 Khalid El Karkouri
Khalid El Karkouri Malgorzata Kowalczewska
Malgorzata Kowalczewska Nicholas ArmstrongSaid AzzaPierre-Edouard Fournier*
Nicholas ArmstrongSaid AzzaPierre-Edouard Fournier* Didier Raoult
Didier Raoult- Unité de Recherche en Maladies Infectieuses et Tropicales Emergentes, UM63, Centre National De La Recherche Scientifique 7278, IRD 198, Institut National De La Santé Et De La Recherche Médicale U1095, Institut Hospitalo-Universitaire Méditerranée-Infection, Aix-Marseille Université, Marseille, France
Arthropod-borne Rickettsia species are obligate intracellular bacteria which are pathogenic for humans. Within this genus, Rickettsia slovaca and Rickettsia conorii cause frequent and potentially severe infections, whereas Rickettsia raoultii and Rickettsia massiliae cause rare and milder infections. All four species belong to spotted fever group (SFG) rickettsiae. However, R. slovaca and R. raoultii cause scalp eschar and neck lymphadenopathy (SENLAT) and are mainly associated with Dermacentor ticks, whereas the other two species cause Mediterranean spotted fever (MSF) and are mainly transmitted by Rhipicephalus ticks. To identify the potential genes and protein profiles and to understand the evolutionary processes that could, comprehensively, relate to the differences in virulence and pathogenicity observed between these four species, we compared their genomes and proteomes. The virulent and milder agents displayed divergent phylogenomic evolution in two major clades, whereas either SENLAT or MSF disease suggests a discrete convergent evolution of one virulent and one milder agent, despite their distant genetic relatedness. Moreover, the two virulent species underwent strong reductive genomic evolution and protein structural variations, as well as a probable loss of plasmid(s), compared to the two milder species. However, an abundance of mobilome genes was observed only in the less pathogenic species. After infecting Xenopus laevis cells, the virulent agents displayed less up-regulated than down-regulated proteins, as well as less number of identified core proteins. Furthermore, their similar and distinct protein profiles did not contain some genes (e.g., ompA/B and rickA) known to be related to rickettsial adhesion, motility and/or virulence, but may include other putative virulence-, antivirulence-, and/or disease-related proteins. The identified evolutionary forces herein may have a strong impact on intracellular expressions and strategies in these rickettsiae, and that may contribute to the emergence of distinct virulence and diseases in humans. Thus, the current multi-omics data provide new insights into the evolution and fitness of SFG virulence and pathogenicity, and intracellular pathogenic bacteria.
Introduction
Rickettsia species (Order Rickettsiales, Family Rickettsiaceae) are obligate intracellular bacteria that diverged into three major phylogenetic groups, with arthropod hosts worldwide (Stothard et al., 1994; Raoult and Roux, 1997). This includes the spotted fever group (SFG) associated with ticks, fleas and mites, the typhus group (TG), including Rickettsia prowazekii and Rickettsia typhi associated with body lice and rat fleas, respectively, and a group containing Rickettsia bellii and R. canadensis, associated with ticks. Recent studies have divided the SFG group into distinct phylogenetic subgroups (Gillespie et al., 2007; Merhej and Raoult, 2011; Merhej et al., 2014). Moreover, it has been reported that other Rickettsia lineages exist, notably associated with amoebas, medusae, ciliates, leeches, or arthropods (Weinert et al., 2009; Merhej and Raoult, 2011; Murray et al., 2016). During their lifecycle, rickettsiae can also infect mammalian hosts, mostly through arthropod bites or feces, causing damage, morbidity, and mortality, as well as a range of mild to severe diseases, such as epidemic typhus and Rocky Mountain spotted fever (RMSF; Parola et al., 2013; Sahni et al., 2013; Portillo et al., 2015). Some Rickettsia spp. have been classified as Category B or C bioterrorism pathogens by the National Institute of Allergy and Infectious diseases (NIAID) and/or the Centers for Disease Control and Prevention (CDC; Chan et al., 2010).
The long-term adaptation of pathogenic bacteria in eukaryotic cells (i.e., in bottleneck ecosystems), allowed them to become allopatric and specialists, and eventually to undergo reductive genome evolution (Merhej et al., 2009; Georgiades and Raoult, 2010). This dominant mode of evolution, in sequestrated intracellular parasites and symbionts from horizontal gene transfers, leads to a pseudogene-riddled genome, loss of non-essential genes and biosynthetic pathway components, as well as survival, by taking advantage of host cell metabolites (Andersson et al., 1998; Ogata et al., 2001; Audia and Winkler, 2006; Blanc et al., 2007a; Darby et al., 2007; Fournier et al., 2009; Sahni and Rydkina, 2009; Wolf and Koonin, 2013). Several intracellular pathogenic bacteria, including Rickettsia, Mycobacterium, and Streptococcus spp., have genomes smaller than less dangerous and cognate species, suggesting that enhanced virulence may be associated with reductive evolution (Demangel et al., 2009; Fournier et al., 2009; Merhej et al., 2014), rather than acquisition of virulence factors (Merhej et al., 2009; Georgiades and Raoult, 2010; Georgiades et al., 2011; Merhej et al., 2013).
Most molecular investigations on rickettsial-host interactions have identified several surface-exposed proteins (e.g., cell surface antigens, Scas), secretome and genes that may play fundamental roles in rickettsial infection pathogenicity and/or virulence (for reviews see Merhej and Raoult, 2011; Gillespie et al., 2015; Merhej et al., 2013; Sahni et al., 2013). However, recent genomic studies have narrowed the field of possible virulence factors of the sca5 (ompB) gene in Rickettsia rickettsii strains that differ in severity of disease (Clark et al., 2015). Moreover, a knockout of the sca0 (cell surface antigens, ompA) gene in the virulent SFG R. rickettsii strain Sheila Smith concluded that this gene is not critical for virulence in the guinea pig model, but may play a role in survival or transmission from the tick vector (Ellison et al., 2008; Noriea et al., 2015). In another example, the rickA gene plays an important role in actin-based bacterial motility (Ogata et al., 2001; Gouin et al., 2004), a phenotype that has been associated with Shigella spp. and Listeria monocytogenes pathogenicity (Frischknecht and Way, 2001; Pollard and Borisy, 2003). However, the relationships between rickettsial pathogenicity and rickA remain questionable. Indeed, it is present in the avirulent and virulent R. rickettsii strains (Ellison et al., 2008), but also is pseudogenized, remnant, mutated or absent as sca0, 1 and/or 2 genes in the most pathogenic and non-motile species R. prowazekii, the less pathogenic and motile R. typhi, and/or in the non-pathogenic Rickettsia peacockii (Ogata et al., 2001; Balraj et al., 2008; Felsheim et al., 2009; Sears et al., 2012). Thus, understanding the mechanisms governing rickettsial pathogenicity and virulence outcomes in human hosts needs to be elucidated, using integrative and modern gel-free omics approaches before for example gel-based or isogenic methods. As an example, in Mycobacterium tuberculosis, the LC-MS/MS proteomic analysis identified high numbers of proteins (from 691 to 983), of which several were up-regulated, down-regulated or unique during the dormancy and reactivation of a virulent strain (Gopinath et al., 2015).
This study focused on four SFG Rickettsia species exhibiting differences in ecologic and biologic features, in which the genetic basis remains sparse. Rickettsia slovaca and R. raoultii, which are mainly associated with Dermacentor ticks, cause scalp eschar and neck lymphadenopathy (SENLAT) in humans (Raoult and Roux, 1997; Parola et al., 2009). Rickettsia conorii and Rickettsia massiliae, which are most often associated with Rhipicephalus ticks (Parola et al., 2013), cause Mediterranean spotted fever (MSF) in humans (Milhano et al., 2014; Bechelli et al., 2015; Portillo et al., 2015). However, R. slovaca and R. conorii share the common characteristics of being both less prevalent in their respective tick vectors and more pathogenic for humans than their counterparts. In Dermacentor ticks, R. slovaca, and R. raoultii exhibit prevalence of 0–4 and 10–82%, respectively (Parola et al., 2009; Milhano et al., 2010; Jiang et al., 2012; Speck et al., 2012; Spitalska et al., 2012; Wen et al., 2014) but in humans, they are detected in 57 and 8% of SENLAT cases, respectively (Parola et al., 2009; Foissac et al., 2013). Similarly, R. conorii and R. massiliae exhibit a prevalence of 0–0.7 and 8–17% in Rhipicephalus ticks (Fernandez-Soto et al., 2006a,b; Marquez et al., 2008), whereas the former is highly virulent and causes severe MSF with a mortality rate up to 30%, and the latter only causes a mild MSF disease (Cascio et al., 2013; Bechelli et al., 2015). Thus, to identify potential genes and protein profiles, as well as to understand the driving forces that could, comprehensively, relate to the differences in virulence and pathogenicity in humans of these four SFG Rickettsia species, we compared their genomic sequences and proteomic profiles and examined their evolutionary relationships.
Materials and Methods
Genomic Analysis
Herein, we studied four rickettsiae, including the Dermacentor-transmitted species R. slovaca strain 13-BT (CSUR R154, Fournier et al., 2012) and R. raoultii strain KhabarovskT (CSUR R3, ATCC VR-1596, El Karkouri et al., 2016) that cause SENLAT, and the Rhipicephalus-associated R. conorii strain Malish 7T (CSUR R41, ATCC VR-613, Ogata et al., 2001) and the R. massiliae strain MTU5 (CSUR R132, Blanc et al., 2007b) causing MSF. For each disease, the former species caused a more severe infection. The four species were obtained from the French “Collection de Souches de l'Unité des Rickettsies” (CSUR).
Genomic sequences of the four species were downloaded from the NCBI FTP server (ftp://ftp.ncbi.nih.gov/Genome/). To avoid potential biases across the originally published data, including unpredicted Open Reading Frames from pseudogenes in the GenBank database that were generated by different gene identification and annotation systems, all genomes were subjected to a standard re-annotation, including CDSs (coding sequences) prediction with the AMIGene software (Bocs et al., 2003). The automatic assignment of protein functions was performed against the RickBase (Blanc et al., 2007a) and non-redundant NR databases using PipRick (an in-house annotation pipeline written in Perl language) and BLASTp algorithm (Altschul et al., 1997). The annotations were then curated and genes that were either complete or altered (split or fragment) were distinguished (Blanc et al., 2007a). Functional classification of gene families (COG ID and Letters) was searched using COGsoft software against the Clusters of Orthologs Groups (COG) database (Kristensen et al., 2010). The pan-genome between the four species was constructed by subjecting predicted proteomes to a reciprocal best BLAST hit (BBH) algorithm with all-against-all search (coverage of the query length ≥60% and E-value < 10−10) using COGsoft software. Each cluster of orthologous groups of rickettsial genes from chromosomes and plasmids was named cRIGs and pRIGs, respectively. The Venn diagrams of pan-genome and pan-proteome were constructed using the Jvenn Javascript library (Bardou et al., 2014). To identify putative virulence factors, a BLASTp search was performed against the virulence factor database, VFDB (Chen et al., 2016). Multiple sequence alignments of the core genes were carried out using MAFFT software (Katoh et al., 2005). Phylogenetic analysis was performed with the maximum likelihood (ML) method under the JTT amino acid substitution matrix, the Nearest-Neighbor-Interchange (NNI), the gamma (Γ) distribution of parameter α to account for substitution rate heterogeneity among sites and complete deletion and the rectangular tree using MEGA software (Tamura et al., 2013). Moreover, a neighbor-joining (NJ) tree was constructed from a gene content distance matrix calculated according to the pan-genome data and Jaccard's dissimilarity coefficient (El Karkouri et al., 2006). The robustness of the nodes in both ML and NJ trees was estimated through Bootstrap (BP) analyses of 100 and 500 replicates with MEGA software and the PHYLIP package (Website: http://evolution.genetics.washington.edu/phylip.html), respectively. To assess the genomic differences in predicted proteins of the core genes between the four species, the percentage of the amino acid identities and the numbers of non-synonymous mutations and insertions/deletions (InDels) were computed using the Smith-Waterman BLASTp search algorithm. For this four-way analysis, the E-value cutoff < 10−10 was used and the false positive matches were removed.
Rickettsia Culture and Purification
A confluent monolayer of the Xenopus laevis cell line (XTC-2 cells) in Leibovitz's L-15 medium supplemented with tryptose phosphate buffer (5%) and fetal bovine serum (4%; Life Technologies) was inoculated with a Rickettsia species, as described by Saisongkorh et al. (2012). For each studied Rickettsia species, about 25 × 104–5 × 105 bacteria were inoculated in X. laevis cells (one 150 cm2 flask each, containing 25 ml of fresh medium) and incubated at 28°C for 5 days. Then, each species was subcultured in 20 flasks prior to being collected, pooled and purified individually on a discontinuous renografin gradient, as previously reported (Eremeeva et al., 1994). Purified rickettsiae counted 1.6 × 107 bacteria for R. slovaca, 5 × 1010 bacteria for R. conorii, 2.4 × 1010 bacteria for R. raoultii and 3.4 × 109 bacteria for R. massiliae. They were then washed in PBS at 10,000 × g at 4°C for 10 min, and then stored at −80°C for further analysis. All infection and purification steps were monitored by Gimenez staining (Gimenez et al., 1964). Bacterial quantities of both steps were assessed using quantitative real-time PCR (qPCR) with the 1,029 system based on the RC0338 hypothetical protein gene of all SFG rickettsiae (Socolovschi et al., 2012).
Preparation of Proteins for Nano-LC/MS/MS
Aliquots of purified bacteria of each species were pelleted, PBS discarded and then resuspended in 200 μl of lysis buffer (7 M urea, 2 M thiourea, 4% w/v CHAPS, 30 mM Tris-HCl, pH 8.0) and lysed by sonication (Vellaiswamy et al., 2011). Soluble proteins were then dialyzed twice using Slide-A-Lyzer Dialysis Cassettes 2K MWCO (Pierce Biotechnology, Rockford, USA) against 1 L of 50 mM ammonium bicarbonate pH 7.4, 1 M urea (7 h and overnight). The total soluble proteins of dialyzed fractions were quantified by Bradford assay (Biorad, Marnes-la-Coquette, France). Disulphide bonds were reduced by treating 50 μg of soluble proteins of each sample with 10 mM DL-dithiothreitol (Euromedex, Souffelweyersheim, France) in 50 mM ammonium bicarbonate (Sigma, Saint-Quentin Fallavier, France) buffer at room temperature for 1 h. The proteins were subsequently alkylated with 20 mM iodoacetamide (Sigma, Saint-Quentin Fallavier, France) in the same buffer at room temperature in the dark. The alkylated proteins were then digested with 2 μg of sequencing-grade trypsin (Promega, Charbonnières, France) overnight at 37°C. The digested peptide solutions were then desalted using Pierce Detergent Removal Spin Columns (Thermo Fisher Scientific, Illkirch, France) and stored at −20°C until the LC/MS/MS analysis.
Liquid Chromatography and Mass Spectrometry
The peptide samples were analyzed using a nano ACQUITY 2D-UPLC system coupled to a Quadrupole Time-of-Flight (Q/TOF) traveling wave ion mobility hybrid mass spectrometer (SYNAPT-G2SI, Waters, Guyancourt, France). Both systems were operated and controlled by MassLynx4.1 software (Waters, Guyancourt, France). All solvents used were ULC-MS grade (Biosolve, Dieuze, France). Each peptide sample was run in three analytical replicates. The digested peptide solutions (equivalent to about 1.4 μg of proteins/μl) were 6-time diluted and spiked with 10 fmol/μl of digested yeast alcohol dehydrogenase (ADH; MassPREP, Waters, Guyancourt, France). Briefly, 3 μl of each peptide sample, equivalent to about 700 ng of proteins was fractionated using a nano 2D chromatography setup and then monitored using a high definition HD-MSE method (Waters, Guyancourt, France), as previously described (Reteno et al., 2015). Briefly, peptides were collected on a first reverse phase column at high pH 10, and then, seven eluting peptide fractions were successively trapped after an online dilution. Each fraction was separated at low pH 2.5 on a second reverse phase column. The mass spectrometry proteomics data have been deposited in the ProteomeXchange Consortium (Vizcaino et al., 2014) via the PRIDE partner repository with the following dataset identifiers: PXD003193, PXD003194, PXD003195, and PXD003197.
Data Processing and Analysis
Raw MSE data from each biological sample were processed using the ProteinLynx Global SERVER v3.0.1 (PLGS, Waters, Guyancourt, France) for protein identification and protein quantification. Noise reduction thresholds for low energy scan ion, high energy scan ion and peptide intensity were set at 1,000, 100, and 800 counts, respectively. A mass correction was applied to all spectra using the Leucine Enkephalin lock mass calibrant at 785.8426 m/z. For each sample, a single mass spectrum file was generated by merging the mass spectra from the seven fractions. To perform the protein sequence database search, we constructed four distinct databases, each containing annotated protein sequences of one Rickettsia species. For each database, we also included protein sequences of the X. laevis species downloaded from the NCBI database, the common contaminants and the yeast ADH sequence (accession number: P00330|ADH1) from the universal protein Knowledgebase, UniProt (UniProt, 2017). The default parameters used for global peptide and protein identifications were set as follows (see Gopinath et al., 2015): at least one fragment ion match per peptide, at least three fragment ion matches per protein, at least one peptide matches for protein identification, mass tolerances was set to automatic with a window of 10 ppm for precursor ions and a window of 20 ppm for fragment ions, at least one positive charge, oxidation of methionine (M) as variable modification and carbamidomethylation (C) of cysteine as the fixed modification, and the trypsin was selected as the enzyme with up to one miss-cleavage. The initial protein false discovery rate (FDR) of the identification algorithm was set at 4% with a randomized database, leading to a peptide FDR that was typically smaller than 1% (Brioschi et al., 2013; Gopinath et al., 2015). Protein quantities were evaluated in the injected solution using the combined intensity of the three most abundant peptides per protein compared to the quantitatively added yeast ADH digest (Hi3 absolute quantification, PLGS, Waters, Guyancourt, France; Silva et al., 2006).
Only proteins identified by at least two matched peptides were considered for proteomic analysis (Treitz et al., 2015). As the three replicates of each sample showed high reproducibility with high significant linear correlations (Spearman's and Pearson's correlations, P < 0.001), we grouped each three replicates and calculated the average abundances (fmol or fmol μg−1) of each protein. The fmol averages of each sample were also normalized by the median using the Perseus Software (v1.5.1.6; www.maxquant.org, Treitz et al., 2015). Moreover, using each protein's molecular weight from the database, the fmol quantity of each protein was converted to nanograms for the triplicates, and by summing the average ng of all proteins we obtained the total average ng in each sample (Saka et al., 2011). To calculate the species protein abundance for each sample, the fmol average for each protein was divided by the average ng sum for only the sample of that species, and scaled by 1,000 to yield fmol μg−1 (Saka et al., 2011). The ratios describing the condition virulent/milder species of two comparative models (R. slovaca/R. raoultii and R. conorii/R. massiliae) were then log2 transformed. Orthologous proteins with stringently defined fold change (FC ≥ 2) were considered to represent up-regulation (log2 ratio ≥ 1) or down-regulation (log2 ratio ≤ −1), whereas orthologous proteins with a FC < 2 were considered as equally regulated (Son et al., 2015). Proteins with FC = 2 included a minimal and a well-represented abundance value of 0.5 fmol. Thus, orthologous proteins with stringent abundance values (≥0.5 fmol) in one species, but with zero abundance values in the second species, were also comprehensively annotated as up-regulated or down-regulated. In contrast, proteins with these abundance values (≥0.5 fmol) in only one species (i.e., the gene is absent in the second species) were annotated as specific or unique (Gopinath et al., 2015). Other proteins that did not match these criteria were annotated as un-classified.
Statistical Analysis
Statistical analyses were processed using the R Commander software (http://r-forge.r-project.org). These included Pearson's and Spearman's correlation coefficients (R and Rho, resp.), Chi-square test (χ2) and Fisher's exact test.
Results
Genomics of SFG Rickettsia spp.
The general features of the four Rickettsia genomes are summarized in Figure 1. The chromosomes of the virulent R. slovaca and R. conorii agents were from 70 to 92 kbp smaller than those of the milder R. raoultii and R. massiliae agents. The former species are plasmidless, whereas the latter harbored one to three plasmid(s) (15–83 kbp). Moreover, about 74–75 and 56–64% of genes in the chromosomes and plasmids were found complete, respectively, whereas the remaining genes were either split or fragments.
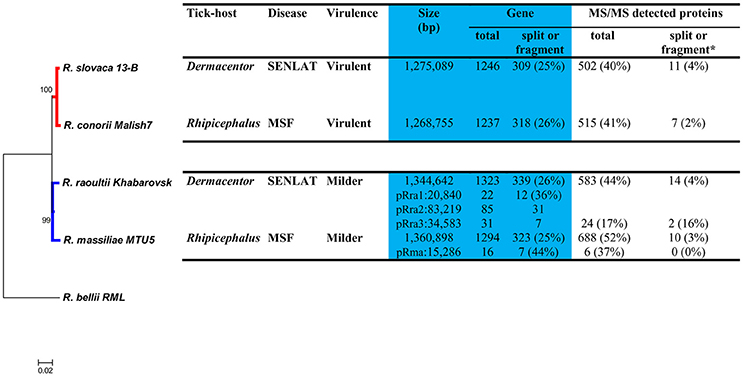
Figure 1. Phylogenomic tree and, biologic, pathogenic, genomic, and proteomic features, of four SFG Rickettsia species. Bootstrap supports higher than 90% are shown at the nodes. *Means that the total of the detected MS/MS proteins corresponded to genes which are either split or fragment.
The pan-genome analysis from the four Rickettsia spp. identified 1,531 and 115 clusters of rickettsial orthologous genes from the chromosomes (cRIGs) and plasmids (pRIGs), respectively (Figure 2). The four species displayed a core-chromosome of 1,084 (71% of 1,531) cRIGs. The pairwise Smith-Waterman similarity search in the 1,084 core genes revealed that the two virulent agents exhibited more conservation in their core genes with a high average aa identity and significant low numbers of non-synonymous (NS) mutations and InDels (Rsl vs. Rco: 98%, 3,335 and 933, resp.) when compared with the two milder species (Rra vs. Rma, 96%, 7,888 and 1,495, P ≤ 0.001, χ2 test, resp.). In the four-way comparisons between one virulent and one milder agent (e.g., Rsl vs. Rra), the conservations in the core genes (average aa identity: 95%) were found to be higher and slightly lower than those obtained between the two virulent agents (98%) and between the two milder agents (96%), respectively. Moreover, the total numbers of NS mutations and InDels observed between one virulent and one milder agent (Rsl vs. Rra: 8,756 and 1,442, resp.; Rsl vs. Rma: 9,126 and 1,659, resp.; Rco vs. Rra: 9,470 and 1,471, resp.; Rco vs. Rma: 9,808 and 1,523) were significantly higher than those found between the two virulent agents (Rsl vs. Rco: 3,335 and 933, resp.) (P ≤ 0.001, χ2 tests) and between the two milder agents (Rra vs. Rma, 7,888 and 1,495, resp.) (P ≤ 0.001, χ2 tests).
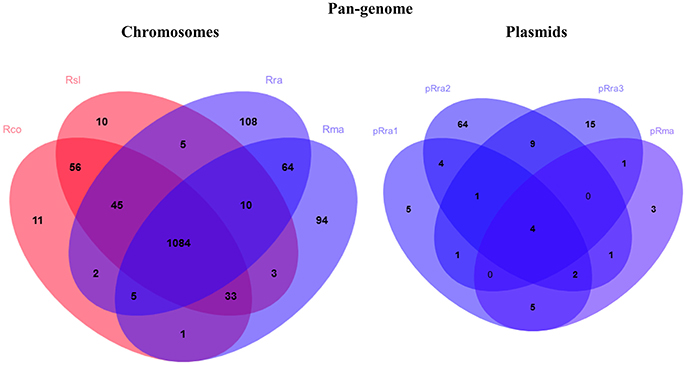
Figure 2. Venn diagrams summarizing pan-genome of the virulent R. slovaca Rsl and the milder R. raoultii Rra causing SENLAT, and the virulent R. conorii Rco and the milder R. massiliae Rma causing MSF.
The phylogenomic tree performed from 330 concatenated core genes revealed that the four pathogenic species diverged from a common ancestor into two major clades that distinguish the virulent species (BP = 100%) from the milder species (BP = 97%; Figure 1). This divergence appears to be more ancient than those which occurred from the common ancestor of the two virulent species and that of the two milder agents. However, this tree did not distinguish the Dermacentor-associated rickettsiae causing SENLAT (R. slovaca and R. raoultii) from the Rhipicephalus-associated rickettsiae causing MSF (R. conorii and R. massiliae). The neighbor-joining tree displayed high gene content dissimilarities (0.20) between the two virulent and the two milder agents (Figure S1). Moreover, the former and the latter agents exhibited very low (0.01) and high (0.20) dissimilarity values, respectively.
Examination of gene degradation and loss revealed that the two virulent agents have altered or lost more genes (92 of 1,161 core genes) than the two milder agents (60 of 1,140 core genes; P = 0.003, χ2 test). This also means that the virulent species shared 60 core genes (with 37–881 aa in sizes) that were deleted or altered in both the milder species (Figure 2, Figure S2A). Of these, comparative genomics followed by hierarchical clustering of 9 complete genes (with ≥60 aa in size) between 29 Rickettsia spp. revealed that they were also either complete, altered or lost in less pathogenic species (e.g., R. peacockii, and Rickettsia africae) and avirulent strains (R. rickettsii Iowa and R. prowazekii ME) as well as in more pathogenic species (e.g., R. rickettsii Sheila, R. australis, R. prowazekii and R. typhi; Figure 3A). None of these genes (except one ankyrin repeat-containing protein which is split, cRIG1027) has found any BLAST match with any putative virulence factor previously described in the genus Rickettsia, and none of them in the virulence factor database of pathogenic bacteria (VFDB). Inversely, the milder species harbored 92 core genes (with 38–2,500 aa in size) that were lost or altered in the virulent species (Figure 2, Figure S2B). Of these, comparative genomics followed by hierarchical clustering of 28 complete genes (with ≥60 aa in size) between 29 Rickettsia spp. showed two major clusters (Figure 3B). The first cluster included 48% of rickettsial species displaying several genes in a gradual degradation process or lost. Among these species, we found the two virulent species (R. conorii and R. slovaca), several more pathogenic species (e.g., R. rickettsii Sheila, R. prowazekii Rp22, and R. typhi), and only three nonpathogenic strains R. peacockii Rustic and the two mutants R. rickettsii Iowa and R. prowazekii ME. In contrast, the second cluster contained 52% of rickettsial species exhibiting several conserved genes. Among these species, we found the two milder species (R. raoultii and R. massiliae) and several other less or non-pathogenic species (e.g., R. africae, R. rhipicephalus, R. montanensis). Moreover, the first cluster also gathered 11 plasmidless species, and only three species with one plasmid, including R. peacockii, R. australis, and Rickettsia felis, whereas the second cluster included 13 species harboring one to four plasmids, but only two plasmidless species, including R. bellii and R. montanensis (P = 0.01, Fisher's exact test). Comparative COG categories showed that the two virulent species mainly lacked mobilome genes relative to the two milder species (5 vs. 29–41 genes per sp.; Figure S3). Moreover, only the latter species exhibited additional genes in several COG categories from plasmids, including for example 2–11 genes from the mobilome. In all, the numbers of genes in the mobilome found in the virulent R. slovaca or R. conorii differed significantly from those of the milder R. raoultii or R. massiliae, respectively (5 of 1,246 vs. 40 of 1,323, P ≤ 0.001, or 5 of 1,237 vs. 43 of 1,294, P ≤ 0.001, resp., χ2 tests).
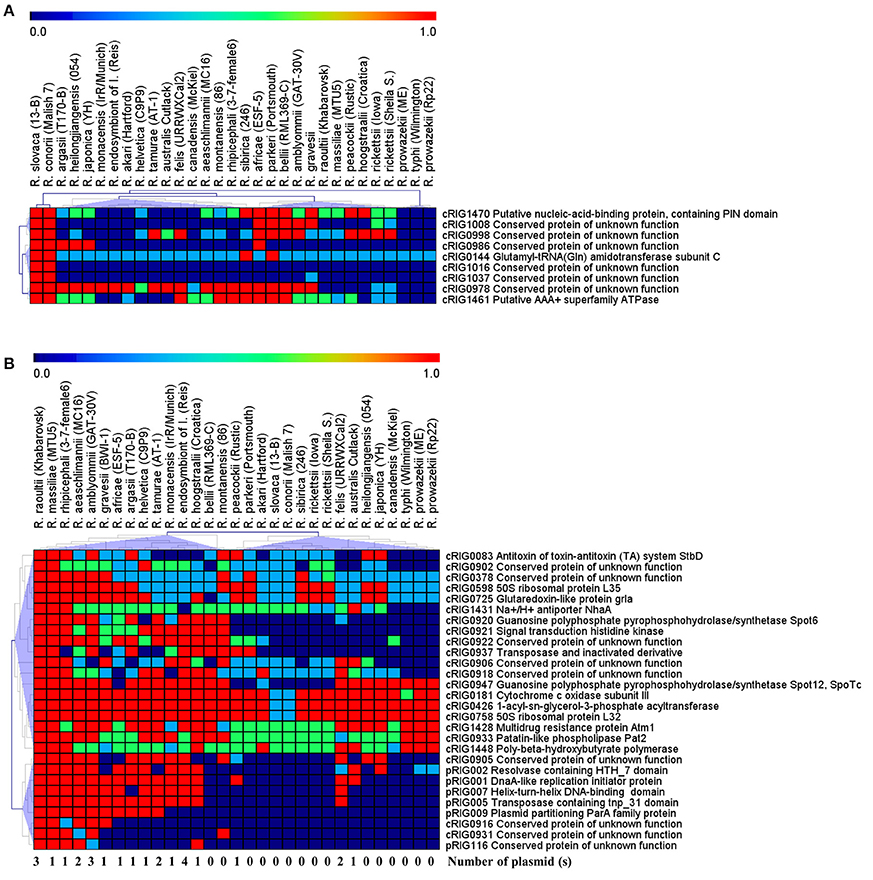
Figure 3. Comparative genomics of 9 complete genes (≥60 aa in sizes) distinguishing the virulent R. slovaca and R. conorii from the milder R. raoultii and R. massiliae (A) and 28 complete genes (≥60 aa in sizes) distinguishing the milder R. raoultii and R. massiliae from the virulent R. slovaca and R. conorii (B), with 25 Rickettsia species. Red, green, light blue, and dark blue colors mean that genes can be either complete, split, fragment and absent/remnant, respectively.
Proteomics of SFG Rickettsia spp.
Proteome analysis of the four SFG species identified 502–688 and 6–24 chromosomes- and plasmids-encoding proteins after infecting X. laevis cells, respectively. These proteins covered 40–52% (of 1,246–1,294) and 17–37% (of 16–138) of the four predicted proteomes, respectively (Figure 1). Although these numbers of identified proteins may appear low, they are similar to those observed strictly intracellular bacteria such as Chlamydia trachomatis (Saka et al., 2011). Expressions from genes, either split or fragment, were rare, counting about 2–4% (7–14 of 302–334 genes split or fragment) from chromosomes and 0–16% (0–2 of 7–12 genes split or fragment) from plasmids.
The pan-proteome of the four Rickettsia spp. clustered into 744 cRIGs and 22 pRIGs (Figure 4). Of these, only 210 (28% of 744 cRIGs) and only 2 (10% of 22 pRIGs) of identified proteins were previously detected in diverse studies using gel-based proteomics, or rarely RT-PCRs (Table S1). However, 534 (72% of 744 cRIGs) and 20 (90% of 22 pRIGs) of expressed proteins are newly identified by the current gel-free proteomics (Table S1). Moreover, 96% (717 of 744 cRIGs) of the pan-proteome represented 66% of genes from the core chromosome (717 of 1,084 cRIGs), while only 4% (27 of 744 cRIGs) were found in up to three species from chromosomes. Only 19% (22 of 115 pRIGs) of genes in plasmids exhibited expressions.
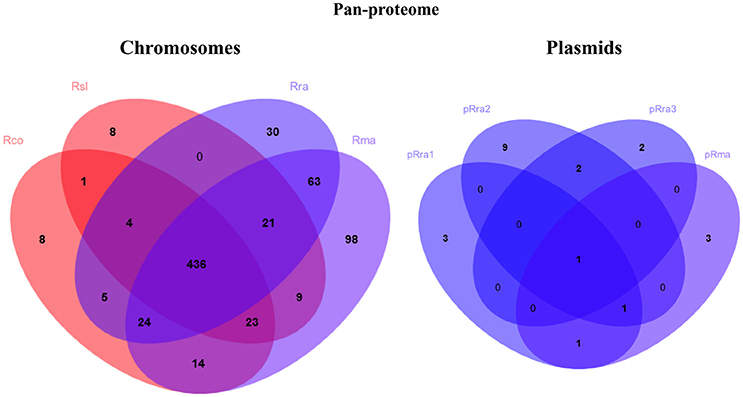
Figure 4. Venn diagrams summarizing pan-proteome of the virulent R. slovaca Rsl and the milder R. raoultii Rra causing SENLAT, and the virulent R. conorii Rco and the milder R. massiliae Rma causing MSF.
The pan-proteome of the four pathogens displayed a number of identified core proteins, significantly lower in the virulent than in the milder species (1 of 437 vs. 66 of 502 c/pRIGs; P < 0.001, χ2 test; Figure 4, see protein list in Table S2). Moreover, the total abundance of these proteins were lower (1.3–2.7 fmol μg−1) in the virulent than in the milder species (230.5–516.4 fmol μg−1; Table S2). Moreover, 90% of these proteins were coded by core genes. All protein abundances of the four species are given in Table S3. Hierarchical clustering analysis of total protein abundances, classified by COG categories, also clearly distinguished the virulent from the milder species (Figure 5). This distinction was found mainly in proteins involved in post-translational modification, protein turnover and chaperones (282–327 vs. 694–1156 fmol μg−1, resp.), cell wall/membrane/envelope biogenesis (317–332 vs. 615–1040 fmol μg−1, resp.), and energy production and conservation (172–206 vs. 331–712 fmol μg−1, resp.).
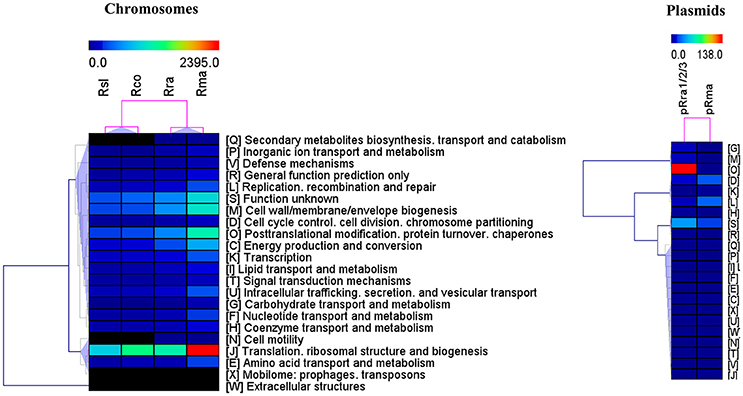
Figure 5. Hierarchical clustering of total protein quantities (fmol μg−1) in the two virulent species R. slovaca Rsl and R. conorii Rco, and, the two milder species R. raoultii Rra and R. massiliae Rma, as classified by COG functional categories.
The protein profile of the virulent compared with the milder SENLAT agents identified 169 core proteins (26% of 643 c/pRIGs) exhibiting abundance changes, in which the virulent R. slovaca up-regulated 50 and down-regulated 119 core proteins (Figure S4). Moreover, only the milder R. raoultii displayed 21 (3.2% of 643 c/pRIGs) specific proteins, including 17 from plasmids. Similarly, the protein profile of the virulent compared with the milder MSF agents resulted in 223 core proteins (31% of 712 c/pRIGs) displaying abundance changes, in which the virulent R. conorii up-regulated 36 and down-regulated 187 core proteins (Figure S4). Moreover, the virulent and the milder agents each exhibited one and 10 specific proteins, including 3 from plasmids (1.5% of 712 c/pRIGs), respectively. Overall, both the virulent compared with both the milder agents exhibited less up- than down-regulated proteins and rarely specific proteins (50–36 vs. 119–187 vs. 21–10, P < 0.001, χ2 test). Examination of the protein profiles by COG categories revealed that both virulent agents displayed large numbers of proteins with changes in abundances and/or specificity in translation, ribosomal structure and biogenesis (26 and 26 proteins), post-translational modification, protein turnover, chaperones (19 and 23 proteins), energy production and conversion (18 and 24) and general function prediction (12 and 13 proteins; Figure 6). However, the virulent SENLAT agent exhibited a number of proteins, with changes in abundances or specificity, slightly lower than those of the virulent MSF agent, mainly in cell wall/membrane/envelope biogenesis (13 vs. 22 proteins) and replication, recombination and repair (11 vs. 17 proteins), intracellular trafficking, secretion, and vesicular transport (4 vs. 12 proteins) and amino acid transport and metabolism (4 vs. 13 proteins; P < 0.6, χ2 test). Comparative analysis of protein profiles between the SENLAT and MSF agents identified 340 c/pRIGs, distinguishing two main patterns. First, the two virulent agents shared 72 proteins (21% of 340 c/pRIGs) exhibiting similar patterns, in which we found 8 up/up-regulated and 61 down/down-regulated proteins, as well as three plasmid-specific proteins of the two milder species (Figure S5). These patterns were found more in proteins associated with post-translational modification, protein turnover, chaperones (12 proteins) and energy production and conversion (8 proteins) than those related to translation, ribosomal structure and biogenesis (6 proteins), cell wall/membrane/envelope biogenesis (5 proteins) and general function prediction only (5 proteins; Figure 7, Table 1, Table S4). In the remaining categories, the similar patterns displayed low numbers of or no proteins. Second, and in contrast, the two virulent agents exhibited 268 proteins (79% of 340 c/pRIGs) displaying distinct protein patterns (Figure S5). As an example, while the virulent SENLAT agent up-regulated 42 proteins, the virulent MSF agent did not up-regulate them, but carried out down-regulations for 8, equal regulations for 25 and un-classified proteins for 9 core proteins. In all, while the virulent SENLAT agent exhibited 42 up-regulated, 58 down-regulated, 91 equally regulated, and 59 un-classified proteins, as well as 18 proteins specifically expressed by the milder agent, the virulent MSF agent displayed 28 up-regulated, 126 down-regulated, 61 equally regulated, and 45 un-classified proteins, as well as 8 specific proteins, including one by the virulent agent and 7 by the milder agent (P < 0.001, χ2 test). These distinct patterns were found more in proteins related to translation, ribosomal structure and biogenesis (38 proteins), cell wall/membrane/envelope biogenesis (25 proteins), energy production and conversion (23 proteins), replication, recombination and repair (20 proteins), post-translational modification, protein turnover, chaperones (16 proteins) and general function prediction only (14 proteins), than in those associated with coenzyme transport and metabolism (13 proteins), intracellular trafficking, secretion, and vesicular transport (12 proteins), amino acid transport and metabolism (11 proteins; Figure 7, Table 1, Table S4). In the remaining categories, the distinct patterns exhibited low numbers of proteins.
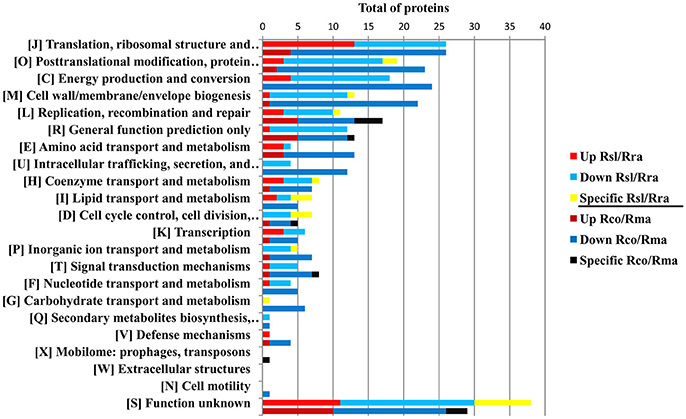
Figure 6. Protein profiles obtained between the SENLAT agents (the virulent R. slovaca Rsl/the milder R. raoultii Rra), and between the MSF agents (the virulent R. conorii Rco/the milder R. massiliae Rma), as classified by COG functional categories. Up, Down and Specific mean up-regulated down-regulated and specific proteins.
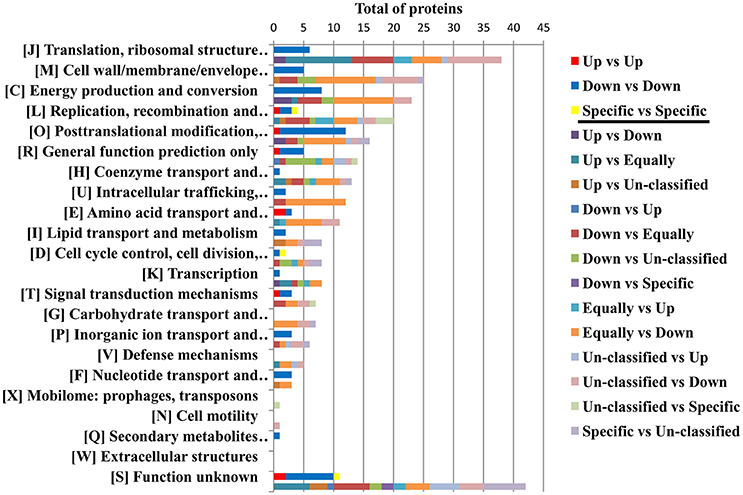
Figure 7. Similar and distinct protein patterns obtained between the SENLAT agents (the virulent R. slovaca Rsl/the milder R. raoultii Rra) and the MSF agents (the virulent R. conorii Rco/the milder R. massiliae Rma), as classified by COG functional categories. Up, Down, Equally, Specific and Un-classified mean up-regulated, down-regulated, equally regulated, specific and un-classified proteins, respectively.
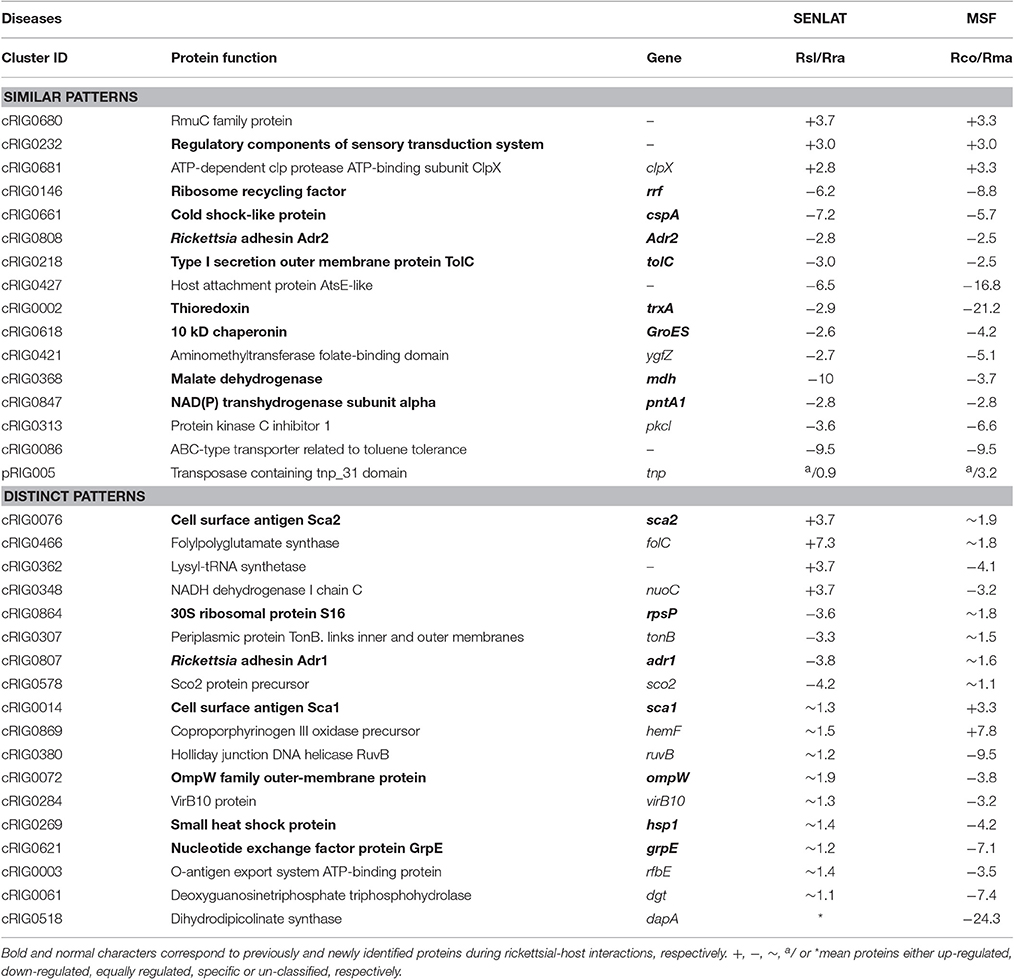
Table 1. Examples of protein patterns obtained between the SENLAT agents (the virulent R. slovaca Rsl/the milder R. raoultii Rra) and between the MSF agents (the virulent R. conorii Rco/the milder R. massiliae Rma).
Discussion
Obligate intracellular bacterial pathogens are constantly evolving, in a bottleneck lifestyle, through various processes, including reductive evolution, selection, mutations, InDels, mobile genetic elements, etc. (Rohmer et al., 2007; Bryant et al., 2012). Members of the Rickettsia genus, which are obligate intracellular bacteria, exhibit diverse ecological and biological features that may involve multiple genes and genetic pathways during pathogen-host colonization and interactions that counteract host homeostasis and/or defense mechanisms. In this study, we performed comparative genomics, phylogenomics and proteomics on four SFG Rickettsia species in order to identify the genomic signature(s) and proteomic profiles, and to understand the evolutionary event(s), that distinguish R. slovaca and R. conorii, two of the virulent human rickettsial pathogens, from R. raoultii and R. massiliae, which cause the milder infections of two distinct diseases; i.e., SENLAT (Raoult and Roux, 1997; Parola et al., 2009; Foissac et al., 2013) and MSF (Cascio et al., 2013; Milhano et al., 2014; Bechelli et al., 2015; Portillo et al., 2015).
Divergent and Convergent Evolution
Phylogenomic analysis revealed a divergent evolution of the virulent and the milder agents into two clades from a common ancestor. This divergence was corroborated by strong differences in structural variations (i.e., non-synonymous mutations and InDels) in the predicted core proteins (i.e., representing 71% of the pan-genome) between the virulent and milder agents. These data suggest that both evolutionary events might have contributed to the differences in virulence between the four SFG species. In a similar study, adaptive mutations were suggested to determine virulence in the highly pathogenic R. prowazekii (Zhang et al., 2006; Bechah et al., 2010). In another example, 115 non-synonymous SNPs were found between the virulent Mycobacterium bovis strains and the attenuated Bacillus Calmette-Guerin BCG strains, affecting important functions such as global regulators, transcriptional factors, and central metabolism, which may impact virulence (Garcia Pelayo et al., 2009). However, the phylogenomic analysis also showed that the virulent and milder agent causing SENLAT or MSF diseases are distantly related, suggesting that both agents of each rickettsiosis may have undergone discrete convergent evolution, as reported for similarities in intracellular strategies between phylogenetically distant microbes (Casadevall, 2008).
Reductive Evolution
The phylogenetic tree based on gene content dissimilarities reflected gene loss between the four taxa. Indeed, during and/or after their evolutionary divergence, the two virulent agents underwent more core gene losses and degradations, including the absence of any plasmid, as compared with the two milder species (three plasmids in R. raoultii and one in R. massiliae). Similar findings were observed in other species causing severe rickettsioses, including the TG R. prowazekii and R. typhi, which exhibit small chromosomes and are plasmidless (McLeod et al., 2004; Bechah et al., 2010; Clark et al., 2015), and other species causing mild or no disease, including the SFG R. helvetica, R. africae, R. felis, and R. peacockii, which have larger chromosomes and harbor one or more plasmids (Ogata et al., 2005; Felsheim et al., 2009; Fournier et al., 2009; Dong et al., 2012). In a recent study, we demonstrated that rickettsial plasmids have undergone a reductive evolution, similar to that observed in rickettsial chromosomes, possibly leading progressively to cryptic plasmids or complete plasmid loss (El Karkouri et al., 2016). Moreover, in the order Rickettsiales, no association was found between virulence and the acquisition of novel genes or the presence of plasmids (Darby et al., 2007). Overall, our data are consistent with the assumption that differences in virulence of the four SFG species in humans may result from reductive evolution (Parish et al., 2003; Parkhill et al., 2003; Moore et al., 2004; Lescot et al., 2008; Fournier et al., 2009; Merhej et al., 2014).
In this study, the core gene set present in the two virulent agents, but lost or altered in the two milder species as well as in other nonpathogenic and pathogenic Rickettsia species, did not include any previously described bacterial virulence factor, and only one core protein of unknown function exhibited a comprehensive protein abundance. This suggests that they cannot be linked to rickettsial pathogenesis and virulence in humans, or are unidentified virulence factors, and hence their roles need to be elucidated. However, the core gene set is conserved in the two milder agents and several other less pathogenic Rickettsia species, but deleted or in a degradation process in the two virulent species and other highly pathogenic Rickettsia species. Moreover, several of these core genes displayed comprehensive protein abundances, including for example those coding for glutaredoxin-like protein GrlA, multidrug resistance protein Atm1, 1-acyl-sn-glycerol-3-phosphate acyltransferase, patatin-like phospholipase Pat2, two transposases, a plasmid partitioning ParA family protein and a conserved protein of unknown function. Thus, the decay of the functions of these genes may contribute to the emergence of highly pathogenic bacteria, but their expressions in the milder agents may reflect antivirulence roles as demonstrated for example in Shigella, Yersinia, and Francisella antivirulence genes (Moore et al., 2004; Maurelli, 2007; Bliven and Maurelli, 2012), and/or may be related to host-adaptation, as was suggested for Bordetella genomes (Parkhill et al., 2003). None of these genes corresponded to any known antivirulence genes (e.g., nadA/B, lacI, lpxL, and pepO genes) identified in other bacterial pathogens (Bliven and Maurelli, 2012), suggesting that they may be unidentified or adaptive genes. Hence, the influence of these genes in increased virulence and/or in adaptation of the examined pathogenic phenotypes needs further functional analysis using, for example, genetic manipulation by the shuttle vector system developed from R. amblyommii plasmids or as demonstrated in Burkholderia pseudomallei and Salmonella enterica (Moore et al., 2004; Burkhardt et al., 2011; Bliven and Maurelli, 2012; Wood et al., 2012).
The Mobilome
It has been proposed that the proliferation of insertion of sequences (IS elements) is the cause of a large number of pseudogenes and genomic rearrangements in emerging or highly virulent pathogens (Parkhill et al., 2003; Wei et al., 2003; Petrosino et al., 2006; Rohmer et al., 2007). For example, in the facultative intracellular bacterium F. tularensis, IS elements and other evolutionary events were correlated with the emergence of strains pathogenic for humans (Rohmer et al., 2007). In contrast, an extraordinary proliferation of mobile genetic elements (≥650 transposases) contributed to a limited synteny in R. endosymbionts of Ixodes scapularis, an SFG species which is not known to invade vertebrate cells (Gillespie et al., 2012). Likewise, the non-pathogenic SFG R. peacockii showed an introduction and a proliferation in 42 copies of the ISRpe1 transposon (Felsheim et al., 2009). These evolutionary events were associated with extensive genome rearrangements and numerous deletions, including deletions of several genes (e.g., ank, dsbA, rickA, protease II, and sca1) and thought to be related to loss of virulence in this species (Felsheim et al., 2009). In the current study, the two milder Rickettsia species harbored more genes related to the mobilome, particularly transposases, integrases and phage sequences, than the virulent Rickettsia agents. This abundance of genes did not disrupt the putative virulence factors deleted in R. peacockii, suggesting that the mobilome in the milder species may have influenced their genomic stability with or without any impact on virulence in humans, or may improve their potential to gain novel genes of adaptation, survival and/or fitness. This study identified one integrase catalytic region and one phage-associated protein specific to the milder MSF agent, suggesting that the mobilome is still active. However, the lack of expression from the remaining genes of the mobilome suggests that they may be in a dormant or inactivated state.
The Pan-Proteome Is Mainly Coded by the Core-Genome
Remarkably, after infecting X. laevis cells, our study revealed that 96% of the pan-proteome was coded by 66% of genes from the core chromosome, whereas 4% of chromosomally-encoded proteins were identified in only up to three species, and 19% of genes in plasmids showed expressions. Although Rickettsia genomes evolved by reductive evolution, 1–2% of the genes, either split or fragments, may still be active, thus corroborating previous studies that demonstrated transcription of several split/fragment genes in R. conorii (Ogata et al., 2001), Mycobacterium leprae (Akama et al., 2009) and Lactobacillus delbrueckii (Zheng et al., 2016). The expressed split genes were thought to conserve functional domains, or proposed to function as a class of non-coding RNAs and act as riboregulators at both the transcriptional and post-transcriptional levels (Erdmann et al., 2001; Zheng et al., 2016). However, the remaining altered genes were not expressed, suggesting that they may have lost their functions (i.e., by becoming pseudogenes). In sum, the proportions of the identified proteins in the current study (40–52 and 17–37% from chromosomes and plasmids, resp.) were higher than those reported in other rickettsial species, including 12% in R. felis (Ogawa et al., 2007), 3 to 19% in R. prowazekii (Renesto et al., 2005; Tucker et al., 2011), and 2.6% in R. conorii (Zhao et al., 2016) using gel-based proteomics, but comparable to those of other intracellular bacteria, such as M. tuberculosis (41%) and C. trachomatis (Saka et al., 2011; Gopinath et al., 2015), using gel–free proteomics.
Examination of the pan-proteome in the four rickettsial agents detected several core genes (e.g., ompA/B, rickA, pld, omp, tlyC, ppcE, tlcD1-3/5, and stbD) that had previously been associated with rickettsial adhesion to and invasion of host cells, motility, survival, and/or virulence (e.g., Blanc et al., 2005; Ellison et al., 2009; Sears et al., 2012; Qi et al., 2013; Rahman et al., 2013; Gong et al., 2014; Gillespie et al., 2015). However, these genes were found to be equally expressed or had un-classified expressions in X. laevis cells (data not shown), suggesting that these known virulence factors may be differentially expressed during the early stage of adhesion to and infection of X. laevis. Furthermore, some of them (e.g., ompA and/or rickA genes) were previously found either altered or lost, for example, in the pathogenic and non-motile R. prowazekii species and the pathogenic and motile R. typhi, as well as in the nonpathogenic R. rickettsii str Iowa and R. peacockii (Ogata et al., 2001; Ellison et al., 2008; Felsheim et al., 2009; Georgiades et al., 2011; Sears et al., 2012). In another study, the pld gene that was shown to be required for virulence of R. prowazekii displayed no defect in the avirulent R. rickettsii strain Iowa (Clark et al., 2015).
However, we cannot rule out the putative role of other known or unidentified proteins that may impact the differences in virulence and/or pathogenicity between the four SFG species. First, the two virulent agents were distinguished from the milder agents by two distinct clusters of protein abundances. Second, they exhibited less up-regulated than down-regulated proteins and nearly no specifically expressed proteins. Authors have reported that Rickettsia spp. survive by taking advantage of host cell nutrients (Andersson et al., 1998; Andersson and Andersson, 1999; Blanc et al., 2007a; Sahni and Rydkina, 2009). Thus, the current data suggest differences in intracellular strategies and biological activities which might reflect mechanisms of pathogenesis between the two virulent and two milder species.
In addition, the two virulent SENLAT and MSF agents displayed 72 proteins with similar patterns, in which 8 were up/up-regulated, 61 were down/down-regulated and three were plasmid-specific of the milder agents. These profiles may include putative virulence/antivirulence-associated proteins, which may have been influenced by the divergent driving forces revealed in this study. Indeed, evolutionary pressures may change or disrupt the structures of genes and their proteins, leading to differences in expressions and loss of function, respectively. In a proteomic study of the pathogenic Shigella flexneri, the down-regulated argT gene was identified as an antivirulence gene which may interfere with the virulence factor of that species (Zhao et al., 2010). The similar patterns identified here mainly involved proteins associated with post-translational modification, protein turnover, chaperones (e.g., ClpX, TrxA, GroES, HtrA, and PrsA) and energy production and conversion (e.g., Mdh, FumC, SdhB, and Ppa), translation, ribosomal structure and biogenesis (e.g., RpmC, RplX′, DksA), cell wall/membrane/envelope biogenesis (e.g., Adr2, TolC and AmpD1) and general function prediction only (e.g., Pat1A, Pat2, and Ybgf). In contrast, the two virulent agents exhibited 268 proteins with distinct patterns. These profiles clearly discriminated the SENLAT agents from the MSF agents, and also the virulent from the milder agent of each rickettsiosis, suggesting that they may contain putative rickettsiosis-related proteins and/or specific virulence/antivirulence-related proteins within each disease. This may reflect convergent evolution related to rickettsial diseases and/or the divergent evolutionary history associated with a specific increase in virulence, respectively. Distinct patterns were found more in proteins related to translation, ribosomal structure and biogenesis (e.g., several tRNA synthetases), cell wall/membrane/envelope biogenesis (e.g., Adr1, LpxA, OmpW, Asma), energy production and conversion (e.g., NuoC/F/B/G, TlcD4, AtpX), replication, recombination and repair (e.g., recA/R, DnaB/N/E/Q/G), post-translational modification, protein turnover, chaperones (e.g., Hsp1/2, TrxB2, GroEL, ClpP, HslV) and general function prediction only (e.g., Uup succinate dehydrogenase iron-sulfur subunit and proteins with unknown functions), than in those involved in coenzyme transport and metabolism (e.g., HemA/B/F and FolC/D), intracellular trafficking, secretion, and vesicular transport (e.g., VirB61/3/4, VirB9-1/2, and VirB10) and amino acid transport and metabolism (e.g., DapF, PepE, IscS), and in lower numbers in proteins of the remaining categories.
Conclusion
In a bottleneck lifestyle associated with genetic drift, the four SFG Rickettsia species have been shaped by distinct evolutionary processes that may have strongly impacted gene conservation and protein profiles, mainly in those of the core genome, as summarized in Figure 8. Thus, these driving forces may have influenced intracellular strategies in these rickettsiae, and that may contribute to the emergence of distinct virulence and rickettsiosis in humans, distinguishing the virulent from the milder agents, and the SENLAT from the MSF diseases. Although the SFG lineage gathered several syntenic genomes, our study suggests that the mechanisms governing virulence and pathogenicity in the examined rickettsiae are more complex than imagined. Recent study has indicated that virulence in SFG rickettsiae is multifactorial (Clark et al., 2015). The current multi-omics data provide new insights into intracellular pathogen-host interactions, and suggest that X. laevis host-cells can be used a tool to clarify genetic determinants underlying rickettsial diseases. Further studies using animal models, small regulatory RNAs (Davids et al., 2002; Schroeder et al., 2015; Narra et al., 2016; Schroeder et al., 2016) and endothelial cell responses to rickettsial infections (Bechelli et al., 2015; Zhao et al., 2016) may improve understanding of the pathogenesis and fitness of the SFG rickettsiae, and intracellular pathogenic bacteria.
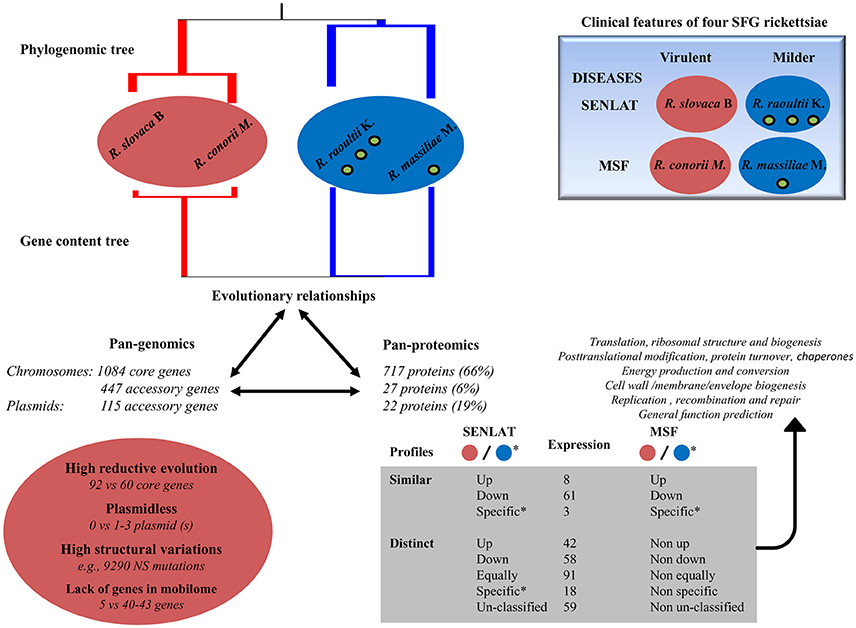
Figure 8. Summary of multi-omics results obtained from comparative analyses between four SFG rickettsiae including the virulent R. slovaca Rsl and the milder R. raoultii Rra which cause SENLAT diseases as well as the virulent R. conorii Rco and the milder R. massiliae Rma which cause MSF diseases. Up, Down, Equally, Specific and Un-classified mean up-regulated, down-regulated, equally regulated, specific and un-classified proteins, respectively. As an example, Non up means that the proteins can be either down-regulated, equally regulated, specific or un-classified proteins. Red and blue colors correspond to the most virulent and the milder agents, respectively. Overall, the two most virulent agents compared with the milder agents exhibited several driving forces that may be associated to differences in virulence and/or plasticity including divergent and reductive evolution, no plasmid, high structural variations and/or a lack of genes in the mobilome. The similarities in the disease (i.e., either SENLAT or MSF) between two distantly related species suggest a convergent evolution. Moreover, the virulent agents also displayed similar and distinct protein profiles mainly in six COG categories. These patterns may include putative virulence- and/or disease-associated proteins as well as putative antivirulence-related proteins of the milder agents.
Author Contributions
DR, KE, and PF conceived the project. KE performed bioinformatic analysis: MK, NA, and SA performed experiments and wrote their corresponding material and methods. KE wrote a draft and edited the manuscript. KE, PF, and DR revised the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The study was funded by the Mediterranée-Infection foundation. We thank Prof. Eric Chabriere for comments on this manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.01363/full#supplementary-material
References
Akama, T., Suzuki, K., Tanigawa, K., Kawashima, A., Wu, H., Nakata, N., et al. (2009). Whole-genome tiling array analysis of Mycobacterium leprae RNA reveals high expression of pseudogenes and noncoding regions. J. Bacteriol. 191, 3321–3327. doi: 10.1128/JB.00120-09
Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Andersson, J. O., and Andersson, S. G. E. (1999). Genome degradation is an ongoing process in Rickettsia. Mol. Biol. Evol. 16, 1178–1191. doi: 10.1093/oxfordjournals.molbev.a026208
Andersson, S. G. E., Zomorodipour, A., Andersson, J. O., Sicheritz-Ponten, T., Alsmark, U. C. M., Podowski, R. M., et al. (1998). The genome sequence of Rickettsia prowazekii and the origin of mitochondria. Nature 396, 133–140. doi: 10.1038/24094
Audia, J. P., and Winkler, H. H. (2006). Study of the five Rickettsia prowazekii proteins annotated as ATP/ADP translocases (Tlc): only Tlc1 transports ATP/ADP, while Tlc4 and Tlc5 transport other ribonucleotides. J. Bacteriol. 188, 6261–6268. doi: 10.1128/JB.00371-06
Balraj, P., El Karkouri, K., Vestris, G., Espinosa, L., Raoult, D., and Renesto, P. (2008). RickA expression is not sufficient to promote actin-based motility of Rickettsia raoultii. PLoS ONE 3:e2582. doi: 10.1371/journal.pone.0002582
Bardou, P., Mariette, J., Escudie, F., Djemiel, C., and Klopp, C. (2014). jvenn: an interactive Venn diagram viewer. BMC Bioinformatics 15:293. doi: 10.1186/1471-2105-15-293
Bechah, Y., El Karkouri, K., Mediannikov, O., Leroy, Q., Pelletier, N., Robert, C., et al. (2010). Genomic, proteomic, and transcriptomic analysis of virulent and avirulent Rickettsia prowazekii reveals its adaptive mutation capabilities. Genome Res. 20, 655–663. doi: 10.1101/gr.103564.109
Bechelli, J., Smalley, C., Milhano, N., Walker, D. H., and Fang, R. (2015). Rickettsia massiliae and Rickettsia conorii israeli spotted fever strain differentially regulate endothelial cell responses. PLoS ONE 10:e0138830. doi: 10.1371/journal.pone.0138830
Blanc, G., Ngwamidiba, M., Ogata, H., Fournier, P. E., Claverie, J. M., and Raoult, D. (2005). Molecular evolution of rickettsia surface antigens: evidence of positive selection. Mol. Biol. Evol. 22, 2073–2083. doi: 10.1093/molbev/msi199
Blanc, G., Ogata, H., Robert, C., Audic, S., Claverie, J. M., and Raoult, D. (2007a). Lateral gene transfer between obligate intracellular bacteria: evidence from the Rickettsia massiliae genome. Genome Res. 17, 1657–1664. doi: 10.1101/gr.6742107
Blanc, G., Ogata, H., Robert, C., Audic, S., Suhre, K., Vestris, G., et al. (2007b). Reductive genome evolution from the mother of Rickettsia. PLoS Genet. 3:e14. doi: 10.1371/journal.pgen.0030014
Bliven, K. A., and Maurelli, A. T. (2012). Antivirulence genes: insights into pathogen evolution through gene loss. Infect. Immun. 80, 4061–4070. doi: 10.1128/IAI.00740-12
Bocs, S., Cruveiller, S., Vallenet, D., Nuel, G., and Medigue, C. (2003). AMIGene: annotation of MIcrobial Genes. Nucleic Acids Res. 31, 3723–3726. doi: 10.1093/nar/gkg590
Brioschi, M., Lento, S., Tremoli, E., and Banfi, C. (2013). Proteomic analysis of endothelial cell secretome: a means of studying the pleiotropic effects of Hmg-CoA reductase inhibitors. J. Proteomics 78, 346–361. doi: 10.1016/j.jprot.2012.10.003
Bryant, J., Chewapreecha, C., and Bentley, S. D. (2012). Developing insights into the mechanisms of evolution of bacterial pathogens from whole-genome sequences. Future Microbiol. 7, 1283–1296. doi: 10.2217/fmb.12.108
Burkhardt, N. Y., Baldridge, G. D., Williamson, P. C., Billingsley, P. M., Heu, C. C., Felsheim, R. F., et al. (2011). Development of shuttle vectors for transformation of diverse Rickettsia species. PLoS ONE 6:511. doi: 10.1371/journal.pone.0029511
Casadevall, A. (2008). Evolution of intracellular pathogens. Annu. Rev. Microbiol. 62, 19–33. doi: 10.1146/annurev.micro.61.080706.093305
Cascio, A., Torina, A., Valenzise, M., Blanda, V., Camarda, N., Bombaci, S., et al. (2013). Scalp eschar and neck lymphadenopathy caused by Rickettsia massiliae. Emerg. Infect. Dis. 19, 836–837. doi: 10.3201/eid1905.121169
Chan, Y. G., Riley, S. P., and Martinez, J. J. (2010). Adherence to and invasion of host cells by spotted Fever group rickettsia species. Front. Microbiol. 1:139. doi: 10.3389/fmicb.2010.00139
Chen, L., Zheng, D., Liu, B., Yang, J., and Jin, Q. (2016). VFDB 2016: hierarchical and refined dataset for big data analysis–10 years on. Nucleic Acids Res. 44, D694–D697. doi: 10.1093/nar/gkv1239
Clark, T. R., Noriea, N. F., Bublitz, D. C., Ellison, D. W., Martens, C., Lutter, E. I., et al. (2015). Comparative genome sequencing of Rickettsia rickettsii strains that differ in virulence. Infect. Immun. 83, 1568–1576. doi: 10.1128/IAI.03140-14
Darby, A. C., Cho, N. H., Fuxelius, H. H., Westberg, J., and Andersson, S. G. (2007). Intracellular pathogens go extreme: genome evolution in the Rickettsiales. Trends Genet. 23, 511–520. doi: 10.1016/j.tig.2007.08.002
Davids, W., Amiri, H., and Andersson, S. G. (2002). Small RNAs in Rickettsia: are they functional? Trends Genet. 18, 331–334. doi: 10.1016/S0168-9525(02)02685-9
Demangel, C., Stinear, T. P., and Cole, S. T. (2009). Buruli ulcer: reductive evolution enhances pathogenicity of Mycobacterium ulcerans. Nat. Rev. Microbiol. 7, 50–60. doi: 10.1038/nrmicro2077
Dong, X., El Karkouri, K., Robert, C., Gavory, F., Raoult, D., and Fournier, P. E. (2012). Genomic comparison of Rickettsia helvetica and other Rickettsia species. J. Bacteriol. 194, 2751. doi: 10.1128/JB.00299-12
El Karkouri, K., Mediannikov, O., Robert, C., Raoult, D., and Fournier, P. E. (2016). Genome sequence of the tick-borne pathogen Rickettsia raoultii. Genome Announc. 4, e00157–16. doi: 10.1128/genomeA.00157-16
El Karkouri, K., Pontarotti, P., Raoult, D., and Fournier, P. E. (2016). Origin and evolution of rickettsial plasmids. PLoS ONE 11:e0147492. doi: 10.1371/journal.pone.0147492
El Karkouri, K., Selosse, M. A., and Mousain, D. (2006). Molecular markers detecting an ectomycorrhizal Suillus collinitus strain on Pinus halepensis roots suggest successful inoculation and persistence in Mediterranean nursery and plantation. FEMS Microbiol. Ecol. 55, 146–158. doi: 10.1111/j.1574-6941.2005.00014.x
Ellison, D. W., Clark, T. R., Sturdevant, D. E., Virtaneva, K., and Hackstadt, T. (2009). Limited transcriptional responses of Rickettsia rickettsii exposed to environmental stimuli. PLoS ONE 4:e5612. doi: 10.1371/journal.pone.0005612
Ellison, D. W., Clark, T. R., Sturdevant, D. E., Virtaneva, K., Porcella, S. F., and Hackstadt, T. (2008). Genomic comparison of virulent Rickettsia rickettsii Sheila Smith and avirulent Rickettsia rickettsii Iowa. Infect. Immun. 76, 542–550. doi: 10.1128/IAI.00952-07
Erdmann, V. A., Barciszewska, M. Z., Hochberg, A., de Groot, N., and Barciszewski, J. (2001). Regulatory RNAs. Cell. Mol. Life Sci. 58, 960–977. doi: 10.1007/PL00000913
Eremeeva, M. E., Balayeva, N. M., and Raoult, D. (1994). Purification of rickettsial cultures contaminated by mycoplasmas. Acta Virol. 38, 231–233.
Felsheim, R. F., Kurtti, T. J., and Munderloh, U. G. (2009). Genome sequence of the endosymbiont Rickettsia peacockii and comparison with virulent Rickettsia rickettsii: identification of virulence factors. PLoS ONE 4:e8361. doi: 10.1371/journal.pone.0008361
Fernandez-Soto, P., Perez-Sanchez, R., Alamo-Sanz, R., and Encinas-Grandes, A. (2006a). Spotted fever group rickettsiae in ticks feeding on humans in northwestern Spain: is Rickettsia conorii vanishing? Ann. N. Y. Acad. Sci. 1078, 331–333. doi: 10.1196/annals.1374.063
Fernandez-Soto, P., Perez-Sanchez, R., Diaz, M. V., Encinas-Grandes, A., and Alamo, S. R. (2006b). Rickettsia massiliae in ticks removed from humans in Castilla y Leon, Spain. Eur. J. Clin. Microbiol. Infect. Dis. 25, 811–813. doi: 10.1007/s10096-006-0217-9
Foissac, M., Socolovschi, C., and Raoult, D. (2013). Update on SENLAT syndrome: scalp eschar and neck lymph adenopathy after a tick bite. Ann. Dermatol. Venereol. 140, 598–609. doi: 10.1016/j.annder.2013.07.014
Fournier, P. E., El Karkouri, K., Leroy, Q., Robert, C., Giumelli, B., Renesto, P., et al. (2009). Analysis of the Rickettsia africae genome reveals that virulence acquisition in Rickettsia species may be explained by genome reduction. BMC Genomics 10:166. doi: 10.1186/1471-2164-10-166
Fournier, P. E., El Karkouri, K., Robert, C., Medigue, C., and Raoult, D. (2012). Complete genome sequence of Rickettsia slovaca, the agent of tick-borne lymphadenitis. J. Bacteriol. 194, 1612. doi: 10.1128/JB.06625-11
Frischknecht, F., and Way, M. (2001). Surfing pathogens and the lessons learned for actin polymerization. Trends Cell Biol. 11, 30–38. doi: 10.1016/S0962-8924(00)01871-7
Garcia Pelayo, M. C., Uplekar, S., Keniry, A., Mendoza, L. P., Garnier, T., Nunez, G. J., et al. (2009). A comprehensive survey of single nucleotide polymorphisms (SNPs) across Mycobacterium bovis strains and M. bovis BCG vaccine strains refines the genealogy and defines a minimal set of SNPs that separate virulent M. bovis strains and M. bovis BCG strains. Infect. Immun. 77, 2230–2238. doi: 10.1128/IAI.01099-08
Georgiades, K., and Raoult, D. (2010). Defining pathogenic bacterial species in the genomic era. Front. Microbiol. 1:151. doi: 10.3389/fmicb.2010.00151
Georgiades, K., Merhej, V., and Raoult, D. (2011). The influence of rickettsiologists on post-modern microbiology. Front. Cell. Infect. Microbiol. 1:8. doi: 10.3389/fcimb.2011.00008
Gillespie, J. J., Beier, M. S., Rahman, M. S., Ammerman, N. C., Shallom, J. M., Purkayastha, A., et al. (2007). Plasmids and rickettsial evolution: insight from Rickettsia felis. PLoS ONE 2:e266. doi: 10.1371/journal.pone.0000266
Gillespie, J. J., Joardar, V., Williams, K. P., Driscoll, T., Hostetler, J. B., Nordberg, E., et al. (2012). A Rickettsia genome overrun by mobile genetic elements provides insight into the acquisition of genes characteristic of an obligate intracellular lifestyle. J. Bacteriol. 194, 376–394. doi: 10.1128/JB.06244-11
Gillespie, J. J., Kaur, S. J., Rahman, M. S., Rennoll-Bankert, K., Sears, K. T., Beier-Sexton, M., et al. (2015). Secretome of obligate intracellular Rickettsia. FEMS Microbiol. Rev. 39, 47–80. doi: 10.1111/1574-6976.12084
Gimenez, D. F. (1964). Staining rickettsiae in yolk-sac cultures. Stain Technol. 39, 135–140. doi: 10.3109/10520296409061219
Gong, W., Xiong, X., Qi, Y., Jiao, J., Duan, C., and Wen, B. (2014). Identification of novel surface-exposed proteins of Rickettsia rickettsii by affinity purification and proteomics. PLoS ONE 9:e100253. doi: 10.1371/journal.pone.0100253
Gopinath, V., Raghunandanan, S., Gomez, R. L., Jose, L., Surendran, A., Ramachandran, R., et al. (2015). Profiling the proteome of Mycobacterium tuberculosis during dormancy and reactivation. Mol. Cell. Proteomics 14, 2160–2176. doi: 10.1074/mcp.M115.051151
Gouin, E., Egile, C., Dehoux, P., Villiers, V., Adams, J., Gertler, F., et al. (2004). The RickA protein of Rickettsia conorii activates the Arp2/3 complex. Nature 427, 457–461. doi: 10.1038/nature02318
Jiang, J., You, B. J., Liu, E., Apte, A., Yarina, T. R., Myers, T. E., et al. (2012). Development of three quantitative real-time PCR assays for the detection of Rickettsia raoultii, Rickettsia slovaca, and Rickettsia aeschlimannii and their validation with ticks from the country of Georgia and the Republic of Azerbaijan. Ticks Tick Borne Dis. 3, 327–331. doi: 10.1016/j.ttbdis.2012.10.004
Katoh, K., Kuma, K., Toh, H., and Miyata, T. (2005). MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 33, 511–518. doi: 10.1093/nar/gki198
Kristensen, D. M., Kannan, L., Coleman, M. K., Wolf, Y. I., Sorokin, A., Koonin, E. V., et al. (2010). A low-polynomial algorithm for assembling clusters of orthologous groups from intergenomic symmetric best matches. Bioinformatics 26, 1481–1487. doi: 10.1093/bioinformatics/btq229
Lescot, M., Audic, S., Robert, C., Nguyen, T. T., Blanc, G., Cutler, S. J., et al. (2008). The genome of Borrelia recurrentis, the agent of deadly louse-borne relapsing fever, is a degraded subset of tick-borne Borrelia duttonii. PLoS Genet. 4:e1000185. doi: 10.1371/journal.pgen.1000185
Marquez, F. J., Rodriguez-Liebana, J. J., Soriguer, R. C., Muniain, M. A., Bernabeu-Wittel, M., Caruz, A., et al. (2008). Spotted fever group Rickettsia in brown dog ticks Rhipicephalus sanguineus in southwestern Spain. Parasitol. Res. 103, 119–122. doi: 10.1007/s00436-008-0938-z
Maurelli, A. T. (2007). Black holes, antivirulence genes, and gene inactivation in the evolution of bacterial pathogens. FEMS Microbiol. Lett. 267, 1–8. doi: 10.1111/j.1574-6968.2006.00526.x
McLeod, M. P., Qin, X., Karpathy, S. E., Gioia, J., Highlander, S. K., Fox, G. E., et al. (2004). Complete genome sequence of Rickettsia typhi and comparison with sequences of other rickettsiae. J. Bacteriol. 186, 5842–5855. doi: 10.1128/JB.186.17.5842-5855.2004
Merhej, V., and Raoult, D. (2011). Rickettsial evolution in the light of comparative genomics. Biol. Rev. Camb. Philos. Soc. 86, 379–405. doi: 10.1111/j.1469-185X.2010.00151.x
Merhej, V., Angelakis, E., Socolovschi, C., and Raoult, D. (2014). Genotyping, evolution and epidemiological findings of Rickettsia species. Infect. Genet. Evol. 25, 122–137. doi: 10.1016/j.meegid.2014.03.014
Merhej, V., Georgiades, K., and Raoult, D. (2013). Postgenomic analysis of bacterial pathogens repertoire reveals genome reduction rather than virulence factors. Brief. Funct. Genomics 12, 291–304. doi: 10.1093/bfgp/elt015
Merhej, V., Royer-Carenzi, M., Pontarotti, P., and Raoult, D. (2009). Massive comparative genomic analysis reveals convergent evolution of specialized bacteria. Biol. Direct. 4:13. doi: 10.1186/1745-6150-4-13
Milhano, N., de Carvalho, I. L., Alves, A. S., Arroube, S., Soares, J., Rodriguez, P., et al. (2010). Coinfections of Rickettsia slovaca and Rickettsia helvetica with Borrelia lusitaniae in ticks collected in a Safari Park, Portugal. Ticks Tick Borne Dis. 1, 172–177. doi: 10.1016/j.ttbdis.2010.09.003
Milhano, N., Popov, V., Vilhena, M., Bouyer, D. H., de, S. R., and Walker, D. H. (2014). Quantitative study of Rickettsia massiliae in Rhipicephalus sanguineus organs. Ticks Tick Borne Dis. 5, 709–714. doi: 10.1016/j.ttbdis.2014.05.009
Moore, R. A., Reckseidler-Zenteno, S., Kim, H., Nierman, W., Yu, Y., Tuanyok, A., et al. (2004). Contribution of gene loss to the pathogenic evolution of Burkholderia pseudomallei and Burkholderia mallei. Infect. Immun. 72, 4172–4187. doi: 10.1128/IAI.72.7.4172-4187.2004
Murray, G. G., Weinert, L. A., Rhule, E. L., and Welch, J. J. (2016). The phylogeny of Rickettsia using different evolutionary signatures: how tree-like is bacterial evolution? Syst. Biol. 65, 265–279. doi: 10.1093/sysbio/syv084
Narra, H. P., Schroeder, C. L., Sahni, A., Rojas, M., Khanipov, K., Fofanov, Y., et al. (2016). Small Regulatory RNAs of Rickettsia conorii. Sci. Rep. 6:36728. doi: 10.1038/srep36728
Noriea, N. F., Clark, T. R., and Hackstadt, T. (2015). Targeted knockout of the Rickettsia rickettsii OmpA surface antigen does not diminish virulence in a mammalian model system. MBio 6:e00323–15. doi: 10.1128/mBio.00323-15
Ogata, H., Audic, S., Renesto-Audiffren, P., Fournier, P. E., Barbe, V., Samson, D., et al. (2001). Mechanisms of evolution in Rickettsia conorii and R. prowazekii. Science 293, 2093–2098. doi: 10.1126/science.1061471
Ogata, H., Renesto, P., Audic, S., Robert, C., Blanc, G., Fournier, P. E., et al. (2005). The genome sequence of Rickettsia felis identifies the first putative conjugative plasmid in an obligate intracellular parasite. PLoS Biol. 3:e248. doi: 10.1371/journal.pbio.0030248
Ogawa, M., Renesto, P., Azza, S., Moinier, D., Fourquet, P., Gorvel, J. P., et al. (2007). Proteome analysis of Rickettsia felis highlights the expression profile of intracellular bacteria. Proteomics 7, 1232–1248. doi: 10.1002/pmic.200600721
Parish, T., Smith, D. A., Kendall, S., Casali, N., Bancroft, G. J., and Stoker, N. G. (2003). Deletion of two-component regulatory systems increases the virulence of Mycobacterium tuberculosis. Infect. Immun. 71, 1134–1140. doi: 10.1128/IAI.71.3.1134-1140.2003
Parkhill, J., Sebaihia, M., Preston, A., Murphy, L. D., Thomson, N., Harris, D. E., et al. (2003). Comparative analysis of the genome sequences of Bordetella pertussis, Bordetella parapertussis and Bordetella bronchiseptica. Nat. Genet. 35, 32–40. doi: 10.1038/ng1227
Parola, P., Paddock, C. D., Socolovschi, C., Labruna, M. B., Mediannikov, O., Kernif, T., et al. (2013). Update on tick-borne rickettsioses around the world: a geographic approach. Clin. Microbiol. Rev. 26, 657–702. doi: 10.1128/CMR.00032-13
Parola, P., Rovery, C., Rolain, J. M., Brouqui, P., Davoust, B., and Raoult, D. (2009). Rickettsia slovaca and R. raoultii in tick-borne Rickettsioses. Emerg. Infect. Dis. 15, 1105–1108. doi: 10.3201/eid1507.081449
Petrosino, J. F., Xiang, Q., Karpathy, S. E., Jiang, H., Yerrapragada, S., Liu, Y., et al. (2006). Chromosome rearrangement and diversification of Francisella tularensis revealed by the type B (OSU18) genome sequence. J. Bacteriol. 188, 6977–6985. doi: 10.1128/JB.00506-06
Pollard, T. D., and Borisy, G. G. (2003). Cellular motility driven by assembly and disassembly of actin filaments. Cell 112, 453–465. doi: 10.1016/S0092-8674(03)00120-X
Portillo, A., Santibanez, S., Garcia-Alvarez, L., Palomar, A. M., and Oteo, J. A. (2015). Rickettsioses in Europe. Microbes Infect. 17, 834–838. doi: 10.1016/j.micinf.2015.09.009
Qi, Y., Xiong, X., Wang, X., Duan, C., Jia, Y., Jiao, J., et al. (2013). Proteome analysis and serological characterization of surface-exposed proteins of Rickettsia heilongjiangensis. PLoS ONE 8:e70440. doi: 10.1371/journal.pone.0070440
Rahman, M. S., Gillespie, J. J., Kaur, S. J., Sears, K. T., Ceraul, S. M., Beier-Sexton, M., et al. (2013). Rickettsia typhi possesses phospholipase A2 enzymes that are involved in infection of host cells. PLoS Pathog. 9:e1003399. doi: 10.1371/journal.ppat.1003399
Raoult, D., and Roux, V. (1997). Rickettsioses as paradigms of new or emerging infectious diseases. Clin. Microbiol. Rev. 10, 694–719.
Renesto, P., Azza, S., Dolla, A., Fourquet, P., Vestris, G., Gorvel, J. P., et al. (2005). Rickettsia conorii and R. prowazekii proteome analysis by 2DE-MS: a step toward functional analysis of rickettsial genomes. Ann. N. Y. Acad. Sci. 1063, 90–93. doi: 10.1196/annals.1355.014
Reteno, D. G., Benamar, S., Khalil, J. B., Andreani, J., Armstrong, N., Klose, T., et al. (2015). Faustovirus, an asfarvirus-related new lineage of giant viruses infecting amoebae. J. Virol. 89, 6585–6594. doi: 10.1128/jvi.00115-15
Rohmer, L., Fong, C., Abmayr, S., Wasnick, M., Larson Freeman, T. J., Radey, M., et al. (2007). Comparison of Francisella tularensis genomes reveals evolutionary events associated with the emergence of human pathogenic strains. Genome Biol. 8:R102. doi: 10.1186/gb-2007-8-6-r102
Sahni, S. K., and Rydkina, E. (2009). Host-cell interactions with pathogenic Rickettsia species. Future Microbiol. 4, 323–339. doi: 10.2217/fmb.09.6
Sahni, S. K., Narra, H. P., Sahni, A., and Walker, D. H. (2013). Recent molecular insights into rickettsial pathogenesis and immunity. Future Microbiol. 8, 1265–1288. doi: 10.2217/fmb.13.102
Saisongkorh, W., El Karkouri, K., Patrice, J. Y., Bernard, A., Rolain, J. M., and Raoult, D. (2012). Tryptose phosphate broth improves Rickettsia felis replication in mammalian cells. FEMS Immunol. Med. Microbiol. 64, 111–114. doi: 10.1111/j.1574-695X.2011.00882.x
Saka, H. A., Thompson, J. W., Chen, Y.-S., Kumar, Y., Dubois, L. G., Moseley, M. A., et al. (2011). Quantitative proteomics reveals metabolic and pathogenic properties of Chlamydia trachomatis developmental forms. Mol. Microbiol. 82, 1185–1203. doi: 10.1111/j.1365-2958.2011.07877.x
Schroeder, C. L., Narra, H. P., Rojas, M., Sahni, A., Patel, J., Khanipov, K., et al. (2015). Bacterial small RNAs in the Genus Rickettsia. BMC Genomics 16, 1075. doi: 10.1186/s12864-015-2293-7
Schroeder, C. L., Narra, H. P., Sahni, A., Rojas, M., Khanipov, K., Patel, J., et al. (2016). Identification and characterization of novel small RNAs in Rickettsia prowazekii. Front. Microbiol. 7:859. doi: 10.3389/fmicb.2016.00859
Sears, K. T., Ceraul, S. M., Gillespie, J. J., Allen, E. D. Jr., Popov, V. L., Ammerman, N. C., et al. (2012). Surface proteome analysis and characterization of surface cell antigen (Sca) or autotransporter family of Rickettsia typhi. PLoS Pathog. 8:e1002856. doi: 10.1371/journal.ppat.1002856
Silva, J. C., Gorenstein, M. V., Li, G. Z., Vissers, J. P., and Geromanos, S. J. (2006). Absolute quantification of proteins by LCMSE: a virtue of parallel MS acquisition. Mol. Cell. Proteomics 5, 144–156. doi: 10.1074/mcp.M500230-MCP200
Socolovschi, C., Kernif, T., Raoult, D., and Parola, P. (2012). Borrelia, Rickettsia, and Ehrlichia species in bat ticks, France, 2010. Emerg. Infect. Dis. 18, 1966–1975. doi: 10.3201/eid1812.111237
Son, M. Y., Kwak, J. E., Kim, Y. D., and Cho, Y. S. (2015). Proteomic and network analysis of proteins regulated by REX1 in human embryonic stem cells. Proteomics 15, 2220–2229. doi: 10.1002/pmic.201400510
Speck, S., Derschum, H., Damdindorj, T., Dashdavaa, O., Jiang, J., Kaysser, P., et al. (2012). Rickettsia raoultii, the predominant Rickettsia found in Mongolian Dermacentor nuttalli. Ticks Tick Borne Dis. 3, 227–231. doi: 10.1016/j.ttbdis.2012.04.001
Spitalska, E., Stefanidesova, K., Kocianova, E., and Boldis, V. (2012). Rickettsia slovaca and Rickettsia raoultii in Dermacentor marginatus and Dermacentor reticulatus ticks from Slovak Republic. Exp. Appl. Acarol. 57, 189–197. doi: 10.1007/s10493-012-9539-8
Stothard, D. R., Clark, J. B., and Fuerst, P. A. (1994). Ancestral divergence of Rickettsia bellii from the spotted fever and typhus groups of Rickettsia and antiquity of the genus Rickettsia. Int. J. Syst. Bacteriol. 44, 798–804. doi: 10.1099/00207713-44-4-798
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Treitz, C., Cassidy, L., Hockendorf, A., Leippe, M., and Tholey, A. (2015). Quantitative proteome analysis of Caenorhabditis elegans upon exposure to nematicidal Bacillus thuringiensis. J. Proteomics 113, 337–350. doi: 10.1016/j.jprot.2014.09.027
Tucker, A. M., Driskell, L. O., Pannell, L. K., and Wood, D. O. (2011). Differential proteomic analysis of Rickettsia prowazekii propagated in diverse host backgrounds. Appl. Environ. Microbiol. 77, 4712–4718. doi: 10.1128/AEM.05140-11
UniProt (2017). The universal protein knowledgebase. Nucleic Acids Res. 45, D158–D169. doi: 10.1093/nar/gkw1099
Vellaiswamy, M., Kowalczewska, M., Merhej, V., Nappez, C., Vincentelli, R., Renesto, P., et al. (2011). Characterization of rickettsial adhesin Adr2 belonging to a new group of adhesins in alpha-proteobacteria. Microb. Pathog. 50, 233–242. doi: 10.1016/j.micpath.2011.01.009
Vizcaino, J. A., Deutsch, E. W., Wang, R., Csordas, A., Reisinger, F., Rios, D., et al. (2014). ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 32, 223–226. doi: 10.1038/nbt.2839
Wei, J., Goldberg, M. B., Burland, V., Venkatesan, M. M., Deng, W., Fournier, G., et al. (2003). Complete genome sequence and comparative genomics of Shigella flexneri serotype 2a strain 2457T. Infect. Immun. 71, 2775–2786. doi: 10.1128/IAI.71.5.2775-2786.2003
Weinert, L. A., Werren, J. H., Aebi, A., Stone, G. N., and Jiggins, F. M. (2009). Evolution and diversity of Rickettsia bacteria. BMC Biol. 7:6. doi: 10.1186/1741-7007-7-6
Wen, J., Jiao, D., Wang, J. H., Yao, D. H., Liu, Z. X., Zhao, G., et al. (2014). Rickettsia raoultii, the predominant Rickettsia found in Dermacentor silvarum ticks in China-Russia border areas. Exp. Appl. Acarol. 63, 579–585. doi: 10.1007/s10493-014-9792-0
Wolf, Y. I., and Koonin, E. V. (2013). Genome reduction as the dominant mode of evolution. Bioessays 35, 829–837. doi: 10.1002/bies.201300037
Wood, D. O., Hines, A., Tucker, A. M., Woodard, A., Driskell, L. O., Burkhardt, N. Y., et al. (2012). Establishment of a replicating plasmid in Rickettsia prowazekii. PLoS ONE 7:715. doi: 10.1371/journal.pone.0034715
Zhang, J. Z., Hao, J. F., Walker, D. H., and Yu, X. J. (2006). A mutation inactivating the methyltransferase gene in avirulent Madrid E strain of Rickettsia prowazekii reverted to wild type in the virulent revertant strain Evir. Vaccine 24, 2317–2323, doi: 10.1016/j.vaccine.2005.11.044
Zhao, G., Zhu, L., Feng, E., Cao, X., Shang, N., Liu, X., et al. (2010). A novel anti-virulence gene revealed by proteomic analysis in Shigella flexneri 2a. Proteome Sci. 8:30. doi: 10.1186/1477-5956-8-30
Zhao, Y., Valbuena, G., Walker, D. H., Gazi, M., Hidalgo, M., DeSousa, R., et al. (2016). Endothelial Cell Proteomic Response to Rickettsia conorii Infection Reveals Activation of the Janus Kinase (JAK)-Signal Transducer and Activator of Transcription (STAT)-Inferferon Stimulated Gene (ISG)15 pathway and reprogramming plasma membrane integrin/cadherin signaling. Mol. Cell. Proteomics 15, 289–304. doi: 10.1074/mcp.M115.054361
Keywords: Rickettsia, intracellular, infectious diseases, virulence, pathogenicity, evolution, genomics, proteomics
Citation: El Karkouri K, Kowalczewska M, Armstrong N, Azza S, Fournier P-E and Raoult D (2017) Multi-omics Analysis Sheds Light on the Evolution and the Intracellular Lifestyle Strategies of Spotted Fever Group Rickettsia spp. Front. Microbiol. 8:1363. doi: 10.3389/fmicb.2017.01363
Received: 19 April 2017; Accepted: 05 July 2017;
Published: 20 July 2017.
Edited by:
Jörg Linde, Leibniz-Institute for Natural Product Research and Infection Biology-Hans-Knoell-Institute, GermanyReviewed by:
Jean Armengaud, Commissariat à l'Energie Atomique et aux Energies Alternatives (CEA), FranceLudovit Skultety, Institute of Virology (SAS), Slovakia
Copyright © 2017 El Karkouri, Kowalczewska, Armstrong, Azza, Fournier and Raoult. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pierre-Edouard Fournier, cGllcnJlLWVkb3VhcmQuZm91cm5pZXJAdW5pdi1hbXUuZnI=