 Carlos A. Rossetti1†
Carlos A. Rossetti1† Kenneth L. Drake2
Kenneth L. Drake2 Sara D. Lawhon1
Sara D. Lawhon1 Jairo S. Nunes1†
Jairo S. Nunes1† Tamara Gull1†
Tamara Gull1† Sangeeta Khare1†
Sangeeta Khare1† Leslie G. Adams1*
Leslie G. Adams1*- 1Department of Veterinary Pathobiology, College of Veterinary Medicine and Biomedical Science, Texas A&M University, College Station, TX, United States
- 2Seralogix, Inc., Austin, TX, United States
To date, fewer than 200 gene-products have been identified as Brucella virulence factors, and most were characterized individually without considering how they are temporally and coordinately expressed or secreted during the infection process. Here, we describe and analyze the in vivo temporal transcriptional profile of Brucella melitensis during the initial 4 h interaction with cattle. Pathway analysis revealed an activation of the “Two component system” providing evidence that the in vivo Brucella sense and actively regulate their metabolism through the transition to an intracellular lifestyle. Contrarily, other Brucella pathways involved in virulence such as “ABC transporters” and “T4SS system” were repressed suggesting a silencing strategy to avoid stimulation of the host innate immune response very early in the infection process. Also, three flagellum-encoded loci (BMEII0150-0168, BMEII1080-1089, and BMEII1105-1114), the “flagellar assembly” pathway and the cell components “bacterial-type flagellum hook” and “bacterial-type flagellum” were repressed in the tissue-associated B. melitensis, while RopE1 sigma factor, a flagellar repressor, was activated throughout the experiment. These results support the idea that Brucella employ a stealthy strategy at the onset of the infection of susceptible hosts. Further, through systems-level in silico host:pathogen protein–protein interactions simulation and correlation of pathogen gene expression with the host gene perturbations, we identified unanticipated interactions such as VirB11::MAPK8IP1; BtaE::NFKBIA, and 22 kDa OMP precursor::BAD and MAP2K3. These findings are suggestive of new virulence factors and mechanisms responsible for Brucella evasion of the host's protective immune response and the capability to maintain a dormant state. The predicted protein–protein interactions and the points of disruption provide novel insights that will stimulate advanced hypothesis-driven approaches toward revealing a clearer understanding of new virulence factors and mechanisms influencing the pathogenesis of brucellosis.
Introduction
Brucella, an aerobic non-motile Gram-negative coccobacillus, is the etiological agent of brucellosis, a worldwide anthropozoonotic infectious disease that causes chronic infections with persistent or recurrent bacteremia in susceptible hosts, and mid- to late gestation abortion in pregnant animals. At present, there are 11 recognized species within the genus Brucella based on preferential host specificity (O'Callaghan and Whatmore, 2011; Whatmore et al., 2014). Goats and sheep are the preferred hosts for Brucella melitensis (Alton, 1990), although this pathogen also infects cattle depending on specific epidemiological conditions (Kalher, 2000; Banai, 2010; Alvarez et al., 2011; Liu et al., 2012; Wareth et al., 2014). B. melitensis is also the most pathogenic and the most frequent causative agent of brucellosis in humans (Traxler et al., 2013). Clinically, human brucellosis can be an incapacitating disease that results in intermittent fever, chills, sweats, weakness, myalgia, osteoarticular complications, endocarditis, depression, and anorexia with low mortality (Dean et al., 2012).
The predominant route for B. melitensis penetration after natural exposure is the alimentary tract (Adams, 2002). Usually B. melitensis enters through the oral mucosa and colonizes the lymph nodes that drain the facial area (Carpenter, 1924; von Bargen et al., 2015); although several studies have isolated Brucella from different sections of the alimentary tract and feces revealing the possibility that brucellae invade in multiple sites of the gastrointestinal tract (Carpenter, 1924; Davis et al., 1988). We previously found that under experimental conditions, B. melitensis was able to invade the bovine host through the domed epithelium of jejuno-ileal Peyer's patches followed by rapid systemic dissemination (Rossetti et al., 2013). The calf ligated jejuno-ileal loop model has been demonstrated to be a very useful model to study in vivo natural host:infectious agent molecular and morphological initial interactions (Santos et al., 2002; Khare et al., 2009, 2012; Winter et al., 2010; Lawhon et al., 2011; Rossetti et al., 2013), a subject that has not been broadly studied in brucellosis.
Brucella lack several classical bacterial virulence factors such as exotoxins, cytolysins, a capsule, fimbriae, plasmids, resistant strains, lysogenic phages, antigenic variation, or endotoxic lipopolysaccharide among others (Moreno and Moriyón, 2002). Brucella has developed a stealthy strategy to avoid being recognized and successfully infect hosts. Succinctly stated, Brucella circumvents strong innate immune responses, obstructs the direct action of bactericidal substances, resists destruction by professional phagocytes and maintains the host cells alive to establish long lasting infections (Barquero-Calvo et al., 2007). Although, significant advances have been made lately (de Figueiredo et al., 2015), the molecular pathogenesis of brucellosis is still incompletely understood. To date, fewer than 200 gene products have been identified as Brucella virulence factors (He, 2012), and very few related to adhesion and invasion function have been characterized. For instance, the heat shock protein 60 (Hsp60) family proteins on the surface of Brucella abortus have been found to bind macrophage and intestinal M cells cellular prion protein (PrPc) before internalization (Watarai et al., 2003; Nakato et al., 2012). The SP41 (UgpB) protein that has significant homology with the glycerol-3-phosphate-binding ABC transporter protein interacts with cellular sialic acid residues, facilitating efficient host invasion (Castaneda-Roldán et al., 2006). Other recently characterized proteins in the brucellae genome, such as, BmaC (Posadas et al., 2012), BtaE (Ruiz-Ranwez et al., 2013a), BtaF (Ruiz-Ranwez et al., 2013b) interact with different components of the extracellular matrix, while novel adhesion-encoding regions inv (Alva-Perez et al., 2014), or BigA (Czibener et al., 2016) have been demonstrated to promote adhesion and invasion, but their target host molecules have not been identified yet.
A well-recognized Brucella virulence factor is the two-component response regulator, BvrR/BvrS, that modulates the host cell cytoskeleton upon Brucella invasion (Sola-Landa et al., 1998) and regulates the Brucella OMP expression (Guzmán-Verri et al., 2002). Dysfunction of BvrR/S response regulator system induces mutant strains with reduced invasiveness and failure to replicate and survive intracellularly. Brucella lipopolysaccharide is also a confirmed virulence factor (Lapaque et al., 2005), that prevents complement-mediated bacterial killing (Allen et al., 1998; Tumurkhuu et al., 2006), provides resistance against antimicrobial peptides such as defensins, lysozyme, and lactorerrin (Martínez de Tejada et al., 1995) and inhibits cell death (Pei and Ficht, 2004; Pei et al., 2006). Additionally, Brucella LPS masks recognition of the pathogen-associated molecular patterns (PAMPs) from immune-receptor recognition, and as a consequence, impedes, or attenuates proinflammatory responses and immune system activation (Forestier et al., 2000; Jiménez de Bagués et al., 2004). Simultaneously, the type four secretion system (T4SS) is also a key virulence factor for Brucella intracellular survival (O'Callaghan et al., 1999), persistent infection in mice and induction of the host immune response (Rolan and Tsolis, 2007; Roux et al., 2007). This virulence factor, encoded by a virB operon, is induced by early phagosome acidification after phagocytosis (Boschiroli et al., 2002; Celli et al., 2003) to translocate effector molecules directly into the host cell cytoplasm. Additional investigations have identified several of these effector proteins, although most of their functions remain undefined (de Jong et al., 2008, 2013; de Barsy et al., 2011; Marchesini et al., 2011, 2016; Salcedo et al., 2013; Ke et al., 2015; Del Giudice et al., 2016). The secretion systems and secretomes of Brucella were recently computationally analyzed, resulting in the prediction of 29 host-pathogen specific interactions between cattle and B. abortus and 36 host-pathogen interactions between sheep and B. melitensis proteins (Sankarasubramanian et al., 2016). The two-component system RegB/A, including the aceA encoding isocitrate lysase, has been found to play a critical role in the persistence and in vivo pathogenicity of Brucella suis (Abdou et al., 2017).
An additional virulence mechanism used by Brucella to survive intracellularly is the periplasmic compound cyclic B-1,2 glucan, that interferes with cellular trafficking and maturation of the Brucella-containing vacuole by disrupting cholesterol-rich lipid rafts present on phagosomal membranes and preventing the phagosome-lysosome fusion (Arellano-Reynoso et al., 2005; Martirosyan et al., 2012). Other virulence elements reported to sustain a chronic infection include: phosphatidylcholine, a phospholipid compound abundant in eukaryotic cell membranes that facilitate Brucella avoidance of host recognition (Comerci et al., 2006; Conde-Alvarez et al., 2006); PrpA, a proline-racemase family compound that elicits B lymphocyte polyclonal activation (Spera et al., 2006); BtpA and BtpB, Brucella TIR-containing effector proteins that suppress innate immunity and modulate host inflammatory responses during infection (Salcedo et al., 2008, 2013); MucR, a transcriptional regulator involved in Brucella metabolism, cell wall/envelope biogenesis, replication, type IV secretion system, quorum sensing system, and stress tolerance (Dong et al., 2013) and a flagellar appendage, required for virulence in a mouse infection model (Fretin et al., 2005).
Most of these virulence factors were characterized individually without considering how they are temporally and coordinately expressed or secreted during the infection process. Recently, several experiments have been performed to more fully understand the sequential expression and coordinated regulation of the infection process (Kohler et al., 2002; Al Dahouk et al., 2008; Rambow-Larsen et al., 2008; Lamontagne et al., 2009; Rossetti et al., 2009; Wang et al., 2009; Viadas et al., 2010; Weeks et al., 2010; Hanna et al., 2013; Tian et al., 2013). These experiments using in vitro culture media, infected cell cultures, or infected mice were successful for generating initial hypotheses to enhance the understanding of the pathogenesis of brucellosis.
Here, we describe an integrative approach of experimentation and computation to analyze the in vivo temporal transcriptional profile of B. melitensis during the first 4 h of the interaction with a naturally susceptible host, using the established calf jejuno-ileal loop model of infection. We then performed a system-level analysis by applying both a traditional statistical differential analysis to determine significance of B. melitensis gene expression and a pathway and gene ontology (GO) analysis that employed a dynamic Bayesian network (DBN) technique (Khare et al., 2016) to identify perturbations trends over time. The fundamental concept of systems biology is to: (1) perturb a system—(time-course B. melitensis infected bovine Peyer's patch), (2) measure systems-wide responses—(B. melitensis and bovine transcriptomes), and (3) integrate measured responses into a model—(host:pathogen::Bovine:B. melitensis interactome model) to understand the observations and iteratively predict novel interactions and perturbations. The system-level analyses aided understanding of the strategies exploited by B. melitensis to invade, survive, and replicate intracellularly; and to identify perturbations of major genes modulating critical cellular pathways in the pathogenesis of brucellosis. Further, through systems-level in silico host-pathogen protein–protein interactions (PPIs) simulation (see File S1), we were able to make inferred predictions of interactions of close apposition with specific B. melitensis expressed genes/proteins to plausible host (bovine) pathway points of disruption or perturbations. The predicted PPIs and the points of disruption provide novel insights that will stimulate advanced iterative hypothesis-driven approaches toward revealing a clearer understanding of new virulence factors and mechanisms contributing to the evasion of the host's protective immune responses.
Materials and Methods
Infection Model
The in vivo infection model for Brucella was described previously (Rossetti et al., 2013). Briefly, five bovine jejuno-ileal segments from four calves were inoculated intraluminally with 3 ml of a suspension containing 1 × 109 CFU of B. melitensis 16 M/ml (total of 3 × 109 CFU) at late-log growth phase cultured in F12K medium (ATCC®) supplemented with 10% heat-inactivated fetal bovine serum (HI-FBS) (ATCC®). One infected segment was removed at every time point (0.25, 0.5, 1, 2, 4 h post-inoculation from each of the four calves.), and six to ten 6 mm biopsy punches were collected from each segment. The mucosal layer of Peyer's patch was immediately dissected, macerated and homogenized in TRI-Reagent® (Ambion, Austin, TX). Subsequently, samples were appropriately contained and transported to an inspected and approved BSL-3 laboratory for immediate RNA extraction. Calves were euthanized with an intravenous bolus of sodium pentobarbital at the completion of the procedures. All animal experiments were approved by the Texas A&M University Institutional Animal Care and Research Advisory Committee (AUP#2003-178). Surgeries were performed under biosecurity laboratory III (BSL-3) conditions in CDC-approved isolation buildings at the Texas A&M University experimental farm (College Station, TX).
RNA Isolation, Labeling, and Hybridization
RNA isolation, labeling, and hybridization procedures were performed as described in previous experiments (Rossetti et al., 2010, 2011b). Total RNA from B. melitensis-infected bovine Peyer's patches was extracted according to the TRI-Reagent manufacturer's instructions. Tissue-associated B. melitensis total RNA was initially enriched (MICROBEnrich®, Ambion) and then amplified from 30 μg of total RNA from B. melitensis-infected bovine Peyer's patches (Rossetti et al., 2010). Briefly, the enriched RNA was precipitated in 100% ethanol at −20°C, washed and re-suspended in 25 μl of DEPC-treated water (Ambion). Immediately, the total amount of RNA was linearly amplified in a 3 step-protocol. First, RNA was reverse transcribed to cDNA using B. melitensis genome direct primers (BmGDPs), T7 promoter-template switching primer (T7-TS) (Sigma Genosys, The Woodland, TX) and Moloney Murine Leukemia Virus Reverse Transcriptase (Clontech, Palo Alto, CA). In the next step, the second-strand cDNA was synthesized and purified (Qiagen, Valencia, CA), followed by concentration in a speed-vac with no heat. In the last step, the in vitro transcription, was performed using the double-stranded cDNA as the template and T7 polymerase (Ambion). Then, 10 μg of each experimental sample (n = 44, i.e., 4 were in vitro-grown cultures of B. melitensis at late-log phase of growth; 20 were enriched and amplified B. melitensis RNA from total RNA from infected bovine Peyer's patches; and an additional 20 were from total RNA of infected bovine Peyer's patches) were reverse transcribed overnight to amino-allyl cDNA using 1.5 ug of B. melitensis genomic directed primers (BmGDPs) (Rossetti et al., 2010), labeled with Cy3 (Amersham Pharmacia Biosciences, Piscataway, NJ), mixed with 0.5 μg of Cy5 labeled B. melitensis gDNA, and applied to a custom 3.2K B. melitensis oligoarray (Weeks et al., 2010). Since the enrichment procedure does not eliminate host RNA, total RNA from B. melitensis-infected bovine Peyer's patches were also reverse transcribed, labeled and hybridized on B. melitensis oligo microarray, due to a potential concern that eukaryotic RNA present in enriched and amplified samples could possibly overlap with sequences of the B. melitensis transcripts and cross hybridized with probes on B. melitensis oligo microarrays, resulting in falsely detected pathogen genes. The isolation and labeling of B. melitensis gDNA has been described in detail elsewhere (Rossetti et al., 2009). Slides were hybridized at 45°C for approximately 20 h in a dark humid chamber (Corning, Corning, NY). Then, washed for 10 min at hybridization temperature with low stringency buffer [1X SSC, 0.2% SDS] followed by two 5-min washes with a higher stringency buffer [0.1X SSC, 0.2% SDS and 0.1X SSC] at room temperature with mild agitation, dried by centrifugation and immediately scanned.
Data Acquisition, Normalization, and Microarray Data Analysis
Immediately after washing, the dried slides were scanned using a commercial laser scanner (GenePix 4100; Axon Instruments Inc., Foster City, CA). Scans were performed using the autoscan feature with the percentage of saturated pixels set at 0.03%. The genes represented on the arrays were adjusted for background and normalized to internal controls using image analysis software (GenePixPro 6.0; Axon Instruments Inc.). Genes with fluorescent signal values below background were disregarded in all analyses. Arrays were initially normalized against B. melitensis genomic DNA, and the resulting data were analyzed and modeled using an integrated platform termed the BioSignature Discovery System (BioSignatureDS®) (Seralogix, LLC, Austin, TX; http://www.seralogix.com) explained in detail elsewhere (Lawhon et al., 2011; Rossetti et al., 2013). The tissue-associated B. melitensis gene expression at every time point (0.25–4 h) was compared to the gene expression of the inoculum (i.e., in vitro-grown cultures of B. melitensis at late-log phase of growth, cultured in F12K medium with 10% HI-FBS; n = 4). Significantly expressed genes were determined with the z-test (p < 0.025) (enhanced with Bayesian methods of variance estimation) after subtracting those genes also expressed at statistically significant levels when total RNA of B. melitensis-infected bovine Peyer's patches was compared to the gene expression of the inoculum. BiosignatureDS tools for statistical z-score gene thresholding, Brucella pathway and gene ontology (GO) perturbation scoring (scored using Bayesian Information Criterion and transformed to z-score), and mechanistic gene identification were used for the comprehensive analysis performed in this study. A specialized application was developed to implement algorithms that integrate multiple sources of prior biological knowledge (PBK) into the inference of host-pathogen protein–protein interaction (PPIs) prediction (see File S1 for complete details). Briefly, we adopted three algorithmic methods for the identification of candidate interaction points for use in network learning between the host and the pathogen from in vivo gene expression data. These algorithmic methods were: (1) a sequence-similarity interaction transference procedure; (2) structural protein domain-based algorithm; and (3) a functional gene-ontology-based algorithm. Gene candidates for inclusion in our interaction prediction process were selected based on interpretation of pathway and GO analyses conducted by our Dynamic Bayesian Network methodology. The B. melitensis gene transcriptome was employed and ≈600 bovine host genes selected from 12 perturbed and immune response relevant pathways and 10 GO terms to form gene sets representing two “unconnected” system models for starting the “interactome model” network learning process. Those algorithms yielded 348, 68, and 295 potential host-pathogen PPIs, respectively, that comprised the set of potential interactions at the interface of the pathogen and host systems. These potential interactions were then included into the Bayesian host-pathogen network structure learning algorithm. The method employs model structures to initialize learning with biologically relevant structures and utilize actual time-course co-expressed gene and other “omic” data from pathogen and host to search for a set of structures in which the data best fit. Microarray data and metadata are deposited in the Gene Expression Omnibus at the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/geo/) Accession #GSE89053.
For microarray results validation, six randomly selected genes with consistently differential expressed from 15 min to 4 h post-infection (p.i.) by microarray results, were analyzed at every time point by quantitative RT-PCR (qRT-PCR) following the protocol described elsewhere (Rossetti et al., 2011b). Primers (Sigma Genosys) of tested genes were designed by Primer Express Software v2.0 (Applied Biosystems) (Table S1). For each gene tested, the individual calculated threshold cycles (Ct) were averaged among each condition and normalized to the Ct of the 16S rRNA gene from the same cDNA samples before calculating the fold change using the ΔΔCt method (Livak and Schmittgen, 2001). For each primer pair, a negative control (water) and an RNA sample without reverse transcriptase (to determine genomic DNA contamination) were included as controls during cDNA quantitation. Statistical significance was determined by Student's t-test and expression differences considered significant when P < 0.05. As gene expression by microarray and qRT-PCR were based on z-score and fold-change, respectively, array data were considered valid if the fold change of each gene tested by qRT-PCR was expressed in the same direction as determined by microarray analysis.
Results
B. melitensis Transcriptome Is Perturbed at the Onset of the Infection Process
We previously reported that the number of B. melitensis 16M organisms after intraluminal inoculation increased from 15 min to 4 h p.i. (Rossetti et al., 2013) in this model. To study pathogen alterations in gene expression, pathway, and GO perturbations during the initial infection process, Brucella RNAs extracted from infected bovine Peyer's patches at different times p.i. were hybridized on B. melitensis microarrays and analyzed. As expected, the traditional z-score analysis (|2.24|, 97.5% confidence) identified a total of 2,356 different B. melitensis genes (1,221 up-regulated vs. 1,135 down-regulated) differentially expressed (DE) at least once over the 4 h time course p.i., compared to the in vitro grown control (Table S2). As opposed to the 1 h-peak of the host gene expression after infection (Rossetti et al., 2013), the total number of perturbed Brucella genes is rather constant in the first 4 h p.i. (15 min: 1,899 genes, 30 min: 1,937 genes, 1 h: 1,968 genes, 2 h: 1,909 genes and 4 h: 1,912 genes; Figure 1). The combined analysis of these results, i.e., the host and pathogen transcriptional profiles during bovine Peyer patch infection, clearly indicate that both host and Brucella gene expression responses are markedly perturbed at a very early time post-interaction. This is in concordance with other results (He et al., 2006; Rossetti et al., 2011b, 2012), which corroborates an initial transcriptionally perturbed period followed by a more quiescent one.
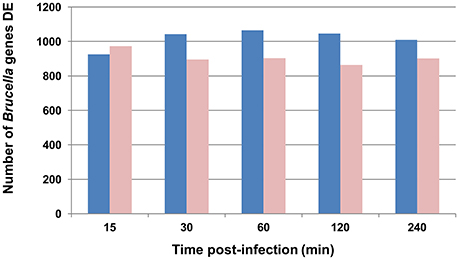
Figure 1. Graphic representation of B. melitensis genes differentially expressed (DE) throughout the experiment. Blue bars represent genes activated; light red bars represent genes repressed. For differential analysis, the in vivo infected loop gene expression is compared to the in vitro log growth phase inoculum as the control.
A group of 1,740 genes (55% of B. melitensis genome) was markedly perturbed in the same direction in at least 4 of 5 time points (Table S3). These genes were considered as the core set of genes associated with the adaptive changes of B. melitensis during the early in vivo bovine Peyer's patch infection, and therefore important in understanding key events in the early modulation of host response. From this set of 1,740 Differentially Expressed (DE) genes, 925 (53%) were activated and 815 (47%) were repressed compared with the in vitro grown culture. Interestingly, genes from the core set located on chromosome I were mainly activated (over 1,174 DE genes: 752 were up- and 422 were down-regulated), while genes located on chromosome II were mainly repressed in higher numbers (566 total DE genes: 173 were up- and 393 were down-regulated). Chromosome I encodes the majority of the core metabolic machinery for transcription, translation and protein synthesis, and Chromosome II is overrepresented in genes involved in pathways for utilization of specific substrates (membrane transport, central intermediary and energy metabolism, and regulation; Paulsen et al., 2002). Altogether, these results suggest that Brucella may restrain metabolic functions while inducing transcriptomic modifications to adapt from an extracellular to an intracellular lifestyle. These results are largely in concordance with previous publications (Lamontagne et al., 2009; Rossetti et al., 2011a,b) even though our study analyzes the complexity of in vivo invasion process in comparison with pathogen gene expression in other in vitro (i.e., one cell line) models of invasion.
Microarray gene expression data were validated by qRT-PCR. Six randomly selected Brucella genes, determined to be significantly affected throughout the first 4 h p.i., were chosen for verification at every time point (i.e., 30 data points). As shown by the representative examples in Figure S1, gene expression changes were consistent between microarray and qRT-PCR for genes with increased expression or genes with decreased expression relative to the control.
Discussion
Major Brucella Virulence Factors Are Down Regulated at the Onset of the Infection
Within 15–30 min of in vivo exposure, B. melitensis adhere and immediately penetrate through the intestinal mucosa and Peyer's patch and rapidly disseminate through systemic circulation (Rossetti et al., 2013). Early stage host gene expression of Syndecan 2, Integrin alpha L and Integrin beta 2 genes coincide with initial Brucella adhesion which is coupled with simultaneous repression of two intestinal barrier-related pathways (Tight Junction and Trefoil Factors Initiated Mucosal Healing), subverting mucosal epithelial barrier function and facilitating Brucella transepithelial migration (Rossetti et al., 2013). To elucidate Brucella virulence mechanisms responsible for this host molecular response, we expanded our analysis on pathogen pathways (Table 1) and GO alterations (Tables S4–S8). Note that pathway molecular interactions and annotations are based on those provided by the Kyoto Encyclopedia of Genes and Genomes (KEGG; Kanehisa et al., 2017). Pathways and Gene Ontology groups are comprised of gene sets which may be either activated or repressed in some combination over time. The Bayesian scoring method computes the log-likelihood of the in vivo expressed data and measures its goodness-of-fit to a model trained with control data (the in vitro inoculum expression data). In this manner, it is possible to determine if a pathway or GO group is activated or repressed. In our computational system biology approach, if the sum of the individual gene scores within a pathway/GO group is positive then the pathway/GO score is considered to be activated. Otherwise, if the sum is negative, the pathway/GO score is considered repressed and assigned a negative score value. Table 1 shows the results of the pathway analysis scoring listed by pathway categories and sorted by activated or repressed state on the 15 min p.i. column. Specific gene expression scores within these pathways are provided in Table S7. Early in the infection process, the pathway category “Environmental Information Processing” has several important pathways involved in B. melitensis pathogenicity which are repressed at 15 min p.i. that include “Type IV secretion system,” “Type III secretion system,” Two-component system,” and ABC transporters. It is interesting to note that the Two-component regulatory systems (TCRSs) and Type III secretion system pathways reverse to an activated state at 30 min. p.i. In the cellular processes category, the “Flagellar assembly” and “Bacterial chemotaxis” pathways are also repressed across all time point p.i.
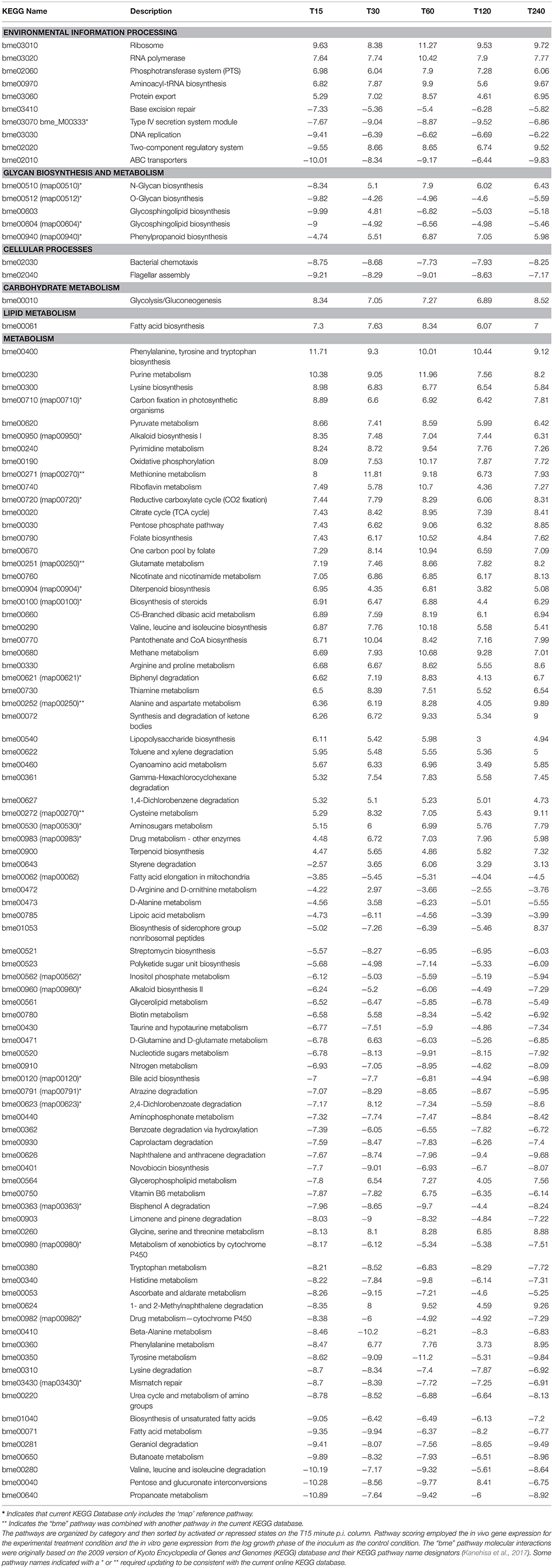
Table 1. Significantly perturbed pathways (Bayesian z-score >|2.24|) of tissue-associated B. melitensis during the first 4 h post-bovine Peyer's patch infection.
The TCRSs are signal transduction mechanisms that allow microorganisms to sense and respond to changes in environmental conditions. Bioinformatic analysis of Brucella genomes has identified 15 predicted bona fide TCRS pairs (Lavin et al., 2010). Several of these systems have been characterized in Brucella species, such as BvrSR (Sola-Landa et al., 1998), FeuQP (Dorrell et al., 1998), NtrBC (Dorrell et al., 1999), NtrXY (Foulongne et al., 2000; Carrica et al., 2012), PrlSR (Mirabella et al., 2012), the flagellar master regulator FtcR (Leonard et al., 2007), the blue-light-activated LOV HKs (Swartz et al., 2007), RegA (Abdou et al., 2017), and CenR (Zhang et al., 2009). Other TCRSs have been identified by transpositional mutagenesis during global screening for virulence factors (Lestrate et al., 2000; Wu et al., 2006) but remain uncharacterized. Our pathway analysis revealed that the TCRSs were initially repressed at 15 min. p.i. and were then activated for the remaining time points (Table 1) providing evidence that in vivo Brucella sense and actively regulate their metabolism through the transition to intracellular lifestyle. Changes in expression between 15 min to 30 min p.i. by the TCRSs genes dctM, glnG, glnL, phoB, phoQ, citE, and divJ resulted in the TCRSs pathway transitioning from a repressed state to an activated state with the exception of aceA which was activated early and then repressed. These genes went from a strongly repressed state to an insignificant expressed state. Contrarily, other Brucella pathways involved in virulence such as “ABC transporters” and “T4SS system” were continuously repressed suggesting a silencing strategy to avoid stimulation of the host's innate immune response very early in the infection process (Table 1). The highly repressed genes associated with the ABC transporters repression included BMEII0196, BMDII0861, PBMII0120, BMEI1138, proW, ybbP, pstB, potB, afuB, rbsC and several others. The T4SS system repression was induced by the repressed genes virB4, virB5, virB6, and virB9 (Table S2). In vitro studies have demonstrated that T4SS is not required for cellular invasion, and its expression begins 15 min after phagocytosis and maximizes at 5 h p.i. (Sieira et al., 2004). It has been shown to be indispensable for intracellular survival of Brucella (O'Callaghan et al., 1999; Sieira et al., 2000; Delrue et al., 2005). Under our in vivo experimental conditions, expression of genes from the virB operon was repressed as was confirmed by qRT-PCR [Figure S1: BMEII0033 (virB9)]. In addition, the transcriptional regulator vjbR (BMEII1116) that positively regulates the expression of B. melitensis virB operon (Delrue et al., 2005) was not differentially expressed in our microarray results (Table S2). These data show that the T4SS was repressed during the first 4 h p.i. of in vivo infection. Collectively, our results, in addition to those reported earlier (Roux et al., 2007; den Hartigh et al., 2008) that failed to detect differences in the number of B. abortus and B. melitensis WT and virB mutant recovered from mice spleens in the first 3 days p.i., suggest that the virB operon may not play a major role in the initial in vivo Brucella pathogenesis. There are likely in vivo environmental signals that modulate the expression of the virB operon differently than reported in in vitro systems of infection. Identification of the host molecule targets of the T4SS will help characterize its expression based on the host cellular response discussed further in the next section “Systems biology in vivo interactome modeling results.”
Systems Biology In vivo Interactome Modeling Results
The simultaneous collection of host and pathogen gene expression data of the bovine host ileal loop infected with B. melitensis WT (Rossetti et al., 2013), provided us with a unique opportunity to examine temporal host pathway perturbations concurrent with those of the pathogen. A computational approach based on DBN machine learning was employed to infer protein–protein interactions (PPIs) and to create a novel in silico host-pathogen interactome model (File S1). To identify plausible PPIs, a specialized application was developed to implement algorithms that integrate multiple sources of PBK such as from KEGG, BIOCARTA, NCBI, PIBASE, and Brucella proteomic analyses (Delvecchio et al., 2002; Wagner et al., 2002; Connolly et al., 2006; Mol et al., 2016), into the inference of host-pathogen protein interactions. Such interactions aid in the identification of mechanisms of host invasion and evasion through manipulation of the host's immune response system. Our application employed Bayesian networks (BNs) (Friedman et al., 2000; Hartemink et al., 2001) that were expanded by others to include PBK (Imoto et al., 2004; Werhli and Husmeier, 2007). We employed methods similar to Werhli for learning PPIs from expression data and PBK (Werhli and Husmeier, 2007). We adopted three algorithmic methods for the identification of candidate interaction points for use in network learning between the host and pathogen from in vivo gene expression data. Through either: (1) protein binding domain, (2) sequence similarity, or (3) Gene Ontology-based functional algorithms of pathogen gene expression with the host gene perturbations, we identified potential B. melitensis interactions with host pathways.
The PPI analysis resulted in identifying the virB gene that encodes the T4SS proteins to have plausible interaction with a number of genes in the host's immune response pathways (Table 2 and Table S8). For example, the significantly perturbed virB11 gene has a high protein domain binding prediction with a negative correlation with the host gene/protein PIK3R2 expression. PIK3R2 is a key regulatory protein in several key pathways including: mTOR signaling, T-cell and B-cell receptor, Integrin-mediated cell adhesion, regulation of actin cytoskeleton, apoptosis, and Toll-like receptor. The host gene PIK3R2 remained activated for all time points p.i. Interestingly, in our host pathway analysis of “Regulation of Actin Cytoskeleton,” the genes ABI2, PPN1, and ARPC5, down-stream from PIK3R2, were repressed suggesting a mechanism of host pathway disruption or highjacking. The VirB11 also had a strong protein domain binding prediction with negative correlation with the host genes MAPK8IP1/2 of the MAPK signaling pathway. The MAPK8IP1/2 host genes were strongly repressed 15 min p.i. and insignificantly expressed thereafter. Down-stream of MAPK8IP1/2, the transcription factor JUND and nuclear factor of activated T-cells, cytoplasmic 3, NFATC2 are both strongly repressed.
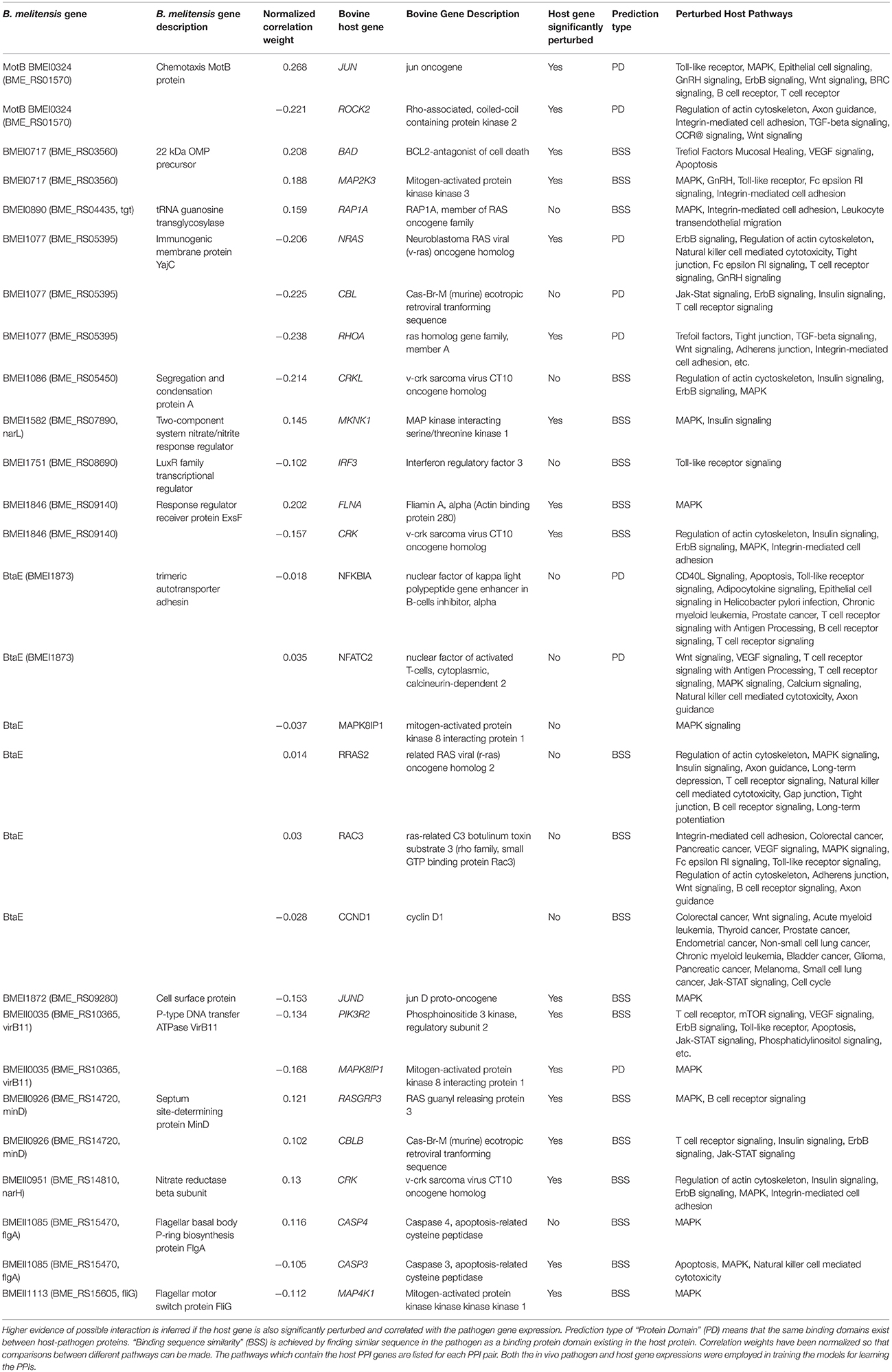
Table 2. Interactome model predicted protein–protein interactions (PPIs) between bovine host and B. melitensis pathogen.
Brucella flagellum is a virulence factor transiently expressed during vegetative growth and required for persistent infection, but not for internalization in vivo (Fretin et al., 2005). In agreement, our results during the first 4 h of infection showed a repression of the three flagellum-encoded loci (BMEII0150-0168, BMEII1080-1089, and BMEII1105-1114) (Table S3), with corresponding repression of the “flagellar assembly” pathway (Table 1), and the cell components “bacterial-type flagellum hook” and “bacterial-type flagellum” (Table S5) in tissue-associated B. melitensis compared with in vitro-grown cultures. In addition, RopE1 sigma factor (BMEI0371), a flagellar repressor (Ferooz et al., 2011), was activated throughout the experiment (Table S3). We further examined potential interaction of the flagella-associated genes with the host. Three highly correlated PPIs were identified which included the flagella genes BMEI0324, BMEII1085 (flgA), and BMEII1113 (fliG-2). The ORF BMEI0324 had strong binding sequence similarity and positive correlation to the host expressed JUN (jun oncogene) which is part of the highly perturbed host pathways: Toll-like Receptor, ErbB Signaling, BRC Signaling, B-cell Signaling, T-cell Signaling, Epithelial Cell Signaling, WNT Signaling, and MAPK Signaling. The flagellum gene flgA also had strong binding sequence similarity and positive gene expression correlation to host CASP2 gene while having a reversed (negative) correlation with the host activated CASP3 gene. Interestingly, the lowly expressed CASP2 is only associated with the highly perturbed MAPK signaling pathway, while CASP3 has several pathway associations that include: Apoptosis, Epithelial Signaling, Natural Killer Cell, and MAPK Signaling. The third flagellum gene fliG-2 had strong binding sequence similarity to the host MAP4K1 gene. The highly activated MAP4K1 is associated with only the host MAPK Signaling pathway. Such interactions may be novel virulence candidates that facilitate circumventing the host immune response. An example of interaction is the pathogen gene flgA with the host gene/protein Casp4/6/7/9 which are MAPK pathway genes. Accordingly, down-stream of Casp genes are the repressed RAC1/2/3 genes of the Rho family of GTPases. RAC1 has been implicated in various downstream cellular functions, including, but not limited to, cellular plasticity, migration and invasion, cellular adhesions, cell proliferation, and apoptosis.
Host cells identify specific pathogen-associated molecular pattern (PAMP) motifs present in the bacteria by pattern-recognition receptors (PRRs), such as Toll-like Receptors (TLRs). These receptors are key to establishing an important network between the innate and adaptive immune systems. TLR5 is the cellular receptor for extracellular flagellin, a major structural protein of Gram-negative flagella. Binding of flagellin to the extracellular domain of TLR5 rapidly induces a signal cascade that culminates in the production of proinflammatory mediators such as cytokines, chemokines, and costimulatory molecules (Honko and Mizel, 2005). Therefore, the absence of flagellum apparatus during extracellular life while inside the host suggests the Brucella strategy is to avoid triggering a host immune response and an initiation of a Brucella persistence mechanism (Terwagne et al., 2013). However, our previous analysis showed that TLR5 pathway is activated in B. melitensis-infected bovine Peyer's patches during the first hour p.i. (Rossetti et al., 2013), which may have been associated with remnants of flagella in the in vitro-growth culture media intraluminally inoculated. More detailed analysis of this pathway showed that down-stream of TLR5 there were several strongly repressed genes including PIK3C2B, PIK3R4, STAT1, AKT3, RAC3, IL6, and TICAM3. This may suggest that the pathogen is manipulating important signaling processes by some other mechanism. PPI analysis indicated that virB genes have predicted interactions with STAT1, PIK3C2B, and IL6 which may also be circumventing the TLR5 response to flagellin stimulation and preventing the host from mounting an effective immune response.
The complexity of a complete system-level host and B. melitensis interaction model (Ginteractome) is illustrated in Figure 2A. This Bayesian network model is comprised of approximately 528 nodes (genes) and 987 arcs that connect gene nodes (relationships). Of the 987 arcs, 101 arcs were learned for host-pathogen points of interaction which are highlighted by the orange colored arcs. The number of host genes were limited to a selected set of perturbed host pathways which included MAPK signaling, ErbB signaling, mTOR signaling, WNT signaling, VEGF signaling, Toll-like Receptor signaling, GnRH signaling, Tight junction, Phosphatidylinositol signaling, Notch signaling, Natural killer cell mediated cytotoxicity, and Apoptosis. The intent for using only perturbed pathways was to look for plausible points of pathogen interactions which could influence the hosts immune response. Although this model is visually complex, the model allows for the computational extraction of potentially important mechanisms of interaction. Of the 101 arcs, the prioritization of these interactions can be analyzed according to the most likely to least likely in the following order: “protein domain,” “sequence similarity,” “GO Functionality,” and “Correlated Data”. The creation of the interactome model employed only PPI relationships based on protein domain or sequence similarity. For example, Figure 2B illustrates the simplification for the interaction between the pathogen's Type IV secretion system and a known cell surface protein, BtaE (Ruiz-Ranwez et al., 2013b), with the host's Toll-like receptor pathway. This demonstrates how predictive information can be employed to interpret host-pathogen responses. The thicker orange arcs are the connections from the pathogen to the host. From this type of analysis, it is possible to understand the state of host's gene expression down-stream from the potential points of interaction/disruption in any of the pathways showing potential manipulation by the pathogen. Table 2 lists a selected subset of predicted interactions representing the pathogen-host pairs based solely on “protein domain” and “sequence similarity” prediction. A complete listing of predicted PPIs based on protein domain or sequence similarity is provided in Table S8. Note that our computational approach did predict interactions of the B. suis gene BtaE with several host proteins listed in Table 2. BtaE belongs to the type II (trimeric) autotransporter family and is an orthologue of B. melitensis BMEI1873. BtaE has been shown to have an active role in host cell adhesion and binding with components of the extracellular matrix such as fibronectin, collagen, and vitronectin (Ruiz-Ranwez et al., 2013b). The interactome model and its predicted PPI list is the analysis output to be employed for further in vitro validation and model refinements. The resulting B. melitensis PPI gene set may represent important new virulence factors with the potential to disrupt or hijack the host immune response. Table 2 also lists the perturbed host pathways in which the host gene PPI is associated, intentionally unfiltered conceptually for subcellular locations so that all PPI are presented. Little is known about the complete secretome of B. melitensis during in vivo host invasion and proliferation although the secretion systems and secretomes of Brucella were recently computationally analyzed which predicted 29 host-pathogen specific interactions between cattle and B. abortus and 36 host-pathogen interactions between sheep and B. melitensis proteins (Sankarasubramanian et al., 2016). The PPI computational approach employed evidence of host pathway perturbation and gene expression disruption as possible indicators of pathogen interaction. If there was plausible potential for a PPI based on binding domain or sequence similarity to known protein interactions between the pathogen and host protein, then it was included in the PPI in list (Table 2 and Table S8). The list of PPIs can be prioritized based on the normalized correlation weights. The larger the normalized weight indicates stronger likelihood of a relationship between the pathogen and host genes. Note that the normalized correlation weight is an output of structure learning and is employed in the acceptance or rejection of an arc (edge) in the final Bayesian network. Arc weight is dependent on the number of incoming arcs to a node and other factors and should not be confused as a true correlation measurement between two gene expression values.
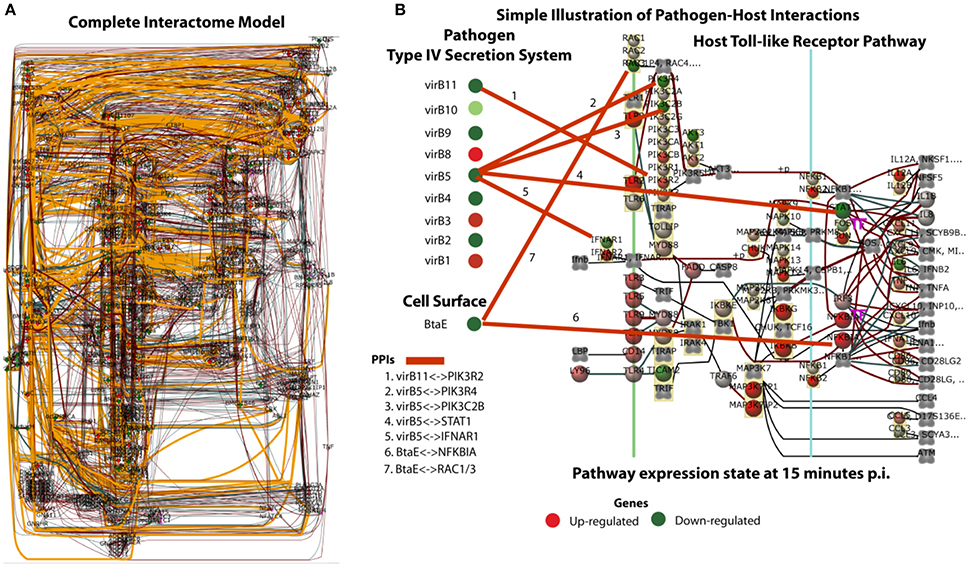
Figure 2. (A) The Bayesian network for host-pathogen interactome model for bovine Peyer's patch challenged with B. melitensis. (B) Simplification of the interactome to illustrate the points of interaction between pathogen's Type IV secretion system and the cell surface gene BtaE with the host's Toll-like receptor pathway. The arcs show the points of predictive interaction which could be possible mechanisms of disrupting the host's effective immune response to B. melitensis.
It is thought that the unfiltered PPI predictions could, in future experiments, employ such information as normalized correlation weights and cellular localization to help prioritize the selection of which PPI to be experimentally validated. Further, it is proposed that the evidence driven computational approach (in vivo host-pathogen responses and machine learning) for predicting bacterial and host cell protein interactions will narrow the focus on likely PPI candidates and will greatly enhance our capacity to design hypothesis-driven experimental approaches to discover which Brucella proteins directly participate in host interactions.
In conclusion, the in silico interactome modeling offers informative insights leading toward new hypotheses regarding host-pathogen mechanisms of invasion and evasion. This modeling infers that B. melitensis has multiple points of host interaction that occur at the early stage post infection. A number of important innate immune response pathways appear to be potential targets of disruption by invading B. melitensis, such as Regulation of Actin Cytoskeleton, mTOR Signaling, MAPK, and Toll-like Receptor Signaling appear to be likely targets of pathogen manipulation that warrant further exploratory research and verification of PPIs. As we have reported and discussed here, identifying interactive host:pathogen PPIs is often the initial step to establish functional significance according to the principle of “guilty by association” (Schauer and Stingl, 2009) that may drive future research to a higher level of understanding of the molecular pathogenesis of brucellosis, thereby facilitating the design of novel immunotherapeutic drugs and vaccines.
Ethics Statement
This study was carried out in accordance with the recommendations of the Texas A&M University Institutional Animal Care and Research Advisory Committee. The protocol (AUP#2003-178) was approved by the Texas A&M University Institutional Animal Care and Research Advisory Committee.
Author Contributions
Conceived and designed the experiments: CR and LA. Performed the experiments: CR, SL, JN, TG, SK, and LA. Analyzed the data: CR, KD, and LA. Writing—original draft: CR, KD, and LA. Writing—review & editing: CR, KD, SL, JN, TG, SK, and LA.
Conflict of Interest Statement
KD is a Chief Technology Officer in Seralogix, LLC. Seralogix is a bioinformatics research and services company commercializing computational systems biology software tools that are being sponsored by the National Institute of Allergy and Infectious Diseases and the National Human Genome Research Institute. KD participated in conducting certain genomic data processing involving pathway analyses and modeling that helped to provide a more system-level perspective of the host-pathogen interaction to the Texas A&M University researchers. Data were processed by KD utilizing Seralogix's proprietary computational pipeline for biological systems analysis. The relation between Seralogix and Texas A&M University, College of Veterinary Medicine and Biomedical Science is strictly on a collaborative (mutually beneficial) research basis with no financial arrangements, commitments or interests. KD's motivation is to see their computational tools produce results that contribute to the improved understanding of host response to pathogen invasions (an objective of his National Health Institute research grants). KD contributed to the interpretation of the analysis results provided to the Texas A&M University researchers. Seralogix has no ownership of the data, nor results produced by their tools.
The other authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Mr. Alan Patranella for the assistance with animal care, and Mrs. Roberta Pugh and Mrs. Doris Hunter for technical support. LA was supported by a grant from the National Institutes of Health (NIH)/National Institute of Allergy and Infectious Diseases 1U54 AI057156-01. LA is also supported by the U.S. Department of Homeland Security National Center of Excellence for Foreign Animal and Zoonotic Disease Defense ONR-N00014-04-1-0 grant. CR was supported by an I.N.T.A.-Fulbright Argentina Fellowship.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.01275/full#supplementary-material
Figure S1. Validation of Brucella melitensis microarray results by quantitative real time PCR. Six randomly selected B. melitensis ORFs that were consistently perturbed in microarray results in the first 4 h p.i. as compared to the inoculum as validated by quantitative RT-PCR. Fold-change was normalized to the expression of B. melitensis 16s rRNA and calculated using the ΔΔCt method. All tested genes at every time point had expression altered in the same direction as microarray. Open bars represent fold-change by microarray analysis and black bars represent fold-change by qRT-PCR.
Table S1. Primers for Real Time PCR analysis of genes in B. melitensis samples.
Table S2. Bayesian z-score of B. melitensis genes in tissue-associated B. melitensis from 15 min to 4 h post-infection. Genes with z-score >|2.24| were considered differentially expressed. Positive numbers in the body of the table indicate activated genes and negative (−) numbers indicate repressed genes. The tissue-associated B. melitensis gene expression at every time point was compared to the gene expression of the inoculum (i.e., in vitro-grown cultures of B. melitensis at late-log phase of growth). Measured time points were 15 (T15), 30 (T30), 60 (T60), 120 (T120), and 240 (T240) min p.i.
Table S3. Core set of B. melitensis genes differentially expressed (>|2.24|) in at least four of five time points in the first 4 h post-infection of bovine Peyer's patch. Genes with z-score >|2.24| were considered differentially expressed. Positive numbers in the body of the table indicate activated genes, negative (−) numbers indicate repressed genes. The tissue-associated B. melitensis gene expression at every time point was compared to the gene expression of the inoculum (i.e. in vitro-grown cultures of B. melitensis at late-log phase of growth). Measured time points were 15 (T15), 30 (T30), 60 (T60), 120 (T120), and 240 (T240) min p.i.
Table S4. Significantly perturbed Biological Processes (BP) (Bayesian z-score >|2.24|) of tissue-associated B. melitensis during the first 4 h post-bovine Peyer's patch infection. Measured time points were 15 (T15), 30 (T30), 60 (T60), 120 (T120), and 240 (T240) min p.i. The Bayesian z-scores represent the degree of perturbation of the group of BP genes in tissue-associated B. melitensis versus the control. Positive z-scores represent activation of the BP (BP score is dominated by more activated genes), while the negative (−) z-score represents BP repression (BP score is dominated repressed genes).
Table S5. Significantly perturbed Cellular Components (CC) (Bayesian z-score >|2.24|) of tissue-associated B. melitensis during the first 4 h post-bovine Peyer's patch infection. Measured time points were 15 (T15), 30 (T30), 60 (T60), 120 (T120), and 240 (T240) minp.i. The Bayesian z-scores represent the degree of perturbation of the group of CC genes in tissue-associated B. melitensis versus the control. Positive z-scores represent activation of the CC (CC score is dominated by activated regulated genes), while the negative (−) z-score represents CC repression (CC score is dominated by repressed genes).
Table S6. Significantly perturbed Molecular Functions (MF) (Bayesian z-score >|2.24|) of tissue-associated B. melitensis during the first 4 h post-bovine Peyer's patch infection. Measured time points were 15 (T15), 30 (T30), 60 (T60), 120 (T120), and 240 (T240) min p.i. The Bayesian z-scores represent the degree of perturbation of the group of MF genes in tissue-associated B. melitensis versus the control. Positive z-scores represent activation of the MF (MF score is dominated by more activated genes), while the negative (−) -score represents MF repression (MF score is dominated by repressed genes).
Table S7. Dynamic Bayesian pathway analysis scores along with the associated individual genes and their Bayesian Scores by time point post-inoculation of tissue-associated B. melitensis during the first 4 h post-bovine Peyer's patch infection. Measured time points were 15 (T15), 30 (T30), 60 (T60), 120 (T120), and 240 (T240) min p.i. Positive z-scores represent activation of genes while negative (−) -scores represent repressed genes).
Table S8. Comprehensive list of predicted host:pathogen tissue-associated B. melitensis:bovine protein–protein interactions (PPI). This list includes only those PPIs which were learned by either known protein domain binding or sequence similarity to known binding domains employing our Bayesian methods for PPI prediction.
File S1. Host-Pathogen Protein-Protein Interaction (PPIs) Prediction.
References
Abdou, E., Jiménez De Bagüés, M. P., Martínez-Abadia, I., Ouahrani-Bettache, S., Pantesco, V., Occhialini, A., et al. (2017). RegA plays a key role in oxygen-dependent establishment of persistence and in isocitrate lyase activity, a critical determinant of in vivo Brucella suis pathogenicity. Front. Cell. Infect. Microbiol. 7:186. doi: 10.3389/fcimb.2017.00186
Adams, L. G. (2002). The pathology of brucellosis reflects the outcome of the battle between the host genome and the Brucella genome. Vet. Microbiol. 90, 553–561. doi: 10.1016/S0378-1135(02)00235-3
Al Dahouk, S., Jubier-Maurin, V., Scholz, H. C., Tomaso, H., Karges, W., Neubauer, H., et al. (2008). Quantitative analysis of the intramacrophagic Brucella suis proteome reveals metabolic adaptation to late stage of cellular infection. Proteomics 8, 3862–3870. doi: 10.1002/pmic.200800026
Allen, C. A., Adams, L. G., and Ficht, T. A. (1998). Transposon-derived Brucella abortus rough mutants are attenuated and exhibit reduced intracellular survival. Infect. Immun. 66, 1008–1016.
Alton, G. G. (1990). “Brucella melitensis,” in Animal Brucellosis, eds K. Nielsen and J. R. Duncan (Boca Raton, FL: CRC Press, Inc.), 383–409.
Alva-Perez, J., Arellano-Reynoso, B., Hernandez-Castro, R., and Suarez-Guemes, F. (2014). The invA gene of Brucella melitensis is involved in intracellular invasion and is required to establish infection in a mouse model. Virulence 5, 563–574. doi: 10.4161/viru.28589
Alvarez, J., Saez, J. L., Garcia, N., Serrat, C., Perez-Sancho, M., Gonzalez, S., et al. (2011). Management of an outbreak of brucellosis due to B. melitensis in dairy cattle in Spain. Res. Vet. Sci. 90, 208–211. doi: 10.1016/j.rvsc.2010.05.028
Arellano-Reynoso, B., Lapaque, N., Salcedo, S., Briones, G., Ciocchini, A. E., Ugalde, R. A., et al. (2005). Cyclic B-1,2-glucan is a Brucella virulence factor required for intracellular survival. Nat. Immunol. 6, 618–625. doi: 10.1038/ni1202
Banai, M. (2010). Insights into the problem of B. Melitensis and rationalizing a vaccination programme in Israel. Prilozi 31, 167–180.
Barquero-Calvo, E., Chaves-Olarte, E., Weiss, D. S., Guzmán-Verri, C., Chacon-Diaz, C., Rucavado, A., et al. (2007). Brucella abortus uses a stealthy strategy to avoid activation of the innate immune system during the onset of infection. PLoS ONE 2:e631. doi: 10.1371/journal.pone.0000631
Boschiroli, M. L., Ouahrani-Bettache, S., Foulongne, V., Michaux-Charachon, S., Bourg, G., Allardet-Servent, A., et al. (2002). The Brucella suis virB operon is induced intracellularly in macrophages. Proc. Natl. Acad. Sci. U.S.A. 99, 1544–1549. doi: 10.1073/pnas.032514299
Carpenter, C. M. (1924). Bacterium abortum invasion of the tissues of calves from the ingestion of infected milk. Cornell Vet. 14, 16–31.
Carrica, M. C., Fernandez, I., Marti, M. A., Paris, G., and Goldbaum, F. A. (2012). The NtrY/X two-component system of Brucella spp. acts as a redox sensor and regulates the expression of nitrogen respiration enzymes. Mol. Microbiol. 85, 39–50. doi: 10.1111/j.1365-2958.2012.08095.x
Castaneda-Roldán, E. I., Ouahrani-Bettache, S., Saldana, Z., Avelino-Flores, F., Rendón, M. A., Dornand, J., et al. (2006). Characterization of SP41, a surface protein of Brucella associated with adherence and invasion of host epithelial cells. Cell. Microbiol. 8, 1877–1887. doi: 10.1111/j.1462-5822.2006.00754.x
Celli, J., De Chastellier, C., Franchini, D. M., Pizarro-Cerdá, J., Moreno, E., and Gorvel, J. P. (2003). Brucella evades macrophages killing via VirB-dependent sustained interactions with the endoplasmic reticulum. J. Exp. Med. 198, 545–556. doi: 10.1084/jem.20030088
Comerci, D. J., Altabe, S., De Mendoza, D., and Ugalde, R. A. (2006). Brucella abortus synthesizes phophatidilcoline from coline provided by the host. J. Bacteriol. 188, 1929–1934. doi: 10.1128/JB.188.5.1929-1934.2006
Conde-Alvarez, R., Grillo, M. J., Salcedo, S. P., De Miguel, M. J., Fugier, E., Gorvel, J. P., et al. (2006). Synthesis of phosphatidylcholine, a typical eukaryotic phospholipid, is necessary for full virulence of the intracellular bacterial parasite Brucella abortus. Cell. Microbiol. 8, 1322–1335. doi: 10.1111/j.1462-5822.2006.00712.x
Connolly, J. P., Comerci, D., Alefantis, T. G., Walz, A., Quan, M., Chafin, R., et al. (2006). Proteomic analysis of Brucella abortus cell envelope and identification of immunogenic candidate proteins for vaccine development. Proteomics 6, 3767–3780. doi: 10.1002/pmic.200500730
Czibener, C., Merwaiss, F., Guaimas, F., Del Giudice, M. G., Serantes, D. A., Spera, J. M., et al. (2016). BigA is a novel adhesin of Brucella that mediates adhesion to epithelial cells. Cell. Microbiol. 18, 500–513. doi: 10.1111/cmi.12526
Davis, D. S., Heck, F. C., Williams, J. D., Simpson, T. R., and Adams, L. G. (1988). Interspecific transmission of Brucella abortus from experimentally infected coyotes (Canis latrans) to parturient cattle. J. Wildl. Dis. 24, 533–537. doi: 10.7589/0090-3558-24.3.533
de Barsy, M., Jamet, A., Filopon, D., Nicolas, C., Laloux, G., Rual, J. F., et al. (2011). Identification of a Brucella spp. secreted effector specifically interacting with human small GTPase Rab2. Cell Microbiol. 13, 1044–1058. doi: 10.1111/j.1462-5822.2011.01601.x
de Figueiredo, P., Ficht, T. A., Rice-Ficht, A., Rossetti, C. A., and Adams, L. G. (2015). Pathogenesis and immunobiology of brucellosis: review of Brucella-host interactions. Am. J. Pathol. 185, 1505–1517. doi: 10.1016/j.ajpath.2015.03.003
de Jong, M. F., Starr, T., Winter, M. G., Den Hartigh, A. B., Child, R., Knodler, L. A., et al. (2013). Sensing of bacterial type IV secretion via the unfolded protein response. MBio 4:e00418–12. doi: 10.1128/mbio.00418-12
de Jong, M. F., Sun, Y. H., Den Hartigh, A. B., Van Dijl, J. M., and Tsolis, R. M. (2008). Identification of VceA and VceC, two members of the VjbR regulon that are translocated into macrophages by the Brucella type IV secretion system. Mol. Microbiol. 70, 1378–1396. doi: 10.1111/j.1365-2958.2008.06487.x
Dean, A. S., Crump, L., Greter, H., Hattendorf, J., Schelling, E., and Zinsstag, J. (2012). Clinical manifestations of human brucellosis: a systematic review and meta-analysis. PLoS Negl. Trop. Dis. 6:e1929. doi: 10.1371/journal.pntd.0001929
Del Giudice, M. G., Dohmer, P. H., Spera, J. M., Laporte, F. T., Marchesini, M. I., Czibener, C., et al. (2016). VirJ is a Brucella virulence factor involved in the secretion of type IV secreted substrates. J. Biol. Chem. 291, 12383–12393. doi: 10.1074/jbc.M116.730994
Delrue, R. M., Deschamps, C., Leonard, S., Nijskens, C., Danese, I., Schaus, J. M., et al. (2005). A quorum-sensing regulator controls expression of both the type IV secretion system and the flagellar apparatus of Brucella melitensis. Cell. Microbiol. 7, 1151–1161. doi: 10.1111/j.1462-5822.2005.00543.x
Delvecchio, V. G., Wagner, M. A., Eschenbrenner, M., Horn, T. A., Kraycer, J. A., Estock, F., et al. (2002). Brucella proteomes–a review. Vet. Microbiol. 90, 593–603. doi: 10.1016/S0378-1135(02)00239-0
den Hartigh, A. B., Rolan, H. G., De Jong, M. F., and Tsolis, R. M. (2008). VirB3 to VirB6 and VirB8 to VirB11, but not VirB7, are essential for mediating persistence of Brucella in the reticuloendothelial system. J. Bacteriol. 190, 4427–4436. doi: 10.1128/JB.00406-08
Dong, H., Liu, W., Peng, X., Jing, Z., and Wu, Q. (2013). The effects of MucR on expression of type IV secretion system, quorum sensing system and stress responses in Brucella melitensis. Vet. Microbiol. 166, 535–542. doi: 10.1016/j.vetmic.2013.06.023
Dorrell, N., Guigue-Talet, P., Spencer, S., Foulongne, V., O'Callaghan, D., and Wren, B. W. (1999). Investigation into the role of the response regulator NtrC in the metabolism and virulence of Brucella suis. Microb. Pathog. 27, 1–11. doi: 10.1006/mpat.1999.0278
Dorrell, N., Spencer, S., Foulongne, V., Guigue-Talet, P., O'Callaghan, D., and Wren, B. W. (1998). Identification, cloning and initial characterisation of FeuPQ in Brucella suis: a new sub-family of two component regulatory systems. FEMS Microbiol. Lett. 162, 143–150. doi: 10.1111/j.1574-6968.1998.tb12991.x
Ferooz, J., Lemaire, J., Delory, M., De Bolle, X., and Letesson, J. J. (2011). RpoE1, an extracytoplasmic function sigma factor, is a repressor of the flagellar system in Brucella melitensis. Microbiology 157, 1263–1268. doi: 10.1099/mic.0.044875-0
Forestier, C., Deleuil, F., Lapaque, N., Moreno, E., and Gorvel, J. P. (2000). Brucella abortus lipopolysaccharide in murine peritoneal macrophages acts as a down-regulator of T cell activation. J. Immunol. 165, 5202–5210. doi: 10.4049/jimmunol.165.9.5202
Foulongne, V., Bourg, G., Cazevieille, C., Michaux-Charachon, S., and O'Callaghan, D. (2000). Identification of Brucella suis genes affecting intracellular survival in an in vitro human macrophage infection model by signature-tagged transposon mutagenesis. Infect. Immun. 68, 1297–1303. doi: 10.1128/IAI.68.3.1297-1303.2000
Fretin, D., Faucommier, A., Kohler, S., Halling, S. M., Léonard, S., Nijskens, C., et al. (2005). The sheathed flagellum of Brucella melitensis is involved in persistence in a murine model of infection. Cell. Microbiol. 7, 687–698. doi: 10.1111/j.1462-5822.2005.00502.x
Friedman, N., Linial, M., Nachman, I., and Pe'er, D. (2000). Using Bayesian networks to analyze expression data. J. Comput. Biol. 7, 601–620. doi: 10.1089/106652700750050961
Guzmán-Verri, C., Manterola, L., Sola-Landa, A., Parra, A., Cloeckaert, A., Garin, J., et al. (2002). The two-component system BvrR/BvrS essential for Brucella abortus virulence regulates the expression of outer membrane proteins with counterparts in members of the Rhizobiaceae. Proc. Natl. Acad. Sci. U.S.A. 99, 12375–12380. doi: 10.1073/pnas.192439399
Hanna, N., Ouahrani-Bettache, S., Drake, K. L., Adams, L. G., Kohler, S., and Occhialini, A. (2013). Global Rsh-dependent transcription profile of Brucella suis during stringent response unravels adaptation to nutrient starvation and cross-talk with other stress responses. BMC Genomics 14:459. doi: 10.1186/1471-2164-14-459
Hartemink, A. J., Gifford, D. K., Jaakkola, T. S., and Young, R. A. (2001). Using graphical models and genomic expression data to statistically validate models of genetic regulatory networks. Pac. Symp. Biocomput. 6, 422–433. doi: 10.1142/9789814447362_0042
He, Y. (2012). Analyses of Brucella pathogenesis, host immunity, and vaccine targets using systems biology and bioinformatics. Front. Cell. Infect. Microbiol. 2:2. doi: 10.3389/fcimb.2012.00002
He, Y., Reichow, S., Ramamoorthy, S., Ding, X., Lathigra, R., Craig, J. C., et al. (2006). Brucella melitensis triggers time-dependent modulation of apoptosis and down-regulation of mitochondrion-associated gene expression in mouse macrophages. Infect. Immun. 74, 5035–5046. doi: 10.1128/IAI.01998-05
Honko, A. N., and Mizel, S. B. (2005). Effects of flagellin on innate and adaptive immunity. Immunol. Res. 33, 83–101. doi: 10.1385/IR:33:1:083
Imoto, S., Higuchi, T., Goto, T., Tashiro, K., Kuhara, S., and Miyano, S. (2004). Combining microarrays and biological knowledge for estimating gene networks via Bayesian networks. J. Bioinform. Comput. Biol. 2, 77–98. doi: 10.1142/S021972000400048X
Jiménez de Bagués, M. P., Terraza, A., Gross, A., and Dornand, J. (2004). Different responses of macrophages to smooth and rough Brucella spp.: relationship to virulence. Infect. Immun. 72, 2429–2433. doi: 10.1128/IAI.72.4.2429-2433.2004
Kalher, S. C. (2000). Brucella melitensis infection discovered in cattle for first time, goats also infected. J. Am. Vet. Med. Assoc. 216:648. Available online at: https://www.avma.org/News/JAVMANews/Pages/s030100b.aspx
Kanehisa, M., Furumichi, M., Tanabe, M., Sato, Y., and Morishima, K. (2017). KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45, D353–D361. doi: 10.1093/nar/gkw1092
Ke, Y., Wang, Y., Li, W., and Chen, Z. (2015). Type IV secretion system of Brucella spp and its effectors. Front. Cell Infect. Microbiol. 5:72. doi: 10.3389/fcimb.2015.00072
Khare, S., Drake, K. L., Lawhon, S. D., Nunes, J. E., Figueiredo, J. F., Rossetti, C. A., et al. (2016). Systems analysis of early host gene expression provides clues for transient Mycobacterium avium ssp avium vs. persistent Mycobacterium avium ssp paratuberculosis intestinal infections. PLoS ONE 11:e0161946. doi: 10.1371/journal.pone.0161946
Khare, S., Lawhon, S. D., Drake, K. L., Nunes, J. E., Figueiredo, J. F., Rossetti, C. A., et al. (2012). Systems biology analysis of gene expression during in vivo Mycobacterium avium paratuberculosis enteric colonization reveals role for immune tolerance. PLoS ONE 7:e42127. doi: 10.1371/journal.pone.0042127
Khare, S., Nunes, J. S., Figueiredo, J. F., Lawhon, S. D., Rossetti, C. A., Gull, T., et al. (2009). Early phase morphological lesions and transcriptional responses of bovine ileum infected with Mycobacterium avium subsp. paratuberculosis. Vet. Pathol. 46, 717–728. doi: 10.1354/vp.08-VP-0187-G-FL
Kohler, S., Foulongne, V., Ouahrani-Bettache, S., Bourg, G., Teyssier, J., Ramuz, M., et al. (2002). The analysis of the intramacrophagic virulome of Brucella suis deciphers the environment encountered by the pathogen inside the macrophage host cell. Proc. Natl. Acad. Sci. U.S.A. 99, 15711–15716. doi: 10.1073/pnas.232454299
Lamontagne, J., Forest, A., Marazzo, E., Denis, F., Butler, H., Michaud, J. F., et al. (2009). Intracellular adaptation of Brucella abortus. J. Proteome Res. 8, 1594–1609. doi: 10.1021/pr800978p
Lapaque, N., Moriyón, I., Moreno, E., and Gorvel, J. P. (2005). Brucella lipopolysaccharide acts as a virulence factor. Curr. Opin. Microbiol. 8, 60–66. doi: 10.1016/j.mib.2004.12.003
Lavin, J. L., Binnewies, T. T., Pisabarro, A. G., Ussery, D. W., Garcia-Lobo, J. M., and Oguiza, J. A. (2010). Differences in two-component signal transduction proteins among the genus Brucella: implications for host preference and pathogenesis. Vet. Microbiol. 144, 478–483. doi: 10.1016/j.vetmic.2010.01.007
Lawhon, S. D., Khare, S., Rossetti, C. A., Everts, R. E., Galindo, C. L., Luciano, S. A., et al. (2011). Role of SPI-1 secreted effectors in acute bovine response to Salmonella enterica serovar Typhimurium: a systems biology analysis approach. PLoS ONE 6:e26869. doi: 10.1371/journal.pone.0026869
Leonard, S., Ferooz, J., Haine, V., Danese, I., Fretin, D., Tibor, A., et al. (2007). FtcR is a new master regulator of the flagellar system of Brucella melitensis 16M with homologs in Rhizobiae. J. Bacteriol. 189, 131–141. doi: 10.1128/JB.00712-06
Lestrate, P., Delrue, R. M., Danese, I., Didembourg, C., Taminiau, B., Mertens, P., et al. (2000). Identification and characterization of in vivo attenuated mutants of Brucella melitensis. Mol. Microbiol. 38, 543–551. doi: 10.1046/j.1365-2958.2000.02150.x
Liu, W., Jing, Z., Ou, Q., Cui, B., He, Y., and Wu, Q. (2012). Complete genome sequence of Brucella melitensis biovar 3 strain NI, isolated from an aborted bovine fetus. J. Bacteriol. 194:6321. doi: 10.1128/jb.01595-12
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔcT method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Marchesini, M. I., Herrmann, C. K., Salcedo, S. P., Gorvel, J. P., and Comerci, D. J. (2011). In search of Brucella abortus type IV secretion substrates: screening and identification of four proteins translocated into host cells through VirB system. Cell. Microbiol. 13, 1261–1274. doi: 10.1111/j.1462-5822.2011.01618.x
Marchesini, M. I., Morrone Seijo, S. M., Guaimas, F. F., and Comerci, D. J. (2016). A T4SS effector targets host cell alpha-enolase contributing to Brucella abortus intracellular lifestyle. Front. Cell. Infect. Microbiol. 6:153. doi: 10.3389/fcimb.2016.00153
Martínez de Tejada, G., Pizarro-Cerdá, J., Moreno, E., and Moriyón, I. (1995). The outer membranes of Brucella spp. are resistant to bactericidal cationic peptides. Infect. Immun. 63, 3054–3061.
Martirosyan, A., Perez-Gutierrez, C., Banchereau, R., Dutartre, H., Lecine, P., Dullaers, M., et al. (2012). Brucella beta 1,2 cyclic glucan is an activator of human and mouse dendritic cells. PLoS Pathog. 8:e1002983. doi: 10.1371/journal.ppat.1002983
Mirabella, A., Yanez Villanueva, R. M., Delrue, R. M., Uzureau, S., Zygmunt, M. S., Cloeckaert, A., et al. (2012). The two-component system PrlS/PrlR of Brucella melitensis is required for persistence in mice and appears to respond to ionic strength. Microbiology 158, 2642–2651. doi: 10.1099/mic.0.060863-0
Mol, J. P., Pires, S. F., Chapeaurouge, A. D., Perales, J., Santos, R. L., Andrade, H. M., et al. (2016). Proteomic profile of Brucella abortus-infected bovine chorioallantoic membrane explants. PLoS ONE 11:e0154209. doi: 10.1371/journal.pone.0154209
Moreno, E., and Moriyón, I. (2002). Brucella melitensis: a nasty bug with hidden credentials for virulence. Proc. Natl. Acad. Sci. U.S.A. 99, 1–3. doi: 10.1073/pnas.022622699
Nakato, G., Hase, K., Suzuki, M., Kimura, M., Ato, M., Hanazato, M., et al. (2012). Cutting edge: Brucella abortus exploits a cellular prion protein on intestinal M cells as an invasive receptor. J. Immunol. 189, 1540–1544. doi: 10.4049/jimmunol.1103332
O'Callaghan, D., and Whatmore, A. M. (2011). Brucella genomics as we enter the multi-genome era. Brief. Funct. Genomics 10, 334–341. doi: 10.1093/bfgp/elr026
O'Callaghan, D., Cazevieille, C., Allardet-Servent, A., Boschiroli, M. L., Bourg, G., Foulongne, V., et al. (1999). A homologue of the Agrobacterium tumefaciens VirB and Bordetella pertussis Ptl type IV secretion systems is essential for intracellular survival of Brucella suis. Mol. Microbiol. 33, 1210–1220. doi: 10.1046/j.1365-2958.1999.01569.x
Paulsen, I. T., Seshadri, R., Nelson, K., Eisen, J. A., and Heidelberg, J. F. (2002). The Brucella suis genome reveals fundamental similarities between animal and plant pathogens and symbionts. Proc. Natl. Acad. Sci. U.S.A. 99, 13148–13153. doi: 10.1073/pnas.192319099
Pei, J., and Ficht, T. A. (2004). Brucella abortus rough mutants are cytopathic for macrophages in culture. Infect. Immun. 72, 440–450. doi: 10.1128/IAI.72.1.440-450.2004
Pei, J., Turse, J. E., Wu, Q., and Ficht, T. A. (2006). Brucella abortus rough mutants induce macrophage oncosis that requires bacterial protein synthesis and direct interaction with the macrophage. Infect. Immun. 74, 2667–2675. doi: 10.1128/IAI.74.5.2667-2675.2006
Posadas, D. M., Ruiz-Ranwez, V., Bonomi, H. R., Martin, F. A., and Zorreguieta, A. (2012). BmaC, a novel autotransporter of Brucella suis, is involved in bacterial adhesion to host cells. Cell. Microbiol. 14, 965–982. doi: 10.1111/j.1462-5822.2012.01771.x
Rambow-Larsen, A. A., Rajashekara, G., Petersoen, E., and Splitter, G. (2008). Putative quorum-sensing regulator BlxR of Brucella melitensis regulates virulence factors including the Type IV Secretion System and flagella. J. Bacteriol. 190, 3274–3282. doi: 10.1128/JB.01915-07
Rolan, H. G., and Tsolis, R. M. (2007). Mice lacking components of adaptive immunity show increased Brucella abortus virB mutant colonization. Infect. Immun. 75, 2965–2973. doi: 10.1128/IAI.01896-06
Rossetti, C. A., Drake, K. L., and Adams, L. G. (2012). Transcriptome analysis of HeLa cells response to Brucella melitensis infection: a molecular approach to understand the role of the mucosal epithelium in the onset of the Brucella pathogenesis. Microbes Infect. 14, 756–767. doi: 10.1016/j.micinf.2012.03.003
Rossetti, C. A., Drake, K. L., Siddavatam, P., Lawhon, S. D., Nunes, J. E., Gull, T., et al. (2013). Systems biology analysis of Brucella infected Peyer's patch reveals rapid invasion with modest transient perturbations of the host transcriptome. PLoS ONE 8:e81719. doi: 10.1371/journal.pone.0081719
Rossetti, C. A., Galindo, C. L., Everts, R. E., Lewin, H. A., Garner, H. R., and Adams, L. G. (2011a). Comparative analysis of the early transcriptome of Brucella abortus–infected monocyte-derived macrophages from cattle naturally resistant or susceptible to brucellosis. Res. Vet. Sci. 91, 40–51. doi: 10.1016/j.rvsc.2010.09.002
Rossetti, C. A., Galindo, C. L., Garner, H. R., and Adams, L. G. (2010). Selective amplification of Brucella melitensis mRNA from a mixed host-pathogen total RNA. BMC Res. Notes 3:244. doi: 10.1186/1756-0500-3-244
Rossetti, C. A., Galindo, C. L., Garner, H. R., and Adams, L. G. (2011b). Transcriptional profile of the intracellular pathogen Brucella melitensis following HeLa cells infection. Microb. Pathog. 51, 338–344. doi: 10.1016/j.micpath.2011.07.006
Rossetti, C. A., Galindo, C. L., Lawhon, S., Garner, H., and Adams, L. G. (2009). Brucella melitensis global gene expression study provides novel information on growth phase-specific gene regulation with potential insights for understanding Brucella:host interaction. BMC Microbiol. 9:81. doi: 10.1186/1471-2180-9-81
Roux, C. M., Roldán, H. G., Santos, R. L., Beremand, P. D., Thomas, T. L., Adams, L. G., et al. (2007). Brucella requires a functional type IV secretion system to elicit innate immune responses in mice. Cell. Microbiol. 9, 1851–1869. doi: 10.1111/j.1462-5822.2007.00922.x
Ruiz-Ranwez, V., Posadas, D. M., Estein, S. M., Abdian, P. L., Martin, F. A., and Zorreguieta, A. (2013a). The BtaF trimeric autotransporter of Brucella suis is involved in attachment to various surfaces, resistance to serum and virulence. PLoS ONE 8:e79770. doi: 10.1371/journal.pone.0079770
Ruiz-Ranwez, V., Posadas, D. M., Van Der Henst, C., Estein, S. M., Arocena, G. M., Abdian, P. L., et al. (2013b). BtaE, an adhesin that belongs to the trimeric autotransporter family, is required for full virulence and defines a specific adhesive pole of Brucella suis. Infect. Immun. 81, 996–1007. doi: 10.1128/IAI.01241-12
Salcedo, S. P., Marchesini, M. I., Degos, C., Terwagne, M., Von Bargen, K., Lepidi, H., et al. (2013). BtpB, a novel Brucella TIR-containing effector protein with immune modulatory functions. Front. Cell. Infect. Microbiol. 3:28. doi: 10.3389/fcimb.2013.00028
Salcedo, S. P., Marchesini, M. I., Lelouard, H., Fugier, E., Jolly, G., Balor, S., et al. (2008). Brucella control of dendritic cell maturation is dependent on the TIR-containing protein Btp1. PLoS Pathog. 4:e21. doi: 10.1371/journal.ppat.0040021
Sankarasubramanian, J., Vishnu, U. S., Dinakaran, V., Sridhar, J., Gunasekaran, P., and Rajendhran, J. (2016). Computational prediction of secretion systems and secretomes of Brucella: identification of novel type IV effectors and their interaction with the host. Mol. Biosyst. 12, 178–190. doi: 10.1039/C5MB00607D
Santos, R. L., Zhang, S., Tsolis, R. M., Baumler, A. J., and Adams, L. G. (2002). Morphologic and molecular characterization of Salmonella typhimurium infection in neonatal calves. Vet. Pathol. 39, 200–215. doi: 10.1354/vp.39-2-200
Schauer, K., and Stingl, K. (2009). ‘Guilty by association’ - protein-protein interactions (PPIs) in bacterial pathogens. Genome Dyn. 6, 48–61. doi: 10.1159/000235762
Sieira, R., Comerci, D. J., Pietrasanta, L. I., and Ugalde, R. A. (2004). Integration host factor is involved in transcriptional regulation of the Brucella abortus virB operon. Mol. Microbiol. 54, 808–822. doi: 10.1111/j.1365-2958.2004.04316.x
Sieira, R., Comerci, D. J., Sánchez, D. O., and Ugalde, R. A. (2000). A homologue of an operon required for DNA transfer in Agrobacterium is required in Brucella abortus for virulence and intracellular multiplication. J. Bacteriol. 182, 4849–4855. doi: 10.1128/JB.182.17.4849-4855.2000
Sola-Landa, A., Pizarro-Cerdá, J., Grilló, M. J., Moreno, E., Moriyón, I., Blasco, J. M., et al. (1998). A two-component regulatory system playing a critical role in plant pathogens and endosymbionts is present in Brucella abortus and controls cell invasion and virulence. Mol. Microbiol. 29, 125–138. doi: 10.1046/j.1365-2958.1998.00913.x
Spera, J. M., Ugalde, J. E., Mucci, J., Comerci, D. J., and Ugalde, R. A. (2006). A B lymphocyte mitogen is a Brucella abortus virulence factor required for persistent infection. Proc. Natl. Acad. Sci. U.S.A. 103, 16514–16519. doi: 10.1073/pnas.0603362103
Swartz, T. E., Tseng, T. S., Frederickson, M. A., Paris, G., Comerci, D. J., Rajashekara, G., et al. (2007). Blue-light-activated histidine kinases: two-component sensors in bacteria. Science 317, 1090–1093. doi: 10.1126/science.1144306
Terwagne, M., Ferooz, J., Rolan, H. G., Sun, Y. H., Atluri, V., Xavier, M. N., et al. (2013). Innate immune recognition of flagellin limits systemic persistence of Brucella. Cell. Microbiol. 15, 942–960. doi: 10.1111/cmi.12088
Tian, M., Qu, J., Han, X., Zhang, M., Ding, C., Ding, J., et al. (2013). Microarray-based identification of differentially expressed genes in intracellular Brucella abortus within RAW264.7 cells. PLoS ONE 8:e67014. doi: 10.1371/journal.pone.0067014
Traxler, R. M., Lehman, M. W., Bosserman, E. A., Guerra, M. A., and Smith, T. L. (2013). A literature review of laboratory-acquired brucellosis. J. Clin. Microbiol. 51, 3055–3062. doi: 10.1128/JCM.00135-13
Tumurkhuu, G., Koide, N., Takahashi, K., Hassan, F., Islam, S., Ito, H., et al. (2006). Characterization of biological activities of Brucella melitensis lipopolysaccharide. Microbiol. Immunol. 50, 421–427. doi: 10.1111/j.1348-0421.2006.tb03810.x
Viadas, C., Rodriguez, M. C., Sangari, F. J., Gorvel, J. P., Garcia-Lobo, J. M., and Lopez-Goni, I. (2010). Transcriptome analysis of the Brucella abortus BvrR/BvrS two-component regulatory system. PLoS ONE 5:e10216. doi: 10.1371/journal.pone.0010216
von Bargen, K., Gagnaire, A., Arce-Gorvel, V., De Bovis, B., Baudimont, F., Chasson, L., et al. (2015). Cervical Lymph nodes as a selective Niche for Brucella during oral infections. PLoS ONE 10:e0121790. doi: 10.1371/journal.pone.0121790
Wagner, M. A., Eschenbrenner, M., Horn, T. A., Kraycer, J. A., Mujer, C. V., Hagius, S., et al. (2002). Global analysis of the Brucella melitensis proteome: Identification of proteins expressed in laboratory-grown culture. Proteomics 2, 1047–1060. doi: 10.1002/1615-9861(200208)2
Wang, Y., Chen, Z., Qiao, F., Ying, T., Yuan, J., Zhong, Z., et al. (2009). Comparative proteomics analyses reveal the virB of B. melitensis affects expression of intracellular survival related proteins. PLoS ONE 4:e5368. doi: 10.1371/journal.pone.0005368
Wareth, G., Melzer, F., Elschner, M. C., Neubauer, H., and Roesler, U. (2014). Detection of Brucella melitensis in bovine milk and milk products from apparently healthy animals in Egypt by real-time PCR. J. Infect. Dev. Ctries. 8, 1339–1343. doi: 10.3855/jidc.4847
Watarai, M., Kim, S., Erdenebaatar, J., Makino, S., Horiuchi, M., Shirahata, T., et al. (2003). Cellular prion protein promotes Brucella infection into macrophages. J. Exp. Med. 198, 5–17. doi: 10.1084/jem.20021980
Weeks, J. N., Galindo, C. L., Drake, K. L., Adams, G. L., Garner, H. R., and Ficht, T. A. (2010). Brucella melitensis VjbR and C12-HSL regulons: contributions of the N-dodecanoyl homoserine lactone signaling molecule and LuxR homologue VjbR to gene expression. BMC Microbiol. 10:167. doi: 10.1186/1471-2180-10-167
Werhli, A., and Husmeier, D. (2007). Reconstructing gene regulatory networks with bayesian networks by combining expression data with multiple sources of prior knowledge. Stat. Appl. Genet. Mol. Biol. 6:15. doi: 10.2202/1544-6115.1282
Whatmore, A. M., Davison, N., Cloeckaert, A., Al Dahouk, S., Zygmunt, M. S., Brew, S. D., et al. (2014). Brucella papionis sp. nov., isolated from baboons (Papio spp.). Int. J. Syst. Evol. Microbiol. 64, 4120–4128. doi: 10.1099/ijs.0.065482-0
Winter, S. E., Thiennimitr, P., Winter, M. G., Butler, B. P., Huseby, D. L., Crawford, R. W., et al. (2010). Gut inflammation provides a respiratory electron acceptor for Salmonella. Nature 467, 426–429. doi: 10.1038/nature09415
Wu, Q., Pei, J., Turse, C., and Ficht, T. A. (2006). Mariner mutagenesis of Brucella melitensis reveals genes with previously uncharacterized roles in virulence and survival. BMC Microbiol. 6:102. doi: 10.1186/1471-2180-6-102
Keywords: interactome model, Bayesian analysis, virulence factors, Peyer's patch
Citation: Rossetti CA, Drake KL, Lawhon SD, Nunes JS, Gull T, Khare S and Adams LG (2017) Systems Biology Analysis of Temporal In vivo Brucella melitensis and Bovine Transcriptomes Predicts host:Pathogen Protein–Protein Interactions. Front. Microbiol. 8:1275. doi: 10.3389/fmicb.2017.01275
Received: 10 March 2017; Accepted: 26 June 2017;
Published: 27 July 2017.
Edited by:
Nieves Vizcaino, University of Salamanca, SpainReviewed by:
Steven Olsen, Agricultural Research Service (USDA), United StatesRoy Martin Roop II, East Carolina University, United States
Juan M. Garcia Lobo, University of Cantabria, Spain
Copyright © 2017 Rossetti, Drake, Lawhon, Nunes, Gull, Khare and Adams. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Leslie G. Adams, Z2FkYW1zQGN2bS50YW11LmVkdQ==
†Present Address: Carlos A. Rossetti, Instituto de Patobiología, CICVyA-CNIA, INTA, CC25 (B1712WAA) Castelar, Buenos Aires, Argentina;
Jairo S. Nunes, Takeda Pharmaceuticals International Co., Cambridge, MA, United States;
Tamara Gull, Department of Veterinary Pathobiology, Oklahoma State University, Stillwater, OK, United States;
Sangeeta Khare, National Center for Toxicological Research, U.S.—Food and Drug Administration, Jefferson, AR, United States