 Diogo Piedade
Diogo Piedade José M. Azevedo-Pereira*
José M. Azevedo-Pereira*- Host-Pathogen Interaction Unit, iMed.ULisboa, Faculdade de Farmácia, Universidade de Lisboa, Lisboa, Portugal
MicroRNAs (miRNAs) are powerful regulators of gene expression and fine-tuning genes in all tissues. Cellular miRNAs can control 100s of biologic processes (e.g., morphogenesis of embryonic structures, differentiation of tissue-specific cells, and metabolic control in specific cell types) and have been involved in the regulation of nearly all cellular pathways. Inherently to their involvement in different physiologic processes, miRNAs deregulation has been associated with several diseases. Moreover, several viruses have been described as either, avoid and block cellular miRNAs or synthesize their own miRNA to facilitate infection and pathogenesis. Adenoviruses genome encodes two non-coding RNAs, known as viral-associated (VA) RNAI and VA RNAII, which seem to play an important role either by blocking important proteins from miRNA pathway, such as Exportin-5 and Dicer, or by targeting relevant cellular factors. Drastic changes in cellular miRNA expression profile are also noticeable and several cellular functions are affected by these changes. This review focuses on the mechanisms underlying the biogenesis and molecular interactions of miRNAs providing basic concepts of their functions as well as in the interplay between miRNAs and human adenoviruses.
Introduction
MicroRNAs (miRNAs) are small non-coding RNAs with approximately 22-nucleoides that regulate gene expression (Doench, 2003; Bartel, 2004; Kloosterman and Plasterk, 2006; Bushati and Cohen, 2007). They were discovered in Lee et al. (1993) and Wightman et al. (1993) in studies about the development of Caenorhabditis elegans. Gene expression control exerted by miRNAs is post-transcriptional as miRNAs regulate mRNA translation and stability in the cytoplasm (Valencia-Sanchez et al., 2006; Nilsen, 2007; Pillai et al., 2007). Human genome encodes more than 1000 miRNAs predicted to regulate over 60% of our genes (Friedman et al., 2009; Kozomara and Griffiths-Jones, 2014). Hence, miRNAs seem to participate in virtually every cellular process and changes in their expression are present in several human pathologies (Esquela-Kerscher and Slack, 2006; Krützfeldt and Stoffel, 2006; Chang and Mendell, 2007; Farazi et al., 2011).
Human adenoviruses were initially isolated from patients with acute respiratory infection but are now associated with many other pathological conditions such as gastroenteritis, keratoconjunctivitis, myocarditis, meningoencephalitis, cystitis, and hepatitis (Lenaerts et al., 2008). Despite its benign and self-limited course in imunocompetent host, adenoviral infections are particular severe in immunocompromised patient where a high morbidity and mortality could be observed (Lion, 2014). Adenoviruses have also been described as potential oncogenic viruses in rodents due do the presence of well-characterized oncogenes in viral genome, although its role in human carcinogenesis still basically unproven. Finally, human adenoviruses have recently been described as being able to establish life-long latent infections particularly in tonsilar and intestinal lymphocytes as well as in lung epithelial cells (Hogg, 2001; Garnett et al., 2009; Roy et al., 2011).
As described for many other viruses, adenoviral infection has a tremendous impact in cellular miRNA. Upon infection, host-cells miRNAs’ expression is severely altered together with viral blockage of key proteins acting in the silencing machinery and miRNA expression by adenoviruses (Carnero et al., 2011).
This review discusses the mechanisms underlying the biogenesis and molecular interactions of miRNAs providing basic concepts of their functions focusing in the interplay between miRNAs and adenoviruses.
MicroRNA Biogenesis
The canonical miRNA biogenesis starts in the nucleus with the transcription of miRNA genes by RNA polymerase II or in some cases RNA polymerase III (Lee et al., 2004). These transcripts – primary-miRNA (pri-miRNAs) – consist of 1–6 hairpin structures containing a stem of about 33-nucleotides, a terminal loop and flanking single stranded RNA sequences (Cai et al., 2004). They contain a 5′-end cap and a poly-A tail sequence and are processed to functional miRNAs in two steps catalyzed by Drosha and Dicer. Both enzymes belongs to the RNase III family and function in complexes with dsRNA-binding proteins (dsRBPs).
The first step takes place in the nucleus and is catalyzed by Drosha and DiGeorge syndrome critical region gene-8 (DGCR8) (Lee et al., 2003). Pri-miRNAs are cropped by the Drosha-DGCR8 complex, also known as Microprocessor Complex, to precursor miRNAs (pre-miRNAs), 70-nucleotide long hairpin structures (Yan et al., 2003; Denli et al., 2004; Gregory et al., 2004; Han, 2004; Landthaler et al., 2004; Zeng et al., 2005). The DGCR8, a dsRBP, specifically recognizes and binds pri-miRNAs, acting as a ruler for Drosha to cleave pre-miRNAs in specific sites to hinder pre-miRNAs (Han et al., 2006). Some miRNAs can bypass processing by Drosha-DGCR8 complex. These are known as mitrons, spliced introns that correspond exactly to pre-miRNAs, both in length and structure, thus not needing the excision step by the Microprocessor Complex (Berezikov et al., 2007; Okamura et al., 2007; Ruby et al., 2007).
The second step of canonical miRNA processing occurs in the cytoplasm, hence the need of nuclear exporting of pre-miRNAs. A RanGTP-dependent dsRNA binding protein known as Exportin-5 binds pre-miRNA in the nucleus and releases it in the cytoplasm upon GTP hydrolysis to GDP (Yi et al., 2003; Bohnsack et al., 2004; Lund et al., 2004). Once in the cytoplasm, pre-miRNAs are processed by Dicer (Bernstein et al., 2001; Hutvágner et al., 2001). This final processing step leads to a 21- to 25- mature dsRNA, known as miRNA::miRNA∗ complex, that is ready for being loaded into a RNA-induced silencing complex (RISC) (Zamore et al., 2000). At this point, the RISC loading complex (RLC) forms and miRNAs are assembled into miRNA-induced silencing complex (miRISC). Beside Dicer, miRISC contains the Argonaute proteins (Ago) and two dsRBPs: TRBP (TAR RNA-binding protein) and PACT (protein activator of PKR), that are implicated in miRNA functions together with pre-miRNA processing (Chendrimada et al., 2005; Gregory et al., 2005; Haase et al., 2005; Lewis Phillips et al., 2008; Lee et al., 2013).
miRNAs Target mRNAs Through Base Pairing
The binding between miRNAs and mRNAs is established through base-pair complementarity. In order to act as post-transcriptional regulators of gene expression, miRNAs must bind to their target(s) mRNA(s) through base-pair complementarity. In general, miRNAs pair imperfectly with target mRNAs in the 3′ untranslated region (3′ UTR). In this case, miRNAs must be perfectly and contiguously complementary in their nucleotides 2–8 at the 5′ end – the so called ‘seed’ region – in order to effectively supress their targets mRNAs (Doench, 2004; Brennecke et al., 2005). However, although important, the ‘seed’ region is not the only determinant for post-transcriptional repression of targeted mRNAs (Lewis et al., 2005; Grimson et al., 2007). In fact, we may define two main categories of miRNAs target sites: in the first category we include those that pair well in both 5′ and 3′ ends and those that require little or no additional 3′ pairing. In the second category, targets have weak 5′ base-pairing that is compensated by a strong pairing to the 3′ end (Brennecke et al., 2005). Apparently, additional 3′ pairing is required to induce miRNA-mediated target repression if complementarity with target mRNA is only based in the 5′ seed region.
Given the short length of the ‘seed’ region of miRNAs, it is obvious that one miRNA can target a large number of different mRNAs. This have been shown by microarray-analysis where the same miRNA can downregulate multiple mRNAs sharing one or more complementary sequences to the ‘seed’ region in their 3′ UTR (Lim et al., 2005). On the other hand, it is also been proved that more than one copy of the same miRNA or even several different miRNAs can pair with a single 3′ UTR from the target mRNA acting in a synergistic way. This synergic effect was demonstrated by adding multiple binding sites into a 3′ UTR: the result inhibition of translation was more efficient that expected from the sum of individual inhibition of each binding site (Doench, 2003). The mechanism of this synergism could be related with the mutual stabilization of different ribonucleoprotein complexes and/or with a more effective inhibition of translation (Doench, 2003). An obvious outcome of this cooperative interaction is that by regulating the degree of miRNA binding to the 3′ UTR of the mRNA will allow a cell to fine-tune mRNA expression.
miRNAs Mediate Gene Repression by Avoiding mRNA Translation and Through mRNA Decay
The mechanisms by which miRNAs modulates mRNA expression are not yet fully understood. Despite the absence of conclusive answers, some aspects of these mechanisms are consensual.
MicroRNAs can modulate mRNA expression in two distinct ways: (i) by inhibiting translation or (ii) by destabilizing mRNAs. Recent studies indicate that these two mechanisms may occur sequentially (Fabian et al., 2009; Guo et al., 2010; Bazzini et al., 2012; Béthune et al., 2012; Djuranovic et al., 2012; Larsson and Nadon, 2013), being the destabilization the last and apparently the predominant step in miRNA-mediated mRNA repression. There are several proposed mechanisms for miRNA-mediated inhibition of translation, ranging from initiation and elongation interference, through ribosomal drop-off. From these, inhibition of translation initiation seems to be the predominant mechanism.
Human miRNAs and their Functions
Considering that miRNAs can regulate up to 60% of human genes it is predictable that miRNAs are able to interfere with virtually all cellular pathways (Friedman et al., 2009). Given that a single miRNA can target 100s of genes (Lim et al., 2005), prediction of the functions of a single miRNA are made by identification of the most targeted pathways. Early reports have shown that miRNAs were able to target genes related with important cellular functions such as binding to nucleic acid, signal transduction and transcription regulation (Lewis et al., 2003). Not surprisingly, miRNA degradation and deregulation has been associated to many human pathologies such as pulmonary diseases (e.g., asthma, allergy, lung cancer) (Tomankova et al., 2010) and liver diseases (e.g., viral hepatitis, hepatocellular carcinoma) (Chen, 2009).
The deregulation of normal miRNAs expression or repression can influence several biologic processes, including carcinogenesis. One good example is hsa-miR-155, a miRNA associated with oncogenic processes when upregulated. However, hsa-miR-155 is important for proper immune system functioning as it regulates immune cells activation, growth and inflammatory cytokines release (Mashima, 2015). It was demonstrated that hsa-miR-155 has critical roles in both innate and adaptive immune responses, as well as in the balance between immune response and immune tolerance. Several studies show that hsa-miR-155 regulates (i) the differentiation of T-CD4+ lymphocytes into different subsets of helper T-cells (i.e., Th1, Th2, and Th17), (ii) the development of regulatory T-cells, (iii) the activation of T-CD8+ lymphocytes, and (iv) the differentiation of B lymphocytes and antibody production (Seddiki et al., 2014). Besides hsa-miR-155, many other miRNAs have been associated with T-cell activation, differentiation and expansion, namely hsa-miR-181a, hsa-miR-146a, hsa-miR-182, hsa-miR-17-92, and hsa-miR-125 (Baumjohann and Ansel, 2013).
Bacterial infections have a tremendous impact in cellular miRNA. For example, upon Mycobacterium tuberculosis (Mtb) infection, host-cells miRNAs’ expression is severely altered with potential implications in survival and pathogenesis of Mycobacterium tuberculosis (Bettencourt et al., 2015). Helicobacter pylori was also referred has being able to interfere with hsa-miRNA-155 expression in gastric epithelial cells (Xiao et al., 2009).
Interestingly, miRNAs can have different and somehow opposite functions depending on the cellular context. The hsa-miR-125 is one of these miRNAs. In fact, it can act as an oncogenic or as tumor suppressive miRNA in prostate and breast cancer, respectively (Sun et al., 2013). Hsa-miR-125 is known to target diverse pathways such as differentiation, apoptosis, immune response, cell proliferation, and metastasis in carcinogenic processes (Sun et al., 2013). Lastly, some miRNA clusters cooperate for the same function. It is the case of hsa-miR17/92 cluster, also known as oncomir1, which is also involved in oncogenic processes. This miRNA cluster encodes six miRNAs that cooperatively target PTEN (phosphatase and tensin homolog), SMAD, TGFBR2 (transforming growth factor, beta-receptor II) and P21 genes, which are related to cell growth and proliferative signaling (Fuziwara and Kimura, 2015).
Role of Cellular miRNAs in Viral Infections
Interactions between virus and host are intricate and complex. Due to their intracellular replication cycle it is no surprise that virus evolved in order to create mechanisms allowing them to use or avoid host-encoded miRNAs to infect, survive and replicate in host cells (Grundhoff and Sullivan, 2011; Harwig et al., 2014).
Mutual interference mechanisms between viruses and host-cell’s miRNA machinery have been described. To generate a more favorable cellular environment or to regulate their own miRNAs, viruses can (i) avoid cellular miRNAs targeting viral mRNAs (Cullen, 2013), (ii) block or impair the miRNA pathway by interacting with some key proteins (Lu and Cullen, 2004; Bennasser et al., 2006), (iii) synthesize their own miRNA (Grundhoff and Sullivan, 2011; Harwig et al., 2014), (iv) or make use of cellular miRNAs to their favor (Luna et al., 2015). Conversely, host-cell’s endogenous miRNAs are also able to target viral mRNAs (Lecellier et al., 2005; Delorme-Axford et al., 2013; Bai and Nicot, 2015).
In general, cellular miRNAs repress viral gene expression resulting in decreased viral replication; this viral repression can contribute to viral latency. However, cellular miRNAs can also increase viral replication and some viruses, such as Hepatitis C Virus (HCV), seem to depend on cellular miRNAs to replicate efficiently. In fact, although some cellular miRNAs (e.g., hsa-miR-181c, hsa-miR-196, hsa-miR-199a, hsa-miR-488, and hsa-miR-let-7b) interact directly with viral genome inhibiting HCV replication (Piedade and Azevedo-Pereira, 2016b), one liver-specific miRNA, hsa-miR-122, binds to 5′ UTR region of HCV RNA enhancing viral genome replication and accumulation in infected cells (Jopling et al., 2005).
Several host-cells’ miRNAs have been described with the capacity to block different viral replication steps. Although some of these data are still controversial, there is a growing body of evidence pointing to an antiviral mechanism mediated by miRNA. These mechanisms could involve a direct interaction with a viral protein or an indirect interaction with some cell protein required to a certain viral replication step. Several examples can be given within viral infections:
(i) In HIV-1 infection, several host miRNAs have been described able to interact with HIV host dependency factors (HDFs). These HDFs are key players in HIV-1 cycle and their repression generally results in repressed viral replication (reviewed in Piedade and Azevedo-Pereira, 2016b). The activation of TLR3 and TLR4 during HIV replication cycle upregulates hsa-miR-155 that targets and repress ADAM10 (disintegrin and metalloproteinase domain-containing protein 10), Nup153 (nucleoporin Mr 153,000), TNPO3 (transportin 3) and LEDGF/p75 (lens epithelium-derived growth factor), all of them important HDFs involved in nuclear import and integration of HIV genome (Swaminathan et al., 2012; Seddiki et al., 2014). Similarly, VprBP (Vpr binding protein), a cellular factor crucial for Vpr-mediated G2 cell-cycle arrest and proper viral replication, is targeted by hsa-miR-1236 (Ma et al., 2014).
Additionally, host cell miRNAs were described targeting HDFs important for HIV-1 Tat-mediated LTR activation: PCAF (P300/CBP-associated factor), a histone acetylase is target by hsa-miR-20a and hsa-miR-17-5p (Triboulet et al., 2007); purine-rich element binding protein alpha (PUR-α), is targeted by hsa-miR-15a, hsa-miR-15b, hsa-miR-16, hsa-miR-20a, hsa-miR-93, and hsa-miR-106b (Shen et al., 2012); and cyclin T1 is repressed by hsa- miR-198 and hsa-miR-27b (Sung and Rice, 2009; Chiang et al., 2011).
(ii) Placental trophoblasts are highly resistant to infection by several and unrelated viruses, including human cytomegalovirus (HCMV) herpes simplex virus-1, poliovirus, coxsackievirus B3, vesicular stomatitis virus, and vaccinia virus (Delorme-Axford et al., 2013). This resistance is conferred by a group of miRNAs from a cluster located in chromosome 19. Apparently these miRNAs are expressed in human placental trophoblasts and transferred to non-placental cells by an exosome-mediated mechanism, rendering recipient cells resistant to viral infection.
(iii) Cellular miRNAs can also interfere with viral infection by regulating innate response after TLR sensing of virus-associated nucleic acid. Hsa-miR-126 was found to regulate the expression of TLR7 and TLR9 in plasmacytoid dendritic cells (pDC), in addition to other molecules involved in signaling pathways crucial for type I IFN-mediated innate immune response (Agudo et al., 2013). This regulation of pDC response could be of paramount importance during HIV interaction with dendritic cells after sexual mucosal transmission (Barroca et al., 2014).
(iv) In herpesviruses infections several cell-encoded miRNAs control latent/lytic cycles. For example, hsa-miR-200b and hsa-miR-429 regulate Epstein–Barr virus (EBV) and HCMV switch from latent to lytic infection (reviewed in Piedade and Azevedo-Pereira, 2016a).
(v) Another example of cell-encoded miRNAs that may promote viral persistence and immune escape by silencing the expression of viral genome is hsa-miR-28-3p. The target sequence of this cellular miRNA is located within the gag/pol gene of human T-lymphotropic virus-1 (HTLV-1) and reduces viral replication and gene expression (Bai and Nicot, 2015). Interestingly, hsa-miR-28 is upregulated by IFN response revealing a mechanism where innate immune response helps viral persistence by reducing virus dissemination to neighboring cells, diminishing local inflammation, and enabling the survival of infected cells.
(vi) Finally, cellular miRNAs were predicted to target several HIV-1 transcripts, thus reducing viral replication (Piedade and Azevedo-Pereira, 2016b). However, these predictions not always translate in effective repression due the multiplicity of factors that are involved in miRNA-mediated gene regulation. For example, HIV-1 encodes RNA interference silencing suppressors that interfere with miRNA pathways: HIV-1 Tat suppresses Dicer function, and Nef viral protein decreases miRNA function by directly binding to Ago2. Noteworthy, Nef stimulates the secretion of several host-cell’s miRNAs in Nef-containing exosomes. Additionally, HIV-1 is able to evade miRNAs by two important mechanisms: the secondary structure of HIV transcripts (Westerhout et al., 2005; Watts et al., 2009), and the outstanding genetic variability both in length and sequence (Bandaranayake et al., 2010). Both strategies render HIV-1 RNAs resistant to host miRNAs.
Role of Virus-Encoded miRNAs in Pathogenesis of Viral Infections
Some viruses encode miRNAs that have an important role in infection and pathology. The functions of these viral miRNAs vary with different viruses and in different tissues but some pathways are often targeted, suggesting that virus evolved separately toward similar purposes. Although the overall influence of miRNAs in the pathogenesis of viral infection is beyond the scope of this review, it is important to highlight that viral miRNAs were reported to regulate genes related with immune response, apoptosis, cell cycle control, differentiation, and intracellular trafficking. As many of these altered pathways are of the uttermost importance for cellular homeostasis, their alterations largely contribute for viral pathology.
One of the most noteworthy pathological events triggered by viral miRNAs is oncogenesis. In fact, oncogenic viruses such as EBV and Kaposi’s sarcoma-associated herpesvirus (KSHV) encode viral miRNAs that can be directly linked with development of malignancies (reviewed in Zhu et al., 2013; Piedade and Azevedo-Pereira, 2016a). For example, different EBV-encoded miRNAs (BART3-5p, BART7, and BART19-3p) target several tumor-suppressors genes facilitating typical B-cell transformation (Wong et al., 2012; Lei et al., 2013), and several KSHV miRNAs cooperatively regulate cytokine expression, cell survival, and cellular transcription factors to facilitate infection and oncogenesis while avoiding the host immune system (Piedade and Azevedo-Pereira, 2016a).
Viral miRNAs can also target viral transcripts, particularly in herpesviruses infections. Repression of viral genes is an efficient strategy to maintain latency and to keep viral loads to minimum, avoiding detection by host defenses. Viral miRNAs can also be considered important switches between lytic and latent infection and their levels in the host cell help to determine the evolution of the viral infection.
Another mentionable feature of viral miRNAs is that they seem to have evolved to target conserved regions of the host genes and even specific sequences targeted by host miRNAs. These strategies avoid potential mutations in the recognition sequences of target mRNAs, ensuring viral miRNA function. Thus, these evolutionary paths are suggestive of the importance of viral miRNAs for establishment and prevalence of viral infection.
Human Adenovirus Encode Virus-Associated RNA with Pro-Viral Functions
Human adenoviruses genome encodes two non-coding RNAs, transcribed by cellular RNA polymerase III known as viral-associated (VA) RNAs (Figure 1). These two VA RNAs have similar lengths, VA RNAI is 157–160 nucleotides long and is found in every adenovirus while VA RNAII is 158–163 nucleotides long and is found in 80% of adenoviruses, including the serotype 5 (Vachon and Conn, 2015). Although the primary nucleotide sequence of VA RNAs may differ between adenoviruses, they show a highly conserved secondary structure (Ma and Mathews, 1996). The secondary structure can be divided in three structural domains (Figure 2): (i) Terminal Stem, (ii) Central Domain, and (iii) Apical Stem (Coventry and Conn, 2008). These different structures are associated with distinct functions of VA RNAs as they interact and bind different cellular factors: the Apical Stem binds to PKR; the Central Domain inhibit PKR; and the Terminal Stem binds to Exportin-5 (Figure 2). These functions are essential for proper viral replication. Deletion of both VA RNAs caused a 60-fold decrease in viral replication rate while deletion of VA RNAI showed a reduction of 10–20 fold (Thimmappaya et al., 1982; Bhat and Thimmappaya, 1984). Interestingly, deletion of VA RNAII did not cause a measurable reduction of viral viability (Thimmappaya et al., 1982) suggesting that VA RNAII absence can be partially compensated by VA RNAI and might exert non-essential functions while VA RNAI is the predominant pro-viral VA RNA (Carnero et al., 2011).
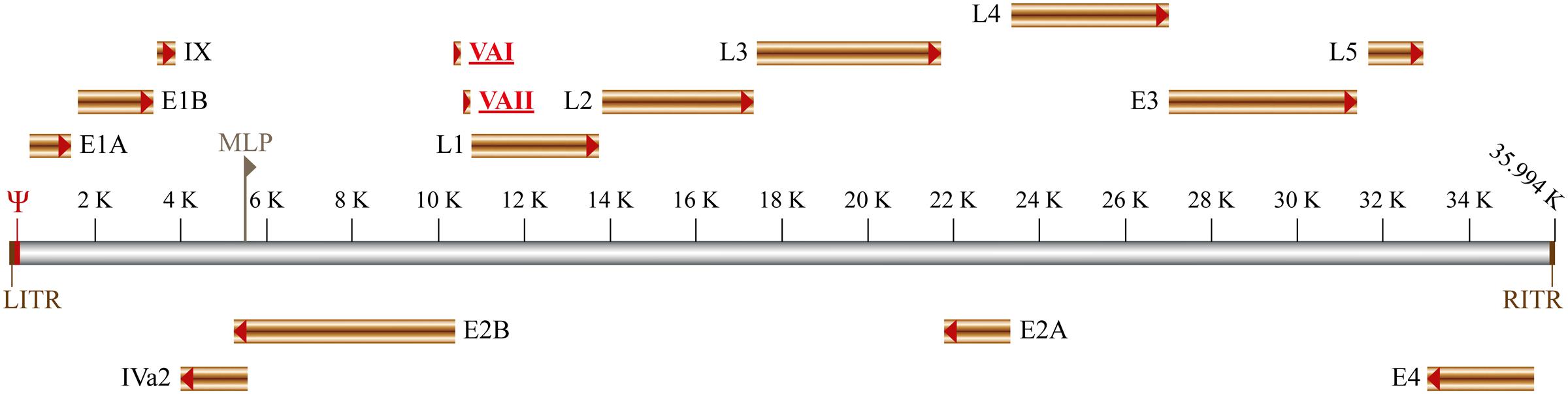
FIGURE 1. Schematic representation of adenovirus genome organization. The early and late genes are shown as well as the left and right Inverted Terminal Repeat (RITR and LITR, respectively). Both viral DNA strands (represented in gray) are transcribed: in the top are indicated the genes transcribed from the ‘rightward’ reading strand (E1A, E1B, IX, L1-L5, and E3), whereas the genes transcribed from the ‘leftward’ reading strand are shown bellow (E4, E2A, E2B, and IVa2). The packaging signal (Ψ) and the major late promoter (MLP) are also indicated. The viral-associated non-coding RNAs I and II (VAI and VAII respectively) are represented in red and underlined.
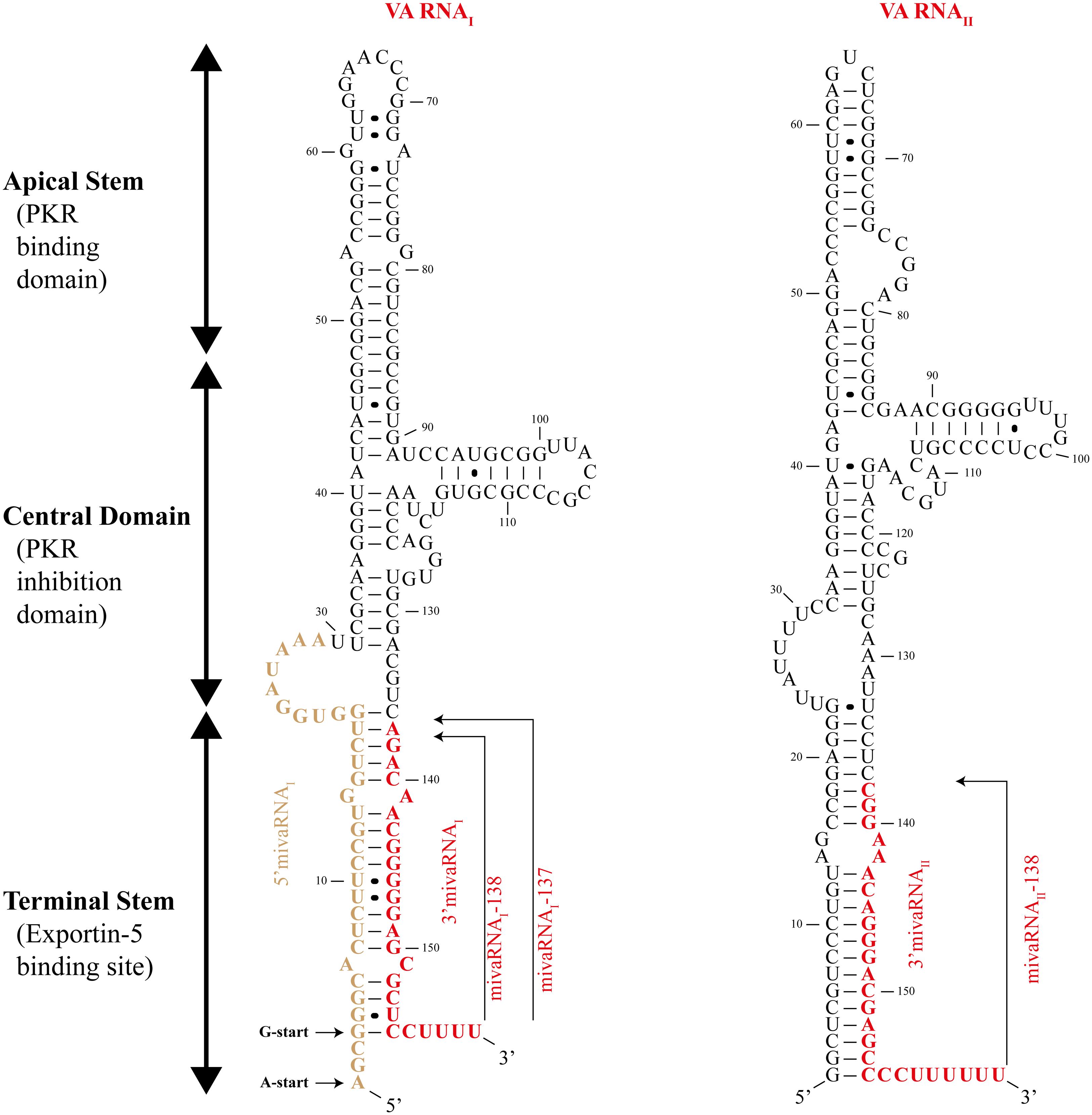
FIGURE 2. Structure of adenovirus virus-associated RNAs. Schematic drawing showing the structure of adenovirus virus-associated (VA) RNAI (left) and VA RNAII (right). The three structural domains – apical stem, central domain, and terminal stem – are indicated. The viral miRNAs (mivaRNAI-138, mivaRNAI-137, and mivaRNAII-138) are represented in red. The VA RNAI could start from two different sites indicated by the A-start and G-start arrows. Black circles between nucleotides represent non-Watson-Crick base pairing.
The first identified function of VA RNAI (Table 1) was the inhibition of innate immune response mediated by the protein double-stranded RNA-activated kinase (PKR) (Kitajewski et al., 1986). This interferon-induced kinase is activated by dsRNA (including viral RNA) and phosphorylates the α subunit of the eukaryotic initiation factor 2 alpha (eIF2α) which in turn prevents the eIF2β of being recharged with GTP, avoiding the formation of the 43S pre-initiation complex following translation initiation (Krishnamoorthy et al., 2001; Cole, 2007). VA RNAI avoids translational blockage by inactivating PKR, allowing the production of viral proteins. The inactivation of PKR is mediated by the central domain of VA RNAI, while the apical domain is required for efficient PKR binding (Launer-Felty and Cole, 2014).
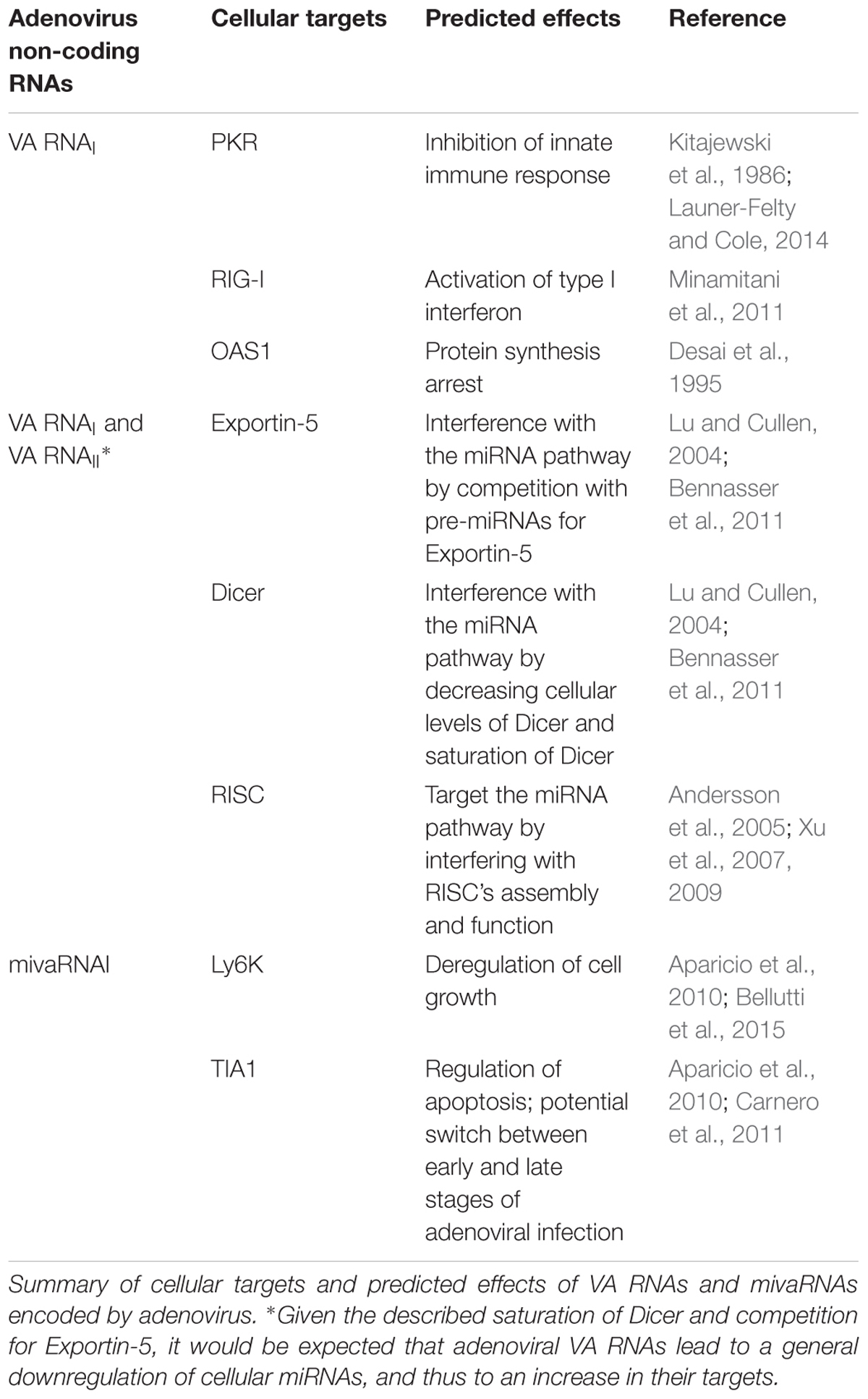
TABLE 1. Adenovirus non-coding RNAs.
Besides PKR, VA RNAI also interacts with other innate immune system factors (Table 1) such as retinoic acid-inducible gene 1 (RIG-I) (Minamitani et al., 2011) and these interactions seem to activate type I interferon. This may seem contradictory with PKR inhibition by VA RNAI and is hypothesized that other viral factors are able to inhibit RIG-I (Punga et al., 2013). Similarly, the 2′-5′ oligoadenylate synthase-1 (OAS1), another dsRNA-sensor, is also activated by VA RNAI (Desai et al., 1995). OAS1 activates RNase L pathway that leads to viral protein synthesis arrest through the degradation of viral RNA (Table 1). Again, this pathway would enhance virus restriction in conflict with PKR inhibition. Some data may help solve this contradiction: first, VA RNAI binding and activation of OAS1 is very inefficient when compared with a dsRNA of the same size (Desai et al., 1995). Furthermore, truncated VA RNAI containing apical stem and central domain – thought to be the result of Dicer cleavage, as will be described – shows an increased affinity for OAS1. These observations might indicate that in the course of adenoviral infection, the truncated form of VA RNAI, acting as a pseudo-inhibitor, will outcompete with full-length VA RNAI leading to a block of OAS1 activation, thus inhibiting several innate immune response pathways (Meng et al., 2012).
VA RNAs Can Overload Exportin-5 and Dicer Disrupting Cellular miRNA Functions
Despite being well-known for its pro-viral activity, VA RNAs can also play an important role in the deregulation of host miRNAs (Table 1). VA RNAs interfere with miRNA pathway on three different levels: (i) competition with pre-miRNAs for Exportin-5, (ii) saturation of Dicer, and (iii) interference with RISC’s assembly and function.
Upon transcription by RNA polymerase III, VA RNAI is exported from the nucleus to the cytoplasm by Exportin-5. This RanGTP-dependent dsRNA binding protein recognizes VA RNAI secondary structure (more precisely, the Terminal Stem; Figure 2) which is similar to pre-miRNAs (Gwizdek et al., 2003). The VA RNAI-Exportin-5 binding is highly efficient and Exportin-5 is thought to be quickly saturated by the VA RNAI leading to the blockage of pre-miRNAs export from the nucleus (Lu and Cullen, 2004). Furthermore, Exportin-5 is also responsible for export Dicer mRNA into the cytoplasm, thus VA RNAI saturation of Exportin-5 can further interfere with miRNA pathway through the decrease of cellular levels of Dicer (Bennasser et al., 2011).
Once in the cytoplasm, VA RNAs bind and sequester Dicer preventing it from maturing cellular pre-miRNAs (Lu and Cullen, 2004). VA RNAs are also processed by Dicer and originate two distinct fragments: Apical Stem-Central Domain (AS-CD), and Terminal Stem which originates viral miRNAs, known as mivaRNAs (Figure 2), that will be loaded into RISC (Andersson et al., 2005; Aparicio et al., 2006; Sano et al., 2006; Xu et al., 2007). AS-CD fragments could not be detected probably due to their instability after Dicer processing (Aparicio et al., 2006). Interestingly, VA RNA is inefficiently processed by Dicer as only 2–5% of total VA RNAI is cleaved (Aparicio et al., 2006). However, there are approximately 108 molecules of VA RNAI per cell, meaning that over 106 molecules of mivaRNAs are perfectly functional and able to act as miRNAs when loaded into RISC (Andersson et al., 2005; Aparicio et al., 2006). Despite having 20-fold less expression than VA RNAI, VA RNAII seems to be the preferential substrate for Dicer processing originating twice the mivaRNAs compared to VA RNAI (Xu et al., 2007).
The unprocessed VA RNAI can give raise to distinct mivaRNAs depending on the cleavage site of Dicer, being mivaRNAI-137 and mivaRNAI-138 (named after their starting nucleotide in the VA RNAI molecule) the two most abundantly produced (Xu et al., 2007). Furthermore, the transcription of VA RNAI could start from two different sites giving rise to VA RNAI(G) and VA RNAI(A) – according to their first nucleotide (Figure 2) – and thus to even more mivaRNAs (Xu et al., 2009).
It was demonstrated that after being processed by Dicer, the 22 nucleotide-long mivaRNAs derived from VA RNAs can incorporate RISC (Lu and Cullen, 2004; Andersson et al., 2005; Aparicio et al., 2006; Sano et al., 2006). Furthermore, mivaRNAII, derived from VA RNAII, was found associated with polyribosomes, indicating the ability to regulate gene expression (Xu et al., 2007). Another compelling evidence pointing to mivaRNAs as functional miRNAs, was the finding that 80% of RISC complexes contain mivaRNAs in late adenoviral infections (Xu et al., 2007), though a more recent report points to a smaller proportion of RISC occupied by mivaRNAs (Bellutti et al., 2015). Despite these somehow contradictory evidences, the sheer numbers of mivaRNAs and their ability to be incorporated into RISC is thought to be enough to interfere with cellular mRNA. As mentioned above, mivaRNAII seems to be the major mivaRNA to be associated with RISC (Andersson et al., 2005; Aparicio et al., 2006), yet the seed sequences of mivaRNA-containing RISC did not match the ones from mivaRNAII (Bellutti et al., 2015) and mivaRNAI was shown to reduce more efficiently the expression of a reporter gene included in constructs containing the mivaRNAI target regions (Bellutti et al., 2015). These findings are consistent with reports of asymmetrical loading of mivaRNAs into RISC. In fact, 5′- and 3′-end strands of mivaRNAI(A) and mivaRNAI(G) are not equally incorporated into RISC as the 5′ strand of mivaRNAI(G) is not found in RISC (Andersson et al., 2005; Aparicio et al., 2006).
Potential mivaRNA targets were evaluated through different approaches (Aparicio et al., 2010; Bellutti et al., 2015). Based on the results obtained, several genes with essential cellular functions were deemed direct or indirect targets of mivaRNAs (Table 1). The cellular functions apparently affected by mivaRNAs are cell signaling (seven genes), cell growth and apoptosis (nine genes), DNA transcription or repair (nine genes) and RNA metabolism (five genes) (Aparicio et al., 2010). One confirmed target of mivaRNA is the protein Ly6K (lymphocyte antigen 6 complex, Locus K) (Bellutti et al., 2015), a protein associated with cell growth and implicated in several cancers such as lung, breast or head and neck (Vachon and Conn, 2015). Another target of mivaRNAs is T-Cell-Restricted Intracellular Antigen-1 (TIA1), a RNA-binding protein, regulator of proapoptotic molecules (Aparicio et al., 2010). This protein is associated with stress granules and binds to U-rich sequences in mRNAs, abundant in adenoviral transcripts, increasing splicing or blocking translation (Carnero et al., 2011). In fact, it is hypothesized that downregulation of this protein may be related with alternative splicing of adenoviral transcripts, such as E1A, being a potential switch between early and late stages of infection (Carnero et al., 2011). Together, these two genes provide an example of pro-viral function of mivaRNAs in infected cells. However, whether the genes targeted by mivaRNAs are essential for viral infection or not is still uncertain (Kamel et al., 2013; Vachon and Conn, 2015). Besides targeting cellular mRNAs, it was expected that mivaRNAs could also target viral transcripts in order to regulate viral life cycle, similar to other viruses, such as herpesviruses (Piedade and Azevedo-Pereira, 2016a). Remarkably, this seem not be the case with adenoviruses since bioinformatics scanning of viral genome for potential mivaRNA targets found no matches (Aparicio et al., 2006).
Adenoviruses code for small RNAs other than VA RNAs and respective mivaRNAs. Interestingly, some of these small RNAs overlap mivaRNA sequences and are synthesized prior to VA RNAs expression. The different patterns of expression between viral small RNAs and mivaRNAs probably signifies different functions as none of the small RNAs detected had the canonical size of miRNAs (Zhao et al., 2013).
Adenovirus Deregulate Cellular miRNAs in Order to Favor Viral Replication
Viral interference with miRNAs goes beyond mivaRNAs effect on putative target expression. In fact, adenoviruses are able to deregulate cellular miRNA levels (Qi et al., 2010; Su et al., 2010; Zhao et al., 2015). Given the large amount of VA RNAs expressed and the subsequent blockade of Dicer and Exportin-5, it would be expected that adenoviral infection lead to a general downregulation of cellular miRNAs, and thus to an increase in their targets. However, this does not occur during the first hours of adenovirus infection since VA RNA accumulation is not immediate and other factors, both cellular and viral, contribute to differential expression of miRNAs (Qi et al., 2010; Zhao et al., 2015). The differential expression of cellular genes appears to follow four different stages upon infection by adenoviruses. The first stage goes from 0 to 12 h post-infection, when the viral gene expression starts and cell growth is inhibited as an host defense against the virus (Granberg et al., 2006; Zhao et al., 2007). The second stage goes from 12 to 24 h post-infection and is marked by expression of E1A viral gene, which contributes largely for changes in cell’s environment that contribute to optimal viral replication. Most of downregulated genes at this stage are related to cell cycle regulation, cell proliferation, and antiviral response. The third stage follows until 36 h post-infection when replication of the viral genome takes place upon full control of cellular metabolism. The fourth and last period is characterized by a general and marked deregulation of cellular genes. At this stage, more than 3700 genes were identified as being deregulated by at least twofold their normal expression (Zhao et al., 2012).
Not surprisingly, deregulation of miRNA also appears to follow this stepped progression, with miRNA expression varying from up to downregulation and vice versa (Qi et al., 2010; Zhao et al., 2015). A recent study reported changes in 175 miRNAs expression above 1.5-fold normal expression (Zhao et al., 2015). This study showed that most deregulated miRNAs during early infection (<24 h post-infection) were upregulated. From those, the most highly expressed were known to target either tumor suppressor or immune response genes. Namely, hsa-miR-22, a cell growth inhibitor, hsa-miR-181b, hsa-miR-320 and hsa-let-7e, all tumor suppressor miRNAs, were all upregulated in the first 6 h post-infection as part of the host-cell immune response to the virus. Another interesting miRNA upregulated during the first 6 h post-infection was hsa-miR-155, a well-studied oncomir, that is hypothesized to play a role in host antiviral response (Zhao et al., 2015). After 12 h of infection a second wave of cellular antiviral miRNAs are expressed. Among these overexpressed miRNAs are hsa-miR-29 and hsa-let-7d, both involved in immune response. Simultaneously, oncogenic miRNAs, such as hsa-miR-21 and miRNAs from hsa-miR-17/92 cluster (oncomir-1) are downregulated. Also at this point, the first miRNAs to be regulated by the virus, opposing cellular efforts against infection, is detected. Oncogenic miRNA hsa-miR-574 is upregulated and tumor suppressor miRNAs such as hsa-let-7i, hsa-miR-34a, hsa-miR-185 and hsa-miR-31 are downregulated (Zhao et al., 2015). The involvement of E1A, VA RNAs and respective mivaRNAs are the probable cause for this, apparently contradictory, pattern of expression.
During the third period of viral infection, deregulation of cellular miRNAs reaches a turning point when viral transcripts completely overcome host defenses. At this stage, 24 h post-infection, the switch between early and late infection occurs with an apparent control of cellular pathways by viral proteins and VA RNAs. The cell-encoded miRNA expression profile changes and almost all cellular miRNAs that were previously overexpressed become downregulated. Among them are hsa-miR-22, hsa-miR-181b and hsa-miR-320 that were overexpressed at 6 and 12 h post-infection as part of the host immune response to the virus. Other cellular miRNAs associated with tumor suppressive and immune-modulating functions, such as hsa-miR-143, hsa-let7a/b and hsa-miR-29a, were downregulated at this point of infection after being highly expressed during early stages. Despite the general downregulation of miRNAs, some were found upregulated at 24 h post-infection. From these, the vast majority were transiently expressed being downregulated soon after, at 36 h post-infection, by late viral transcripts. Two cell-encoded miRNAs, hsa-miR-27a and hsa-miR-125b (as well as hsa-miR-27b and hsa-miR-125a, although in less extent), are particularly important to refer in this context given their role as oncogenic or tumor suppressive miRNAs, depending on the tissue type, and their interference with viral replication during infection by other viruses such as HCV, HCMV, and HPV. These cellular miRNAs are upregulated at 24 h post-infection and are suppressed during late stages of adenoviral infection as mentioned above. Together with hsa-miR-199a and hsa-miR-140, known for their impairment of tumor growth, they may stand as host cell last line of antiviral defense. Interestingly some oncomirs were found downregulated at this time point, something not expected given the miRNA profile. Whether the suppression of these miRNAs, such as hsa-miR-193 and hsa-miR-221, is a consequence of viral activity or stands as yet another cellular defense against the virus remains to be clarified (Zhao et al., 2015).
After 36 h of infection, only five miRNAs were upregulated, all of them known as being oncogenic. From the remaining deregulated miRNAs, the most significant were hsa-miR-23/27 cluster and hsa-let-7, hsa-miR-30, and hsa-miR-376 families. These combination of miRNAs have established or predicted properties as tumor suppressors, cell proliferation inhibitors or apoptosis inducers (Zhao et al., 2015). Interestingly, a previous study found only 80 differentially expressed miRNAs after 72 h of infection (Qi et al., 2010). The most noteworthy miRNAs referred in both studies were hsa-miR-27a/b, hsa-miR-30a/b/c, hsa-miR-125a/b, hsa-miR-181b, and hsa-let-7e (Qi et al., 2010; Zhao et al., 2015). All of them have important roles as antiviral, tumor suppressive or oncogenic miRNAs. However, since there are several possible targets for these deregulated cellular miRNAs, and some are differentially expressed depending the tissue or the cell type (e.g., cancer cell vs. non-transformed cell), it is difficult to anticipate the outcome of their decreased or increased expression.
Effect of Host Cell miRNAs on Adenoviral Infection
As for many other viruses, adenovirus replication could also be inhibited by cell-encoded miRNA. Considering the basic mechanisms of viral evolution, those cellular miRNA that are specifically deregulated during adenovirus infection should constitute those that have more pronounced effects on viral replication. Not surprisingly, hsa-miR-27 was recently described as a potent adenovirus inhibitor (Machitani et al., 2017). Adenovirus replication seems to be inhibited via the suppression of SNAP25 (synaptosomal-associated protein, 25 kDa) – associated with membrane fusion (Sudhof and Rothman, 2009) – and TXN2 (thioredoxin, mitochondrial, also known as thioredoxin-2) – a redox-active protein playing important roles in the regulation of mitochondrial redox and the production of reactive oxygen species with direct effects on redox balance, cell growth, and apoptosis (Tanaka, 2002; Conrad et al., 2004).
The post-transcriptional silencing of both genes by hsa-miR-27, lead to efficient suppression of adenovirus replication by two distinct mechanisms: silencing of SNAP25 interferes with adenovirus entry into target cells, while TXN2 suppression hampers adenovirus replication through a G1 arrest of cell cycle (Machitani et al., 2017). Due to the role of SNAP25 in endocytic intracellular trafficking and since adenovirus enter the cell through endocytosis (Meier and Greber, 2003) it is conceivable that suppression of SNAP25 expression will affect virus entry. Conversely, adenovirus replication relies on the induction of cell cycle transition from G0 or G1 to S phase in order to create optimal conditions for viral replication (Ben-Israel, 2002). Therefore, the arrest on G1 phase due to TXN2 suppression predictably imposes a non-productive adenoviral infection.
Conversely, cellular miRNAs can also induce adenovirus replication. Hsa-miR-26b has been identified as a potent stimulator of human adenovirus serotype 5 (Ad5) replication and propagation in prostate cancer cells (Hodzic et al., 2017). Apparently, an hsa-miR-26b-dependent NF-kB inhibition is one of the mechanisms underlying the enhancement of adenovirus replication. The identification of a cell miRNA promoting adenovirus replication in cancer cells is particularly important since adenoviruses have been used as tumor-lysing therapeutics (oncolytic virotherapy) as well as immune-stimulating vectors aiming to induce an immunogenic tumor cell death (Lichty et al., 2014; Rosewell Shaw and Suzuki, 2016). Although it is difficult to fully anticipate all the consequences of miRNA-mediated gene suppression, any mechanism that may potentiate the replication of these adenovirus-based vectors should be considered for the improvement of such therapies.
Conclusion
Given the complexity of the interaction between host-cell and virus is difficult to realize all the players during viral infection. Adenoviruses evolved complex mechanisms to overcome host defenses. One of the most remarkable of these mechanisms is the ability to interfere with host miRNAs (Carnero et al., 2011; Vachon and Conn, 2015). VA RNAs and derived mivaRNAs seem to play an important role either by blocking important proteins from miRNA pathway, such as Exportin-5 and Dicer, or by targeting relevant cellular factors, such as TIA1 and cell-growth protein Ly6K (Vachon and Conn, 2015). Drastic changes in cellular miRNA expression profile are also noticeable and several cellular functions are affected by these changes as confirmed by transcriptome analysis and miRNA target prediction (Zhao et al., 2012, 2015). Most of the differentially expressed miRNAs are related to cell proliferation, apoptosis, cell signaling and immune response, contributing to a more favorable host environment for viral replication (Zhao et al., 2015). It will be interesting to understand if mivaRNAs and VA RNAs have an important role in this deregulation of cellular miRNAs or if the effect of viral miRNAs is restricted to gene suppression. In addition, it is important to clarify the importance of mivaRNAs in viral pathogenesis. The expression of miRNA and miRNA-mediated regulation of gene expression is relatively slow, being only noticeable after 24 h of infection (Qi et al., 2010; Zhao et al., 2015). Moreover, it is conceivable that miRNA regulation by adenoviruses could be more important for persistent rather than lytic infection, as it has been suggested by the association of VA RNAs with latent infection by adenoviruses (Xu et al., 2007). Finally, some cell miRNAs impact adenovirus replication cycle, either with pro-viral or antiviral activities. As the complex interactions between adenovirus and miRNAs are being uncovered, a better understanding should be achieved to fully elucidate the role of cell-encoded miRNAs in the regulation of adenovirus infection, particularly in the context of oncolytic therapy using adenovirus-based vectors.
Author Contribution
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Agudo, J., Ruzo, A., Tung, N., Salmon, H., Leboeuf, M., Hashimoto, D., et al. (2013). The miR-126–VEGFR2 axis controls the innate response to pathogen-associated nucleic acids. Nat. Immunol. 15, 54–62. doi: 10.1038/ni.2767
Andersson, M. G., Haasnoot, P. C. J., Xu, N., Berenjian, S., Berkhout, B., and Akusjärvi, G. (2005). Suppression of RNA interference by adenovirus virus-associated RNA. J. Virol. 79, 9556–9565. doi: 10.1128/JVI.79.15.9556-9565.2005
Aparicio, O., Carnero, E., Abad, X., Razquin, N., Guruceaga, E., Segura, V., et al. (2010). Adenovirus VA RNA-derived miRNAs target cellular genes involved in cell growth, gene expression and DNA repair. Nucleic Acids Res. 38, 750–763. doi: 10.1093/nar/gkp1028
Aparicio, O., Razquin, N., Zaratiegui, M., Narvaiza, I., and Fortes, P. (2006). Adenovirus virus-associated RNA is processed to functional interfering RNAs involved in virus production. J. Virol. 80, 1376–1384. doi: 10.1128/JVI.80.3.1376-1384.2006
Bai, X. T., and Nicot, C. (2015). miR-28-3p is a cellular restriction factor that inhibits human T cell leukemia virus, type 1 (HTLV-1) replication and virus infection. J. Biol. Chem. 290, 5381–5390. doi: 10.1074/jbc.M114.626325
Bandaranayake, R. M., Kolli, M., King, N. M., Nalivaika, E. A., Heroux, A., and Kakizawa, J. (2010). The effect of clade-specific sequence polymorphisms on HIV-1 protease activity and inhibitor resistance pathways. J. Virol. 84, 9995–10003. doi: 10.1128/JVI.00505-10
Barroca, P., Calado, M., and Azevedo-Pereira, J. M. (2014). HIV/dendritic cell interaction: consequences in the pathogenesis of HIV infection. AIDS Rev. 16, 223–235.
Bartel, D. P. (2004). MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116, 281–297. doi: 10.1016/S0092-8674(04)00045-5
Baumjohann, D., and Ansel, K. M. (2013). MicroRNA-mediated regulation of T helper cell differentiation and plasticity. Nat. Rev. Immunol. 13, 666–678. doi: 10.1038/nri3494
Bazzini, A. A., Lee, M. T., and Giraldez, A. J. (2012). Ribosome profiling shows that miR-430 reduces translation before causing mRNA decay in zebrafish. Science 336, 233–237. doi: 10.1126/science.1215704
Bellutti, F., Kauer, M., Kneidinger, D., Lion, T., and Klein, R. (2015). Identification of RISC-associated adenoviral microRNAs, a subset of their direct targets, and global changes in the targetome upon lytic adenovirus 5 Infection. J. Virol. 89, 1608–1627. doi: 10.1128/JVI.02336-14
Bennasser, Y., Chable-Bessia, C., Triboulet, R., Gibbings, D., Gwizdek, C., Dargemont, C., et al. (2011). Competition for XPO5 binding between dicer mRNA, pre-miRNA and viral RNA regulates human dicer levels. Nat. Struct. Mol. Biol. 18, 323–327. doi: 10.1038/nsmb.1987
Bennasser, Y., Yeung, M. L., and Jeang, K.-T. (2006). HIV-1 TAR RNA subverts rna interference in transfected cells through sequestration of TAR RNA-binding protein, TRBP. J. Biol. Chem. 281, 27674–27678. doi: 10.1074/jbc.C600072200
Berezikov, E., Chung, W., Willis, J., Cuppen, E., and Lai, E. C. (2007). Mammalian mirtron genes. Mol. Cell 28, 328–336. doi: 10.1016/j.molcel.2007.09.028
Bernstein, E., Caudy, A. A., Hammond, S. M., and Hannon, G. J. (2001). Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 409, 363–366. doi: 10.1038/35053110
Béthune, J., Artus-Revel, C. G., and Filipowicz, W. (2012). Kinetic analysis reveals successive steps leading to miRNA-mediated silencing in mammalian cells. EMBO Rep. 13, 716–723. doi: 10.1038/embor.2012.82
Bettencourt, P., Pires, D., and Anes, E. (2015). Immunomodulating microRNAs of mycobacterial infections. Tuberculosis. 97, 1–7. doi: 10.1016/j.tube.2015.12.004
Bhat, R. A., and Thimmappaya, B. (1984). Adenovirus mutants with DNA sequence perturbations in the intragenic promoter of VAI RNA gene allow the enhanced transcription of VAII RNA gene in HeLa cells. Nucleic Acids Res. 12,k7377–7388.
Bohnsack, M. T., Czaplinski, K., and Gorlich, D. (2004). Exportin 5 is a RanGTP-dependent dsRNA-binding protein that mediates nuclear export of pre-miRNAs. RNA 10, 185–191. doi: 10.1261/rna.5167604
Brennecke, J., Stark, A., Russell, R. B., and Cohen, S. M. (2005). Principles of MicroRNA–target recognition. PLoS Biol. 3:e85. doi: 10.1371/journal.pbio.0030085
Bushati, N., and Cohen, S. M. (2007). microRNA functions. Annu. Rev. Cell Dev. Biol. 23, 175–205. doi: 10.1146/annurev.cellbio.23.090506.123406
Cai, X., Hagedorn, C. H., and Cullen, B. R. (2004). Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA 10, 1957–1966. doi: 10.1261/rna.7135204
Carnero, E., Sutherland, J. D., and Fortes, P. (2011). Adenovirus and miRNAs. Biochim. Biophys. Acta 1809, 660–667. doi: 10.1016/j.bbagrm.2011.05.004
Chang, T.-C., and Mendell, J. T. (2007). microRNAs in vertebrate physiology and human disease. Annu. Rev. Genomics Hum. Genet. 8, 215–239. doi: 10.1146/annurev.genom.8.080706.092351
Chen, X. M. (2009). MicroRNA signatures in liver diseases. World J. Gastroenterol. 15, 1665–1672. doi: 10.3748/wjg.15.1665
Chendrimada, T. P., Gregory, R. I., Kumaraswamy, E., Norman, J., Cooch, N., Nishikura, K., et al. (2005). TRBP recruits the dicer complex to Ago2 for microRNA processing and gene silencing. Nature 436, 740–744. doi: 10.1038/nature03868
Chiang, K., Sung, T.-L., and Rice, A. P. (2011). Regulation of cyclin T1 and HIV-1 replication by MicroRNAs in resting CD4+ T lymphocytes. J. Virol. 86, 3244–3252. doi: 10.1128/JVI.05065-11
Cole, J. L. (2007). Activation of PKR: an open and shut case? Trends Biochem. Sci. 32, 57–62. doi: 10.1016/j.tibs.2006.12.003
Conrad, M., Jakupoglu, C., Moreno, S. G., Lippl, S., Banjac, A., Schneider, M., et al. (2004). Essential role for mitochondrial thioredoxin reductase in hematopoiesis, heart development, and heart function. Mol. Cell. Biol. 24, 9414–9423. doi: 10.1128/MCB.24.21.9414-9423.2004
Coventry, V. K., and Conn, G. L. (2008). Analysis of adenovirus VA RNAI structure and stability using compensatory base pair modifications. Nucleic Acids Res. 36, 1645–1653. doi: 10.1093/nar/gkn020
Cullen, B. R. (2013). How do viruses avoid inhibition by endogenous cellular MicroRNAs? PLoS Pathog. 9:e1003694. doi: 10.1371/journal.ppat.1003694
Delorme-Axford, E., Donker, R. B., Mouillet, J.-F., Chu, T., Bayer, A., Ouyang, Y., et al. (2013). Human placental trophoblasts confer viral resistance to recipient cells. Proc. Natl. Acad. Sci. U.S.A. 110, 12048–12053. doi: 10.1073/pnas.1304718110
Denli, A. M., Tops, B. B. J., Plasterk, R. H. A., Ketting, R. F., and Hannon, G. J. (2004). Processing of primary microRNAs by the microprocessor complex. Nature 432, 231–235. doi: 10.1038/nature03049
Desai, S. Y., Patel, R. C., Sen, G. C., Malhotra, P., Ghadge, G. D., and Thimmapaya, B. (1995). Activation of interferon-inducible 2’-5’ oligoadenylate synthetase by adenoviral VAI RNA. J. Biol. Chem. 270, 3454–3461.
Djuranovic, S., Nahvi, A., and Green, R. (2012). miRNA-mediated gene silencing by translational repression followed by mRNA deadenylation and decay. Science 336, 237–240. doi: 10.1126/science.1215691
Doench, J. G. (2003). siRNAs can function as miRNAs. Genes Dev. 17, 438–442. doi: 10.1101/gad.1064703
Doench, J. G. (2004). Specificity of microRNA target selection in translational repression. Genes Dev. 18, 504–511. doi: 10.1101/gad.1184404
Esquela-Kerscher, A., and Slack, F. J. (2006). Oncomirs - microRNAs with a role in cancer. Nat. Rev. Cancer 6, 259–269. doi: 10.1038/nrc1840
Fabian, M. R., Mathonnet, G., Sundermeier, T., Mathys, H., Zipprich, J. T., Svitkin, Y. V., et al. (2009). Mammalian miRNA RISC recruits CAF1 and PABP to affect PABP-dependent deadenylation. Mol. Cell 35, 868–880. doi: 10.1016/j.molcel.2009.08.004
Farazi, T. A., Spitzer, J. I., Morozov, P., and Tuschl, T. (2011). miRNAs in human cancer. J. Pathol. 223, 102–115. doi: 10.1002/path.2806
Friedman, R. C., Farh, K. K.-H., Burge, C. B., and Bartel, D. P. (2009). Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 19, 92–105. doi: 10.1101/gr.082701.108
Fuziwara, C. S., and Kimura, E. T. (2015). Insights into regulation of the miR-17-92 cluster of miRNAs in cancer. Front. Med. 2:64. doi: 10.3389/fmed.2015.00064
Garnett, C. T., Talekar, G., Mahr, J. A., Huang, W., Zhang, Y., Ornelles, D. A., et al. (2009). Latent species C adenoviruses in human tonsil tissues. J. Virol. 83, 2417–2428. doi: 10.1128/JVI.02392-08
Granberg, F., Svensson, C., Pettersson, U., and Zhao, H. (2006). Adenovirus-induced alterations in host cell gene expression prior to the onset of viral gene expression. Virology 353, 1–5. doi: 10.1016/j.virol.2006.06.019
Gregory, R. I., Chendrimada, T. P., Cooch, N., and Shiekhattar, R. (2005). Human RISC couples microRNA biogenesis and posttranscriptional gene silencing. Cell 123, 631–640. doi: 10.1016/j.cell.2005.10.022
Gregory, R. I., Yan, K.-P., Amuthan, G., Chendrimada, T., Doratotaj, B., Cooch, N., et al. (2004). The microprocessor complex mediates the genesis of microRNAs. Nature 432, 235–240. doi: 10.1038/nature03120
Grimson, A., Farh, K. K.-H., Johnston, W. K., Garrett-Engele, P., Lim, L. P., and Bartel, D. P. (2007). MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol. Cell 27, 91–105. doi: 10.1016/j.molcel.2007.06.017
Grundhoff, A., and Sullivan, C. S. (2011). Virus-encoded microRNAs. Virology 411, 325–343. doi: 10.1016/j.virol.2011.01.002
Guo, H., Ingolia, N. T., Weissman, J. S., and Bartel, D. P. (2010). Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 466, 835–840. doi: 10.1038/nature09267
Gwizdek, C., Ossareh-Nazari, B., Brownawell, A. M., Doglio, A., Bertrand, E., Macara, I. G., et al. (2003). Exportin-5 mediates nuclear export of minihelix-containing RNAs. J. Biol. Chem. 278, 5505–5508. doi: 10.1074/jbc.C200668200
Haase, A. D., Jaskiewicz, L., Zhang, H., Lainé, S., Sack, R., Gatignol, A., et al. (2005). TRBP, a regulator of cellular PKR and HIV-1 virus expression, interacts with dicer and functions in RNA silencing. EMBO Rep. 6, 961–967. doi: 10.1038/sj.embor.7400509
Han, J. (2004). The drosha-DGCR8 complex in primary microRNA processing. Genes Dev. 18, 3016–3027. doi: 10.1101/gad.1262504
Han, J., Lee, Y., Yeom, K.-H., Nam, J.-W., Heo, I., Rhee, J.-K., et al. (2006). Molecular basis for the recognition of primary microRNAs by the drosha-DGCR8 complex. Cell 125, 887–901. doi: 10.1016/j.cell.2006.03.043
Harwig, A., Das, A. T., and Berkhout, B. (2014). Retroviral microRNAs. Curr. Opin. Virol. 7, 47–54. doi: 10.1016/j.coviro.2014.03.013
Hodzic, J., Sie, D., Vermeulen, A., and van Beusechem, V. W. (2017). Functional screening identifies human miRNAs that modulate adenovirus propagation in prostate cancer cells. Hum. Gene Ther. doi: 10.1089/hum.2016.143 [Epub ahead of print].
Hogg, J. C. (2001). Role of latent viral infections in chronic obstructive pulmonary disease and asthma. Am. J. Respir. Crit. Care Med. 164, 71–75. doi: 10.1164/rccm2106063
Hutvágner, G., McLachlan, J., Pasquinelli, A. E., Bálint, E., Tuschl, T., and Zamore, P. D. (2001). A cellular function for the RNA-interference enzyme dicer in the maturation of the let-7 small temporal RNA. Science 293, 834–838. doi: 10.1126/science.1062961
Jopling, C. L., Yi, M., Lancaster, A. M., Lemon, S. M., and Sarnow, P. (2005). Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science 309, 1577–1581. doi: 10.1126/science.1113329
Kamel, W., Segerman, B., Öberg, D., Punga, T., and Akusjärvi, G. (2013). The adenovirus VA RNA-derived miRNAs are not essential for lytic virus growth in tissue culture cells. Nucleic Acids Res. 41, 4802–4812. doi: 10.1093/nar/gkt172
Kitajewski, J., Schneider, R. J., Safer, B., Munemitsu, S. M., Samuel, C. E., Thimmappaya, B., et al. (1986). Adenovirus VAI RNA antagonizes the antiviral action of interferon by preventing activation of the interferon-induced eIF-2 alpha kinase. Cell 45, 195–200.
Kloosterman, W. P., and Plasterk, R. H. A. (2006). The diverse functions of MicroRNAs in animal development and disease. Dev. Cell 11, 441–450. doi: 10.1016/j.devcel.2006.09.009
Kozomara, A., and Griffiths-Jones, S. (2014). miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 42, D68–D73. doi: 10.1093/nar/gkt1181
Krishnamoorthy, T., Pavitt, G. D., Zhang, F., Dever, T. E., and Hinnebusch, A. G. (2001). Tight binding of the phosphorylated alpha subunit of initiation factor 2 (eIF2alpha) to the regulatory subunits of guanine nucleotide exchange factor eIF2B is required for inhibition of translation initiation. Mol. Cell. Biol. 21, 5018–5030. doi: 10.1128/MCB.21.15.5018-5030.2001
Krützfeldt, J., and Stoffel, M. (2006). MicroRNAs: a new class of regulatory genes affecting metabolism. Cell Metab. 4, 9–12. doi: 10.1016/j.cmet.2006.05.009
Landthaler, M., Yalcin, A., and Tuschl, T. (2004). The human DiGeorge syndrome critical region gene 8 and its D. melanogaster homolog are required for miRNA biogenesis. Curr. Biol. 14, 2162–2167. doi: 10.1016/j.cub.2004.11.001
Larsson, O., and Nadon, R. (2013). Re-analysis of genome wide data on mammalian microRNA-mediated suppression of gene expression. Translation 1, e24557/1–e24557/9. doi: 10.4161/trla.24557
Launer-Felty, K., and Cole, J. L. (2014). Domain interactions in adenovirus VAI RNA mediate high-affinity PKR binding. J. Mol. Biol. 426, 1285–1295. doi: 10.1016/j.jmb.2013.12.019
Lecellier, C. H., Dunoyer, P., Arar, K., Lehmann-Che, J., Eyquem, S., Himber, C., et al. (2005). A cellular microRNA mediates antiviral defense in human cells. Science 308, 557–560. doi: 10.1126/science.1108784
Lee, H. Y., Zhou, K., Smith, A. M., Noland, C. L., and Doudna, J. A. (2013). Differential roles of human dicer-binding proteins TRBP and PACT in small RNA processing. Nucleic Acids Res. 41, 6568–6576. doi: 10.1093/nar/gkt361
Lee, R. C., Feinbaum, R. L., and Ambros, V. (1993). The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75, 843–854. doi: 10.1016/0092-8674(93)90529-Y
Lee, Y., Ahn, C., Han, J., Choi, H., Kim, J., Yim, J., et al. (2003). The nuclear RNase III drosha initiates microRNA processing. Nature 425, 415–419. doi: 10.1038/nature01957
Lee, Y., Kim, M., Han, J., Yeom, K.-H., Lee, S., Baek, S. H., et al. (2004). MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 23, 4051–4060. doi: 10.1038/sj.emboj.7600385
Lei, T., Yuen, K.-S., Xu, R., Tsao, S. W., Chen, H., Li, M., et al. (2013). Targeting of DICE1 tumor suppressor by epstein-barr virus-encoded miR-BART3∗ microRNA in nasopharyngeal carcinoma. Int. J. Cancer 133, 79–87. doi: 10.1002/ijc.28007
Lenaerts, L., De Clercq, E., and Naesens, L. (2008). Clinical features and treatment of adenovirus infections. Rev. Med. Virol. 18, 357–374. doi: 10.1002/rmv.589
Lewis, B. P., Burge, C. B., and Bartel, D. P. (2005). Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120, 15–20. doi: 10.1016/j.cell.2004.12.035
Lewis, B. P., Shih, I., Jones-Rhoades, M. W., Bartel, D. P., and Burge, C. B. (2003). Prediction of mammalian microRNA targets. Cell 115, 787–798. doi: 10.1016/S0092-8674(03)01018-3
Lewis Phillips, G. D., Li, G., Dugger, D. L., Crocker, L. M., Parsons, K. L., Mai, E., et al. (2008). Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 68, 9280–9290. doi: 10.1158/0008-5472.CAN-08-1776
Lichty, B. D., Breitbach, C. J., Stojdl, D. F., and Bell, J. C. (2014). Going viral with cancer immunotherapy. Nat. Rev. Cancer 14, 559–567. doi: 10.1038/nrc3770
Lim, L. P., Lau, N. C., Garrett-Engele, P., Grimson, A., Schelter, J. M., Castle, J., et al. (2005). Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 433, 769–773. doi: 10.1038/nature03315
Lion, T. (2014). Adenovirus infections in immunocompetent and immunocompromised patients. Clin. Microbiol. Rev. 27, 441–462. doi: 10.1128/CMR.00116-13
Lu, S., and Cullen, B. R. (2004). Adenovirus VA1 noncoding RNA can inhibit small interfering RNA and MicroRNA biogenesis. J. Virol. 78, 12868–12876. doi: 10.1128/JVI.78.23.12868-12876.2004
Luna, J. M., Scheel, T. K. H., Danino, T., Shaw, K. S., Mele, A., Fak, J. J., et al. (2015). Hepatitis C virus RNA functionally sequesters miR-122. Cell 160, 1099–1110. doi: 10.1016/j.cell.2015.02.025
Lund, E., Güttinger, S., Calado, A., Dahlberg, J. E., and Kutay, U. (2004). Nuclear export of microRNA precursors. Science 303, 95–98. doi: 10.1126/science.1090599
Ma, L., Shen, C.-J., Cohen,É. A., Xiong, S.-D., and Wang, J.-H. (2014). miRNA-1236 inhibits HIV-1 infection of monocytes by repressing translation of cellular factor VprBP. PLoS ONE 9:e99535. doi: 10.1371/journal.pone.0099535
Ma, Y., and Mathews, M. B. (1996). Structure, function, and evolution of adenovirus-associated RNA: a phylogenetic approach. J. Virol. 70, 5083–5099. doi: 10.1006/viro.1995.1146
Machitani, M., Sakurai, F., Wakabayashi, K., Nakatani, K., Tachibana, M., and Mizuguchi, H. (2017). MicroRNA miR-27 inhibits adenovirus infection by suppressing the expression of SNAP25 and TXN2. J. Virol. 91, e159–e117. doi: 10.1128/JVI.00159-17
Meier, O., and Greber, U. F. (2003). Adenovirus endocytosis. J. Gene Med. 5, 451–462. doi: 10.1002/jgm.409
Meng, H., Deo, S., Xiong, S., Dzananovic, E., Donald, L. J., van Dijk, C. W., et al. (2012). Regulation of the interferon-inducible 2’-5’-oligoadenylate synthetases by adenovirus VA(I) RNA. J. Mol. Biol. 422, 635–649. doi: 10.1016/j.jmb.2012.06.017
Minamitani, T., Iwakiri, D., and Takada, K. (2011). Adenovirus virus-associated RNAs induce type I interferon expression through a RIG-I-mediated pathway. J. Virol. 85, 4035–4040. doi: 10.1128/JVI.02160-10
Nilsen, T. W. (2007). Mechanisms of microRNA-mediated gene regulation in animal cells. Trends Genet. 23, 243–249. doi: 10.1016/j.tig.2007.02.011
Okamura, K., Hagen, J. W., Duan, H., Tyler, D. M., and Lai, E. C. (2007). The mirtron pathway generates microRNA-class regulatory RNAs in Drosophila. Cell 130, 89–100. doi: 10.1016/j.cell.2007.06.028
Piedade, D., and Azevedo-Pereira, J. (2016a). The role of microRNAs in the pathogenesis of herpesvirus infection. Viruses 8:156. doi: 10.3390/v8060156
Piedade, D., and Azevedo-Pereira, J. M. (2016b). MicroRNAs, HIV and HCV: a complex relation towards pathology. Rev. Med. Virol. 26, 197–215. doi: 10.1002/rmv.1881
Pillai, R. S., Bhattacharyya, S. N., and Filipowicz, W. (2007). Repression of protein synthesis by miRNAs: how many mechanisms? Trends Cell Biol. 17, 118–126. doi: 10.1016/j.tcb.2006.12.007
Punga, T., Kamel, W., and Akusjärvi, G. (2013). Old and new functions for the adenovirus virus-associated RNAs. Future Virol. 8, 343–356. doi: 10.2217/fvl.13.19
Qi, Y., Tu, J., Cui, L., Guo, X., Shi, Z., Li, S., et al. (2010). High-throughput sequencing of MicroRNAs in adenovirus type 3 infected human laryngeal epithelial cells. J. Biomed. Biotechnol. 2010, 1–8. doi: 10.1155/2010/915980
Rosewell Shaw, A., and Suzuki, M. (2016). Recent advances in oncolytic adenovirus therapies for cancer. Curr. Opin. Virol. 21, 9–15. doi: 10.1016/j.coviro.2016.06.009
Roy, S., Calcedo, R., Medina-Jaszek, A., Keough, M., Peng, H., and Wilson, J. M. (2011). Adenoviruses in lymphocytes of the human gastro-intestinal tract. PLoS ONE 6:e24859. doi: 10.1371/journal.pone.0024859
Ruby, J. G., Jan, C. H., and Bartel, D. P. (2007). Intronic microRNA precursors that bypass Drosha processing. Nature 448, 83–86. doi: 10.1038/nature05983
Sano, M., Kato, Y., and Taira, K. (2006). Sequence-specific interference by small RNAs derived from adenovirus VAI RNA. FEBS Lett. 580, 1553–1564. doi: 10.1016/j.febslet.2006.01.085
Seddiki, N., Brezar, V., Ruffin, N., Lévy, Y., and Swaminathan, S. (2014). Role of miR-155 in the regulation of lymphocyte immune function and disease. Immunology 142, 32–38. doi: 10.1111/imm.12227
Shen, C.-J., Jia, Y.-H., Tian, R.-R., Ding, M., Zhang, C., and Wang, J.-H. (2012). Translation of Pur- is targeted by cellular miRNAs to modulate the differentiation-dependent susceptibility of monocytes to HIV-1 infection. FASEB J. 26, 4755–4764. doi: 10.1096/fj.12-209023
Su, J.-L., Chen, P. B., Chen, Y.-H., Chen, S.-C., Chang, Y.-W., Jan, Y.-H., et al. (2010). Downregulation of microRNA miR-520h by E1A contributes to anticancer activity. Cancer Res. 70, 5096–5108. doi: 10.1158/0008-5472.CAN-09-4148
Sudhof, T. C., and Rothman, J. E. (2009). Membrane fusion: grappling with SNARE and SM proteins. Science 323, 474–477. doi: 10.1126/science.1161748
Sun, Y.-M., Lin, K.-Y., and Chen, Y.-Q. (2013). Diverse functions of miR-125 family in different cell contexts. J. Hematol. Oncol. 6:6. doi: 10.1186/1756-8722-6-6
Sung, T.-L., and Rice, A. P. (2009). miR-198 inhibits HIV-1 gene expression and replication in monocytes and its mechanism of action appears to involve repression of cyclin T1. PLoS Pathog. 5:e1000263. doi: 10.1371/journal.ppat.1000263
Swaminathan, S., Murray, D. D., and Kelleher, A. D. (2012). The role of microRNAs in HIV-1 pathogenesis and therapy. AIDS 26, 1325–1334. doi: 10.1097/QAD.0b013e328352adca
Tanaka, T. (2002). Thioredoxin-2 (TRX-2) is an essential gene regulating mitochondria-dependent apoptosis. EMBO J. 21, 1695–1703. doi: 10.1093/emboj/21.7.1695
Thimmappaya, B., Weinberger, C., Schneider, R. J., and Shenk, T. (1982). Adenovirus VAI RNA is required for efficient translation of viral mRNAs at late times after infection. Cell 31, 543–551.
Tomankova, T., Petrek, M., and Kriegova, E. (2010). Involvement of microRNAs in physiological and pathological processes in the lung. Respir. Res. 11:159. doi: 10.1186/1465-9921-11-159
Triboulet, R., Mari, B., Lin, Y.-L., Chable-Bessia, C., Bennasser, Y., Lebrigand, K., et al. (2007). Suppression of MicroRNA-silencing pathway by HIV-1 during virus replication. Science 315, 1579–1582. doi: 10.1126/science.1136319
Vachon, V. K., and Conn, G. L. (2015). Adenovirus VA RNA: an essential pro-viral non-coding RNA. Virus Res. 212, 39–52. doi: 10.1016/j.virusres.2015.06.018
Valencia-Sanchez, M. A., Liu, J., Hannon, G. J., and Parker, R. (2006). Control of translation and mRNA degradation by miRNAs and siRNAs. Genes Dev. 20, 515–524. doi: 10.1101/gad.1399806
Watts, J. M., Dang, K. K., Gorelick, R. J., Leonard, C. W., Bess, J. W., Swanstrom, R., et al. (2009). Architecture and secondary structure of an entire HIV-1 RNA genome. Nature 460, 711–716. doi: 10.1038/nature08237
Westerhout, E. M., Ooms, M., Vink, M., Das, A. T., and Berkhout, B. (2005). HIV-1 can escape from RNA interference by evolving an alternative structure in its RNA genome. Nucleic Acids Res. 33, 796–804. doi: 10.1093/nar/gki220
Wightman, B., Ha, I., and Ruvkun, G. (1993). Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 75, 855–862. doi: 10.1016/0092-8674(93)90530-4
Wong, A. M. G., Kong, K. L., Tsang, J. W. H., Kwong, D. L. W., and Guan, X.-Y. (2012). Profiling of epstein-barr virus-encoded microRNAs in nasopharyngeal carcinoma reveals potential biomarkers and oncomirs. Cancer 118, 698–710. doi: 10.1002/cncr.26309
Xiao, B., Liu, Z., Li, B. S., Tang, B., Li, W., Guo, G., et al. (2009). Induction of microRNA-155 during Helicobacter pylori infection and its negative regulatory role in the inflammatory response. J. Infect. Dis. 200, 916–925. doi: 10.1086/605443
Xu, N., Gkountela, S., Saeed, K., and Akusjärvi, G. (2009). The 5’-end heterogeneity of adenovirus virus-associated RNAI contributes to the asymmetric guide strand incorporation into the RNA-induced silencing complex. Nucleic Acids Res. 37, 6950–6959. doi: 10.1093/nar/gkp764
Xu, N., Segerman, B., Zhou, X., and Akusjärvi, G. (2007). Adenovirus virus-associated RNAII-derived small RNAs are efficiently incorporated into the rna-induced silencing complex and associate with polyribosomes. J. Virol. 81, 10540–10549. doi: 10.1128/JVI.00885-07
Yan, K. S., Yan, S., Farooq, A., Han, A., Zeng, L., and Zhou, M.-M. (2003). Structure and conserved RNA binding of the PAZ domain. Nature 426, 468–474. doi: 10.1038/nature02252
Yi, R., Qin, Y., Macara, I. G., and Cullen, B. R. (2003). Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev. 17, 3011–3016. doi: 10.1101/gad.1158803
Zamore, P. D., Tuschl, T., Sharp, P. A., and Bartel, D. P. (2000). RNAi: double-stranded RNA directs the ATP-dependent cleavage of mRNA at 21 to 23 nucleotide intervals. Cell 101, 25–33. doi: 10.1016/S0092-8674(00)80620-0
Zeng, Y., Yi, R., and Cullen, B. R. (2005). Recognition and cleavage of primary microRNA precursors by the nuclear processing enzyme Drosha. EMBO J. 24, 138–148. doi: 10.1038/sj.emboj.7600491
Zhao, H., Chen, M., and Pettersson, U. (2013). Identification of adenovirus-encoded small RNAs by deep RNA sequencing. Virology 442, 148–155. doi: 10.1016/j.virol.2013.04.006
Zhao, H., Chen, M., Tellgren-Roth, C., and Pettersson, U. (2015). Fluctuating expression of microRNAs in adenovirus infected cells. Virology 478, 99–111. doi: 10.1016/j.virol.2015.01.033
Zhao, H., Dahlö, M., Isaksson, A., Syvänen, A.-C., and Pettersson, U. (2012). The transcriptome of the adenovirus infected cell. Virology 424, 115–128. doi: 10.1016/j.virol.2011.12.006
Zhao, H., Granberg, F., and Pettersson, U. (2007). How adenovirus strives to control cellular gene expression. Virology 363, 357–375. doi: 10.1016/j.virol.2007.02.013
Keywords: microRNA, adenoviruses, pathogenesis, gene regulation, mRNA translation, oncogenesis
Citation: Piedade D and Azevedo-Pereira JM (2017) MicroRNAs as Important Players in Host–Adenovirus Interactions. Front. Microbiol. 8:1324. doi: 10.3389/fmicb.2017.01324
Received: 13 April 2017; Accepted: 30 June 2017;
Published: 17 July 2017.
Edited by:
Akio Adachi, Tokushima University, JapanReviewed by:
Fuminori Sakurai, Osaka University, JapanCristina Fillat, Institut d’Investigacions Biomèdiques August Pi i Sunyer, Spain
Copyright © 2017 Piedade and Azevedo-Pereira. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: José M. Azevedo-Pereira, bWlndWVsLnBlcmVpcmFAZmYudWwucHQ=