 Sagar M. Utturkar
Sagar M. Utturkar Dawn M. Klingeman2,3
Dawn M. Klingeman2,3 Richard A. Hurt Jr.
Richard A. Hurt Jr. Steven D. Brown
Steven D. Brown- 1Graduate School of Genome Science and Technology, University of Tennessee, Knoxville, TN, United States
- 2Biosciences Division, Oak Ridge National Laboratory, Oak Ridge, TN, United States
- 3BioEnergy Science Center, Oak Ridge, TN, United States
This study characterized regions of DNA which remained unassembled by either PacBio and Illumina sequencing technologies for seven bacterial genomes. Two genomes were manually finished using bioinformatics and PCR/Sanger sequencing approaches and regions not assembled by automated software were analyzed. Gaps present within Illumina assemblies mostly correspond to repetitive DNA regions such as multiple rRNA operon sequences. PacBio gap sequences were evaluated for several properties such as GC content, read coverage, gap length, ability to form strong secondary structures, and corresponding annotations. Our hypothesis that strong secondary DNA structures blocked DNA polymerases and contributed to gap sequences was not accepted. PacBio assemblies had few limitations overall and gaps were explained as cumulative effect of lower than average sequence coverage and repetitive sequences at contig termini. An important aspect of the present study is the compilation of biological features that interfered with assembly and included active transposons, multiple plasmid sequences, phage DNA integration, and large sequence duplication. Our targeted genome finishing approach and systematic evaluation of the unassembled DNA will be useful for others looking to close, finish, and polish microbial genome sequences.
Introduction
Since the first Next-Generation Sequencing (NGS) platform was released by 454 Life science (Margulies et al., 2005), there has been a remarkable increase in sequencing efficiency, throughput, and read lengths (Koren and Phillippy, 2014). Sequencing costs continue to drop dramatically and whole genome sequencing is within reach for small-scale laboratories on relatively modest budgets. During the past decade, the sequencing industry has been largely dominated by the second generation, sequencing by synthesis platforms such as Illumina which are characterized by the low-cost, high-throughput, and short reads with high accuracy (van Dijk et al., 2014). Short sequencing reads have limited power to resolve large repetitive regions even within small microbial genomes (Chain et al., 2009; Nagarajan and Pop, 2013). Short read technologies are generally able to resolve microbial genomes up to the high-quality draft standard (Treangen and Salzberg, 2012), which is sufficient for many applications such as understanding gene-coding potential, strain typing, or pan-genome analysis (Roberts et al., 2013). However, draft genomes are fragmented assemblies that can contain misassembled regions, incorrect gene calls, and other artifacts. Fragmented assemblies are often attributed to repetitive DNA regions (such as rRNA operons) which are abundant in microbial genomes and present the greatest technical challenge to the assembly process especially when the repetitive region is longer than the read lengths (Treangen and Salzberg, 2012; Brown S. et al., 2014). Finished genome sequences are high quality by definition, represent more accurate genomic information and are often desirable for model organisms and industrially important microbes (Fraser et al., 2002; Thomma et al., 2015).
The application of new protocols (e.g., use of complementary paired and mate-pair libraries) and algorithm developments have facilitated improved genome assemblies. Progress in next-generation sequencing platforms, metrics, and performances has been reviewed (Liu et al., 2012; Quail et al., 2012; van Dijk et al., 2014), assessments for various assembly methods conducted (Salzberg et al., 2012; Magoc et al., 2013; Koren and Phillippy, 2014; Utturkar et al., 2014), and various applications (Buermans and den Dunnen, 2014; Rhoads and Au, 2015) have been discussed elsewhere. Development of so-called third-generation sequencing platforms for single-molecule sequencing is a more recent development for producing long sequence reads which facilitate assembly. Pacific Biosciences (PacBio) RS-II instrument outputs are characterized by long reads and average read lengths are reported in the range of 10–11 kb (Hua and Hua, 2016). Relatively high rate of random errors within individual reads can be overcome by error-correction algorithms given sufficient sequencing depth (Chin et al., 2013). The longest reported PacBio reads from the RS-II instrument extend well beyond 20 kb. A key aspect of longer reads is their ability to span large repetitive regions, which greatly aids the assembly process (Brown S. et al., 2014; Koren and Phillippy, 2014; Utturkar et al., 2015) when sufficient coverage (>100X) is available (Chin et al., 2013; Koren et al., 2013). In 2014, Oxford Nanopore Technologies released a nanopore-based sequencer for long single molecule DNA reads (Feng et al., 2015). In the time since its release, hybrid and de novo assembly strategies have also been developed and tested using Oxford Nanopore datasets (Risse et al., 2015; Deschamps et al., 2016).
The application of longer sequencing reads facilitated finished genome assemblies for many bacterial genomes (Koren et al., 2013). The utility of long reads is demonstrated by the increasing number of finished genomes obtained using PacBio technology (Koren et al., 2013; Brown S. D. et al., 2014; Eckweiler et al., 2014; Harhay et al., 2014; Mehnaz et al., 2014; Satou et al., 2014; Kanda et al., 2015; Nakano et al., 2015). However, examples exist where genomes are only resolved into 10 or fewer contigs despite high (>100X) PacBio sequence coverage (Hoefler et al., 2013; Dunitz et al., 2014; Bishnoi et al., 2015; Okutani et al., 2015; Shapiro et al., 2015; The NCTC 3000 Project, 2016), and manual finishing is necessary to obtain complete genome sequences. Substantial developments for long read assembly methods and analysis are reported, but information is lacking on the nature of unassembled DNA regions or gaps within unfinished PacBio assemblies. Therefore, a systematic evaluation of draft, near-finished (containing up to 10 contigs) and finished genome assemblies would be useful to reveal the features and properties of the unassembled DNA regions from Illumina and/or PacBio platforms.
In the present study, seven bacterial genomes were sequenced using Illumina Paired-End (PE) and PacBio RS-II platforms. De novo and hybrid genome assemblies were created using platform specific or hybrid datasets from Illumina and PacBio platforms with various assembly programs and parameter optimizations. In this focused study, manual genome finishing was performed for two genomes, generating up to finished grade assemblies and permitted further analysis of prior gap sequences for which there is a dearth of data. Additional genome polishing was performed on PacBio assemblies with the recently described Pilon software (Walker et al., 2014). The impact of improving genome assemblies and polishing was assessed by several metrics that included gene models. This study offers insights into the nature of gaps associated with Illumina and PacBio assemblies of microbial genomes, describes bioinformatics and manual steps for assembly improvement and underlines the importance of post-assembly polishing steps for genome refinement.
Materials and Methods
Whole Genome Sequencing
Whole genome sequencing data for seven microorganisms (Clostridium pasteurianum ATCC 6013 (Pyne et al., 2014), Clostridium paradoxum JW-YL-7 (Lancaster et al., 2016), Clostridium thermocellum AD2 (Utturkar et al., 2016), Pelosinus fermentans UFO1 (Brown S. D. et al., 2014), P. fermentans JBW45 (De Leon et al., 2015), Halomonas sp. KO116 (O'Dell et al., 2015) and Bacteroides cellulosolvens DSM 2933) (Dassa et al., 2015) using Illumina MiSeq (Illumina, San Diego, CA, USA) (Quail et al., 2012) and PacBio RS-II (Pacific Biosciences, Menlo Park, CA, USA) (Korlach et al., 2010) platforms have been reported. The bacteria were chosen for the availability of Illumina and PacBio sequence data, with most having relevance to bioenergy applications, and in the case of P. fermentans species they are fermentative metal-reducing bacteria. For all genomes in current study, Illumina paired-end library preparation, PacBio SMRTbell library preparation, and sequencing protocols are performed as described previously (Utturkar et al., 2015). GenBank and SRA sequence accession numbers for each genome are provided in Table S1.
Data Quality Control, Genome Assembly, and Annotation
Quality based trimming of raw Illumina data was performed using CLC Genomics workbench software (CLC) to remove bases having PHRED quality score <30 and any reads shorter than 20 bp. Adapter trimming and filtering of raw PacBio data was performed through SMRT analysis software to obtain “filtered subreads” with default parameters (Utturkar et al., 2015). De novo genome assembly of Illumina data was performed using SPAdes version 3.5.0 (Bankevich et al., 2012) and ABySS version 1.5.2 (Simpson et al., 2009) with parameter optimization (Utturkar et al., 2014). Hybrid assembly of Illumina and PacBio data was performed using SPAdes hybrid assembler version 3.5.0 with default parameters. Exact commands used for SPAdes and ABySS assemblies are provided in Section S1. Long read PacBio data were assembled using the SMRT Analysis software and the HGAP protocol (Chin et al., 2013). In the HGAP protocol, the “Target Coverage” parameter was updated to 15X as recommended for microbial genomes (Pacific-Biosciences, 2014a). The specific versions of SMRT Analysis software used for each genome are provided in the results section. Assembly summary statistics were determined using Quast software version 2.3 (Gurevich et al., 2013). Gene-calling and genome annotation were performed through the Prodigal algorithm and microbial genome annotation pipeline at Oak Ridge National Laboratory (Hyatt et al., 2010; Woo et al., 2014).
Manual Genome Finishing
Manual genome finishing was performed using bioinformatics tools and PCR/Sanger sequencing. During bioinformatics steps, contigs from different draft and hybrid genome assemblies were mapped to PacBio-only assemblies using Geneious software version 8.1.6 (Biomatters, Auckland, New Zealand) (Kearse et al., 2012) with default parameters. Mapping results were manually inspected to identify a possible extension (or overhang) relative to reference contigs. Supported extensions were added to the reference contigs and assembly of contigs (super-assembly) was created through Geneious software to derive a longer consensus sequence. See Section S1 for details of the Geneious software modules used in each step. Bioinformatically derived contig extensions and super-assembly derived consensus sequences were verified by PCR and Sanger sequencing. Bioinformatics finishing steps, designing of PCR/Sanger sequencing based validations and various experimental modifications of standard PCR protocol are described in detail in Section S1 with examples of two manually finished genomes (Figures S1, S2, and S3).
Analysis of Unassembled (Gap) DNA
Mapping of Illumina draft contigs to finished/near-finished assemblies was performed using the “Map to Reference” module in the Geneious software, followed by manual inspection to reveal Illumina gaps and associated annotations. PacBio gaps were revealed through manual finishing of two genomes and the resulting sequences were submitted to the mfold web server (Zuker, 2003) to determine DNA folding properties and secondary structures. Default DNA folding parameters in mfold software were modified to mimic the PCR conditions (folding temperature = 550 C, [Na+] concentration = 50 mM, [Mg++] concentration = 2.5 mM). Positional preference was determined using PerPlot and PerScan tools (Mrazek et al., 2011) with default parameters, and genes with periodicity intensity cutoff higher than 2.5 were determined.
Post-assembly Polishing and Validation Steps
PacBio-only assemblies were polished by running one additional round of the Quiver algorithm (Chin et al., 2013), followed by basecall correction through Pilon software (Walker et al., 2014) (version 1.13) with default parameters. Quiver uses PacBio reads while Pilon uses Illumina reads to perform base corrections and derive an accurate consensus sequence. The circular nature of HGAP derived contigs was assessed via the dot-plotting tool Gepard (Krumsiek et al., 2007) and circular genome sequences were derived through an alignment approach described in PacBio training manual (Pacific-Biosciences, 2015). The presence of non-chromosomal DNA such as a plasmid or phage-DNA elements was tested by evaluation of any singleton sequences and/or “deg.fasta” files (which may contain high copy number sequences such as plasmids or phage DNA) generated during the HGAP protocol. For assemblies containing fewer than 5 contigs, each contig was individually tested for circularity. The presence of plasmid DNA was further analyzed by searching for the annotated plasmid related genes such as “RepA—plasmid replication protein.” Additionally, DNA base modification analysis was performed for complete genomes using SMRT analysis software and methylation profiles (Pacific-BioSciences, 2014b) were determined for incorporation into the REBASE database (Roberts et al., 2015). REBASE is a database for information on recognition and cleavage sites for both restriction enzymes and methyltransferases and methylation sensitivity. PacBio data generates data on modified bases, which may be useful for related studies. Pilon corrections and comparison of Illumina and PacBio assemblies were further assessed by measuring the impact of nucleotide changes on protein coding potential and positive/negative influence on gene calling accuracy (See Section S1 for details).
Results and Discussion
Sequencing and Assembly Overview
Illumina sequence coverage for each genome is >200X, sufficient to derive high-quality draft genome assemblies (Haridas et al., 2011; Utturkar et al., 2014). PacBio sequence coverage for each genome is >100X except for the isolates of Pelosinus sp. UFO1 (97x) and B. cellulosolvens DSM 2933 (48X). Post-trimming and filtering statistics for Illumina and PacBio data including the number of reads, average read lengths and genome coverage and total bases are summarized in Tables S1, S2, respectively. Genome assemblies were performed using combinations of Illumina and PacBio platforms and various assembly programs. Consistent with previous results (Brown S. et al., 2014), most of the genomes in the current study have superior PacBio-only assemblies (based on assembly statistics) followed by hybrid and Illumina-only assemblies, respectively. Out of seven genomes, three were assembled as complete circular chromosomes, manual finishing was performed for two genomes and remaining two were reported as near-finished assemblies. Details of the assembly results and manual finishing approaches are described in later sections. Using these seven genomes as a case study, we describe the best practices to obtain high-quality genome assembly using long sequence reads, post-assembly polishing steps, and gap-closure strategies for automated near-finished assemblies. The finishing approach outlined in this study includes the use of super-assemblies and supporting Illumina data to determine contig order followed by PCR and Sanger sequencing to validate contig joining. Post-finishing data were used to determine the characteristics of the unassembled DNA regions within Illumina and PacBio assembly.
Unassembled DNA Regions in PacBio-Only Assemblies
Inspection of unassembled DNA regions within PacBio assemblies was performed using five gap sequences generated through manual finishing of C. thermocellum AD2 and B. cellulosolvens DSM 2933 genomes. The unassembled DNA from PacBio assemblies were analyzed for GC content, read coverage, gap length, ability to form strong secondary structures, and corresponding annotations (Table 1). GC content of gap sequences does not diverge markedly from the genome sequence. Four of five PacBio gaps were associated with lower than the recommended coverage for HGAP assembly (100x). Gaps AD2_overlap1, AD2_Gap1 and BC_Gap1 were the most difficult to resolve by PCR and Sanger sequencing and had low sequence coverages (36X, 82X, and 4X, respectively), while high average sequence coverage values were present across the genomes (see Section S1 for details).
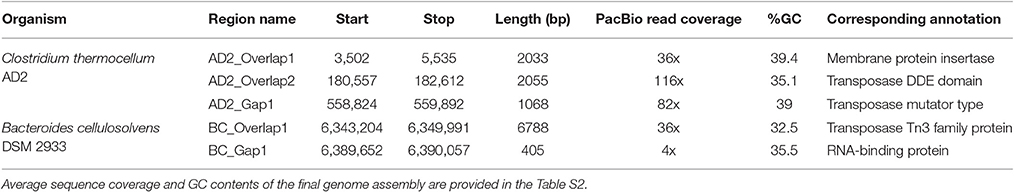
Table 1. Properties of gap sequences present within PacBio assembly.
Considering the low sequence coverage values and challenges associated with PCR amplification for the closed gap sequences, we hypothesized that PacBio gap sequences might form strong hairpin loop structures that would prevent DNA polymerase from being able to unwind and extend through the DNA region. To test our hypothesis, structural properties of gap sequences were analyzed using the mfold web server, which predicts the secondary structures or ability to form hairpin loops and associated minimum free energy (ΔG) values. Mfold analysis of PacBio gap sequences revealed the potential to form small stem-loop structures but large and/or strong secondary structural loops that might interfere with DNA polymerase and result in low sequence coverage were not identified. Significant differences were not observed between minimum free energies and secondary structures of PacBio gaps and 20 randomly selected regions from the AD2 and DSM 2933 genomes (Table S3). In addition, we utilized DNA periodicity criteria to determine any associations between PacBio gaps and other structural features of DNA. Regular spacing of short runs of A or T nucleotides with DNA helical period of ~10.5 bp (termed as a positional preference) has been associated with DNA curvature, supercoiling and nucleosome positioning. Relatively rigid sections of the prokaryotic DNA (characterized by short intrinsically bent DNA segments) are proposed to be associated with strong periodic patterns while structurally flexible regions are associated with weak periods (Mrazek et al., 2011; Tong and Mrazek, 2014). Positional preference was determined for all the genomes in current study and regions which correspond to Illumina gaps and also have positional preference higher than 2.50 are highlighted in orange color (Tables S4–S9). However, gaps appear to be randomly distributed as compared to strong/weak positional preference and a specific trend was not observed for this metric. An example of positional preference locations and Illumina/PacBio gaps in AD2 genome is presented (Figure 1). Post-finishing, we determined Illumina reads have uniform coverage across the gap regions. However, short read length and repetitive nature of these regions may have prevented the accurate assembly. Therefore, our initial hypothesis that resilient PacBio gaps resulted from the inability of DNA polymerase to sequence through strong hairpin loop structures was rejected.
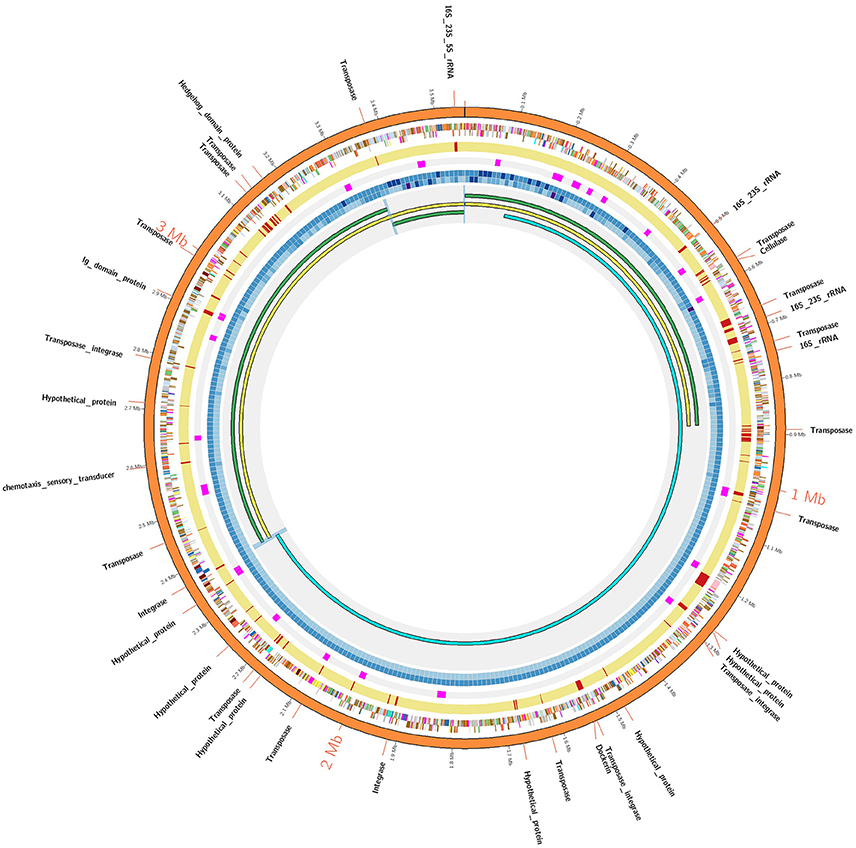
Figure 1. AD2 genome assembly comparisons. The outermost orange colored circle corresponds to finished genome assembly. The next two circles show genes on positive and negative strands and using color coded by standards for COG categories. The next yellow colored circle corresponds to Illumina assembly and gaps within Illumina assembly are denoted by red strokes. The next circle denotes the strong positional preference marked in pink color. The next two concentric circles denote the sequence coverage for Illumina and PacBio technologies respectively as heatmap (lowest: light blue, highest: dark blue). The innermost circle: AD2_SC1 (yellow) was generated by super assembly of draft contigs (green). AD2_HC1 (sky blue) share 780 kb overlap with AD2_SC1. Blue-highlighted region denotes sequence overlaps validated using PCR/Sanger approach. A detailed Illustration is provided in Figure S1.
For further characterization, we analyzed 1 kb DNA sequences flanking PacBio gaps (i.e., contig termini regions) from three near-finished genomes in this study. A self-blast was performed using 1 kb regions as a query against the entire genome using Geneious software with default parameters. The grade score from Geneious software (i.e., a cumulative score generated by combining the % pairwise identity, % query coverage, e-value) for the top blast hits for gap termini regions are described in Table S10. In each genome, except AD2, sequences flanking the gap regions showed high similarity (grade: >95%) with another region within the same genome indicating repetitive DNA sequences could have contributed to assembly challenges. Sequences flanking AD2_Gap1 have a low (grade: 72%) similarity score within the genome, consistent with the finding that the AD2 was comparatively easier to finish using standard PCR/Sanger sequencing approaches. To further validate this observation, we repeated the flanking DNA sequence analysis steps for an independent dataset (Koren et al., 2013). In three incomplete genome assemblies, most of the sequences flanking the gaps were determined to have high similarity (grade: >95%) within the same genome (Table S14) that may contribute to the fragmented PacBio assemblies.
Various biological aspects of seven genomes within this study, as well as for the C. thermocellum LQRI (LQRI), and P. fermentans DSM 17108 genomes (Utturkar et al., 2016) were further analyzed for gaps within PacBio assemblies. Specific biological features of the genomes that likely interfered with overall assembly process are summarized in Figure 2. The complete genome sequence of strain JBW45 was characterized by the presence of an active transposon element which interfered with the genome circularization process (De Leon et al., 2015). The C. paradoxum genome was reported to contain multiple rRNA operons with heterogeneous intervening sequences (15 different sequences in the variable region I) of 16S rRNA (Rainey et al., 1996) which could contribute to the fragmented assembly. Assembly analysis of strain ATCC 6013 and P. fermentans DSM 17108 revealed a possible phage integration and presence of large sequence duplication. The KO116 genome was characterized by the presence of two megaplasmids. For a previously uncharacterized bacterium, multiple contigs could lead to the impression of having a near-finished genome assembly instead of megaplasmid sequences in the absence of manual inspection. We expect new tools such as plasmidSPAdes (Antipov et al., 2016) will be useful in assembling and assessing plasmid DNA content from whole genome sequencing data. Automated HGAP assembly of LQRI obtained a near-finished assembly containing two contigs. After careful evaluation, one of the contigs was found to represent a complete circular genome while smaller 12 kb contig was determined as duplicated sequence artifact. B. cellulosolvens DSM 2933 contig termini were characterized by the presence of transposon-related genes (Dassa et al., 2015). In summary, our initial hypothesis that structural features of DNA (hairpin-loops, secondary structures, supercoiling, and nucleosome positioning) might affect the PacBio coverage in certain regions leading to assembly gaps was not accepted. In terms of assembly, it is likely that the HGAP software did not have sufficient read coverage to support automatic closure of these sequences and resulted in assembly gaps. Analysis of gap sequences revealed that in many cases, DNA sequences flanking the gaps have more than one copy within the genome, and some were corresponding to long repetitive elements such as “Transposon-related proteins” (De Leon et al., 2015). Further analysis revealed specific biological features such as the presence of active mobile genetic elements, plasmid sequences and phage integration which can lead to fragmented PacBio assemblies. Hence, although we could not determine one specific trend, PacBio gaps sequences were associated with a cumulative effect of number of repeats and their sizes, sequence depth, and various biological features associated with specific genomes.
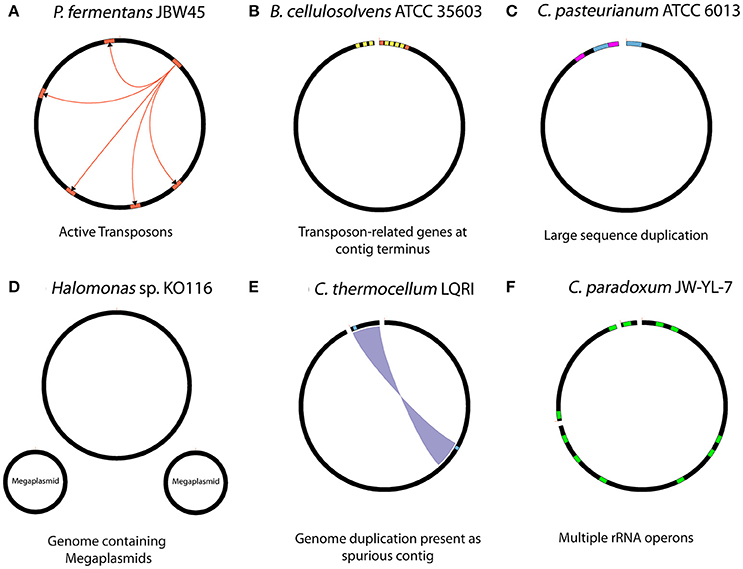
Figure 2. Summary of biological features with potential to interfere with the assembly process. (A) Presence of active transposon elements in strain JBW45 (B) repetitive transposon sequences at the contig terminus region of B. cellulosolvens (C) large sequence duplications in C. pasteurianum (D) presence of megaplsmids in stain KO116 (E) genome duplication assembled as spurious contig in C. thermocellum LQRI (F) multiple copies of rRNA operons in C. paradoxum JW-YL-7. The figures are illustration only and not drawn to scale.
Unassembled DNA Regions in Illumina-Only Assemblies
Unassembled DNA or assembly breakpoints in Illumina assemblies were revealed by mapping the contigs from Illumina-only assembly against the final (finished or near-finished) genome assemblies. Short reads from Illumina technology have limited power to resolve longer repetitive regions (Salzberg et al., 2012; Utturkar et al., 2014) and rRNA operons are considered among the most difficult regions to assemble (Brown S. et al., 2014). Our comparisons demonstrate at least half the total rRNA operons were completely missing (unassembled) from the Illumina assembly while from remaining half, most could only be assembled partially (i.e., missing one of the 5S, 16S, or 23S elements). These findings are consistent with our previous results suggesting that rRNA operons correspond to many of breakpoints within short-read assemblies (Brown S. et al., 2014; Utturkar et al., 2014). On the other hand, longer PacBio reads resolved the majority of rRNA operons as evident through circular genome assemblies and comparison of finished vs. draft assemblies (Tables S4–S9). The total number of rRNA operons present in each genome, number of missing rRNA operons, and number of partially assembled rRNA operons in Illumina assembly are provided in Table 2. Illumina-only assemblies often also lacked tRNA due to their physical linkage to incomplete rRNA operons, as well as other genes encoding putative functions for transposase and hypothetical proteins. The average size of rRNA operons is ~5–7 kb and constituted the longest gaps within Illumina assemblies. Apart from rRNA operons, other regions that contributed to fragmented Illumina assemblies include transposon sequences, ABC-type transporters (which number in the double digits for most genomes), RNA-directed DNA polymerases (which have long sequences and share high homology), as well hypothetical proteins. A complete table describing the draft vs. finished assembly comparison details (gap coordinates, length, associated annotation, and locus tags) for each genome are provided (Tables S4–S9) and graphical representation of Illumina/PacBio gaps within AD2 genome is shown (Figure 1). The genome of C. pasteurianum was the only exception where two large contigs from Illumina assembly were accurate and contained all the rRNA operons and no other gaps were detected.
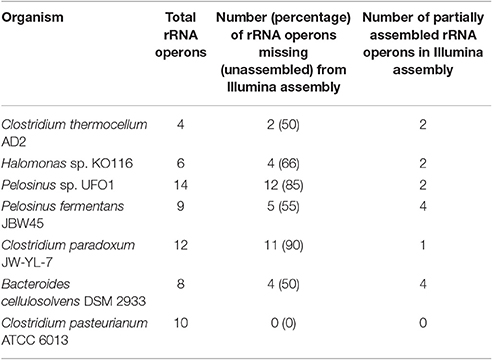
Table 2. Summary of rRNA operons present within Illumina assembly.
Insights into Assembly and Polishing Improvement Approaches
A variety of assembly algorithms are available for de novo and hybrid assembly (Salzberg et al., 2012; Magoc et al., 2013; Koren and Phillippy, 2014), read error correction (Lin and Liao, 2015), scaffolding (Bashir et al., 2012; English et al., 2012), and genome finishing (Swain et al., 2012) with different NGS data types. Our aim was to perform an assessment of gaps rather than an evaluation of assemblers and we chose SPAdes and ABySS to assemble Illumina data and HGAP to assemble PacBio data based on previous success (Brown S. et al., 2014; Utturkar et al., 2014). Consistent with previous findings (Brown S. D. et al., 2014), PacBio-only assemblies have the best statistics followed by hybrid and Illumina-only assemblies. Assembly summary statistics for de novo and hybrid assemblies are described in Table 3. It is worth mentioning that using the latest versions of assembly algorithms had significant impacts on overall assembly statistics. For example, B. cellulosolvens DSM 2933 (Dassa et al., 2015) and C. pasteurianum ATCC 6013 (Pyne et al., 2014) genomes assembled through SMRT analysis v2.2 obtained substantial improvement over v2.0 assembly (Table 3). The field of bioinformatics is rapidly evolving with the novel, efficient assembly algorithms such as Canu (Koren et al., 2017), HINGE (Kamath et al., 2017) for long reads, and integrated pipelines (Coil et al., 2015; Page et al., 2016) for short reads. For future assembly projects, it is recommended to use multiple assembly programs to obtain the optimal assembly and use our rRNA analysis approach for an additional verification of assembly accuracy (Utturkar et al., 2014). It is also important to perform a careful analysis of contigs to check for the presence of plasmid content, which could be misinterpreted as near-finished assemblies.
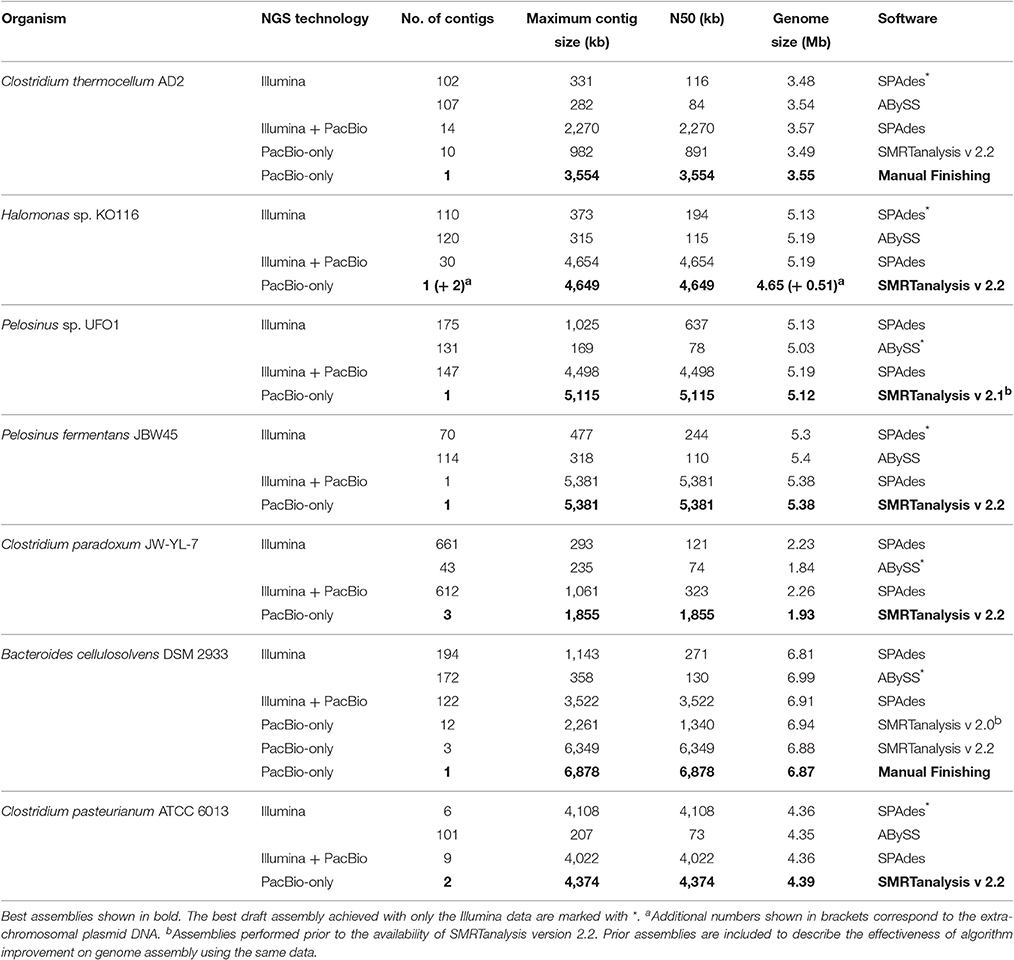
Table 3. Assembly summary statistics for de novo and hybrid assemblies.
Long read sequencing platforms are criticized for their frequent (~15%), but random errors in the PacBio platform can be corrected by using high (>100x) sequence coverage and/or Illumina data. However, uniform sequence coverage across the entire genome is not guaranteed and low coverage regions are prone to base-call errors. Assembly polishing is a crucial step to obtain accurate consensus sequence and facilitate downstream applications. Two assembly base-call correction algorithms applied in this study are Quiver (correction using PacBio reads) and Pilon (correction using Illumina reads) while iCORN (Otto et al., 2010) is another alternative. The default HGAP protocol is implemented with a single round of Quiver polishing and we applied additional rounds of Pilon correction for further assembly quality improvements. The majority of the base-call errors corrected by Pilon were insertions-deletions (indels) (Table 4 and Tables S11–S14), which were responsible for the frameshift mutations and correspond to altered Open Reading Frame (ORF) predictions. To validate the accuracy of Pilon calls, 47 random Pilon corrections across four finished genomes were verified by PCR and Sanger sequencing. Our results indicate that 40 of the 47 (~85%) tested corrections by Pilon were accurate and supported by two (forward and reverse) high quality Sanger reads. The remaining seven suggested Pilon modifications were ruled out based on lack of support from analysis of Sanger data.
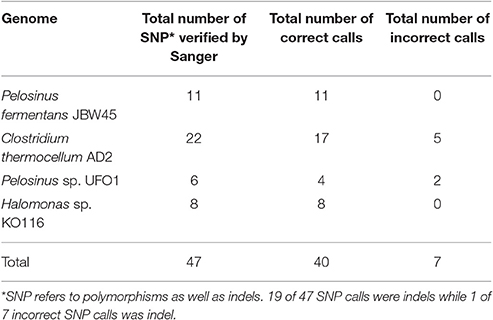
Table 4. Summary of Pilon call verification by Sanger sequencing.
Further evaluation of Pilon corrections was performed by measuring the changes in the protein coding potential and positive/negative influence on gene calling accuracy (Section S1). In most cases, Pilon corrections improved the protein coding potential by predicting longer ORFs, joined ORFs (previously split genes were joined together to represent single long ORF), and few novel ORFs (Tables S11–S14). Certain Pilon corrections resulted in split ORFs (previously longer ORF were split into two ORFs), but such cases were comparatively fewer than the number of improved ORFs. Moreover, most of the changed ORFs were associated with improved BLASTN results (e-value, percent similarity, percent identity, and subject length) suggesting enhanced gene-calling accuracy. To summarize, there were total 314 modifications suggested by Pilon across four finished genomes, of which 154 (49%) have resulted in improved protein coding potential (longer/joined/novel ORFs), 38 (12%) were associated with split/shorter ORFs while 122 (38%) had no change. Considering the BLASTN results, 183 (58%) corrections have a positive influence on gene calling accuracy, 35 (11%) corrections deteriorated the BLASTN results while 96 (31%) had no changes. Pilon is a useful tool for in silico genome refinement and recommended when Illumina data is available.
Another important aspect of finished genome sequences is an accurate representation of a circular chromosome. Automatically finished assemblies generated through HGAP often have (duplicated) overlapping ends which need to be trimmed off for the final assembly. This could be achieved using the circulator (Hunt et al., 2015) software which performs automated assembly circularization and sets the dnaA gene as the starting position. In this study, assembly circularization was performed manually through a read mapping and alignment approach before the availability of circlator software. Later, a comparison of circlator and manual assemblies was performed and results were similar (data not shown). Therefore, for future projects, the application of circlator software followed by a careful inspection of the trimmed regions is recommended.
Conclusions
In this study, we present an effective manual finishing approach targeted toward near-finished microbial genome assemblies. The importance of genome polishing steps is demonstrated through its positive influence on gene calling accuracy and improved protein coding potential, which will be useful to others looking to improve long-read assemblies. Assessment of Illumina gaps confirmed previous findings that repetitive rRNA operons are major contributors to fragmented short-read assemblies. For PacBio assemblies, our initial hypothesis that structural features of DNA might affect the PacBio sequence coverage leading to assembly gap was not accepted. However, we demonstrated that certain biological features such as presence of active transposons, plasmid sequences, and phage integration are possible reasons for assembly fragmentation. Additionally, DNA regions flanking the PacBio gap sequences showed high degrees of similarity with other loci and are likely contributors to incomplete PacBio assemblies in this dataset. The PacBio gap sequences in this study are attributed to a cumulative effect of various aspects of repetitive DNA content and biological features for specific genomes. Despite a few limitations, long reads from third-generation sequencing, in this case from the PacBio platform, are particularly advantageous for generating de novo microbial genome assemblies. Our datasets and analyses will aid future efforts to better understand and overcome unassembled DNA from PacBio assemblies.
Author Contributions
SU designed the study, performed, and contributed to all the experiments and analyses and wrote the manuscript draft; DK extracted genomic DNA, performed Illumina sequencing, and assisted with PCR and Sanger sequencing; RH contributed to study design and edited the manuscript; SB contributed to study design, assisted with draft writing, and editing. All authors reviewed and approved the manuscript.
Funding
This work was supported by the Plant-Microbe Interfaces Scientific Focus Area and the BioEnergy Science Center, a U.S. DOE Bioenergy Research Center, in the Genomic Science Program, the Office of Biological and Environmental Research in the U.S. Department of Energy Office of Science. Oak Ridge National Laboratory is managed by UT-Battelle, LLC, for the United States Department of Energy under contract DE-AC05-00OR22725.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Submitted as a partial requirement for SU's PhD thesis and we thank his thesis committee (Mitch Doktycz, Dale Pelletier, Chris Schadt, ORNL, and Gladys Alexandre UT) for helpful suggestions and guidance. Miriam Land (ORNL) is acknowledged for her support maintaining the Microbial Genome Annotation Pipeline, which facilitated annotations and gene model comparisons. Some sequence data analyzed in this study were published earlier in collaboration with the U.S. Department of Energy Joint Genome Institute, a DOE Office of Science User Facility, which is supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. Sanger sequencer data was generated at the Molecular Biology Resource Facility at the University of Tennessee, Knoxville.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.01272/full#supplementary-material
References
Antipov, D., Hartwick, N., Shen, M., Raiko, M., Lapidus, A., and Pevzner, P. A. (2016). plasmidSPAdes: assembling plasmids from whole genome sequencing data. Bioinformatics 32, 3380–3387. doi: 10.1093/bioinformatics/btw493
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Bashir, A., Klammer, A. A., Robins, W. P., Chin, C. S., Webster, D., Paxinos, E., et al. (2012). A hybrid approach for the automated finishing of bacterial genomes. Nat. Biotechnol. 30, 701–707. doi: 10.1038/nbt.2288
Bishnoi, U., Polson, S. W., Sherrier, D. J., and Bais, H. P. (2015). Draft genome sequence of a natural root isolate, Bacillus subtilis UD1022, a potential plant growth-promoting biocontrol agent. Genome Announc. 3:e00696-15. doi: 10.1128/genomeA.00696-15
Brown, S. D., Utturkar, S. M., Magnuson, T. S., Ray, A. E., Poole, F. L., Lancaster, W. A., et al. (2014). Complete genome sequence of Pelosinus sp. strain UFO1 assembled using Single-Molecule Real-Time DNA sequencing technology. Genome Announc. 2:e00881-14. doi: 10.1128/genomeA.00881-14
Brown, S., Nagaraju, S., Utturkar, S., De Tissera, S., Segovia, S., Mitchell, W., et al. (2014). Comparison of single-molecule sequencing and hybrid approaches for finishing the genome of Clostridium autoethanogenum and analysis of CRISPR systems in industrial relevant Clostridia. Biotechnol. Biofuels 7:40. doi: 10.1186/1754-6834-7-40
Buermans, H. P., and den Dunnen, J. T. (2014). Next generation sequencing technology: advances and applications. Biochim. Biophys. Acta 1842, 1932–1941. doi: 10.1016/j.bbadis.2014.06.015
Chain, P. S., Grafham, D. V., Fulton, R. S., Fitzgerald, M. G., Hostetler, J., Muzny, D., et al. (2009). Genomics. Genome project standards in a new era of sequencing. Science 326, 236–237. doi: 10.1126/science.1180614
Chin, C. S., Alexander, D. H., Marks, P., Klammer, A. A., Drake, J., Heiner, C., et al. (2013). Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 10, 563–569. doi: 10.1038/nmeth.2474
Coil, D., Jospin, G., and Darling, A. E. (2015). A5-miseq: an updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics 31, 587–589. doi: 10.1093/bioinformatics/btu661
Dassa, B., Utturkar, S., Hurt, R. A., Klingeman, D. M., Keller, M., Xu, J., et al. (2015). Near-complete genome sequence of the cellulolytic bacterium Bacteroides (Pseudobacteroides) cellulosolvens ATCC 35603. Genome Announc. 3. doi: 10.1128/genomeA.01022-15
De Leon, K. B., Utturkar, S. M., Camilleri, L. B., Elias, D. A., Arkin, A. P., Fields, M. W., et al. (2015). Complete genome sequence of Pelosinus fermentans JBW45, a member of a remarkably competitive group of negativicutes in the firmicutes phylum. Genome Announc. 3:e01090-15. doi: 10.1128/genomeA.01090-15
Deschamps, S., Mudge, J., Cameron, C., Ramaraj, T., Anand, A., Fengler, K., et al. (2016). Characterization, correction and de novo assembly of an Oxford Nanopore genomic dataset from Agrobacterium tumefaciens. Sci. Rep. 6:28625. doi: 10.1038/srep28625
Dunitz, M. I., Coil, D. A., Jospin, G., Eisen, J. A., and Adams, J. Y. (2014). Draft genome sequences of Escherichia coli strains isolated from septic patients. Genome Announc. 2:e01278-14. doi: 10.1128/genomeA.01278-14
Eckweiler, D., Bunk, B., Sproer, C., Overmann, J., and Haussler, S. (2014). Complete genome sequence of highly adherent Pseudomonas aeruginosa small-colony variant SCV20265. Genome Announc. 2:e01232-13. doi: 10.1128/genomeA.01232-13
English, A. C., Richards, S., Han, Y., Wang, M., Vee, V., Qu, J., et al. (2012). Mind the gap: upgrading genomes with Pacific Biosciences RS long-read sequencing technology. PLoS ONE 7:e47768. doi: 10.1371/journal.pone.0047768
Feng, Y., Zhang, Y., Ying, C., Wang, D., and Du, C. (2015). Nanopore-based fourth-generation DNA sequencing technology. Genomics Proteomics Bioinformatics 13, 4–16. doi: 10.1016/j.gpb.2015.01.009
Fraser, C. M., Eisen, J. A., Nelson, K. E., Paulsen, I. T., and Salzberg, S. L. (2002). The value of complete microbial genome sequencing (you get what you pay for). J. Bacteriol. 184, 6403–6405. doi: 10.1128/JB.184.23.6403-6405.2002
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Harhay, G. P., McVey, D. S., Koren, S., Phillippy, A. M., Bono, J., Harhay, D. M., et al. (2014). Complete closed genome sequences of three Bibersteinia trehalosi nasopharyngeal isolates from cattle with shipping fever. Genome Announc. 2:e00084-14. doi: 10.1128/genomeA.00084-14
Haridas, S., Breuill, C., Bohlmann, J., and Hsiang, T. (2011). A biologist's guide to de novo genome assembly using next-generation sequence data: A test with fungal genomes. J. Microbiol. Methods 86, 368–375. doi: 10.1016/j.mimet.2011.06.019
Hoefler, B. C., Konganti, K., and Straight, P. D. (2013). De Novo assembly of the Streptomyces sp. strain Mg1 genome using PacBio single-molecule sequencing. Genome Announc. 1:e00535-13. doi: 10.1128/genomeA.00535-13
Hua, X., and Hua, Y. (2016). Improved complete genome sequence of the extremely radioresistant bacterium Deinococcus radiodurans R1 obtained using PacBio single-molecule sequencing. Genome Announc. 4:e00886-16. doi: 10.1128/genomeA.00886-16
Hunt, M., Silva, N. D., Otto, T. D., Parkhill, J., Keane, J. A., and Harris, S. R. (2015). Circlator: automated circularization of genome assemblies using long sequencing reads. Genome Biol. 16, 294. doi: 10.1186/s13059-015-0849-0
Hyatt, D., Chen, G. L., Locascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119
Kamath, G. M., Shomorony, I., Xia, F., Courtade, T. A., and Tse, D. N. (2017). HINGE: long-read assembly achieves optimal repeat resolution. Genome Res. 27, 747–756. doi: 10.1101/gr.216465.116
Kanda, K., Nakashima, K., and Nagano, Y. (2015). Complete genome sequence of Bacillus thuringiensis serovar tolworthi strain Pasteur Institute Standard. Genome Announc. 3:e00710-15. doi: 10.1128/genomeA.00710-15
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Koren, S., and Phillippy, A. M. (2014). One chromosome, one contig: complete microbial genomes from long-read sequencing and assembly. Curr. Opin. Microbiol. 23C, 110–120. doi: 10.1016/j.mib.2014.11.014
Koren, S., Harhay, G., Smith, T., Bono, J., Harhay, D., Mcvey, S., et al. (2013). Reducing assembly complexity of microbial genomes with single-molecule sequencing. Genome Biol. 14:R101. doi: 10.1186/gb-2013-14-9-r101
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., and Phillippy, A. M. (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736. doi: 10.1101/gr.215087.116
Korlach, J., Bjornson, K. P., Chaudhuri, B. P., Cicero, R. L., Flusberg, B. A., Gray, J. J., et al. (2010). Real-time DNA sequencing from single polymerase molecules. Methods Enzymol. 472, 431–455. doi: 10.1016/S0076-6879(10)72001-2
Krumsiek, J., Arnold, R., and Rattei, T. (2007). Gepard: a rapid and sensitive tool for creating dotplots on genome scale. Bioinformatics 23, 1026–1028. doi: 10.1093/bioinformatics/btm039
Lancaster, W. A., Utturkar, S. M., Poole, F. L., Klingeman, D. M., Elias, D. A., Adams, M. W., et al. (2016). Near-complete genome sequence of Clostridium paradoxum strain JW-YL-7. Genome Announc. 4:e00229-16. doi: 10.1128/genomeA.00229-16
Lin, H. H., and Liao, Y. C. (2015). Evaluation and validation of assembling corrected PacBio long reads for microbial genome completion via hybrid approaches. PLoS ONE 10:e0144305. doi: 10.1371/journal.pone.0144305
Liu, L., Li, Y., Li, S., Hu, N., He, Y., Pong, R., et al. (2012). Comparison of next-generation sequencing systems. J. Biomed. Biotechnol. 2012:251364. doi: 10.1155/2012/251364
Magoc, T., Pabinger, S., Canzar, S., Liu, X., Su, Q., Puiu, D., et al. (2013). GAGE-B: an evaluation of genome assemblers for bacterial organisms. Bioinformatics 29, 1718–1725. doi: 10.1093/bioinformatics/btt273
Margulies, M., Egholm, M., Altman, W. E., Attiya, S., Bader, J. S., Bemben, L. A., et al. (2005). Genome sequencing in microfabricated high-density picolitre reactors. Nature 437, 376–380. doi: 10.1038/nature03959
Mehnaz, S., Bauer, J. S., and Gross, H. (2014). Complete genome sequence of the sugar cane endophyte Pseudomonas aurantiaca PB-St2, a disease-suppressive bacterium with antifungal activity toward the plant pathogen Colletotrichum falcatum. Genome Announc. 2:e01108-13. doi: 10.1128/genomeA.01108-13
Mrazek, J., Chaudhari, T., and Basu, A. (2011). PerPlot & PerScan: tools for analysis of DNA curvature-related periodicity in genomic nucleotide sequences. Microb. Inform. Exp. 1:13. doi: 10.1186/2042-5783-1-13
Nagarajan, N., and Pop, M. (2013). Sequence assembly demystified. Nat. Rev. Genet. 14, 157–67. doi: 10.1038/nrg3367
Nakano, K., Terabayashi, Y., Shiroma, A., Shimoji, M., Tamotsu, H., Ashimine, N., et al. (2015). First complete genome sequence of Clostridium sporogenes DSM 795T, a nontoxigenic surrogate for Clostridium botulinum, determined using PacBio Single-Molecule Real-Time Technology. Genome Announc. 3:e00832-15. doi: 10.1128/genomeA.00832-15
O'Dell, K. B., Woo, H. L., Utturkar, S., Klingeman, D., Brown, S. D., and Hazen, T. C. (2015). Genome sequence of Halomonas sp. strain KO116, an Ionic liquid-tolerant marine bacterium isolated from a lignin-enriched seawater microcosm. Genome Announc. 3:e00402-15. doi: 10.1128/genomeA.00402-15
Okutani, A., Osaki, M., Takamatsu, D., Kaku, Y., Inoue, S., and Morikawa, S. (2015). Draft genome sequences of Bacillus anthracis strains stored for several decades in Japan. Genome Announc. 3:e00633-15. doi: 10.1128/genomeA.00633-15
Otto, T. D., Sanders, M., Berriman, M., and Newbold, C. (2010). Iterative Correction of Reference Nucleotides (iCORN) using second generation sequencing technology. Bioinformatics 26, 1704–1707. doi: 10.1093/bioinformatics/btq269
Pacific-Biosciences (2014a). HGAP in SMRT Analysis. Available online at: https://github.com/PacificBiosciences/Bioinformatics-Training/wiki/HGAP-in-SMRT-Analysis
Pacific-BioSciences (2014b). SMRT Analysis Release Notes v2.2.0. Available online at: https://github.com/PacificBiosciences/SMRT-Analysis/wiki/SMRT-Analysis-Release-Notes-v2.2.0
Pacific-Biosciences (2015). Circularizing and Trimming. Available online at: https://github.com/PacificBiosciences/Bioinformatics-Training/wiki/Circularizing-and-trimming
Page, A. J., De Silva, N., Hunt, M., Quail, M. A., Parkhill, J., Harris, S. R., et al. (2016). Robust high-throughput prokaryote de novo assembly and improvement pipeline for Illumina data. Microbial Genomics 2:e000083. doi: 10.1099/mgen.0.000083
Pyne, M. E., Utturkar, S., Brown, S. D., Moo-Young, M., Chung, D. A., and Chou, C. P. (2014). Improved draft genome sequence of Clostridium pasteurianum strain ATCC 6013 (DSM 525) using a hybrid Next-Generation Sequencing approach. Genome Announc. 2:e00790-14. doi: 10.1128/genomeA.00790-14
Quail, M. A., Smith, M., Coupland, P., Otto, T. D., Harris, S. R., Connor, T. R., et al. (2012). A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genomics 13:341. doi: 10.1186/1471-2164-13-341
Rainey, F. A., Ward-Rainey, N. L., Janssen, P. H., Hippe, H., and Stackebrandt, E. (1996). Clostridium paradoxum DSM 7308T contains multiple 16S rRNA genes with heterogeneous intervening sequences. Microbiology 142 (Pt. 8), 2087–2095. doi: 10.1099/13500872-142-8-2087
Rhoads, A., and Au, K. F. (2015). PacBio sequencing and its applications. Genomics Proteomics Bioinformatics 13, 278–289. doi: 10.1016/j.gpb.2015.08.002
Risse, J., Thomson, M., Patrick, S., Blakely, G., Koutsovoulos, G., Blaxter, M., et al. (2015). A single chromosome assembly of Bacteroides fragilis strain BE1 from Illumina and MinION nanopore sequencing data. Gigascience 4, 1–7. doi: 10.1186/s13742-015-0101-6
Roberts, R. J., Carneiro, M. O., and Schatz, M. C. (2013). The advantages of SMRT sequencing. Genome Biol. 14:405. doi: 10.1186/gb-2013-14-6-405
Roberts, R. J., Vincze, T., Posfai, J., and Macelis, D. (2015). REBASE–a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res. 43(Database issue):D298–D299. doi: 10.1093/nar/gku1046
Salzberg, S. L., Phillippy, A. M., Zimin, A., Puiu, D., Magoc, T., Koren, S., et al. (2012). GAGE: a critical evaluation of genome assemblies and assembly algorithms. Genome Res. 22, 557–567. doi: 10.1101/gr.131383.111
Satou, K., Shiroma, A., Teruya, K., Shimoji, M., Nakano, K., Juan, A., et al. (2014). Complete genome sequences of eight Helicobacter pylori strains with different virulence factor genotypes and methylation profiles, isolated from patients with diverse gastrointestinal diseases on Okinawa Island, Japan, determined using PacBio Single-Molecule Real-Time Technology. Genome Announc. 2:e00286-14. doi: 10.1128/genomeA.00286-14
Shapiro, L. R., Scully, E. D., Roberts, D., Straub, T. J., Geib, S. M., Park, J., et al. (2015). Draft genome sequence of Erwinia tracheiphila, an economically important bacterial pathogen of cucurbits. Genome Announc. 3:e00482-15. doi: 10.1128/genomeA.00482-15
Simpson, J. T., Wong, K., Jackman, S. D., Schein, J. E., Jones, S. J., and Birol, I. (2009). ABySS: a parallel assembler for short read sequence data. Genome Res. 19, 1117–1123. doi: 10.1101/gr.089532.108
Swain, M. T., Tsai, I. J., Assefa, S. A., Newbold, C., Berriman, M., and Otto, T. D. (2012). A post-assembly genome-improvement toolkit (PAGIT) to obtain annotated genomes from contigs. Nat. Protoc. 7, 1260–1284. doi: 10.1038/nprot.2012.068
The NCTC 3000 Project (2016). The NCTC 3000 Project: Public Health England Reference Collections - Wellcome Trust Sanger Institute (Accessed July 25, 2016). Available online at: http://www.sanger.ac.uk/resources/downloads/bacteria/nctc/
Thomma, B. P., Seidl, M. F., Shi-Kunne, X., Cook, D. E., Bolton, M. D., van Kan, J. A., et al. (2015). Mind the gap; seven reasons to close fragmented genome assemblies. Fungal Genet. Biol. 90, 24–30. doi: 10.1016/j.fgb.2015.08.010
Tong, H., and Mrazek, J. (2014). Investigating the interplay between nucleoid-associated proteins, DNA curvature, and CRISPR elements using comparative genomics. PLoS ONE 9:e90940. doi: 10.1371/journal.pone.0090940
Treangen, T. J., and Salzberg, S. L. (2012). Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat. Rev. Genet. 13, 36–46. doi: 10.1038/nrg3164
Utturkar, S. M., Bayer, E. A., Borovok, I., Lamed, R., Hurt, R. A., Land, M. L., et al. (2016). Application of long sequence reads to improve genomes for Clostridium thermocellum AD2, Clostridium thermocellum LQRI, and Pelosinus fermentans R7. Genome Announc. 4:e01043-16. doi: 10.1128/genomeA.01043-16
Utturkar, S. M., Klingeman, D. M., Bruno-Barcena, J. M., Chinn, M. S., Grunden, A. M., Kopke, M., et al. (2015). Sequence data for Clostridium autoethanogenum using three generations of sequencing technologies. Sci Data 2, 150014. doi: 10.1038/sdata.2015.14
Utturkar, S. M., Klingeman, D. M., Land, M. L., Schadt, C. W., Doktycz, M. J., Pelletier, D. A., et al. (2014). Evaluation and validation of de novo and hybrid assembly techniques to derive high quality genome sequences. Bioinformatics 30, 2709–2716. doi: 10.1093/bioinformatics/btu391
van Dijk, E. L., Auger, H., Jaszczyszyn, Y., and Thermes, C. (2014). Ten years of next-generation sequencing technology. Trends Genet. 30, 418–426. doi: 10.1016/j.tig.2014.07.001
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9:e112963. doi: 10.1371/journal.pone.0112963
Woo, H. L., Utturkar, S., Klingeman, D., Simmons, B. A., DeAngelis, K. M., Brown, S. D., et al. (2014). Draft genome sequence of the lignin-degrading Burkholderia sp. strain LIG30, isolated from wet tropical forest soil. Genome Announc. 2:e00637-14. doi: 10.1128/genomeA.00637-14
Keywords: PacBio, Illumina, genome assembly, next-generation sequencing (NGS), repetitive DNA, Pilon, circlator
Citation: Utturkar SM, Klingeman DM, Hurt RA Jr. and Brown SD (2017) A Case Study into Microbial Genome Assembly Gap Sequences and Finishing Strategies. Front. Microbiol. 8:1272. doi: 10.3389/fmicb.2017.01272
Received: 08 May 2017; Accepted: 26 June 2017;
Published: 18 July 2017.
Edited by:
Angel Angelov, Technische Universität München, GermanyReviewed by:
Gwenael Piganeau, FR3724 Observatoire Océanologique de Banyuls sur Mer (OOB), FranceHilary G. Morrison, Marine Biological Laboratory, United States
Copyright © 2017 Utturkar, Klingeman, Hurt and Brown. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Steven D. Brown, YnJvd25zZEBvcm5sLmdvdg==
†Present Address: Sagar M. Utturkar, Bioinformatics Core, Purdue University, West Lafayette, IN, United States