 Xi Zhang
Xi Zhang Ya L. Shi
Ya L. Shi Zhong Q. Wang
Zhong Q. Wang Ruo D. Liu
Ruo D. Liu Jing Cui
Jing Cui- Department of Parasitology, School of Basic Medical Sciences, Zhengzhou University, Zhengzhou, China
Thelazia callipaeda, also called the oriental eyeworm, is the major etiological agent of human thelaziasis. Cases of thelaziasis have increased in recent years in China. Although this species is of medical importance, the genetics and phylogenetic systematics of T. callipaeda are poorly understood. In this study, we first reported three cases of thelaziasis in central China. All clinical isolates were identified as T. callipaeda according to morphological characteristics by light microscopy and scanning electron microscopy. Next, complete mitochondrial (mt) genomes for the three T. callipaeda isolates from different geographical locations were fully characterized using an Illumina sequencing platform. In addition, all available mt genomes of spirurid nematodes in GenBank were included to reconstruct the phylogeny and to explore the evolutionary histories of the isolates. The genome features of the T. callipaeda isolates contained 12 PCGs, 22 transfer RNA genes, two ribosomal RNA genes and a major non-coding region. The mtDNA nucleotide sequences of the T. callipaeda isolates from different hosts and different locations were similar. The nad6 gene showed high sequence variability among all isolates, which is worth considering for future population genetic studies of T. callipaeda. Phylogenetic analyses based on maximum parsimony and Bayesian inference methods revealed close relationships among Thelaziidae, Onchocercidae, Setariidae, Gongylonematidae, Physalopteridae, Dracunculidae, and Philometridae. The monophyly of the T. callipaeda isolates from different hosts and distinct geographical locations was confirmed. The entire mt genomes of T. callipaeda presented in this study will serve as a useful dataset for studying the population genetics and phylogenetic relationships of Thelazia species.
Introduction
Thelazia callipaeda Railliet and Henry, 1910 (Nematoda: Spirurida: Thelaziidae), also known as the oriental eyeworm, is the major etiological agent of human thelaziasis (Fuentes et al., 2012). T. callipaeda infective larvae, which are transmitted by zoophilic insects of the genus Phortica, can parasitize the orbital cavity and associated tissues of humans, causing mild to severe signs and symptoms (Otranto et al., 2005a; Shen et al., 2006). Thelaziasis has a worldwide distribution, but most cases occur in eastern and southeastern Asia (Otranto and Dutto, 2008; Nguyen et al., 2012). Human cases are usually associated with poor health and socioeconomic settings, in which heavily affected domestic and wild animals live in close vicinity with humans (Hodžić et al., 2014). China probably has the largest number of cases of thelaziasis in the world with more than 600 cases reported to date (Shen et al., 2006; Wang et al., 2014). In addition, the cases of thelaziasis in China appear to have increased in recent years (Wang et al., 2014).
In clinical diagnostics, it is difficult to differentiate thelaziasis from allergic conjunctivitis when some of the adult or larval stages of T. callipaeda are present in the eyes of infected patients (Liu et al., 2013a). Fortunately, the DNA barcoding approach has been useful for Thelazia species (Otranto and Traversa, 2004). Several DNA markers have been applied in the taxonomy and genetic studies of T. callipaeda, such as cytochrome c oxidase subunit 1 gene, cox1 (Otranto et al., 2005b; Nguyen et al., 2012; Wang et al., 2014; Mihalca et al., 2015), and the first internal transcribed spacer of ribosomal DNA, ITS1 (Otranto and Traversa, 2004). However, in comparison with a single gene, the complete mitochondrial (mt) genome can provide more markers for molecular identification, and provide the potential to discover population variants or cryptic species (Ramesh et al., 2012; Zhang et al., 2016). In recent years, several remarkable advances have been made in the different taxonomic levels of spirurid nematodes using complete mt genomes (Yatawara et al., 2010; Park et al., 2011; McNulty et al., 2012). The first complete mtDNA sequence of T. callipaeda has been sequenced and characterized (Liu et al., 2013a). These available mtDNA datasets offer opportunities to compare the diversity of mt genomes among closely related Thelazia taxa. In this study, we first describe three cases of thelaziasis in central China. Additionally, the worms collected from patients were sequenced using next generation sequencing (NGS) technology to perform a mitogenomics comparative analysis of spirurid nematodes and to discuss the implications of the new dataset as a new resource for future genetic studies of T. callipaeda populations.
Materials and Methods
Ethics Statement
This study was approved by the Life Science Ethics Committee of Zhengzhou University (No. 2016-0096). All worms were collected from eyes of children after their legal guardians provided permission. These worm samples were collected not with the purpose of the present study but were removed from patients to treat the thelaziasis.
Sampling and Data Collection
Three samples of T. callipaeda named HeN-ZZ1, HeN-LY1, and HeN-PDS1 were harvested from three patients from different geographical locations of central China (details are shown in the ‘cases report’ part). In addition, all related complete mt genomes available in the GenBank database were included in this study (Table 1). To date (January 1, 2017), GenBank contains 24 mitogenomes of spirurid nematodes representing the following eight families: Dracunculidae (1 mtDNAs), Philometridae (1), Onchocercidae (13), Setariidae (1), Gnathostomatidae (3), Physalopteridae (1), Gongylonematidae (1) and Thelaziidae (3). Two Ascaris nematodes, Ascaris lumbricoides (HQ704900) and A. suum (HQ704901), were used as the outgroup.
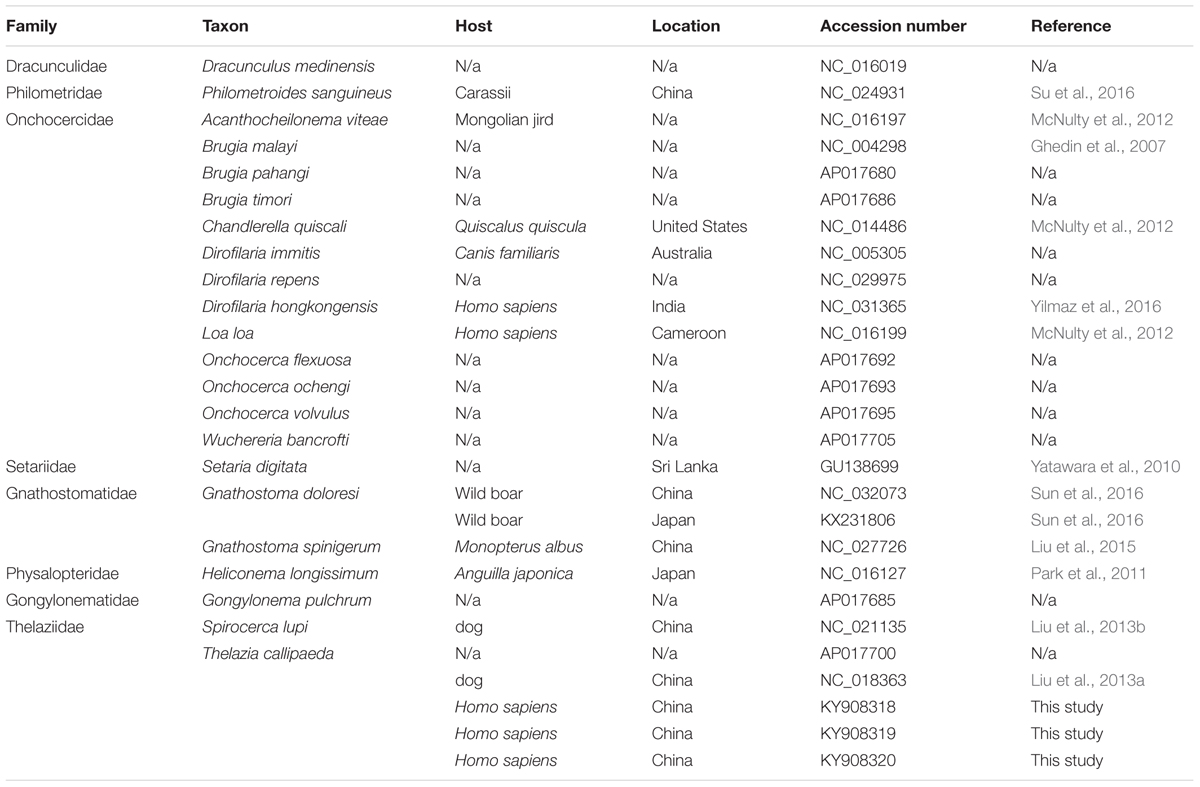
TABLE 1. The taxa of complete mitochondrial genomes of spirurid nematodes used for this study.
Morphological Identification
The nematodes collected from the patients were transferred to Petri dishes containing physiological saline (0.9% NaCl). The eyeworms were fixed as previously described (Otranto et al., 2004) and examined by light microscopy (Leica Micro system, German) at different magnifications, as well as under the scanning electron microscopy (SEM). All important morphological characteristics of T. callipaeda were evaluated for adult forms as reported in Otranto et al. (2003).
Extraction of Genomic DNA and Library Preparation
Genomic DNA of each specimen was extracted and purified using EasyPure Genomic DNA Kit (Transgen, China) following the manufacturer’s instructions. The complete mt genome was amplified by long-PCR as two overlapping amplicons from the genomic DNA using primers described by Liu et al. (2013a). Next, these two overlapping products were mixed in approximately equal amounts after determining the concentration of each amplicon. The normalized mixed DNA was fragmented to an average size of 500 bp using Covaris M220 system (Covaris, Woburn, MA, United States). The library was prepared using TruSeq DNA PCR-Free Sample Preparation Kit (Illumina, United States) following the manufacturer’s protocols. After these libraries were purified, the DNA concentration of all samples were quantified and 30 ng of each were pooled together. Finally, the oligonucleotide mix was sequenced on an Illumina HiSeq 2000 at the Genewiz Company (Beijing, China).
Mitogenome Assembly and Annotation
The quality of original sequencing reads was evaluated using the FastQC v.0.11.5 (Andrews, 2010). All ambiguous nucleotides and reads with an average quality value (lower than Q20) were excluded from further analysis. The trimmed sequences were mapped against two reference mt genomes of T. callipaeda worms (NC_018363 and AP017700) using the CLC Genomic Workbench v.7.0.4 (Qiagen, Germany). The mapped sequences were later subjected to de novo assembly. Contigs with hits to mitochondrial genes or genomes were identified and extracted from the CLC Genomic Workbench. A contig identified as mt genome was manually examined for repeats at the beginning and end of the sequence to establish a circular mtDNA. It was then annotated with MITOS (Bernt et al., 2013) followed by manual validation of the coding regions using the NCBI ORF Finder1. The sequin file generated from MITOS was edited and submitted to GenBank with the following accession numbers: KY908318, KY908319, and KY908320.
Phylogenetic Analysis
Nucleotide sequences of the 12 protein-coding genes (PCGs) were separately aligned using MEGA v.6.06 (Tamura et al., 2013). The sequences of rrnS, rrnL, and mt-tRNA genes were aligned in MAFFT v.7 (Katoh and Standley, 2013). The program DAMBE v5.2 (Xia and Xie, 2001) was used to measure the nucleotide substitution saturation using the method of Xia et al. (2003) as the substitution saturation masked the phylogenetic signal. Phylograms were constructed using the maximum parsimony (MP) and Bayesian inference (BI) methods, respectively. The MP analyses were performed in PAUP∗4b10 (Swofford, 2003) using heuristic searches with TBR branch swapping and 10,000 random addition sequences. Confidence in each node was assessed by boot-strapping (1000 pseudo-replicates, heuristic search of 20 random addition replicates with TBR option). BI analysis was performed in MrBayes v.3.2 (Ronquist et al., 2012) with 10, 000, 000 generations and sampling trees every 100 generations. The best-fit nucleotide substitution model for each data partition was selected by jModelTest 2 (Darriba et al., 2012) under the Akaike Information Criterion (AIC). Stationarity was assessed using a convergence diagnostic. An average standard deviation of the split frequencies (ASDSF) < 0.01 was used as the criteria of convergence between both runs. The consensus tree was drawn after removing the first 20 000 trees (20%) as the burn-in phase. In addition, the data used in the phylogenetic analyses were partitioned under three schemes: partitioned among PCGs (12 partitions, nad5, cox3, cytb, nad4L, nad4, atp6, nad2, nad1, nad3, cox1, cox2, and nad6), partitioned by rRNA genes (2 partitions, rrnL and rrnS), and partitioned by PCGs and rRNA (14 partitions).
Results
Cases Descriptions and Parasite Identification
The first case was a 6-year-old boy from Lushan county of Pingdingshan city, Henan province (33°36′ N, 112°25′ E) in central China. In September 2014, the boy was admitted to the First Affiliated Hospital of Zhengzhou University with a 7-day history of creeping sensation, itching, and increased secretions in his right eye, but without any systemic or visual symptoms. His medical history was notable only for an unknown insect flying into his right eye approximately 2 months earlier. On ophthalmological examination, several thread-like worms were seen moving in the nasal upper eyelid of the eyes above the conjunctiva. The worms nested in the subconjunctiva tissue were completely cleared with intraocular forceps. Four worms were removed, 1 in the left eye and 3 in the right eye. The second case was a 2.5-year-old boy who lived in the suburbs of Zhengzhou city, Henan province (34°40′ N, 113°12′ E). He visited his doctor in August 2015 with an approximately 2 weeks history of itching, conjunctival hyperemia and increased secretions in eyes. Ophthalmological examination found six slender worms on the conjunctival sac of the left eye. No additional abnormal manifestations were noted on physical examination. The symptoms disappeared after the worms were removed with forceps. The third case was a 1.5-year-old child from a village in Luoyang, Henan province (34°43′ N, 112°42′ E). The clinical manifestation was similar to the second case, and five worms were removed in her conjunctival sac, four in the left eye and one in the right eye.
All worms collected from the patients were identified as T. callipaeda according to the morphological characteristics described by Otranto et al. (2003). More specifically, for the females, the following characteristics were noted: (1) T. callipaeda was characterized by the presence of a buccal capsule (Figure 1Fa); (2) in the posterior half of the body, immature eggs or germ cells filled the uterine tubules (Figure 1Fb); (3) an anus was present on the tip of the tail (Figure 1Fc); (4) the transversally striated cuticle was more corrugated on the anterior of the body than on the caudal end (Figure 1Fd); and (5) the vulva with a short vulvar flap was located in the anterior region of the body (Figure 1Fe). For the males, the following characteristics were noted: (1) in contrast with the female, the buccal capsule was inconspicuous (Figure 1Ma), and the mouth opening showed a cycle or ellipse profile (Figure 1Mc); (2) two dissimilar spicules were present in the males, one was the anterior extremity of the longer left spicule and other was the typical crescent shape of the shorter right spicule (Figure 1Mb); (3) the caudal end was ventrally curved (Figure 1Md); and (4) there were five pairs of postcloacal papillae (Figure 1Me).

FIGURE 1. Photomicrographs and scanning electron micrographs (SEMs) of female and male Thelazia callipaeda. (Fa) Light micrograph of female anterior head and buccal capsule. (Fb) Light micrograph of immature eggs or germ cells filling the uterine tubules of female. (Fc) SEM of the tip of the female tail and the femal anus. (Fd) SEM of the transversally striated cuticle on the anterior of female. (Fe) SEM of the anterior region of female, vulva with a short vulvar flap. (Ma) Light micrograph of male anterior head. (Mb) Light micrograph of the male caudal end and detail of the shorter right spicule. (Mc) SEM of anterior region of the male. (Md) SEM of caudal end of male. (Me) SEM of caudal end of male, five pairs of postcloacal papillae.
Assembly of the mt Genome
A total of 961 424, 961 094, and 970 104 sequence reads with a total of 162 140 093, 166 174 524, and 162 301 339 bases were mapped to full mitochondrial genomes for the T. callipaeda isolates of HeN-LY1, HeN-PDS1, and HeN-ZZ1, respectively (Supplementary Table S1). All three novel mt genomes were fully annotated and each encoded an identical set of 12 PCGs, 22 tRNA genes, and 2 rRNA genes. All genes were encoded in an identical order and direction, and all PCGs annotated across the three representatives of T. callipaeda were of the same length (Figure 2). Several genes overlapped at the boundaries, such as nad6/trnR (9 bp), trnL1/cox3 (3 bp), trnL2/trnN (11 bp), nad1/trnF (32 bp), cox2/trnH (10 bp), rrnL/nad3 (8 bp), trnS1/nad2 (3 bp), and trnT/nad4 (6 bp) (Supplementary Table S2). All PCGs had ATG, TTT, ATA, and TTG as the initiation codons and TAA or TAG as the termination codons. However, the TAT termination codon was used only in the nad1 gene. The ribosomal RNA genes rrnL and rrnS were 965 and 666 bp long, respectively. The rrnL was located between trnH and nad3, and rrnS was between nad4L and trnY. The lengths of the 22 tRNA genes ranged from 53 to 66 nucleotides (nt). A major non-coding region located between cox3 and trnA was interspersed in T. callipaeda isolates.
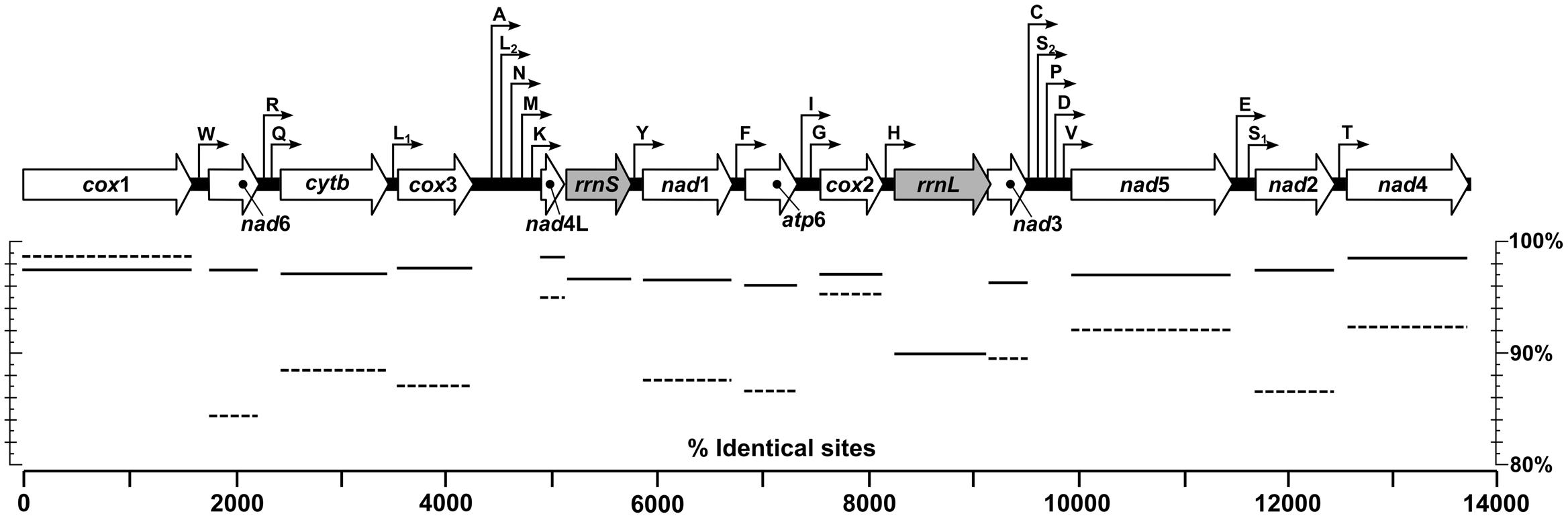
FIGURE 2. Linear maps of the mitochondrial genomes of T. callipaeda. Outline arrows indicate the positions and direction of transcription of protein- and rRNA-coding genes, hairline arrows indicate the position of tRNA encoding genes (trn). Solid and dotted horizontal lines below depict average nucleotide and amino acid identities of aligned protein- and rRNA-coding regions of specimens from Luoyang (HeN-LY1), Pingdingshan (HeN-PDS1) and Zhengzhou (HeN-ZZ1) of central China and Guangdong (NC_018363) and Japan (AP017700), respectively. atp6, adenosine triphosphatase subunit 6; cox, cytochrome c oxidase complex; cytb, cytochrome b; nad, nicotinamide dehydrogenase complex; rrnL, large subunit rDNA; rrnS, small subunit rDNA.
The mtDNA nucleotide sequences from the T. callipaeda isolates from different locations were conspicuously similar. The full-length mt genomes were identical in 98.22% nucleotides of the 13 668 bp long alignment of the three isolates. When published T. callipaeda mt sequences from Guangdong of China (isolated from dog, No. NC_018363) and Japan (No. AP017700) were added, the similarities of the mt genomes declined to 96.89%. Pairwise genetic distances between individual pairs of these isolates measured as a percentage of identical nucleotides ranged from 98.06% (Guangdong and HeN-LY1 isolates) to 99.53% (HeN-ZZ1 and Japan isolates). Comparing the protein-coding gene regions (Figure 2), T. callipaeda displayed high levels of sequence conservation ranging from 96.05% of identical nucleotides in atp6 to 98.74% in nad4L. The amino acid sequence similarities ranged from 84.31% (nad6) to 98.73% (cox1). The most divergent amino acid sequence was nad6 between the HeN-PDS1 and Japan isolates. The mt large and small subunit rDNAs (rrnL, rrnS) shared 89.95 and 96.71% identical nucleotides, respectively.
Phylogeny Reconstruction
The test of substitution saturation showed that the observed indexes of substitution saturation (Iss) for all alignments were significantly lower than the corresponding critical index substitution saturation (Iss.c), indicating that there was little saturation in the present sequences (Table 2). The likelihood models identified by the jModelTest (AIC) suggested that the GTR model was most suitable for included data partitions (Supplementary Table S3). A total of six analyses by two phylogenetic inference methods (MP and BI) under three partition schemes (partitioned among PCGs, partitioned by rRNA genes, and PCGs + rRNA) were completed in this study. Generally, the trees generated from different methods with different partition schemes shared similar topologies (Figure 3 and Supplementary Figures S1–S5). Among spirurid nematodes, the family Gnathostomatidae was a sister group to the remaining families: Dracunculidae, Philometridae, Physalopteridae, Thelaziidae, Gongylonematidae, Setariidae, and Onchocercidae (Figure 3). The earliest diversifications among these remaining families gave rise to Dracunculidae and Philometridae and then to Physalopteridae. In addition, Dracunculidae and Philometridae composed clade with high support value (posterior probabilities = 1.0). The next diversification event would have separated Thelaziidae, Gongylonematidae, Setariidae, and Onchocercidae, in which the Thelaziidae and Gongylonematidae were basal. The Onchocercidae as a monophyletic group (pp = 1.0) was the last diverged. However, the monophyly of family Thelaziidae was unresolved: the T. callipaeda isolates from different geographical locations generated a single group (pp = 1.0), and the other species Spirocerca lupi plus Gongylonematidae made up a separate clade.
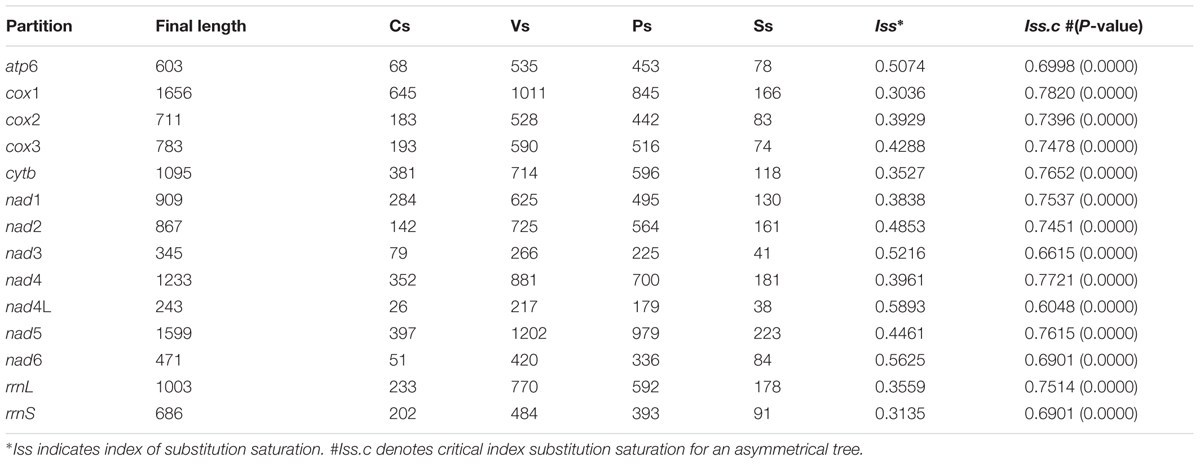
TABLE 2. Results of conserved sites (Cs), variable sites (Vs), parsim-info sites (Ps), singleton sites (Ss) and test of substitution saturation of aligned sequences of spirurid nematodes.
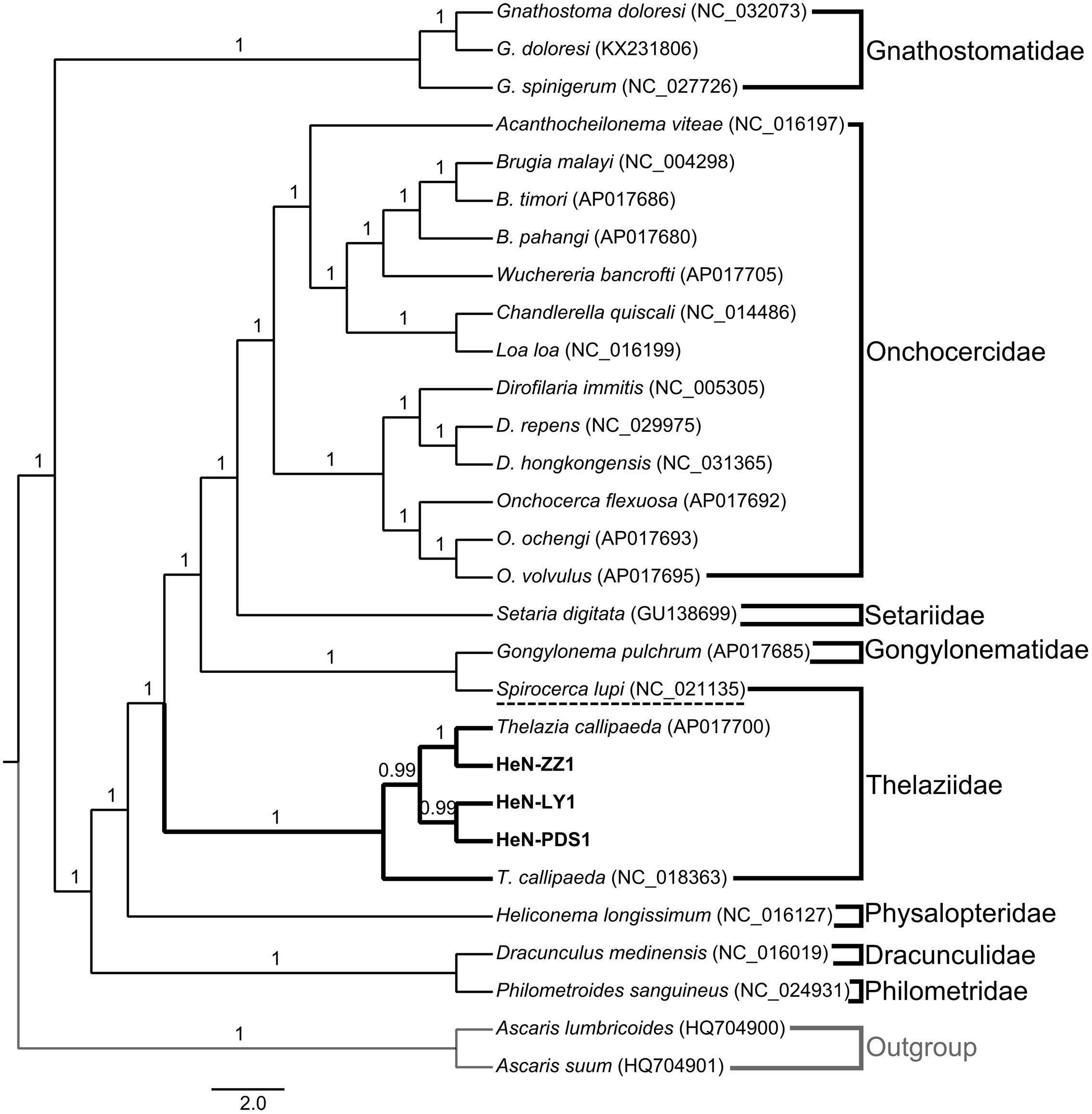
FIGURE 3. Bayesian phylogenetic tree of collected spirurid nematodes based on the analysis of 12 protein-coding genes (PCGs) and 2 rRNA genes. Numbers above branches represent the Bayesian posterior probabilities. Only posterior probabilities above 0.6 are shown.
Discussion
Thelaziasis can occur when infected flies of the genus Phortica feed on the ocular secretions, tears, and conjunctiva of hosts (Fuentes et al., 2012). In China, since the first case with human thelaziasis was described in 1917, more than 600 cases scattered across almost the entire country have been reported (Wang et al., 2014). Human cases typically occur in rural villages associated with poor health and socioeconomic settings (Shen et al., 2006). Until now, the etiological agent of all human thelaziasis reported in China was identified as T. callipaeda, but few cases reported detailed morphological characteristics. In this study, we firstly reported three cases with clinical eyeworms in central China, and we examined these worms under light microscopy and scanning electron microscopy. The key characteristics of the T. callipaeda isolates in our study were similar to those of T. callipaeda isolates from dogs in southern Italy (Otranto et al., 2003). In addition, the mouth profile of the T. callipaeda male was first described in this study. In comparison with the isolates from Italy, the sizes of the parasites collected here were relatively smaller; the males measured 6.4–10.3 mm in length and 307–389 μm in width (7.7–12.8 mm in length and 338–428 μm in width for the Italy isolates), and the females measured 11.2–15.3 mm in length and 311–459 μm in width (12–18.5 mm in length and 370–510 μm for the Italy isolates).
In recent years, the mitochondrial genomes of spirurid nematodes have been extensively studied (Ghedin et al., 2007; Yatawara et al., 2010; Park et al., 2011; McNulty et al., 2012; Liu et al., 2015; Yilmaz et al., 2016). These whole mt genomes will serve as a useful dataset for studying the genetics, systematics and phylogenetic relationships of spirurid nematodes. In accordance with the two known mt genomes of T. callipaeda (NC_018363 and AP017700), all T. callipaeda isolates from central China contained 12 PCGs, 22 transfer RNA genes, two ribosomal RNA genes and a non-coding region but lacked an atp8 gene (Liu et al., 2013a). The gene content and arrangement are the same as those of Acanthocheilonema viteae, Brugia species, Chandlerella quiscali, Dirofilaria species, Loa loa, Onchocerca species, Wuchereria bancrofti, Gongylonema pulchrum, Setaria digitata (Ghedin et al., 2007; Yatawara et al., 2010; McNulty et al., 2012; Yilmaz et al., 2016), but distinct from those of Dracunculus medinensis, Philometroides sanguineus, Gnathostoma doloresi, and Heliconema longissimum (Su et al., 2016; Sun et al., 2016). The mt genome sequence of T. callipaeda has high a A+T content (above 70%), which is consistent with the mt genomes of nematodes from the order Spirurida (Yatawara et al., 2010; McNulty et al., 2012). All PCGs had ATG, TTT, ATA, or TTG as their initiation codons, and TAA or TAG as their termination codons, which was consistent with a previous study (Liu et al., 2013a). The sizes of the 22 tRNA genes identified here were similar to those of other spirurid nematodes, indicating their similar functions (Liu et al., 2013b; Yilmaz et al., 2016). The locations of rrnL and rrnS were also identical to previously published mt genomes of Thelazia. The nucleotide sequences of the mtDNA of five T. callipaeda isolated from different geographical locations were conspicuously similar, which indicated that the influence of genetic variations on the phylogenetic distances in T. callipaeda is relatively small (Zhang et al., 2015). The mt genome of the T. callipaeda isolate from Guangdong (NC_018363) was the most divergent of the entire set. The results of sequence divergence of PCGs suggested that nad4L is the most conserved protein-coding gene. Nad6 showed high sequence variability among the five isolates, indicating that this gene is suitable as a genetic marker for population genetic studies of T. callipaeda from different geographic origins.
Although nuclear ribosomal genes (rDNA) have been verified for phylogenetic studies of a wide range of organisms (Nadler et al., 2007; Zhang et al., 2017), in comparison with a single gene or a single region of rDNA, the complete mt genome sequences have significantly improved the power of phylogenetic analysis and made possible more accurate resolution of taxonomic relationships even at deep levels (Brabec et al., 2016). For spirurid nematodes, the mt genomes have been applied particularly to studies regarding phylogeny and evolution (Park et al., 2011; Liu et al., 2013a, 2015; Yilmaz et al., 2016). To date, there are 24 mitogenomes of the order Spirurida in GenBank representing eight families. Our study brings a new perspective by presenting additional mt genomic data of T. callipaeda from three clinical isolates. These mtDNA sequences will be useful for current molecular taxonomy studies focused on Thelazia species. In our phylogenetic analyses, the taxonomic relationships of Spirurida based on different data partitions and different inference methods were generally congruent. In general, the tree topology of selected spirurid nematodes can be divided into two main clades: one small clade only contained the family Gnathostomatidae, and the other large one included Onchocercidae, Setariidae, Gnathostomatidae, Thelaziidae, Physalopteridae, Dracunculidae, and Philometridae. The position of Gnathostomatidae was also supported by Liu et al. (2015). Within the large clade, Dracunculidae, Philometridae, and Physalopteridae were in the basal, which was consistent with previous publications (Park et al., 2011; Liu et al., 2013a, 2015). The close relationships among Onchocercidae, Setariidae, and Gnathostomatidae was verified in studies of Park et al. (2011); McNulty et al. (2012), and Liu et al. (2015). In this study, these relationships have also been supported via the addition of more representative sequences. For Thelaziidae, the monophyly of T. callipaeda isolates from different geographical locations was undoubted. However, the position of Spirocerca lupi was ambiguous. In the previous studies by Liu et al. (2013a, 2015) and Yilmaz et al. (2016), S. lupi was placed in the family Spiruridae. However, in the NCBI taxonomy browser2, Spirocerca lupi is included within the family Thelaziidae. Increased sampling and molecular marker analysis should be conducted to determine the exact phylogenetic position of S. lupi. Our results also supported Spirocerca lupi as a single clade with a close relationship to the family Gongylonematidae.
Conclusion
In this study, three cases of thelaziasis were reported in central China, and all clinical worms were identified as T. callipaeda according to several key characteristics examined under light microscopy and scanning electron microscopy. In addition, the whole mitochondrial genomes of three T. callipaeda isolates from different geographical locations were sequenced using next-generation sequencing methods. The genome featured 12 PCGs, 22 transfer RNA genes, two ribosomal RNA genes and a non-coding region, which was similar to that of most of other spirurid nematodes. The mtDNA nucleotide sequences of these isolates from different locations were conspicuously similar. The nad6 gene showed high sequence variability among all isolates, which is worth considering for future population genetic studies of T. callipaeda. Phylogenetic analyses support the monophyly of T. callipaeda isolates from different hosts and distinct geographical locations. However, the taxonomic position of Spirocerca lupi is still ambiguous.
Author Contributions
Conceived and designed the experiments: XZ, ZW, and JC. Performed the experiments: XZ, YS, JD, PJ, and RL. Analyzed the data: XZ. Contributed reagents/materials/analysis tools: JC, XZ, and ZW. Wrote the paper: XZ, ZW, and JC. All authors read and approved the final version of the manuscript.
Funding
This work was supported by the Outstanding Young Talent Research Fund of Zhengzhou University (No. 1521328001).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.01335/full#supplementary-material
Footnotes
References
Andrews, S. (2010). FastQC: A Quality Control Tool for High Throughput Sequence Data. Available at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc
Bernt, M., Donath, A., Jühling, F., Externbrink, F., Florentz, C., Fritzsch, G., et al. (2013). MITOS: improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 69, 313–319. doi: 10.1016/j.ympev.2012.08.023
Brabec, J., Kuchta, R., Scholz, T., and Littlewood, D. T. (2016). Paralogues of nuclear ribosomal genes conceal phylogenetic signals within the invasive Asian fish tapeworm lineage: evidence from next generation sequencing data. Int. J. Parasitol. 46, 555–562. doi: 10.1016/j.ijpara.2016.03.009
Darriba, D., Taboada, G. L., Doallo, R., and Posada, D. (2012). jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 9:772. doi: 10.1038/nmeth.2109
Fuentes, I., Montes, I., Saugar, J. M., Latrofa, S., Gárate, T., and Otranto, D. (2012). Thelaziosis in humans, a zoonotic infection, Spain, 2011. Emerg. Infect. Dis. 18, 2073–2075. doi: 10.3201/eid1812.120472
Ghedin, E., Wang, S., Spiro, D., Caler, E., Zhao, Q., Crabtree, J., et al. (2007). Draft genome of the filarial nematode parasite Brugia malayi. Science 317, 1756–1760. doi: 10.1126/science.1145406
Hodžić, A., Latrofa, M. S., Annoscia, G., Aliæ, A., Beck, R., Lia, R. P., et al. (2014). The spread of zoonotic Thelazia callipaeda in the Balkan area. Parasit. Vectors 7:352. doi: 10.1186/1756-3305-7-352
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Liu, G. H., Gasser, R. B., Otranto, D., Xu, M. J., Shen, J. L., Mohandas, N., et al. (2013a). Mitochondrial genome of the eyeworm, Thelazia callipaeda (Nematoda: Spirurida), as the first representative from the family Thelaziidae. PLoS Negl. Trop. Dis. 7:e2029. doi: 10.1371/journal.pntd.0002029
Liu, G. H., Shao, R., Cai, X. Q., Li, W. W., and Zhu, X. Q. (2015). Gnathostoma spinigerum mitochondrial genome sequence: a novel gene arrangement and its phylogenetic position within the class chromadorea. Sci. Rep. 5:12691. doi: 10.1038/srep12691
Liu, G. H., Wang, Y., Song, H. Q., Li, M. W., Ai, L., Yu, X. L., et al. (2013b). Characterization of the complete mitochondrial genome of Spirocerca lupi: sequence, gene organization and phylogenetic implications. Parasit. Vectors 6, 45. doi: 10.1186/1756-3305-6-45
McNulty, S. N., Mullin, A. S., Vaughan, J. A., Tkach, V. V., Weil, G. J., and Fischer, P. U. (2012). Comparing the mitochondrial genomes of Wolbachia-dependent and independent filarial nematode species. BMC Genomics 13:145. doi: 10.1186/1471-2164-13-145
Mihalca, A. D., D’Amico, G., Scurtu, I., Chirilã, R., Matei, I. A., and Ionicã, A. M. (2015). Further spreading of canine oriental eyeworm in Europe: first report of Thelazia callipaeda in Romania. Parasit. Vectors 8:48. doi: 10.1186/s13071-015-0663-2
Nadler, S. A., Carreno, R. A., Mejía-Madrid, H., Ullberg, J., Pagan, C., Houston, R., et al. (2007). Molecular phylogeny of clade III nematodes reveals multiple origins of tissue parasitism. Parasitology 134, 1421–1442. doi: 10.1017/S0031182007002880
Nguyen, V. D., Thanh, H. L., and Chai, J. Y. (2012). The first human case of Thelazia callipaeda infection in Vietnam. Korean J. Parasitol. 50, 221–223. doi: 10.3347/kjp.2012.50.3.221
Otranto, D., and Dutto, M. (2008). Human thelaziasis, Europe. Emerg. Infect. Dis. 14, 647–649. doi: 10.3201/eid1404.071205
Otranto, D., Lia, R. P., Buono, V., Traversa, D., and Giangaspero, A. (2004). Biology of Thelazia callipaeda (Spirurida, Thelaziidae) eyeworms in naturally infected definitive hosts. Parasitology 129, 627–633. doi: 10.1017/S0031182004006018
Otranto, D., Lia, R. P., Cantacessi, C., Testini, G., Troccoli, A., Shen, J. L., et al. (2005a). Nematode biology and larval development of Thelazia callipaeda (Spirurida, Thelaziidae) in the drosophilid intermediate host in Europe and China. Parasitology 131, 847–855.
Otranto, D., Lia, R. P., Traversa, D., and Giannetto, S. (2003). Thelazia callipaeda (Spirurida, Thelaziidae) of carnivores and humans: morphological study by light and scanning electron microscopy. Parassitologia 45, 125–133.
Otranto, D., Testini, G., De Luca, F., Hu, M., Shamsi, S., and Gasser, R. B. (2005b). Analysis of genetic variability within Thelazia callipaeda (Nematoda: Thelazioidea) from Europe and Asia by sequencing and mutation scanning of the mitochondrial cytochrome c oxidase subunit 1 gene. Mol. Cell Probes. 19, 306–313.
Otranto, D., and Traversa, D. (2004). Molecular characterization of the first internal transcribed spacer of ribosomal DNA of the most common species of eyeworms (Thelazioidea: Thelazia). J. Parasitol. 90, 185–188. doi: 10.1645/GE-115R1
Park, J. K., Sultana, T., Lee, S. H., Kang, S., Kim, H. K., Min, G. S., et al. (2011). Monophyly of clade III nematodes is not supported by phylogenetic analysis of complete mitochondrial genome sequences. BMC Genomics 12:392. doi: 10.1186/1471-2164-12-392
Ramesh, A., Small, S. T., Kloos, Z. A., Kazura, J. W., Nutman, T. B., Serre, D., et al. (2012). The complete mitochondrial genome sequence of the filarial nematode Wuchereria bancrofti from three geographic isolates provides evidence of complex demographic history. Mol. Biochem. Parasitol. 183, 32–41. doi: 10.1016/j.molbiopara.2012.01.004
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Shen, J., Gasser, R. B., Chu, D., Wang, Z. X., Yuan, X., Cantacessi, C., et al. (2006). Human thelaziosis—a neglected parasitic disease of the eye. J. Parasitol. 92, 872–875. doi: 10.1645/GE-823R.1
Su, Y. B., Kong, S. C., Wang, L. X., Chen, L., and Fang, R. (2016). Complete mitochondrial genome of Philometra carassii (Nematoda: Philometridae). Mitochondrial DNA A DNA Mapp. Seq. Anal. 27, 1397–1398. doi: 10.3109/19401736.2014.947598
Sun, M. M., Ma, J., Sugiyama, H., Ando, K., Li, W. W., Xu, Q. M., et al. (2016). The complete mitochondrial genomes of Gnathostoma doloresi from China and Japan. Parasitol. Res. 115, 4013–4020. doi: 10.1007/s00436-016-5171-6
Swofford, D. L. (2003). PAUP∗. Phylogenetic Analysis Using Parsimony (∗and Other Methods). Version 4.0b10. Sunderland, MA: Sinauer Associates.
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Wang, X. L., Guo, J. Y., Wang, X. L., Ma, X. L., Wang, Y., and An, C. L. (2014). Two cases of human thelaziasis as confirmed by mitochondrial cox1 sequencing in China. Pathog. Glob. Health 108, 298–301. doi: 10.1179/2047773214Y.0000000153
Xia, X., and Xie, Z. (2001). DAMBE: software package for data analysis in molecular biology and evolution. J. Hered. 92, 371–373. doi: 10.1093/jhered/92.4.371
Xia, X., Xie, Z., Salemi, M., Chen, L., and Wang, Y. (2003). An index of substitution saturation and its application. Mol. Phylogenet. Evol. 26, 1–7. doi: 10.1016/S1055-7903(02)00326-3
Yatawara, L., Wickramasinghe, S., Rajapakse, R. P., and Agatsuma, T. (2010). The complete mitochondrial genome of Setaria digitata (Nematoda: Filarioidea): mitochondrial gene content, arrangement and composition compared with other nematodes. Mol. Biochem. Parasitol. 173, 32–38. doi: 10.1016/j.molbiopara.2010.05.004
Yilmaz, E., Fritzenwanker, M., Pantchev, N., Lendner, M., Wongkamchai, S., Otranto, D., et al. (2016). The mitochondrial genomes of the zoonotic canine filarial parasites Dirofilaria (Nochtiella) repens and Candidatus Dirofilaria (Nochtiella) honkongensis provide evidence for presence of cryptic species. PLoS Negl. Trop. Dis. 10:e0005028. doi: 10.1371/journal.pntd.0005028
Zhang, X., Duan, J. Y., Wang, Z. Q., Jiang, P., Liu, R. D., and Cui, J. (2017). Using the small subunit of nuclear ribosomal DNA to reveal the phylogenetic position of the plerocercoid larvae of Spirometra tapeworms. Exp. Parasitol. 175, 1–7. doi: 10.1016/j.exppara.2017.01.006
Zhang, X., Wang, H., Cui, J., Jiang, P., Fu, G. M., Zhong, K., et al. (2015). Characterisation of the relationship between Spirometra erinaceieuropaei and Diphyllobothrium species using complete cytb and cox1 genes. Infect. Genet. Evol. 35, 1–8. doi: 10.1016/j.meegid.2015.07.025
Keywords: Thelazia callipaeda, thelaziasis, mitochondrial genome, next generation sequencing, phylogeny
Citation: Zhang X, Shi YL, Wang ZQ, Duan JY, Jiang P, Liu RD and Cui J (2017) Morphological and Mitochondrial Genomic Characterization of Eyeworms (Thelazia callipaeda) from Clinical Cases in Central China. Front. Microbiol. 8:1335. doi: 10.3389/fmicb.2017.01335
Received: 20 March 2017; Accepted: 30 June 2017;
Published: 13 July 2017.
Edited by:
Wei Hu, Fudan University, ChinaReviewed by:
David Blair, James Cook University Townsville, AustraliaDonald McManus, QIMR Berghofer Medical Research Institute, Australia
Copyright © 2017 Zhang, Shi, Wang, Duan, Jiang, Liu and Cui. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhong Q. Wang, d2FuZ3pxQHp6dS5lZHUuY24= Jing Cui, Y3VpakB6enUuZWR1LmNu