 Daniel I. Poppleton1
Daniel I. Poppleton1 Magalie Duchateau2
Magalie Duchateau2 Véronique Hourdel2Mariette Matondo2
Véronique Hourdel2Mariette Matondo2 Jennifer Flechsler3
Jennifer Flechsler3 Andreas Klingl3
Andreas Klingl3 Christophe Beloin4*
Christophe Beloin4* Simonetta Gribaldo1*
Simonetta Gribaldo1*- 1Unité de Biologie Moléculaire du Gène chez les Extrêmophiles, Département de Microbiologie, Institut Pasteur, Paris, France
- 2Unité de Spectrométrie de Masse Structurale et Protéomique, Plateforme Protéomique, Départment de Biologie Structurale et Chime, Institut Pasteur, USR 2000 Centre National de la Recherche Scientifique, Paris, France
- 3Pflanzliche Entwicklungsbiologie und Elektronenmikroskopie, Department I. Botanik, Biozentrum der LMU München, Planegg-Martinsried, Germany
- 4Unité de Génétique des Biofilms, Département de Microbiologie, Institut Pasteur, Paris, France
Veillonella parvula is a biofilm-forming commensal found in the lungs, vagina, mouth, and gastro-intestinal tract of humans, yet it may develop into an opportunistic pathogen. Furthermore, the presence of Veillonella has been associated with the development of a healthy immune system in infants. Veillonella belongs to the Negativicutes, a diverse clade of bacteria that represent an evolutionary enigma: they phylogenetically belong to Gram-positive (monoderm) Firmicutes yet maintain an outer membrane (OM) with lipopolysaccharide similar to classic Gram-negative (diderm) bacteria. The OMs of Negativicutes have unique characteristics including the replacement of Braun's lipoprotein by OmpM for tethering the OM to the peptidoglycan. Through phylogenomic analysis, we have recently provided bioinformatic annotation of the Negativicutes diderm cell envelope. We showed that it is a unique type of envelope that was present in the ancestor of present-day Firmicutes and lost multiple times independently in this phylum, giving rise to the monoderm architecture; however, little experimental data is presently available for any Negativicutes cell envelope. Here, we performed the first experimental proteomic characterization of the cell envelope of a diderm Firmicute, producing an OM proteome of V. parvula. We initially conducted a thorough bioinformatics analysis of all 1,844 predicted proteins from V. parvula DSM 2008's genome using 12 different localization prediction programs. These results were complemented by protein extraction with surface exposed (SE) protein tags and by subcellular fractionation, both of which were analyzed by liquid chromatography tandem mass spectrometry. The merging of proteomics and bioinformatics results allowed identification of 78 OM proteins. These include a number of receptors for TonB-dependent transport, the main component of the BAM system for OM protein biogenesis (BamA), the Lpt system component LptD, which is responsible for insertion of LPS into the OM, and several copies of the major OmpM protein. The annotation of V. parvula's OM proteome markedly extends previous inferences on the nature of the cell envelope of Negativicutes, including the experimental evidence of a BAM/TAM system for OM protein biogenesis and of a complete Lpt system for LPS transport to the OM. It also provides important information on the role of OM components in the lifestyle of Veillonella, such as a possible gene cluster for O-antigen synthesis and a large number of adhesins. Finally, many OM hypothetical proteins were identified, which are priority targets for further characterization.
Introduction
Veillonella parvula, an anaerobic coccus, was discovered and described 120 years ago when Veillon and Zuber isolated it from an appendicitis abscess (Veillon and Zuber, 1898). Sixty years later the same microbe was used in the first observation of an outer membrane (OM) (Bladen and Mergenhagen, 1964) thereby demonstrating a key difference between the Gram-negative and Gram-positive cell envelope architecture. Since then, 13 other Veillonella species have been isolated and described from both humans and rodents (Euzeby, 1997). Veillonella strains are found in several niches of the human body including the mouth (Do et al., 2015), lungs, gastrointestinal tract (Rosen et al., 2014), and vagina (Africa et al., 2014). V. parvula may serve an important role in many of these environments, however its dominant niche is as a secondary colonizer in the mouth (Griffen et al., 2012). This normal component of the human microbiome may be an opportunistic pathogen; its presence has been associated with several disease states and was the primary infectious agent in at least fifty cases since the 1970's, both with and without an underlying condition (Hirai et al., 2016). In direct opposition to its role as an opportunistic pathogen, recent data suggests that Veillonella may perform a protective role and aid in early childhood immune system development. Epidemiological studies of infants have demonstrated that presence of Veillonella is negatively correlated with asthma (Arrieta et al., 2015), bronchiolitis (Hasegawa et al., 2016), and autism (Strati et al., 2017).
In addition to being an important component of the human microbiome, Veillonella, and other members of the Negativicutes are evolutionary enigmas. They phylogenetically belong to the Firmicutes (low GC Gram-positives or monoderms), yet possess an OM with lipopolysaccharides (LPS) similar to Gram-negative (diderm) bacteria (Zeikus et al., 1983; Tocheva et al., 2011; Campbell et al., 2014). We recently showed that the genomes of Negativicutes contain genes encoding typical diderm OM machinery including flagellar rings typical of diderms, Type 4 pili (T4P) secretin, and an ancestral form of the BAM/TAM system for OM β-barrel assembly (Antunes et al., 2016). Furthermore, most OM-related genes were found to reside in a single genomic cluster (Antunes et al., 2016), hereafter referred to as the “diderm cluster,” that included genes responsible for LPS biosynthesis. Through phylogenomic analysis, we proposed that the diderm cell envelope of Negativicutes represents an ancestral characteristic that was present in the ancestor of all Firmicutes, and was lost multiple times independently to give rise to the monoderm cell envelope in this phylum (Antunes et al., 2016). Although this work presented important perspectives on the OM of Veillonella, it was performed in silico and cannot tell us which proteins are true components of the OM and whether they are expressed. These predictions can be validated using experimental proteomic approaches, such as those performed on Bacteroides fragilis (Wilson et al., 2015) and Actinobacillus pleuropneumoniae (Chung et al., 2007).
Only two outer membrane proteins from the Negativicutes have been cloned and studied: OmpM and hemagglutinin-like adhesins. OmpM is an alternative method for tethering the OM to the peptidoglycan by binding of polyamine-modified peptidoglycan through an S-Layer homology (SLH) domain and a transmembrane β-barrel for OM attachment (Kojima and Kamio, 2012). This contrasts with Escherichia coli's Braun's lipoprotein (Lpp), which covalently binds PG and integrates into the OM via a lipid moiety (Braun and Rehn, 1969). In the case of adhesins, eight were found in Veillonella atypica; one of which, Hag1, was shown to bind human buccal cells and oral streptococci (Zhou et al., 2015).
In order to extend these bioinformatic and experimental data, we have performed the first proteomic analysis of the OM of V. parvula. By combining bioinformatic prediction and subcellular fractionation, we have obtained fundamental insight into the nature of these unique cell envelopes and the lifestyle of Veillonella, which will help future work on this important component of the human microbiome.
Materials and Methods
Bioinformatic Prediction
To perform in silico prediction, we used 12 distinct bioinformatic programs on all 1,844 proteins encoded in the V. parvula DSM 2008 genome. Initial prediction was performed using three general prediction programs for IM (Inner Membrane), cytoplasmic, periplasmic, secreted, and OM: PSORTb 3.0.2 (Yu et al., 2010) using default input parameters for Gram-negative bacteria, CELLO 2.5 (Yu et al., 2004) with default input parameters for Gram-negative bacteria, and SOSUI-GramN (Imai et al., 2008) with default parameters. These analyses were complemented with prediction of transmembrane helices by TMHMM 2.0 (Krogh et al., 2001). β-Barrels were predicted using BOMP (Berven et al., 2004) with the additional BLAST option. LipoP 1.0 (Juncker et al., 2003) was used to refine cytoplasmic and IM prediction. Positive lipoprotein prediction was defined as a consensus from PRED-LIPO (Bagos et al., 2008) and LipoP. TAT secreted proteins were identified as a consensus from PRED-TAT (Bagos et al., 2010) and TatP (Bendtsen et al., 2005). Positive SEC signal sequences were defined as a two out of three or greater concurrent result of SignalP, PRED-LIPO, and Phobius (Kall et al., 2007). Extended signal peptides (ESP) were queried using HMMER (Mistry et al., 2013) with the ESPR domain (PF13018) from PFAM within an E-value cutoff of 0.001.
Positive prediction for cytoplasmic proteins was determined by a three out of four or greater concurrent results of PSORT, CELLO, LipoP, and SOSUI. IM prediction was determined by three out of five or greater concurrent results of PSORT, CELLO, TMHMM, LipoP, and SOSUI. Positive OM prediction was determined by three out of four or greater concurrent results of PSORT, CELLO, BOMP, and SOSUI. Positive periplasmic prediction and secreted proteins were determined by a two out of three or greater concurrent results of PSORT, CELLO, and SOSUI.
Search for Conserved Domains and Homologs
To identify homologs of experimentally identified proteins we ran a BLAST 2.2.30+ (Camacho et al., 2009) search against two local databases. The first contains a selection of 255 representative Firmicutes and the second contains all 112 currently available Negativicutes genomes. Default settings were used except for an E-value cutoff of 0.0001. Protein domains were searched using the conserved domain database (CDD) from NCBI (Marchler-Bauer et al., 2014), PFAM (Finn et al., 2015), InterPro (Finn et al., 2017), and PANTHER 10.0 (Mi et al., 2015) with the required E-value cutoff of 0.00001. Protein folds were predicted with Phyre2 (Kelley et al., 2015).
Outer Membrane Protein Extraction
The extraction protocol was modified from Thein et al. (2010). Fifty milliliters of V. parvula DSM 2008 were grown anaerobically in triplicate to an optical density at 600 nm of 0.4 (10 h after a 1:100 dilution) in BHIL (BHI (Bacto) + 0.6% sodium L-lactate). The bacteria were harvested by centrifugation at 15,000 × g for 20 min at 4°C and resuspended in an equal volume of Tris-HCl (0.1 M pH 7.3 supplemented with 7 mg of DNAse). Cells were washed two additional times and suspended in 6 ml of the same buffer. Cells were lysed by French press at 10,000 kPa for four passes in Tris-HCl (20 mM pH 7.3). Cellular debris were pelleted by centrifugation at 15,000 × g for 20 min at 4°C. The supernatant was collected and the pellet was discarded. This step was repeated an additional time. Supernatant was then diluted with ice cold 0.1 M Na2CO3 pH 11 to a volume of 60 ml and stirred for 1 h at 4°C. The suspension was separated at 120,000 × g for 1 h at 4°C and the pellet washed in an equal volume of Tris-HCl (0.1 M pH 7.3) and spun at 85,000 × g for 20 min at 4°C. The wash was repeated twice. The pellet was resuspended in ddH2O and proteins were TCA precipitated before MS analysis.
Surface Exposed and Control Sample Extraction
For extraction of surface exposed (SE) fraction, as well as for control samples, a protocol was modified from Voss et al. (2014). Bacteria were grown in triplicate and harvested as in the OM protein extraction. Five milliliters of bacterial cells were washed three times in PBS (0.1 M PO4, 0.15 M NaCl pH 8.0). Cells were resuspended in PBS containing 20 mM of NHS-PEG4-Biotin (Thermo Fisher Scientific) and incubated on ice for 30 min. Reaction was quenched by washing cells in quench buffer (PBS + 100 mM Glycine) three times. Control samples (WC) were treated identically except that they were incubated in PBS without labeling substrate. Cells were lysed by French press at 10,000 kPa for four passes in radioimmunoprecipitation (RIPA) buffer [25 mM Tris-HCl, pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, and 0.1% sodium dodecyl sulfate (SDS) containing a 1:100 dilution of protease inhibitors] and cellular debris were removed by centrifugation at 15,000 × g for 20 min at 4°C twice. Control sample was then TCA precipitated. For SE fraction 1 ml high Capacity Streptavidin Resin columns (Thermo Fisher Scientific) and reagents were equilibrated to room temperature. PBS and elution buffer (8 M guanidine-HCl, pH 1.5) were filtered and degassed. Column was equilibrated with five column volumes of PBS at a flow rate of 0.2 mL/min on an AKTA-FPLC. Sample was then applied to the column using the same rate and washed with 10 column volumes of PBS before elution in a single column volume of elution buffer. Eluate then underwent MS analysis.
Trichloroacetic Acid (TCA) Precipitation
TCA was added to OM samples and WC samples to a final concentration of 20%. The precipitate was spun at 6,500 × g for 1 h at 4°C. The pellet was washed with 800 μl of −20°C acetone overnight and then spun at 6,500 g for 1 h at 4°C. The washing procedure was performed two additional times before resuspension or storage at −20°C.
LPS Extraction and Visualization
E. coli strains were kindly provided by Laurent Debarbieux. E. coli (81009, 81009ΔwaaF, E47a, E47aΔrfb3; Szijarto et al., 2014) and V. parvula DSM 2008 were grown in triplicate to an OD of 0.4. LPS was isolated using hot phenol extraction without modification as previously described (Davis et al., 2012). LPS was then resolved onto either a 0.1% SDS 13% or 17% PAGE gel and visualized with Pro-Q Emerald 300 (ThermoFisher Scientific) as per the manufacturer's instructions.
In-Gel Protein Digestion
Protein samples were loaded on a 0.1% SDS 12% PAGE gel. After the electrophoretic migration, the gel was stained with Coomassie Blue, each band of interest was cut, and in-gel tryptic digestion was performed as described previously (Wilm et al., 1996). Briefly, gel slices were washed in 100 mM ammonium bicarbonate for 15 min, followed by several washing steps to eliminate the stain in 100 mM ammonium bicarbonate/acetonitrile (1:1). Samples were reduced (10 mM DTT in 100 mM ammonium bicarbonate, 30 min at 56°C) and alkylated (55 mM iodoacetamide in 100 mM ammonium bicarbonate, 30 min at room temperature in the dark). Proteins were digested by 250 ng Sequencing Grade Modified Trypsin (Promega, Madison, WI, USA) in 10 mM ammonium bicarbonate overnight at 37°C. Resulting peptides were extracted, dried in the Speed-Vac and then resuspended in water/acetronitrile/formic acid (98:2:0.1).
Liquid Protein Digestion
Tryptic digestion was performed by eFASP (enhanced Filter-Aided Sample Preparation) as described previously (Erde et al., 2014). Samples were buffer-exchanged into buffer containing 8 M urea, 100 mM ammonium bicarbonate, and 0.2% deoxycholic acid using Tween passivated 30-kDa cut-off Amicon filters (Millipore Inc., USA). Samples were reduced with 5 mM tris (2-carboxyethyl) phosphine (TCEP) for 30 min at room temperature, alkylated in iodoacetamide (50 mM final, 30 min room temperature in the dark), treated with 1 μg of sequencing-grade modified trypsin (Promega, USA) overnight at 37°C under agitation. Peptides were recovered by centrifugation.
Mass Spectrometry Analysis
Trypsin-digested peptides coming from gel samples were analyzed by nano LC-MS/MS using an Ultimate 3000 system (Dionex, Amsterdam, The Netherlands) coupled to an LTQ-Orbitrap Velos. File microliters of each sample were loaded on a C18 pre-column (300 μm inner diameter × 5 mm; Dionex) at 30 μL/min in 2% ACN, 0.1% FA. After 5 min of desalting, the pre-column was switched online an in-house packed 15 cm nano-HPLC column (75 μm inner diameter) with C18 resin (3 μm particles, 100 Å pore size, ReproSil-Pur Basic C18, Dr. Maisch GmbH, Ammerbuch-Entringen, Germany) and equilibrated in 95% solvent A (2% ACN, 0.1% FA) and 5% solvent B (80% ACN, 0.08% FA). The peptides were eluted using a 2–50% gradient of solvent B during 60 min at 300 nL/min flow rate. The LTQ-Orbitrap Velos (Thermo Fisher Scientific, Bremen) was operated in data-dependent acquisition mode with the XCalibur software (Thermo Fisher Scientific, Bremen). Survey scan MS were acquired in the Orbitrap on the 300–2,000 m/z range with the resolution set to a value of 60,000 at m/z = 400. The 10 most intense ions per survey scan were selected for CID fragmentation, and the resulting fragments were analyzed in the linear trap (LTQ). The dynamic exclusion was enabled with the following settings: repeat count, 1; repeat duration, 30 s; exclusion list size, 500; and exclusion duration, 20 s.
Digests coming from liquid samples were analyzed on an Orbitrap Q Exactive Plus mass spectrometer (Thermo Fisher Scientific, Bremen) coupled with an EASY nLC 1000 chromatography system (Thermo Fisher Scientific). Sample was loaded on an in-house packed 40 cm nano-HPLC column (75 μm inner diameter) with C18 resin (1.9 μm particles, 100 Å pore size, Reprosil-Pur Basic C18-HD resin, Dr. Maisch GmbH, Ammerbuch-Entringen, Germany) and equilibrated in 98% solvent A (H2O, 0.1% FA) and 2% solvent B (ACN, 0.1% FA). Peptides were eluted using a 2–45% gradient of solvent B during 120 or 240 min at 250 nL/min flow rate. The instrument method for the Q Exactive Plus was set up in the data dependent acquisition mode using XCalibur 2.2 software (Thermo Fisher Scientific, Bremen). After a survey scan in the Orbitrap (resolution 70,000 at m/z 400), the 10 most intense precursor ions were selected for higher-energy collision dissociation (HCD) fragmentation with a normalized collision energy set to 28. Charge state screening was enabled, and precursors with unknown charge state or a charge state of 1 and >7 were excluded. Dynamic exclusion was enabled for 35 s.
In order to increase throughput and sensitivity methods used to acquire the data with these two mass spectrometer have been optimized in accordance with recommended settings from Thermo Fisher Scientific using HeLa extract sample (from Thermo) and an in-house complex quality control sample.
Data Processing and Analysis
Raw data were searched using MaxQuant (version 1.4.1.2) (Cox and Mann, 2008; Cox et al., 2014; with the Andromeda search engine) against V. parvula Uniprot database (1,844 entries, version 10-2015) and concatenated with known MS contaminants and reversed sequences of all entries. Andromeda searches were performed choosing trypsin as specific enzyme with a maximum number of two missed cleavages. Possible modifications included carbamidomethylation (Cys, fixed), oxidation (Met, variable), and Nter acetylation (variable). Maximum peptide charge was set to seven and five amino acids were required as minimum peptide length. Most peptides (~89%) were identified with charge states < +3. Less than 10% of peptides were identified with a charge state > +4. As you may see on the graph, peptides (~0.1%) were identified with a charge state of +6 (for details see PSM charges Pourcentage file deposited in PRIDE). Andromeda default settings were used; an initial search was performed using a mass tolerance of 20 ppm, followed by mass recalibration and a main search with a mass tolerance of 5 ppm for parent ions (Tyanova et al., 2016a). In the MS/MS search, mass tolerance was set to 10 ppm for the Q Exactive Plus and to 0.5 Da for the LTQ-Orbitrap Velos. Additional peptides were identified by the “match between run” option with a maximal retention time window of 1 min. One unique peptide to the protein group was required for the protein identification. Only unique peptides were used to distinguish isoforms. If no unique peptide was identified, isoforms were combined into the same “protein group” and the quantification was done for this protein group. A false discovery rate (FDR) cutoff of 1% was applied at the peptide and protein levels. MaxLFQ, Maxquant's label-free quantification (LFQ) algorithm was used to calculate protein intensity profiles across samples (Cox et al., 2014). Data were filtered by requiring a minimum peptide ratio count of two in MaxLFQ. Absolute protein amounts were calculated in Maxquant as the sum of all peptide peak intensities divided by the number of theoretically observable tryptic peptides (intensity-based absolute quantification or iBAQ (Schwanhäusser et al., 2011).
For statistical and bioinformatics analysis, as well as for visualization, Perseus environment was used, which is part of Maxquant (Tyanova et al., 2016b).
The “proteinGroup.txt” file generated with Maxquant was used by Perseus to identify proteins enriched in Biotin samples. Protein identifications were filtered, removing hits to the reverse decoy database as well as proteins only identified by modified peptides or considered as potential contaminant. Protein LFQ intensities were logarithmized. Two valid values out of three were required for each protein for the confident quantification across all replicates and missing values imputed by values simulating low abundance values close to the noise level. For pairwise comparison and identification of enriched proteins, a modified t-test were applied with permutation-based FDR statistics set to 1% and a S0 of 1 (Tusher et al., 2001).
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Vizcaíno et al., 2016) partner repository with the dataset identifier PXD005929.
Transmission Electron Microscopy
A densely grown culture of V. parvula DSM 2008 was used for the preparation for transmission electron microscopy (TEM). After concentration of the cells via centrifugation, the cells were either chemically fixed with glutaraldehyde or cryo-fixed with a high-pressure freezer and subsequent freeze substitution.
For chemical fixation, the cell pellet was resuspended in a fixation buffer consisting of 100 mM cacodylate including 2 mM MgCl2, 2.5% glutaraldehyde (final concentration; 25% stock solution), and 270 mM NaCl to adjust the osmolarity of the buffer to the original growth medium. After fixation, the cells were washed five times with buffer, post-fixed for 30 min in 1% osmium tetroxide, washed again two times with buffer and three times with double distilled water. Afterwards, the cells were dehydrated in a graded acetone series, infiltrated with Spurr‘s resin and polymerized for 72 h at 63°C. The embedded samples were then ultrathin sectioned (50 nm sections), post-stained with lead citrate and visualized in a TEM.
High-pressure freezing was performed with 2 μl of the resuspended concentrated cells in the respective aluminum platelets using a Leica HPM 100 high-pressure freezer (Leica Microsystems GmbH, Wetzlar, Germany). Freeze substitution was performed in a Leica AFS 2 (Leica Microsystems GmbH, Wetzlar, Germany) according to the following protocol (substitution solution): −90°C for 20 h (2% OsO4/acetone), heating to −60°C within 3 h (2% OsO4/acetone), −60°C for 4 h (2% OsO4/acetone), −60°C for 4 h (2% OsO4/0. 2% uranyl acetate/acetone), heating to −30°C within 3 h (2% OsO4/0.2% uranyl acetate/acetone), −30°C for 8 h (2% OsO4/0.2% uranyl acetate/acetone), heating to 0°C (2% OsO4/0.2% uranyl acetate/acetone). After washing three times with ice cold acetone, the samples were infiltrated with epoxy resin (Spurr‘s resin) and polymerized for 72 h at 63°C. The following steps were identical to the chemical fixation protocol.
Transmission electron microscopy was performed on a Zeiss EM 912 (Carl Zeiss AG, Oberkochen, Germany), operated at 80 kV in the zero-loss mode and equipped with a 2 k × 2 k dual-speed CCD camera (Tröndle Restlichtverstärkersysteme, Moorenweis, Germany).
Results and Discussion
Bioinformatic Localization Prediction
We started our study with a comprehensive in silico analysis of all 1,844 annotated protein-coding genes within the genome of V. parvula DSM 2008 (see Section Materials and Methods). The localization prediction of these 1,844 proteins are shown in Figure 1A. We used three general prediction programs (PSORT, CELLO, and SOSUI) for inner membrane (IM), cytoplasmic, periplasmic, secreted, and OM, and specific programs for OM (BOMP), IM (TMHMM and LipoP), and cytoplasmic (LipoP) localization. With this strategy, we managed to robustly predict the localization of 85% (1559/1844) of the proteins: 63% (1,171) cytoplasmic, 17% (309) IM, 2% (44) periplasmic, 1% (23) OM, 0.5% (12) secreted, whereas 15% (285) proteins remained with undefined localization (Figure 1A and Table S1).
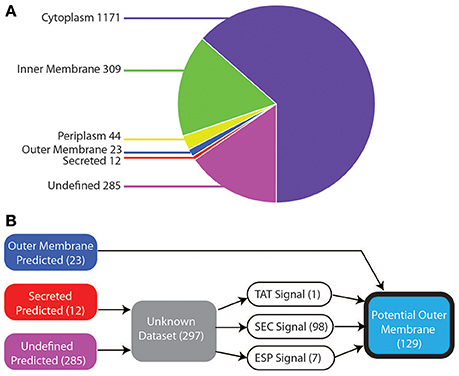
Figure 1. Bioinformatic localization prediction. (A) Localization of V. parvula proteome predicted by PSORT, CELLO, SOSUI, LipoP, TMHMM, and BOMP (see text for details). (B) Strategy for refining prediction for the sequences with undefined localization (see text for details).
The 23 proteins robustly predicted to be in the OM include many known OM components such as TonB-dependent receptors, OmpM, components of the BAM/TAM machinery, TolC, and OmpA (Table S1). Although localization prediction methods were able to identify these OM components, they have limitations due to training on datasets of known OM proteins, most of which are from the phylogenetically distant Proteobacteria. This could prevent proper prediction of many proteins and fail to find novel OM proteins. Indeed, they failed to correctly identify the experimentally characterized trimeric autotransporters, which were predicted as secreted or had undefined localization (Table S1).
As such, we more thoroughly investigated the 297 proteins with undefined prediction or predicted as secreted, as they might contain additional OM proteins. As shown in Figure 1B, to do so, we constructed a bioinformatic filter to sort these 297 proteins. Briefly, we identified the proteins that contained a signal sequence for translocating through the IM, by checking for the three IM transport mechanisms: SEC, ESP (Extended signal peptide), and TAT with seven different programs (Figure 1B; see Section Materials and Methods). Hundred and ninety-one proteins lacked any recognizable signal sequence and were excluded. The remaining 106, which included all of the previously missed trimeric autotransporters, were combined with the 23 predicted OM proteins to provide a final dataset of 129 potential outer membrane proteins (Table S2).
Subcellular Fractionation
To validate our in silico prediction and to firmly identify OM proteins, we carried out a proteomic analysis of the OM of V. parvula DSM 2008 (see Section Materials and Methods for details). We performed three extractions: an outer membrane (OM) extract, a surface exposed (SE) extract, and a control whole cell (WC) protein extract. For all three extractions, V. parvula was grown anaerobically in BHIL medium in triplicate and the extraction was performed during the exponential growth phase. For OM protein extraction, we performed French press lysis to produce vesicles and isolated the OM vesicles with a chaotropic agent. To identify SE proteins we used a cell impermeable substrate, PEG-Biotin, which would label any primary amine exposed to the external environment of the cell. For each of the three extractions, only proteins that were present in all three biological replicates were considered for further analysis.
To identify proteins that were unique or shared by the different fractions we constructed a Venn diagram that is depicted in Figure 2. The WC control extract contained 1,342 proteins, while the OM fraction contained 990 proteins, and the SE fraction contained 849 proteins. Many false positives, including ribosomal proteins, were present in all three fractions (Table S1). This is a known product of the high sensitivity of MS analysis (Pocsfalvi et al., 2016).
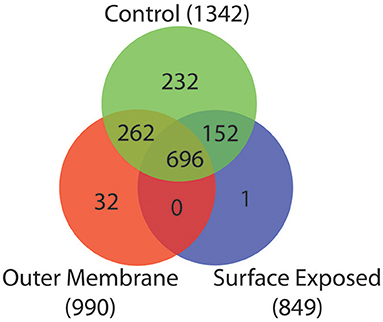
Figure 2. Venn diagram of peptides detected from the three extraction methods. Overlap of peptides detected in the control sample (Green), OM extraction (Red), and SE extraction (Blue).
Among the 1,342 proteins present in the WC extract, 696 were also detected in both the OM and SE fractions (Figure 2). Two hundred and sixty-two were detected in the WC and OM fractions but not in the SE. These proteins probably lack an exposed primary amine that would make them available to the biotinylation substrate. Similarly, 152 proteins were detected in the WC and SE fractions but not the OM fraction. These included many cytoplasmic or IM components and are therefore possible contaminants (Table S1). Finally, we found 32 proteins unique to the OM extract, and one protein unique to the SE extract. These discrepancies may be due to the enrichment of specific proteins by the OM and SE extraction protocols, combined with a too low concentration for detection in the WC control sample.
For the remainder of the analysis we only considered proteins as potential OM if they were detected in both the OM and the WC control samples, while we considered proteins as potential SE if they were present in all three samples, leading to 958 proteins (WC + OM = 262, WC + OM + SE = 696). In order to reduce false positives from this dataset, we crossed it with the 129 potential OM proteins obtained by our bioinformatics prediction (Table S2). This led to a final list of 78 OM proteins (Table 1 and Table S3) that we annotated and created a graphical schematic representation in a V. parvula cell as shown in Figure 3. Identification of these OM proteins markedly extends previous inferences on the nature of the cell envelope of Negativicutes (Antunes et al., 2016) and provides information on the role of OM systems in the lifestyle of Veillonella.
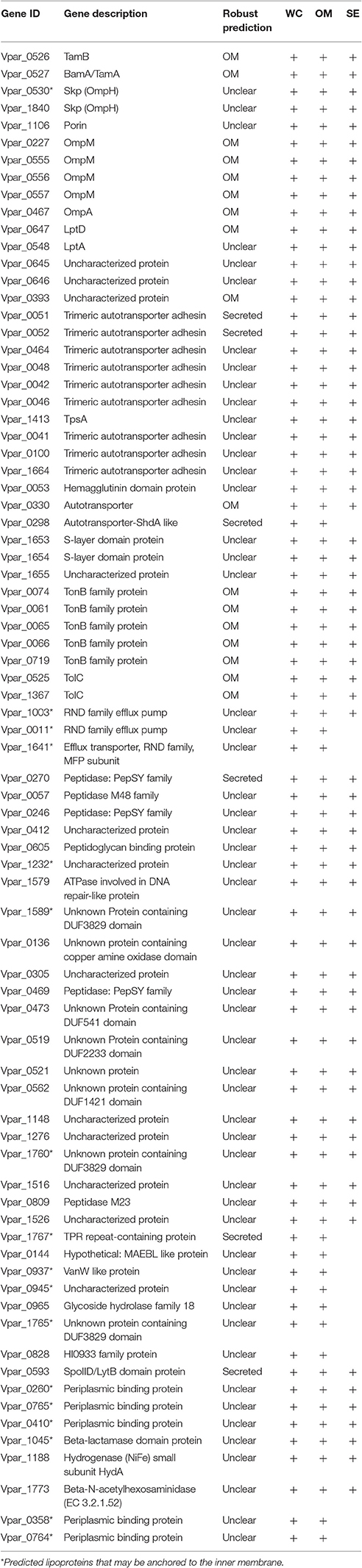
Table 1. 78 OM proteins identified by bioinformatic localization and proteomic analysis. Order follows discussion in text.
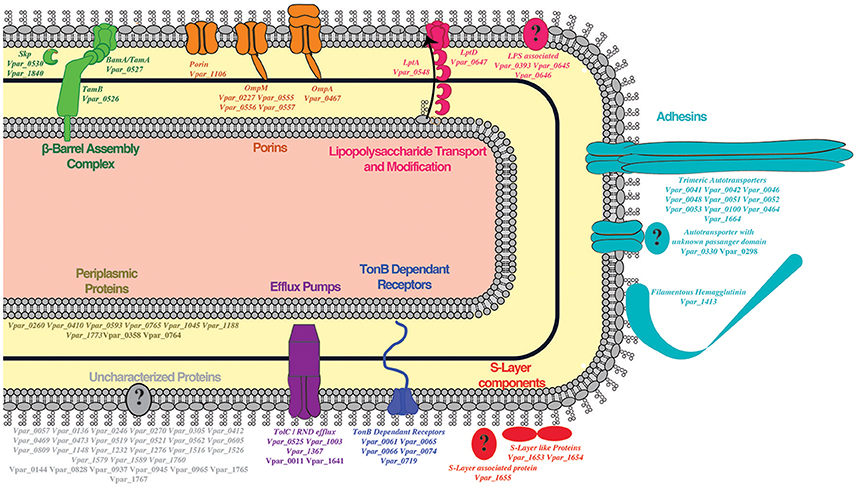
Figure 3. Outer membrane proteome schematic demonstrating the different systems detected by our methods. Proteins found in the OM are in bold while SE detection is indicated by bold and italics.
Additionally, among the 1,844 predicted proteins of V. parvula roughly one quarter of them (502) were not expressed in our growth conditions or were not detected by MS due to their low concentration. As an example, we previously detected a conserved genomic locus coding for all components of a Type 4 pilus (T4P), including an OM secretin (Antunes et al., 2016). This cluster is similar to characterized Type 4A pili, which are known to be responsible for twitching motility (Pelicic, 2008) and natural competence (Knapp et al., 2017). However, we could only detect three peptides of PilM in one of the three WC control extraction replicates and three peptides of the component PilA in one of the three OM extraction replicates (Table S4). All remaining T4P components were absent in all of our samples, demonstrating the absence of their production in our growth conditions. Nonetheless, a T4P and the other undetected proteins may be produced in other conditions such as in vivo or during biofilm formation. Indeed, other species of V. parvula have shown differential competence capabilities in different media; this is likely related to T4P expression (Knapp et al., 2017).
An Ancestral β-Barrel Assembly Machinery, BAM/TAM, Is Present and Functional in the OM of V. parvula
The detection of OM proteins implies the existence of a functional system to insert them in the OM. The presence of a peculiar and probably ancestral BAM/TAM machinery has been inferred in Negativicutes and other bacteria (Webb et al., 2012; Heinz et al., 2015; Antunes et al., 2016). This system putatively contains three components, the BamA/TamA assembly barrel (Omp85), the TAM IM/periplasmic chaperone (TamB), and the BAM periplasmic chaperone (Skp). It has been hypothesized that this ancestral BAM/TAM machinery functions as both BAM and TAM in diderm Firmicutes (Heinz et al., 2015; Antunes et al., 2016). This hypothesis is supported by the detection of only a single Omp85 homolog in each genome and the fact all these genes form a conserved genomic cluster, suggesting functional linkage. Indeed, a recent study on TamB from Borrelia burgdorferi demonstrated that it interacts with BamA composing a BAM/TAM system (Iqbal et al., 2016).
Twenty-one of the 23 robustly predicted OM proteins were predicted as β-barrels, of which 17 were detected in our OM. This provides the first experimental evidence that the BAM/TAM system is functional in V. parvula. Moreover, we detected the major OM component BamA/TamA (Vpar_0527), as well as TamB (Vpar_0526) in both our SE and OM fractions (Figure 3, Table 1, and Table S3). While the presence of the OM component BamA/TamA was expected, that of TamB is surprising, as TamB in other species is known to be periplasmic and attached to the IM. One can speculate that in V. parvula an entire complex containing BamA/TamA and TamB may be captured in the OM fraction. However, this fails to account for the strong OM prediction of TamB by localization software PSORT, CELLO, and SOSUI (Table S1), which indicates that some OM characteristics must be present within the V. parvula TamB; unfortunately we cannot determine what these characteristics are, due to the machine learning nature of the programs. Interestingly, TamB of E. coli (NP_418642.1) is also predicted to be in the OM by two out of the three programs (not shown), yet its IM/periplasmic localization has been firmly established through protease shaving (Selkrig et al., 2012). Discrepancies also exist in previous fractionation mass-spectrometry studies, as TamB was found in higher concentration in the OM than in the IM in E. coli (Martorana et al., 2014). Together, these data suggest that TamB may actually be both in the OM and the IM, or span the two membranes.
Four genes potentially encode the chaperone Skp in the V. parvula DSM 2008 genome (vpar_0528-30, vpar_1840), three of which are present in the diderm cluster immediately downstream of the BamA/TamA gene (Antunes et al., 2016). All four Skp proteins were detected in our OM fraction. However, two of them were excluded by our localization prediction procedure because of their strong prediction as periplasmic proteins (Table S1). Presence of Skp in the OM fraction is not unusual as Skp in Salmonella was originally thought to be in the OM, hence the old name OmpH (Thome and Müller, 1991). Furthermore, it has been shown experimentally that this protein interacts strongly with both the BAM system and OmpA, by functioning as a chaperone helping insertion of proteins in the OM (Selkrig et al., 2014).
Porins Are a Fundamental Component of Veillonella's Outer Membrane
A key component of bacterial OM are porins, which are necessary for passive diffusion of small molecules (Galdiero et al., 2012). The genome of V. parvula contains genes encoding a typical porin (vpar_1106) consisting entirely of a β-barrel, four copies of OmpM (vpar_0227, vpar_0555-7) which contains a porin domain and a periplasmic SLH domain, and a homolog of the pathogenicity factor OmpA (vpar_0467) containing typical OmpA and beta-barrel domains (Confer and Ayalew, 2013).
We previously described the presence of three OmpM encoding genes in tandem within the diderm cluster of V. parvula (vpar_0555-7) (Antunes et al., 2016). During the course of this analysis we discovered a fourth (vpar_0227) at a different locus. The three OmpM proteins found within the diderm cluster (Vpar_0555-7) are among the most abundant proteins in the OM fraction by both quantification of our LC-MS/MS data (Table S5) and SDS-PAGE band excision MS (Bands 4 and 5) (Table S6). This correlates well with the function as an OM tether, as in E. coli Braun's lipoprotein is the most numerous protein in the cell (Braun, 1975). Furthermore, this supports the hypothesis that all Negativicutes utilize OmpM as a form of attachment (Kojima and Kamio, 2012). Moreover, this is consistent with a transcriptomics study of three Veillonella species from the mouth, which found all three copies of OmpM to be the most abundant OM transcripts (Do et al., 2015), further highlighting the importance of these proteins (Kojima et al., 2016).
We detected all four copies of OmpM in both the OM and the SE fractions (Figure 3, Table 1, and Table S3). Because of OmpM's abundance and dominant role in the OM, an exposed area of Veillonella's OmpM may be involved in adhesion and biofilm formation. Previous bioinformatics studies have suggested that OmpM in the Negativicute Selenomonas ruminantium may be SE and interact with other bacteria (Kojima et al., 2011). We previously wondered why there are multiple copies of the OmpM tether in diderm Firmicutes (Antunes et al., 2016); indeed, Braun's lipoprotein is usually in single copy, with the exception of Salmonella's from a recent gene duplication event (Sha et al., 2004). Because the four OmpM proteins are not identical, it is possible that they each provide a different surface for adhesion and/or immune evasion.
We also detected an OmpA homolog (Vpar_0467) in the OM and SE fractions. OmpA is an important protein in the OM of bacteria as it has strong pathogenic roles involving cellular invasion, adhesion, and host cell evasion (Confer and Ayalew, 2013) and therefore may be involved specifically in pathogenicity of V. parvula. Consistent with this hypothesis, a search for OmpA homologs in other Negativicutes revealed its presence in a few other human-related members of the family Veillonellaceae (Figure 4).

Figure 4. Distribution of selected protein-coding genes in select proteins among Negativicutes. Schematic tree based on phylogeny of the Negativicutes from previous analysis (Antunes et al., 2016). Presence and absence of key genes are indicated by + and − symbols, respectively. The dominant niche of each strain taken from the available literature is also given. For discussion, see main text.
A Complete LPS Transport System
As we previously reported (Antunes et al., 2016), Negativicutes have the genetic potential to make and transport LPS to the OM. Most LPS biosynthesis genes are embedded in the diderm cluster in Negativicutes genomes, including that of V. parvu (Figure 5A). We were previously unable to identify the OM flippase components LptD or LptE, nor any component of potential O-antigen biosynthesis (Antunes et al., 2016). Here, an LptD homolog (Vpar_0647) was detected in the OM and SE fractions and identified as being an OM protein by localization prediction software (Figure 3, Table 1, and Table S1). This potential LptD encoding gene is located outside of the diderm cluster in a different region of the genome (Figure 5B). Interestingly, two proteins encoded by genes upstream of lptD (vpar_0645 & 0646) were both found in the OM and SE fractions (Figure 5B and Table S1). In addition, vpar_0646 and lptD are conserved in sequence and synteny among all Negativicutes members (Table S3). We were unable to ascertain the function of Vpar_0645 & 0646 as we could find no conserved domains or clear homologs in sequence databases. It is possible that these proteins function with the Lpt complex of V. parvula or are functional equivalents of LptE, which caps the transport pore of LptD.
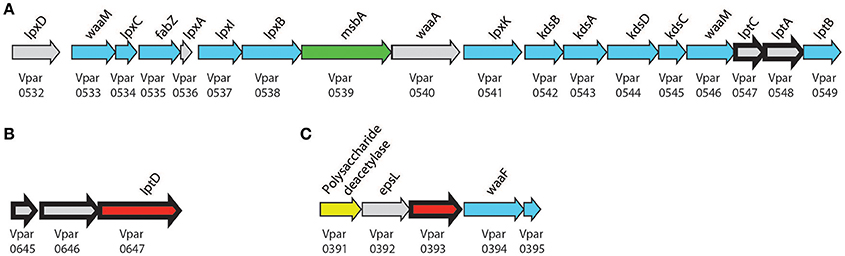
Figure 5. Lipopolysaccharide biosynthesis clusters. Biosynthesis clusters found within V. parvula of (A) core biosynthetic and transport machinery, (B) conserved lptD cluster, (C) proposed LPS modification cluster. Bold arrows represent peptides detected in the OM. Localization is presented by color: Gray, unclear; Blue, cytoplasmic; Green, IM; Yellow, Periplasmic; and Red, OM. Genomic coordinates are (A), CP001820.1:650916–670828 (+); (B) CP001820.1:780878–784136 (+); (C), CP001820.1:477425–481382 (+).
Finally, we detected the periplasmic components LptA (Vpar_0548) and LptC (Vpar_0547) in our OM and surface-exposed fractions. LptC was excluded from our final OM dataset because it did not pass the bioinformatics filter due to a lack of any discernible signal sequence. While we cannot exclude a contamination of periplasmic components in our OM fraction, it is possible that LptA and LptC were dragged to the OM fraction by strong interaction with LptD, thereby suggesting the existence of a complete Lpt complex in V. parvula similar to other studied bacteria (Villa et al., 2013).
O-Antigen in V. parvula?
Despite the presence of a complete pathway to make the lipid-A core of LPS, we previously could not predict if Negativicutes make O-antigen or not (Antunes et al., 2016), and the structure of Veillonella's LPS is currently unknown. Here, we detected a protein in the OM fraction that may be used for further LPS processing (Vpar_0393). Although this protein has no conserved domains, we could tentatively infer its function based on the surrounding genomic region (Figure 5C). This region contains three genes probably involved in sugar anabolism (vpar_0391, 0392, 0394) and is well conserved only among the Negativicutes. The first protein, Vpar_0391, contains a CE4_SF domain; this domain is characteristic of polysaccharide deacetylases, such as PgaB or IcaB. The second protein, Vpar_0392, has the highest similarity to EpsL from a plasmid of Lactococcus lactis, which has no known function, yet is the final gene in the eps operon of exopolysaccharide biosynthesis (Forde and Fitzgerald, 2003). Although Vpar_0392 and EpsL are clear homologs, none of the surrounding genes bear any remarkable similarity. Vpar_0394 is annotated as WaaF, an enzyme that is known to be specific of diderm bacteria. It is responsible for O-antigen attachment to LPS by means of transferring a L-glycero-D-manno-heptose residue to the core oligosaccharide moiety of LPS. Mutation of this gene in Salmonella results in increase in sensitivity to antibiotics, detergents, and bile salts (Brooke and Valvano, 1996). Together, these data suggest that this could represent a new gene cluster responsible for O-antigen biosynthesis. To confirm this hypothesis and evaluate whether V. parvula LPS contains O-antigen we extracted and visualized its LPS on SDS PAGE (Figure S1). The extraction showed the characteristic ladder-banding pattern of O-antigen, however further characterization is necessary to determine V. parvula LPS structure and function.
V. parvula Has Many Potential Surface Exposed Adhesins
One of the most prevalent and important aspects of Veillonella's lifestyle is its ability to form biofilms and interact with host cells or other members of the microbiota where it has been identified. Such interactions are mediated by adhesins and Veillonella has many of them (Hughes et al., 1992); these adhesins represent prolonged adaptation and evolution to the biofilm niche of Veillonella. By controlling the type and quantity of adhesins expressed it could prefer one substrate or cell and bind with high affinity. The presence of eight putative hemagglutinins belonging to the YadA-like family of trimeric autotransporters was recently reported in V. atypica OK5, and one of them (Hag1) was shown to be responsible for co-aggregation with resident flora in the oral cavity (Zhou et al., 2015).
Among the 12 trimeric autotransporters containing a typical YadA anchor domain in V. parvula DSM 2008 (Figure S2), we found 10 in all three samples (Table 1). These 10 trimeric autotransporters possess typical Yad_Head, Yad_Stalk, and Cter Beta-barrel Yad _anchor domains. They are likely adhesins of the hemagglutinin family (Bassler et al., 2015). Strikingly, six of these 10 trimeric autotransporters are located in a massive gene cluster (56 kb) (Figures S2, S3A). It is difficult to ascertain if this cluster is conserved in all Negativicutes and how many trimeric autotransporters it contains, as most of these genomes are in scaffolds. Trimeric autotransporters contain many repeated regions, are modular in nature, contain many recent duplication events, and may get to be very large; as such, many Negativicutes scaffolds terminate in these genes thereby preventing synteny analysis. Two trimeric ATs are notable (Figure S2A): Vpar_0450 was not included in our final list of 78 OM proteins due to no detection in the OM fraction. It probably had too low of a concentration to be detected. Vpar_0045 is composed almost entirely of a Yad_anchor domain and thus is probably not functional and was not detected in any of our samples.
Interestingly, three trimeric autotransporter adhesins of V. parvula (Vpar_0041, Vpar_0100, Vpar_1664) possess a C-terminal SLH motif that is located downstream of the Beta-barrel YadA_anchor domain (Figure S2). This suggests a topology where the SLH domain is periplasmic and interacts with the peptidoglycan, similar to OmpM, to stabilize the trimeric configuration of these adhesins. We found this type of architecture between SLH and YadA anchor domains present in other autotransporters. However, it is restricted to the Negativicutes pointing to a unique feature potentially linked to their lifestyle (data not shown).
Upon further analysis, we noticed a distinct pattern of distribution for the presence of flagella and trimeric autotransporters in Negativicutes (Figure 4). The Veillonellaceae and Acidaminococcaceae contain YadA-like trimeric autotransporters and lack flagella, while the Selenomonadaceae and Sporomusaceae typically possess flagella and generally lack the YadA-like proteins, with only two exceptions (Figure 4). This peculiar distribution of flagella or adhesins between strains has not been observed previously and presents a unique case of evolution driven by lifestyle with the distinct presence of adhesion or motility. This observation provides a unique opportunity to study the transition between adhesion and motile life style, especially in Mitsuokella multacida DSM 20544, which represents an interesting specimen: it belongs to the Selenomonadaceae, which are typically flagellated, however it has lost its flagellum cluster and has acquired adhesins (Figure 4).
The V. parvula genome also contains two potentially functional filamentous hemagglutinin (Fha)-like proteins transported by the two-partner system (TPS) (Figure S2B): Vpar_0979/Vpar_0980 and Vpar_1413/Vpar_1414 as TpsA adhesin/TpsB transporter pair. We detected only the TpsA adhesin Vpar_1413 in the OM and SE fractions (Figure 3, Table 1, and Table S3). Its transporter Vpar_1414 was detected in the WC fraction but may have been present at too low concentration to be detected in the OM or SE fractions. The other system, Vpar_0979-Vpar_0980, was not detected in any fraction, including the whole protein fraction, suggesting an absence of its expression in our growing conditions. It may only be expressed upon contact with a host cell or when interacting with other microbial species. Furthermore, Vpar_0979 SEC secretion signal was only detected by one out of three programs and may represent a degenerate coding sequence.
In addition to trimeric and TPS systems that almost exclusively correspond to adhesins, V. parvula encodes classical autotransporters that contain a C-terminus β-barrel domain for insertion in the OM and an N-terminus passenger domain for function. Although some classical autotransporters are adhesins, within others the passenger domain may carry additional virulence functions such as protease or lipase domains (van Ulsen et al., 2014). We found four genes encoding such classical autotransporters within the genome (vpar_0037, vpar_0298, vpar_0330, vpar_1322) (Figure S2C). Vpar_0037 seems to be composed solely of a Beta-Barrel domain without any passenger domain or signal sequence; as such it is probably not functional. Vpar_1322 has a passenger domain with no detectable secondary or tertiary structural homology to known functional domains. Neither of these two autotransporters were detected in any of our samples suggesting they are not produced in our growth conditions. One that was found in the OM fraction, Vpar_0298, has a detectable homology to ShdA, the AidA-like adhesin of Salmonella typhimurium. Vpar_0330 is an autotransporter with no clear functional identification of its passenger domain, however we could find weak homology to a polysaccharide lyase-like protein using Phyre2 (see Section Materials and Methods); we detected Vpar_0330 both in our OM and SE fractions. Interestingly, vpar_0330 was recently shown to be upregulated in V. parvula in a mouse tumor colonization model compared to in vitro growth, both with and without co-culture of Pseudomonas aeruginosa (Pustelny et al., 2015). Furthermore, it was highly upregulated in caries when compared to healthy teeth (Do et al., 2015). These data suggest that this autotransporter may be an important colonization factor in Veillonella and a priority target for further investigation.
Other Outer Membrane Features
Lipoproteins
Lipoproteins are found in most diderm bacteria and can be targeted to the outer membrane by the LOL transport machinery (Sutcliffe, 2010; Dowdell et al., 2017). We previously noted the absence of any LOL transport components (lolA-E) in any Negativicute genome (Antunes et al., 2016), however we find that 17 out of 78 OM proteins contain a lipoprotein signal sequence as predicted by LipoP and PRED-LIPO software (Table 1 and Table S3). To resolve this incongruence, we further analyzed the MS data of OM proteins that contained a cysteine residue. We searched for any mono, di, or triacylglycerol with a saturated C16 or C18 fatty acid as a modification (Data not shown). No such modification was found on any of the proteins, supporting the hypothesis that Veillonella does not have OM-targeted lipoproteins. These data are not conclusive though, as our MS analysis was not designed specifically for this purpose, and further studies should be performed. If V. parvula is shown to possess OM lipoproteins, it may use an alternative pathway, which remains to be identified. Another possibility is that these proteins may be tethered to the IM with a lipid moiety and span the periplasmic space in addition to being in the OM.
Cell Ultrastructure
Although images of Veillonella are available in the literature, high quality electron microscopy has not been performed. We carried out ultrastructural analysis of V. parvula cells in two different ways: via chemical fixation (Figures 6A,C,E) and via high-pressure freezing of cells (Figures 6B,D,F). Subsequent resin embedding and ultrathin sectioning of V. parvula cells delivered a clear insight into the cell wall composition of this organism. Within the homogenous cytoplasm, electron dense particles are visible especially after high-pressure freezing (Figures 6B,D,F). In chemically fixed cells, an electron brighter area in the central part of the electron dense cytoplasm might show the DNA (genome). In both fixation variants, the cytoplasm is surrounded by the cytoplasmic membrane, which is again surrounded by a periplasm. Within this periplasm, peptidoglycan can sometimes be detected as electron dense line (Figure 6C). At the outermost part of the cell envelope, an outer membrane covers chemically fixed as well as high-pressure frozen cells. As the wavy appearance of the outer membrane is present after both fixation methods, this might either represent a special structural feature or a preparation artifact due to imperfect adjustment of the osmolarity of the post-fixation solutions to the original growth medium of the organism. The relative high thickness and in some cases fluffy appearance of the outer moiety of the outer membrane (Figure S4) is a strong indication for presence of LPS, although an S-layer cannot completely be ruled out (see below).
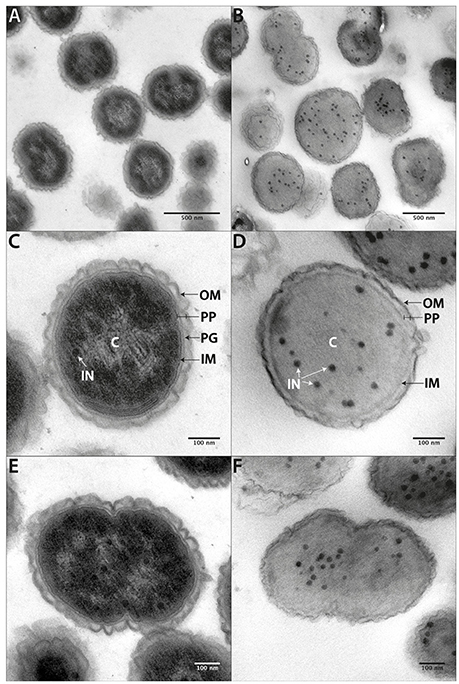
Figure 6. Cell ultrastructure. Ultrathin sections of chemically fixed (A,C,E) or high-pressure frozen (B,D,F) V. parvula cells. At higher magnification the inner membrane (IM), the peptidoglycan (PG) containing periplasm (PP), and the slightly waved outer membrane (OM) become apparent. In chemically fixed cells, an electron brighter area within the periplasm most likely represents DNA. Electron dense circular structures (IN) can be seen within the homogenous cytoplasm of high-pressure frozen cells and, to a less extent also in chemically fixed cells.
An S-Layer in V. parvula?
Surface layer proteins (S-layer proteins) are important cellular factors, in both monoderms and diderms. They are assembled as paracrystalline mono-layers on the cell surface of bacteria and archaea, are attachment sites for enzymes and/or substrates, or participate in interactions with abiotic surfaces or bacteria (Sara and Sleytr, 2000; Gerbino et al., 2015). We found two proteins containing three SLH contiguous domains (Vpar_1653 & Vpar_1654) in our OM and SE fractions (Figure 3, Table 1, and Table S3). Such domain architecture can be found in typical S-layer glycoproteins yet may also be associated to other functional domains such as hydrolases. Unfortunately, no other functional domains could be detected in Vpar_1653 and Vpar_1654. A classical S-layer has not been documented in Veillonella. A fluffy outer leaflet of the outer membrane was often visible, especially in high-pressure frozen cells (Figure S4). This may show the S-layer on top of the outer membrane, however it is more likely that this structure represents the LPS moiety. It is unlikely that a complete S-layer is produced, as these proteins are normally extremely abundant, but were 100 fold less abundant than OmpM in the OM fraction (Table S5).
Interestingly, vpar_1653 and vpar_1654 genes are in cluster along with a third gene (vpar_1655), which was also found in our OM and SE fractions (Table 1, Table S3). This third gene does not code for an S-layer protein and has no obvious function or conserved domains, other than one of unknown function (DUF4163). We could not find this three-gene cluster outside of Veillonella (Table S3), suggesting that it may have a genus-specific function. Further functional analysis is required to determine if Veillonella utilizes this gene cluster to make a typical S-layer or if it is used for enzymatic activity, interaction with surfaces and/or biofilm formation.
TonB-Dependent Transport
The TonB system is an OM transport mechanism present in most diderm bacteria, which has never been described in any Firmicute. Bacteria generally contain multiple TonB transport systems specifically associated with the acquisition of small molecules including vitamin B12 or metals such as iron (Krewulak and Vogel, 2011). We identified nine such TonB systems in the V. parvula genome, of which six were present in the OM and SE fractions (Figure 3, Table 1, and Table S3). Some of these systems must be functional to provide iron for the V. parvula haem cluster which has recently been characterized in V. atypica (Zhou et al., 2016). Interestingly, almost all of the operons encoding these TonB systems are located within a single genomic cluster downstream of the previously mentioned adhesion cluster (Figure S3B). Two other tonB operons are located at distant loci but were not found in our samples; specific molecules that were not present in our growing conditions might induce their expression.
Efflux Pumps
TolC is an outer membrane channel responsible for export of antibiotics and other toxic compounds from the cell. These systems are important in antibiotic resistance and in infection (Zgurskaya et al., 2011). Among the two TolC-ABC transporters and three similar RND type efflux pumps that are encoded in the genome of V. parvula DSM2008, we detected three in our OM and SE fractions (Vpar_0525, Vpar_1003, Vpar_1367) and two in only the OM fraction (Vpar_0011, Vpar_1641) (Figure 3, Table 1, and Tables S1, S3). Intriguingly, it was previously shown that the RND pump Vpar_1367 is upregulated in an infectious community responsible for caries (Do et al., 2015), pointing to the importance of such systems to scavenge essential molecules in in vivo conditions in a multispecies context.
Putative OM Proteins of Unknown Function
Among the list of 78 OM proteins, 28 proteins have no clear function (Figure 3, Table 1, and Table S3), many of which may correspond to previously undescribed systems and novel OM-related functions. We represented these unknown function proteins, as well as six unknown proteins previously described in other sections, with their detected functional domains in Figure 7.

Figure 7. Domain structure of unknown OM proteins. Protein domains identified in all poorly annotated OM proteins at the NCBI CDD database, and colored according to proposed function based on CDD annotation.
Of special interest is Vpar_0521 (COG3064). We previously found it is well-conserved within the diderm Firmicute, and hypothesized its possible involvement in a yet unknown OM-related function (Antunes et al., 2016). Vpar_0521 contains two domains (Figure 7): an N-terminus SpoIVB and a C-terminal DUF1090. The N-terminus domain, SpoIVB, is an autoprotease (S5 peptidase) involved in sporulation in the Firmicutes; it is normally synthesized in the forespore and transported to the interspace between the forespore membrane and the outer spore membrane, where it functions in signal transduction (Campo and Rudner, 2007). Because V. parvula is a non-sporulating Firmicute, the role of this SpoIVB domain remains to be determined. The other domain, DUF1090, is an uncharacterized domain found in all three domains of life, and predominantly present in Bacteria. For example, the E. coli YqjC protein only contains this domain. Only one study has been performed on the corresponding gene and it was found to be down regulated when the sigma factor rpoS gene was mutated (Zhang et al., 2012). In the Stepdb database that details the localization of all E. coli proteins (Orfanoudaki and Economou, 2014), YqjC is annotated as periplasmic. In some bacteria, this domain constitutes the C-terminal hydrophobic substrate-binding domain of the chaperone DnaK and it may have a similar function in Vpar_0521. These indications, and the conservation of Vpar_0521 in diderm Firmicutes, make this protein an important target of future study.
We found three proteins corresponding to the DUF3829 domain (Vpar_1589, Vpar_1760, Vpar_1765) (Figure 7). Although this domain is found in the distantly related Proteobacteria using a CDD search, the only close BLAST hits were from human-associated Negativicutes (Figure 4), more specifically the Dialister, Centipeda, Selenomonas, and Veillonella. We could not detect any homologs in V. ratti or any of the other rodents-associated Veillonella (Whitman, 2011). These unstudied proteins may be involved with interactions in the human niche, both for pathogenic and commensal lifestyles.
Three proteins contain one (Vpar_0246, Vpar_0469, Vpar_1597) or two (Vpar_0270) PepSY domains, and no other conserved domains. All proteins containing a PepSY domain were found in all three extractions except for Vpar_1597, which was not detected in any sample. This domain has not been extensively studied, yet it is known to possess a peptidase inhibitory function and is found throughout bacteria and some archaeal species (Yeats et al., 2004). The dominantly studied member of this family is YpeB, an inhibitor of the spore cortex lytic enzyme SleB in sporulating Firmicutes (Yeats et al., 2004). The four V. parvula OM PepSY proteins may function similarly to YpeB and inhibit peptidases of the M4 family or possess some unknown function.
Conclusions
In recent years we have learnt how important Veillonella is to the human microbiome, infection, and immune development (Whitman, 2011; Arrieta et al., 2015; Hirai et al., 2016). Moreover, these bacteria deserve to be studied not only for these characteristics, but also for the evolutionary questions they pose. This work represents the first proteomic characterization of a diderm Firmicute cell envelope and provides important information to guide further characterization. It confirms that the Negativicutes cell envelope has many aspects of the classical and well-studied Proteobacterial Gram-negative cell envelopes, such as LPS, TolC, OmpA, and other components, yet it also possesses many unique and potentially ancestral characteristics, such as the peculiar OM-PG attachment system, the BAM/TAM complex, as well as potentially new OM systems. The function of these systems needs to be further characterized through mutational and expression studies. Finally, our results are instrumental in increasing our understanding of the lifestyle of Veillonella, including its potential for biofilm formation and its role in infection and host-interaction.
Author Contributions
DP prepared all samples and carried out bioinformatics analysis of genomic and proteomic data. MD, VH, and MM carried out mass spectrometry analysis. JF and AK carried out electron microscopy. CB supervised the study and analyzed the data. SG conceived and supervised the study, and analyzed the data. All authors participated in writing the manuscript.
Funding
This work was supported by grant ANR-16-CE12-0010-02 FIR-OM from “Agence Nationale de la Recherche,” and PTR 39-16 from the Institut Pasteur “Programmes Transversaux de Recherche.” DP is the recipient of a Ph.D. fellowship from the Institute Pasteur PPU program. CB is supported by the Institut Pasteur, the French Government Investissement d'Avenir Program Laboratoire d'Excellence “Integrative Biology of Emerging Infectious Diseases” (grant no. ANR-10-LABX-62-IBEID), Fondation pour la Recherche Médicale grant “Equipe FRM DEQ20140329508.” SG acknowledges support by Investissement d'Avenir Grant “Ancestrome” (ANR-10-BINF-01-01).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.01215/full#supplementary-material
Figure S1. Glycostain of LPS. Thirteen and seventeen percentage SDS-PAGE loaded with identical samples and visualized with Pro-Q emerald glycostain. O-antigen producing positive control E. coli were loaded in lanes 1 and 3, while O-antigen negative extractions were loaded in lanes 2–4. Three biological replicates of V. parvula were loaded in lanes 5–7.
Figure S2. Adhesins domain structure. Figure demonstrating the domain structure of all trimeric autotransporters (A), two partner systems (B), and autotransporters (C).
Figure S3. Trimeric autotransporter (A) and TonB (B) genomic clusters. Bold arrows represent peptides detected in the OM. Localization is presented by color: Gray, Unclear; Blue, Cytoplasmic; Green, IM; Yellow, Periplasmic; Purple, Secreted; and Red, OM.
Figure S4. High-pressure frozen cell illustrating LPS. The ultrathin section of a high-pressure frozen cell shows the cell wall of V. parvula (A). Beside the inner membrane (IM), the outer membrane (OM), and the peptidoglycan (PG) within the periplasm, the fluffy outer leaflet of the outer membrane is visible at higher magnification (B). This might represent the LPS moiety. Bold arrows represent peptides detected in the OM.
Table S1. Localization prediction and Mass spectrometry results for all proteins of V. parvula DSM 2008. Proteins predicted to be in the OM are in red.
Table S2. Localization prediction and Mass spectrometry results for 129 potential OM proteins of V. parvula DSM 2008.
Table S3. Localization prediction and Mass spectrometry results for 78 OM proteins of V. parvula DSM 2008.
Table S4. Type 4 pili components in silico digestion and detection.
Table S5. OM extraction gel excision Mass spectrometry results. Top 5 hits for each band are shown; Proteins predicted to be in the OM are highlighted.
Table S6. IBAQ results for the OM extraction.
References
Africa, C. W. J., Nel, J., and Stemmet, M. (2014). Anaerobes and bacterial vaginosis in pregnancy: virulence factors contributing to vaginal colonisation. Int. J. Environ. Res. Public Health 11, 6979–7000. doi: 10.3390/ijerph110706979
Antunes, L. C., Poppleton, D., Klingl, A., Dupuy, B., Brochier-Armanetd, C., Beloin, C., et al. (2016). Phylogenomic analysis supports the ancestral presence of LPS outer membranes in the Firmicutes. Elife 5:e14589. doi: 10.7554/eLife.14589
Arrieta, M.-C., Stiemsma, L. T., Dimitriu, P. A., Thorson, L., Russell, S., Yurist-Doutsch, S., et al. (2015). Early infancy microbial and metabolic alterations affect risk of childhood asthma. Sci. Transl. Med. 7:307ra152. doi: 10.1126/scitranslmed.aab2271
Bagos, P. G., Nikolaou, E. P., Liakopoulos, T. D., and Tsirigos, K. D. (2010). Combined prediction of Tat and Sec signal peptides with hidden Markov models. Bioinformatics 26, 2811–2817. doi: 10.1093/bioinformatics/btq530
Bagos, P. G., Tsirigos, K. D., Liakopoulos, T. D., and Hamodrakas, S. J. (2008). Prediction of lipoprotein signal peptides in Gram-positive bacteria with a Hidden Markov Model. J. Proteome Res. 7, 5082–5093. doi: 10.1021/pr800162c
Bassler, J., Hernandez Alvarez, B., Hartmann, M. D., and Lupas, A. N. (2015). A domain dictionary of trimeric autotransporter adhesins. Int. J. Med. Microbiol. 305, 265–275. doi: 10.1016/j.ijmm.2014.12.010
Bendtsen, J., Nielsen, H., Widdick, D., Palmer, T., and Brunak, S. (2005). Prediction of twin-arginine signal peptides. BMC Bioinformatics 6:167. doi: 10.1186/1471-2105-6-167
Berven, F. S., Flikka, K., Jensen, H. B., and Eidhammer, I. (2004). BOMP: a program to predict integral beta-barrel outer membrane proteins encoded within genomes of Gram-negative bacteria. Nucleic Acids Res. 32, W394–W399. doi: 10.1093/nar/gkh351
Bladen, H. A., and Mergenhagen, S. E. (1964). Ultrastructure of Veillonella and morphological correlation of an outer membrane with particles associated with endotoxic activity. J. Bacteriol. 88, 1482–1492.
Braun, V. (1975). Covalent lipoprotein from the outer membrane of Escherichia coli. Biochim. Biophys. Acta 415, 335–377. doi: 10.1016/0304-4157(75)90013-1
Braun, V., and Rehn, K. (1969). Chemical characterization, spatial distribution and function of a lipoprotein (murein-lipoprotein) of the E. coli cell wall. The specific effect of trypsin on the membrane structure. Eur. J. Biochem. 10, 426–438. doi: 10.1111/j.1432-1033.1969.tb00707.x
Brooke, J. S., and Valvano, M. A. (1996). Biosynthesis of inner core lipopolysaccharide in enteric bacteria identification and characterization of a conserved phosphoheptose isomerase. J. Biol. Chem. 271, 3608–3614. doi: 10.1074/jbc.271.7.3608
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421
Campbell, C., Sutcliffe, I. C., and Gupta, R. S. (2014). Comparative proteome analysis of Acidaminococcus intestini supports a relationship between outer membrane biogenesis in Negativicutes and Proteobacteria. Arch. Microbiol. 196, 307–310. doi: 10.1007/s00203-014-0964-4
Campo, N., and Rudner, D. Z. (2007). SpoIVB and CtpB are both forespore signals in the activation of the sporulation transcription factor sigmaK in Bacillus subtilis. J. Bacteriol. 189, 6021–6027. doi: 10.1128/JB.00399-07
Chung, J. W., Ng-Thow-Hing, C., Budman, L. I., Gibbs, B. F., Nash, J. H. E., Jacques, M., et al. (2007). Outer membrane proteome ofActinobacillus pleuropneumoniae: LC-MS/MS analyses validate in silico predictions. Proteomics 7, 1854–1865. doi: 10.1002/pmic.200600979
Confer, A. W., and Ayalew, S. (2013). The OmpA family of proteins: roles in bacterial pathogenesis and immunity. Vet. Microbiol. 163, 207–222. doi: 10.1016/j.vetmic.2012.08.019
Cox, J., Hein, M. Y., Luber, C. A., Paron, I., Nagaraj, N., and Mann, M. (2014). Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteomics 13, 2513–2526. doi: 10.1074/mcp.M113.031591
Cox, J., and Mann, M. (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372. doi: 10.1038/nbt.1511
Davis, M. R., Goldberg, J. B., and Goldberg, J. B. (2012). Purification and visualization of lipopolysaccharide from Gram-negative bacteria by hot aqueous-phenol extraction. J. Vis. Exp. 2012:3916. doi: 10.3791/3916
Do, T., Sheehy, E. C., Mulli, T., Hughes, F., and Beighton, D. (2015). Transcriptomic analysis of three Veillonella spp. present in carious dentine and in the saliva of caries-free individuals. Front. Cell. Infect. Microbiol. 5:25. doi: 10.3389/fcimb.2015.00025
Dowdell, A. S., Murphy, M. D., Azodi, C., Swanson, S. K., Florens, L., Chen, S., et al. (2017). Comprehensive spatial analysis of the Borrelia burgdorferi lipoproteome reveals a compartmentalization bias toward the bacterial surface. J. Bacteriol. 199:e00658–16. doi: 10.1128/jb.00658-16
Erde, J., Loo, R. R. O., and Loo, J. A. (2014). Enhanced FASP (eFASP) to increase proteome coverage and sample recovery for quantitative proteomic experiments. J. Proteome Res. 13, 1885–1895. doi: 10.1021/pr4010019
Euzeby, J. (1997). List of bacterial names with standing in nomenclature: a folder available on the internet. Int. J. Syst. Bacteriol. 590–592. doi: 10.1099/00207713-47-2-590
Finn, R. D., Attwood, T. K., Babbitt, P. C., Bateman, A., Bork, P., Bridge, A. J., et al. (2017). InterPro in 2017-beyond protein family and domain annotations. Nucleic Acids Res. 45, D190–D199. doi: 10.1093/nar/gkw1107
Finn, R. D., Coggill, P., Eberhardt, R. Y., Eddy, S. R., Mistry, J., Mitchell, A. L., et al. (2015). The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 44, D279–D285. doi: 10.1093/nar/gkv1344
Forde, A., and Fitzgerald, G. F. (2003). Molecular organization of exopolysaccharide (EPS) encoding genes on the lactococcal bacteriophage adsorption blocking plasmid, pCI658. Plasmid 49, 130–142. doi: 10.1016/S0147-619X(02)00156-7
Galdiero, S., Falanga, A., Cantisani, M., Tarallo, R., Della Pepa, M. E., D'Oriano, V., et al. (2012). Microbe-host interactions: structure and role of Gram-negative bacterial porins. Curr. Protein Pept. Sci. 13, 843–854. doi: 10.2174/1389203711213080012
Gerbino, E., Carasi, P., Mobili, P., Serradell, M. A., and Gómez-Zavaglia, A. (2015). Role of S-layer proteins in bacteria. World J. Microbiol. Biotechnol. 31, 1877–1887. doi: 10.1007/s11274-015-1952-9
Griffen, A. L., Beall, C. J., Campbell, J. H., Firestone, N. D., Kumar, P. S., Yang, Z. K., et al. (2012). Distinct and complex bacterial profiles in human periodontitis and health revealed by 16S pyrosequencing. ISME J. 6, 1176–1185. doi: 10.1038/ismej.2011.191
Hasegawa, K., Linnemann, R. W., Mansbach, J. M., Ajami, N. J., Espinola, J. A., Petrosino, J. F., et al. (2016). The fecal microbiota profile and bronchiolitis in infants. Pediatrics 51:474. doi: 10.1016/j.jemermed.2016.08.033
Heinz, E., Selkrig, J., Belousoff, M., and Lithgow, T. (2015). Evolution of the Translocation and Assembly Module (TAM). Genome Biol. Evol. 7:evv097. doi: 10.1093/gbe/evv097
Hirai, J., Yamagishi, Y., Kinjo, T., Hagihara, M., Sakanashi, D., Suematsu, H., et al. (2016). Osteomyelitis caused by Veillonella species: case report and review of the literature. J. Infect. Chemother. 22, 417–420. doi: 10.1016/j.jiac.2015.12.015
Hughes, C., Andersen, R., and Kolenbrander, P. (1992). Characterization of Veillonella atypica PK1910 adhesin-mediated coaggregation with oral Streptococcus spp. Infect. Immun. 60:1178.
Imai, K., Asakawa, N., Tsuji, T., Akazawa, F., Ino, A., Sonoyama, M., et al. (2008). SOSUI-GramN: high performance prediction for sub-cellular localization of proteins in gram-negative bacteria. Bioinformation 2, 417–421. doi: 10.6026/97320630002417
Iqbal, H., Kenedy, M. R., Lybecker, M., and Akins, D. R. (2016). The TamB ortholog of Borrelia burgdorferi interacts with the β-barrel assembly machine (BAM) complex protein BamA. Mol. Microbiol. 102, 757–774. doi: 10.1111/mmi.13492
Juncker, A. S., Willenbrock, H., Von Heijne, G., Brunak, S., Nielsen, H., and Krogh, A. (2003). Prediction of lipoprotein signal peptides in Gram-negative bacteria. Protein Sci. 12, 1652–1662. doi: 10.1110/ps.0303703
Kall, L., Krogh, A., and Sonnhammer, E. L. L. (2007). Advantages of combined transmembrane topology and signal peptide prediction–the Phobius web server. Nucleic Acids Res. 35, W429–W432. doi: 10.1093/nar/gkm256
Kelley, L. A., Mezulis, S., Yates, C. M., Wass, M. N., and Sternberg, M. J. E. (2015). The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858. doi: 10.1038/nprot.2015.053
Knapp, S., Brodal, C., Peterson, J., Qi, F., Kreth, J., and Merritt, J. (2017). Natural competence is common among clinical isolates of Veillonella parvula and is useful for genetic manipulation of this key member of the oral microbiome. Front. Cell. Infect. Microbiol. 7:139. doi: 10.3389/fcimb.2017.00139
Kojima, S., Hayashi, K., Tochigi, S., Kusano, T., Kaneko, J., and Kamio, Y. (2016). Peptidoglycan-associated outer membrane protein Mep45 of rumen anaerobe Selenomonas ruminantium forms a non-specific diffusion pore via its C-terminal transmembrane domain. Biosci. Biotechnol. Biochem. 80, 1954–1959. doi: 10.1080/09168451.2016.1194185
Kojima, S., and Kamio, Y. (2012). Molecular basis for the maintenance of envelope integrity in Selenomonas ruminantium: cadaverine biosynthesis and covalent modification into the peptidoglycan play a major role. J. Nutr. Sci. Vitaminol. (Tokyo). 58, 153–160. doi: 10.3177/jnsv.58.153
Kojima, S., Kaneko, J., Abe, N., Takatsuka, Y., and Kamio, Y. (2011). Cadaverine covalently linked to the peptidoglycan serves as the correct constituent for the anchoring mechanism between the outer membrane and peptidoglycan in Selenomonas ruminantium. J. Bacteriol. 193, 2347–2350. doi: 10.1128/JB.00106-11
Krewulak, K. D., and Vogel, H. J. (2011). TonB or not TonB: is that the question? Biochem. Cell Biol. 89, 87–97. doi: 10.1139/O10-141
Krogh, A., Larsson, B., von Heijne, G., and Sonnhammer, E. L. (2001). Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305, 567–580. doi: 10.1006/jmbi.2000.4315
Marchler-Bauer, A., Derbyshire, M. K., Gonzales, N. R., Lu, S., Chitsaz, F., Geer, L. Y., et al. (2014). CDD: NCBI's conserved domain database. Nucleic Acids Res. 43, D222–D226. doi: 10.1093/nar/gku1221
Martorana, A. M., Motta, S., Di Silvestre, D., Falchi, F., Dehò, G., Mauri, P., et al. (2014). Dissecting Escherichia coli outer membrane biogenesis using differential proteomics. PLoS ONE 9:e100941. doi: 10.1371/journal.pone.0100941
Mi, H., Poudel, S., Muruganujan, A., Casagrande, J. T., and Thomas, P. D. (2015). PANTHER version 10: expanded protein families and functions, and analysis tools. Nucleic Acids Res. 44, D336–D342. doi: 10.1093/nar/gkv1194
Mistry, J., Finn, R. D., Eddy, S. R., Bateman, A., and Punta, M. (2013). Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 41:e121. doi: 10.1093/nar/gkt263
Orfanoudaki, G., and Economou, A. (2014). Proteome-wide subcellular topologies of E. coli polypeptides database (STEPdb). Mol. Cell. Proteomics 13, 3674–3687. doi: 10.1074/mcp.O114.041137
Pelicic, V. (2008). Type IV pili: e pluribus unum? Mol. Microbiol. 68, 827–837. doi: 10.1111/j.1365-2958.2008.06197.x
Pocsfalvi, G., Stanly, C., Vilasi, A., Fiume, I., Capasso, G., Turiák, L., et al. (2016). Mass spectrometry of extracellular vesicles. Mass Spectrom. Rev. 35, 3–21. doi: 10.1002/mas.21457
Pustelny, C., Komor, U., Pawar, V., Lorenz, A., Bielecka, A., Moter, A., et al. (2015). Contribution of Veillonella parvula to Pseudomonas aeruginosa-mediated pathogenicity in a murine tumor model system. Infect. Immun. 83:417. doi: 10.1128/IAI.02234-14
Rosen, R., Amirault, J., Liu, H., Mitchell, P., Hu, L., Khatwa, U., et al. (2014). Changes in gastric and lung microflora with acid suppression. JAMA Pediatr. 168:932. doi: 10.1001/jamapediatrics.2014.696
Sara, M., and Sleytr, U. B. (2000). S-layer proteins. J. Bacteriol. 182, 859–868. doi: 10.1128/JB.182.4.859-868.2000
Schwanhäusser, B., Busse, D., Li, N., Dittmar, G., Schuchhardt, J., Wolf, J., et al. (2011). Global quantification of mammalian gene expression control. Nature 473, 337–342. doi: 10.1038/nature10098
Selkrig, J., Leyton, D. L., Webb, C. T., and Lithgow, T. (2014). Assembly of β-barrel proteins into bacterial outer membranes. Biochim. Biophys. Acta 1843, 1542–1550. doi: 10.1016/j.bbamcr.2013.10.009
Selkrig, J., Mosbahi, K., Webb, C. T., Belousoff, M. J., Perry, A. J., Wells, T. J., et al. (2012). Discovery of an archetypal protein transport system in bacterial outer membranes. Nat. Struct. Mol. Biol. 19, 506–510. doi: 10.1038/nsmb.2261
Sha, J., Fadl, A. A., Klimpel, G. R., Niesel, D. W., Popov, V. L., and Chopra, A. K. (2004). The two murein lipoproteins of Salmonella enterica serovar Typhimurium contribute to the virulence of the organism. Infect. Immun. 72, 3987–4003. doi: 10.1128/IAI.72.7.3987-4003.2004
Strati, F., Cavalieri, D., Albanese, D., De Felice, C., Donati, C., Hayek, J., et al. (2017). New evidences on the altered gut microbiota in autism spectrum disorders. Microbiome 5:24. doi: 10.1186/s40168-017-0242-1
Sutcliffe, I. C. (2010). A phylum level perspective on bacterial cell envelope architecture. Trends Microbiol. 18, 464–470. doi: 10.1016/j.tim.2010.06.005
Szijarto, V., Lukasiewicz, J., Gozdziewicz, T. K., Magyarics, Z., Nagy, E., and Nagy, G. (2014). Diagnostic potential of monoclonal antibodies specific to the unique O-antigen of multidrug-resistant epidemic Escherichia coli clone ST131-O25b:H4. Clin. Vaccine Immunol. 21, 930–939. doi: 10.1128/CVI.00685-13
Thein, M., Sauer, G., Paramasivam, N., Grin, I., and Linke, D. (2010). Efficient subfractionation of gram-negative bacteria for proteomics studies. J. Proteome Res. 9, 6135–6147. doi: 10.1021/pr1002438
Thome, B. M., and Müller, M. (1991). Skp is a periplasmic Escherichia coli protein requiring SecA and SecY for export. Mol. Microbiol. 5, 2815–2821. doi: 10.1111/j.1365-2958.1991.tb01990.x
Tocheva, E. I., Matson, E. G., Morris, D. M., Moussavi, F., Leadbetter, J. R., and Jensen, G. J. (2011). Peptidoglycan remodeling and conversion of an inner membrane into an outer membrane during sporulation. Cell 146, 799–812. doi: 10.1016/j.cell.2011.07.029
Tusher, V. G., Tibshirani, R., and Chu, G. (2001). Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. U.S.A. 98, 5116–5121. doi: 10.1073/pnas.091062498
Tyanova, S., Temu, T., and Cox, J. (2016a). The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 11, 2301–2319. doi: 10.1038/nprot.2016.136
Tyanova, S., Temu, T., Sinitcyn, P., Carlson, A., Hein, M. Y., Geiger, T., et al. (2016b). The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740. doi: 10.1038/nmeth.3901
van Ulsen, P., Rahman, S., Jong, W. S. P., Daleke-Schermerhorn, M. H., and Luirink, J. (2014). Type V secretion: from biogenesis to biotechnology. Biochim. Biophys. Acta 1843, 1592–1611. doi: 10.1016/j.bbamcr.2013.11.006
Veillon, A., and Zuber, A. (1898). Sur Quelques microbes strictment anaerobies et leur role en pathologie. Arch. Med. Exp. 517–545.
Villa, R., Martorana, A. M., Okuda, S., Gourlay, L. J., Nardini, M., Sperandeo, P., et al. (2013). The Escherichia coli Lpt transenvelope protein complex for lipopolysaccharide export is assembled via conserved structurally homologous domains. J. Bacteriol. 195, 1100–1108. doi: 10.1128/JB.02057-12
Vizcaíno, J. A., Csordas, A., del-Toro, N., Dianes, J. A., Griss, J., Lavidas, I., et al. (2016). 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 44, D447–D456. doi: 10.1093/nar/gkw880
Voss, B., Gaddy, J., McDonald, W., and Cover, T. (2014). Analysis of surface-exposed outer membrane proteins in Helicobacter pylori. J. Bacteriol. 196, 2455–2471. doi: 10.1128/JB.01768-14
Webb, C. T., Heinz, E., and Lithgow, T. (2012). Evolution of the ß-barrel assembly machinery. Trends Microbiol. 20, 612–620. doi: 10.1016/j.tim.2012.08.006
Whitman, W. B. (2011). “Veillonellaceae” in Bergey's Manual of Systematic Bacteriology, Vol. 3, The Firmicutes, 2nd Edn., eds. D. Vos, G. Garrity, D. Jones, N. Krieg, W. Ludwig, F. A. Rainey, K.-H. Schleifer, and W. Whitman (New York, NY: Springer), 1059–1119.
Wilm, M., Shevchenko, A., Houthaeve, T., Breit, S., Schweigerer, L., Fotsis, T., et al. (1996). Femtomole sequencing of proteins from polyacrylamide gels by nano-electrospray mass spectrometry. Nature 379, 466–469. doi: 10.1038/379466a0
Wilson, M. M., Anderson, D. E., Bernstein, H. D., McInnes, P., and Wang, L. (2015). Analysis of the outer membrane proteome and secretome of Bacteroides fragilis reveals a multiplicity of secretion mechanisms. PLoS ONE 10:e0117732. doi: 10.1371/journal.pone.0117732
Yeats, C., Rawlings, N. D., and Bateman, A. (2004). The PepSY domain: a regulator of peptidase activity in the microbial environment? Trends Biochem. Sci. 29, 169–172. doi: 10.1016/j.tibs.2004.02.004
Yu, C. S., Lin, C. J., and Hwang, J. K. (2004). Predicting subcellular localization of proteins for Gram-negative bacteria by support vector machines based on n-peptide compositions. Protein Sci. 13, 1402–1406. doi: 10.1110/ps.03479604
Yu, N. Y., Wagner, J. R., Laird, M. R., Melli, G., Rey, S., Lo, R., et al. (2010). PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics 26, 1608–1615. doi: 10.1093/bioinformatics/btq249
Zeikus, J. G., Hegge, P. W., Thompson, T. E., Phelps, T. J., and Langworthy, T. A. (1983). Isolation and description of Haloanaerobium praevalens gen. nov. and sp. nov., an obligately anaerobic halophile common to Great Salt Lake sediments. Curr. Microbiol. 9, 225–233. doi: 10.1007/BF01567586
Zgurskaya, H. I., Krishnamoorthy, G., Ntreh, A., and Lu, S. (2011). Mechanism and function of the outer membrane channel TolC in multidrug resistance and physiology of enterobacteria. Front. Microbiol. 2:189. doi: 10.3389/fmicb.2011.00189
Zhang, Y., Shi, C., Yu, J., Ren, J., and Sun, D. (2012). RpoS regulates a novel type of plasmid DNA transfer in Escherichia coli. PLoS ONE 7:e33514. doi: 10.1371/journal.pone.0033514
Zhou, P., Liu, J., Merritt, J., and Qi, F. (2015). A YadA-like autotransporter, Hag1 in Veillonella atypica is a multivalent hemagglutinin involved in adherence to oral streptococci, Porphyromonas gingivalis, and human oral buccal cells. Mol. Oral Microbiol. 30, 269–279. doi: 10.1111/omi.12091
Keywords: Negativicutes, proteomics, BAM/TAM complex, adhesins, OmpM, LPS, Lpt system, O-antigen
Citation: Poppleton DI, Duchateau M, Hourdel V, Matondo M, Flechsler J, Klingl A, Beloin C and Gribaldo S (2017) Outer Membrane Proteome of Veillonella parvula: A Diderm Firmicute of the Human Microbiome. Front. Microbiol. 8:1215. doi: 10.3389/fmicb.2017.01215
Received: 17 March 2017; Accepted: 14 June 2017;
Published: 30 June 2017.
Edited by:
Mickael Desvaux, Institut National de la Recherche Agronomique (INRA), FranceReviewed by:
Iain Sutcliffe, Northumbria University, United KingdomJean Armengaud, Commissariat à l'Energie Atomique et aux Energies Alternatives (CEA), France
Copyright © 2017 Poppleton, Duchateau, Hourdel, Matondo, Flechsler, Klingl, Beloin and Gribaldo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christophe Beloin, Y2hyaXN0b3BoZS5iZWxvaW5AcGFzdGV1ci5mcg==
Simonetta Gribaldo, c2ltb25ldHRhLmdyaWJhbGRvQHBhc3RldXIuZnI=