 Stefanie Böhnke
Stefanie Böhnke Mirjam Perner
Mirjam Perner- Molecular Biology of Microbial Consortia, Biocenter Klein Flottbek, University of Hamburg, Hamburg, Germany
Ribulose-1,5-bisphosphate carboxylase/oxygenase (RubisCO) catalyzes the first major step of carbon fixation in the Calvin-Benson-Bassham (CBB) cycle. This autotrophic CO2 fixation cycle accounts for almost all the assimilated carbon on Earth. Due to the primary role that RubisCO plays in autotrophic carbon fixation, it is important to understand how its gene expression is regulated and the enzyme is activated. Since the majority of all microorganisms are currently not culturable, we used a metagenomic approach to identify genes and enzymes associated with RubisCO expression. The investigated metagenomic DNA fragment originates from the deep-sea hydrothermal vent field Nibelungen at 8∘18′ S along the Mid-Atlantic Ridge. It is 13,046 bp and resembles genes from Thiomicrospira crunogena. The fragment encodes nine open reading frames (ORFs) which include two types of RubisCO, form I (CbbL/S) and form II (CbbM), two LysR transcriptional regulators (LysR1 and LysR2), two von Willebrand factor type A (CbbO-m and CbbO-1), and two AAA+ ATPases (CbbQ-m and CbbQ-1), expected to function as RubisCO activating enzymes. In silico analyses uncovered several putative LysR binding sites and promoter structures. Functions of some of these DNA motifs were experimentally confirmed. For example, according to mobility shift assays LysR1’s binding ability to the intergenic region of lysR1 and cbbL appears to be intensified when CbbL or LysR2 are present. Binding of LysR2 upstream of cbbM appears to be intensified if CbbM is present. Our study suggests that CbbQ-m and CbbO-m activate CbbL and that LysR1 and LysR2 proteins promote CbbQ-m/CbbO-m expression. CbbO-1 seems to activate CbbM and CbbM itself appears to contribute to intensifying LysR’s binding ability and thus its own transcriptional regulation. CbbM furthermore appears to impair cbbL expression. A model summarizes the findings and predicts putative interactions of the different proteins influencing RubisCO gene regulation and expression.
Introduction
Ribulose-1,5-bisphosphate carboxylase/oxygenase (RubisCO, EC 4.1.1.39) is believed to be the most abundant enzyme on Earth (Ellis, 1979; Raven, 2009). It is the key enzyme of the autotrophic Calvin-Benson-Bassham (CBB) cycle and catalyzes the carboxylation of ribulose-1,5-bisphosphate (RuBP) to 3-phosphoglycerate (3-PGA) (Berg, 2011). Since the CBB cycle is estimated to account for most of Earth’s net primary production (>99.5% of 105 × 109 tons/year) (Field et al., 1998; Raven, 2013), it is important to understand RubisCO expression and its activation in many different organisms.
The RubisCO enzyme is widespread and can be found in plants, algae, cyanobacteria, many autotrophic bacteria (phototrophs and chemolithotrophs), and archaea (Tabita et al., 2007; Hauser et al., 2015). Although four types of structural RubisCOs are known, only the RubisCO form I (CbbLS) and the form II (CbbM) are evidenced to operate in the classical CBB cycle (Berg, 2011). For the expression and activation of a catalytically active form I and form II RubisCO distinct transcriptional regulators and activases are essential (Maddocks and Oyston, 2008; Dangel and Tabita, 2015; Tsai et al., 2015). LysR-type transcriptional regulators (LTTRs) have been found adjacent to the structural RubisCO genes in several genomes and are evidenced to regulate their transcription (Dangel and Tabita, 2015). LTTRs can function as an activator and/or as a repressor for their target genes (Maddocks and Oyston, 2008 and references therein), but can also positively autoregulate their own transcription (Axler-DiPerte et al., 2006). Indeed, LTTR associated regulation can be highly complex as is indicated by LTTRs which need to interact with other transcriptional regulators (Joshi et al., 2013; Dangel et al., 2014). Since RubisCO forms inhibited complexes with its substrate RuBP but also with other sugar phosphates (Tsai et al., 2015), the removal of the active site inhibitor is essential for proceeding with the RubisCO catalyzed carboxylation reaction. In case of plant green-type and α-proteobacterial red-type form I RubisCOs, this is done by the RubisCO activase (rca) and CbbX, respectively (Parry et al., 2008; Mueller-Cajar et al., 2011). CbbQ (AAA+ATPase) and CbbO (von Willebrand factor type A) represent a third class of RubisCO activases and were shown to act on green-type form I RubisCOs of chemoautotrophic bacteria (Tsai et al., 2015).
Given that the majority of microorganisms are currently unculturable (Amann et al., 1995), we recently developed an activity-based screen, which enables us to seek RubisCO active clones from metagenomic fosmid libraries (Böhnke and Perner, 2015). One of these newly discovered RubisCO active metagenomic clones stems from a fosmid library constructed with DNA from the Nibelungen vent field (8°18′S on the Mid-Atlantic Ridge): It exhibited similarities to genes from the gammaproteobacterial Thiomicrospira crunogena XCL-2 (96%). Our metagenomic fragment encodes a 13 kb RubisCO gene cluster and flanking DNA regions of additional 22.2 kb. The 13 kb DNA fragment encodes two divergently directed reading frames: (i) lysR1, lysR2, cbbM, cbbQ-m, and cbbO-m, and (ii) cbbL, cbbS, cbbQ-1, and cbbO-1. To date, only one study has ever investigated regulatory mechanisms in metagenome derived RubisCO gene clusters (Böhnke and Perner, 2015). Here, total RubisCO activity was significantly influenced when cbbL and cbbM neighboring genes were knocked out (Böhnke and Perner, 2015), but it remained unclear which of the two RubisCOs was primarily affected by these mutations. While most of the studies on RubisCO regulation investigate the regulation of alphaproteobacterial RubisCOs (Paoli et al., 1998; Dubbs and Tabita, 2003; van Keulen et al., 2003; Dubbs et al., 2004; Joshi et al., 2013; Dangel et al., 2014), little work exists on the regulatory machinery behind gammaproteobacterial RubisCO transcription (Kusano and Sugawara, 1993). The arrangement of alphaproteobacterial RubisCOs and their associated genes as well as the location of the RubisCO gene clusters on the genome are very different to what is observed on our metagenomic fragment. For example, while the alphaproteobacterial Rhodobacter capsulatus RubisCO form I gene cluster is arranged like our RubisCO form I gene cluster (lysR1 cbbLSQO), the RubisCO form II gene cluster is considerably different to that on our metagenomic fragment (cbbFPTGAM versus cbbMQO, respectively) (Paoli et al., 1998) suggesting different interactions with respect to regulatory processes. Also, our metagenomic RubisCO form I and form II gene clusters are located within each other’s vicinity on a 13 kb DNA fragment. In contrast, the RubisCO gene clusters of the so far investigated Alphaproteobacteria are either encoded on different chromosomes (Rhodobacter sphaeroides) or on distant regions of the genome (separated by 2 Mb or 1.4 Mb, Rhodobacter capsulatus and Rhodopseudomonas palustris, respectively) (Paoli et al., 1998; Dubbs and Tabita, 2004; Joshi et al., 2013). The here investigated metagenome derived form I and form II RubisCOs, thus, represent a unique opportunity to investigate the role that genes and respective products have on the expression and activation of two forms of RubisCOs from an uncultured Gammaproteobacterium colonizing a chemically dynamic environment.
Materials and Methods
Bacterial Strains, Vectors, and Constructs, Media, and Growth Conditions
The bacterial strains, vectors, and constructs used in this study are summarized in Table 1. Escherichia coli cultures were routinely grown on lysogeny broth (LB) medium (Bertani, 1951). For cloning procedures cultures were incubated at 37°C. If cultivated for measuring recombinant RubisCO activities, the growth temperature was lowered to 28°C, while cultures grown as part of over expression experiments were incubated at 17°C or 22°C. If required, the following supplements were added: ampicillin, 100 μg ml-1; 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-gal), 50 μg ml-1, chloramphenicol, 12.5 μg ml-1; isopropyl β-D-1-thiogalactopyranoside (IPTG), 100 μg ml-1 (cloning) or 0.1–1 mM (expression); kanamycin, 50 μg ml-1, and tetracycline, 10 μg ml-1.
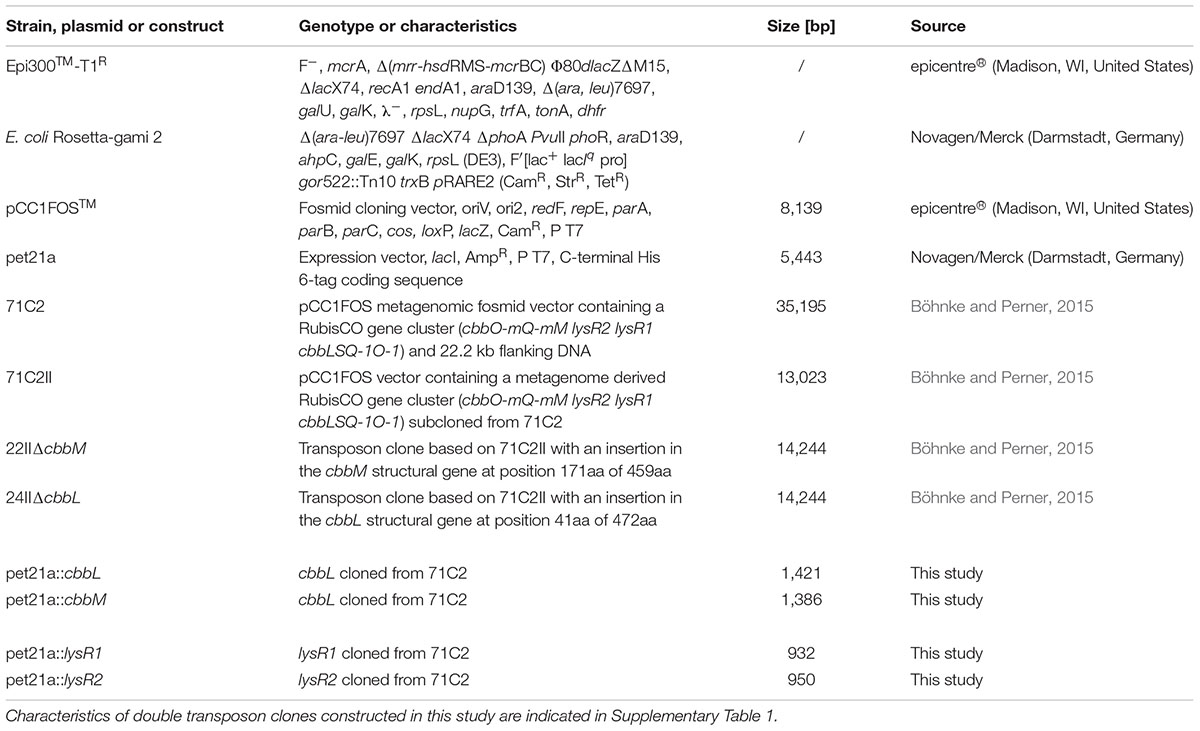
TABLE 1. Strains, vectors, and constructs used in this study.
Construction of Double Transposon Mutant Libraries
Two double transposon mutant libraries were constructed from two versions of the 13 kb metagenomic fragment consisting of the RubisCO gene cluster (cbbO-mQ-mM lysR2 lysR1 cbbLSQ-1O-1; accession: KJ639815.1) using the EZ-Tn5TM <TET-1> Tnp TransposomeTM Kit (epicentre®, Madison, WI, United States) according to manufacturer’s instructions, with chemically competent Epi300TM – T1R (epicentre®) as the host. One library was constructed with transposon clone 22II, where the cbbM structural gene was deleted (ΔcbbM). The second library was constructed using transposon clone 24II, where the cbbL structural gene was impaired (ΔcbbL). Clones containing fosmids with <TET-1> insertions were selected on LB agar plates using the following antibiotic additions: (i) chloramphenicol (12.5 μg ml-1) for selecting the fosmid vector, (ii) kanamycin (50 μg ml-1) to verify the presence of the first insertion, i.e., ΔcbbM or ΔcbbL, and (iii) tetracycline (100 μg ml-1) to verify the insertion of the second transposon element. Fosmids of double transposon clones were isolated from autoinduced cultures (for detailed information on autoinduction procedure see the manual for the CopyControlTM Fosmid Library Production Kit, epicentre®) using the High-Speed Plasmid Mini Kit (Geneaid, New Taipei City, Taiwan) according to manufacturer’s instruction. Isolated fosmids were sequenced starting from the <TET-1> insertion using the TET-1 FP-1 forward and TET-1 RP-1 reverse primers (see manual of the EZ-Tn5TM <TET-1> Insertion Kit, epicentre®) to identify the exact insertion position. Selected clones were tested for their RubisCO activities.
RubisCO Activity Assay
For RubisCO activity measurements double transposon clones were cultivated at 28°C on 200 ml pre-heated LB medium supplemented with chloramphenicol (12.5 μg ml-1), kanamycin (50 μg ml-1), tetracycline (10 μg ml-1), and autoinduction solution [1x final concentration (epicentre®)] in 1 l flasks with shaking (130 rpm) and harvested after 18 h by centrifugation (9,800 × g, 10 min, and 4°C). Subsequently crude extracts were prepared. For this purpose, cell pellets were washed twice with buffer A [100 mM Tris-HCl (pH 7.8), 10 mM MgCl2, 1 mM EDTA, 25 mM NaHCO3 and 1 mM DTT] before resuspension in 2 ml of the same buffer. Cells were disrupted by the French pressure cell press method, followed by centrifugation (19,580 × g, 20 min, and 4°C), as described before (Böhnke and Perner, 2015). The generated crude extracts were finally used as template to perform the RubisCO activity assay, where the concentrations of the reactant (RuBP) and the product (3-PGA) of RubisCO reaction were quantified over time using High-Performance Liquid Chromatography (HPLC) (Böhnke and Perner, 2015). At least two biological replicates and three technical replicates were used for the RubisCO activity assay. Mean values of technical replicates were used to calculate the overall mean. Errors of RubisCO activity measurements were calculated with the Gaussian propagation of error. Standard derivations of technical replicates were propagated forward and are thus entered into the equation. Significant differences were calculated using an unpaired t-test with equal variance and two-tailed distribution. For each performed HPLC run different controls were tested additionally to the measured samples. The crude extract of the metagenome derived fosmid clone 71C2 containing the RubisCO gene cluster (cbbO-mQ-mM lysR2 lysR1 cbbLSQ-1O-1) and 22.2 kb flanking DNA serves as positive control and the crude extract of an E. coli fosmid clone without RubisCO genes encoded on its fosmid insert serves as a negative control. A protein free reference sample with 5 mM RuBP and 5 mM 3-PGA dissolved in buffer A were furthermore applied through the assay and used (i) for sample peak assignment and (ii) to gather the non-enzymatic degradation of educts and products. The latter was used to calculate the pseudo-activity which is subtracted from each sample activity.
Quantitative Reverse Transcriptase PCR
Clones were cultivated in 100 ml flasks on 20 ml LB media supplemented with autoinduction solution [1x final concentration (epicentre®)] and the following antibiotics: chloramphenicol (12.5 μg ml-1) for the fosmid subclone 71C2II, chloramphenicol (12.5 μg ml-1) and kanamycin (50 μg ml-1) for transposon clones 22II (ΔcbbM), 24II (ΔcbbL), 6II (ΔlysR1), and 149II (ΔlysR2), and chloramphenicol (12.5 μg ml-1), kanamycin (50 μg ml-1), and tetracycline (10 μg ml-1) for the double transposon clones 22II2B2 (ΔcbbM ΔlysR1), 22II3A3 (ΔcbbM ΔlysR2), 24II1H1 (ΔcbbL ΔlysR1), and 24II1H7 (ΔcbbL ΔlysR2). Cultures were allowed to grow until an optical density (λ = 600 nm) between 2.0 and 3.0 was reached [for clone 6II (ΔlysR1) 24 h, all other clones 16 h]. Total RNA was isolated with the UltraClean® Microbial RNA Isolation Kit (MO BIO Laboratories, Inc., Carlsbad, CA, United States) according to manufacturer’s instructions with the exception that only 1 ml cell culture was harvested instead of the recommended 2 ml. Subsequently, genomic DNA was removed by using the RTS DNaseTM Kit (MO BIO Laboratories, Inc.) following the provided protocol, but with the modification that after half an hour an additional microliter RTS DNase was added. The reaction was incubated at 37°C for further 30 min followed by RTS DNA removal using 10 μl instead of 5 μl RTS DNase removal resins. One thousand two hundred microgram isolated RNA was used to synthesize cDNA with Invitrogen’s SuperScript® VILOTM cDNA Synthesis Kit (Life TechnologiesTM, Darmstadt, Germany), according to manufacturer’s instructions. The generated cDNA was used to examine the fold change of RubisCO form I (cbbL) and form II (cbbM) structural genes during expression in above mentioned transposon and double transposon clones relative to the intact version 71C2II. The expression data were normalized to the transcripts of three different genes, namely (i) the chloramphenicol-acetyltransferase (cat) gene, which is encoded on the fosmid vector and reflects its copy number, (ii) the RNA polymerase sigma factor rpoD, which is a housekeeping gene, and (iii) the 16S rRNA encoding gene. For this purpose, cDNA was diluted 1–10 and the cDNA that was derived from transcripts was used as a template for the amplification of cbbL and cbbM genes as well as the three different housekeeping genes. The SYBR® Select Master Mix, CFX (Applied Biosystems® by Life TechnologiesTM) and the following primer pairs were used: for (i) cbbL – cbbL_810F and cbbL_1115R, for (ii) cbbM – cbbM_647F and cbbM_976R, for (iii) cat – ChlR_821F and ChlR_1104R, for (iv) rpoD – rpoD_416F and rpoD_720R, and for (v) the 16S rRNA gene – 16S_280F and 16S_564R (for details on primer characteristics see Table 2). The qRT-PCR on the MJ MiniTM Gradient Thermal Cycler (Bio-Rad, Hercules, CA, United States) was performed under the following conditions: 95°C for 2 min followed by 40 cycles of 98°C for 15 s, 51°C for 20 s, and 72°C for 30 s. Each run contains, next to the samples, various controls like (i) the non-template controls, (ii) the no reverse transcriptase control as well as (iii) an inter run calibrator to ensure comparability between different runs, i.e., one reaction from the previous plate was repeated on the new plate. At least two biological and three technical replicates were measured and used to calculate fold changes (2-ΔΔCt). Technical replicates were arithmetically averaged and resulting mean values were used to calculate an overall mean. Errors were calculated with the Gaussian propagation of error. Standard derivations of technical replicates were entered into the equation and thus propagated forward. Significant differences were calculated from log transformed values using an unpaired t-test with equal variance and two-tailed distribution.
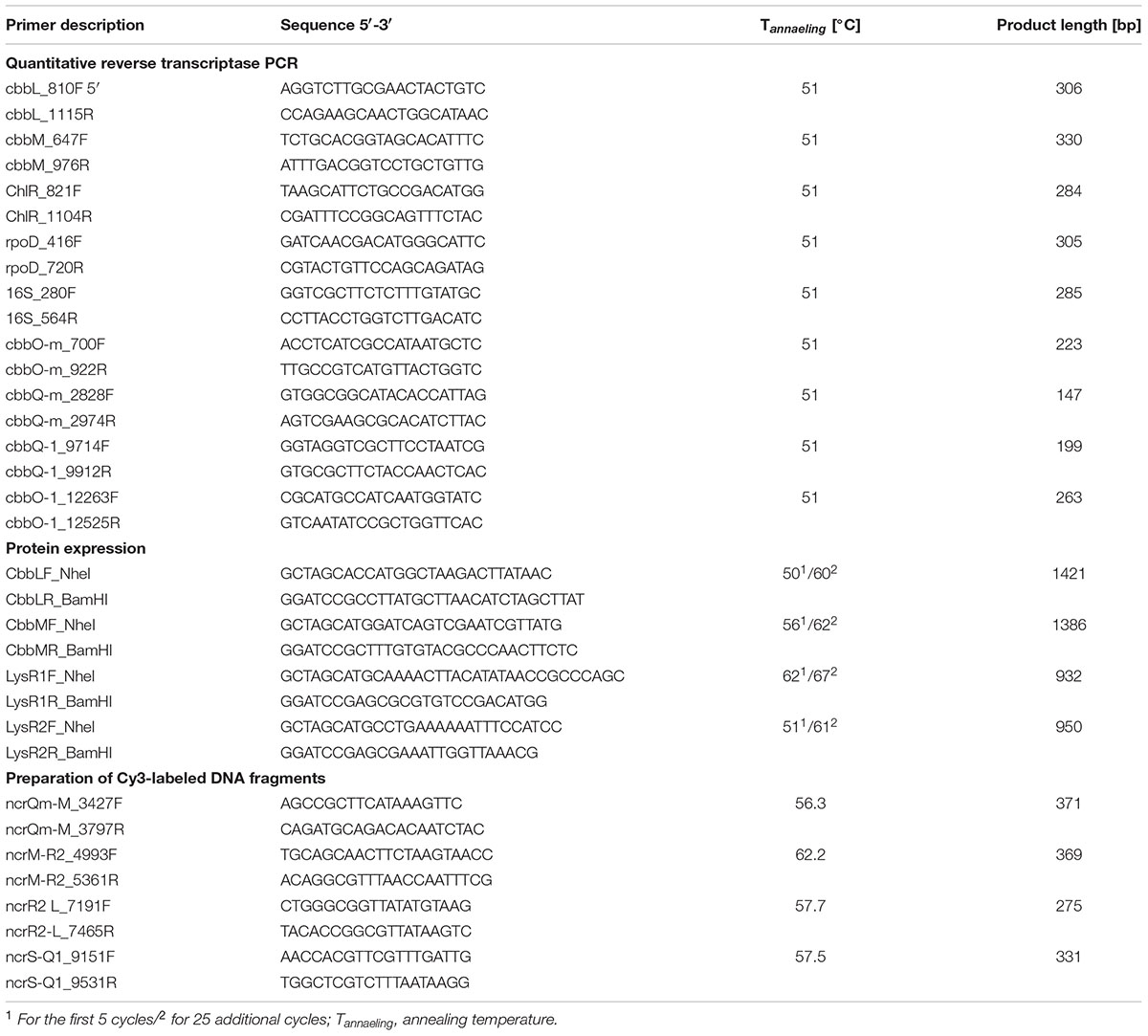
TABLE 2. Primers used in this study.
Polar Effects
Polar effects were investigated to determine whether transposon insertions have an impact on transcript abundances of genes located downstream of an insertion site. Therefore, transcript abundances of genes located downstream of cbbM and cbbL were measured for ΔcbbM (22II) and for ΔcbbL (24II), and compared with the transcript abundances in the intact version 71C2II. Investigated genes were (i) cbbO-m and (ii) cbbQ-m in ΔcbbM and (iii) cbbQ-1 and (iv) cbbO-1 in ΔcbbL. The cDNA used as template was the same as that isolated before for qRT-PCR of cbbL and cbbM. The qRT-PCR conditions were the same as mentioned above for the amplification of cbbL and cbbM, but with different primers: (i) cbbO-m – cbbO-m_700F and cbbO-m_922R, for (ii) cbbQ-m – cbbQ-m_2828F and cbbQ-m_2974R, for (iii) cbbQ-1 – cbbQ-1_9714F and cbbQ-1_9912R, and for (iv) cbbO-1 – cbbO-1_11263F and cbbO-1_12525R (for details on primer characteristics see Table 2). Three biological and three technical replicates were measured and used to calculate fold changes (2-ΔΔCt). Statistics were calculated in the same way as has been described for qRT-PCR data of cbbL and cbbM.
Overexpression and Protein Purification
The four genes encoding CbbL, CbbM, LysR1, and LysR2 were cloned in the expression vector pet21a (Novagen/Merck, Darmstadt, Germany). The coding regions of the targeted genes were amplified from the fosmid DNA of the metagenome derived clone 71C2, whereby restriction sites for NheI and BamHI were inserted using following primer pairs: (i) for cbbL – CbbLF_NheI and CbbLR_BamHI, (ii) for cbbM – cbbMF_NheI and cbbMR_BamH1, (iii) for lysR1 – LysR1F_NheI and LysR1R_BamHI, and (iv) for lysR2 – LysR2F_NheI 5′- and LysR2R_BamHI (primer sequences, annealing temperatures and product length are listed in Table 2). Amplification was done with the Pfu DNA Polymerase (Thermo Fisher Scientific, Waltham, MA, United States), following manufacturer’s instructions. The amplified fragments were ligated in the pet21a expression vector (Novagen/Merck) using the previously inserted restriction sites (NheI and BamHI). This vector has a His-tag coding sequence for the C-terminus of the cloned protein. The constructed plasmids (i) pet21a::cbbL, (ii) pet21a::cbbM, (iii) pet21a::lysR1, and (iv) pet21a::lysR2 were transformed into E. coli Rosetta-gami2 host strains. Verified clones were cultured at 17°C (CbbM) or at 22°C (CbbL, LysR1, and LysR2) in 200 ml LB supplemented with ampicillin (100 μg ml-1), tetracycline (10 μg ml-1), and chloramphenicol (12.5 μg ml-1) to an optical density (λ = 600 nm) of 0.7–0.8. IPTG was added to a final concentration of 0.1 mM for CbbL, 1 mM for CbbM, 1 mM for LysR1, and 0.1 mM for LysR2. The cultures were then grown over night at 17°C (CbbM) or at 22°C (CbbL, LysR1, and LysR2). Cells were harvested by centrifugation (7,600 × g, 8 min, and 8°C) and washed twice with 1x PBS buffer (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 2 mM KH2PO4, pH = 7.4). Cell pellets were stored at -20°C until proceeding with His-tag purification using Ni-NTA agarose (Qiagen, Hilden, Germany) as described in protocol 14 of TheQiaexpressionist (Qiagen, 2003), with some modifications: Initially cell pellets were resuspended in 2 ml lysis buffer (containing 10 mM imidazole). Each lysate was passed through the French press in order to disrupt cells. Cellular debris were removed by centrifugation (19,580 × g, 20 min, and 4°C) and supernatant volumes were brought to 20 ml with lysis buffer. Afterward 2 ml Ni-NTA agarose was added to the diluted lysates, which were incubated for 2 h on ice (shaking). After pelleting (1,000 × g, 30 s, and 8°C) Ni-NTA resins were washed twice with washing buffer (containing 20 mM imidazole). The protein was eluted from the column with five volumes of elution buffer (containing 250 mM imidazole). The concentration of the total purified protein was measured by performing the Bradford assay as described previously (Bradford and Williams, 1976) using bovine serum albumin as a standard. The proteins were further analyzed by SDS polyacrylamide gel electrophoresis using 12% (w/v) gels and Western-immunoblotting using 6-His-specific antibodies (see Supplementary Figure 1).
Preparation of Cy3-Labeled DNA Fragments
In preparation for the mobility shift assay four non-coding regions located within the metagenome derived RubisCO gene cluster were Cy3-labeled, namely the non-coding regions between: (i) cbbQ-m and cbbM, (ii) cbbM and lysR2, (iii) lysR1 and cbbL as well as (iv) cbbS and cbbQ-1. Labeling was done during amplification using Cy3-labeled dCTP’s (1 mM, GE Healthcare, Little Chalfont, United Kingdom), a mixture of dATP, dTTP, and dGTP (2 mM), the Phusion DNA Polymerase (Thermo Fisher Scientific) and the following primer pairs: (i) ncrQm-M_3427F and ncrQm-M_3797R for the non-coding region between cbbQ-m and cbbM, (ii) ncrM-R2_4993F and ncrM-R2_5361R for the non-coding region between cbbM and lysR2, (iii) ncrR1-L_7191F and ncrR1-L_7465R for the non-coding region between lysR1 and cbbL, and (iv) ncrS-Q1_9151F and ncrS-Q1_9531R for the non-coding region between cbbS and cbbQ-1 (see Table 2 for primer sequences). PCR conditions were: Denaturation at 98°C for 10 s, primer annealing for 30 s at appropriated annealing temperatures (see Table 2), and elongation at 72°C for 12 s (32 cycles).
Electrophoretic Mobility Shift Assay (EMSA)
The mobility shift assay was based on a previously published protocol (Charoenpanich et al., 2013) but with modifications: Purified His6-CbbL, His6-CbbM, His6-LysR1, and His6-LysR2 were investigated for the ability to bind at the four non-coding regions amplified from the metagenome derived RubisCO gene cluster. For this purpose, proteins were tested (i) individually but also (ii) pairwise in combination with each other. The protein concentrations used for approaches with individual proteins ranged from 0 to 1,000 ng per 30 μl reaction mixture. For approaches with two different proteins up to 2,000 ng total protein per 30 μl were used in one reaction, which corresponds to a maximum of 1,000 ng of each protein and thus ensures the comparability with the single protein approaches. Regardless of whether one or two proteins were used for the assay, protein(s) was/were firstly incubated with a total of 200 ng salmon sperm DNA for 5 min at room temperature in binding buffer (50 mM Tris-HCl, 250 mM KCl, pH = 8.5) to prevent unspecific DNA shifts. After this Cy3-labeled DNA fragments were added (200 ng per 30 μl reaction) and reaction mixture was incubated at room temperature in the dark for further 20 min. Subsequently 5 μl loading dye (20% TBE buffer and 80% glycerol) was added and samples were loaded on a 5% TBE-polyacrylamide gel. Following electrophoresis at 50 mV for 3 h in cooled TBE-buffer, gels were visualized on a VersaDocTM MP4000 (Bio-Rad) at 550 nm and an exposure time of 300 ms.
Computational Analyses
Distinct regulatory features were predicted for the DNA of the metagenome derived RubisCO gene cluster using different online tools. Promoter regions were predicted for all non-coding regions with the SoftBerry program BProm (Solovyev and Salamov, 2011). We also searched for inverted repeats, which are putatively able to fold into stem-loop structures using Emboss Palindrome (Rice et al., 2000), with a minimum length for repeats of 8 nt and a maximum gap between repeated elements of 100 nt. With respect to the formation of stem-loop structures, inverted repeats with loops less than three bases were not taken into account, because they are thought to be sterically impossible and thus are believed not to be formed (Bon and Orland, 2011). LysR binding sites were identified manually by searching for the typical LysR binding motif TnA-n7/8-AnT which has been identified in other RubisCO harboring organisms before (van Keulen et al., 2003; Maddocks and Oyston, 2008 and references therein).
Results
To understand the processes involved in expression of a fully active RubisCO form I (CbbLS) and form II (CbbM) enzyme, we constructed two double mutant libraries using a 13 kb metagenomic fragment encoding the RubisCO gene cluster. In one case, the ΔcbbM fragment of transposon clone 22II, and in the other case, the ΔcbbL fragment of transposon clone 24II provided the base for the second mutant library. These double mutants were used to study how gene deletions influence cbbL and cbbM transcription and respective enzyme activities. We also searched through the metagenomic DNA sequence in silico for putative LysR binding sites, promoter regions or structures capable of forming stem-loops – possibly affecting transcription – and determined experimentally whether RubisCO and LysR proteins and protein combinations bind to non-coding regions in the metagenomic fragment.
cbbL and cbbM Transcription after Gene Deletions
cbbL and cbbM transcription abundances were tested for eight mutants and normalized to three different reference genes (cat, rpoD, and 16S rRNA) (Figure 1). Generally, transcript levels of cbbL in ΔcbbM and of cbbM in ΔcbbL remained unchanged relative to the undeleted metagenomic fragment (71C2II). cbbL gene transcription was only significantly downregulated in ΔlysR1 if cbbM was expressed, since no changes in cbbL transcript levels were observed in ΔlysR1 ΔcbbM. In contrast, cbbM gene transcription was significantly upregulated in ΔcbbL ΔlysR1 double transposon clone 24II1H1 (3-fold) and in ΔcbbL ΔlysR2 double transposon clone 24II1H7 (15-fold).
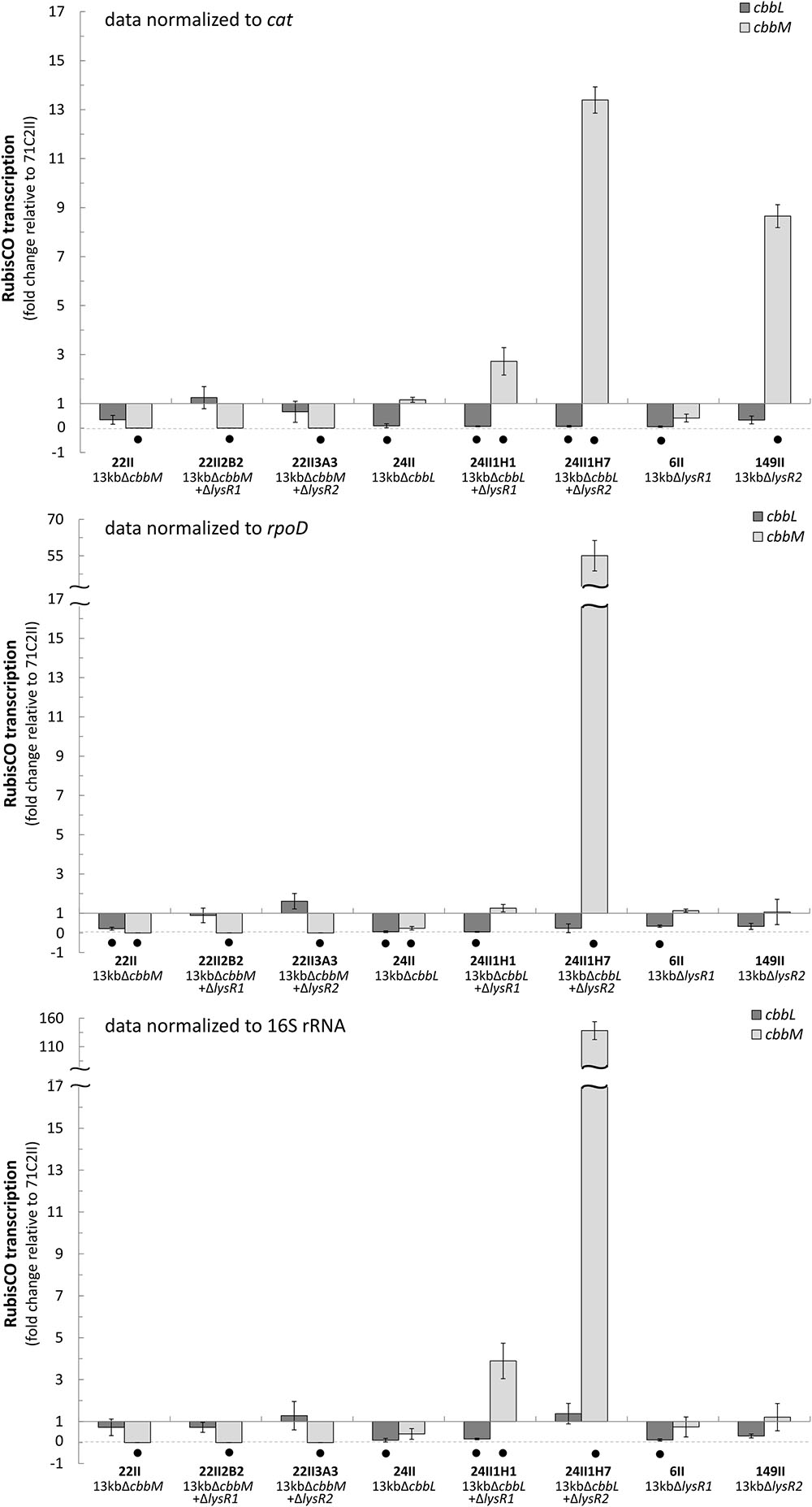
FIGURE 1. Fold change of cbbL and cbbM transcription. Fold change (2- Δ ΔCt) of cbbL (light-gray) and cbbM (dark-gray) transcripts expressed from selected transposon and double transposon clones normalized to the cat (chloramphenicol acetyltransferase), rpoD, and 16S rRNA genes. All data is relative to the 13 kb metagenomic fragment (clone 71C2II) encoding the RubisCO gene cluster (cbbO-mQ-mM lysR2 lysR1 cbbLSQ1O1). Bars and error bars indicate mean values and +/– standard error. Black dots denote significantly different values (p-value ≤ 0.05).
cbbL and cbbM Activity after Gene Deletions
All RubisCO activities of clones from the double mutant libraries where either ΔcbbM (22II) or ΔcbbL (24II) was used for the construction of the double mutants can be viewed in Figures 2A,B, respectively. Total RubisCO activity of the undeleted 13 kb fragment (71C2II) increased considerably in ΔcbbM (22II) (5-fold). Additional deletions in lysR1, lysR2, cbbQ-m, and cbbO-m resulted in a significant decrease of RubisCO activity.
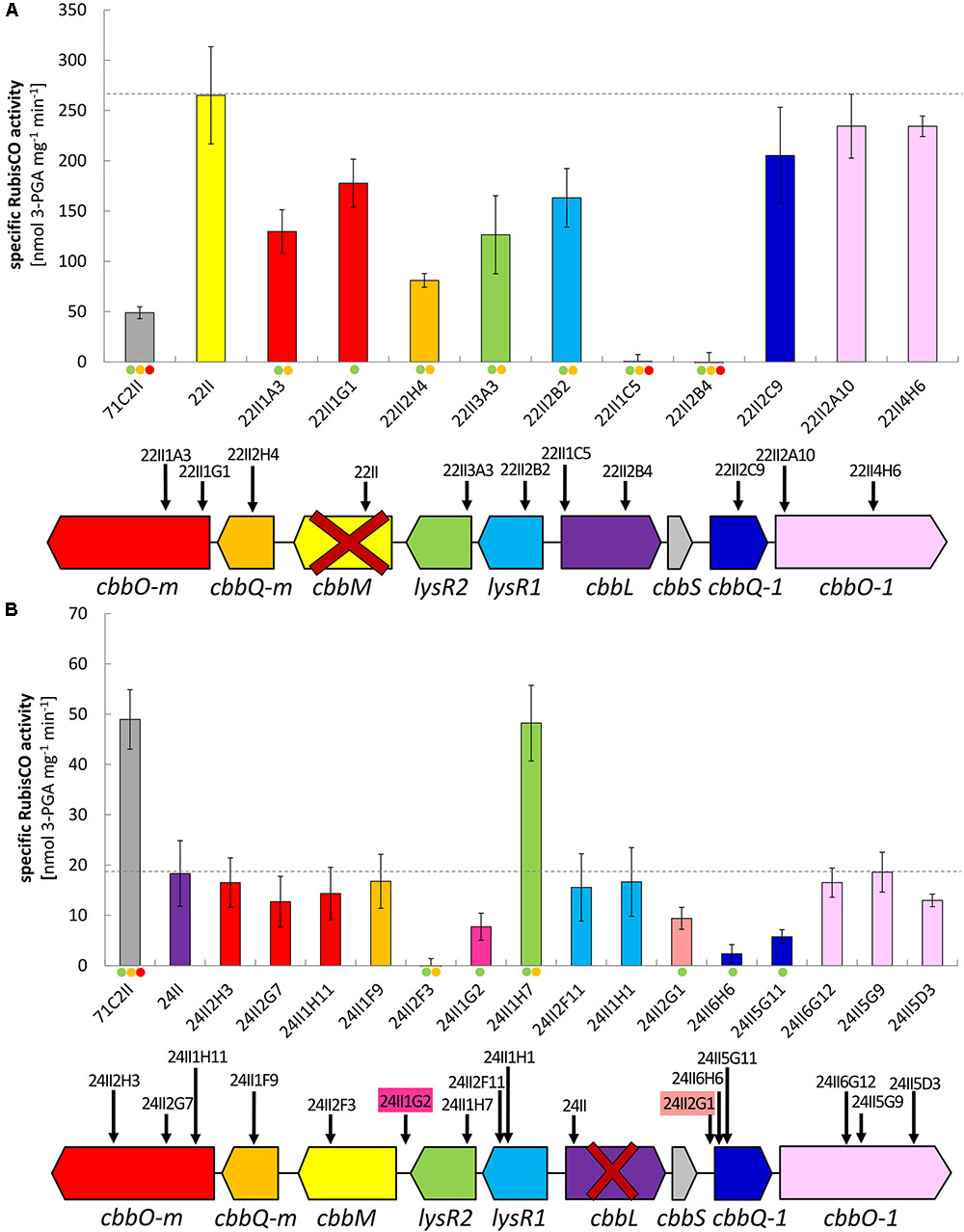
FIGURE 2. Specific RubisCO activities and insertion positions of tested double transposon clones. Specific RubisCO activities and schematic gene arrangement of (A) double transposon clones constructed on the basis of the ΔcbbM transposon clone 22II and (B) double transposon clones constructed on the basis of the ΔcbbL transposon clone 24II. Identified open reading frames (ORFs) are indicated as arrows in the direction of transcription. Insertion sites are denoted by vertical black arrows and transposon clone numbers. Genes are color coded according to the bars indicating corresponding RubisCO activity. Gene abbreviations are as follows: cbbO-m – von Willebrand factor type A; cbbQ-m – ATPase AAA-type; cbbM – ribulose-1,5-bisphosphate carboxylase/oxygenase large subunit, form II; lysR2 – transcriptional regulator, LysR family; lysR1 – transcriptional regulator, LysR family; cbbL – ribulose-1,5-bisphosphate carboxylase/oxygenase large subunit, form I; cbbS – ribulose-1,5-bisphosphate carboxylase small subunit; cbbQ-1 – ATPase, AAA-type; cbbO-1 – von Willebrand factor. Bars and error bars indicate mean values and +/– standard error. The level of significant differences is denoted by dots where green is ≤0.05, yellow is ≤0.01, and red is ≤0.001.
Total RubisCO activity of the undeleted 13 kb fragment (71C2II) was significantly reduced in ΔcbbL (24II). When additionally deleting lysR2 (24II1H7), the RubisCO activity increased (3.5-fold), restoring the original activity of clone 71C2II. In four of the tested double mutant ΔcbbL clones the RubisCO activity was considerably reduced. These were clone 24II1G2, where parts of the intergenic region of cbbM and lysR2 were deleted, clone 24II2G1, where parts of the non-coding region between cbbS and cbbQ-1 were deleted, and the two ΔcbbL ΔcbbQ-1 clones (24II6H6 and 24II5G11). As expected, no RubisCO activity was measured for ΔcbbL ΔcbbM.
Putative Promoters, LysR Binding Sites and Stem-Loop Forming Structures
We searched the intergenic regions of our metagenomic fragment for structures which encode putative promoter regions that provide potential LysR binding sites or that may form stem-loops (Figure 3). We found 6 putative promoter binding sites, 15 putative LysR binding sites, and 18 putative stem-loop forming structures (for exact positions on the metagenomic fragment see Supplementary Figure 2).
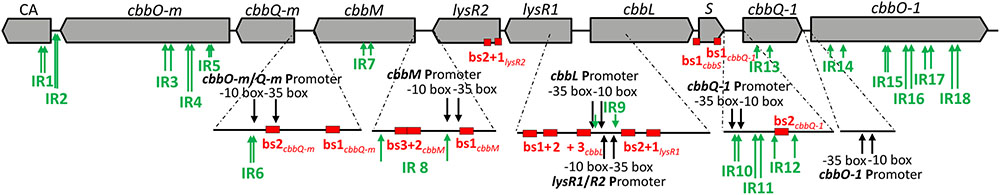
FIGURE 3. Regulatory features predicted for the DNA of the metagenome derived RubisCO gene cluster. ORFs are displayed as gray arrows in the direction of transcription. The same gene abbreviations as specified in Figure 2 are used. Predicted promoters are indicated by black arrows, inverted repeats (IR) are denoted by green arrows and putative LysR binding sites (bs) are represented by red boxes.
We also performed mobility shift assays to test whether LysR1, LysR2, CbbL, and CbbM or a combination of these proteins bind to the non-coding regions cbbQ-m and cbbM, cbbM and lysR2, lysR1 and cbbL or cbbS and cbbQ-1 (Figure 4). LysR1 binds to all tested non-coding regions (Figure 4A). Its binding ability is enhanced for the intergenic region lysR1 and cbbL if CbbL or LysR2 are additionally present (Figure 4B). LysR2 alone appears to only bind to two non-coding regions: between cbbQ-m and cbbM and between cbbM and lysR2 (Figure 4A). CbbM addition intensifies the binding ability to the cbbM and lysR2 intergenic region (Figure 4B). The presence of CbbM also enables LysR2 to bind to two further non-coding regions, namely lysR1 and cbbL as well as cbbS and cbbQ-1 (Figure 4B). Other protein combinations likely reflect binding of one of the proteins alone.

FIGURE 4. Binding of CbbL, CbbM, LysR1, and LysR2 to four different non-coding regions of the metagenome derived RubisCO gene cluster. Mobility shift assays with CbbL, CbbM, LysR1, and LysR2 individually (A) as well as in (B) combination are shown. Corresponding semi-quantitative data is depicted below each gel where the y-axis denotes intensity in percent relative to the unshifted band in lane 1. DNA fragments contained the non-coding regions between cbbQ-m and cbbM, cbbM and lysR2, lysR1 and cbbL, and cbbS and cbbQ-1. The protein concentrations used for approaches with individual proteins, given per 30 μl reaction mixture: 0 ng (1), 10 ng (2), 25 ng (3), 50 ng (4), 100 ng (5), 250 ng (6), 500 ng (7), and 1000 ng (8). For approaches with two different proteins tested in one reaction the following protein concentrations per 30 μl reaction mixture were used: 0 ng (1), 25 ng (2), 50 ng (3), 100 ng (4), 250 ng (5), 500 ng (6), 1000 ng (7) and 2000 ng (8). Gene abbreviations are the same as described in Figure 2. Other abbreviation: ncr, non-coding region.
Discussion
Possible CbbL Expression and Regulation
In the intergenic region of lysR1 and cbbL two promoters were predicted: (i) one could be for cbbL transcription (with the -10 box ‘AGGAATCAT’ at position 7,271 bp and the -35 box ‘TTGATA’ at position 7,250 bp) and (ii) the other for lysR1/lysR2 transcription with the -10 box at position 7,275 bp ‘ATCATATAC’ and with the -35 box at position 7,302 bp ‘TAACAA’ (Supplementary Figure 2). This is in line with previous predicted functions for the non-coding region between lysR and cbbL in other organisms, where promoters for both directions were identified (Kusano and Sugawara, 1993; Wei et al., 2004). Additionally, three and two putative LTTR binding sites upstream of the putative cbbL and lysR1/lysR2 promoters, respectively, were recognized (Figure 3). These sites may be involved in LysR1 and/or LysR2 regulated cbbL transcription as well as autoregulation of their own transcription, as has been commonly demonstrated for enzymes of the LysR family (Schell, 1993; Maddocks and Oyston, 2008). The mobility shift assay verified that binding sites are located in this non-coding region (Figure 4). Here, DNA binding of LysR1 is intensified by the presence of LysR2 or CbbL (Figure 4B). LysR2 is also capable of binding to this region, but only when CbbM proteins are available. Promiscuous heterotypic interactions between different LTTRs in E. coli have been shown before, but the relevance of such cross-interactions remains unknown (Knapp and Hu, 2010). However, since our experiment showed that LysR1’s DNA binding ability is increased by LysR2, one may conclude that LysR1 and LysR2 are also able to cross-interact and form heteromultimers with its non-cognate partner. The heteromultimer (LysR1+LysR2) may cause different regulatory effects relative to the homomultimers (LysR1+LysR1 or LysR2+LysR2). The role that CbbL and CbbM play for intensified LysR binding currently remains unclear but may be related to DNA or RNA stability. Mobility shift assays with RNA and the large RubisCO subunit of Chlamydomonas reinhardtii demonstrated CbbL’s ability to bind to RNA in a sequence-independent manner under certain conditions (Yosef et al., 2004).
Although cbbL transcription levels remained unchanged in ΔcbbM ΔlysR1 and ΔcbbM ΔlysR2 (Figure 1), these clones exhibited reduced CbbL activity (Figure 2A). This discrepancy may be explained if LysR proteins also act on the transcription of genes encoding proteins, which influence CbbL activity. Likely candidates encoded on this metagenomic fragment are CbbO-m, CbbQ-m, CbbQ-1, and CbbO-1, previously shown to be involved in post-translational activation of RubisCO enzymes (Tsai et al., 2015). Indeed LysR1 proteins are demonstrated to bind upstream of cbbQ-m/cbbO-m and cbbQ-1/cbbO-1 regions (Figure 4A). LysR2 can also bind upstream of cbbQ-m/cbbO-m and if CbbM is present can bind upstream of cbbQ-1/cbbO-1, too (Figures 4A,B, respectively). While the deletion of cbbQ-m and cbbO-m in ΔcbbM illustrated a RubisCO activity loss, CbbQ-1 and CbbO-1 did not have an effect on CbbL activity (Figure 2A). Based on the available information, we posit that CbbQ-m/CbbO-m activates CbbL and that LysR1 and LysR2 proteins promote CbbQ-m/CbbO-m expression (compare model in Figure 5).
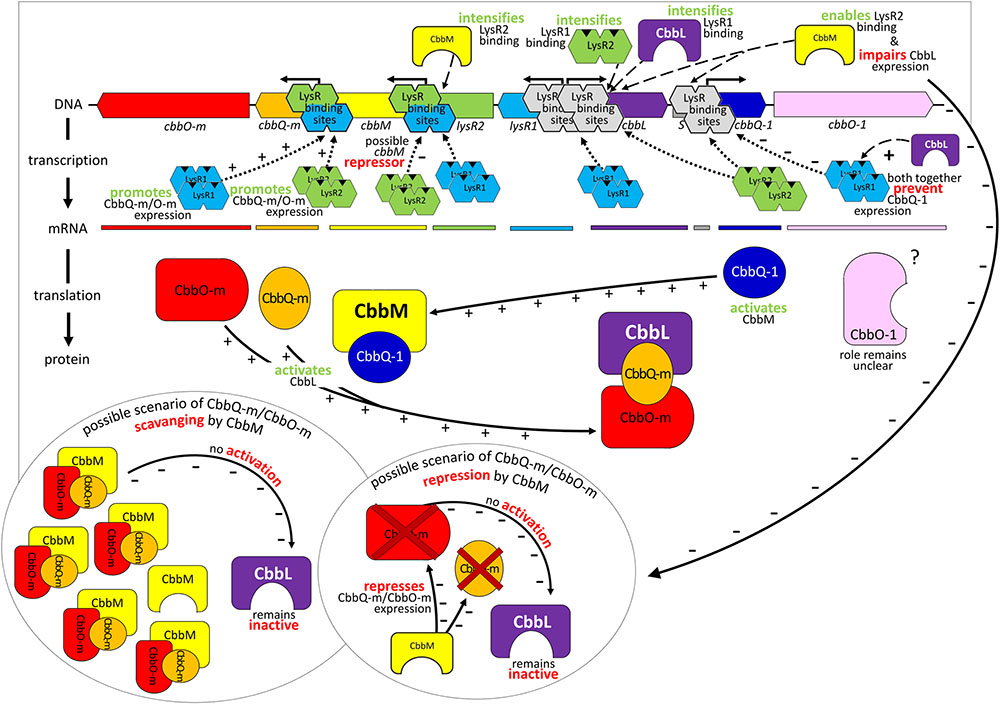
FIGURE 5. Model of gene regulation and possible protein interaction. Relevant transcriptional and post-translational regulatory processes hypothesized for the 13 kb comprising metagenome derived RubisCO gene cluster. Identified genes are indicated as arrows in the direction of transcription and marked in the same color as the corresponding mRNA and resulting gene products. Abbreviations used are the same as described in Figure 2.
Intriguingly, CbbM also appears to play a role for cbbL expression, which has not been observed in any other study before. The deletion of cbbM leads to a 5-fold RubisCO activity increase (Figure 2A) indicative of CbbM’s repressive nature for cbbL expression. However, in ΔcbbM cbbL transcript levels are not elevated (Figure 1). Possible scenarios include that CbbM scavenges post-translational activators (CbbQ-m/CbbO-m), which are then not available for CbbL activation or that CbbM is involved in repressing CbbQ-m/CbbO-m, which may be needed for CbbL activation (Figure 5). For information on putative polar effects for cbbQ-m/cbbO-m transcript abundance caused by transposon insertion in the upstream neighboring cbbM gene see Supplementary Results and Discussion and Supplementary Figure 3.
Possible CbbM Expression and Regulation
A putative promoter was identified upstream of cbbM (Figure 3), confirming recent results indicative of cbbM being transcribed alone in this fragment (Böhnke and Perner, 2015). In the intergenic region of cbbM and lysR2 three putative LysR binding sites were predicted, two of which partially overlap (‘LysR bs1cbbM,’ ‘LysR bs2/3cbbM,’ see Supplementary Figure 2). In Xanthobacter flavus the same arrangement of a single LysR binding site followed by two overlapping binding sites, between a RubisCO structural gene and a LysR transcriptional regulator was also identified and all three sites were evidenced to be functional (van Keulen et al., 2003). In our fragment LysR binding sites upstream of cbbM exist, which allow binding of LysR1 and LysR2 proteins (Figure 4A). LysR2 binding was even intensified if CbbM was present (compare Figure 4B). Upregulation of the cbbM transcript (Figure 1) and increasing RubisCO activity in the double mutant ΔcbbL ΔlysR2 (Figure 2B) strongly suggest that LysR2 acts as a repressor for cbbM gene expression where CbbM itself contributes to intensified LysR binding ability and thus its own transcriptional regulation (Figure 5). The combination of CbbL and LysR1 also appears to result in a repressive cbbM transcriptional regulation but single mutations in cbbL and lysR1 did not cause upregulation of the cbbM gene (see Figure 1). However, despite higher cbbM transcript levels in ΔcbbL ΔlysR1, this clone did not demonstrate an increase in RubisCO activity (Figure 2B), contrasting the transcriptional data at first glance. These results can be explained though if LysR1 also controls the expression of post-translational activators, which here only seems applicable if CbbL is also present. And indeed LysR1 appears to be able to bind upstream of cbbQ-m/cbbQ-m and of cbbQ-1/cbbO-1 (Figure 4), where also putative LysR binding sites were recognized (Figure 3). Of these likely post-translational activators only the deletion of cbbQ-1 caused a CbbM activity loss and thus makes its LysR regulated role in CbbM activation under the provided conditions highly likely. We thus suggest that CbbQ-1 activates CbbM and that LysR1 proteins may prevent CbbQ-1 expression if CbbL is present. Additionally, an insertion at position 9,463 bp, i.e., 15 bp upstream of the cbbQ-1 transcription start in the ΔcbbL clone 24II, resulted in significantly reduced CbbM activity (see Figure 2B, clone 24II2G1). Even though this insertion is located downstream of putative regulatory features, the 1,674 bp comprising <TET> insertion represents a barrier the RNA polymerase most likely cannot simply overcome, which would result in an impaired cbbQ-1 transcription and thus no CbbQ-1 would be present. Unexpectedly, the deletion in cbbO-1, downstream of cbbQ-1, did not alter RubisCO activity (Figure 2B). However, the elevated number of inverted repeats (5/18 identified on the metagenomic fragment) in the cbbO-1 gene, which are theoretically capable of forming stem-loop structures, may indicate some fine-tuned transcriptional regulation (Treangen et al., 2009 and references therein). Under the provided conditions cbbO-1 may be downregulated and under other environmental conditions may well be important for RubisCO activation.
In the intergenic region of cbbM and lysR2 a large inverted repeat flanking 86 nt (IR8, see Supplementary Figure 2) also exists. Such inverted repeats often form stem-loop structures that are important for controlling transcription initiation and termination, mRNA stabilization or genome plasticity (Treangen et al., 2009 and references therein). They also play roles in supporting DNA binding proteins in finding their binding sites (Frost et al., 1994). The IR8 in our metagenomic fragment may well represent such a signaling stem-loop structure that guides LysR to the LysR binding sites. An insertion of a tetracycline cassette in the left arm of the IR8 (clone 24II1G2), directly between the LysR binding sites ‘bs1cbbM’ and ‘bs2/3cbbM’ (see position 5,232 nt in Supplementary Figure 2) resulted in a significant RubisCO activity loss (Figure 2B). One explanation could be that this structure is necessary for the expression of a functional CbbM. Since this insertion also separates the putative -10 from the -35 promoter box, the RubisCO activity loss may be due to the impaired promoter region.
Benefits and Drawbacks of Working with a Metagenomic Fragment in a Non-native System
Restricting the work to culture-depended approaches, neglects the large majority of RubisCO gene clusters from uncultured organisms. However, working with metagenomes in non-native systems holds both promise and pitfalls. The benefits of using E. coli as a host organism are well-known: E. coli has an unrivaled fast growth on inexpensive media and the genetics are very well-known, making transformations with exogenous DNA simple and straightforward (Rosano and Ceccarelli, 2014). In contrast, expression of a metagenomic fragment in a surrogate host may also entail cross-talks, inhibitions and unspecific reactions. Recombinant gene expression in E. coli and other surrogate hosts might be troublesome due to, e.g., unrecognized intrinsic promotors and associated factors, a diverging codon usage or problems with correct protein folding (Perner et al., 2011).
One major advantage of working with a metagenomic fragment in fosmid clones (besides gaining access to the world of the unculturables) is its relatively small size (in this case: 13 kb metagenomic fragment). The small size infers clear gene arrangements and a limited number of possible gene and/or protein interactions relative to the (hardly tangible) complexity in a native system. The same work in a cultured representative is considerably more difficult and time consuming, because genes with yet unknown functions, which are not necessarily located in the vicinity of the gene cluster under investigation, may well participate in/contribute to the gene regulation and activation of the enzyme (indirectly), as has recently been shown for orf06 (Böhnke and Perner, 2015). Although the metagenomic approach with a defined number of genes can simplify first insights into regulatory mechanisms, it can also hinder the understanding of the mechanisms given that some vital genes/respective products cannot be expressed/synthesized as they are located on parts of the genome not present on the captured fraction of the metagenome.
To overcome such limitations the use of a host with the genomic inventory to operate the CBB cycle may be a viable option. In our case a cultured Thiomicrospira strain could be used and the genes under investigation deleted. However, deleting gene clusters in T. crunogena which are comparable to our 13 kb metagenomic fragment and expressing the latter heterologously in the Thiomicrospira host or alternatively constructing double mutants as we did in our metagenomic fragment (i.e., nine deletions in ΔcbbL and eight deletions in ΔcbbM) to investigate regulatory mechanisms is hardly feasible, given that Thiomicrospira’s genetic accessibility is not understood and thus any transformation with exogenous DNA becomes challenging. An alternative host that operates the CBB cycle and where mutations have been successfully constructed is R. capsulatus (Alphaproteobacteria) (Paoli et al., 1998; Witte et al., 2010; Dangel et al., 2014; Varaljay et al., 2016). However, this potential host encodes different types of RubisCOs and has other RubisCO gene cluster arrangements and likely different gene regulation mechanisms than the organism encoding our metagenomic fragment.
For future work one may consider combining studies in a genetically accessible surrogate host such as E. coli, naturally incapable of operating the CBB cycle, with subsequent investigations in a closely related cultured representative. Thus, the first insights of complex RubisCO regulatory mechanisms obtained through studies dealing with RubisCO gene expression in a non-native system could be used in further studies where, e.g., the role of external factors could be studied in a native system.
Conclusion
The intense interactions between the different proteins suggest the complex, but fine-tuned nature of the RubisCO regulatory machinery. This fine-tuned regulatory machinery reflects the highly dynamic nature of hydrothermal vent environments from which this metagenomic fragment was extracted. Albeit the CBB cycle has a much higher energy requirement than other autotrophic CO2 fixation pathways (Berg et al., 2010), it can operate when O2 is present, while many enzymes of other CO2 fixation pathways are highly O2 sensitive (Berg, 2011). Given that RubisCO form I and form II have different capabilities to discriminate between CO2 and O2 (Berg, 2011) and both CO2 and O2 concentrations can be highly variable in hydrothermal vent habitats (Perner et al., 2013 and references therein), the ability to rapidly react to environmental CO2 and O2 changes may pose a benefit for local organisms with both forms of RubisCO. A quick response to increasing O2 levels may be the key to successfully colonizing dynamic hydrothermal environments. Having understood some of the possible interactions between the proteins encoded by our metagenomic fragment, this knowledge could now be transferred to a closely related cultured representative. Distinct experiments under different environmental conditions such as high/low CO2 or O2 concentrations could be performed and changes in the transcriptome investigated.
Author Contributions
SB planned and performed experiments, performed computational analyses, and wrote the paper. MP designed the research project, planned experiments, and wrote the paper.
Funding
SB was supported by grants from the Deutsche Forschungsgemeinschaft (DFG) (PE1549/5 1).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Birte Claßen and Laura Lindloff for helping us to construct the double mutant transposon libraries and Constantin König for excellent technical support with the CbbM mobility shift assays.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.01303/full#supplementary-material
References
Amann, R. I., Ludwig, W., and Schleifer, K. H. (1995). Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev. 59, 143–169.
Axler-DiPerte, G. L., Miller, V. L., and Darwin, A. J. (2006). YtxR, a conserved LysR-like regulator that induces expression of genes encoding a putative ADP-ribosyltransferase toxin homologue in Yersinia enterocolitica. J. Bacteriol. 188, 8033–8043. doi: 10.1128/JB.01159-06
Berg, I. A. (2011). Ecological aspects of the distribution of different autotrophic CO2 fixation pathways. Appl. Environ. Microbiol. 77, 1925–1936. doi: 10.1128/AEM.02473-10
Berg, I. A., Kockelkorn, D., Ramos-Vera, W. H., Say, R. F., Zarzycki, J., Hügler, M., et al. (2010). Autotrophic carbon fixation in archaea. Nat. Rev. Microbiol. 8, 447–460. doi: 10.1038/nrmicro2365
Bertani, G. (1951). Studies on lysogenesis. I. The mode of phage liberation by lysogenic Escherichia coli. J. Bacteriol. 62, 293–300.
Böhnke, S., and Perner, M. (2015). A function-based screen for seeking RubisCO active clones from metagenomes: novel enzymes influencing RubisCO activity. ISME J. 9, 735–745. doi: 10.1038/ismej.2014.163
Bon, M., and Orland, H. (2011). TT2NE: a novel algorithm to predict RNA secondary structures with pseudoknots. Nucleic Acids Res. 39:e93. doi: 10.1093/nar/gkr240
Bradford, M. M., and Williams, W. L. (1976). New, rapid, sensitive method for protein determination. Fed. Proc. 35, 274–274.
Charoenpanich, P., Meyer, S., Becker, A., and McIntosh, M. (2013). Temporal expression program of quorum sensing-based transcription regulation in Sinorhizobium meliloti. J. Bacteriol. 195, 3224–3236. doi: 10.1128/JB.00234-13
Dangel, A. W., Luther, A., and Tabita, F. R. (2014). Amino acid residues of RegA important for interactions with the CbbR-DNA complex of Rhodobacter sphaeroides. J. Bacteriol. 196, 3179–3190. doi: 10.1128/JB.01842-14
Dangel, A. W., and Tabita, F. R. (2015). CbbR, the master regulator for microbial carbon dioxide fixation. J. Bacteriol. 197, 3488–3498. doi: 10.1128/JB.00442-15
Dubbs, J. M., and Tabita, F. R. (2003). Interactions of the cbbII promoter-operator region with CbbR and RegA (PrrA) regulators indicate distinct mechanisms to control expression of the two cbb operons of Rhodobacter sphaeroides. J. Biol. Chem. 278, 16443–16450. doi: 10.1074/jbc.M211267200
Dubbs, J. M., and Tabita, F. R. (2004). Regulators of nonsulfur purple phototrophic bacteria and the interactive control of CO2 assimilation, nitrogen fixation, hydrogen metabolism and energy generation. FEMS Microbiol. Rev. 28, 353–376. doi: 10.1016/j.femsre.2004.01.002
Dubbs, P., Dubbs, J. M., and Tabita, F. R. (2004). Effector-mediated interaction of CbbRI and CbbRII regulators with target sequences in Rhodobacter capsulatus. J. Bacteriol. 186, 8026–8035. doi: 10.1128/JB.186.23.8026-8035.2004
Ellis, R. J. (1979). Most abundant protein in the world. Trends Biochem. Sci. 4, 241–244. doi: 10.1016/0968-0004(79)90212-3
Field, C. B., Behrenfeld, M. J., Randerson, J. T., and Falkowski, P. (1998). Primary production of the biosphere: integrating terrestrial and oceanic components. Science 281, 237–240. doi: 10.1126/science.281.5374.237
Frost, L. S., Ippen-Ihler, K., and Skurray, R. A. (1994). Analysis of the sequence and gene products of the transfer region of the F sex factor. Microbiol. Rev. 58, 162–210.
Hauser, T., Popilka, L., Hartl, F. U., and Hayer-Hartl, M. (2015). Role of auxiliary proteins in Rubisco biogenesis and function. Nat. Plants 1:15065. doi: 10.1038/nplants.2015.65
Joshi, G. S., Zianni, M., Bobst, C. E., and Tabita, F. R. (2013). Regulatory twist and synergistic role of metabolic coinducer- and response regulator-mediated CbbR-cbbI interactions in Rhodopseudomonas palustris CGA010. J. Bacteriol. 195, 1381–1388. doi: 10.1128/JB.02060-12
Knapp, G. S., and Hu, J. C. (2010). Specificity of the E. coli LysR-type transcriptional regulators. PLoS ONE 5:e15189. doi: 10.1371/journal.pone.0015189
Kusano, T., and Sugawara, K. (1993). Specific binding of Thiobacillus ferrooxidans RbcR to the intergenic sequence between the rbc operon and the rbcR gene. J. Bacteriol. 175, 1019–1025. doi: 10.1128/jb.175.4.1019-1025.1993
Maddocks, S. E., and Oyston, P. C. F. (2008). Structure and function of the LysR-type transcriptional regulator (LTTR) family proteins. Microbiology 154, 3609–3623. doi: 10.1099/mic.0.2008/022772-0
Mueller-Cajar, O., Stotz, M., Wendler, P., Hartl, F. U., Bracher, A., and Hayer-Hartl, M. (2011). Structure and function of the AAA+ protein CbbX, a red-type Rubisco activase. Nature 479, 194–199. doi: 10.1038/nature10568
Paoli, G. C., Vichivanives, P., and Tabita, F. R. (1998). Physiological control and regulation of the Rhodobacter capsulatus cbb operons. J. Bacteriol. 180, 4258–4269.
Parry, M. A., Keys, A. J., Madgwick, P. J., Carmo-Silva, A. E., and Andralojc, P. J. (2008). Rubisco regulation: a role for inhibitors. J. Exp. Bot. 59, 1569–1580. doi: 10.1093/jxb/ern084
Perner, M., Gonnella, G., Hourdez, S., Böhnke, S., Kurtz, S., and Girguis, P. (2013). In situ chemistry and microbial community compositions in five deep-sea hydrothermal fluid samples from Irina II in the Logatchev field. Environ. Microbiol. 15, 1551–1560. doi: 10.1111/1462-2920.12038
Perner, M., Ilmberger, N., Köhler, H. U., Chow, J., and Streit, W. R. (2011). “Emerging fields in functional metagenomics and its industrial relevance: overcoming limitations and redirecting the search for novel biocatalysts,” in Handbook of Moleculare Microbial Ecology II: Metagenomics in Different Habitats, ed. F. J. de Bruijn (Hoboken, NJ: Blackwell), 484–485.
Qiagen (2003). TheQiaexpressionistTM, A Handbook for High-Level Expression and Purifiction of 6xHis-Tagged Protein, 5th Edn. Hilden: Qiagen.
Raven, J. A. (2009). Contributions of anoxygenic and oxygenic phototrophy and chemolithotrophy to carbon and oxygen fluxes in aquatic environments. Aquat. Microb. Ecol. 56, 177–192. doi: 10.3354/ame01315
Raven, J. A. (2013). Rubisco: still the most abundant protein of Earth? New Phytol. 198, 1–3. doi: 10.1111/nph.12197
Rice, P., Longden, I., and Bleasby, A. (2000). EMBOSS: the European molecular biology open software suite. Trends Genet. 16, 276–277. doi: 10.1016/S0168-9525(00)02024-2
Rosano, G. L., and Ceccarelli, E. A. (2014). Recombinant protein expression in Escherichia coli: advances and challenges. Front. Microbiol. 5:172. doi: 10.3389/fmicb.2014.00172
Schell, M. A. (1993). Molecular biology of the LysR family of transcriptional regulators. Annu. Rev. Microbiol. 47, 597–626. doi: 10.1146/annurev.mi.47.100193.003121
Solovyev, V., and Salamov, A. (2011). “Automatic annotation of microbial genomes and metagenomic sequences,” in Metagenomics and Its Applications in Agriculture, Biomedicine and Environmental Studies, ed. R. W. Li (Hauppauge, NY: Nova Science Publishers), 61–78.
Tabita, F. R., Hanson, T. E., Li, H., Satagopan, S., Singh, J., and Chan, S. (2007). Function, structure, and evolution of the RubisCO-like proteins and their RubisCO homologs. Microbiol. Mol. Biol. Rev. 71, 576–599. doi: 10.1128/MMBR.00015-07
Treangen, T. J., Abraham, A. L., Touchon, M., and Rocha, E. P. (2009). Genesis, effects and fates of repeats in prokaryotic genomes. FEMS Microbiol. Rev. 33, 539–571. doi: 10.1111/j.1574-6976.2009.00169.x
Tsai, Y. C., Lapina, M. C., Bhushan, S., and Mueller-Cajar, O. (2015). Identification and characterization of multiple rubisco activases in chemoautotrophic bacteria. Nat. Commun. 6:8883. doi: 10.1038/ncomms9883
van Keulen, G., Ridder, A. N., Dijkhuizen, L., and Meijer, W. G. (2003). Analysis of DNA binding and transcriptional activation by the LysR-type transcriptional regulator CbbR of Xanthobacter flavus. J. Bacteriol. 185, 1245–1252. doi: 10.1128/JB.185.4.1245-1252.2003
Varaljay, V. A., Satagopan, S., North, J. A., Witte, B., Dourado, M. N., Anantharaman, K., et al. (2016). Functional metagenomic selection of ribulose 1, 5-bisphosphate carboxylase/oxygenase from uncultivated bacteria. Environ. Microbiol. 18, 1187–1199. doi: 10.1111/1462-2920.13138
Wei, X., Sayavedra-Soto, L. A., and Arp, D. J. (2004). The transcription of the cbb operon in Nitrosomonas europaea. Microbiology 150, 1869–1879. doi: 10.1099/mic.0.26785-0
Witte, B., John, D., Wawrik, B., Paul, J. H., Dayan, D., and Tabita, F. R. (2010). Functional prokaryotic RubisCO from an oceanic metagenomic library. Appl. Environ. Microbiol. 76, 2997–3003. doi: 10.1128/AEM.02661-09
Keywords: autotrophic CO2 fixation, Calvin-Benson-Bassham (CBB) cycle, RubisCO gene regulation, LysR, CbbQ, CbbO, heterologous gene expression, non-native system
Citation: Böhnke S and Perner M (2017) Unraveling RubisCO Form I and Form II Regulation in an Uncultured Organism from a Deep-Sea Hydrothermal Vent via Metagenomic and Mutagenesis Studies. Front. Microbiol. 8:1303. doi: 10.3389/fmicb.2017.01303
Received: 12 April 2017; Accepted: 28 June 2017;
Published: 12 July 2017.
Edited by:
Ivan Berg, Universität Münster, GermanyReviewed by:
Kathleen Scott, University of South Florida, United StatesMarina G. Kalyuzhanaya, San Diego State University, United States
Copyright © 2017 Böhnke and Perner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mirjam Perner, bWlyamFtLnBlcm5lckB1bmktaGFtYnVyZy5kZQ==