 Xiaoting Hua1,2†Zhihui Zhou1,2†Qing Yang3
Xiaoting Hua1,2†Zhihui Zhou1,2†Qing Yang3 Qiucheng Shi1,2Qingye Xu1,2Jianfeng Wang4Keren Shi1,2Feng Zhao5Long Sun6
Qiucheng Shi1,2Qingye Xu1,2Jianfeng Wang4Keren Shi1,2Feng Zhao5Long Sun6 Zhi Ruan5Yan Jiang1,2
Zhi Ruan5Yan Jiang1,2 Yunsong Yu1,2*
Yunsong Yu1,2*- 1Department of Infectious Diseases, Sir Run Run Shaw Hospital, College of Medicine, Zhejiang University, Hangzhou, China
- 2Key Laboratory of Microbial Technology and Bioinformatics of Zhejiang Province, Hangzhou, China
- 3State Key Laboratory for Diagnosis and Treatment of Infectious diseases, Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases, The First Affiliated Hospital, College of Medicine, Zhejiang University, Hangzhou, China
- 4Department of Respiratory Diseases, The Affiliated Hospital of Hangzhou Normal University, Hangzhou, China
- 5Department of Clinical Laboratory, Sir Run Run Shaw Hospital, School of Medicine, Zhejiang University, Hangzhou, China
- 6Department of Clinical Laboratory, Hangzhou General Hospital of Zhejiang Provincial Corps, Chinese People’s Armed Police Forces, Hangzhou, China
Acinetobacter baumannii is an important nosocomial pathogen worldwide. A more comprehensive understanding of the within-host genomic evolution of A. baumannii would provide a molecule basis for improving treatment of A. baumannii infection. To understand the evolutionary mechanism facilitating A. baumannii survived in human body, we here reported the genomic analysis of A. baumannii isolated sampled from Chinese patients. We used whole-genome sequence of A. baumannii isolates from the same patient to determine single-nucleotide variants, insertion sequence mapping, and gene change. The MICs for 10 antimicrobial agents were determined. Motility assay and microscopy were performed on the isolated pairs harboring ptk mutations. The gene ptk encoded a putative protein tyrosine kinase involved in the production of capsular polysaccharide. Approximately half (39/86) of the strains isolated from the same patient harbored the same MLST patterns, and during the replacement of international clonal lineage II (ICL-II) and non-ICL-II strains, most of the alteration was that non-ICL-II strain was replaced by ICL-II strain (10/12). A. baumannii was resistant to major antimicrobial agents, whereas the strains were more resistant to ceftazidime, azithromycin, and sulfonamides after within-host evolution. Isolates from the ICL-II lineage displayed greater resistance to antimicrobial agents than non-ICL-II isolates. Isolates from ICL-II harbored more resistance genes and mobile elements than non-ICL-II strains. Several lineages evolved a more mucoid phenotype. Genome sequencing revealed that the phenotype was achieved by genetic changes in the ptk gene. ICL-II (especially ST195 and ST208) was the terminal destination for bacteria after within-host evolution. These results indicate that the molecular basis and the treatment for ICL-II strains needed further investigation.
Introduction
It is important to achieve comprehensive understanding of how bacteria evolve during human host infections to be able to treat infections effectively. Recent advances in DNA sequencing technologies have made it possible to identify genetic changes between the genome of the same strains over shorter or longer periods of time to reveal important adaptations to the host environment (Wilson, 2012). The comparison of bacterial pathogens’ genome over time from individual patients during infection and therapy will advance the understanding of the bacteria’s evolution, epidemiology, and antibiotic resistance (Marvig et al., 2013). It could determine the relationship among infecting bacteria isolates and gain insight in genetic adaptation in infected host and response to antibiotic treatment. Several studies have focused on the genomic evolution of pathogens causing acute infections, such as Yersinia pestis (Morelli et al., 2010), Neisseria meningitidis (Klughammer et al., 2017) and Vibrio cholera (Mutreja et al., 2011), or causing long-term infections, such as Pseudomonas aeruginosa (Marvig et al., 2013) and Acinetobacter baumannii (Wright et al., 2016).
Acinetobacter baumannii has a greater correlation with resistance to antibiotics and higher mortality among bacteremic patients, than does A. nosocomialis (Chuang et al., 2011). International clonal lineage II (ICL-II) represents the largest and most geographically diverse CC (Zarrilli et al., 2013; Kamolvit et al., 2015; Chen et al., 2017). High-virulence, healthcare-associated A. baumannii were also isolated, which altered the general consideration that A. baumannii was a low-virulence organism (Jones et al., 2015). Many virulence mechanisms have been identified in A. baumannii, including capsule formation and siderophore-mediated iron acquisition systems (Russo et al., 2010; Cerqueira and Peleg, 2011). Previous genomic analysis of A. baumannii isolates from the same patient over time showed the enrichment in mutation associated with antibiotic and host response (Wright et al., 2016).
To understand the evolutionary mechanism facilitating A. baumannii survived in human body, we here reported the genomic analysis of A. baumannii isolated sampled from Chinese patients. In total, we sequenced the genomes of 172 A. baumannii isolates sampled from 86 different patients between 2011 and 2015. Genomic analyses and MIC assays were performed to provide a comprehensive view of within-host evolution in A. baumannii.
Materials and Methods
Bacterial Strains and Genome Sequencing
This study encompassed 172 isolates of the A. baumannii strains that were sampled from 86 patients in three hospitals in Hangzhou, China in a pair-wise longitudinal manner. The criterion of isolate selection was listed followed: (1) The bacteria were isolated from the same patient; (2) The time interval between two isolates is longer than 1 month. The isolation and identification of A. baumannii were performed as previously described (Ruan et al., 2013). All 172 isolates were sequenced at Zhejiang Tianke (Hangzhou, China) on an Illumina HiSeq2000 platform (Illumina, San Diego, CA, United States). In general, more than 300-fold coverage was obtained for each genome sequences. Sequence data from all isolates were deposited in GenBank under the accession numbers indicated in the Supplementary Table S1.
Ethics Statement
This study was approved by each ethics committee of Sir Run Run Shaw Hospital College of Medicine Zhejiang University, The First Affiliated Hospital College of Medicine Zhejiang University and Hospital of Zhejiang Corps, Chinese People’s Armed Police Forces. Written informed consent was waived by all Committees. The need for a written consent by the Ethical Committees was waived and this was approved by all committees, due to the fact that only the bacterial isolates were taken from the patients and the study with confidentially fully guaranteed.
Antimicrobial Susceptibility Testing
Antimicrobial susceptibility testing was performed in Muller Hinton (MH) broth using the two-fold serial agar dilution method according to the guidelines of the Clinical and Laboratory Standards Institute (CLSI) (Patel et al., 2015). The following antimicrobial agents were tested: amikacin (AK), ciprofloxacin (CIP), chloramphenicol (C), streptomycin (SM), sulfonamides (S), meropenem (MEM), tetracycline (TET), ceftazidime (CAZ), azithromycin (AZM), and kanamycin (KANA). The concentration of the sulfonamides ranged from 1.25 to 320 mg/L, and the concentration of the other antimicrobial agents ranged from 0.125 to 256 mg/L. The Tigecycline MIC was determined by using the CLSI broth dilution method, and E. coli ATCC 25922 was used as Quality control strain. Shapiro–Wilk normality test was used to detect whether the data is parametric. Statistical analysis was performed by using pairwise student t-test for parametric variables and Mann–Whitney U test for non-parametric variables.
Mutation Detection and Analysis
Sequence types (STs) and resistance genes were derived directly from short reads via SRST21 using both the Oxford scheme and the Institute Pasteur scheme (Inouye et al., 2014). To detect the presence of transposon carrying blaOXA-23, the genome sequences of all the strains was blasted with the sequences of Tn2006, Tn2008, and Tn2009. The minimum spanning tree was calculated by Prim’s algorithm using the MLST plugin in the BioNumerics 6.6.4 software (Applied Maths, Sint-Martens-Latem, Belgium) incorporated with the eBURST algorithm. Clonal complexes (CCs) were defined as containing at least three STs sharing the same allele numbers in at least six of seven loci. The number and position of insertion sequence were identified by ISseeker via annotating IS elements in draft genome assemblies with the reference genome (XH386, ST208) (Adams et al., 2016). XH386 was belonged to ICL-II isolate and isolated from China in 2014, and was selected as a reference genome for the majority of isolates in this study are ICL-II.
For the comparison between the strain pairs with the same STs, the sequence reads of the former isolate were de novo assembled using CLC Genomic Workbench 8.5.1 (Qiagen, Aarhus, Denmark). The assembled contigs were annotated with Prokka 1.11 (Seemann, 2014). The copy number of resistance genes were detected by CNV-seq via mapping the reads to the sequence of resistance genes (Xie and Tammi, 2009). The identification of transposon harbored blaOXA-23 was based on blast the genome sequences of the sequenced strains with the sequence of Tn2006, Tn2008, and Tn2009. The sequence read mappings of the latter isolate against annotated contigs and mutation prediction were all performed with Breseq (Deatherage and Barrick, 2014). The pan genome analysis was performed using Roary (Page et al., 2015), and the core genes were identified. The genes harboring mutations were mapped to the core genes to obtain the mutant gene frequency.
Microscopy
Bacterial capsules were detected using the India ink method (Geisinger and Isberg, 2015). Briefly, three bacterial colonies were mixed with 1 ml of 0.9% NaCl solution, and India ink was added. Finally, the bacteria were analyzed by light microscopy (1000×).
Motility Assay
The motility of all isolates was tested on MH medium supplemented with 0.3% (wt/vol) agarose as described previously (McQueary et al., 2012). Briefly, the MH plates were inoculated with single colonies in the middle of the plates to measure swarming motility. The measurement was performed after incubation at 37°C for 24 h and the motility zones were measured triplicate. Statistical analysis was performed by using student t-test.
Results
Clinical Collection of Bacterial Isolates for Sequencing
In this study, we sequenced the whole genomes of 172 isolates of A. baumannii sampled from 86 patients in sputum. Two groups of isolates for each patients were chosen for sequencing to gain insight into the evolution of A. baumannii in vivo. The first group of isolates was designated as Group A, and the second group was designated as Group B. The average time span between obtaining the A. baumannii isolates from each patients was 46 days (range = 31–394 days) (Supplementary Figure S1).
MLST and Clonal Complexes
The MLST profiles were generated from the WGS data. Using the Pasteur MLST scheme, a total of 12 different STs were identified, including eight existing STs and four novel STs (Supplementary Table S1). During the process of MLST identification, XH551, XH653, XH654, XH762, and XH796 were A. nosocomialis, while XH765 was A. pittii. ST2 (using the Pasteur scheme) was the predominant ST, comprising 80% of the isolates. Using the Oxford MLST scheme, there were 24 different ST, nine of which were novel. Furthermore, 139 isolates belonging to ST2 (using the Pasteur scheme) were divided into types including ST195, ST208, ST191 using the Oxford MLST scheme. Among them, 59 isolates were ST195, 24 isolates were ST208 and 22 isolates were ST191. A minimum spanning tree analysis indicated that 12 of the 24 STs were clustered into two CCs which were ICL-II and CC462. ST704 (A. nosocomialis) and ST642 were two locus variants of ST1343 (A. nosocomialis) and ST447, respectively. They were clustered into CC10. And ST1342, ST1344, ST1345, and ST1346 were singletons.
Of the 86 isolate pairs, 39 were the same STs (one isolate pairs were A. nosocomialis: XH653 and XH654). In the pairs harboring the same STs, 33 pairs belonged to ICL-II. The isolate pairs were separated into three categories: (i) different STs; (ii) the same STs and belonging to ICL-II; or (iii) the same STs and not belonging to ICL-II (non-ICL-II).
For the harboring different STs isolate pairs, it was common that the strain from ICL-II was replaced by another strain from ICL-II, and ST195, ST208 and ST191 played an important role in within-host evolution for they represent the STs that were most frequently switched to during within-host evolution (Figure 1). During the replacement of ICL-II and non-ICL-II strains, most of the alteration was that non-ICL-II strain was replaced by ICL-II strain (10/12). It might cause by the competition of different types of strains in vivo.
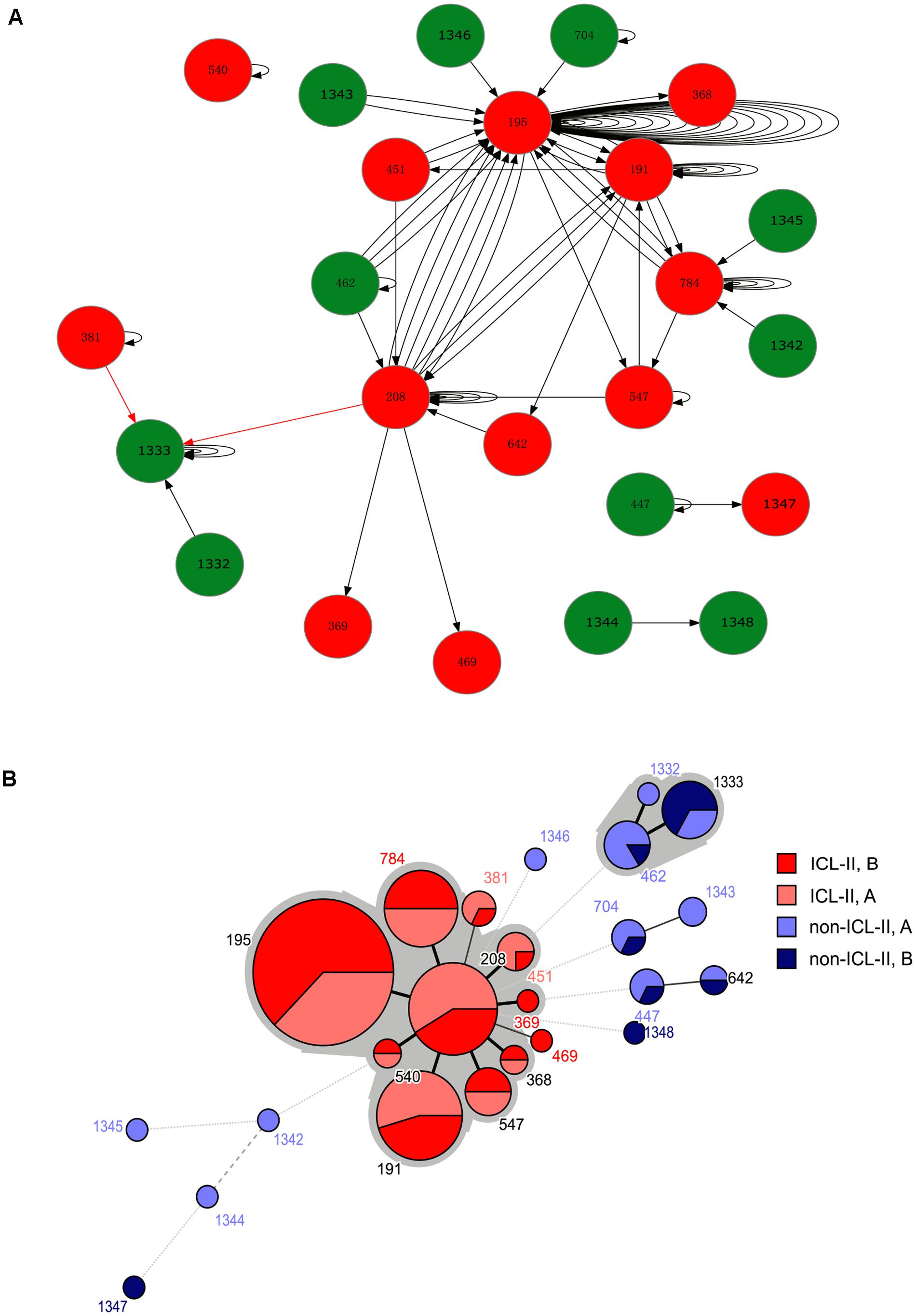
FIGURE 1. (A) A schematic of A. baumannii within-host evolution. The arrow indicated the alteration of two isolates in one patient. A circle at the head of the arrow indicates that the first isolates in the patients belonged to Group A. A circle at the end of the arrow indicates that the second isolates in the patients belonged to Group B. The black arrow indicates an alteration between the same STs or from non-ICL-II to ICL-II. The red arrow indicated an alteration from ICL-II to non-ICL-II. The number within the circles is the MLST. The color of the circle indicates whether the isolate belonged to ICL-II: Red: ICL-II; Green: non-ICL-II. (B) Minimum spanning tree analysis of all genome-sequenced A. baumannii isolates based on MLST data. Each circle represents an independent ST. The size of each circle indicates the number of isolates. The lines connecting the circles indicate the relationship between different STs. Different types of lines represent the difference in one allele (solid line), two alleles (dashed line), and three or more alleles (dotted lines). Two colored zones surrounding the STs indicate that they belong to the same clonal complex.
Antibiotic Resistance
To explore whether the evolution of A. baumannii would alter the antibiotic resistance profiles, the MICs for 10 antibiotics were determined and compared between Groups A and B by Mann–Whitney U tests. For the total sample, Group B demonstrated higher resistance than Group A did for CAZ (P = 0.006), AZM (P = 0.035) and S (P = 0.046) (Figure 2A). For the isolate pairs harboring different STs, three antibiotics resistances were significantly increased when comparing Groups A and B, including CAZ (P = 0.002), AZM (P = 0.011) and S (P = 0.026) (Figure 2B). No significant differences were observed between isolate pairs harboring the same STs (ICL-II and non-ICL-II) (Figures 2C,D). Furthermore, ICL-II isolates exhibited a higher resistance to antibiotics compared to non-ICL-II isolates (Figures 2C,D).
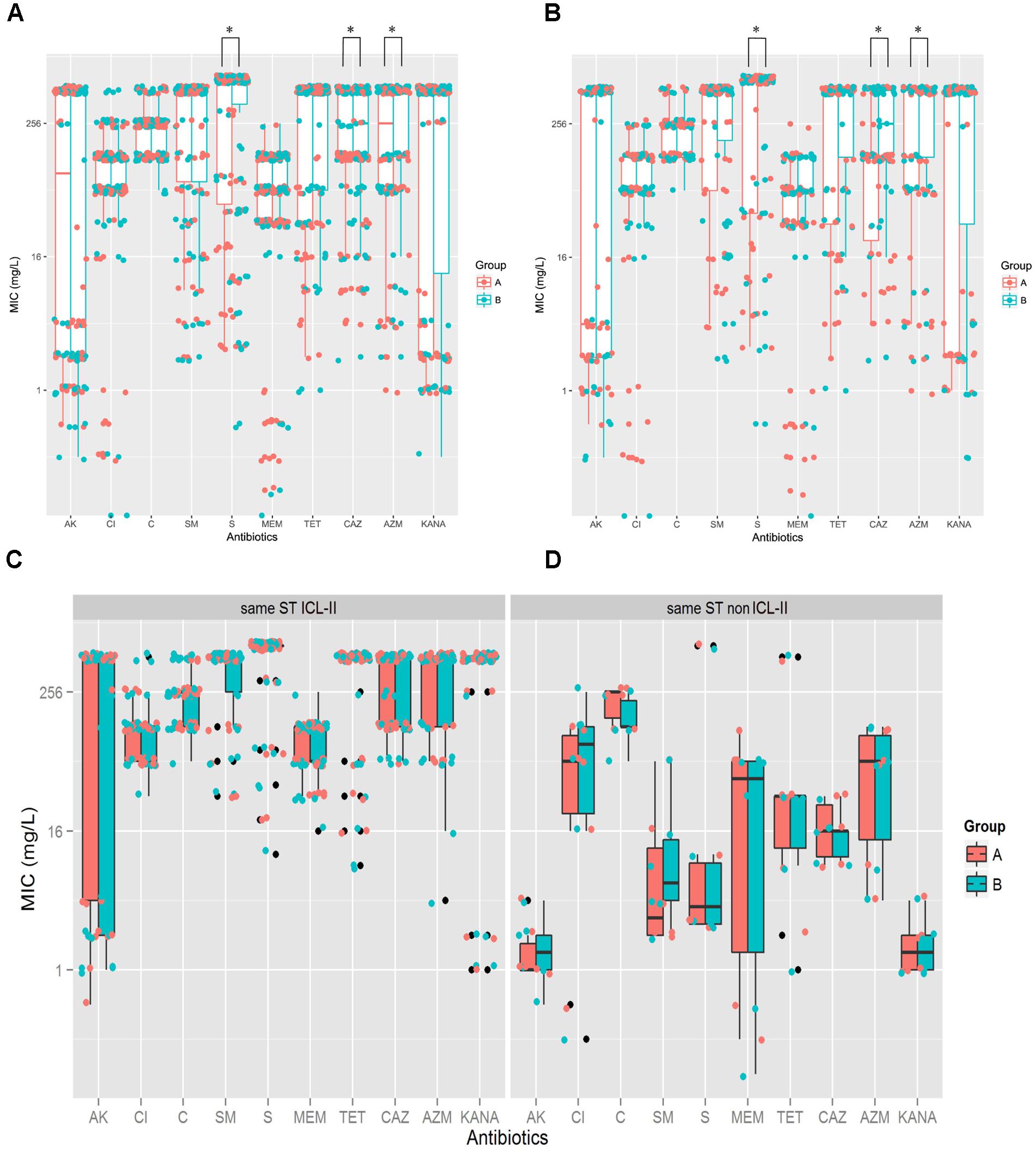
FIGURE 2. A. baumannii exhibited more resistance after within-host evolution, among the isolates, ICL-II strain demonstrated more resistance than non-ICL-II strains did. (A). For total sample, boxplots for Groups A and B showing the mean ratio of MIC for CAZ between Groups A and B. Dots indicate the detailed MIC values ratio for CAZ. (B) For the isolate pairs harboring different STs, boxplots for Groups A and B showing the mean ratio of MIC for AK, CAZ, MEM, and AZM between Groups A and B. Dots indicate the detailed MIC values ratio for that antibiotic. (C) MIC distributions for 10 antibiotics in whole genome-sequenced A. baumannii isolate pairs harboring the same ICL-II STs. Colors indicate the isolate in Groups A and B. Boxplots for each group indicate the mean MICs against the respective antibiotic. Dots indicate the detailed MIC value for that antibiotic. (D) MIC distribution for 10 antibiotics in all genome-sequenced A. baumannii isolate pairs harboring the same non-ICL-II STs.
To elucidate the relationship between antibiotic resistance and the resistance genes, we detected the distribution of resistance genes among the isolates via SRST2 (Supplementary Figure S2). ICL-II isolates contained more resistance genes than non-ICL-II isolates did (Figure 3A). To estimate the role of IS elements in mobilizing resistance genes, the number and the position of insertion sequence were identified by ISSeeker. ICL-II isolates harbored higher copy number of ISAba1, ISAba33 and IS26 than non-ICL-II isolates did (Figure 3B). Combining the data of resistance genes and MICs of A. baumannii, resistance gene loss and acquisition were detected in three pairs of isolates (Figure 4A). For example, armA conferred high-level resistance of AK. The presence and absence of armA were correlated with the MICs for AK in the three pairs of isolates (Supplementary Table S1). We identified resistance gene acquisition and loss events during the within-host evolution. After mapping the reads of three pairs of isolates to the reference genome XH386, a 50 kb chromosomal fragment containing multiple resistance genes was absent in XH550 compared to XH549 (Figure 4B). Further, a 38 kb chromosomal fragment that included resistance genes was absent in XH850 compared to XH608. Additionally, there was a 18 kb fragment insertion at the same site in XH820 compared to XH819. To investigate whether the copy number of resistance gene changed during within-host evolution, the copy number of resistance gene was detected by CNV-seq in Groups A and B. The result showed that only blaOXA-23 displayed copy number dynamic during within-host evolution (Figure 5). We also detected the copy number or position change in Groups A and B. For copy numbers, IS26, ISAba1, and ISAba33 were the most frequency changed IS element between Groups A and B (Figure 6A). For the position of IS elements, the mobilization of IS elements were common observed in within host evolution (Figures 6B–D). The genome sequences of all the strains was blasted with Tn2006, Tn2008, and Tn2009. Among all strains, 64 strains harbored Tn2006, and Tn2009 was appeared in 93 strains. For STs, Tn2006 presented in ST195, ST469, and ST547, while Tn2009 showed in all STs except ST469 and ST547 (Figure 7).
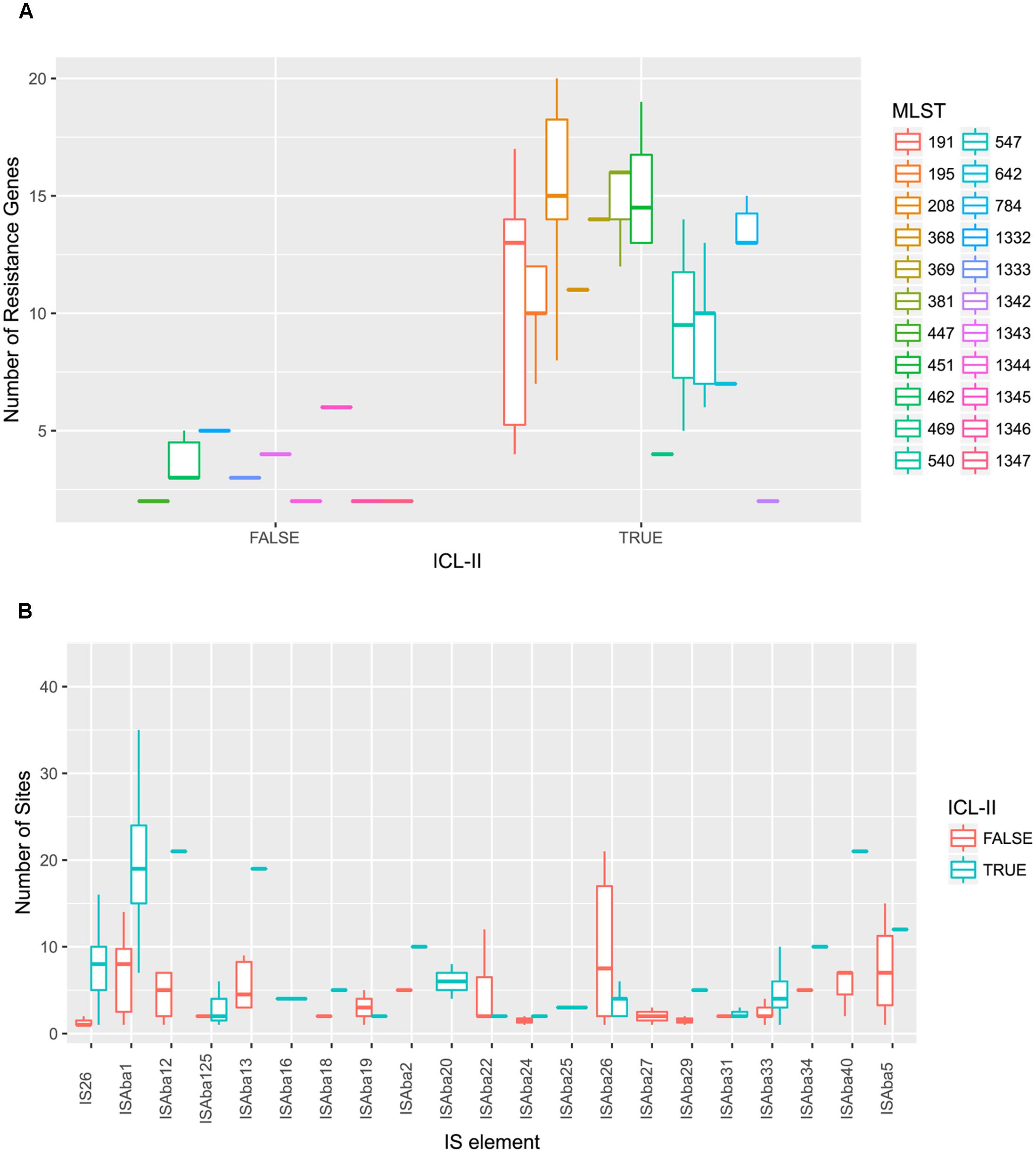
FIGURE 3. A. baumannii ICL-II presented a higher number of resistance genes and mobile element. (A) A. baumannii ICL-II harbored more resistance gene than non-ICL-II isolates. Colors are indicated for isolates belonging to different MLST. Boxplots for each MLST display the mean number of resistance genes. Dots indicate the number of resistance gene for isolate. (B) A. baumannii ICL-II harbored more ISAba1 than non-ICL-II isolates. Colors are indicated for isolates belonging to different MLST. Boxplots for each MLST display the mean number of mobile element. Dots indicate the number of ISAba1 for each isolate.
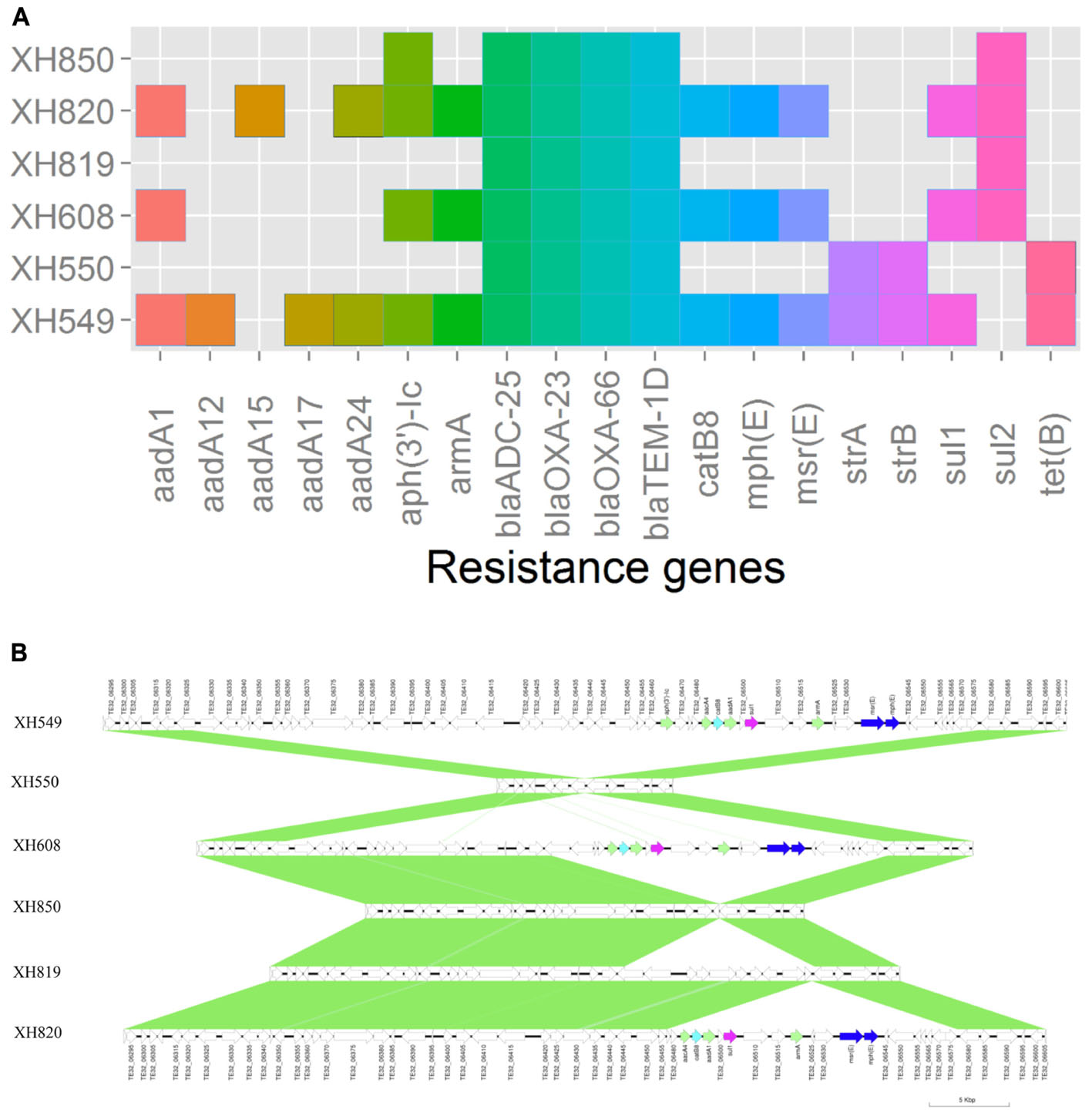
FIGURE 4. Gene acquire and loss in three isolates pairs of A. baumannii. (A) Heatmap of resistance genes in three isolates pairs. Both XH549 and XH608 represent resistance gene loss. XH819 was shown to acquire resistance genes. (B) Comparative analysis of the genomes of three isolates pairs generated by Easyfig. The arrows for resistance genes are shown in different colors.
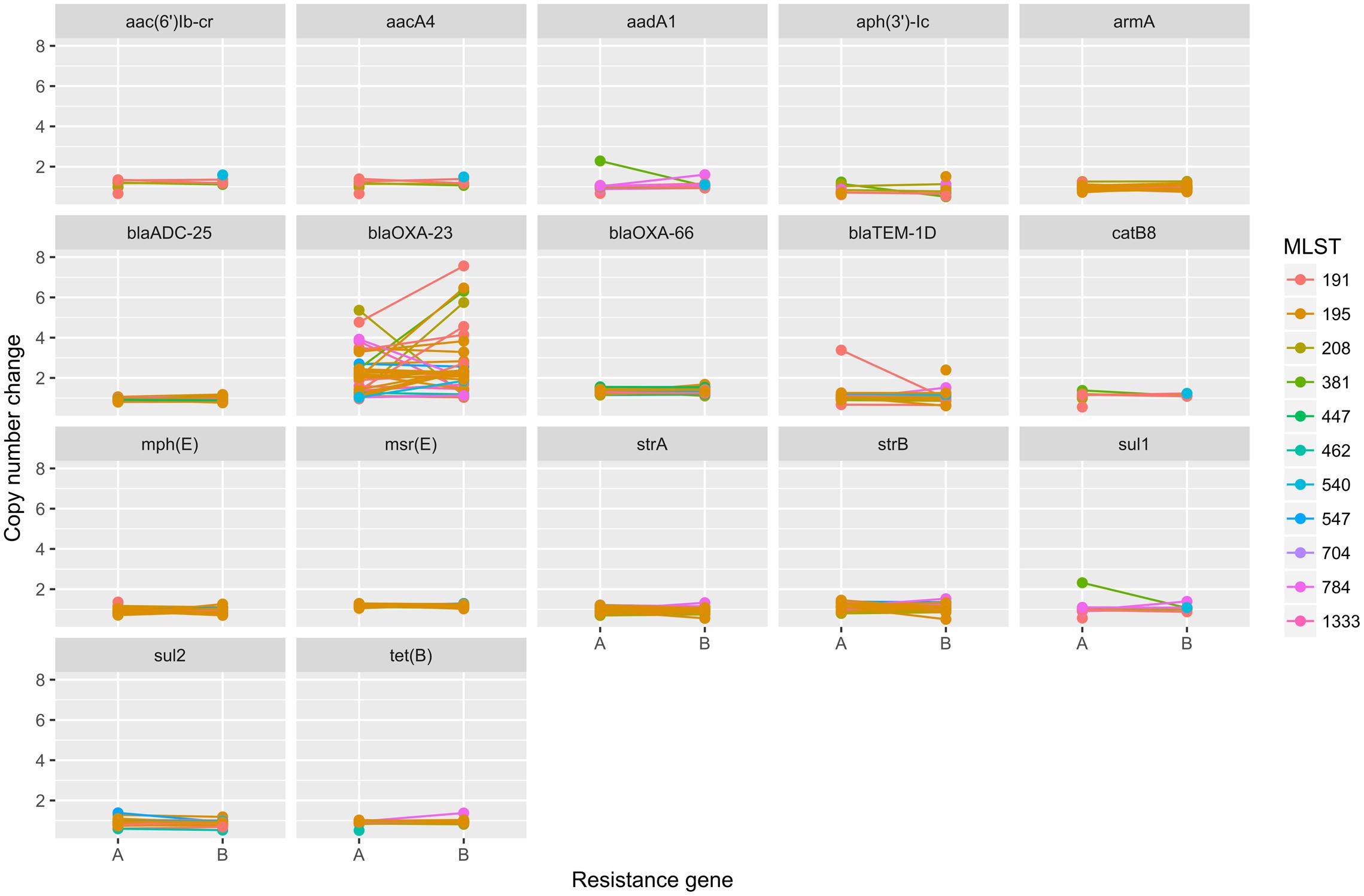
FIGURE 5. The copy number change of resistance genes in Groups A and B in A. baumannii. The copy number of resistance genes were detected by cnv-seq via mapped the WGS data to the sequence of resistance genes.
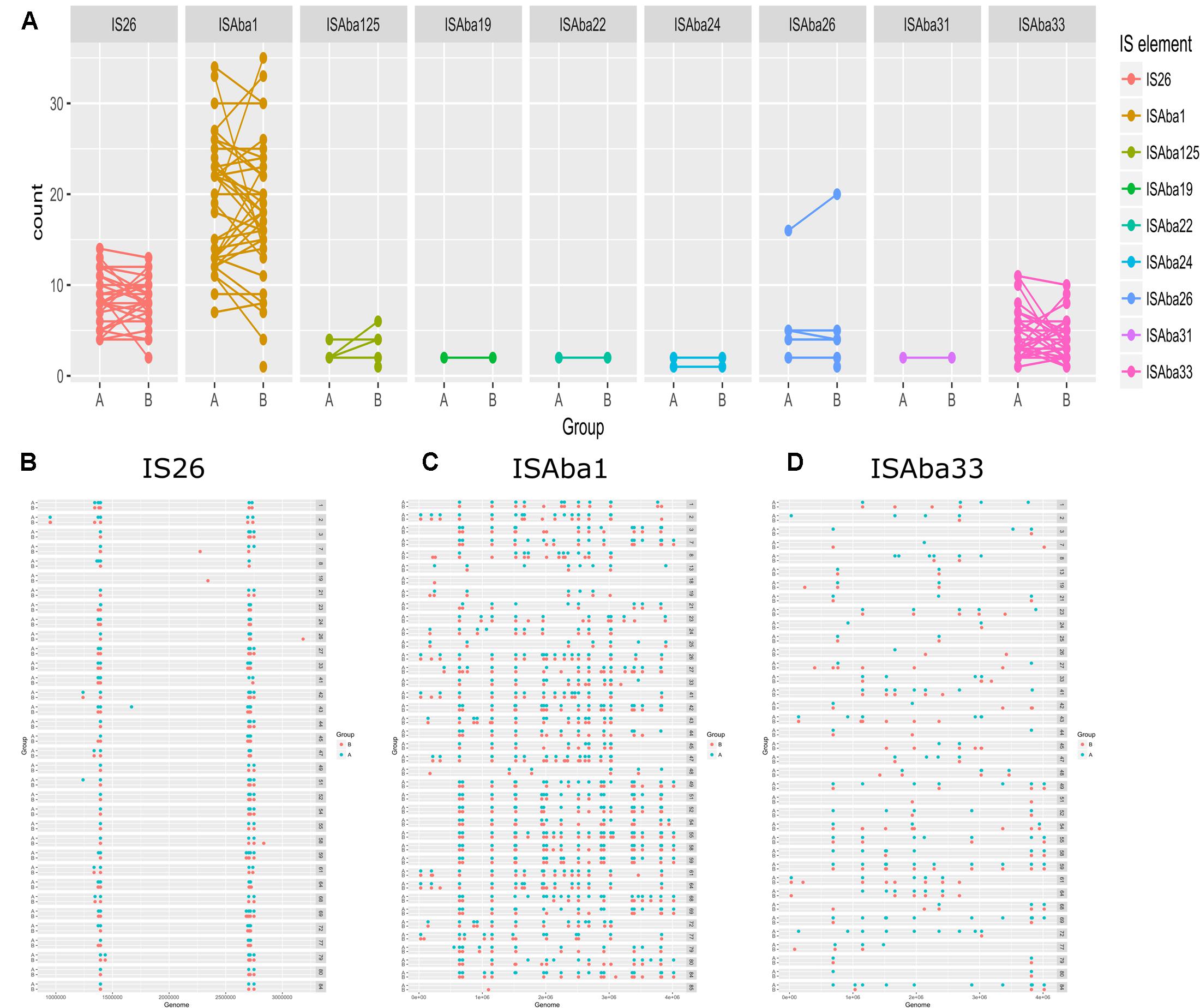
FIGURE 6. The distribution of IS elements in Groups A and B in A. baumannii. (A) The copy number of IS elements in Groups A and B. The position of IS26 (B), ISAba1 (C), ISAba33 (D) in Group A and Group B.
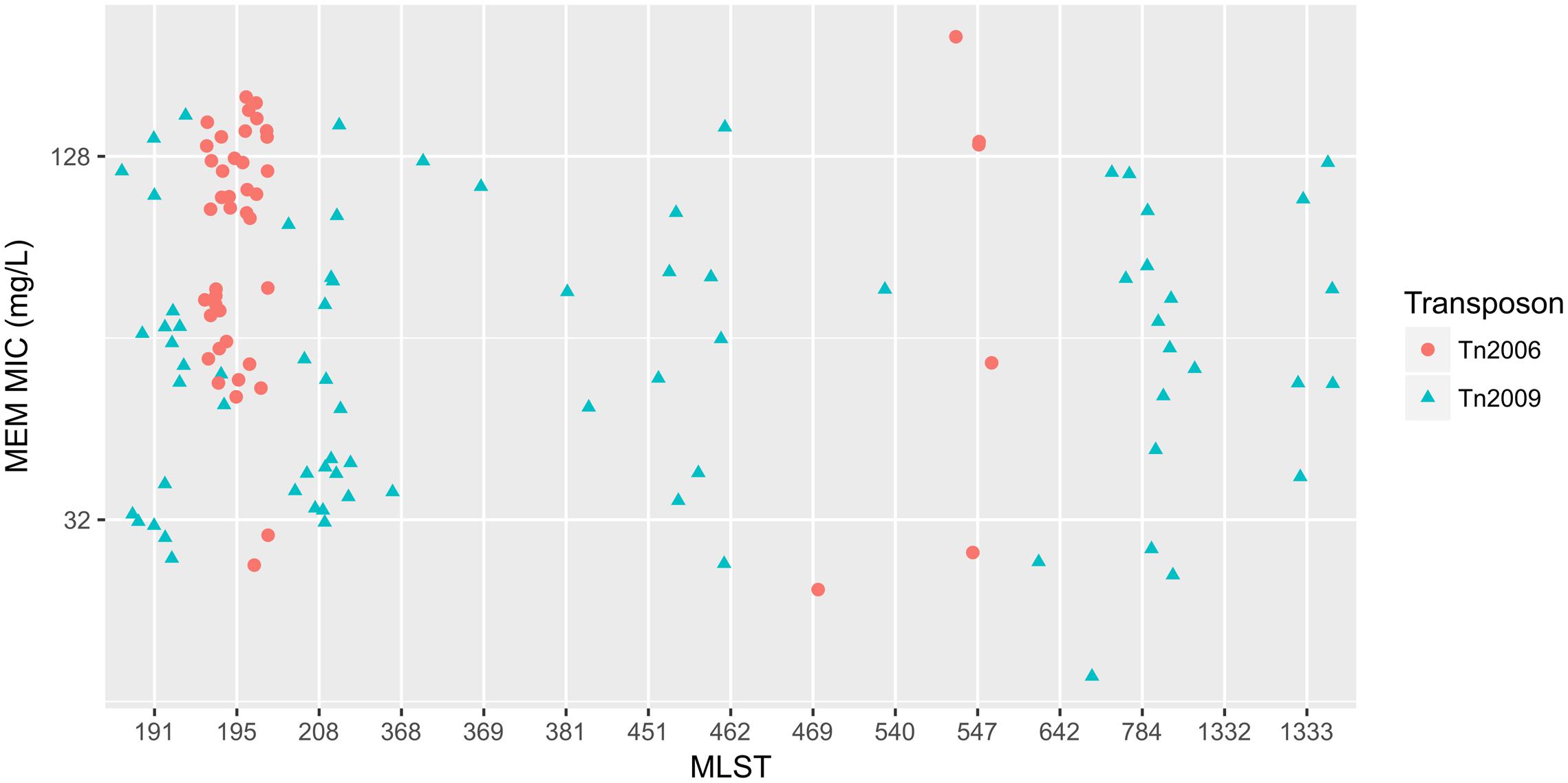
FIGURE 7. The relationship between meropenem MICs and Transposons carrying blaOXA-23 in A. baumannii. The x-axis showed the MLST of strains in A. baumannii. The y-axis presented meropenem MICs. Red circle: Tn2006; cyan triangle: Tn2009.
SNP Analysis
To detect the within-patient diversity of the clonal population, we counted the number of SNPs that each isolate had accumulated based on the first isolate of the pair. The greatest pairwise genome SNP counts from the same patients was less than 20 SNPs (Supplementary Figure S3A). We identified a total of 15 genes that were mutated more than one time (Supplementary Figure S3B), and the detailed SNPs are shown in Table 1. Among them, the ptk (also referred as wzc) mutation occurred in nine isolate pairs. The gene product of ptk has a predicted role in capsule biosynthesis. For example, XH507 generated a mucoviscidose colony phenotype compared with XH506 for it harbored G667D mutation in ptk (Figure 8A). XH507 also displayed capsules with increased thickness when inspected with India ink (Figures 8B,C). Genome sequencing showed that the ptk mutations appeared in ptk conserved domains (Supplementary Figure S4). To investigate whether the genes required for the production of the K1 capsule are required for motility, we performed a motility assay for these isolates. These ptk mutants demonstrated higher motility levels compared to those of the parental strain (student t-test, P = 0.003) (Figure 8D). The result suggested that the capsular polysaccharide contributed to the motility phenotype.
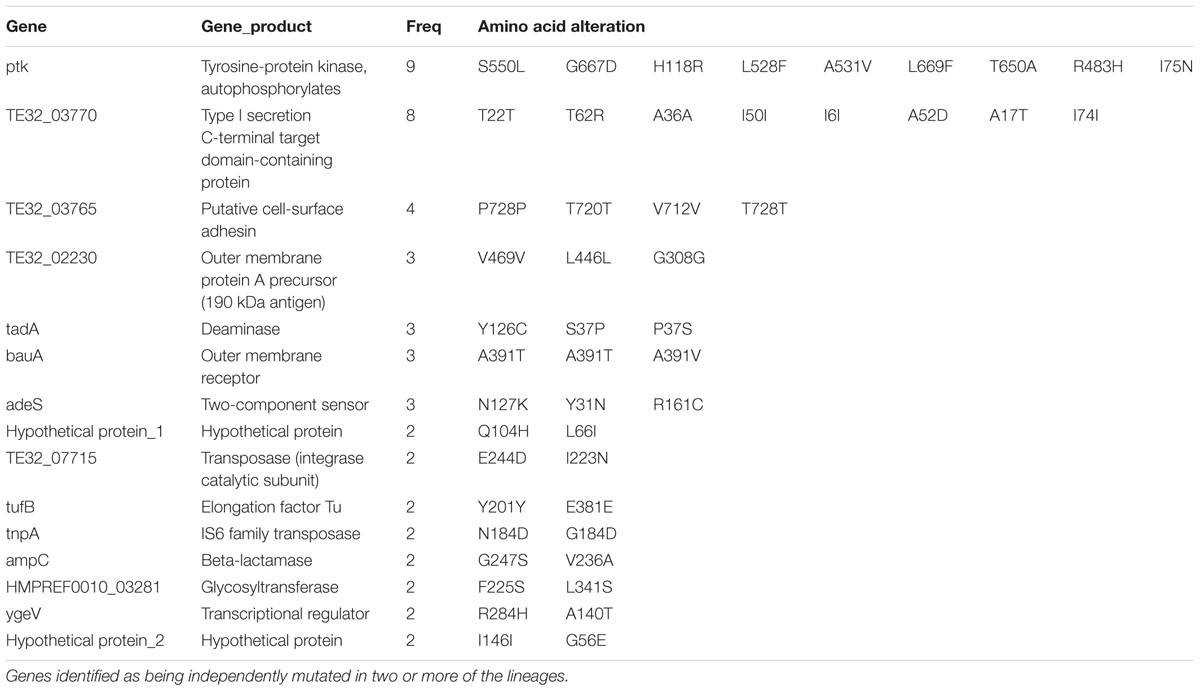
TABLE 1. The SNPs identified in A. baumannii during infection.
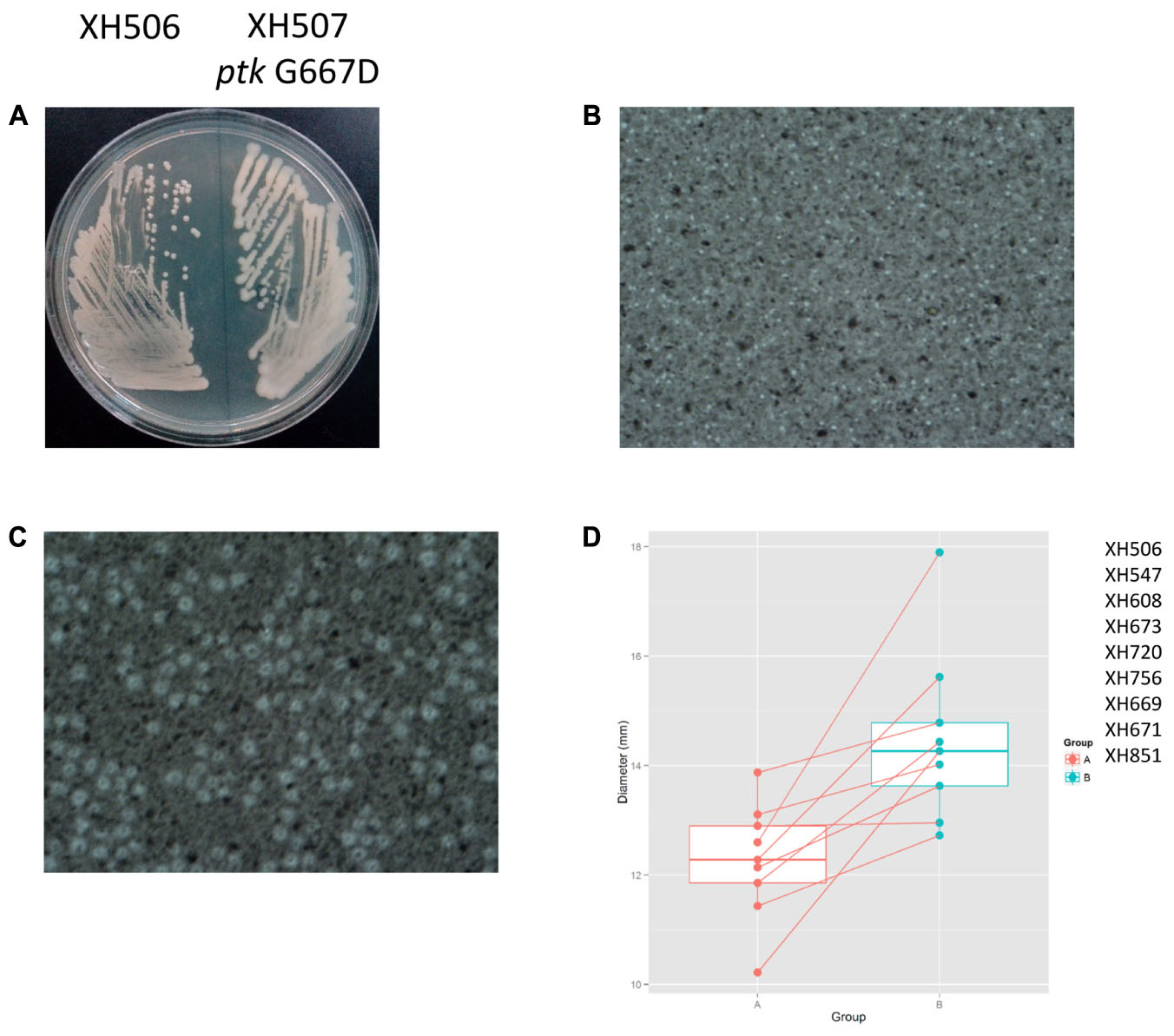
FIGURE 8. Spontaneous point mutations in ptk resulted the misregulation of capsular exopolysaccharide production and higher motility. (A) Colony morphology on MH plates. (B,C) Capsules of XH506 and XH507 (ptk G667D) were analyzed by India ink staining. (D) Spontaneous ptk mutants showed higher motility than the wild type. The y-axis indicates the measurement of diameter (mm) of the cell halos of each isolate pairs harbored ptk mutations in Group B. The data are represented as the means ± SD of experiments that were performed in triplicate.
The mutation in adeRS appeared four times. adeRS was involved in the regulation of efflux pump adeABC. The result of MICs for tigecycline showed the mutation in adeRS increased the tigecycline resistance in A. baumannii (Table 2). The result illustrated the evolution steps of tigecycline resistance in vivo in A. baumannii.
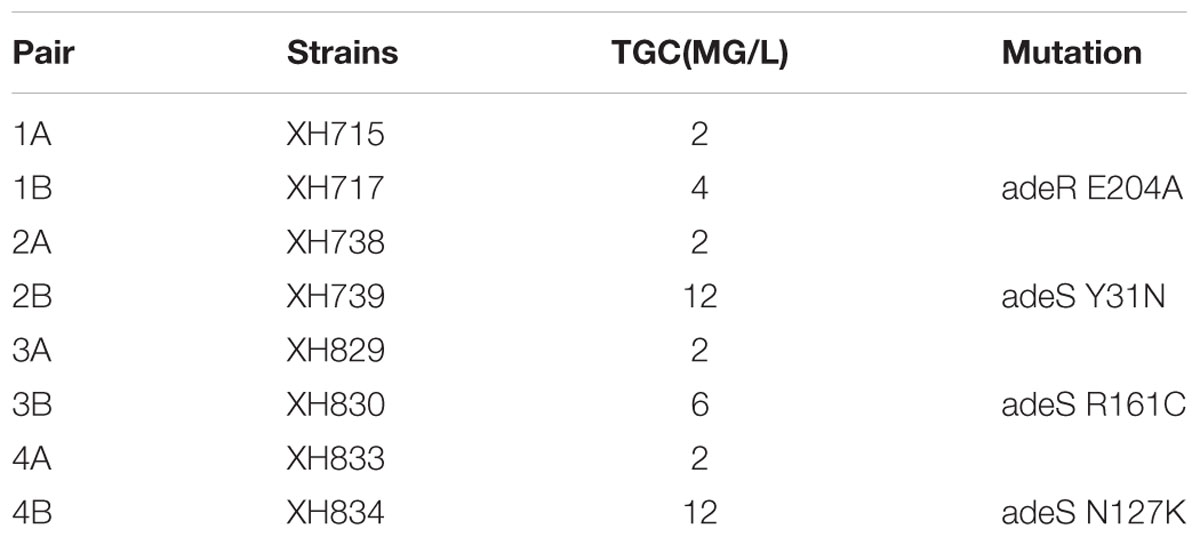
TABLE 2. adeRS mutation conferred tigecycline resistance.
Discussion
In this study, to better understand the genotypic and phenotypic properties underlying the worldwide distribution and evolution of a successful lineage of A. baumannii, we explored the evolution pathway of A. baumannii in vivo. A. baumannii comprises three major international lineages: EC I, II and III (Dijkshoorn et al., 1996; van Dessel et al., 2004). ICL-II, a subgroup of EC II, has spread globally and is the dominant CC of carbapenem-resistant A. baumannii (Ruan et al., 2013). Our result are also consistent with those of a recent study by Ruan et al. (2013), who found that isolates from ICL-II exhibited greater resistance to antibiotics than did non-ICL-II isolates. The explanation of the greater resistance might be that isolates from ICL-II harbored more resistance genes than non-ICL-II isolates did. To assess the contribution of mobile elements to the development of antibiotic resistance, we detected the insertion of IS in all of the genome-sequencing isolates to determine the role of IS in the expression of antibiotic resistance genes in A. baumannii. ISAba1 and IS26 appeared more frequently in ICL-II isolates than in non-ICL-II isolates, which might provide a higher potential to acquire and express resistance gene in the ICL-II isolates.
Two major factors might contribute to the persistence of A. baumannii in the host: more resistance and more mucoid. Our data showed that A. baumannii demonstrated resistance to major antimicrobial agents. Antibiotic resistance might provide A. baumannii with a selective advantage in an environment where bacteria would be exposed to antibiotics (Visca et al., 2011). Furthermore, our data showed that A. baumannii became more resistant to CAZ, AZM, and S after within-host evolution. It might cause by ST switching that the former bacteria was replaced by ICL-II strains. Moreover, four isolate pairs presented tigecycline resistance evolution in vivo. They harbored mutations in adeRS, and the MICs for tigecycline increased (Ruzin et al., 2007). Thus, A. baumannii was able to become more resistant to eradication by antibiotics.
A more mucoid phenotype indicated an overproduction of capsular exopolysaccharides, which was caused by the mutations in ptk (Geisinger and Isberg, 2015). The overproduction of capsular exopolysaccharides would help bacteria colonize newly available spaces by producing adhesins, and then protect the colonized niche from encroachment by competitors. Especially at the air–liquid interface, the overproduction of capsular exopolysaccharides would push the bacteria themselves into more nutrient and oxygen-rich regions, while smothering their competitors (Hibbing et al., 2010). Our results showed that the overproduction of capsular exopolysaccharides also resulted in higher motility in A. baumannii (McQueary et al., 2012). The capsular exopolysaccharides also have demonstrated roles on biofilm modulation (Geisinger and Isberg, 2015). It was proposed that A. baumannii would use motility to outcompete other bacteria for more suitable regions in vivo and to increase resistance against desiccation and detergents via biofilm. As a consequence, becoming more mucoid would promote the persistence of A. baumannii within the host.
Previous studies in Burkholderia dolosa and other organisms have identified convergent evolution patterns, including decreased virulence and increased antimicrobial resistance (Lieberman et al., 2011). The convergent evolution indicated that bacteria undergo special genetic changes in vivo, which helped the bacteria keep a substantial and sustained niche shift away from becoming pathogenic. We observed point mutations, insertions, deletions and copy number dynamics during within-host evolution of A. baumannii in this study. The point mutation in adeRS led to increased antimicrobial resistance, which confirmed previous findings (Lieberman et al., 2011). We also detected the amino acid changes in AmpC, which were located in the AmpC Ω-loop domain. The amino acid alteration in AmpC might result in the extension of the antibiotic-resistance spectrum (Tian et al., 2011), although the more common mechanism to increase AmpC activity is through the hyper-production of AmpC via inserting ISAba1 into the promoter of the ampC gene (Heritier et al., 2006). OmpA is a β-barrel porin in A. baumannii (Smith et al., 2007). OmpA played a role in biofilm formation and in interaction with eukaryotic cells (Gaddy et al., 2009). The overexpression of ompA was a risk factor associated with pneumonia, bacteremia and mortality in A. baumannii (Sanchez-Encinales et al., 2017). OmpA also played a role in antimicrobial resistance in A. baumannii (Smani et al., 2014). Bacterial tRNA adenosine deaminase (TadA) would convert adenosine to inosine at the wobble position of tRNAArg2. This process enabled the single tRNA to recognize three different arginine codons in mRNA (Losey et al., 2006). It was shown that tadA is an essential gene in E. coli, highlighting the importance of inosine at the wobble position in prokaryotes (Wolf et al., 2002). Elongation factor Tuf is a conserved protein acted as a surface exposed plasminogen-binding protein (Kunert et al., 2007). A. baumannii EF-Tu was associated with the bacterial cell surface, OMVs and fibronectin (Dallo et al., 2012). EF-Tu would active plasminogen to active plasmin, then active plasmin proteolytically degrade fibrinogen and the key complement component C3b. EF-Tu was considered as multifunctional protein which play a role in virulence of A. baumannii via aiding evasion of the complement system (Koenigs et al., 2015). As mentioned above, the ICL-II strains harbored large number of ISAba1. The high activity of ISAba1 led to copy number dynamics in OXA-23, which was observed in within-host evolution in this study. It was reported that multiplication of blaOXA-23 was common in clinical A. baumannii (Hua et al., 2016). Another explanation might be the usage of carbapenem in a clinical context. With increased exposure to the selective pressure of carbapenem, the strain with high copy number of OXA-23 might be then selected for. The OXA-23 gene had been mobilized to form Tn2008, Tn2008B, Tn2006, and Tn2009. Tn2006 was most frequently observed worldwide (Nigro and Hall, 2016). It was reported that Tn2008 were detected in 96.4% isolates and Tn2006 in 4.6% isolates in carbapenem-resistant A. baumannii in Shanghai, China (Chen et al., 2017). In this study, Tn2006 presented in 37% isolates, while 54% isolates harbored Tn2009. The transposons contributed significantly to the dissemination of OXA-23, although the isolation rate of transposons was different. It was reported that the commonly mutated genes included pmrAB, adeRS, transporters, iron acquisition gene, motility and adhesion related genes and the tyrosine kinase wzc (ptk) gene during in vivo evolution in A. baumannii (Wright et al., 2016). The gene adeRS and ptk were shared between Wright et al. (2016) and our study, which demonstrated the existence of mutation hotspot in within-host evolution in A. baumannii.
Approximately half of the isolates from the same patients harbored different STs. The significance of this observation is equivocal and may indicate that the latter isolate was a re-infection or that it gained a growth advantage within the host. We could not differentiate between these possibilities for the numbers of isolates from the same patients were limited. The resistance genes in the isolates were predicted by in silico profiles using WGS data against the ResFinder database (Zankari, 2014). Only some antibiotic resistance profiles were predicted, which indicated that further experiments are required to fully understand the resistance mechanism in A. baumannii, although the resistance database was useful for some predictions.
The strains used in this study were isolated from sputum. This is a limitation of this study that only single isolate was sequenced from each sample, then we were not able to detect the population of the strains that were presented in a patient. To figure out the population of the strains, multiple isolates at multiple isolated time need to be analyzed. Our study focused on single isolate of A. baumannii evolved in host. Meanwhile, multiple studies already used isolates from sputum to research in vivo evolution. Marvig et al. (2015) sequenced the whole genomes of a total of 474 isolates of Pseudomonas aeruginosa isolated from sputum. Wright et al. (2016) detected the genome dynamics of MDR A. baumannii during infection and treatment. The 54% (74/136) strains were isolated from sputum in Wright et al. (2016) study. Overall, this study explored the within-host evolution in A. baumannii in real time and highlighted the ability of NGS to explore evolutionary events within resistant organisms.
Nucleotide Sequence Accession Numbers
The whole-genome shotgun projects of A. baumannii strains have been deposited at DDBJ/EMBL/GenBank under the accession numbers LYEX00000000- LYLM00000000, respectively. The versions described in this paper are versions LYEX00000000- LYLM00000000.
Author Contributions
XH analyzed the genomic data and drafted the manuscript. ZZ, QY, QS, QX, JW, KS, FZ, LS, ZR, and YJ performed the experiment. XH and YY designed the study and drafted the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the National Natural Science of China (81230039, 81371858, 31670135, 81401698), the Natural Science Foundation of Zhejiang Province, China (LY15H190004) and Zhejiang Province Medical Platform Backbone Talent Plan (2016DTA003). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
We thank the team of curators of the Acinetobacter MLST system for curating the data and making them publicly available at http://pubmlst.org/abaumannii/.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.01256/full#supplementary-material
FIGURE S1 | The overview of the 172 genome-sequenced A. baumannii isolates. (A) Each symbol represents a genome-sequenced A. baumannii isolate. The y-axis indicates the patient from whom the isolate was obtained, while x-axis indicates the sampling time for each isolate. Colors are indicated for isolates belonging to different MLST patterns. The line connecting two symbols indicated the time between two isolate samplings from the same patient. (B) The distribution of the isolation range for all 172 genome-sequenced isolates. Colors are indicated for isolates belonging to different MLST patterns.
FIGURE S2 | Resistance genes detected in these genome-sequencing isolates of A. baumannii.
FIGURE S3 |(A) Distribution of SNPs in Group B compared with Group A in A. baumannii. (B) Pathoadaptive genes mutated in Group B compare with Group A. The blue squares in the large matrix denote whether the genes underwent non-synonymous mutation. The analysis was performed based only on non-synonymous mutated genes that were altered independently in two of the lineages.
FIGURE S4 | Schematic of the ptk domains indicates the location of point mutations in the conserved motifs. White: conserved motif.
Footnotes
References
Adams, M. D., Bishop, B., and Wright, M. S. (2016). Quantitative assessment of insertion sequence impact on bacterial genome architecture. Microb. Genomics 2:e000062. doi: 10.1099/mgen.0.000062
Cerqueira, G. M., and Peleg, A. Y. (2011). Insights into Acinetobacter baumannii pathogenicity. IUBMB Life 63, 1055–1060. doi: 10.1002/iub.533
Chen, Y., Gao, J., Zhang, H., and Ying, C. (2017). Spread of the blaOXA-23-containing Tn2008 in carbapenem-resistant Acinetobacter baumannii isolates grouped in CC92 from China. Front. Microbiol. 8:163. doi: 10.3389/fmicb.2017.00163
Chuang, Y. C., Sheng, W. H., Li, S. Y., Lin, Y. C., Wang, J. T., Chen, Y. C., et al. (2011). Influence of genospecies of Acinetobacter baumannii complex on clinical outcomes of patients with acinetobacter bacteremia. Clin. Infect. Dis. 52, 352–360. doi: 10.1093/cid/ciq154
Dallo, S. F., Zhang, B., Denno, J., Hong, S., Tsai, A., Haskins, W., et al. (2012). Association of Acinetobacter baumannii EF-Tu with cell surface, outer membrane vesicles, and fibronectin. ScientificWorldJournal 2012:128705. doi: 10.1100/2012/128705
Deatherage, D. E., and Barrick, J. E. (2014). Identification of mutations in laboratory-evolved microbes from next-generation sequencing data using breseq. Methods Mol. Biol. 1151, 165–188. doi: 10.1007/978-1-4939-0554-6_12
Dijkshoorn, L., Aucken, H., Gerner-Smidt, P., Janssen, P., Kaufmann, M. E., Garaizar, J., et al. (1996). Comparison of outbreak and nonoutbreak Acinetobacter baumannii strains by genotypic and phenotypic methods. J. Clin. Microbiol. 34, 1519–1525.
Gaddy, J. A., Tomaras, A. P., and Actis, L. A. (2009). The Acinetobacter baumannii 19606 OmpA protein plays a role in biofilm formation on abiotic surfaces and in the interaction of this pathogen with eukaryotic cells. Infect. Immun. 77, 3150–3160. doi: 10.1128/IAI.00096-09
Geisinger, E., and Isberg, R. R. (2015). Antibiotic modulation of capsular exopolysaccharide and virulence in Acinetobacter baumannii. PLoS Pathog. 11:e1004691. doi: 10.1371/journal.ppat.1004691
Heritier, C., Poirel, L., and Nordmann, P. (2006). Cephalosporinase over-expression resulting from insertion of ISAba1 in Acinetobacter baumannii. Clin. Microbiol. Infect. 12, 123–130. doi: 10.1111/j.1469-0691.2005.01320.x
Hibbing, M. E., Fuqua, C., Parsek, M. R., and Peterson, S. B. (2010). Bacterial competition: surviving and thriving in the microbial jungle. Nat. Rev. Microbiol. 8, 15–25. doi: 10.1038/nrmicro2259
Hua, X., Shu, J., Ruan, Z., Yu, Y., and Feng, Y. (2016). Multiplication of blaOXA-23 is common in clinical Acinetobacter baumannii, but does not enhance carbapenem resistance. J. Antimicrob. Chemother. 71, 3381–3385. doi: 10.1093/jac/dkw310
Inouye, M., Dashnow, H., Raven, L. A., Schultz, M. B., Pope, B. J., Tomita, T., et al. (2014). SRST2: rapid genomic surveillance for public health and hospital microbiology labs. Genome Med. 6, 90. doi: 10.1186/s13073-014-0090-6
Jones, C. L., Clancy, M., Honnold, C., Singh, S., Snesrud, E., Onmus-Leone, F., et al. (2015). Fatal outbreak of an emerging clone of extensively drug-resistant Acinetobacter baumannii with enhanced virulence. Clin. Infect. Dis. 61, 145–154. doi: 10.1093/cid/civ225
Kamolvit, W., Sidjabat, H. E., and Paterson, D. L. (2015). Molecular epidemiology and mechanisms of carbapenem resistance of Acinetobacter spp. in Asia and Oceania. Microb. Drug Resist. 21, 424–434. doi: 10.1089/mdr.2014.0234
Klughammer, J., Dittrich, M., Blom, J., Mitesser, V., Vogel, U., Frosch, M., et al. (2017). Comparative genome sequencing reveals within-host genetic changes in Neisseria meningitidis during invasive disease. PLoS ONE 12:e0169892. doi: 10.1371/journal.pone.0169892
Koenigs, A., Zipfel, P. F., and Kraiczy, P. (2015). Translation elongation factor Tuf of Acinetobacter baumannii is a plasminogen-binding protein. PLoS ONE 10:e0134418. doi: 10.1371/journal.pone.0134418
Kunert, A., Losse, J., Gruszin, C., Huhn, M., Kaendler, K., Mikkat, S., et al. (2007). Immune evasion of the human pathogen Pseudomonas aeruginosa: elongation factor Tuf is a factor H and plasminogen binding protein. J. Immunol. 179, 2979–2988.
Lieberman, T. D., Michel, J. B., Aingaran, M., Potter-Bynoe, G., Roux, D., Davis, M. R. Jr., et al. (2011). Parallel bacterial evolution within multiple patients identifies candidate pathogenicity genes. Nat. Genet. 43, 1275–1280. doi: 10.1038/ng.997
Losey, H. C., Ruthenburg, A. J., and Verdine, G. L. (2006). Crystal structure of Staphylococcus aureus tRNA adenosine deaminase TadA in complex with RNA. Nat. Struct. Mol. Biol. 13, 153–159. doi: 10.1038/nsmb1047
Marvig, R. L., Johansen, H. K., Molin, S., and Jelsbak, L. (2013). Genome analysis of a transmissible lineage of Pseudomonas aeruginosa reveals pathoadaptive mutations and distinct evolutionary paths of hypermutators. PLoS Genet. 9:e1003741. doi: 10.1371/journal.pgen.1003741
Marvig, R. L., Sommer, L. M., Molin, S., and Johansen, H. K. (2015). Convergent evolution and adaptation of Pseudomonas aeruginosa within patients with cystic fibrosis. Nat. Genet. 47, 57–64. doi: 10.1038/ng.3148
McQueary, C. N., Kirkup, B. C., Si, Y., Barlow, M., Actis, L. A., Craft, D. W., et al. (2012). Extracellular stress and lipopolysaccharide modulate Acinetobacter baumannii surface-associated motility. J. Microbiol. 50, 434–443. doi: 10.1007/s12275-012-1555-1
Morelli, G., Song, Y., Mazzoni, C. J., Eppinger, M., Roumagnac, P., Wagner, D. M., et al. (2010). Yersinia pestis genome sequencing identifies patterns of global phylogenetic diversity. Nat. Genet. 42, 1140–1143. doi: 10.1038/ng.705
Mutreja, A., Kim, D. W., Thomson, N. R., Connor, T. R., Lee, J. H., Kariuki, S., et al. (2011). Evidence for several waves of global transmission in the seventh cholera pandemic. Nature 477, 462–465. doi: 10.1038/nature10392
Nigro, S. J., and Hall, R. M. (2016). Structure and context of Acinetobacter transposons carrying the oxa23 carbapenemase gene. J. Antimicrob. Chemother. 71, 1135–1147. doi: 10.1093/jac/dkv440
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Patel, J., Cockerill, F., Alder, J., Bradford, P., and Eliopoulus, G. (2015). Performance Standards for Antimicrobial Susceptibility Testing. M100-M125. Wayne, PA: CLSI.
Ruan, Z., Chen, Y., Jiang, Y., Zhou, H., Zhou, Z., Fu, Y., et al. (2013). Wide distribution of CC92 carbapenem-resistant and OXA-23-producing Acinetobacter baumannii in multiple provinces of China. Int. J. Antimicrob. Agents 42, 322–328. doi: 10.1016/j.ijantimicag.2013.06.019
Russo, T. A., Luke, N. R., Beanan, J. M., Olson, R., Sauberan, S. L., MacDonald, U., et al. (2010). The K1 capsular polysaccharide of Acinetobacter baumannii strain 307-0294 is a major virulence factor. Infect. Immun. 78, 3993–4000. doi: 10.1128/IAI.00366-10
Ruzin, A., Keeney, D., and Bradford, P. A. (2007). AdeABC multidrug efflux pump is associated with decreased susceptibility to tigecycline in Acinetobacter calcoaceticus-Acinetobacter baumannii complex. J. Antimicrob. Chemother. 59, 1001–1004. doi: 10.1093/jac/dkm058
Sanchez-Encinales, V., Alvarez-Marin, R., Pachon-Ibanez, M. E., Fernandez-Cuenca, F., Pascual, A., Garnacho-Montero, J., et al. (2017). Overproduction of outer membrane protein A (OmpA) by Acinetobacter baumannii is a risk factor for nosocomial pneumonia, bacteremia and mortality increase. J. Infect. Dis. doi: 10.1093/infdis/jix010 [Epub ahead of print].
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Smani, Y., Fabrega, A., Roca, I., Sanchez-Encinales, V., Vila, J., and Pachon, J. (2014). Role of OmpA in the multidrug resistance phenotype of Acinetobacter baumannii. Antimicrob. Agents Chemother. 58, 1806–1808. doi: 10.1128/AAC.02101-13
Smith, S. G., Mahon, V., Lambert, M. A., and Fagan, R. P. (2007). A molecular Swiss army knife: OmpA structure, function and expression. FEMS Microbiol. Lett. 273, 1–11. doi: 10.1111/j.1574-6968.2007.00778.x
Tian, G. B., Adams-Haduch, J. M., Taracila, M., Bonomo, R. A., Wang, H. N., and Doi, Y. (2011). Extended-spectrum AmpC cephalosporinase in Acinetobacter baumannii: ADC-56 confers resistance to cefepime. Antimicrob. Agents Chemother. 55, 4922–4925. doi: 10.1128/AAC.00704-11
van Dessel, H., Dijkshoorn, L., van der Reijden, T., Bakker, N., Paauw, A., van den Broek, P., et al. (2004). Identification of a new geographically widespread multiresistant Acinetobacter baumannii clone from European hospitals. Res. Microbiol. 155, 105–112. doi: 10.1016/j.resmic.2003.10.003
Visca, P., Seifert, H., and Towner, K. J. (2011). Acinetobacter infection–an emerging threat to human health. IUBMB Life 63, 1048–1054. doi: 10.1002/iub.534
Wilson, D. J. (2012). Insights from genomics into bacterial pathogen populations. PLoS Pathog. 8:e1002874. doi: 10.1371/journal.ppat.1002874
Wolf, J., Gerber, A. P., and Keller, W. (2002). tadA, an essential tRNA-specific adenosine deaminase from Escherichia coli. EMBO J. 21, 3841–3851. doi: 10.1093/emboj/cdf362
Wright, M. S., Iovleva, A., Jacobs, M. R., Bonomo, R. A., and Adams, M. D. (2016). Genome dynamics of multidrug-resistant Acinetobacter baumannii during infection and treatment. Genome Med. 8, 26. doi: 10.1186/s13073-016-0279-y
Xie, C., and Tammi, M. T. (2009). CNV-seq, a new method to detect copy number variation using high-throughput sequencing. BMC Bioinformatics 10:80. doi: 10.1186/1471-2105-10-80
Zankari, E. (2014). Comparison of the web tools ARG-ANNOT and ResFinder for detection of resistance genes in bacteria. Antimicrob. Agents Chemother. 58, 4986. doi: 10.1128/AAC.02620-14
Keywords: Acinetobacter baumannii, within-host evolution, whole-genome sequencing, mucoid, ICL-II
Citation: Hua X, Zhou Z, Yang Q, Shi Q, Xu Q, Wang J, Shi K, Zhao F, Sun L, Ruan Z, Jiang Y and Yu Y (2017) Evolution of Acinetobacter baumannii In Vivo: International Clone II, More Resistance to Ceftazidime, Mutation in ptk. Front. Microbiol. 8:1256. doi: 10.3389/fmicb.2017.01256
Received: 27 January 2017; Accepted: 23 June 2017;
Published: 10 July 2017.
Edited by:
Dongsheng Zhou, Beijing Institute of Microbiology and Epidemiology, ChinaReviewed by:
Raffaele Zarrilli, University of Naples Federico II, ItalyBenjamin Andrew Evans, University of East Anglia, United Kingdom
Copyright © 2017 Hua, Zhou, Yang, Shi, Xu, Wang, Shi, Zhao, Sun, Ruan, Jiang and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yunsong Yu, eXZ5czExOUAxNjMuY29t
†These authors have contributed equally to this work.