 Phuong N. Tran
Phuong N. Tran Michael A. Savka
Michael A. Savka Han Ming Gan
Han Ming Gan- 1Genomics Facility, Tropical and Medicine Biology Platform, Monash University Malaysia, Bandar Sunway, Malaysia
- 2School of Science, Monash University Malaysia, Bandar Sunway, Malaysia
- 3Thomas H. Gosnell School of Life Sciences, Rochester Institute of Technology, Rochester, NY, United States
- 4Centre for Integrative Ecology, School of Life and Environmental Sciences, Deakin University, Geelong, VIC, Australia
The genus Pseudomonas has one of the largest diversity of species within the Bacteria kingdom. To date, its taxonomy is still being revised and updated. Due to the non-standardized procedure and ambiguous thresholds at species level, largely based on 16S rRNA gene or conventional biochemical assay, species identification of publicly available Pseudomonas genomes remains questionable. In this study, we performed a large-scale analysis of all Pseudomonas genomes with species designation (excluding the well-defined P. aeruginosa) and re-evaluated their taxonomic assignment via in silico genome-genome hybridization and/or genetic comparison with valid type species. Three-hundred and seventy-three pseudomonad genomes were analyzed and subsequently clustered into 145 distinct genospecies. We detected 207 erroneous labels and corrected 43 to the proper species based on Average Nucleotide Identity Multilocus Sequence Typing (MLST) sequence similarity to the type strain. Surprisingly, more than half of the genomes initially designated as Pseudomonas syringae and Pseudomonas fluorescens should be classified either to a previously described species or to a new genospecies. Notably, high pairwise average nucleotide identity (>95%) indicating species-level similarity was observed between P. synxantha-P. libanensis, P. psychrotolerans–P. oryzihabitans, and P. kilonensis- P. brassicacearum, that were previously differentiated based on conventional biochemical tests and/or genome-genome hybridization techniques.
Introduction
The genus Pseudomonas is considerably diverse and consists of more than 100 characterized species to date (Mulet et al., 2013). Some species of Pseudomonas are well-known and characterized such as P. aeruginosa (Oliver et al., 2000). Some are plant-associated such as Pseudomonas fluorescens and Pseudomonas syringae and define or are present in numerous plant-microbe system (Pieterse et al., 2014). Pseudomonas fluorescens in general, is known for its ability to colonize plant rhizospheres and produce antimicrobial compounds targeting pathogens; thus, protecting plants from diseases (Hol et al., 2013). On the other hand, P. syringae is commonly associated with plant disease and has been known to invade a variety of plant species with its extracellular flagella and pili appendages. Infection with P. syringae can also predispose the host plant to environmental stresses such as frost damage (Maki et al., 1974).
The genetic threshold level for bacterial species definition has seen various changes in the past 20 years due to the development and availability of new bioinformatics tools and genetic resources. For example, Hagström et al. (2002) reported a threshold of 97% or lower in homology of the 16S rRNA DNA sequence to be sufficient to characterize two bacteria as different species. Later this value was raised and is now more widely accepted at 99% (Buckley and Roberts, 2007). However, 16S rRNA has also been previously suggested to be efficient at delineating bacterial strains to a genera but not for species identification (Moore et al., 1996; Anzai et al., 2000; Yamamoto et al., 2000).
Based on pairwise comparisons of amino acid identity (AAI) with cutoff of 95% such that members within one genomic cluster have AAI >95%, Jun et al. (2016) reported nine major groups corresponding to the major Pseudomonas species groups including seven well-described species P. aeruginosa, P. fluorescens, P. syringae, P. putida, P. stuzeri, P. fragi, P. fusovaginae and two mixtures of unidentified species. P. aeruginosa contributes to more than half of the 1,073 genomes used in the study and forms a single well delineated genomic cluster suggesting that it is well-characterized. On the contrary, 29 genomes deposited as P. fluorescens in public database were found in 20 different genomic clusters indicating potential mislabeling of these genomes. In addition, many suggestions for redefinition of some Pseudomonas species such as P. avellanae and P. putida have been raised (Jun et al., 2016).
In this study, we inferred the phylogenomic placement of 373 Pseudomonas genomes identified to the species level representing 79 unique species and evaluated their species validity based on in silico genome-genome hybridization. The re-classification of Pseudomonas strains based on whole genome sequences will assist future comparative genomics analysis study and more importantly highlights the need for a more robust classification of Pseudomonas strains in the future especially with the availability of new genomic resources.
Materials and Methods
Datasets
Whole genome sequences of Pseudomonas strains with species designation excluding those of P. aeruginosa were downloaded from the NCBI FTP server (ftp.ncbi.nih.gov) in February, 2016. All genomes were filtered for assemblies with contig number no greater than 300 to avoid the inclusion of overly fragmented genome into the analysis. In addition, ten Acinetobacter, four Cellvibrio species and two Azotobacter species whole genomes were also included as the outgroup for phylogenomic analysis.
Phylogenomic Inference
Whole proteome was predicted using Prodigal V2.6.2 (Hyatt et al., 2010) and piped into PhyloPhlAn 0.99 (Segata et al., 2013) to construct a maximum likelihood tree using FastTree2 (–bionj –slownni –mlacc 2 –pseudo –spr 4 setting) (Price et al., 2010) based on the identification, alignment and concatenation of 400 universally conserved proteins (Edgar, 2004, 2010). Local branch support values were computed by FastTree2 using the Shimodaira-Hasegawa test. The tree was subsequently visualized and manually collapsed based on genospecies (Supplemental Table 1) using MEGA6 (Tamura et al., 2013).
Average Nucleotide Identity (ANI) Calculation
Pairwise ANIm was calculated with Jspecies v1.2.1 using the standard MUMmer algorithm (Richter and Rosselló-Móra, 2009). To reduce computational calculation, we separated the ANIm calculation into 4 groups based on phylogenomic clustering e.g., Clades 1, 2, 3 and strains that are not part of the labeled clade (Figure 1). The matrix obtained from Jspecies for each major group was clustered and visualized using Rstudio 0.99.902, pheatmap package.
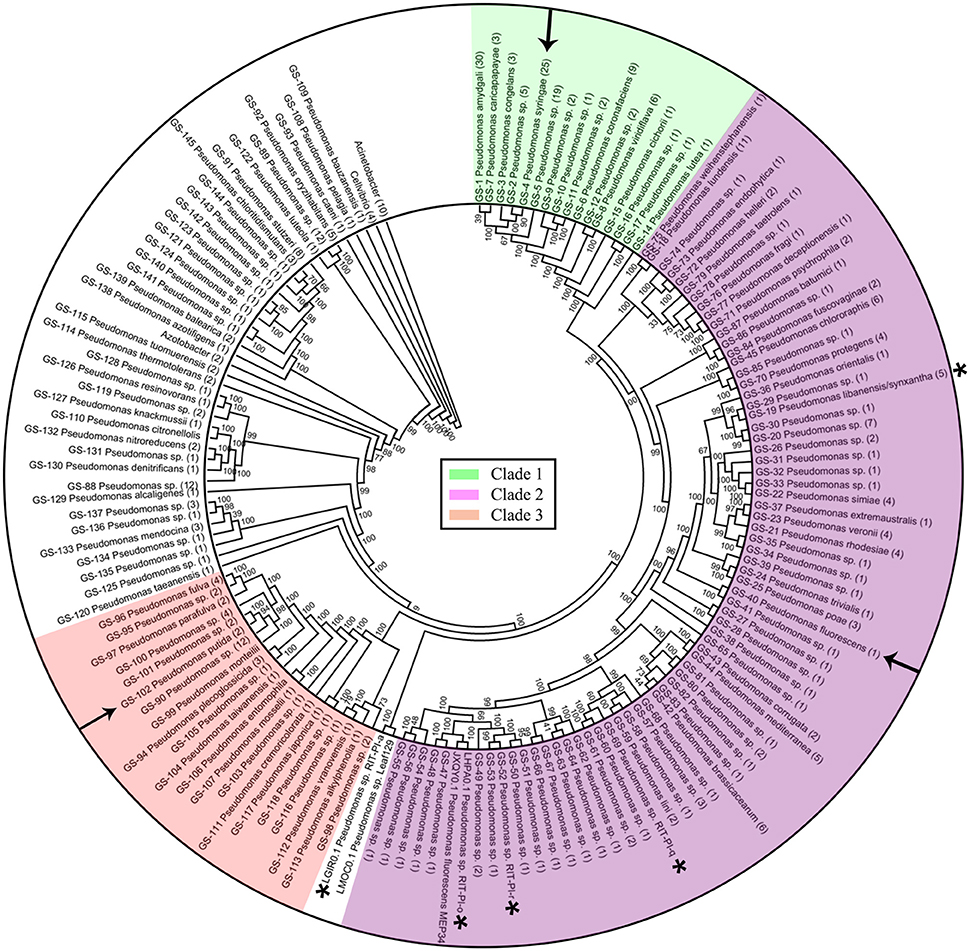
Figure 1. Phylogenomic tree of 373 Pseudomonas strains with corrected taxonomic assignments. Three major clades were colored accordingly with arrows indicating the type strains of 3 common Pseudomonas species e.g., P. putida, P. fluorescens, and P. syringae. Node labels indicate Shimodaira-Hasegawa(SH)-like bootstrap values in percentage. Branches classified as the same genospecies (GS) were collapsed with the number in bracket at the end of each lineage representing the number of whole genome sequences in the corresponding GS. Whole genome sequence of P. syringae, P. fluorescens, and P. putida type strains are indicated with arrows while asterisks-labeled genomes are the five Pseudomonas species isolated from poison ivy vine tissue.
Housekeeping Gene Similarity Calculation
Nucleotide similarity search on housekeeping genes (rpoB, recA, rpoD, and/or gyrB) was performed with basic local alignment search tool (BLASTN) using an e-value cutoff of 1 × e−10 (Altschul et al., 1990).
Results and Discussion
High Genomic Diversity among Pseudomonas Strains
A total of 373 Pseudomonas genome sequences were selected for phylogenomic tree reconstruction. The genome size among Pseudomonas strains is highly variable, ranging from 3,022,325 bp (P. caeni DSM 24390) to 8,608,769 bp (P. bauzanesis W13Z2). Similarly, the GC content of their genomes ranges from 56.51% (P. endophytica BSTT44) to 67.43% (P. syringae pv tomato T1). Based on current classification, the most commonly deposited non-P. aeruginosa pseudomonad species is P. syringae (N = 67), followed by P. fluorescens (N = 62), P. putida (N = 28), P. stuzeri (N = 17), P. amygdali (N = 15), P. psychrotolerans (N = 14), P. savastanoi (N = 13), and P. denitrificans (N = 11). Out of a total of 79 species outcomes, surprisingly 43 species have only one whole genome sequence representative in the database.
Maximum likelihood tree as inferred from 400 universal conserved proteins clustered the Pseudomonas strains into several major clades with maximal SH-like branch support (Figure 1 and Supplemental Figure 1). Support values of some inner clades are low suggesting that shallow relationships between strains within the same genospecies are not resolved, presumably due to the lack of divergence at the amino acid level among closely related strains. A wgMLST analysis restricted to strains from closely related Pseudomonas species will likely identify more conserved loci which can then be used to refine their evolutionary relationships which unfortunately is beyond the scope of this study. Such wgMLST analysis can be performed using existing bioinformatic tools such as BIGSdb, Roary and PGAdb-builder (Jolley and Maiden, 2010; Page et al., 2015; Liu et al., 2016). Strikingly, several whole genome sequences labeled as the same Pseudomonas species were located in different major clades or distantly apart taxa (Supplemental Figure 1). For example, while P. protegens CHA0T clustered within Clade 2, P. protegens 231 PPRO formed a monophyletic group with other sequences labeled as P. denitrificans in a distant clade. Similarly, P. marginalis ICMP 11289 belongs to Clade 1 and clusters with other P. viridiflava whereas P. marginalis ICMP 9505 belongs to Clade 2 (Supplemental Figure 1). The lack of taxonomic congruence as reflected by the inconsistent phylogenetic clustering among Pseudomonas strains with the same species designation raises suspicion about the species validity of Pseudomonas genomes submitted to public database.
Disentangling the Taxonomy of Pseudomonas Using In silico Genome-Genome Hybridization
Using 95% ANIm as the cutoff point for species delineation, a total of 145 genomic clusters were formed and when possible, assigned to a valid Pseudomonas species (Figure 1 and Supplemental Table 1). Two-hundred and seven genomes were wrongly assigned at the species level as evidence by the lack of genomic clustering with their expected type strain genome and/or low nucleotide similarity (<95%) to the type strain Multilocus Sequence Typing (MLST) sequences.
All of the major species contain at least one member with incorrect species classification except for P. amygdali (Figure 2). Surprisingly, all 13 P. savastanoi genomes, majority of which originated from a single study (Mott et al., 2016) may have been misclassified as they were clustered into GS-1 containing P. amygdali ICMP3918T (Figure 2 and Supplemental Table 1). ANIm matrix of 113 whole genome sequences within Clade 1 (Figure 1) containing a majority of P. syringae identified at least 14 different genomic clusters (Supplemental Figure 2). The presence of whole genome sequence for the P. syringae type strain KTCC 12500T = DSM 10604T in GS-4 suggests that only 23 out of 67 P. syringae genomes were correctly assigned (Supplemental Figure 2), indicating that more than 50% of the currently deposited P. syringae genomes should be taxonomically revised. At least 10 P. syringae genomes have to be assigned to other valid Pseudomonas species (Supplemental Table 1). For example, P. syringae CC1513 should be re-classified as P. coronafaciens given its high pairwise ANIm of 99.2% to P. coronafaciens LMG5060T.
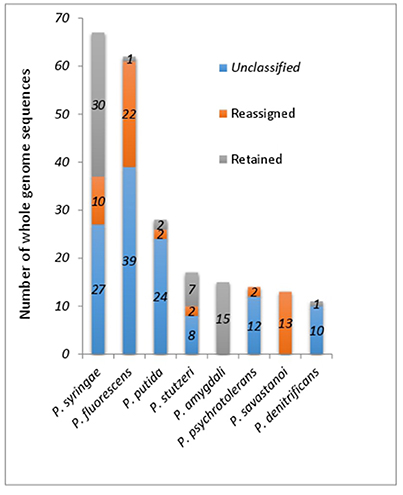
Figure 2. Proportion of retained, reassigned and unclassified whole genome sequences within 8 most common Pseudomonas species excluding P. aeruginosa.
Prior to the genome availability of P. fluorescens DSM 50090T, P. fluorescens strain WH6 has served as the reference genome for the taxonomic assignment of P. fluorescens (Duan et al., 2013; Feng et al., 2015). Unfortunately, ANIm calculation shows that strain WH6 belongs to GS-35 with a pairwise ANIm of only 87.94% to the recently available P. fluorescens DSM 50090T genome (Supplemental Table 1 and Supplemental Figure 3). In another study, strain WH6 was also shown to be more similar to P. azotofomans LMG 21611T than the representative species P. fluorescens DSM 50090T using the MLSA method (Gomila et al., 2015). This finding along with our results here, suggest that the designation of P. fluorescens WH6 genome as the reference genome might have led to the subsequent mislabeling of newly-sequenced Pseudomonas genomes. As expected, out of a total of 62 whole genome sequences that were assigned as P. fluorescens, none of them belongs to the same genospecies as P. fluorescens DSM 50090T. By re-evaluating the species designation based on ANIm clustering against other type strains of Pseudomonas, 22 of the 61 wrongly identified P. fluorescens strains were successfully re-assigned to the correct valid species name leaving 39 pseudomonad genospecies in Clade 2 (Figure 1) unassigned at the species level.
Only 1 strain of the total 28 P. putida strains belongs to the same genospecies with the P. putida type strain NBRC 14164T (GS-102) which was similarly supported by their monophyletic clustering in the phylogenomic tree (Clade 3 in Supplemental Figure 1). Some of the mislabeled P. putida strains might be novel species given its independent formation of genospecies e.g., GS-103 in P. putida MTCC5279 (Supplemental Table 1 and Supplemental Figure 4).
Species Validity of Closely Related Pseudomonas Species
Pseudomonas libanensis and Pseudomonas synxantha
Pseudomonas libanensis DSM 17149T was isolated from Lebanon spring water whereas P. synxantha DSM 18928T was isolated from milk cream in Iowa, USA (De Vos et al., 1989). Despite a pairwise 16S rDNA similarity of up to 99.5% between P. libanensis DSM 17149T and P. synxantha DSM 18928T, these two strains were considered as different species on the basis that strikingly high 16S rDNA similarity does not corroborate their reported low relative binding ratio of DNAs (RBR) (Dabboussi et al., 1999). The difference between P. libanensis and P. synxantha was further substantiated by other phenotypes such as levan formation and assimilation of histidine and erythritol (Dabboussi et al., 1999). Since 2005, both species have been included in the Bergey's Manual (Palleroni, 2005). However, it is worth noting that the difference in biochemical property can be due to single nucleotide polymorphism or presence/absence of horizontally transfer gene(s) that does not contribute significantly to the overall nucleotide difference at the genomic level (Carnoy and Moseley, 1997; Boddicker et al., 2002; Monk et al., 2017). Furthermore, conventional DNA-DNA hybridization (DDH) procedures are technically demanding yet error-prone and often result in different outcomes (Rosselló-Mora, 2006; Johnson and Whitman, 2007). On the other hand, digital DDH is considered a more robust, pragmatic and an accurate replacement method for conventional DDH procedures (Richter and Rosselló-Móra, 2009; Auch et al., 2010). Contrary to their low RBR result, the whole genome sequences of P. synxantha DSM 18928T and P. libanensis DSM 17149T exhibit a pairwise ANIm of 96.7% clustering placing them both into GS-19 (Figure 3). In other words, genomic evidence does not support the classification of P. synxantha and P. libanensis as two separate species, warranting additional taxonomic investigations.
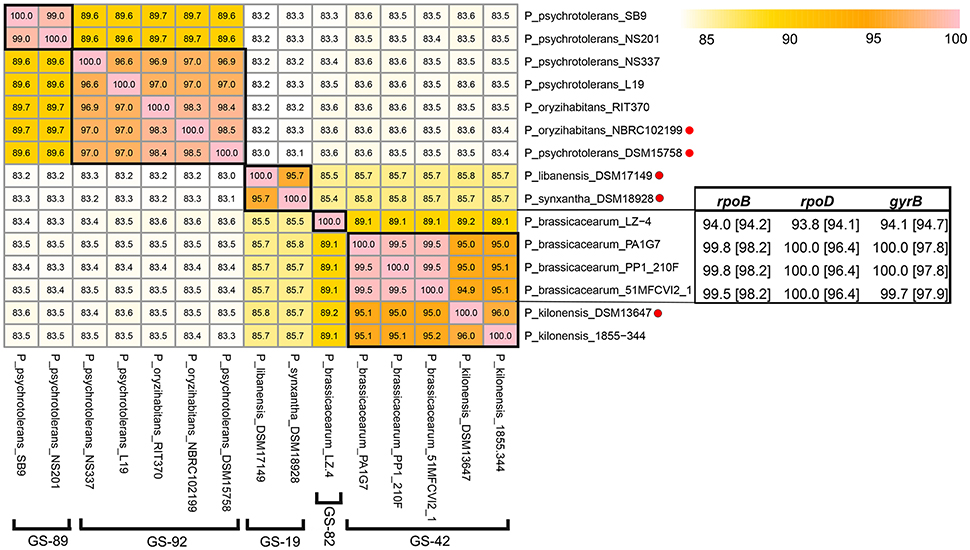
Figure 3. ANIm matrix of 15 Pseudomonas genomes from closely-related species including P. psychrotolerans-P. oryzihanbitans, P. brassicacearum-P. kilonensis, and P. libanensis-P. synxantha. Type strains were indicated with red dots and the genospecies corresponding to each cluster was specified in bracket. Blastn results were expressed in percentage with values in and outside the bracket show percentage identity to the housekeeping genes (rpoB, rpoD, and gyrB) of P. kilonensis DSM 13647T and P. brassicacearum CIP 107059T, respectively. These two genomes along with whole genome of P. psychrotolerans DSM 15758T (downloaded in April 2017) were only used to generate the ANIm matrix and were excluded from other analyses.
Pseudomonas brassicacearum and Pseudomonas kilonensis
Similarly, Pseudomonas brassicacearum and Pseudomonas kilonensis were described as two distinct species (Sikorski et al., 2001) on the basis of substantial dissimilarity in metabolic properties and a surprisingly borderline DNA-DNA hybridization similarity for species delineation. P. kilonensis was isolated from agricultural soil of northern Germany and described as a distinct species from P. brassicacearum by 10-12 different metabolic properties with convential DNA-DNA hybridization similarity of 93% (Sikorski et al., 2001). Pseudomonas brassicacearum was firstly described in 2000 when these strains were isolated from the rhizoplane of Arabidopsis thaliana and Brassica napus growing on different soils (Achouak et al., 2000). Since whole genome sequence of P. brassicacearum type strain is not yet available, blastn was conducted against housekeeping genes (rpoB, rpoD and gyrB) of P. brassicacearum CFBP 11706T = CIP 107059T and P. kilonensis 520-20T = DSM 13647T to validate the correct identification of 3 P. brassicacearum genomes belonging to GS-42 (Figure 3). P. kilonensis DSM 13647T also show strikingly high pairwise ANIm to these three P. brassicacearum genomes leading to the clustering of all strains to GS-42, indicating that P. kilonensis should be considered as the junior synonym to P. brassicacearum as P. brassicacearum was described earlier than P. kilonensis.
Pseudomonas psychrotolerans and Pseudomonas oryzihabitans
Fourteen whole genome sequences labeled as P. psychrotolerans were distributed into two separate genospecies: GS-89 and GS-92. The presence of both P. oryzihabitans NBRC 102199T and P. psychrotolerans DSM 15758T in GS-92 implies that the two species are closely-related (ANIm similarity of 98.5%). P. psychrotolerans strain NS337 and L19 were correctly labeled but the other 12 sequences in GS-89 including strain SB9 and NS201 were incorrectly identified (Figure 3). P. psychrotolerans was firstly isolated from small European ungulates (Hauser et al., 2004) whereas P. oryzihabitans was isolated from soil in rice paddies (Kodama et al., 1985). It is suggested that P. psychrotolerans can be a junior synonym of P. oryzihabitans for such high similarity in digital DNA-DNA hybridization.
Taxonomic Re-Evaluation of Pseudomonas Strains Isolated from Poison Ivy (Toxicodendron radicans) Vine Tissue
With the current availability of defined genospecies within the genus Pseudomonas, we re-evaluated the taxonomic status of 5 Pseudomonas strains previously isolated from interior vine tissue of poison ivy by our group (Tran et al., 2015). Strain RIT-PI-g could be assigned to the species Pseudomonas libanensis forming GS-19 along with the P. libanensis DSM 17149T corroborating our previous species assignment based on similarity to various housekeeping genes. Pseudomonas. sp. RIT-PI-q and P. sp. RIT-PI-r both belong to a single-member genospecies (GS-69 and GS-52, respectively), further underscoring the high genomic diversity of plant-associated Pseudomonas that remains to be explored and described with extensive taxon sampling effort. On the contrary, Pseudomonas sp. RIT-PI-o and P. sp. RIT-PI-a were placed in the same genomic cluster (GS-13) as the incorrectly labeled P. fluorescens MEP34 and Pseudomonas sp. leaf129 (Supplemental Table 1 and Supplemental Figure 5). Both P. fluorescens MEP34 and P.s sp. leaf129 were isolated from leaf of Arabidopsis thaliana, a popular flowering plant in North America that has been adopted as the flowering plant genetic model species (Bai et al., 2015; Perisin et al., 2015). Such similar plant origins, e.g., poision ivy vine and Arabidopsis, poses interesting links between bacterial genomic characteristics and plant-hosting capacity.
Conclusions
The bacterial community has waited many years for the whole genome of P. fluorescens type strain to be sequenced (Flury et al., 2016). It is also surprising to note that whole genome sequence of some well-known Pseudomonas species such as P. brassicacearum is still unavailable despite the dramatic reduction in sequencing cost. Usually, a traditional BLASTn against 16S rRNA database was performed with varying degree of analysis depth to taxonomically assign newly-sequenced bacterial genomes which has potentially resulted in taxonomic misclassifications of several Pseudomonas genomes. Our study shows that in the future, a phylogenomic inference coupled with ANIm calculation could be a practical and a more reproducible method for inferring accurate genomic relatedness among Pseudomonas strains.
Author Contributions
PT and HG conceived the experiments. PT performed the analysis of the data. PT, HG, and MS interpret the results and contributed to the manuscript write-up.
Funding
Funding for this study is provided by the Monash University Malaysia Tropical and Medicine Biology Platform. We also thank the Monash University Malaysia Genomics Facility for the provision of computational resources.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.01296/full#supplementary-material
Supplemental Figure 1. Phylogenomic tree of 373 Pseudomonas strains with original taxonomic assignments. Three major clades were colored accordingly with arrows indicating the type strains of 3 common Pseudomonas species e.g., P. putida, P. fluorescens, P. syringae. Whole genome sequence of P. syringae, P. fluorescens and P. putida type strains are indicated with arrows while asterisks-labeled genomes are the five Pseudomonas species isolated from poison ivy vine tissue.
Supplemental Figure 2. Genomic clustering of Clade 1 using ANIm calculation.
Supplemental Figure 3. Genomic clustering of Clade 2 using ANIm calculation.
Supplemental Figure 4. Genomic clustering of Clade 3 using ANIm calculation.
Supplemental Figure 5. Genomic clustering of other Pseudomonas strains using ANIm calculation.
Supplemental Table 1. Genomic clusters of Clade 1, Clade 2, Clade 3 and other Pseudomonas strains with suggested assignation. Mislabeled genomes are indicated with asterisk whereas type strains are labeled “T.”
References
Achouak, W., Sutra, L., Heulin, T., Meyer, J.-M., Fromin, N., Degraeve, S., et al. (2000). Pseudomonas brassicacearum sp. nov. and Pseudomonas thivervalensis sp. nov., two root-associated bacteria isolated from Brassica napus and Arabidopsis thaliana. Int. J. Syst. Evol. Microbiol. 50, 9–18. doi: 10.1099/00207713-50-1-9
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Anzai, Y., Kim, H., Park, J. Y., Wakabayashi, H., and Oyaizu, H. (2000). Phylogenetic affiliation of the pseudomonads based on 16S rRNA sequence. Int. J. Syst. Evol. Microbiol. 50(Pt 4), 1563–1589. doi: 10.1099/00207713-50-4-1563
Auch, A. F., Jan, M., Klenk, H.-P., and Göker, M. (2010). Digital DNA-DNA hybridization for microbial species delineation by means of genome-to-genome sequence comparison. Stand. Genomic Sci. 2, 117–134. doi: 10.4056/sigs.531120
Bai, Y., Müller, D. B., Srinivas, G., Garrido-Oter, R., Potthoff, E., Rott, M., et al. (2015). Functional overlap of the Arabidopsis leaf and root microbiota. Nature. 528, 364–369. doi: 10.1038/nature16192
Boddicker, J. D., Ledeboer, N. A., Jagnow, J., Jones, B. D., and Clegg, S. (2002). Differential binding to and biofilm formation on, HEp-2 cells by Salmonella enterica serovar Typhimurium is dependent upon allelic variation in the fimH gene of the fim gene cluster. Mol. Microbiol. 45, 1255–1265. doi: 10.1046/j.1365-2958.2002.03121.x
Buckley, M., and Roberts, R. J. (2007). Reconciling Microbial Systematics and Genomics. Washington, DC: American Academy of Microbiology.
Carnoy, C., and Moseley, S. L. (1997). Mutational analysis of receptor binding mediated by the Dr family of Escherichia coli adhesins. Mol. Microbiol. 23, 365–379. doi: 10.1046/j.1365-2958.1997.2231590.x
Dabboussi, F., Hamze, M., Elomari, M., Verhille, S., Baida, N., Izard, D., et al. (1999). Pseudomonas libanensis sp. nov., a new specie isolated from Lebanese spring waters. Int. J. Syst. Evol. Microbiol. 49, 1091–1101.
De Vos, P., Van Landschoot, A., Segers, P., Tytgat, R., Gillis, M., Bauwens, M., et al. (1989). Genotypic relationships and taxonomic localization of unclassified Pseudomonas and Pseudomonas-like strains by deoxyribonucleic acid: ribosomal ribonucleic acid hybridizations. Int. J. Syst. Evol. Microbiol. 39, 35–49.
Duan, J., Jiang, W., Cheng, Z., Heikkila, J. J., and Glick, B. R. (2013). The complete genome sequence of the plant growth-promoting bacterium Pseudomonas sp. UW4. PLoS ONE 8:e58640. doi: 10.1371/journal.pone.0058640
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Feng, K., Li, R., Chen, Y., Zhao, B., and Yin, T. (2015). Sequencing and analysis of the Pseudomonas fluorescens GcM5-1A genome: a pathogen living in the surface coat of Bursaphelenchus xylophilus. PLoS ONE 10:e0141515. doi: 10.1371/journal.pone.0141515
Flury, P., Aellen, N., Ruffner, B., Péchy-Tarr, M., Fataar, S., Metla, Z., et al. (2016). Insect pathogenicity in plant-beneficial pseudomonads: phylogenetic distribution and comparative genomics. ISME. J. 10, 2527–2542. doi: 10.1038/ismej.2016.5
Gomila, M., Peña, A., Mulet, M., Lalucat, J., and García-Valdés, E. (2015). Phylogenomics and systematics in Pseudomonas. Front. Microbiol. 6:214. doi: 10.3389/fmicb.2015.00214
Hagström, Å., Pommier, T., Rohwer, F., Simu, K., Stolte, W., Svensson, D., et al. (2002). Use of 16S ribosomal DNA for delineation of marine bacterioplankton species. Appl. Environ. Microbiol. 68, 3628–3633. doi: 10.1128/AEM.68.7.3628-3633.2002
Hauser, E., Kampfer, P., and Busse, H. J. (2004). Pseudomonas psychrotolerans sp. nov. Int. J. Syst. Evol. Microbiol. 54(Pt.5), 1633–1637. doi: 10.1099/ijs.0.03024-0
Hol, W. H. G., Bezemer, T. M., and Biere, A. (2013). Getting the ecology into interactions between plants and the plant growth-promoting bacterium Pseudomonas fluorescens. Front. Plant Sci. 4:81. doi: 10.3389/fpls.2013.00081
Hyatt, D., Chen, G. L., Locascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119
Johnson, J. L., and Whitman, W. B. (2007). “Similarity analysis of DNAs,” in Methods for General and Molecular Microbiology, 3rd Edn., eds C. A. Reddy, T. J. Beveridge, J. A. Breznak and G. Marzluf (American Society of Microbiology), 624–652.
Jolley, K. A., and Maiden, M. C. J. (2010). BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics 11:595. doi: 10.1186/1471-2105-11-595
Jun, S.-R., Wassenaar, T. M., Nookaew, I., Hauser, L., Wanchai, V., Land, M., et al. (2016). Diversity of Pseudomonas genomes, including populus-associated isolates, as revealed by comparative genome analysis. Appl. Environ. Microbiol. 82, 375–383. doi: 10.1128/AEM.02612-15
Kodama, K., Kimura, N., and Komagata, K. (1985). Two new species of Pseudomonas: P. oryzihabitans isolated from rice paddy and clinical specimens and P. luteola isolated from clinical specimens. Int. J. Syst. Evol. Microbiol. 35, 467–474.
Liu, Y.-Y., Chiou, C.-S., and Chen, C.-C. (2016). PGAdb-builder: a web service tool for creating pan-genome allele database for molecular fine typing. Sci. Rep. 6:36213. doi: 10.1038/srep36213
Maki, L. R., Galyan, E. L., Chang-Chien, M. M., and Caldwell, D. R. (1974). Ice nucleation induced by pseudomonas syringae. Appl. Microbiol. 28, 456–459.
Monk, I. R., Howden, B. P., Seemann, T., and Stinear, T. P. (2017). Correspondence: Spontaneous secondary mutations confound analysis of the essential two-component system WalKR in Staphylococcus aureus. Nat. Commun. 8:14403. doi: 10.1038/ncomms14403
Moore, E. R. B., Mau, M., Arnscheidt, A., Böttger, E. C., Hutson, R. A., Collins, M. D., et al. (1996). The determination and comparison of the 16S rRNA gene sequences of species of the genus Pseudomonas (sensu stricto and estimation of the natural intrageneric relationships). Syst. Appl. Microbiol. 19, 478–492. doi: 10.1016/S0723-2020(96)80021-X
Mott, G. A., Thakur, S., Smakowska, E., Wang, P. W., Belkhadir, Y., Desveaux, D., et al. (2016). Genomic screens identify a new phytobacterial microbe-associated molecular pattern and the cognate Arabidopsis receptor-like kinase that mediates its immune elicitation. Genome Biol. 17:98. doi: 10.1186/s13059-016-0955-7
Mulet, M., García-Valdés, E., and Lalucat, J. (2013). Phylogenetic affiliation of Pseudomonas putida biovar A and B strains. Res. Microbiol. 164, 351–359. doi: 10.1016/j.resmic.2013.01.009
Oliver, A., Cantón, R., Campo, P., Baquero, F., and Blázquez, J. (2000). High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science 288, 1251–1253. doi: 10.1126/science.288.5469.1251
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T. G., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Palleroni, N. J. (2005). “Genus I. Pseudomonas Migula 1894.,” in Bergey's Manual of Systematic Bacteriology, 2nd Edn., eds D. J. Brenner, N. R. Krieg, and J. T. Staley (New York, NY: Springer), 323–379.
Perisin, M., Vetter, M., Gilbert, J. A., and Bergelson, J. (2015). 16Stimator: statistical estimation of ribosomal gene copy numbers from draft genome assemblies. ISME. J. 10, 1020–1024. doi: 10.1038/ismej.2015.16
Pieterse, C. M. J., Zamioudis, C., Berendsen, R. L., Weller, D. M., Van Wees, S. C. M., and Bakker, P. A. H. M. (2014). Induced systemic resistance by beneficial microbes. Annu. Rev. Phytopathol. 52, 347–375. doi: 10.1146/annurev-phyto-082712-102340
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE 5:e9490. doi: 10.1371/journal.pone.0009490
Richter, M., and Rosselló-Móra, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Nat. Acad. Sci. U.S.A. 106, 19126–19131. doi: 10.1073/pnas.0906412106
Rosselló-Mora, R. (2006). “DNA-DNA reassociation methods applied to microbial taxonomy and their critical evaluation,” in Molecular Identification, Systematics, and Population Structure of Prokaryotes, ed E. Stackebrandt (Berlin: Springer), 23–50.
Segata, N., Bornigen, D., Morgan, X. C., and Huttenhower, C. (2013). PhyloPhlAn is a new method for improved phylogenetic and taxonomic placement of microbes. Nat. Commun. 4:2304. doi: 10.1038/ncomms3304
Sikorski, J., Stackebrandt, E., and Wackernagel, W. (2001). Pseudomonas kilonensis sp. nov., a bacterium isolated from agricultural soil. Int. J. Syst. Evol. Microbiol. 51, 1549–1555. doi: 10.1099/00207713-51-4-1549
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Tran, P. N., Tan, N. E. H., Lee, Y. P., Gan, H. M., Polter, S. J., Dailey, L. K., et al. (2015). Whole-genome sequence and classification of 11 endophytic bacteria from poison ivy (Toxicodendron radicans). Genome Announc. 3, e01319–e01315. doi: 10.1128/genomeA.01319-15
Keywords: Pseudomonas, taxonomy, phylogenomics, in-silico genome-genome hybridization, comparative genomics, average nucleotide identity, genospecies
Citation: Tran PN, Savka MA and Gan HM (2017) In-silico Taxonomic Classification of 373 Genomes Reveals Species Misidentification and New Genospecies within the Genus Pseudomonas. Front. Microbiol. 8:1296. doi: 10.3389/fmicb.2017.01296
Received: 26 April 2017; Accepted: 27 June 2017;
Published: 12 July 2017.
Edited by:
Jesus L. Romalde, Universidade de Santiago de Compostela, SpainReviewed by:
Siomar De Castro Soares, Universidade Federal do Triângulo Mineiro, BrazilSe-Ran Jun, University of Arkansas for Medical Sciences, United States
Copyright © 2017 Tran, Savka and Gan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Han Ming Gan, aGFuLmdhbkBkZWFraW4uZWR1LmF1