 Wycliff M. Kinoti
Wycliff M. Kinoti Fiona E. Constable1
Fiona E. Constable1 Kim M. Plummer
Kim M. Plummer- 1Biosciences Research Division, AgriBio, La Trobe University, Melbourne, VIC, Australia
- 2AgriBio, School of Applied Systems Biology, La Trobe University, Melbourne, VIC, Australia
- 3Department of Animal, Plant and Soil Sciences, AgriBio, La Trobe University, Melbourne, VIC, Australia
The distribution of Ilarvirus species populations amongst 61 Australian Prunus trees was determined by next generation sequencing (NGS) of amplicons generated using a genus-based generic RT-PCR targeting a conserved region of the Ilarvirus RNA2 component that encodes the RNA dependent RNA polymerase (RdRp) gene. Presence of Ilarvirus sequences in each positive sample was further validated by Sanger sequencing of cloned amplicons of regions of each of RNA1, RNA2 and/or RNA3 that were generated by species specific PCRs and by metagenomic NGS. Prunus necrotic ringspot virus (PNRSV) was the most frequently detected Ilarvirus, occurring in 48 of the 61 Ilarvirus-positive trees and Prune dwarf virus (PDV) and Apple mosaic virus (ApMV) were detected in three trees and one tree, respectively. American plum line pattern virus (APLPV) was detected in three trees and represents the first report of APLPV detection in Australia. Two novel and distinct groups of Ilarvirus-like RNA2 amplicon sequences were also identified in several trees by the generic amplicon NGS approach. The high read depth from the amplicon NGS of the generic PCR products allowed the detection of distinct RNA2 RdRp sequence variant populations of PNRSV, PDV, ApMV, APLPV and the two novel Ilarvirus-like sequences. Mixed infections of ilarviruses were also detected in seven Prunus trees. Sanger sequencing of specific RNA1, RNA2, and/or RNA3 genome segments of each virus and total nucleic acid metagenomics NGS confirmed the presence of PNRSV, PDV, ApMV and APLPV detected by RNA2 generic amplicon NGS. However, the two novel groups of Ilarvirus-like RNA2 amplicon sequences detected by the generic amplicon NGS could not be associated to the presence of sequence from RNA1 or RNA3 genome segments or full Ilarvirus genomes, and their origin is unclear. This work highlights the sensitivity of genus-specific amplicon NGS in detection of virus sequences and their distinct populations in multiple samples, and the need for a standardized approach to accurately determine what constitutes an active, viable virus infection after detection by molecular based methods.
Introduction
The genus Ilarvirus is the largest in the Bromoviridae family, includes more than 20 recognized and tentative Ilarvirus species which are divided into six subgroups, and are characterized by a positive-sense, single-stranded tripartite RNA genome (Bujarski et al., 2012). Genomic RNA1 and RNA2 of all Ilarvirus species harbor genes that encode conserved proteins involved in viral replication. Genes on RNA3 encodes a movement protein (MP) and a coat protein (CP), which is expressed via sub-genomic RNA4 (Codoner and Elena, 2008; Pallas et al., 2012a). Many ilarviruses are transmitted by seed and pollen, and all are transmitted by vegetative propagation. Several Ilarvirus species also infect a wide plant host range within the family Rosaceae, including Prunus species, and can cause diseases of economic importance (Card et al., 2007; Pallas et al., 2012b).
Specific reverse transcription-polymerase chain reaction (RT-PCR) tests have been used widely for routine diagnosis of individual Ilarvirus species (Parakh et al., 1994; MacKenzie et al., 1997; Pallás et al., 1998; Scott et al., 2003). However, the infection of some plant hosts by several Ilarvirus species complicates species identification and necessitates the use of several different specific RT-PCR tests for detection. Furthermore, sequence diversity exists within Ilarvirus species (Kinoti et al., 2017), which can make the design of specific primers difficult and impact their detection by RT-PCR tests.
Broad spectrum degenerate primers based on conserved sequences within virus taxonomic groups such as family, genera and species, offer an alternative to the use of multiple species-specific RT-PCR tests for simultaneous detection of related viruses and unknown virus species (Compton, 1990; James et al., 2006; Maliogka et al., 2007). Sanger sequencing of the cloned generic PCR amplicons can then be used to identify the diversity of these virus groups. However, this approach to study virus diversity is usually limited to the sequencing of a few clones due to time, labor and cost constraints (Beerenwinkel and Zagordi, 2011). This limitation would hamper the detection of some low titers virus species and/or strains present in mixed infection when compared to higher titre virus species and/or strains.
NGS enable parallel sequencing of DNA from multiple samples at very high-throughput and at a high degree of sequence coverage generating large amounts of sequence data compared to Sanger sequencing of cloned PCR amplicons (Adams et al., 2009; Wu et al., 2015). However, most applications of NGS in plant virology are designed for virus discovery and full genome sequencing which frequently results only in consensus sequences of high occurring virus sequence variants and/or virus strains genomes (Adams et al., 2009; Radford et al., 2012; Prabha et al., 2013). In addition, most sample preparation methods lead to high background levels of host sequences compared to virus sequences associated during NGS (Marston et al., 2013; Hall et al., 2014). The consensus sequence and high levels of non-viral sequence associated with NGS offers limited resolution of low occurring sequence variant and/or strains within a virus isolate and also limits the number of samples that can sequenced in a single NGS run.
Next generation amplicon sequencing offers an alternative approach of an in-depth estimation of virus diversity and has been previously used to study the diversity of the Ilarvirus Prunus necrotic ringspot virus (PNRSV), within Prunus trees (Kinoti et al., 2017). An advantage of this approach is that it enables sequencing of virus amplicons from a high number of plant samples in a single run and can detect populations of both high and low-titre viruses within each sample (Wang et al., 2007; Eriksson et al., 2008). Until now, virus amplicon NGS has only been used to study population diversity within specific virus species (Eriksson et al., 2008; Beerenwinkel and Zagordi, 2011; Mancuso et al., 2011; Kinoti et al., 2017), unlike in bacteria where 16S ribosomal RNA gene amplicons are regularly deep sequenced to identify the diversity of bacterial species within an environmental sample (Sogin et al., 2006; Sanschagrin and Yergeau, 2014).
In this study, the novel approach of deep sequencing of genus-specific RT-PCR amplicons from a conserved region of the RNA2 encoded RNA-dependent RNA polymerase (RdRp) gene of Ilarvirus species was applied to 61 Ilarvirus-positive Prunus tree samples. Sequence analysis of resulting generic amplicon NGS data was used to determine the diversity of Ilarvirus species and strains infecting Prunus species in Australia.
Materials and Methods
Sample Extraction
Leaf tissue from 105 Prunus trees were collected in spring (2014–15) from five states of Australia (Table 1). Symptoms characteristic of virus infections were not noted at the time of sample collection. Total RNA was extracted from 0.3 g leaf tissue of each sample using the RNeasy® Plant Mini Kit (Qiagen) with a modified lysis buffer (MacKenzie et al., 1997).
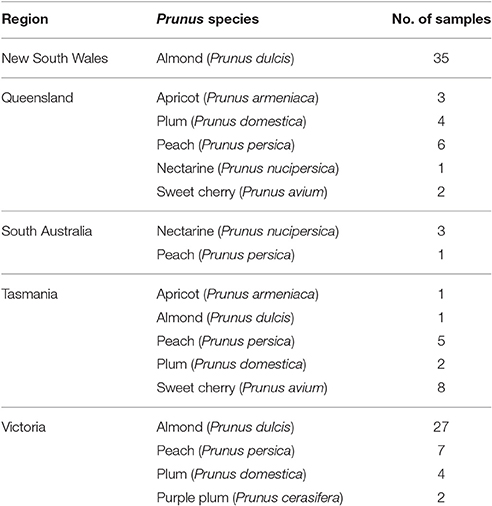
Table 1. The number and Australian region of origin of Prunus samples used in this study.
PCR Amplification
A 371 nucleotide (nt) region of the RNA2-encoded RNA-dependent RNA polymerase (RdRp) gene of ilarviruses was amplified from each sample using a previously published generic ramped annealing RT-PCR and nested PCR using Ilarvirus genus-specific degenerate primers (Maliogka et al., 2007). The RT-PCR was carried out using SuperScript® III One-Step RT-PCR System with Platinum® Taq High Fidelity (Invitrogen) for amplification of a 381 nt segment of the RdRp gene according to the manufacturer's instructions. The nested PCR reaction was performed using 1 μL of the first RT-PCR product with Platinum® Taq DNA Polymerase High Fidelity for amplification of a 371 nt segment of the RdRp gene according to the manufacturer's instructions. The PCR products were visualized by electrophoresis in 1.5% agarose gels stained with SYBR® Safe DNA gel stain (Invitrogen).
Amplicon NGS Library Preparation
The Ilarvirus RNA2 RdRp amplicons were gel-purified using the Wizard® PCR clean-up kit (Promega) according to the manufacturer's instructions. Amplicon libraries were prepared and sequenced using the Illumina MiSeq as described previously (Kinoti et al., 2017). Briefly, in-house dual indexing adapter mpxPE2 consisting of unique barcodes for each amplicon were ligated to the 3′-terminus to incorporate the sequencing primer site, while an adaptor containing one of eight indexing sequences 5 bp in length (mpxPE1), was ligated to the 5′-terminus of each of the amplicons using the NEBNext® T4 ligase (New England BioLabs). PCR enrichment was carried out using an in-house multiplex PE barcode primer mix (2.5 μM) which were unique for each of the amplicons and Phusion® high fidelity PCR mastermix (New England BioLabs). PCR cycling conditions consisted of: one cycle at 98°C for 30 s, 15 cycles at 98°C for 10 s, 65°C for 30 s, 72°C for 30 s; and a final extension step at 72°C for 5 min.
After the PCR, excess primers and any primer dimer present were removed using Ampure XP® system (Beckman Coulter) according to the manufacturer's instructions. The size distribution and concentration of the amplicon libraries were determined using the 2200 TapeStation® system (Agilent technologies) and Qubit® Fluorometer 2.0 (Invitrogen), respectively, and the resulting quantification values were used to pool the amplicon libraries at equal concentration. The amplicon library was sequenced using the Illumina MiSeq with a paired read length of 301 base pairs. The amplicon sequence read data for this study have been submitted to the NCBI Sequence Read Archive (SRA) database under the Bioproject accession PRJNA380730 and SRA study accession SRP102631.
Amplicon Sequence Reads Analysis
The generated raw amplicon sequence reads were quality trimmed with a quality score of >20 and minimum read length of 200 nt using Trim Galore! (version 0.4.0) (Krueger, 2012) and paired using PEAR (version 0.9.4), with default parameters (Zhang et al., 2014). Cutadapt (version 1.4.1) (Martin, 2011) was used to trim each sample amplicon read to a similar size of 350 bp and the resulting amplicon sequence reads clustered/merged at 100% identity into distinct sequence variants clusters using Usearch (version 7.0.1090) (Edgar, 2010). To filter off any potential RT-PCR and sequencing error associated reads, sequence variants comprising of <10 individual reads were removed from the amplicon NGS dataset (Kinoti et al., 2017). Additionally, non-coding sequence variants were also removed because the generic-amplicons were each derived from Ilarvirus RdRp gene region.
In order to identify the Ilarvirus species that were detected by the Ilarvirus genus-specific PCR, the sequence variants were searched against published Ilarvirus RNA2 type isolate reference sequences in a local database (Table 2) using BLASTn with default parameters. Only sequence variants BLASTn matches with the highest identity and the highest bit score, were retained for sequence variants that had BLASTn matches to multiple Ilarvirus species.
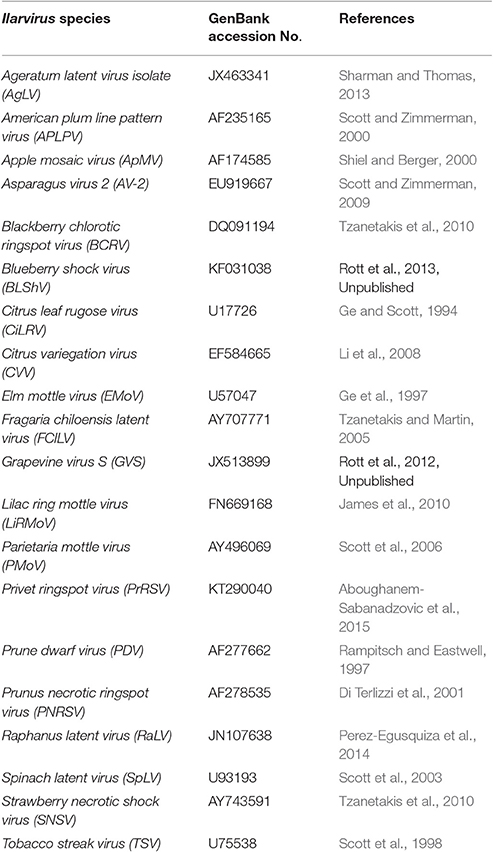
Table 2. Ilarvirus RNA2 type isolate reference sequences to which the generic amplicon next generation sequencing data were mapped using BLASTn.
All Ilarvirus species variant sequences that were identified by the BLASTn analysis were pooled and aligned with all available RNA2 sequences (Table S3) for each Ilarvirus species available on GenBank, which were trimmed according to the corresponding RdRp RNA2 region amplified by the genus-specific degenerate primers using Muscle (version 3.8.31) (Edgar, 2004). A neighbor-joining phylogenetic tree was constructed using Phylip version 3.6 (Felsenstein, 2005). The resulting trees were visualized in FigTree version 1.4.2 (Andrew, 2009) and branches that had <90% bootstrap support were collapsed. Sequence pairwise identity comparison analysis was then carried out using the sequence demarcation tool (SDT) (version 1.2) (Muhire et al., 2014) on the aligned amplicon sequence variants of each sample.
Sanger Sequencing and Metagenomics NGS of Selected Samples for Validation of Ilarvirus Species Detected by the RNA2-Generic Amplicon NGS
To confirm the presence of the Ilarvirus species sequences detected by the generic amplicon NGS analysis, amplicons of ten samples selected were cloned using the pGEM®-T Easy vector system (Promega). Five clones of each of the 10 samples were sequenced using the SP6 and T7 promoter primers and ABI BigDye Terminator Version 3.1 kit on an AB3730xl sequencing machine (Applied Biosystems). The resulting sequences were subjected to a BLASTn search of NCBI database with default parameters (Altschul et al., 1997).
Specific PCR primers were designed to detect the same region of the RdRp gene amplified using the generic Ilarvirus nested PCR for each of the viruses that were identified by NGS (Table 3). Previously published and new PCR primer pairs were also used for RT-PCR amplification of either RNA1 and/or RNA3 segment of each Ilarvirus species/subgroup detected by the generic amplicon NGS (Table 3). RT-PCR was carried using the SuperScript™ III One-Step RT-PCR System (Invitrogen) as previously described with the appropriate temperature for each primer pair (Table 3). The RT-PCR amplicons were cloned, Sanger sequenced and the resulting sequences subjected to a BLASTn search as previously described.
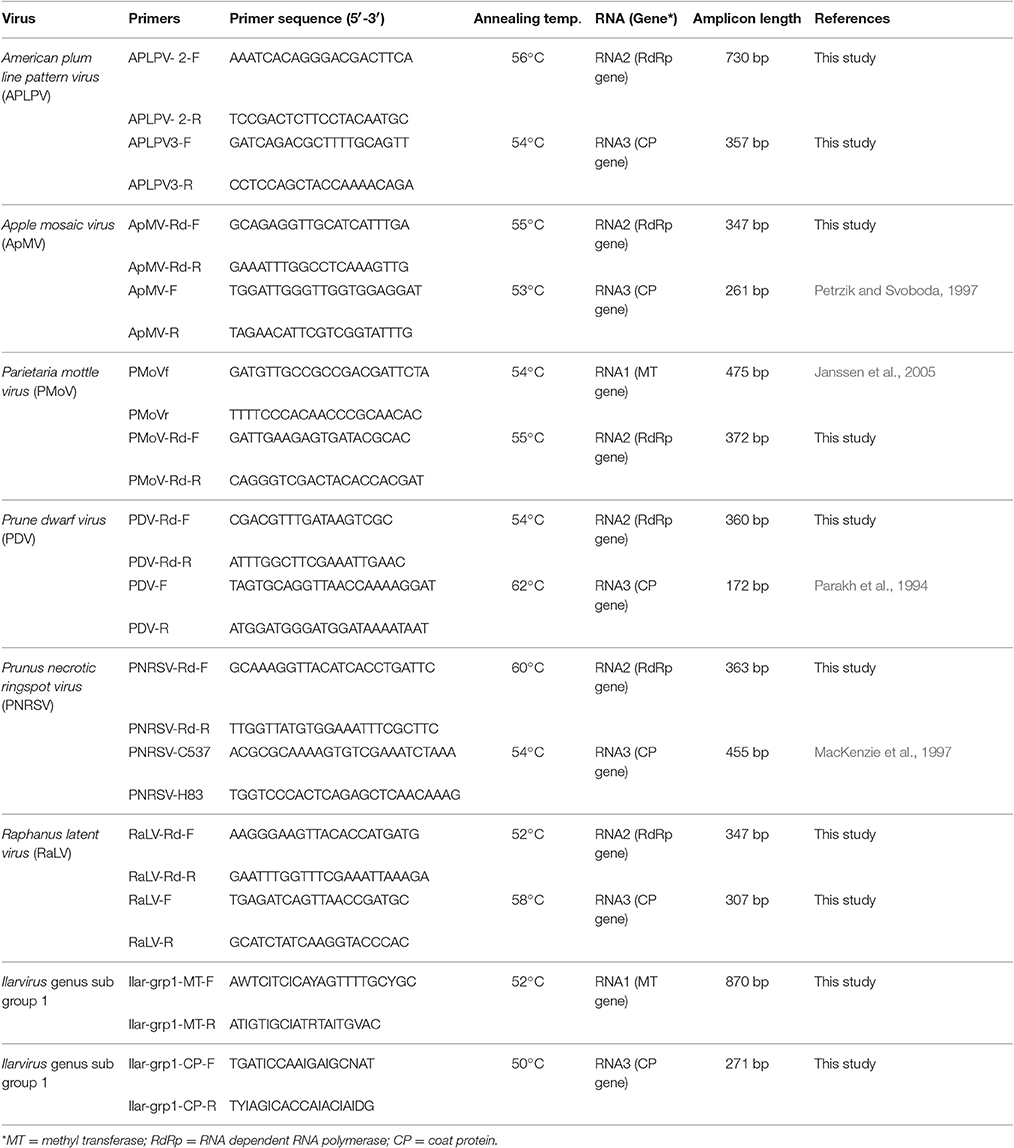
Table 3. Specific and degenerate primers used for PCR amplification of either RNA1, RNA2 and/or RNA3 segment of each Ilarvirus species.
To sequence the full genome of the ilarviruses that were detected by generic amplicon NGS, 5 μl aliquots of total RNA extract from each of the 10 selected samples were used to prepare 10 metagenomic NGS libraries using NEBNext® Ultra™ RNA Library Prep Kit (New England BioLabs) following the manufacturer's instructions. The 10 libraries were sequenced using the Illumina MiSeq with a paired read length of 2 × 300 bp. De novo assembly of the resulting sequence reads into contigs using CLC Genomics Workbench and BLASTn analysis of the assembled contigs was carried out. The assembled virus contig sequences were submitted to GenBank and the accession number of each virus RNA sequences is included as supplementary information (Table S5).
Results
Ilarvirus RNA2 (RdRp) Generic Amplicon NGS Data and BLASTn Analyses
In total, amplicons of the expected size were obtained for 61 out of 105 samples tested with the Ilarvirus genus RNA2-specific nested PCR. A total of 10,381,728 total raw reads were generated following NGS of the 61 amplicon samples. After quality trimming, there were a total of 8,083,442 reads used for analysis, with an average of 132,515 reads per sample (Table S1). The quality trimmed amplicon reads were clustered at 100% identity, which resulted in an average of 33,819 sequence variants clusters per sample. Filtering/removal of singletons and sequence variant clusters comprising of <10 reads resulted in averages of 66,532 reads and 554 sequence variants per amplicon samples (Table S1).
BLASTn analyses of the clustered RNA2 amplicon sequence variants revealed the presence of four Ilarvirus species (PNRSV, PDV, APLPV, and ApMV) that have been previously reported in Prunus species and two potentially new Ilarvirus species amongst the 61 Prunus samples (Table 4; Table S2). PNRSV was the most frequently detected Ilarvirus, occurring in 48 of the 61 Ilarvirus RNA2-positive samples (Table 4; Table S2). PDV and APLPV were detected in three samples and ApMV was detected in one sample (Table 4; Table S2).

Table 4. Generic Ilarvirus species amplicon sequences detection summary.
RNA2 sequence variants of a putative Ilarvirus species that was detected in 13 peach tree samples of the 61 Prunus species samples had a most similar BLASTn match, with a sequence identity range of 77–82% and sequence query coverage of 81–87%, to the RNA2 of Parietaria mottle virus (PMoV) type isolate (GenBank accession AY496069) (Table 4; Table S2). Due to the low BLASTn identity and coverage to the RNA2 of the PMoV type isolate, the putative Ilarvirus sequences detected in these 13 samples have been tentatively named Ilarvirus-S1. Similarly, RNA2 sequences which had 92–98% identity and coverage of 90–96%, with Raphanus latent virus (RaLV) type isolate (GenBank accession JN107638), were detected in one cherry sample (Table 4; Table S2) and this putative Ilarvirus was tentatively named Ilarvirus-S2.
Mixed infections of PNRSV and APLPV was detected in three Prunus samples and one sample had a mixed infection of PNRSV and PDV. Ilarvirus-S2 which was only detected in one sample, also occurred in a mixed infection with Ilarvirus-S1. In samples with mixed Ilarvirus infections, one Ilarvirus species always occurred at a higher frequency, based on read numbers, compared to the other Ilarvirus species that were present in the sample (Table S2).
Phylogenetic and Sequence Identity Analysis
Phylogenetic analysis of the Ilarvirus species sequences detected in the 61 samples and GenBank isolate sequences, revealed that the Australian ApMV and APLPV sequence variants each formed one phylogenetic group and PNRSV, PDV, Ilarvirus-S1 and Ilarvirus-S2 sequence variants each formed two distinct phylogenetic groups. All phylogenetic groups had >90% bootstrap support which was used, along with a ≥96% identity criterion, as the minimum support required to define a distinct Ilarvirus phylogenetic group (Figure 1; Table 5). Phylogroups from Ilarvirus-S2 and APLPV phylogroup 1 were the only groups that included sequences from both GenBank and this study (Figure 1).
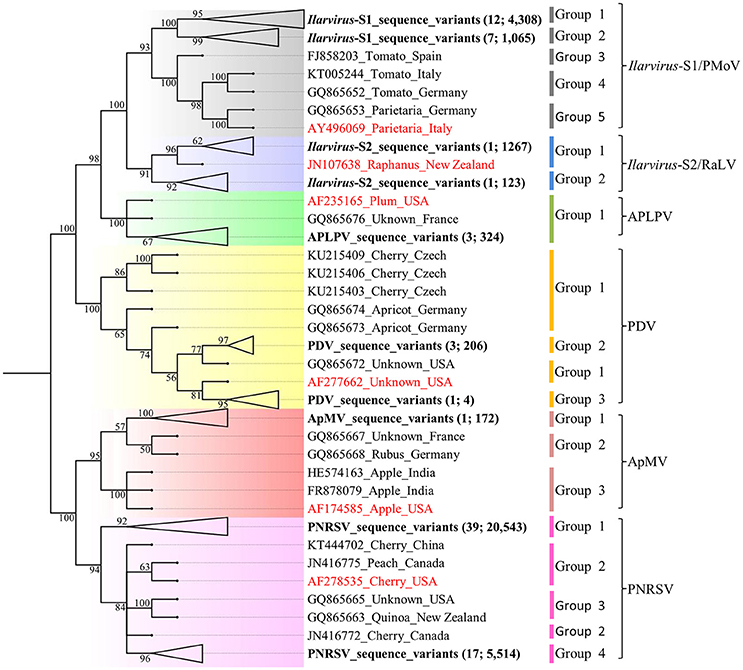
Figure 1. Neighbor-joining phylogenetic relationship of 324 American plum line pattern virus (APLPV), 172 Apple mosaic virus (ApMV), 210 Prune dwarf virus (PDV), 26,057 Prunus necrotic ringspot virus (PNRSV), 5,634 Ilarvirus-S1 and 1,390 Ilarvirus-S2 pooled sequence variants and the corresponding GenBank sequences of isolates of each virus (Table S3). The phylogenetic tree was constructed using Phylip version 3.6 with 1,000 bootstrap replicates and branches with <50% bootstrap support were collapsed. The branch position of the sequence variants of each Ilarvirus species from this study are in bold letters, the number of samples and variants are indicated in brackets and their branches collapsed for ease of presentation (Table 5). Each of the Ilarvirus type species is indicated in red font.
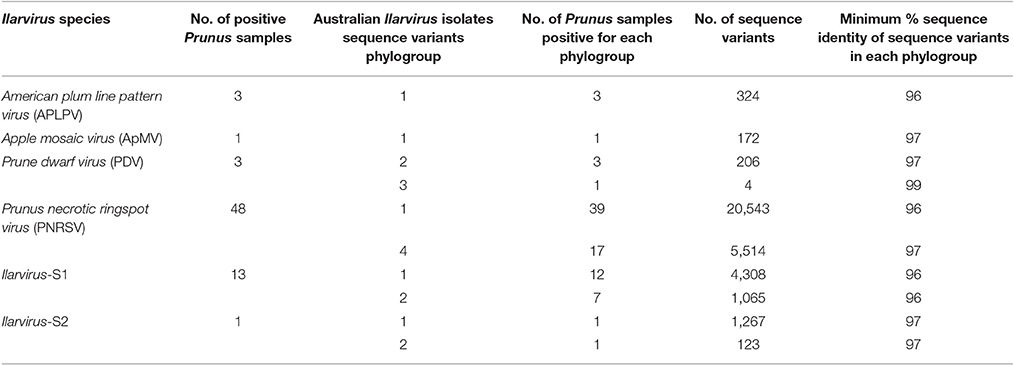
Table 5. The RNA2 phylogroups identified from phylogenetic analysis of pooled generic amplicon variant sequences from Ilarvirus species that were detected in 61 Prunus samples and the minimum percentage (%) sequence identity of sequence variants within each phylogroup.
PNRSV, PDV, Ilarvirus-S1 and Ilarvirus-S2 each had one major phylogroup that was represented by more sequence variants and occurred in more of the plant samples compared to the other minor phylogroups for each virus (Figure 1; Table 5; Table S4). Seven of the 39 PNRSV-positive plant samples, one of the three PDV positive plant samples and seven of the 13 Ilarvirus-S1-positive plant samples had sequence variants occurring in two phylogenetic groups (Table S4). Ilarvirus-S2 was detected in a single cherry sample and its sequence variants diverged into two phylogroups (Figure 1; Table S4).
SDT identity analysis of amplicon sequence variants showed similar groupings as observed in the phylogenetic analysis (Figure S1). The sequence variants within each Ilarvirus species phylogroup had an identity of ≥96% (Table 5). However, in most samples, Ilarvirus sequence variants within a phylogroup had a higher percentage identity than the specified identity cut-off (Table S4). The distribution of pairwise identity between Ilarvirus sequence variants ranged between 90 and 99% for PNRSV, 94–99% for PDV, 97–99% for ApMV, and 96–99% for APLPV. Ilarvirus-S1 sequence variants had the widest distribution of pairwise identity with a range of 89–99% whereas the identity of Ilarvirus-S2 sequence variants ranged between 92 and 99% (Figure S1).
Sanger Sequencing and Metagenomics NGS of Selected Samples for Validation of Ilarvirus Species Detected by the RNA2-Generic Amplicon NGS
Sanger sequencing of the cloned Ilarvirus RNA2 generic amplicons confirmed the presence of some of the Ilarvirus species detected by the generic amplicon NGS in each of the ten samples. However, the PNRSV and PDV sequences in sample Pch4 and Ilarvirus-S1 and PNRSV sequences in sample Q15 that were identified by the Ilarvirus generic amplicon NGS, were not detected by cloning and sequencing of the Ilarvirus generic amplicon (Table 6).
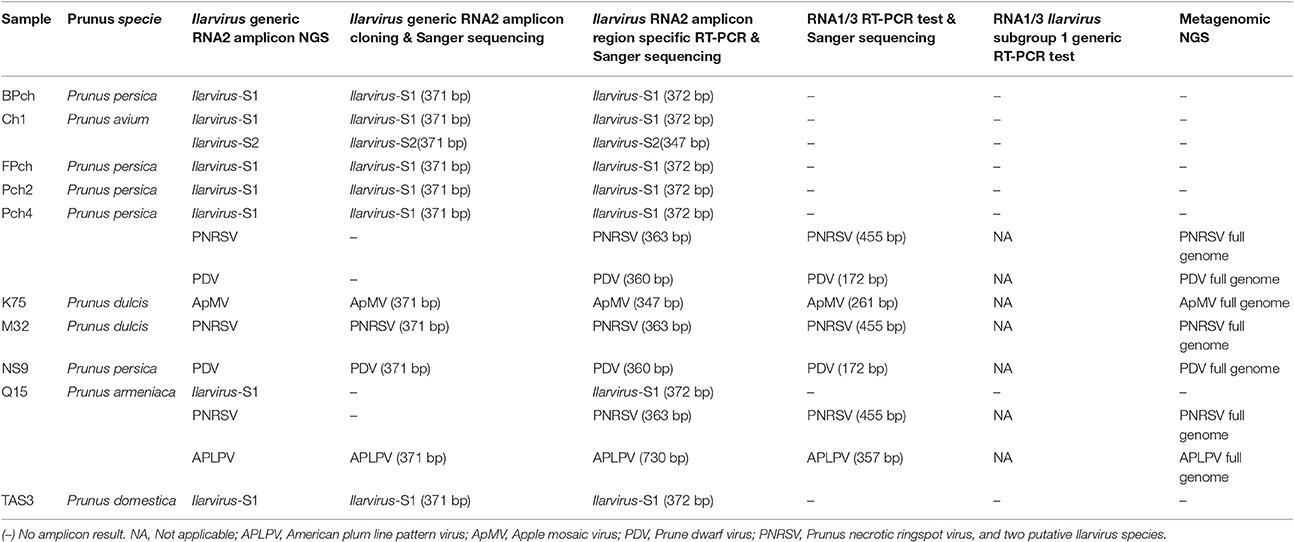
Table 6. The viruses that were detected in each sample by amplicon NGS, cloning and sanger sequencing of Ilarvirus generic or virus specific PCRs and by metagenomics NGS in the ten Prunus samples that were analyzed further for the presence of ilarviruses.
PNRSV, PDV, ApMV, APLPV, Ilarvirus-S1 and Ilarvirus-S2, were also detected using the specific RT-PCR tests. These tests were designed to detect the same RNA2 RdRp region as the generic Ilarvirus test in each of the samples in which they were detected by generic RNA2 amplicon NGS, and direct Sanger sequencing confirmed their identity (Table 6). Specific RT-PCR tests of the RNA1 and/or the RNA3 segment also detected PNRSV, PDV, ApMV and APLPV in each sample, in which these viruses had been detected by generic RNA2 amplicon NGS, and direct Sanger sequencing confirmed their identity (Table 6). However, Ilarvirus-S1 and Ilarvirus-S2 were not detected by the specific RT-PCR for PMoV RNA1 and RaLV RNA3, respectively (Table 6). Additionally, degenerate primers that were designed in this study to detect RNA1 and RNA 3 of subgroup 1 Ilarvirus species did not detect Ilarvirus sequences in any of the samples in which Ilarvirus-S1 and Ilarvirus-S2 RNA2 sequences were detected by generic amplicon NGS (Table 6).
The total raw reads obtained from the 10 plant samples by metagenomic NGS ranged from 1,192,869–5,489,161 and these numbers were reduced to 1,183,552–5,417,537 reads after quality trimming (Table S5). De novo assembly of reads from each individual plant sample resulted in 2,512–12,287 contigs across the 10 samples (Table S5). A BLASTn search of the GenBank database (Altschul et al., 1997) revealed contigs covering full genomes of PNRSV, PDV, ApMV and APLPV in samples in which they were detected by generic amplicon NGS. Ilarvirus-S1 and Ilarvirus-S2 RNA1, RNA2 and/or RNA3 sequences were not detected by metagenomic NGS in any of the ten plant samples despite the detection of their sequences by Ilarvirus RNA2 generic amplicon NGS (Table 6; Table S5).
Discussion
NGS of Ilarvirus genus-specific PCR amplicons was used in this study to identify the diversity of Ilarvirus species detected in 61 Prunus trees in Australia. PNRSV, PDV, and ApMV which are known to infect Australian Prunus tree species, and APLPV, which has not been previously reported in Australia, were detected by this approach. Two novel and distinct groups of Ilarvirus-like RNA2 amplicon sequences were also identified in several trees by the generic amplicon NGS approach and these Ilarvirus-like sequences were tentatively named Ilarvirus-S1 and Ilarvirus-S2. The generic amplicon NGS approach used in this study was also sensitive enough to identify mixed infections of PNRSV and APLPV or PNRSV and PDV in four Prunus samples.
PNRSV was the most frequently detected Ilarvirus, occurring in 48 of the 61 Ilarvirus positive samples, but no particular Prunus host specificity was observed as described in other studies (Aparicio et al., 1999; Cui et al., 2015). In contrast, PDV was only detected in three peach tree samples and ApMV was only detected in one almond tree sample. Phylogenetic analysis identified distinct populations of sequence variants that formed two or more phylogroups within sequences representing PNRSV, PDV and ApMV (Figure 1; Table 5). A high bootstrap value of ≥90% was adopted as the lowest threshold required to confidently define a distinct RNA2 phylogroup for each Ilarvirus sequence variant population and these phylogroups were further confirmed by pairwise sequence identity comparison analysis (Kinoti et al., 2017). Phylogroups of PNRSV, PDV and ApMV sequence variants from this study did not include isolates from other countries that have been published in GenBank, which could indicate that Australian PNRSV, PDV, and ApMV isolates are distinct populations that may have evolved separately from isolates from other geographical regions. However, the limited number of Ilarvirus sequences available in GenBank for comparison with the Australian PNRSV, PDV, and ApMV sequence variants from this study, makes it difficult to explicitly conclude the observed phylogroup separation is associated with geographical origin.
APLPV was detected in three samples, representing the first report of APLPV detection in Australia. APLPV has been previously reported in North America, Europe and the Mediterranean region (Paulsen and Fulton, 1968; Myrta et al., 2002; Alayasa et al., 2003) but very little sequence information is available for comparison and analysis. Australian APLPV sequence variants and sequences of isolates from France and the USA formed a single phylogroup which indicated a low genetic variability of APLPV RNA2 RdRp sequences. Similar low genetic variability of the APLPV coat protein and movement protein gene regions on RNA3 was observed in other studies and did not differentiate between American and Mediterranean isolates nor plant host origins (Herranz et al., 2008).
The lack of APLPV sequence data and the lack of diversity observed in this study and in a study by Herranz et al. (2008), might reflect the low incidence and limited distribution of APLPV worldwide. However, symptoms associated with APLPV are similar to ApMV in Prunus hosts and it maybe that the virus is more widely distributed but remains undiagnosed due to the assumption that disease is caused by other viruses (Desvignes et al., 1999). The occurrence of distinct phylogroups in ApMV, PNRSV, PDV, Ilarvirus-S1 and Ilarvirus-S2 was further supported by pairwise identity analysis, with all Ilarvirus species phylogroups detected in this study having a general identity cut-off of ≥96%. Therefore, it is possible that these phylogroups represent putative genetic strains based on RNA2 and made up of a population of variants with ≥96% identity within the RNA2 component of ilarviruses detected in this study. Based on this assumption, this study identified two strains of PNRSV and PDV and only one strain for each of ApMV and APLPV occurring in Australia. The partial RNA2 RdRp segment used for analysis in this study is the most conserved region across all ilarviruses and it is possible more strains of these ilarviruses would be identified if similar analysis was carried out on the whole sequence of RNA2 component or the other genomic RNAs (Kinoti et al., 2017).
Other molecular based methods used in this study did not always confirm the presence of Ilarvirus species detected by the generic amplicon NGS. Sanger sequencing of the cloned Ilarvirus-generic RT-PCR amplicons only detected Ilarvirus species that occurred as a single infection or the Ilarvirus species with the highest amplicon NGS read numbers in mixed infections. For example, PNRSV and PDV occurred at a lower frequency (22.9 and 1.8% of amplicon NGS total read numbers, respectively) when compared to Ilarvirus-S1 sequences in sample Pch4 and these low frequency amplicon sequences were not detected when cloned generic amplicons were sequenced (Table 6). However, full genomes of PDV and PNRSV were assembled during metagenomic NGS, confirming their presence in Pch4. Therefore, it is possible that sequencing of cloned generic PCR amplicons may be limited in its detection of some virus species or strains with low frequency genomes in mixed infections compared to amplicon NGS approaches. This observed variation in species-specific numbers of the generic amplicons may be a result of varied PCR efficiency associated with the degenerate primer binding specificity to different virus species or variants which have previously been shown to negatively impact PCR amplicon copy numbers (Whiley and Sloots, 2005; Stadhouders et al., 2010).
The degeneracy associated with generic PCR primers make them more prone to non-specific amplification and formation of PCR artifacts due to mis-priming and diversity of nucleic acid extracts used for PCR (Qiu et al., 2001; Huber et al., 2009). For this reason, all the Ilarvirus species in each sample that were detected by NGS were tested by specific RT-PCR tests designed to amplify the same RdRp region as the generic Ilarvirus PCR (Maliogka et al., 2007). The detection of all expected Ilarvirus species confirmed that the generic amplicon NGS sequences were not from non-specific PCR products or artifacts. Virus species-specific RT-PCR also detected the other RNA components (RNA1 and 3) of all the Ilarvirus species expected in each sample with the exception of Ilarvirus-S1 and Ilarvirus-S2. Ilarvirus-S1 and Ilarvirus-S2 that were detected by NGS of the generic Ilarvirus RNA2 amplicons may represent two putative and novel Prunus infecting Ilarvirus species or strains. Ilarvirus-S1 and Ilarvirus-S2 sequences had greatest BLAST sequence identity to PMoV and RaLV, respectively, which belong to Ilarvirus subgroup 1 (Pallas et al., 2012b). However, the presence of a novel PMoV and RaLV related Ilarvirus could not be confirmed by the amplification of another region of the Ilarvirus RNA2 component nor by the amplification of RNA1 or RNA3. Nevertheless, RNA2 sequences of both putative ilarviruses, Ilarvirus-S1 and Ilarvirus-S2, underwent similar phylogenetic analysis as the known ilarviruses. Ilarvirus-S1 sequence variants formed two phylogroups that diverged from PMoV type isolate and Ilarvirus-S2 sequence variants formed two phylogroups.
Metagenomic NGS is widely used for plant virus detection and full genome characterization due to its sensitivity and lack of specificity compared to other molecular techniques (Petrosino et al., 2009; Mokili et al., 2012). Therefore, metagenomic NGS was used to confirm the presence of the Ilarvirus species detected by generic amplicon NGS in this study. Full genomes were assembled for PNRSV, PDV, ApMV, and APLPV. However, full genomes of Ilarvirus-S1 and Ilarvirus-S2, which have not been previously reported infecting Prunus species, were not assembled by the metagenomic NGS. The reason for the absence of Ilarvirus-S1 and Ilarvirus-S2 sequences when analyzed using the metagenomic NGS approach is not known, despite Illavirus-S1 and Illarvirus-S2 sequences being detected in large number based on generic amplicon NGS read numbers (Table S2). It is possible that Ilarvirus-S1 and Ilarvirus-S2 occurred in low titre in the plant, and the multiple PCR amplification steps during the generic nested PCR (Maliogka et al., 2007) coupled with preferential degenerate primer binding to Ilarvirus-S1 and Ilarvirus-S2 sequence may have resulted in their high generic amplicon NGS read numbers. It is also possible that the high background levels of host sequences compared to virus sequences associated with NGS (Hall et al., 2014), could have a negative impact on the detection of low titre virus infection by metagenomic NGS.
There is also a possibility that Ilarvirus-S1 and Ilarvirus-S2 virus sequences detected by generic amplicon NGS in this study may be derived from endogenous viral elements (EVEs). EVEs are composed of fragments or complete genome sequences of plant RNA viruses that are integrated in some plant and insect host genomes (Ndowora et al., 1999; Chiba et al., 2011; Cui and Holmes, 2012). These integrated viral sequences can have high sequence divergence from their related extant virus species that may have continued to evolve rapidly whilst the EVE has only evolved at the same rate as the host (Aiewsakun and Katzourakis, 2015). The Ilarvirus-S1 sequences detected in this study occurred mainly in peach trees and had a higher sequence divergence to PMoV type isolate, but their presence could not be confirmed by metagenomic NGS and therefore these Ilarvirus-S1 sequences could represent EVEs.
Ilarvirus-S2 sequences which were detected in only one sample were not highly divergent from RaLV (92–98% similarity) could also represent an EVE, especially as a full genome of this virus was not obtained via metagenomic NGS and no other sequences were amplified with RaLV specific PCR tests. Therefore, in this study it is difficult to conclusively determine whether the detection of Ilarvirus-S1 and Ilarvirus-S2 by generic NGS represent two potential new ilarviruses infecting Prunus species in Australia or are in fact EVEs. Further work is required to elucidate the origin of Ilarvirus-S1 and Ilarvirus-S2 amplicon sequences.
The lack of detection of Ilarvirus-S1 and Ilarvirus-S2 by metagenomic NGS in this study and also recently reported lack of NGS detection of some human viruses which were known to be present in reference material (Mee et al., 2016), raise serious concerns in the use of metagenomic and amplicon NGS as a “universal” virus diagnostic tool without understanding its detection limits and appropriate interpretation of NGS data. Similar concerns have also been raised by other authors regarding metagenomic NGS virus diagnostic sensitivity and what constitutes a virus infection (Martin et al., 2016). Currently, there is no standardized approach to accurately determine what constitutes an extant and active virus infection when analyzing an NGS dataset, especially where varied results and partial virus detection by molecular based methods is concerned, as in the case of Ilarvirus-S1 and Ilarvirus-S2 detection in this study.
This study highlights the potential of nested generic PCR to give false positive results for a virus, based on the detection of only a fraction of the genome, especially when an unknown virus or virus-related sequence is concerned. It is paramount that partial detection of virus sequences by molecular based methods be correlated to the presence of sequence from other RNA segments or region of the virus genome. Nevertheless, the findings of this study established that generic amplicon NGS approach is a highly suitable method for the rapid detection and identification of specific virus species from multiple plant samples and multiple virus infections in a single plant sample, all within one NGS run. Amplicon NGS allowed for the detection of known endemic and exotic Ilarvirus species in Australian Prunus species. The generic amplicon NGS analysis approach used in this study was also able to identify distinct RNA2 sequence variant populations, within and between plant samples, which could be identified as possible genetic strains of an Ilarvirus species.
Author Contributions
WK participated in the design of the study, collected and screened the samples for virus infection, carried out the library preparation and sequencing, performed computational analysis, and drafted the manuscript. FC participated in the design of the study, sample collection, data analysis, and contributed to drafting the manuscript. NN participated in collection and screening of samples for virus infection. BR participated in the design of the study, data analysis, and drafting the manuscript. KP participated in the design of the study, data analysis, and contributed to drafting the manuscript. All authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to acknowledge and thank Almond Board Australia, Horticulture Innovation Australia Limited, Agriculture Victoria and La Trobe University School of Life Science Postgraduate Publication Award and Postgraduate research scholarship for financial support. This research work was undertaken using the facilities of Agriculture Victoria, Australia. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.01219/full#supplementary-material
References
Aboughanem-Sabanadzovic, N., Tzanetakis, I. E., Lawrence, A., Stephenson, R. C., and Sabanadzovic, S. (2015). A novel ilarvirus is associated with privet necrotic ringspot disease in the Southern United States. Phytopathology 106, 87–93. doi: 10.1094/PHYTO-12-14-0387-R
Adams, I. P., Glover, R. H., Monger, W. A., Mumford, R., Jackeviciene, E., Navalinskiene, M., et al. (2009). Next-generation sequencing and metagenomic analysis: a universal diagnostic tool in plant virology. Mol. Plant Pathol. 10, 537–545. doi: 10.1111/j.1364-3703.2009.00545.x
Aiewsakun, P., and Katzourakis, A. (2015). Endogenous viruses: connecting recent and ancient viral evolution. Virology 479, 26–37. doi: 10.1016/j.virol.2015.02.011
Alayasa, N., Abbadi, H., Al Rwahnih, M., Herranz, M., Myrta, A., Minafra, A., et al. (2003). Detection and partial characterization of different isolates of American plum line pattern virus in the Mediterranean. CIHEAM 45, 43–45.
Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Andrew, R. (2009). FigTree. Available online at: http://tree.bio.ed.ac.uk/software/ (Accessed 20 March 2017).
Aparicio, F., Myrta, A., Di Terlizzi, B., and Pallás, V. (1999). Molecular variability among isolates of Prunus necrotic ringspot virus from different Prunus spp. Phytopathology 89, 991–999. doi: 10.1094/PHYTO.1999.89.11.991
Beerenwinkel, N., and Zagordi, O. (2011). Ultra-deep sequencing for the analysis of viral populations. Curr. Opin. Virol. 1, 413–418. doi: 10.1016/j.coviro.2011.07.008
Bujarski, J., Figlerowicz, M., Gallittelli, D., Roossinck, M., and Scott, S. (2012). “Family Bromoviridae,” in Virus Taxonomy, Ninth Report of the International Committee on Taxonomy of Viruses, eds A. King, M. J. Adams, E. B. Carstens, and E. Lefkowitz (Amsterdam: Elsevier-Academic Press), 965–976.
Card, S., Pearson, M., and Clover, G. (2007). Plant pathogens transmitted by pollen. Australas. Plant Pathol. 36, 455–461. doi: 10.1071/AP07050
Chiba, S., Kondo, H., Tani, A., Saisho, D., Sakamoto, W., Kanematsu, S., et al. (2011). Widespread endogenization of genome sequences of non-retroviral RNA viruses into plant genomes. PLoS Pathog. 7:e1002146. doi: 10.1371/journal.ppat.1002146
Codoner, F. M., and Elena, S. F. (2008). The promiscuous evolutionary history of the family Bromoviridae. J. Gen. Virol. 89, 1739–1747. doi: 10.1099/vir.0.2008/000166-0
Compton, T. (1990). “Degenerate primers for DNA amplification,” in PCR Protocols: A Guide to Methods and Applications, Vol. 21, eds M. A. Innis, D. H. Gelfand, J. J. Sninsky, and T. J. White (London: Academic Press), 39–45. doi: 10.1016/b978-0-12-372180-8.50009-3
Cui, H., Liu, H., Chen, J., Zhou, J., Qu, L., Su, J., et al. (2015). Genetic diversity of Prunus necrotic ringspot virus infecting stone fruit trees grown at seven regions in China and differentiation of three phylogroups by multiplex RT-PCR. Crop Protect. 74, 30–36. doi: 10.1016/j.cropro.2015.04.001
Cui, J., and Holmes, E. C. (2012). Endogenous RNA viruses of plants in insect genomes. Virology 427, 77–79. doi: 10.1016/j.virol.2012.02.014
Desvignes, J. C., Boyé, R., Cornaggia, D., Grasseau, N., Hurtt, S., and Waterworth, H. (1999). Virus Diseases of Fruit Trees. Paris: CTIFL.
Di Terlizzi, B., Skrzeczkowski, L., Mink, G., Scott, S., and Zimmerman, M. (2001). The RNA 5 of Prunus necrotic ringspot virus is a biologically inactive copy of the 3′-UTR of the genomic RNA 3. Arch. Virol. 146, 825–833. doi: 10.1007/s007050170151
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Eriksson, N., Pachter, L., Mitsuya, Y., Rhee, S.-Y., Wang, C., Gharizadeh, B., et al. (2008). Viral population estimation using pyrosequencing. PLoS Comp. Biol. 4:e1000074. doi: 10.1371/journal.pcbi.1000074
Ge, X., and Scott, S. (1994). The nucleotide sequence of citrus leaf rugose ilarvirus RNA-2. J. Gen. Virol. 75, 2841–2846. doi: 10.1099/0022-1317-75-10-2841
Ge, X., Scott, S., and Zimmerman, M. (1997). The complete sequence of the genomic RNAs of spinach latent virus. Arch. Virol. 142, 1213–1226. doi: 10.1007/s007050050153
Hall, R. J., Wang, J., Todd, A. K., Bissielo, A. B., Yen, S., Strydom, H., et al. (2014). Evaluation of rapid and simple techniques for the enrichment of viruses prior to metagenomic virus discovery. J. Virol. Methods 195, 194–204. doi: 10.1016/j.jviromet.2013.08.035
Herranz, M., Al Rwahnih, M., Sánchez-Navarro, J., Elena, S., Choueiri, E., Myrta, A., et al. (2008). Low genetic variability in the coat and movement proteins of American plum line pattern virus isolates from different geographic origins. Arch. Virol. 153, 367–373. doi: 10.1007/s00705-007-1100-4
Huber, J. A., Morrison, H. G., Huse, S. M., Neal, P. R., Sogin, M. L., and Mark Welch, D. B. (2009). Effect of PCR amplicon size on assessments of clone library microbial diversity and community structure. Environ. Microbiol. 11, 1292–1302. doi: 10.1111/j.1462-2920.2008.01857.x
James, D., Varga, A., Leippi, L., Godkin, S., and Masters, C. (2010). Sequence analysis of RNA 2 and RNA 3 of lilac leaf chlorosis virus: a putative new member of the genus Ilarvirus. Arch. Virol. 155, 993–998. doi: 10.1007/s00705-010-0673-5
James, D., Varga, A., Pallas, V., and Candresse, T. (2006). Strategies for simultaneous detection of multiple plant viruses. Can. J. Plant Pathol. 28, 16–29. doi: 10.1080/07060660609507267
Janssen, D., Saez, E., Segundo, E., Martin, G., Gil, F., and Cuadrado, I. (2005). Capsicum annuum–a new host of Parietaria mottle virus in Spain. Plant Pathol. 54, 567–567. doi: 10.1111/j.1365-3059.2005.01195.x
Kinoti, W., Constable, F., Nancarrow, N., Plummer, K., and Rodoni, B. (2017). Analysis of intra-host genetic diversity of Prunus necrotic ringspot virus (PNRSV) using amplicon next generation sequencing. PLoS ONE 12:e0179284. doi: 10.1371/journal.pone.0179284
Krueger, F. (2012). Trim Galore! Available online at: www.bioinformatics.babraham.ac.uk/projects/trim_galore (Accessed 12 March 2017).
Li, W., Adkins, S., and Hilf, M. (2008). Characterization of complete sequences of RNA 1 and RNA 2 of Citrus variegation virus. Arch. Virol. 153, 385–388. doi: 10.1007/s00705-007-1090-2
MacKenzie, D. J., McLean, M. A., Mukerji, S., and Green, M. (1997). Improved RNA extraction from woody plants for the detection of viral pathogens by reverse transcription-polymerase chain reaction. Plant Dis. Rep. 81, 222–226. doi: 10.1094/PDIS.1997.81.2.222
Maliogka, V., Dovas, C., and Katis, N. (2007). Demarcation of ilarviruses based on the phylogeny of RNA2-encoded RdRp and a generic ramped annealing RT-PCR. Arch. Virol. 152, 1687–1698. doi: 10.1007/s00705-007-0995-0
Mancuso, N., Tork, B., Skums, P., Măndoiu, I., and Zelikovsky, A. (2011). “Viral quasispecies reconstruction from amplicon 454 pyrosequencing reads,” in Bioinformatics and Biomedicine Workshops (BIBMW), 2011 IEEE International Conference on: IEEE (Orlando, FL), 94–101.
Marston, D. A., McElhinney, L. M., Ellis, R. J., Horton, D. L., Wise, E. L., Leech, S. L., et al. (2013). Next generation sequencing of viral RNA genomes. BMC Genomics 14, 1. doi: 10.1186/1471-2164-14-444
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17, 10–12. doi: 10.14806/ej.17.1.200
Martin, R. R., Constable, F., and Tzanetakis, I. (2016). Quarantine regulations and the impact of modern detection methods. Annu. Rev. Phytopathol. 54, 189–205. doi: 10.1146/annurev-phyto-080615-100105
Mee, E. T., Preston, M. D., Minor, P. D., Schepelmann, S., and Participants, C. S. (2016). Development of a candidate reference material for adventitious virus detection in vaccine and biologicals manufacturing by deep sequencing. Vaccine 34, 2035–2043. doi: 10.1016/j.vaccine.2015.12.020
Mokili, J. L., Rohwer, F., and Dutilh, B. E. (2012). Metagenomics and future perspectives in virus discovery. Curr. Opin. Virol. 2, 63–77. doi: 10.1016/j.coviro.2011.12.004
Muhire, B. M., Varsani, A., and Martin, D. P. (2014). SDT: a virus classification tool based on pairwise sequence alignment and identity calculation. PLoS ONE 9:e108277. doi: 10.1371/journal.pone.0108277
Myrta, A., Abbadi, H., Herranz, M., Al Rwahnih, M., Di Terlizzi, B., Minafra, A., et al. (2002). First report of American plum line pattern virus (APLPV) in Albania, Italy and Tunisia. J. Plant Pathol. 84, 188.
Ndowora, T., Dahal, G., LaFleur, D., Harper, G., Hull, R., Olszewski, N. E., et al. (1999). Evidence that badnavirus infection inmusacan originate from integrated pararetroviral sequences. Virology 255, 214–220. doi: 10.1006/viro.1998.9582
Pallas, V., Aparicio, F., Herranz, M., Amari, K., Sanchez-Pina, M., Myrta, A., et al. (2012a). Ilarviruses of Prunus spp.: a continued concern for fruit trees. Phytopathology 102, 1108–1120. doi: 10.1094/PHYTO-02-12-0023-RVW
Pallas, V., Aparicio, F., Herranz, M. C., Sanchez-Navarro, J. A., and Scott, S. W. (2012b). The molecular biology of ilarviruses. Adv. Virus Res. 87, 139–181. doi: 10.1016/B978-0-12-407698-3.00005-3
Pallás, V., Sánchez-Navarro, J., and Canizares, M. (1998). Molecular diagnostic techniques and their potential role in stone fruit certification schemes. CIHEAM 19, 191–208.
Parakh, D., Shamloul, A., Hadidi, A., Waterworth, H., Scott, S., Howell, H., et al. (1994). Detection of Prune dwarf ilarvirus from infected stone fruits using reverse transcription-polymerase chain reaction. Acta Hortic. 386, 421–430.
Paulsen, A. Q., and Fulton, R. (1968). Hosts and properties of a Plum line pattern virus. Phytopathology 58, 766–772.
Perez-Egusquiza, Z., Ward, L., and Clover, G. (2014). Detection of a new ilarvirus infecting wild radish in New Zealand. Australas. Plant Dis. Notes 9:133. doi: 10.1007/s13314-014-0133-2
Petrosino, J. F., Highlander, S., Luna, R. A., Gibbs, R. A., and Versalovic, J. (2009). Metagenomic pyrosequencing and microbial identification. Clin. Chem. 55, 856–866. doi: 10.1373/clinchem.2008.107565
Petrzik, K., and Svoboda, P. (1997). Screening of Apple mosaic virus in hop cultivars in the Czech Republic by reverse transcription-polymerase chain reaction. Acta Virol. 41, 101–103.
Prabha, K., Baranwal, V., and Jain, R. (2013). Applications of next generation high throughput sequencing technologies in characterization, discovery and molecular interaction of plant viruses. Indian J. Virol. 24, 157–165. doi: 10.1007/s13337-013-0133-4
Qiu, X., Wu, L., Huang, H., McDonel, P. E., Palumbo, A. V., Tiedje, J. M., et al. (2001). Evaluation of PCR-generated chimeras, mutations, and heteroduplexes with 16S rRNA gene-based cloning. Appl. Environ. Microbiol. 67, 880–887. doi: 10.1128/AEM.67.2.880-887.2001
Radford, A. D., Chapman, D., Dixon, L., Chantrey, J., Darby, A. C., and Hall, N. (2012). Application of next-generation sequencing technologies in virology. J. Gen. Virol. 93, 1853–1868. doi: 10.1099/vir.0.043182-0
Rampitsch, C., and Eastwell, K. (1997). The complete nucleotide sequenceof Prune dwarf ilarvirus RNA-1. Arch. Virol. 142, 1911–1918. doi: 10.1007/s007050050210
Sanschagrin, S., and Yergeau, E. (2014). Next-generation sequencing of 16S ribosomal RNA gene amplicons. J. Vis. Exp. 90:e51709. doi: 10.3791/51709
Scott, S., and Zimmerman, M. (2000). American plum line pattern virus is a distinct Ilarvirus. Acta Hortic. 505, 221–225. doi: 10.17660/ActaHortic.2001.550.31
Scott, S., and Zimmerman, M. (2009). The nucleotide sequences of the RNA 1 and RNA 2 of asparagus virus 2 show a close relationship to citrus variegation virus. Arch. Virol. 154, 719. doi: 10.1007/s00705-009-0355-3
Scott, S., Zimmerman, M., and Ge, X. (1998). The sequence of RNA 1 and RNA 2 of tobacco streak virus: additional evidence for the inclusion of alfalfa mosaic virus in the genus Ilarvirus. Arch. Virol. 143, 1187–1198. doi: 10.1007/s007050050366
Scott, S., Zimmerman, M., and Ge, X. (2003). Viruses in subgroup 2 of the genus Ilarvirus share both serological relationships and characteristics at the molecular level. Arch. Virol. 148, 2063–2075. doi: 10.1007/s00705-003-0148-z
Scott, S., Zimmerman, M., and Rankin, D. (2006). Complete sequence of the RNA 1 and RNA 2 of Parietaria mottle virus. Arch. Virol. 151, 1895–1898. doi: 10.1007/s00705-006-0803-2
Sharman, M., and Thomas, J. (2013). Genetic diversity of subgroup 1 ilarviruses from eastern Australia. Arch. Virol. 158, 1637–1647. doi: 10.1007/s00705-013-1628-4
Shiel, P., and Berger, P. (2000). The complete nucleotide sequence of Apple mosaic virus (ApMV) RNA 1 and RNA 2: ApMV is more closely related to Alfalfa mosaic virus than to other ilarviruses. J. Gen. Virol. 81, 273–278. doi: 10.1099/0022-1317-81-1-273
Sogin, M. L., Morrison, H. G., Huber, J. A., Welch, D. M., Huse, S. M., Neal, P. R., et al. (2006). Microbial diversity in the deep sea and the underexplored “rare biosphere.” Proc. Natl. Acad. Sci. U.S.A. 103, 12115–12120. doi: 10.1073/pnas.0605127103
Stadhouders, R., Pas, S. D., Anber, J., Voermans, J., Mes, T. H., and Schutten, M. (2010). The effect of primer-template mismatches on the detection and quantification of nucleic acids using the 5′ nuclease assay. J. Mol. Diagn. 12, 109–117. doi: 10.2353/jmoldx.2010.090035
Tzanetakis, I. E., and Martin, R. R. (2005). New features in the genus Ilarvirus revealed by the nucleotide sequence of Fragaria chiloensis latent virus. Virus Res. 112, 32–37. doi: 10.1016/j.virusres.2005.02.010
Tzanetakis, I., Martin, R., and Scott, S. (2010). Genomic sequences of blackberry chlorotic ringspot virus and strawberry necrotic shock virus and the phylogeny of viruses in subgroup 1 of the genus Ilarvirus. Arch. Virol. 155, 557–561. doi: 10.1007/s00705-010-0601-8
Wang, C., Mitsuya, Y., Gharizadeh, B., Ronaghi, M., and Shafer, R. W. (2007). Characterization of mutation spectra with ultra-deep pyrosequencing: application to HIV-1 drug resistance. Genome Res. 17, 1195–1201. doi: 10.1101/gr.6468307
Whiley, D. M., and Sloots, T. P. (2005). Sequence variation in primer targets affects the accuracy of viral quantitative PCR. J. Clin. Virol. 34, 104–107. doi: 10.1016/j.jcv.2005.02.010
Wu, Q., Ding, S.-W., Zhang, Y., and Zhu, S. (2015). Identification of viruses and viroids by next-generation sequencing and homology-dependent and homology-independent algorithms. Annu. Rev. Phytopathol. 53, 425–444. doi: 10.1146/annurev-phyto-080614-120030
Keywords: Ilarvirus species, generic amplicon next-generation sequencing, virus genetic diversity, metagenomic next-generation sequencing
Citation: Kinoti WM, Constable FE, Nancarrow N, Plummer KM and Rodoni B (2017) Generic Amplicon Deep Sequencing to Determine Ilarvirus Species Diversity in Australian Prunus. Front. Microbiol. 8:1219. doi: 10.3389/fmicb.2017.01219
Received: 06 April 2017; Accepted: 16 June 2017;
Published: 30 June 2017.
Edited by:
Arvind Varsani, Arizona State University, United StatesReviewed by:
Philippe Roumagnac, Agricultural Research Centre For International Development, FrancePierre Lefeuvre, Agricultural Research Centre For International Development, France
Copyright © 2017 Kinoti, Constable, Nancarrow, Plummer and Rodoni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wycliff M. Kinoti, d2tpbm90aUBzdHVkZW50cy5sYXRyb2JlLmVkdS5hdQ==