 3. Host-associated microbial repertoire and respective rarefaction curve. Holobionts consist of an individual host and associated microbiota that varies due to the interactions between the host genome, microbial metagenome, and environment (GH x GM x E; Bordenstein and Theis, 2015). In principle, the composition of the hologenome reflects the ecological niche in which the holobiont inhabits, such that each environment may correspond with a unique hologenome. By enumerating the total unique microbial associates within an environment and then summing them across all environmental conditions in which a given host naturally encounters, we predict that the holobiont compiles its hologenome from a grander network of associated microbes, which we term the “host-associated microbial repertoire” (H).
The environment experienced by the host, whether laboratory vs. field or the presence/absence of a stressor(s), may largely define the composition and structure of the associated microbiome and corresponding physiological function (Turnbaugh et al., 2009; Chandler et al., 2011; Webster et al., 2011a; Gauthier et al., 2015; Har et al., 2015). Based on the conceptual framework above, of which we view as a further development of an existing component of the hologenome theory of evolution (Zilber-Rosenberg and Rosenberg, 2008; Rosenberg et al., 2009; Bordenstein and Theis, 2015), we identify three “signatures” for applying our hypothesis: diet, indirect life cycles, and seasonality (but see Kohl and Carey, 2016 for others). In doing so, we use examples from “model” and “non-model” animals across the animal kingdom with unique life-histories that may serve to answer sets of questions pertaining to evolutionary and ecological functions of the microbiome.
Signatures of H: Diet
Feeding history and diet composition have a significant impact on the gut microbial communities of many animals (e.g., Ren et al., 2016) because the ability of the host to metabolize specific dietary biomolecules is dependent on the composition of these consortiums. Host species that feed on food sources that cannot be metabolized by the host or would be toxic in the absence of certain symbionts are more fit when associated with microbes that have these metabolic pathways. Thus, microbes can facilitate shifts in the permissible food sources from otherwise inaccessible energy sources (e.g., Hehemann et al., 2010), which may be a driving force in evolution of some groups (e.g., mammals; Ley et al., 2008; Alberdi et al., 2016). Microbial communities also respond to, and perhaps facilitate the host response to, prolonged periods of food deprivation (Kohl et al., 2014a) by providing the physiological toolkit that facilitates survival of the host (Turnbaugh et al., 2006, 2008, 2009; Mueller and Sachs, 2015; Kohl and Carey, 2016). In the context of the host-associated microbial repertoire, the sum of unique OTUs associated with the gut for each diet and feeding history along with the shared OTUs between them, enumerate Hgut for an individual at a point in time.
One of the most comprehensive systems outlining the environmental influence of feeding on the host-associated microbiome is the gut microbiota of humans. Changes in diet can result in shifts in the gut microbiome that favor a lean or obese phenotype (e.g., Ley et al., 2005; Turnbaugh et al., 2006, 2008, 2009; Ley R. et al., 2006; Spor et al., 2011). The microbiome of individuals exhibiting an obese phenotype have a higher metabolic efficiency, in part, because of a higher Firmicutes to Bacteroidetes ratio (Turnbaugh et al., 2006, 2009). These microbes plus several others make up a portion of the core gut microbiota (Turnbaugh and Gordon, 2009; Turnbaugh et al., 2009); however, the complete composition of gut flora microbes in these states extends beyond these major players. We reanalyzed the 16S metagenomic data from Turnbaugh et al. (2009) to determine how microbial OTUs are distributed between these two phenotypes. Our analysis shows that there are 427 OTUs (with four or more reads) between the gut microbiome of lean and obese phenotypes. Of these, 254 OTUs (59.5%) were shared while 81 OTUs (19.0%) were specific to the lean phenotype and 92 OTUs (21.5%) were obese-specific (Figure 2C). In a related study, Yatsunenko et al. (2012) reported that adults in the United States have upwards of 1,200 associated OTUs while Amerindian and Malwian adults have in excess of 1,400 OTUs and 1,600 OTUs, respectively, implying that Amerindian and Malwian adults have approximately 200 and 400 unique microbes in comparison to adults in the United States on a Western diet. Therefore, the gut microbiota of humans corresponds physiologically with the environmental (feeding) conditions, implying a change in associated microbiota derived from a repertoire of microbial partners. This example emphasizes the additional importance of longitudinal and regional variation in microbiomes in the evolution of hologenomes (further discussed in Zilber-Rosenberg and Rosenberg, 2008; Rosenberg and Zilber-Rosenberg, 2013, 2016).
Signatures of H: Indirect Life Cycles
Life-history strategies are diverse and many animals have successive stages occurring in unique ecological niches. Developing embryos and larvae often experience a different environment from juveniles and adults (e.g., Strathmann, 1985). Animals with biphasic life-histories provide experimental systems to discern how ecological experience influences two inter-related facets of the associated microbial community: (i) colonization of developmental stages and impacts of these on microbial communities of subsequent developmental stages, and (ii) developmental stage-specific microbial communities for different ecological niches.
The colonization of sexually reproduced offspring by microbes is dependent on mode of microbe transmission as well as the mechanisms for selection of microbial symbionts. First, the classic dichotomy of vertical and horizontal transmission of symbionts from parent to offspring results in different probabilities for successful establishment of microbes in successive generations (Bright and Bulgheresi, 2010). Whether beneficial or not, vertically transmitted microbes are more likely to contribute to the offspring's microbial community than those that are selected for from the surrounding environment. However, these transmission strategies more likely reflect a continuum dependent on maternal or paternal behaviors that may influence the likelihood of offspring being exposed to particular microbial species (Funkhouser and Bordenstein, 2013), as seen in dung beetles (Schwab et al., 2016). Second, studies using diverse animals are beginning to show how individuals initially acquire their microbiome. Two divergent strategies can occur: (i) little apparent selection for microbes followed by a winnowing in later stages (Nyholm and Mcfall-Ngai, 2004) or (ii) stricter selection for colonizers (Funkhouser and Bordenstein, 2013; Lema et al., 2014), with the latter sometimes being independent of the environmental microbial community (Apprill et al., 2012). Because the initial colonizing microbes can significantly influence the ability for later colonizing species to successfully establish on a host (Cho and Blaser, 2012), the influence of the host can be modulated by inter-microbial interactions in community establishment.
Species with biphasic life cycles, including many marine invertebrates, insects, and amphibians, would be predicted to have altered associated microbiota in response to shifts in life stage and corresponding environmental niche. Many marine invertebrates, for example, release eggs into the water column that are fertilized and develop into either planktotrophic (feeding) or lecithotrophic (non-feeding) larvae that remain in the plankton for weeks to months (Levin, 2006; Shanks, 2009) or, in some cases, more than a year (Strathmann, 1978; Strathmann and Strathmann, 2007). In terrestrial habitats, larvae are predominately in- or epifaunal and adults may be aerial and potentially more mobile (e.g., winged insects). In both environments, it is common that these life cycle stages differ in exposure to abiotic and biotic stressors. Moreover, a single life cycle stage (e.g., larvae) often experiences spatial and temporal stochasticity of abiotic factors, and specifically for feeding larvae, large shifts in the composition and availability of food (Olson and Olson, 1989).
An example of the magnitude that pre- and post-metamorphosis environments differ comes from the deep-sea mussel “Bathymodiolus” childressi. Larvae of these mussels utilize ocean currents to migrate from deep-sea methane seeps to the surface waters to feed (Sibuet and Olu, 1998; Arellano et al., 2014). Similarly, invertebrate taxa at hydrothermal vents, such as the tubeworm Riftia pachyptila and crab Bythograea thermydron disperse 10s–100s of km in search of a habitable vent site (Marsh et al., 2001; Adams et al., 2012). These examples outline the substantial difference in environments that life-history stages experience, and, in doing so, are projected to have a diverse repertoire of symbiotic microbiota corresponding to abiotic and biotic variation in each respective habitat type. If microbes were significant players in life-history evolution, then we would expect that changes in microbial species associated with animals evolving specializations in habitat, prey, etc. should have a signature when compared with other species representative of the ancestral condition.
The host-associated microbial repertoire for taxa with complex life-histories can be sub-grouped based on developmental stage, and transitioning between stages may make divisions of the host-associated microbial repertoire at one stage different than at another (Hlarva vs. Hadult; e.g., Wang et al., 2011). Furthermore, exposure to an environmental stress during an early life stage may later shape the initial colonizers at a later life stages and corresponding repertoire of microbial associates (e.g., amphibians: Kohl et al., 2013; insects: Hroncova et al., 2015; Chen et al., 2016; cnidarians: Fraune et al., 2016). These stage-specific influences on the physiology and survival of subsequent stages are referred to as carryover or latent effects (Pechenik et al., 1998; Pechenik, 2006). Thus, microbial communities may similarly constitute a carryover or latent effect when determining how the microbial community changes over life histories in different ecological niches and why conspecifics may vary despite experiencing similar environments.
Signatures of H: Seasonal Variation
The diversity of ecological scenarios a host faces in its natural setting may also be driven by changes in the seasonal environment. Animals have diverse physiological and behavioral responses to the time of year that corresponds with seasonal changes, including periods elevating (e.g., reproduction and migration) and suppressing (e.g., hibernation and diapause) activity during life-history stages (Kohl and Carey, 2016). The associated microbes with a host that experience seasonal events change either in response to a new environment or in preparation for physiological shifts (Davenport et al., 2014; Kohl and Carey, 2016). One group that experiences an array of seasonally-driven ecological stressors that are also a well-studied system for animal-bacteria interactions in the marine environment (e.g., Lokmer and Wegner, 2015) are the bivalve mollusks.
Bivalves, such as mussels, clams, and oysters, inhabit the benthos throughout the world oceans, where the primary mode of energy acquisition is actively feeding on phytoplankton via pumping water from its surrounding and concentrating suspended particles (Jørgensen, 1996). The phytoplankton community changes throughout the year, particularly following the onset of the spring phytoplankton bloom, where the concentration of these particles increases by an order of magnitude of more (Evans and Parslow, 1985; Townsend et al., 1994). On a seasonal time scale, the phytoplankton community shifts from being dominated by large diatoms to small, mobile flagellates, with the zooplankton bloom lagging that of the phytoplankton bloom. As a consequence of dynamic shifts in the phytoplankton community during a bloom, it is predicted that the gut microbiota of marine bivalves will change and be constituted by more microbes with complementary metabolic and physiologic profiles for the shifted feeding regime (King et al., 2012). In the context of the host-associated microbial repertoire, we would predict that the bivalve microbial community would respond to shifts in phytoplankton composition as well as abundance, as specifically sub-divided by season (e.g., diatoms vs. dinoflagellates) and food source (e.g., phytoplankton vs. zooplankton).
The phytoplankton community does not, however, consist solely of phytoplankton beneficial for host growth and reproduction. Harmful phytoplankton produce toxins and secondary metabolites that are detrimental to health of bivalves and other animals (Hallegraeff, 1993; Anderson, 1994; Landsberg, 2002), and thus are harmful to the host and potentially the associated microbiota (Kohl and Dearing, 2012). Hosts may evolve resistance to these toxins if there is sufficient selection: in some populations of the soft-shell clam Mya aneraria, such as those in the Bay of Fundy, Canada, a mutation in a sodium channel that saxitoxins of the harmful dinoflagellate Alexandrium fundyense block arose, thereby stochastically developing a sub-population resistant to these toxins (Bricelj et al., 2005). An alternate solution to this is that bivalves acquire microbial symbionts able to metabolically utilize these potent neurotoxins. This mutation has independently arisen in at least two species of bivalves, the blue mussel Mytilus edulis and Mya aneraria (Stewart et al., 1998), through associations with Moraxella, Alteromonas, and Pseudomonus.
Association with microbes able to utilize or degrade natural toxins is not unique to marine bivalves. For example, sub-populations of woodrats (Neotoma bryanti and N. lepida) have specialized to consume the toxic creosote bush Larrea tridentate while other individuals in the same geographical location have not. When digesting the phenolic-rich leaves, woodrats populations that consume the toxin exhibited a marked increase in the diversity of their gut microbiota that further remained distinct from a non-toxic diet (Kohl and Dearing, 2012). Moreover, populations having had no previous exposure to phenol exhibited a decline in the diversity of gut microbiota. This, similar to the bivalve example above, suggests that microbial symbionts enable the host to adapt to consuming toxic biomaterials (Kohl and Dearing, 2012).
Expanding Theory on Holobionts
Nearly a decade ago, Zilber-Rosenberg and Rosenberg (2008) proposed the hologenome theory of evolution, which has been summarized, expanded (Figure 4), and clarified in recent years, providing novel hypotheses for the evolution of holobionts (Bosch and McFall-Ngai, 2011; Gilbert et al., 2012, 2015; Theis et al., 2016). Current theory and research on holobionts are now beginning to explicitly consider the variation in a natural environmental context, in part, because it is a necessary factor to understand the evolution of holobionts and hologenomes (Zilber-Rosenberg and Rosenberg, 2008; Theis et al., 2016). Studies of the host-associated microbial repertoire are useful in the effort to highlight the importance of microbes in host biology (e.g., physiology, development, immunology, behavior, population genetics; Jaenike, 2012; Eisthen and Theis, 2016; Kohl and Carey, 2016; Shropshire and Bordenstein, 2016) and ecology. Furthermore, this concept may aid in advancing our understanding of how these associations may have driven or directed the evolutionary trajectory of the holobiont, and their inheritable repertoire of symbiotic microbiota (van Opstal and Bordenstein, 2015; Gilbert, 2016).
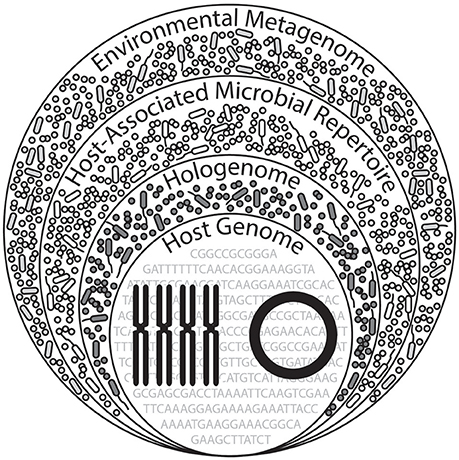
Figure 4. Proposed expansion of hologenomic organization. A hologenome comprises the total genomic components of the host (nuclear and mitochondrial genomes) and associated microbiota (bacterial, viral, archaeal, and fungal). Across diverse environments experienced by the holobiont, contents of these hologenomes exceed that of a hologenome in a single, unique environmental setting, implying an additional layer of hologenomic complexity. We term this the “host-associated microbial repertoire,” which may alternatively be viewed as the collection hologenomes associated with the host, likely holding a biological or ecological importance in the context of the environment. (This Figure was inspired by Theis et al., 2016 and subsequently expanded here).
Our Hypothesis and Theory article has primarily focused on animal-associated microbiota but numerous studies suggest that plants, fungi, and other eukaryotes would have similar associations. Plant-associated microbiota (primarily bacteria and fungi) clearly have important roles in nutrient acquisition and buffering against both abiotic and biotic stressors, such that the plants are associated with particular microbiota that aid in acclimating to the local environment (Vandenkoornhuyse et al., 2015). For example, the evergreen tree Metrosideros polymorpha inhabits environments that broadly range in annual precipitation as well as mean temperature. In profiling the fungal endophyte communities across such abiotic gradients, the composition of these communities is directly related to temperature and rainfall (Zimmerman and Vitousek, 2012), implying that like animal examples presented through this article, plants may also associate with a larger network of microbial partners as a product of environmental variation.
As microbial taxa have been linked to specific evolutionary processes, such as Wolbachia and reproductive compatibility (e.g., Bordenstein et al., 2001) or Vibrio fischeri and countershading (e.g., Nyholm and Mcfall-Ngai, 2004), clusters of microbial associates as part of the host-associated microbial repertoire may be linked to ecological functions (Krause et al., 2006; Langille et al., 2013) including tolerance to salinity (e.g., Schmidt et al., 2015) and/or temperature (e.g., Webster et al., 2011a; Kohl and Yahn, 2016). Characterizing the host-associated microbial repertoire and subsequent function for clusters of associated microbiota may, therefore, serve as an conceptual framework that links the ecological role of microbes to the evolution of the host with which they associate. Furthermore, although these microbial associates have an ecologically important function for the holobiont, the environment in which they have the potential to associate with a holobiont may, in turn, be physiologically stressful to the microbes; thus, it is equally important to study the physiological parameters of the members of the host-associated microbial repertoire.
Author Contributions
All authors listed, have made substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Holly Moeller (Woods Hole Oceanographic Institute and University of California at Santa Barbara, USA) for discussing the mathematical framework of this conceptual model, Jason Macrander (UNC Charlotte, USA) for computational analysis, Sally Leys (University of Alberta, Canada) for providing the image of a sponge larva in Figure 2A, Seth Bordenstein (Vanderbilt University, USA), members of the Reitzel Laboratory, and reviewers for providing critical comments on earlier drafts of this manuscript. TC was supported by a National Science Foundation (NSF) Graduate Research Fellowship and a Sigma Xi GIAR grant. AR was supported by award #1545539 from the NSF and a Young Investigator Award RGY0079 from the Human Frontier Science Program.
References
Adams, D. K., Arellano, S. M., and Govenar, B. (2012). Larval dispersal: vent life in the water column. Oceanography 25, 256–268. doi: 10.5670/oceanog.2012.24
Alberdi, A., Aizpurau, O., Bohmann, K., Zepeda-Mendoze, M. L., and Gilbert, M. T. P. (2016). Do vertebrate gut metagenomes confer rapid ecological adaptation? Trends Ecol. Evol. 311, 689–699. doi: 10.1016/j.tree.2016.06.008
Apprill, A., Marlow, H. Q., Martindale, M. Q., and Rappe, M. S. (2012). Specificity of associations between bacteria and the coral Pocillopora meandrina during early development. Appl. Environ. Microbiol. 78, 7467–7475. doi: 10.1128/AEM.01232-12
Arellano, S. M., Van Gaest, A. L., Johnson, S. B., Vrijenhoek, R. C., and Young, C. M. (2014). Larvae from deep-sea methane seeps disperse in surface waters. Proc. R. Soc. B Biol. Sci. 281:20133276. doi: 10.1098/rspb.2013.3276
Arumugam, M., Raes, J., Pelletier, E., Le Paslier, D., Yamada, T., Mende, D. R., et al. (2011). Enterotypes of the human gut microbiome. Nature 473, 174–180. doi: 10.1038/nature09944
Bansal, R., Mian, M. A. R., and Michel, A. P. (2014). Microbiome diversity of Aphis glycines with extensive superinfection in native and invasive populations. Environ. Microbiol. Rep. 6, 57–69. doi: 10.1111/1758-2229.12108
Belda, E., Pedrola, L., Pereto, J., Martinez-Blanch, J. F., Montagud, A., Navarro, E., et al. (2011). Microbial diversity in the midguts of field and lab-reared populations of the european corn borer ostrinia nubilalis. PLoS Biol. 6:e21751. doi: 10.1371/journal.pone.0021751
Berg, M., Stenuit, B., Ho, J., Wang, A., Parke, C., Knight, M., et al. (2016). Assembly of the Caenorhabditis elegans gut microbiota from diverse soil microbial environments. ISME J. 10, 1998–2009. doi: 10.1038/ismej.2015.253
Bletz, M. C., Goedbloed, D. J., Sanchez, E., Reinhardt, T., Tebbe, C. C., Bhuju, S., et al. (2016). Amphibian gut microbiota shifts differentially in community structure but converges on habitatspecific predicted functions. Nat. Commun. 7:13699. doi: 10.1038/ncomms13699
Bordenstein, S. R., O'Hara, F. P., and Werren, J. H. (2001). Wolbachia-induced incompatibility precedes other hybrid incompatibilities in Nasonia. Nature 409, 707–710. doi: 10.1038/35055543
Bordenstein, S. R., and Theis, K. R. (2015). Host biology in light of the microbiome: ten principles of holobionts and hologenomes. PLoS Biol. 13:e1002226. doi: 10.1371/journal.pbio.1002226
Bosch, T. C. G., and McFall-Ngai, M. J. (2011). Metaorganisms as the new frontier. Zoology 114, 185–190. doi: 10.1016/j.zool.2011.04.001
Bosch, T. C. G., and Miller, D. J. (2016). The Holobiont Imperative: Perspectives from Early Emerging Animals. Vienna: Springer.
Bricelj, V. M., Connell, L., Konoki, K., Macquarrie, S. P., Scheuer, T., Catterall, W. A., et al. (2005). Sodium channel mutation leading to saxitoxin resistance in clams increases risk of PSP. Nature 434, 763–767. doi: 10.1038/nature03415
Bright, M., and Bulgheresi, S. (2010). A complex journey: transmission of microbial symbionts. Nat. Rev. Microbiol. 8, 218–230. doi: 10.1038/nrmicro2262
Brucker, R. M., and Bordenstein, S. R. (2012). Speciation by symbiosis. Trends Ecol. Evol. 27, 443–451. doi: 10.1016/j.tree.2012.03.011
Brucker, R. M., and Bordenstein, S. R. (2013a). The capacious hologenome. Zoology 116, 260–261. doi: 10.1016/j.zool.2013.08.003
Brucker, R. M., and Bordenstein, S. R. (2013b). The hologenomic basis of speciation: gut bacteria cause hybrid lethality in the genus Nasonia. Science 341, 667–669. doi: 10.1126/science.1240659
Cabreiro, F., and Gems, D. (2013). Worms need microbes too: microbiota, health and aging in Caenorhabditis elegans. EMBO Mol. Med. 5, 1300–1310. doi: 10.1002/emmm.201100972
Callens, M., Macke, E., Muylaert, K., Bossier, P., Leievens, B., Waud, M., et al. (2016). Food availability affects the strength of mutualistic host-microbiota interactions in Daphnia magna. ISME J. 10, 911–920. doi: 10.1038/ismej.2015.166
Casey, J. M., Connolly, S. R., and Ainsworth, T. (2015). Coral transplantation triggers shift in microbiome and promotion of coral disease associated potential pathogens. Sci. Rep. 5:11903. doi: 10.1038/srep11903
Chandler, J. A., Lang, J. M., Bhatnagar, S., Eisen, J. A., and Kopp, A. (2011). Bacterial communities of diverse Drosophila species: ecological context of a host-microbe model system. PLoS Genet. 7:e1002272. doi: 10.1371/journal.pgen.1002272
Chen, B., Teh, B.-S., Sun, C., Hu, S., Lu, X., Boland, W., et al. (2016). Biodiversity and activity of the gut microbiota across the life history of the insect herbivore Spodoptera littoralis. Sci. Rep. 6:29505. doi: 10.1038/srep29505
Cho, I., and Blaser, M. J. (2012). The human microbiome: at the interface of health and disease. Nat. Rev. Genet. 13, 260–270. doi: 10.1038/nrg3182
Chong, R. A., and Moran, N. A. (2016). Intraspecific genetic variation in hosts affects regulation of obligate heritable symbionts. Proc. Natl. Acad. Sci. U.S.A. 113, 13114–13119. doi: 10.1073/pnas.1610749113
Clayton, J. B., Vangay, P., Huang, H., Ward, T., Hillmann, B. M., Al-Ghalith, G. A., et al. (2016). Captivity humanizes the primate microbiome. Proc. Natl. Acad. Sci. U.S.A. 113, 10376–10381. doi: 10.1073/pnas.1521835113
Darling, J. A., Reitzel, A. M., Burton, P. M., Mazza, M. E., Ryan, J. F., Sullivan, J. C., et al. (2005). Rising starlet: the starlet sea anemone, Nematostella vectensis. Bioessays 27, 211–221. doi: 10.1002/bies.20181
Davenport, E. R., Mizrahi-Man, O., Michelini, K., Barreiro, L. B., Ober, C., and Gilad, Y. (2014). Seasonal variation in human gut microbiome composition. PLoS Biol. 9:e90731. doi: 10.1371/journal.pone.0090731
Dirksen, P., Marsh, S. A., Braker, I., Heitland, N., Wagner, S., Nakad, R., et al. (2016). The native microbiome of the nematode Caenorhabditis elegans: gateway to a new host-microbiome model. BMC Biol. 14:38. doi: 10.1186/s12915-016-0258-1
Eichmiller, J. J., Hamilton, M. J., Staley, C., Sadowsky, M. J., and Sorensen, P. W. (2016). Environment shapes the fecal microbiome of invasive carp species. Microbiome 4:44. doi: 10.1186/s40168-016-0190-1
Eisthen, H. L., and Theis, K. R. (2016). Animal–microbe interactions and the evolution of nervous systems. Philos. Trans. R. Soc. B Biol. Sci. 371:20150052. doi: 10.1098/rstb.2015.0052
Evans, G. T., and Parslow, J. S. (1985). A model of annual plankton cycles. Biol. Oceanogr. 3, 327–347.
Fraune, S., Augustin, R., Anton-Erxleben, F., Wittlieb, J., Gelhaus, C., Klimovich, V. B., et al. (2010). In an early branching metazoan, bacterial colonization of the embryo is controlled by maternal antimicrobial peptides. Proc. Natl. Acad. Sci. U.S.A. 107, 18067–18072. doi: 10.1073/pnas.1008573107
Fraune, S., and Bosch, T. C. G. (2007). Long-term maintenance of species-specific bacterial microbiota in the basal metazoan Hydra. Proc. Natl. Acad. Sci. U.S.A. 104, 13146–13151. doi: 10.1073/pnas.0703375104
Fraune, S., Forêt, S., and Reitzel, A. M. (2016). Using Nematostella vectensis to study the interactions between genome, epigenome, and bacteria in a changing environment. Front. Microbiol. 3:148. doi: 10.3389/fmars.2016.00148
Freeman, C. J., Baker, D. M., Easson, C. G., and Thacker, R. W. (2015). Shifts in sponge-microbe mutualisms across an experimental irradiance gradient. Mar. Ecol. Prog. Ser. 526, 41–53. doi: 10.3354/meps11249
Funkhouser, L., and Bordenstein, S. R. (2013). Mom knows best: the universitality of maternal microbial transmission. PLoS Biol. 11:e1001631. doi: 10.1371/journal.pbio.1001631
Gauthier, J. P., Outreman, Y., Mieuzet, L., and Simon, J. C. (2015). Bacterial communities associated with host-adapted populations of pea aphids revealed by deep sequencing of 16S ribosomal DNA. PLoS Biol. 10:e0120664. doi: 10.1371/journal.pone.0120664
Gilbert, S. F. (2016). Developmental plasticity and developmental symbiosis: the return of Evo-Devo. Curr. Top. Dev. Biol. 116, 415–433. doi: 10.1016/bs.ctdb.2015.12.006
Gilbert, S. F., Bosch, T. C. G., and Ledon-Rettig, C. (2015). Eco-Evo-Devo: developmental symbiosis and developmental plasticity as evolutionary agents. Nat. Rev. Genet. 16, 611–622. doi: 10.1038/nrg3982
Gilbert, S. F., Sapp, J., and Tauber, A. I. (2012). A symbiotic view of life: we have never been individuals. Q. Rev. Biol. 87, 325–341. doi: 10.1086/668166
Gomez, A., Petrzelkova, K., Yeoman, C. J., Vlckova, K., Mrazek, J., Koppova, I., et al. (2015). Gut microbiome composition and metabolomic profiles of wild western lowland gorillas (Gorilla gorilla gorilla) reflect host ecology. Mol. Ecol. 24, 2551–2565. doi: 10.1111/mec.13181
Hallegraeff, G. M. (1993). A review of harmful algal blooms and their apparent global increase. Phycologia 32, 79–99. doi: 10.2216/i0031-8884-32-2-79.1
Har, J. Y., Helbig, T., Lim, J. H., Fernando, S. C., Reitzel, A. M., Penn, K., et al. (2015). Microbial diversity and activity in the Nematostella vectensis holobiont: insights from 16S rRNA gene sequencing, isolate genomes, and a pilot-scale survey of gene expression. Front. Microbiol. 6:818. doi: 10.3389/fmicb.2015.00818
He, H., Wei, C., and Wheeler, D. E. (2014). The gut bacterial communities associated with lab-raised and field-collected ants of Camponotus fragilis (Formicidae: Formicinae). Curr. Microbiol. 69, 292. doi: 10.1007/s00284-014-0586-8
Hehemann, J. H., Correc, G., Barbeyron, T., Helbert, W., Czjzek, M., and Michel, G. (2010). Transfer of carbohydrate-active enzymes from marine bacteria to Japanese gut microbiota. Nature 464, 908–912. doi: 10.1038/nature08937
Hroncova, Z., Havlik, J., Killer, J., Doskocil, I., Tyl, J., Kamler, M., et al. (2015). Variation in honey bee gut microbial diversity affected by ontogenetic stage, age and geographic location. PLoS ONE 10:e0118707. doi: 10.1371/journal.pone.0118707
Hurrell, J., Kushnir, Y., Ottersen, G., and Visbeck, M. (eds.). (2003). An Overview of the North Atlantic Oscillation. Washington, DC: American Geophysical Union.
Husnik, F., Nikoh, N., Koga, R., Ross, L., Duncan, R. P., Fujie, M., et al. (2013). Horizontal gene transfer from diverse bacteria to an insect genome enables a tripartite nested mealybug symbiosis. Cell 153, 1567–1578. doi: 10.1016/j.cell.2013.05.040
Jaenike, J. (2012). Population genetics of beneficial heritable symbionts. Trends Ecol. Evol. 27, 226–232. doi: 10.1016/j.tree.2011.10.005
Jørgensen, C. B. (1996). Bivalve Filter Feeding: Hydrodynamics, Bioenergetics, Physiology and Ecology. Fredensborg: Olsen & Olsen.
Kiers, E. T., Hutton, M. G., and Denison, R. F. (2007). Human selection and the relaxation of legume defences against ineffective rhizobia. Proc. R. Soc. B Biol. Sci. 274, 3119–3126. doi: 10.1098/rspb.2007.1187
Kikuchi, Y., Tada, A., Musolin, D., Hari, N., Hosokawa, T., Fujisaki, K., et al. (2016). Collapse of insect gut symbiosis under simulated climate change. MBio 7, e01578–e01516. doi: 10.1128/mBio.01578-16
King, G. M., Judd, C., Kuske, C. R., and Smith, C. (2012). Analysis of stomach and gut microbiomes of the eastern oyster (Crassostrea virginica) from Coastal Louisiana, USA. PLoS ONE 7:e51475. doi: 10.1371/journal.pone.0051475
Kline, D. I., Kuntz, N. M., Breitbart, M., Knowlton, N., and Rohwer, F. (2006). Role of elevated organic carbon levels and microbial activity in coral mortality. Mar. Ecol. Prog. Ser. 314, 119–125. doi: 10.3354/meps314119
Kohl, K. D., Amaya, J., Passement, C. A., Dearing, M. D., and McCue, M. D. (2014a). Unique and shared responses of the gut microbiota to prolonged fasting: a comparative study across five classes of vertebrate hosts. FEMS Microbiol. Ecol. 90, 883–894. doi: 10.1111/1574-6941.12442
Kohl, K. D., Brun, A., Magallanes, M., Brinkerhoff, J., Laspiur, A., Acousta, J. C., et al. (2017). Gut microbial ecology of lizards: insights into diversity in the wild, effects of captivity, variation across gut regions, and transmission. Mol. Ecol. 26, 1175–1189. doi: 10.1111/mec.13921
Kohl, K. D., and Carey, H. V. (2016). A place for host-microbe symbiosis in the comparative physiologist's toolbox. J. Exp. Biol. 219, 3496–3504. doi: 10.1242/jeb.136325
Kohl, K. D., Cary, T. L., Karasov, W. H., and Dearing, M. D. (2013). Restructuring of the amphibian gut microbiota through metamorphosis. Environ. Microbiol. Rep. 5, 899–903. doi: 10.1111/1758-2229.12092
Kohl, K. D., and Dearing, M. D. (2012). Experience matters: prior exposure to plant toxins enhances diversity of gut microbes in herbivores. Ecol. Lett. 15, 1008–1015. doi: 10.1111/j.1461-0248.2012.01822.x
Kohl, K. D., and Dearing, M. D. (2014). Wild-caught rodents retain a majority of their natural gut microbiota upon entrance into captivity. Environ. Microbiol. Rep. 6, 191–195. doi: 10.1111/1758-2229.12118
Kohl, K. D., Skopec, M. M., and Dearing, M. D. (2014b). Captivity results in disparate loss of gut microbial diversity in closely related hosts. Conserv. Physiol. 2:cou009. doi: 10.1093/conphys/cou009
Kohl, K. D., and Yahn, J. (2016). Effects of environmental temperature on the gut microbial communities of tadpoles. Environ. Microbiol. 18, 1561–1565. doi: 10.1111/1462-2920.13255
Kooperman, N., Ben-Dov, E., Kramarsky-Winter, E., Barak, Z., and Kushmaro, A. (2007). Coral mucus-associated bacterial communities from natural and aquarium environments. FEMS Microbiol. Lett. 276, 106–113. doi: 10.1111/j.1574-6968.2007.00921.x
Krause, L., Diaz, N. N., Bartels, D., Edwards, R. A., Puhler, A., Rohwer, F., et al. (2006). Finding novel genes in bacterial communities isolated from the environment. Bioinformatics 22, E281–E289. doi: 10.1093/bioinformatics/btl247
Kueneman, J. G., Parfrey, L. W., Woodhams, D. C., Archer, H. M., Knight, R., and McKenzie, V. J. (2014). The amphibian skin-associated microbiome across species, space and life history stages. Mol. Ecol. 23, 1238–1250. doi: 10.1111/mec.12510
Landsberg, J. H. (2002). The effects of harmful algal blooms on aquatic organisms. Rev. Fish. Sci. 10, 113–390. doi: 10.1080/20026491051695
Langille, M. G. I., Zaneveld, J., Caporaso, J. G., McDonald, D., Knights, D., Reyes, J. A., et al. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821. doi: 10.1038/nbt.2676
Lema, K. A., Bourne, D. G., and Willis, B. L. (2014). Onset and establishment of diazotrophs and other bacterial associates in the early life history stages of the coral Acropora millepora. Mol. Ecol. 23, 4682–4695. doi: 10.1111/mec.12899
Levin, L. A. (2006). Recent progress in understanding larval dispersal: new directions and digressions. Integr. Comp. Biol. 46, 282–297. doi: 10.1093/icb/icj024
Ley, R. E., Backhed, F., Turnbaugh, P., Lozupone, C. A., Knight, R. D., and Gordon, J. I. (2005). Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. U.S.A. 102, 11070–11075. doi: 10.1073/pnas.0504978102
Ley, R. E., Hamady, M., Lozupone, C., Turnbaugh, P. J., Ramey, R. R., Bircher, J. S., et al. (2008). Evolution of mammals and their gut microbes. Science 320, 1647–1651. doi: 10.1126/science.1155725
Ley, R. E., Petersen, D. A., and Gordon, J. I. (2006). Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell 124, 837–848. doi: 10.1016/j.cell.2006.02.017
Ley, R., Turnbaugh, P., Klein, S., and Gordon, J. I. (2006). Microbial ecology: human gut microbes associated with obesity. Nature 444, 1022–1023. doi: 10.1038/4441022a
Littman, R., Willis, B. L., and Bourne, D. G. (2011). Metagenomic analysis of the coral holobiont during a natural bleaching event on the Great Barrier Reef. Environ. Microbiol. Rep. 3, 651–660. doi: 10.1111/j.1758-2229.2010.00234.x
Lokmer, A., and Wegner, K. M. (2015). Hemolymph microbiome of Pacific oysters in response to temperature, temperature stress, and infection. ISME J. 9, 670–682. doi: 10.1038/ismej.2014.160
Lynch, M. D., and Neufeld, J. D. (2015). Ecology and exploration of the rare biosphere. Nat. Rev. Microbiol. 13, 217–229. doi: 10.1038/nrmicro3400
Macke, E., Tasiemski, A., Massol, F., Callens, M., and Decaestecker, E. (2016). Life history and eco-evolutionary dynamics in light of the gut microbiota. Oikos 126, 508–531. doi: 10.1111/oik.03900
Margulis, L. (1970). Origin of Eukaryotic Cells: Evidence and Research Implications for a Theory of the Origin and Evolution of Microbial, Plant, and Animal Cells on the Precambrian Earth. Yale University Press (New Haven, CT).
Marsh, A. G., Mullineaux, L. S., Young, C. M., and Manahan, D. T. (2001). Larval dispersal potential of the tubeworm Riftia pachyptila at deep-sea hydrothermal vents. Nature 411, 77–80. doi: 10.1038/35075063
McFall-Ngai, M. J. (2002). Unseen forces: the influence of bacteria on animal development. Dev. Biol. 242, 1–14. doi: 10.1006/dbio.2001.0522
McFall-Ngai, M., and Ruby, E. G. (2000). Developmental biology and marine invertebrate symbioses. Curr. Opin. Microbiol. 3, 603–607. doi: 10.1016/S1369-5274(00)00147-8
Meron, D., Rodolfo-Metalpa, R., Cunning, R., Baker, A. C., Fine, M., and Banin, E. (2012). Changes in coral microbial communities in response to a natural pH gradient. ISME J. 6, 1775–1785. doi: 10.1038/ismej.2012.19
Moczek, A. P., Sears, K. E., Stollewerk, A., Wittkopp, P. J., Diggle, P., Dworkin, I., et al. (2015). The significance and scope of evolutionary developmental biology: a vision for the 21st century. Evol. Dev. 17, 198–219. doi: 10.1111/ede.12125
Morrow, J. L., Frommer, M., Shearman, D. C. A., and Riegler, M. (2015). The Microbiome of field-caught and laboratory-adapted Australian Tephritid Fruit Fly Species with different host plant use and specialisation. Microb. Ecol. 70, 498–508. doi: 10.1007/s00248-015-0571-1
Mortzfeld, B. M., Urbanski, S., Reitzel, A. M., Kunzel, S., Technau, U., and Fraune, S. (2015). Response of bacterial colonization in Nematostella vectensis to development, environment and biogeography. Environ. Microbiol. 18, 1764–1781. doi: 10.1111/1462-2920.12926
Mueller, U. G., and Sachs, J. L. (2015). Engineering microbiomes to improve plant and animal health. Trends Microbiol. 23, 606–617. doi: 10.1016/j.tim.2015.07.009
Mushegian, A. A., and Ebert, D. (2015). Rethinking “mutualism” in diverse host-symbiont communities. Bioessays 38, 100–108. doi: 10.1002/bies.201500074
Nyholm, S. V., and Mcfall-Ngai, M. J. (2004). The winnowing: establishing the squid-Vibrio symbiosis. Nat. Rev. Microbiol. 2, 632–642. doi: 10.1038/nrmicro957
Olson, R. R., and Olson, M. H. (1989). Food limitation of planktotrophic marine invertebrate larvae: does it control recruitment success? Annu. Rev. Ecol. Syst. 20, 225–247. doi: 10.1146/annurev.es.20.110189.001301
Pechenik, J. A. (2006). Larval experience and latent effects–metamorphosis is not a new beginning. Integr. Comp. Biol. 46, 323–333. doi: 10.1093/icb/icj028
Pechenik, J. A., Wendt, D. E., and Jarrett, J. N. (1998). Metamorphosis is not a new beginning. Bioscience 48, 901–910. doi: 10.2307/1313294
Ren, T. T., Grieneisen, L. E., Alberts, S. C., Archie, E. A., and Wu, M. (2016). Development, diet and dynamism: longitudinal and cross-sectional predictors of gut microbial communities in wild baboons. Environ. Microbiol. 18, 1312–1325. doi: 10.1111/1462-2920.12852
Reveillaud, J., Maignien, L., Eren, A. M., Huber, J. A., Apprill, A., Sogin, M. L., et al. (2014). Host-specificity among abundant and rare taxa in the sponge microbiome. ISME J. 8, 1198–1209. doi: 10.1038/ismej.2013.227
Roeselers, G., Mittge, E. K., Stephens, W. Z., Parichy, D. M., Cavanaugh, C. M., Guillemin, K., et al. (2011). Evidence for a core gut microbiota in the zebrafish. ISME J. 5, 1595–1608. doi: 10.1038/ismej.2011.38
Rosenberg, E., Sharon, G., and Zilber-Rosenberg, I. (2009). The hologenome theory of evolution contains Lamarckian aspects within a Darwinian framework. Environ. Microbiol. 11, 2959–2962. doi: 10.1111/j.1462-2920.2009.01995.x
Rosenberg, E., and Zilber-Rosenberg, I. (2013). The Hologenome Concept: Human, Animal and Plant Microbiota. Berlin: Springer.
Rosenberg, E., and Zilber-Rosenberg, I. (2016). Microbes drive evolution of animals and plants: the hologenome concept. MBio 7, e01395–e01315. doi: 10.1128/mBio.01395-15
Samuel, B. S., Rowedder, H., Braendle, C., Felix, M. A., and Ruvkun, G. (2016). Caenorhabditis elegans responses to bacteria from its natural habitats. Proc. Natl. Acad. Sci. U.S.A. 113, E3941–E3949. doi: 10.1073/pnas.1607183113
Schmidt, V. T., Smith, K. F., Melvin, D. W., and Amaral-Zettler, L. A. (2015). Community assembly of a euryhaline fish microbiome during salinity acclimation. Mol. Ecol. 24, 2537–2550. doi: 10.1111/mec.13177
Schwab, D., Riggs, H., Newton, I., and Moczek, A. (2016). Developmental and ecological benefits of the materally transmitted microbiota in a dung beetle. Am. Nat. 188, 679–692. doi: 10.1086/688926
Shanks, A. L. (2009). Pelagic larval duration and dispersal distance revisited. Biol. Bull. 216, 373–385. doi: 10.1086/BBLv216n3p373
Shapira, M. (2016). Gut microbiotas and host evolution: scaling up symbiosis. Trends Ecol. Evol. 31, 539–549. doi: 10.1016/j.tree.2016.03.006
Shin, S. C., Kim, S. H., You, H., Kim, B., Kim, A. C., Lee, K. A., et al. (2011). Drosophila microbiome modulates host developmental and metabolic homeostasis via insulin signaling. Science 334, 670–674. doi: 10.1126/science.1212782
Shropshire, J., and Bordenstein, S. R. (2016). Speciation by symbiosis: the microbiome and behavior. MBio 7:e01785. doi: 10.1128/mBio.01785-15
Sibuet, M., and Olu, K. (1998). Biogeography, biodiversity and fluid dependence of deep-sea cold-seep communities at active and passive margins. Deep Sea Res. II Top. Stud. Oceanogr. 45, 517–567. doi: 10.1016/S0967-0645(97)00074-X
Soen, Y. (2014). Environmental disruption of host-microbe co-adaptation as a potential driving force in evolution. Front. Genet. 5:168. doi: 10.3389/fgene.2014.00168
Sonnenburg, E. D., Smits, S. A., Tikhonov, M., Higginbottom, S. K., Wingreen, N. S., and Sonnenburg, J. L. (2016). Diet-induced extinctions in the gut microbiota compound over generations. Nature 529, U212–U208. doi: 10.1038/nature16504
Spor, A., Koren, O., and Ley, R. (2011). Unravelling the effects of the environment and host genotype on the gut microbiome. Na. Rev. Microbiol. 9, 279–290. doi: 10.1038/nrmicro2540
Steinert, M., Hentschel, U., and Hacker, J. (2000). Symbiosis and pathogenesis: evolution of the microbe-host interaction. Naturwissenschaften 87, 1–11. doi: 10.1007/s001140050001
Stewart, J. E., Marks, L. J., Gilgan, M. W., Pfeiffer, E., and Zwicker, B. M. (1998). Microbial utilization of the neurotoxin domoic acid: blue mussels (Mytilus edulis) and soft shell clams (Mya arenaria) as sources of the microorganisms. Can. J. Microbiol. 44, 456–464. doi: 10.1139/cjm-44-5-456
Strathmann, M. F., and Strathmann, R. R. (2007). An extrafordinarily long larval duration of 4.5 years from hatching to metamorphosis for teleplanic veligers of Fusitriton oregonenis. Biol. Bull. 213, 152–159. doi: 10.2307/25066631
Strathmann, R. R. (1978). Length of pelagic period in echinoderms with feeding larvae from the Northeast Pacific. J. Exp. Mar. Biol. Ecol. 34, 23–27. doi: 10.1016/0022-0981(78)90054-0
Strathmann, R. R. (1985). Feeding and nonfeeding larval development and life-history evolution in marine invertebrates. Annu. Rev. Ecol. Syst. 16, 339–361. doi: 10.1146/annurev.es.16.110185.002011
Tarrant, A. M., Gilmore, T. D., Reitzel, A. M., Levy, O., Technau, U., and Martindale, M. Q. (2015). Current directions and future perspectives from the third Nematostella research conference. Zoology 118, 135–140. doi: 10.1016/j.zool.2014.06.005
Thaiss, C. A., Levy, M., Korem, T., Dohnalova, L., Shapiro, H., Jaitin, D. A., et al. (2016). Microbiota diurnal rhythmicity programs host transcriptome oscillations. Cell 167, 1495–1510. doi: 10.1016/j.cell.2016.11.003
Theis, K. R., Dheilly, N. M., Klassen, J. L., Brucker, R. M., Baines, J. F., Bosch, T. C. G., et al. (2016). Getting the hologenome concept right: an eco-evolutionary framework for hosts and their microbiomes. mSystems 1, e00028–e00016. doi: 10.1128/mSystems.00028-16
Townsend, D. W., Cammen, L. M., Holligan, P. M., Campbell, D. E., and Pettigrew, N. R. (1994). Causes and consequences of variability in the timing of spring phytoplankton blooms. Deep Sea Res. I Oceanogr. Res. Pap. 41, 747–765. doi: 10.1016/0967-0637(94)90075-2
Tung, J., Barreiro, L. B., Burns, M. B., Grenier, J.-C., Lynch, J., Grieneisen, L. E., et al. (2015). Social networks predict gut microbiome composition in wild baboons. Elife 4:e05224. doi: 10.7554/eLife.05224
Turnbaugh, P., and Gordon, J. I. (2009). The core gut microbiome, energy balance and obesity. J. Physiol. 587, 4153–4158. doi: 10.1113/jphysiol.2009.174136
Turnbaugh, P. J., Baeckhed, F., Fulton, L., and Gordon, J. I. (2008). Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe 3, 213–223. doi: 10.1016/j.chom.2008.02.015
Turnbaugh, P. J., Hamady, M., Yatsunenko, T., Cantarel, B. L., Duncan, A., Ley, R. E., et al. (2009). A core gut microbiome in obese and lean twins. Nature 457, 480–484. doi: 10.1038/nature07540
Turnbaugh, P. J., Ley, R. E., Mahowald, M. A., Magrini, V., Mardis, E. R., and Gordon, J. I. (2006). An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444, 1027–1031. doi: 10.1038/nature05414
Vandenkoornhuyse, P., Quaiser, A., Duhamel, M., Le Van, A., and Dufresne, A. (2015). The importance of the microbiome of the plant holobiont. New Phytol. 206, 1196–1206. doi: 10.1111/nph.13312
van Opstal, E. J., and Bordenstein, S. R. (2015). Rethinking heritablity of the microbiome. Science 349, 1172–1173. doi: 10.1126/science.aab3958
Wang, J., Linnenbrink, M., Kunzel, S., Fernandes, R., Nadeau, M. J., Rosenstiel, P., et al. (2014). Dietary history contributes to enterotype-like clustering and functional metagenomic content in the intestinal microbiome of wild mice. Proc. Natl. Acad. Sci. U.S.A. 111, E2703–E2710. doi: 10.1073/pnas.1402342111
Wang, Y., Gibreath, T. M. III., Kukutla, P., Yan, G., and Xu, J. (2011). Dynamic gut microbiome across life history of the malaria mosquito Anopheles gambiae in Kenya. PLoS ONE 6:e24767. doi: 10.1371/journal.pone.0024767
Webster, N. S., Botte, E. S., Soo, R. M., and Whalan, S. (2011a). The larval sponge holobiont exhibits high thermal tolerance. Environ. Microbiol. Rep. 3, 756–762. doi: 10.1111/j.1758-2229.2011.00296.x
Webster, N. S., Negri, A. P., Botte, E. S., Laffy, P. W., Flores, F., Noonan, S., et al. (2016). Host-associated coral reef microbes respond to the cumulative pressures of ocean warming and ocean acidification. Sci. Rep. 6:19324. doi: 10.1038/srep19324
Webster, N. S., Soo, R., Cobb, R., and Negri, A. P. (2011b). Elevated seawater temperature causes a microbial shift on crustose coralline algae with implications for the recruitment of coral larvae. ISME J. 5, 759–770. doi: 10.1038/ismej.2010.152
Webster, N. S., and Thomas, T. (2016). The sponge hologenome. MBio 7, e00135–e00116. doi: 10.1128/mBio.00135-16
Wegner, K. M., Volkenborn, N., Peter, H., and Eiler, A. (2013). Disturbance induced decoupling between host genetics and composition of the associated microbiome. BMC Microbiol. 13:252. doi: 10.1186/1471-2180-13-252
Weldon, L., Abolins, S., Lenzi, L., Bourne, C., Riley, E. M., and Viney, M. (2015). The gut microbiota of wild mice. PLoS ONE 10:e0134643. doi: 10.1371/journal.pone.0134643
White, J. R., Patel, J., Ottesen, A., Arce, G., Blackwelder, P., and Lopez, J. V. (2012). Pyrosequencing of bacterial symbionts within axinella corrugata sponges: diversity and seasonal variability. PLoS ONE 7:e38204. doi: 10.1371/journal.pone.0038204
Xiang, H., Wei, G.-F., Jia, S., Huang, J., Miao, X.-X., Zhou, Z., et al. (2006). Microbial communities in the larval midgut of laboratory and field populations of cotton bollworm (Helicoverpa armigera). Can. J. Microbiol. 52, 1085–1092. doi: 10.1139/w06-064
Yatsunenko, T., Rey, F. E., Manary, M. J., Trehan, I., Dominguez-Bello, M. G., Contreras, M., et al. (2012). Human gut microbiome viewed across age and geography. Nature 486, 222–227. doi: 10.1038/nature11053
Zilber-Rosenberg, I., and Rosenberg, E. (2008). Role of microorganisms in the evolution of animals and plants: the hologenome theory of evolution. FEMS Microbiol. Rev. 32, 723–735. doi: 10.1111/j.1574-6976.2008.00123.x
Keywords: ecology, holobiont ecology, hologenome theory, microbial repertoire, evolution
Citation: Carrier TJ and Reitzel AM (2017) The Hologenome Across Environments and the Implications of a Host-Associated Microbial Repertoire. Front. Microbiol. 8:802. doi: 10.3389/fmicb.2017.00802
Received: 03 December 2016; Accepted: 19 April 2017;
Published: 11 May 2017.
Edited by:
Mike Taylor, University of Auckland, New ZealandReviewed by:
Peter Deines, University of Kiel, GermanyKevin R. Theis, Wayne State University School of Medicine, USA
Copyright © 2017 Carrier and Reitzel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tyler J. Carrier, dGNhcnJpZTFAdW5jYy5lZHU=