 Lassaad Belbahri1,2*
Lassaad Belbahri1,2* Ali Chenari Bouket2,3,4
Ali Chenari Bouket2,3,4 Imen Rekik2
Imen Rekik2 Faizah N. Alenezi2
Faizah N. Alenezi2 Armelle Vallat5Lenka Luptakova2,6
Armelle Vallat5Lenka Luptakova2,6 Eva Petrovova7
Eva Petrovova7 Tomasz Oszako8Semcheddine Cherrad9Sébastien Vacher10
Tomasz Oszako8Semcheddine Cherrad9Sébastien Vacher10 Mostafa E. Rateb11
Mostafa E. Rateb11- 1Laboratory of Soil Biology, University of Neuchatel, Neuchatel, Switzerland
- 2NextBiotech, Agareb, Tunisia
- 3Graduate School of Life and Environmental Sciences, Osaka Prefecture University, Sakai, Japan
- 4Young Researchers and Elite Club, Tabriz Branch, Islamic Azad University, Tabriz, Iran
- 5Neuchâtel Platform of Analytical Chemistry, Institute of Chemistry, University of Neuchâtel, Neuchâtel, Switzerland
- 6Department of Biology and Genetics, Institute of Biology, Zoology and Radiobiology, University of Veterinary Medicine and Pharmacy, Kosice, Slovakia
- 7Institute of Anatomy, University of Veterinary Medicine and Pharmacy, Kosice, Slovakia
- 8Forest Research Institute, Raszyn, Poland
- 9CONIPHY, Parc d'activités en Chuel, Quincieux, France
- 10CONIDIA, Parc d'activités en Chuel, Quincieux, France
- 11School of Science and Sport, University of the West of Scotland, Paisley, United Kingdom
The Gram positive, non-pathogenic endospore-forming soil inhabiting prokaryote Bacillus amyloliquefaciens is a plant growth-promoting rhizobacterium. Bacillus amyloliquefaciens processes wide biocontrol abilities and numerous strains have been reported to suppress diverse bacterial, fungal and fungal-like pathogens. Knowledge about strain level biocontrol abilities is warranted to translate this knowledge into developing more efficient biocontrol agents and bio-fertilizers. Ever-expanding genome studies of B. amyloliquefaciens are showing tremendous increase in strain-specific new secondary metabolite clusters which play key roles in the suppression of pathogens and plant growth promotion. In this report, we have used genome mining of all sequenced B. amyloliquefaciens genomes to highlight species boundaries, the diverse strategies used by different strains to promote plant growth and the diversity of their secondary metabolites. Genome composition of the targeted strains suggest regions of genomic plasticity that shape the structure and function of these genomes and govern strain adaptation to different niches. Our results indicated that B. amyloliquefaciens: (i) suffer taxonomic imprecision that blurs the debate over inter-strain genome diversity and dynamics, (ii) have diverse strategies to promote plant growth and development, (iii) have an unlocked, yet to be delimited impressive arsenal of secondary metabolites and products, (iv) have large number of so-called orphan gene clusters, i.e., biosynthetic clusters for which the corresponding metabolites are yet unknown, and (v) have a dynamic pan genome with a secondary metabolite rich accessory genome.
Introduction
As public pressure mounts to protect the environment, biological control strategies of phytopathogens including viruses, bacteria, fungi, and oomycetes (Lara and Belbahri, 2011; Olson et al., 2012; Luchi et al., 2013; Prospero et al., 2013; Abad et al., 2014) are more considered as ecologically sound and economically viable alternatives to pesticide usage strategies (Gurr and You, 2016; Mefteh et al., 2017). Plant-associated B. amyloliquefaciens strains colonize plant rhizosphere, promote plant growth and suppress competing phytopathogens. Therefore, they have been widely used as biofertilizers and biopesticides (Wu et al., 2015). Abilities to compete with pathogens are linked to the production of secondary metabolites (Chen et al., 2007; Boottanun et al., 2017) that possess antimicrobial activity (Alenezi et al., 2015a,b; Belbahri et al., 2015; Alenezi et al., 2016a,b, 2017; Mefteh et al., 2017) or host plant immune system stimulation (Chowdhury et al., 2015). Secondary metabolites have been widely documented in the fields of food processing (Chang et al., 2015; Chaves-Lopez et al., 2015), pharmaceuticals (Prazdnova et al., 2015) and environmental engineering (Alvarez et al., 2015; Mlaik et al., 2015; Sellami et al., 2016).
Bacillus amyloliquefaciens promotes plant growth using diverse mechanisms including indole-3-acetic acid (IAA) synthesis (Shao et al., 2015; Liu et al., 2016), phosphorus solubilisation (Ravari and Heidarzadeh, 2014) and potassium solubilisation (Shakeel et al., 2015). Extracellular phytase, for instance, is considered as a plant growth promoting factor for improvement of phosphorus-use efficiency by plants (Shao et al., 2015). Bacillus amyloliquefaciens has also been used as biocontrol of numerous plant diseases caused by soil-borne microorganisms (Islam et al., 2016; Tan et al., 2016), post-harvest pathogens (Chen et al., 2016), insects (Aziz et al., 2016), nematodes (Castaneda-Alvarez et al., 2016), and aphids (Gadhave and Gange, 2016). Moreover, B. amyloliquefaciens has been reported to directly antagonize plant pathogens by competing for essential nutrients (Wu et al., 2016), producing antibiotic compounds (Srivastava et al., 2016) and inducing systemic acquired resistance (Ng et al., 2016). Volatile components such as acetoin have shown to be a potent inducer of systemic acquired resistance in plants (Magno-Perez-Bryan et al., 2015). Cyclic dipeptide such as cyclo(L-leucyl-L-prolyl) mitigates virulence in pathogenic bacteria (Gowrishankar et al., 2016). Additionally, biofilm-producing bacteria on the plant-root surfaces show promise for the use in the control of soil-borne pathogens (Tan et al., 2016). Therefore, it is currently regarded as promising environmental friendly means for crop protection (Wei et al., 2015). Recently, using comparable concentrations of B. amyloliquefaciens to those expected when the bacteria are used as Plant growth-promoting rhizobacteria (PGPR) and biocontrol agent (107 cells ml−1) proved non harmful to the non-target soil dwelling earthworms (Lagerlof et al., 2015). Therefore, B. amyloliquefaciens could be safely used to optimize ecosystem services and resilience toward the development of sustainable agricultural systems.
Besides, being used as PGPR bacteria with wide metabolic capabilities, B. amyloliquefaciens is used for new applications such as degradation of crude oil from oil-contaminated soils (Zhang, J. H. et al., 2016), feather degradation (Yang et al., 2016), production of proteases (Wang et al., 2016), feruloyl esterases (Wang et al., 2017), and phytases (Verma et al., 2016) for industrial and food applications. Moreover, it is widely used for extraction of lipases for biodiesel production (Saengsanga et al., 2016), biosorbent for the removal of pollutants (Sun et al., 2016) and their degradation (Zuhlke et al., 2016), production of biosurfactants and antimicrobial lipopeptides (Perez et al., 2017; Zhi et al., 2017), probiotics (Gowrishankar et al., 2016), and food preservation (Eom and Choi, 2016; Calvo et al., 2017).
Comparative genomic analysis in B. amylioliquefaciens is made possible by the recent sequencing of multiple strains of the species. Similar to other bacterial groups the conserved “core” genome is defined as the shared genetic material among nearly all the strains of the species. The core genome contains majority of housekeeping genes and is interspersed with “accessory” genomic parts. It is believed that accessory genome is present in some strains while being absent in the rest of the species strains (Ozer et al., 2014).
In the current study, genomes of 48 strains of B. amylioliquefaciens available in GenBank (genomes submitted until December, 2016) have been mined for genes contributing to plant-beneficial functions and therefore, plant growth promotion potential and secondary metabolite arsenal. The contribution of core and accessory genome to plant growth promotion and secondary metabolite biosynthesis are also discussed.
Materials and Methods
Selection of Genomes and Genome Phylogeny
Genomes of B. amyloliquefaciens used in the study were selected among those submitted until December, 2016 in GenBank DNA database. They all have been deposited under the nomination B. amyloliquefaciens. The genomes and their corresponding strains have been described in Table 1. Nucleotide as well as the amino acid sequences of the whole genomes and the deduced coding sequences were retrieved from the GenBank DNA database for all strains (Table 1). Whole genome alignments have been conducted using REALPHY (The Reference sequence alignment based phylogeny builder, available at http://realphy.unibas.ch; Bertels et al., 2014). A Maximum Likelihood (ML) algorithm (Felsenstein, 1981) as implemented in MEGA v. 6 (Tamura et al., 2013) with evolutionary distances computed using the Kimura 2-parameter model (Kimura, 1980) was used to build the phylogenetic tree. Validity of branches in the resulting tree was evaluated by bootstrap re-sampling support of the data sets with 1,000 replications. Average nucleotide identity (ANI) values of B. amyloliquefaciens strains were estimated using the algorithm developed by Goris et al. (2007) combined with the 95~96% cut-off for species boundary proposed by Richter and Rosselló-Móra (2009), as implemented in the server EzBioCloud available at http://www.ezbiocloud.net/tools/ani (Yoon et al., 2017). In silico genome-to-genome distance values were calculated using the web-based DSMZ service available at http://ggdc.dsmz.de (Meier-Kolthoff et al., 2013). Species and sub-species cut-off were those suggested by default analysis (70%).
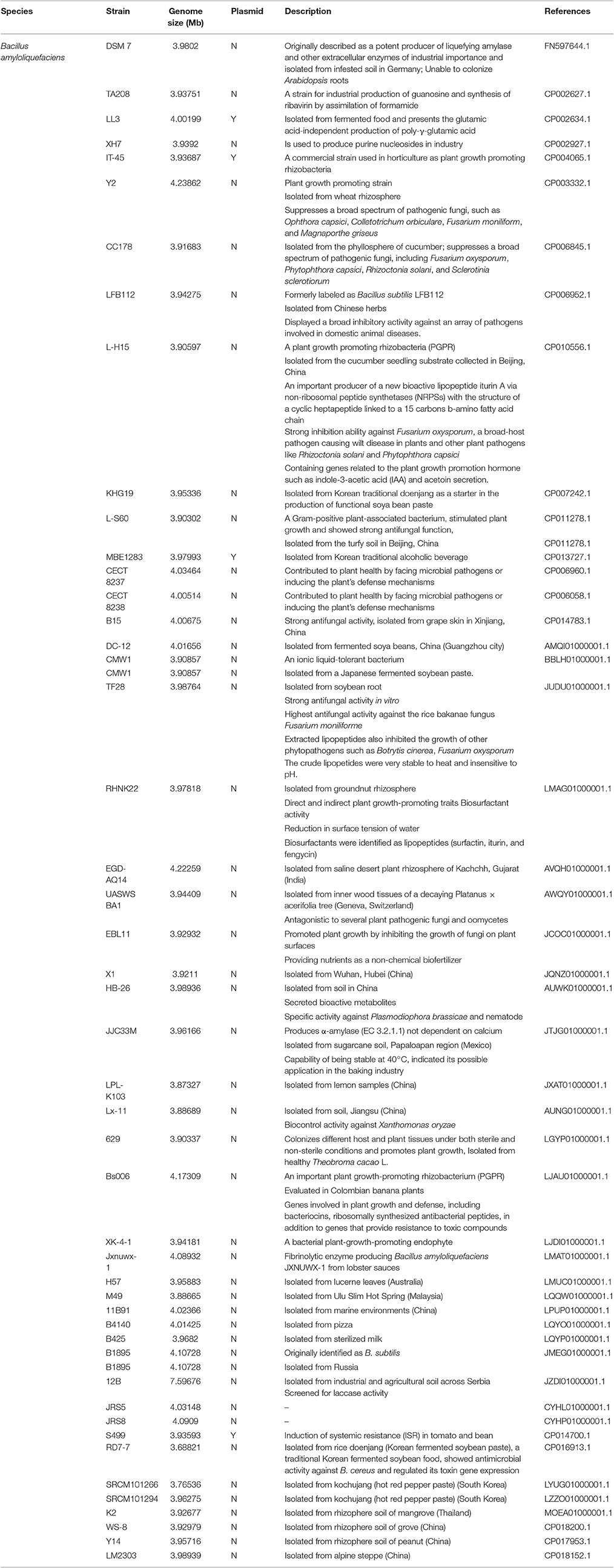
Table 1. List and description of the strains used in the study.
Homology Based Mining of Genes Contributing to Plant-Beneficial Functions
Nutrient Acquisition
The nitrogenase-encoding genes nifHDK, nifS, and nifU responsible for nitrogen fixation in proteobacterial PGPR from Azospirillum, Burkholderia, and Bacillus were used as bait to search for similar sequences (Bruto et al., 2014). The pyrroloquinoline quinone-encoding genes pqqBCDEFG in the PGPR Pseudomonas fluorescens F113, Erwinia herbicola, and Enterobacter intermedium (Liu et al., 1992 and Kim et al., 2003; Miller et al., 2010) were used to mine the studied genomes. The gene encoding the B. velezensis SQR9 3-phytase was selected to mine B. amyloliquefaciens genomes for phytase production (Shao et al., 2015). Genes encoding ureABC of Bacillus subtilis (strain 168) was used in blast searches to recover urease genes in B. amyloliquefaciens studied genomes (Niazi et al., 2014). Exoenzyme genome mining was carried out using either keyword search in the different genomes followed by checking of secretion using SignalP 4.1 (Petersen et al., 2011) or by blasting exoenzyme sequences described in closely related species (Niazi et al., 2014). Enzymes targeted were proteases, lipases, cellulases, pectinases, amylases, laccases, xylanases, and lichenases. Heat-shock protein genes dnaJ, dnaK, and groE, cold shock protein genes cspA, cspC, cspD, and cspE (Gupta et al., 2014), osmoprotectant glycine betaine synthesis genes gbsAB (Boch et al., 1996). Genes encoding phenazine (phzADEFG) were also mined since phenazine aid in long term survival and ability to compete with the resident microflora (Mazzola et al., 1992).
Root Colonization and Growth Promotion Factors
The presence of gene clusters (flgBCDEGKLMN, flhABFOP) and the swrABC gene cluster have been searched in the genomes of the different B. amyloliquefaciens targeted genomes (Ghelardi et al., 2012). che/fla/fli/tlp/mcp operons involved in the regulation of B. subtilis chemotactic response and their relatives in the genome of Bacillus velezensis UCMB5113, motABPS cluster responsible for cell-envelope and cellular processes motility and chemotaxis, have been mined in the different genomes studied (Niazi et al., 2014). The xerCD genes, site recombinase, are critical for the PGPRs to be effective rhizosphere colonizers (Shen et al., 2013) have been mined. Annotation and homology-based searches were conducted in the Bacillus genomes for genes encoding exopolysaccharide using B. subtilis epsA-O operon genes, tapA, tasA, sipW, pgsB, and bslA (Vlamakis et al., 2013).
Plant Growth-Promoting Traits: Hormones
The genes involved in the tryptophan-dependent pathways for synthesis of the auxinic phytohormone indole acetic acid (IAA) in the closely related B. velezensis FZB42 and B. velezensis SQR9 (Idris et al., 2007; Shao et al., 2015) were selected. The different pathways mined were: (i) indole-3-pyruvate (IPyA) pathway involving the tryptophan transaminase (patB), indole-3-pyruvate decarboxylase (YclC and YclB) and indole-3-acetaldehyde dehydrogenase (DhaS) genes, (ii) indole-3-acetonitrile (IAN) involving the nitrilase gene (yhcX), (iii) uncharacterized IAA biosynthesis pathway involving tryptophan acetyltransferase gene (ysnE) and (Zimmer et al., 1991; Idris et al., 2007; Shao et al., 2015). Additionally, the ywkB gene involved in the transport of auxin out of the bacterial cell, its redistribution to the plant roots, and encoding a putative auxin efflux carrier protein was also mined in the different genomes (Niazi et al., 2014).
The Agrobacterium tumefaciens trans-zeatin synthase, tzs gene and the miaA gene encoding tRNA dimethylallyl transferase that removes zeatin precursor from tRNA were used to query the collected genomes (Vacheron et al., 2013).
The IpdC gene directs the production of phenylacetic acid (PAA), having weak auxin activity and antimicrobial against both bacteria and fungi in Azospirillum brasilense (Somers et al., 2005). As in Azospirillum, the B. simplex genome has the paa operon (data not shown), which is important for the degradation of PAA.
Genes encoding ACC deaminase structural genes (acdS) and leucine responsive regulatory protein (LRP) gene (acdR) of Pseudomonas putida GR12-2 were selected to mine B. amyloliquefaciens analyzed genomes (Glick et al., 1994).
The gene of A. brasilense Sp245 nirK copper nitrite reductase and Bacillus nitric oxide synthase (nos) genes leading to formation of NO and hence root branching was used to mine the B. amyloliquefaciens genomes (Bruto et al., 2014).
In B. subtilis OKB105 polyamines such as spermine, spermidine, and putrescine have PGP properties (Xie et al., 2014). Genes involved in polyamine synthesis such as speA (agmatine synthesis), speB (putrescine synthesis); speD and speE (spermidine synthesis) and metK, responsible for the conversion of methionine to S-adenosyl-methionine were mined. Genes for various binding proteins, permeases, and transporters for polyamines have also been mined by keyword searches in the different genomes.
Plant Protection from Oxidative Stress (Antioxidant Enzymes)
The battery of enzymes produced by Bacillus spp. in response to oxidative stress has been fetched in the different B. amyloliquefaciens genomes. In B. velezensis UCMB5113 superoxide dismutases (SodA, SodC, and SodF), three hydrogen peroxide decomposing catalases (KatA, KatE, and KatX), manganese catalase (YdbD), three alkyl hydroperoxide reductases (AhpC, AhpF, and BASU_0830), thiol peroxidase (tpx), glutathione peroxidase (gpo), bacillopeptidase F (bpr), gamma-glutamyl transpeptidase (ggt), and an operon (ohrARB) for resistance to organic peroxides have been described by Niazi et al. (2014) and included in our genome mining efforts. The flavohemoprotein nitric oxide dioxygenase encoded by the B. velezensis UCMB5113 genes hmp and BASU_2738, that protect the bacterium from nitrosative stress have also been included in our study. The genes gacS, soxS, soxR, and oxyR involved in plant protection against oxidative stress were also mined (Whistler et al., 1998; Ochsner et al., 2000).
Plant Induction of Disease Resistance
The P. aeruginosa genes have been mined in the different genomes. Genes selected for homology-based searches involved the B. velezensis SQR9 genes encoding acetoin biosynthesis: acetolactate synthase alsS (E.C. 2.2.1.6), acetolactate decarboxylase alsD (E.C. 4.1.1.5) and the regulatory gene alsR as well as the gene bdhA encoding 2,3-butanediol dehydrogenase encoding 2,3-butandiol biosynthesis (Shao et al., 2015).
Antibiotics and Related Compounds
hcnABC genes directing production of HCN in Pseudomonas spp. have been used to mine B. amyloliquefaciens genomes (Bruto et al., 2014). phlACBD genes were used in blast searches to discover similar sequences in the genomes of the mined B. amyloliquefaciens strains (Bruto et al., 2014). gabD and gabT involved in production of pest/disease suppressing γ-aminobutyric acid (GABA) (Loper et al., 2012) have been used as baits in genome mining.
Resistance to Drugs
Homologues of the tetB protein that contributes to tetracycline resistance and the tetR tetracycline operon transcriptional regulator tetR in B. subtilis have been searched in the different genomes (Sakaguchi et al., 1988). Multifunctional tetracycline-metal/H+ antiporter (tetA) have also been mined (Someya et al., 1995). The operon yyaACDEHJKLRST encoding a streptothricin acetyltransferase (Jacob et al., 1994) was used as a bait in the screening of homologs in the different genomes. Fosfomycin resistance gene fosB from B. cereus was used to search for homologs in the B. amyloliquefaciens genomes (Fu et al., 2016). The homolog of the B. licheniformis glyoxalase/bleomycin resistance gene ykcA have been used as a bait in the blast search against mined genomes (Rey et al., 2004). The homolog of the B. subtilis (strain 168) β-lactamase gene penP have been used as a bait in the blast search against mined genomes (Barbe et al., 2009). Quinolone resistance norA homology have been searched in the different B. amyloliquefaciens genomes (Neyfakh et al., 1993). The E. coli gene floR have been mined in the B. amyloliquefaciens genome collection (Doublet et al., 2005). Bacillus subtilis 168 aadK gene, which encodes aminoglycoside 6-adenylyltransferase, a streptomycin-modifying enzyme, was mined in the different strains (Noguchi et al., 1993). Bacillus subtilis ycbJ gene encoding an aminoglycoside phosphotransferase has been used to search homologs in the genomes of the mined strains (Hosoya et al., 2002). Bacillus subtilis vmlR encoding antibiotic efflux ATP-binding transport protein has been used to mine the different genomes (Ohki et al., 2005). Genes encoding putative multidrug exporters have been mined from the different genomes according to Niazi et al. (2014).
Resistance to Heavy Metals
The genes arsABC and ywrK were used as a bait to detect any putative arsenic detoxification ability (Duan et al., 2013). We have mined the copYZAB operon formed by four genes: copA and copB that encode ATPases for influx and efflux of copper, respectively; copZ that encodes a copper chaperone; and copY, a copper responsive repressor. CopA encodes a major copper resistance mechanism. One-component regulators CueR, CopY, and CsoR, identified in E. coli, E. hirae, and M. tuberculosis, respectively, have also been mined (Rademacher and Masepohl, 2012). CtpAB and ycnJ genes encoding copper resistance proteins (Zhang et al., 2015) were also mined. Homologs of the B. subtilis (strain 168) ynbB gene have been mined in the different genomes (Barbe et al., 2009). Homology of crcA, cspE, crcB,yhdV has been mined in all the genomes (Hu et al., 1996). Homologs of the yceGH and yaaN have been searched in all genomes (Franks et al., 2014). CzcD encodes a cadmium, cobalt and zinc/H(+)-K(+) antiporter in B. subtilis and protects the cell against elevated levels of Zn(II), Cu, Co(II), and Ni(II) (Moore et al., 2005). GenendoA (ydcE) and antitoxin gene, ndoAI (ydcD) have been mined (Wu et al., 2011). Sensors for metals; Fur, ArsR, MerR, NikR, DtxR, mtnR, and yfmP family of metalloregulators of the B. subtilis genome were mined from the different B. amyloliquefaciens genomes (Osman and Cavet, 2010).
Degradation of Aromatic Compounds
Vanillate, 4-hydroxybenzoate, salicylic, ferulic, p-coumaric acids are considered as natural toxins and cause specific stress responses in microorganisms that have developed resistance against phenolic acids. Both phenolic acid decarboxylases padC and bsdBCD (yclBCD) of B. subtilis were mined. The putative LysR-type regulator encoded by bsdA (yclA) gene upstream of the bsdBCD operon revealed is the transcriptional activator of bsdBCD expression in response to phenolic acids were also mined (Graf et al., 2016). Dibenzothiophene (DBT) is the model compound for this class of molecules. The operon dszABC of Rhodococcus sp. (Piddington et al., 1995) was used to mine the genomes. Genes encoding homologs of the B. velezenzis FZB42 azoR2, mhqADNOPE have been mined in the genome of the different strains (Nguyen et al., 2007).
Secondary Metabolite Clusters Identification using Antismash, Prism, Napdos, NP.Search, and Bagel3
The annotated draft genome sequence files, which included information for both contigs and ORFs (Table 1) were subjected to secondary metabolite gene cluster analysis using antiSMASH 3.0 (Weber et al., 2015), prediction informatics for secondary metabolomes (PRISM) (Skinnider et al., 2015), NapDos (Ziemert et al., 2012), NP.search (Li et al., 2009), and the bacteriocin specific software BAGEL3 (Van Heel et al., 2013).
Identification of Core Genome and Accessory Genomes of the Strain Collection
Spine, used to determine the core genome, defined as those sequences present in nearly all genomes from bacteria of a given species, from the sequences of all B. amyloliquefaciens isolates collection (Ozer et al., 2014). Identification of accessory genomic sequences in the different B. amyloliquefaciens isolates genomes was performed using Agent (Ozer et al., 2014).
Results
Species Status of B. amyloliquefaciens
In total, 48 strains of the species (submitted until December 2016) have been selected for genome mining. Their genome size varied between 3.60 and 7.60 mega base pairs (MB) (Table 1). GGDC analysis revealed the presence of three species lumped together in the strains collection sensu Meier-Kolthoff et al. (2013), where 70 % similarity between two genomes was established as the gold standard threshold for species boundaries (Figure 1A), ANI analysis revealed also three putative species sensu Richter and Rosselló-Móra (2009), where 95–96% cut-off was set up to delimit species boundaries. In both analysis, a set of 10 strains represented probably the “true” B. amyloliquefaciens species termed “B. amyloliquefaciens sensu stricto” while a set of 37 strains matched B. velezensis and a single isolate represented new species, yet to be described (Figures 1A,B). The proposed threshold for species discrimination (70%) clearly delimit species boundaries because strain pairs were found to be between 50 and 70% GGDC distance. GGDC values plotted against ANI values (Figure 1C) showed agreement between the two technologies for species discrimination and no discontinuity in the graph could be observed. Finally, whole genome phylogeny confirmed results using GGDC and ANI values, with three sister branches representing the three species (Figure 1D).
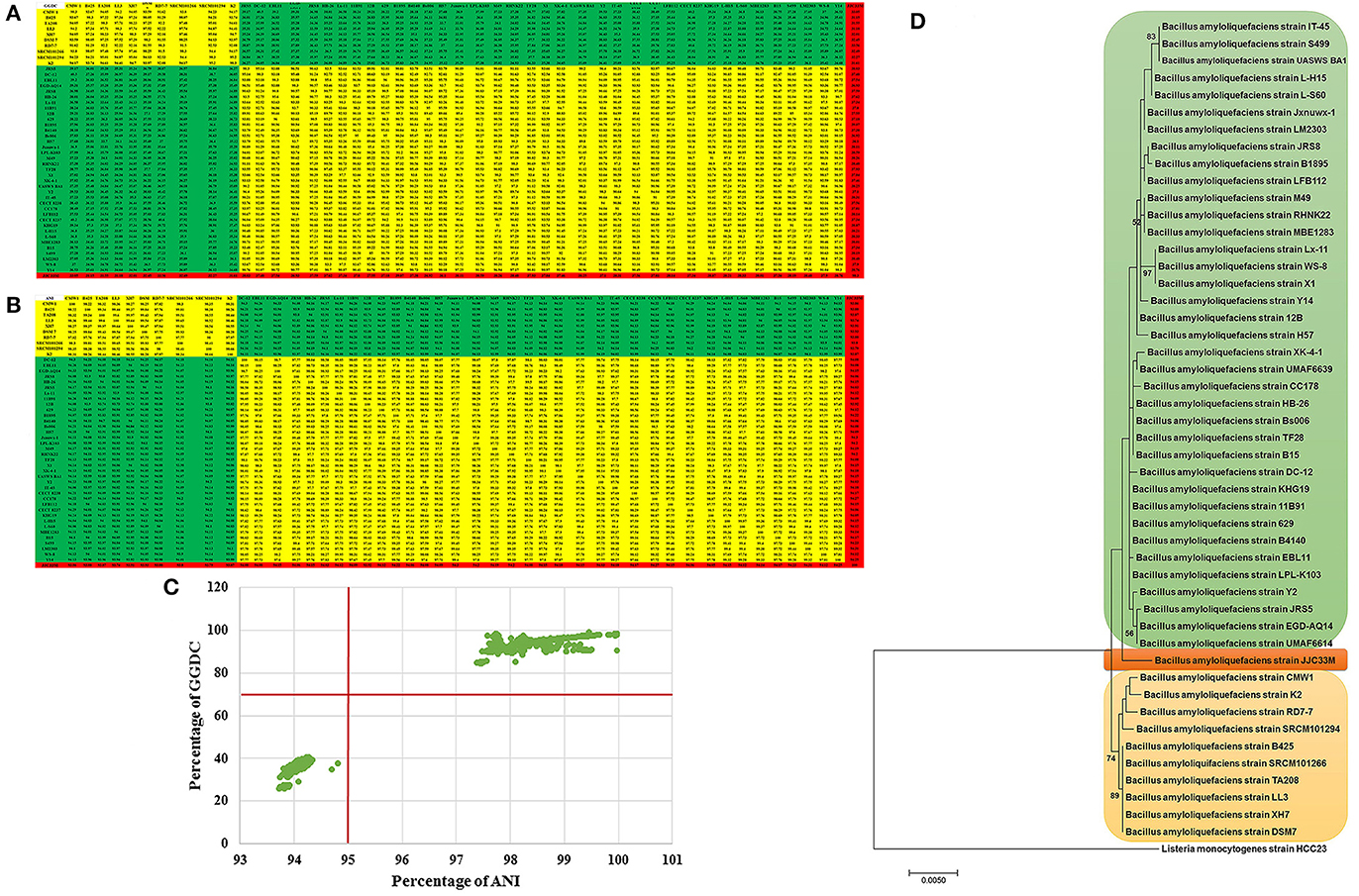
Figure 1. (A,B) Genome-to-Genome Distance Calculation (GGDC) and Average nucleotide identity (ANI) values between each indicated strains were calculated with GGDC 2 and EzBiocloud web-based programs showed 3 species candidates based on 70% and 95% similarity thresholds. (C) Scatter plot of ANI and GGDC values of B. amyloliquefaciens strains. (D) Maximum Likelihood phylogenomic tree of G-positive bacteria B. amyloliquefaciens strains. L. monocytogenes strain HCC23 was used as outgroup. Supports for branches were assessed by bootstrap resampling of the data set with 1,000 replications.
Bioinformatic Evaluation of Plant Growth Promotion Potential of B. amyloliquefaciens Strains
Bioinformatic evaluation of plant growth promotion potential of B. amyloliquefaciens strains collection has been performed through homology-based mining of genes contributing to plant-beneficial functions. As unambiguously shown in Figure 2, large majority of B. amyloliquefaciens strains show presence of mined genes independently of whether these strains are represented by a complete coverage of the genome or their association to plant rhizosphere.
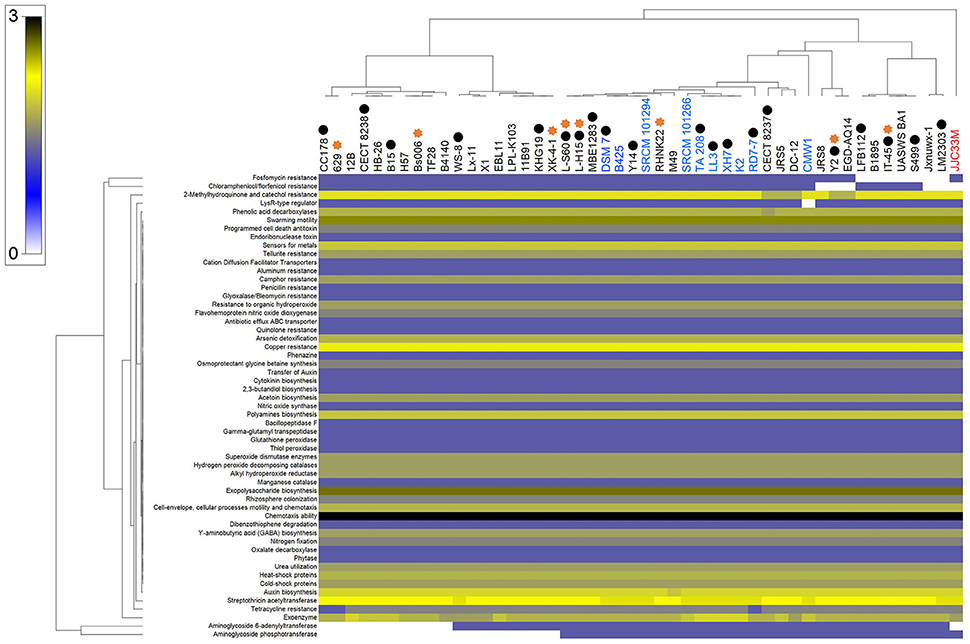
Figure 2. Heat map of mining of genes contributing to plant-beneficial functions in B. amyloliquefaciens strains. Bacterial strains belonging to the same species are highlighted with the same colors. Bacterial strains indicated with asterisk sign are related to strains that are emphasized in the literatures as plant growth promoting (PGP) bacteria. Black circles show completely sequenced strains.
Secondary Metabolites from B. amyloliquefaciens
Secondary metabolite clusters present in the genome of the B. amyloliquefaciens collection have been evaluated using antiSMASH 3.0 (Weber et al., 2015), prediction informatics for secondary metabolomes (PRISM) (Skinnider et al., 2015), NapDos (Ziemert et al., 2012), NP.search (Li et al., 2009), and the bacteriocin specific software BAGEL3 (Van Heel et al., 2013). As shown in Figure 3 and Supplementary Table S1, different strains showed high levels of diverse secondary metabolite clusters using all implied programs. Rarefaction analysis of secondary metabolite clusters from the results of genome sequencing progress clearly attested that saturation could not be reached using all genome collection analyzed (Figure 3B). A very clear correlation between genome size and number of gene clusters known to be involved in secondary metabolite biosynthesis and mined by antiSMASH was found. Approximately 65% of the variance in the number of secondary metabolite clusters can be explained by genome size (Figure 3C). However, for PRISM only 41% of the variance in the number of secondary metabolite clusters can be explained by genome size (Figure 3D).
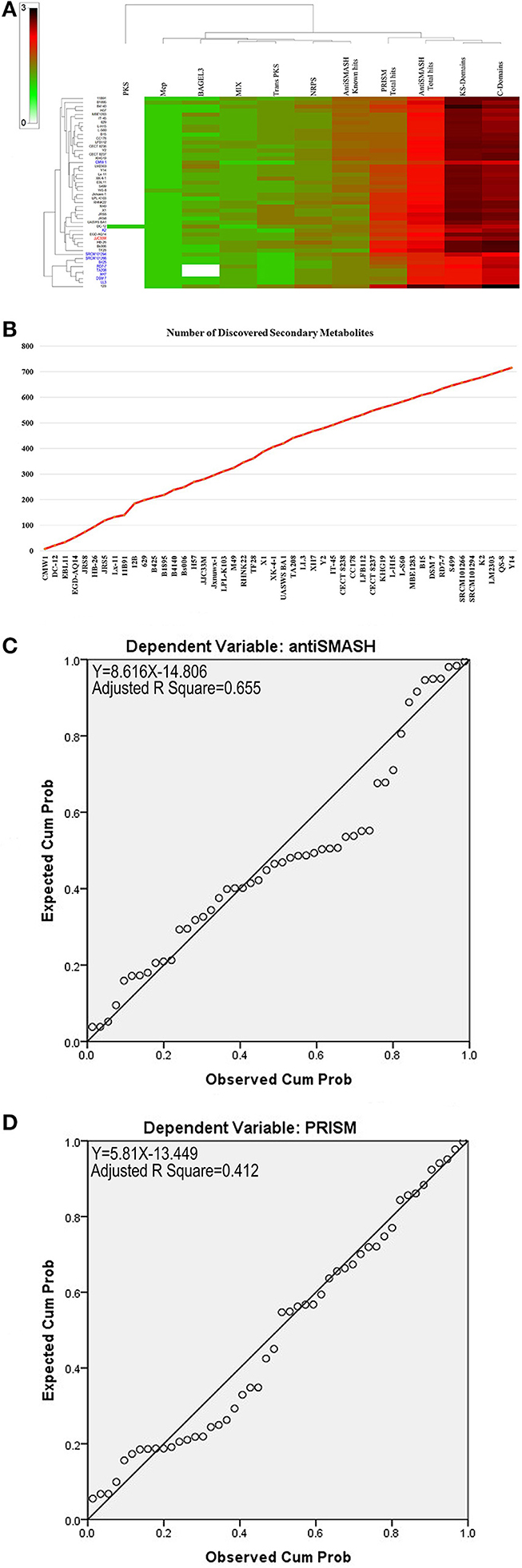
Figure 3. (A) Heat map of mining of genes contributing to secondary metabolite clusters. (B) Number of discovered secondary metabolites. (C) Statistically significant linear relationship between genome sizes and antiSmash total hits (p < 0.05). (D) Statistically significant linear relationship between genome sizes and PRISM total hits.
Genomes to Natural Products Prediction in B. amyloliquefaciens
Natural products prediction in the core genome and the accessory genomes of the B. amyloliquefaciens collection revealed high numbers of unknown secondary metabolites across the strains analyzed (Figure 4A and Supplementary Table S1). Only bacillibactin could be found in all the strains and in the core genome of B. amyloliquefaciens (Figure 4A). All remaining known secondary metabolites such as surfactin, difficidin, fengycin, macrolactin, bacillaene, bacilysin, and mersacidin are harbored by the accessory genome of the different strains. Only 3% of the variance in the number of secondary metabolite clusters can be explained by accessory genome size (Figure 4B).
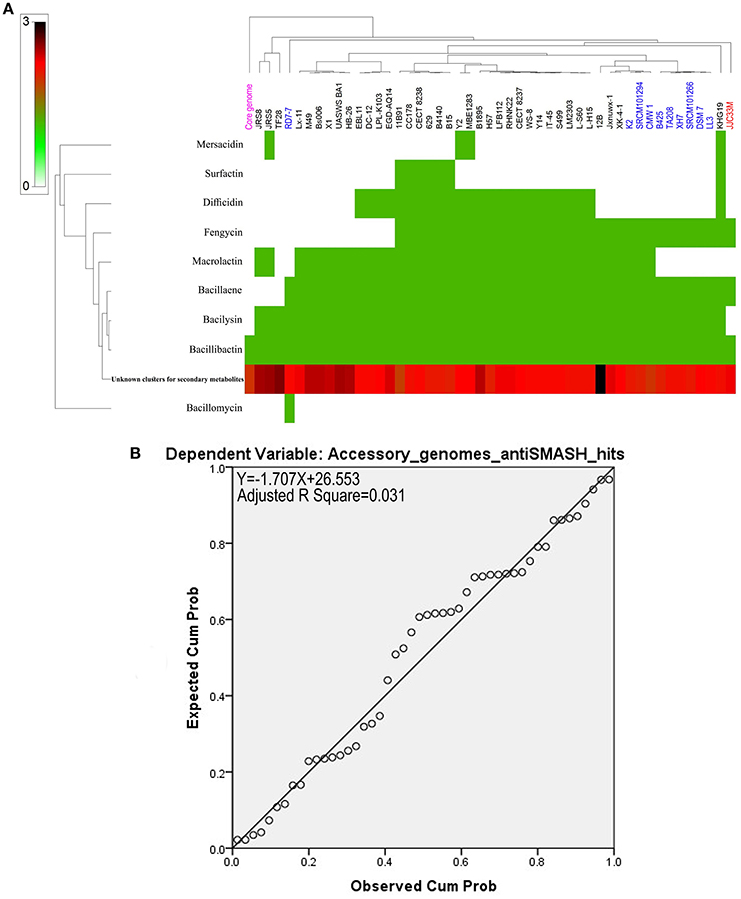
Figure 4. (A) Heat map of B. amyloliquefaciens accessory genome secondary metabolites. (B) Non-significant linear relationship between genome sizes and accessory genome antiSmash total hits (p > 0.1).
Discussion
Species Status of B. amyloliquefaciens
Given the high phenotypic similarity of B. amyloliquefaciens to B. subtilis and other closely related Bacillus spp. such as B. velezensis, it is not possible to distinguish these organisms solely on the basis of conventional assays (Dunlap et al., 2015 and 2016). Sequencing of 16S rRNA gene, while has historically been used in defining bacterial taxonomy and phylogeny, proved difficult and controversial that lead to well-documented misidentifications (Hahnke et al., 2016). Therefore, considerable taxonomic confusion blurs biotechnological applications of this highly relevant group. Recently, genome based approaches such as Average Nucleotide Identity (ANI) and digital DNA-DNA hybridization (DDH) calculated using the Genome-to-Genome Distance Calculation (GGDC) complemented with genome comparisons, alignments and phylogenetic reconstructions have been suggested as alternative methods for species discrimination (Goris et al., 2007; Richter and Rosselló-Móra, 2009; Meier-Kolthoff et al., 2013). Using these accurate tools, several later heterotypic synonyms were documented in this group such as B. methyltrophicus, B. amyloliquefaciens subsp. plantarum, and B. oryzicola that have been shown, using phylogenomics, later heterotypic synonyms of B. velezensis (Dunlap et al., 2016). Therefore, phylogenomic approaches are urgently required to resolve outstanding problems in the phylogenetic systematics of the B. subtilis group (Dunlap et al., 2016). Phylogenomic analysis of all sequenced genomes of B. amyloliquefaciens strains available in GenBank, the National Centre for Biotechnology Information (NCBI) database (Table 1), allowed us to check taxonomic validity of these isolates, determine the extent of inter-species genome variability within B. amyloliquefaciens and reconstruct their phylogenetic relationships. Figures 1A–D clearly showed that at least three Bacillus spp. were lumped under the name B. amyloliquefaciens along with B. amyloliquefaciens sensu stricto. While isolates DC12, EBL11, EGD-AQ14, JRS8, HB26, JRS5, LX-11, 11B91, 12B, 629, B1895, B4140, Bs006, H57, Jxnuwx-1, LPL-K103, M49, RHNK22, TF28, UASWS BA1, Y2, IT-45, CECT 8238, CC178, LFB112, CECT 8237, KHG 19, L-H15, L-S60, MBE 1283, B15, S499, LM2303, WS-8, and Y14 matched B. velezensis in ANI and GGDC analysis (data not shown), JJC33M failed to match known species and should be described as a new species. Bacillus isolates CMW1, B425, TA208, LL3, XH7, DSM7, RD7-7, SRCM101266, SRCM101294, and K2, should therefore be regarded as B. amyloliquefaciens sensu stricto. Phylogenomic tree based on the core genome of all isolates of B. amyloliquefaciens showed consistent results with earlier observations using either ANI or GGDC values. Our findings suggest that despite the pivotal role of microbial taxonomy in industrial exploitation of microbes and their products, classification and accurate identification have often been a neglected task. We recommend inclusion of phylogenomic studies as a prerequisite gold standard to the use of the name B. amyloliquefaciens in new reports.
Bioinformatic Evaluation of Plant Growth Promoting Potential of B. amyloliquefaciens Strains
Genome mining of the different strains of B. amyloliquefaciens allowed the discovery of numerous features documented in earlier studies as efficient factors of the interaction between host plants and the associated B. amyloliquefaciens strains (Niazi et al., 2014; Zhang, N. et al., 2016). These features allow nutrient acquisition, PGPR fitness, root colonization and growth promotion factors, plant growth promoting traits (hormones), plant protection from oxidative stress, plant induction of disease resistance, antibiotics and related compounds, resistance to drugs and heavy metals and degradation of aromatic compounds (Bruto et al., 2014; Niazi et al., 2014; Chen et al., 2016; Zhang, N. et al., 2016; Rekik et al., 2017). All these features were present in approximately all the genomes analyzed independently of whether these strains are represented by a complete coverage of the genome or their association to the plant rhizosphere. All these features could be also found in the core genome of the B. amyloliquefaciens sensu-stricto or the three-conserved species core genome. We speculate that plant growth promoting features could be considered as evolutional traits for adaptation to plant-associated habitats as suggested by Zhang, N. et al. (2016).
Secondary Metabolites from B. amyloliquefaciens
Bacillus amyloliquefaciens strains proved a prolific source of diverse secondary metabolite classes including polyketides (PKs) such as macrolactins and difficidins, peptides such as bacteriocins, lanthipeptides such as cerecidins, and lipopeptides (LPs) such as surfactins and iturins (Cimermancic et al., 2014; Wang et al., 2014; Aleti et al., 2015). PKs and LPs are the key inhibitors of plant pathogens and strains bearing these metabolites have been widely used in agriculture (Cochrane and Vederas, 2014). Despite the exponential increase of the number of B. amyliquefaciens genomes sequenced and the description of efficient analysis tools for secondary metabolite prediction, cursory investigation of these genome's wealth is available for describing the novelties and predicting uncharacterized metabolites (Aleti et al., 2015). In our study using recently described bioinformatic tools designed for the identification of clusters involved in secondary metabolism such as PRISM (Skinnider et al., 2015), antiSMASH 3.0 (Weber et al., 2015), NapDos (Ziemert et al., 2012), NP.searcher (Li et al., 2009), and the bacteriocin specific software BAGEL3 (Van Heel et al., 2013) and the B. amyloliquefaciens genomes available in databases, we documented high structural and functional diversity of secondary products in the species and their underlying gene clusters. Our data clearly showed high variety of secondary metabolites suggested by the high number of matches using five different programs for their prediction.
Rarefaction analysis of secondary metabolite clusters from the results of genome sequencing progress demonstrated clearly that saturation could not be reached using all genomes available and more sequencing effort of new strains is necessary to tackle the wide diversity of secondary metabolites potentially harbored by the species. This result confirmed the observations of Alenezi et al. (2016b) using the genus Aneurinibacillus. A very clear correlation between genome size and number of gene clusters known to be involved in secondary metabolite biosynthesis and mined by antiSMASH and PRISM was found. About 65% of the variance in the number of secondary metabolite clusters can be explained by genome size for antiSMASH for instance. This confirmed the results established by Jeske et al. (2013) while contrasted those conducted by Machado et al. (2015) and Alenezi et al. (2016b).
Genomes to Natural Products Prediction in B. amyloliquefaciens
Genome mining was also used to predict uncharacterized gene clusters and evaluate their potential to produce new yet to be characterized secondary metabolites. We found that while few known secondary metabolites such as surfactin, difficidin, bacilysin, fengycin, macrolactin, bacuillaene, and bacillibactin were identified, hundreds of secondary products still await for accurate molecular identification and the assignment of subsequent biological function. Similar finding has been reported by Jeske et al. (2013), Machado et al. (2015), and Alenezi et al. (2016b). Dynamics of evolution of the clusters was also investigated using comparative genomics across all known core and accessory genomes of B. amyloliquefaciens strains. Our findings unambiguously suggested that except bacillomycin, all remaining known or unknown secondary metabolites were harbored by the strains specific accessory genomes. This finding highlights the extraordinary potential offered by these plants associated Bacillus spp.
Summary and Outlook
Our findings clearly suggest plant growth promoting features as evolutional traits for adaptation of B. amyloliquefaciens sensu lato to plant-associated habitats. They also document large repertoire of secondary metabolites harbored by a dynamic accessory genome that warrants more genome sequencing efforts of B. amyloliquefaciens sensu lato in order to shed the light on the wealth of these natural products offered by these bacteria.
Ethics Statement
This research did not involve any work with human participants or animals by any of the authors.
Author Contributions
Conceived and designed the experiments: LB and AC. Performed the experiments: LB, FA, LL, IR, and AC. Analyzed the data: LB and AC. Contributed reagents/materials/analysis tools: LB. Wrote the manuscript and enriched the literature: LB. Corrected the manuscript: LB, MR, TO, LL, EP, FA, AV, SC, SV, and AC.
Funding
This project was funded by the Government of Kuwait (to FA) and the European Union Seventh Framework Programme under grant agreement 245268 (ISEFOR; to LB). Further support came from the SwissBOL project, financed by the Swiss Federal Office for the Environment (grant holder LB) and the Sciex–Scientific Exchange Program (https://www.swissuniversities.ch/en/topics/sciex/) (NMS.CH; to LL and LB). LL and EP are indebted to the Ministry of Education, Science, Research and Sport of the Slovak Republic for financial support in the frame of the project “VEGA 1/0046/16.” Part of the study was financially supported by the Life Plus project HESOFF, Life 11 ENV/PL/459 financed by the European Union and the National Fund for Environmental Protection and Water Management in Warsaw (Grant to TO).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.01438/full#supplementary-material
Supplementary Table S1. Predicted secondary metabolites (antiSmash cluster hits) of Bacillus amyloliquefaciens strains.
References
Abad, Z. G., Abad, J. A., Cunnington, J. H., Smith, I. W., Blomquist, C., Balci, Y., et al. (2014). Phytophthora niederhauserii sp. nov. a new polyphagous species mostly isolated from ornamentals potted plants in twelve countries of five continents. Mycologia 106, 431–447. doi: 10.3852/12-119
Alenezi, F. N., Fraser, S., Bełka, M., Doǧmuş, T. H., Hečkova, Z., Oskay, F., et al. (2016a). Biological control of Dothistroma needle blight on pine with Aneurinibacillus migulanus. Forest Pathol. 46, 555–558. doi: 10.1111/efp.12237
Alenezi, F. N., Rekik, I., Bełka, M., Ibrahim, A. F., Luptakova, L., Jaspars, M., et al. (2016b). Strain-level diversity of secondary metabolism in the biocontrol species Aneurinibacillus migulanus. Microbiol. Res. 182, 116–124. doi: 10.1016/j.micres.2015.10.007
Alenezi, F. N., Rekik, I., Chenari Bouket, A., Luptakova, L., Weitz, H. J., Rateb, M. E., et al. (2017). Increased biological activity of Aneurinibacillus migulanus strains correlates with the production of new gramicidin secondary metabolites. Front. Microbiol. 8:517. doi: 10.3389/fmicb.2017.00517
Alenezi, F. N., Weitz, H. J., Belbahri, L., Ben Rebah, H., Luptakova, L., Jaspars, M., et al. (2015a). Draft genome sequence of Aneurinibacillus migulanus strain Nagano. Genome Announc. 3:e00232–15. doi: 10.1128/genomeA.00232-15
Alenezi, F. N., Weitz, H. J., Belbahri, L., Nidhal, J., Luptakova, L., Jaspars, M., et al. (2015b). Draft genome sequence of Aneurinibacillus migulanus NCTC 7096. Genome Announc. 3:e00234–15. doi: 10.1128/genomeA.00234-15
Aleti, G., Sessitsch, A., and Brader, G. (2015). Genome mining: prediction of lipopeptides and polyketides from Bacillus and related Firmicutes. Comput. Struct. Biotechnol. J. 13, 192–203. doi: 10.1016/j.csbj.2015.03.003
Alvarez, V. M., Jurelevicius, D., Marques, J. M., de Souza, P. M., de Araújo, L. V., Barros, T. G., et al. (2015). Bacillus amyloliquefaciens TSBSO 3.8, a biosurfactant-producing strain with biotechnological potential for microbial enhanced oil recovery. Colloids Surf. B. Biointerfaces. 136, 14–21. doi: 10.1016/j.colsurfb.2015.08.046
Aziz, M., Nadipalli, R., Xie, X. T., Sun, Y., Surowiec, K., Zhang, J. L., et al. (2016). Augmenting sulfur metabolism and herbivore defense in Arabidopsis by bacterial volatile signaling. Front. Plant Sci. 7:458. doi: 10.3389/fpls.2016.00458
Barbe, V., Cruveiller, S., Kunst, F., Lenoble, P., Meurice, G., Sekowska, A., et al. (2009). From a consortium sequence to a unified sequence: the Bacillus subtilis 168 reference genome a decade later. Microbiology 155, 1758–1775. doi: 10.1099/mic.0.027839-0
Belbahri, L., Alenezi, F. N., Luptakova, L., Rateb, M. E., and Woodward, S. (2015). Complete genome sequence of Aneurinibacillus migulanus E1, a Gramicidin S- and D-phenylalanyl-l-propyl diketopiperazine-deficient mutant. Genome Announc. 3, e01441–e01415. doi: 10.1128/genomeA.01441-15
Bertels, F., Silander, O. K., Pachkov, M., Rainey, P. B., and van Nimwegen, E. (2014). Automated reconstruction of whole-genome phylogenies from short-sequence reads. Mol. Biol. Evol. 31, 1077–1088. doi: 10.1093/molbev/msu088
Boch, J., Kempf, B., Schmid, R., and Bremer, E. (1996). Synthesis of the osmoprotectant glycine betaine in Bacillus subtilis: characterization of the gbsAB genes. J. Bacteriol. 178, 5121–5129. doi: 10.1128/jb.178.17.5121-5129.1996
Boottanun, P., Potisap, C., Hurdle, J. G., and Sermswan, H. R. (2017). Secondary metabolites from Bacillus amyloliquefaciens isolated from soil can kill Burkholderia pseudomallei. AMB Express. 7:16. doi: 10.1186/s13568-016-0302-0
Bruto, M., Prigent-Combaret, C., Muller, D., and Moënne-Loccoz, Y. (2014). Analysis of genes contributing to plant-beneficial functions in plant growth-promoting rhizobacteria and related proteobacteria. Sci. Rep. 4:6261. doi: 10.1038/srep06261
Calvo, H., Marco, P., Blanco, D., Oria, R., and Venturini, M. E. (2017). Potential of a new strain of Bacillus amyloliquefaciens BUZ-14 as a biocontrol agent of postharvest fruit diseases. Food Microbiol. 63, 101–110. doi: 10.1016/j.fm.2016.11.004
Castaneda-Alvarez, C., Prodan, S., Rosales, I. M., and Aballay, E. (2016). Exoenzymes and metabolites related to the nematicidal effect of rhizobacteria on Xiphinema index Thorne & Allen. J. Appl. Microbiol. 120, 413–424. doi: 10.1111/jam.12987
Chang, X. J., Wu, Z. D., Wu, S. L., Dai, Y. S., and Sun, C. P. (2015). Degradation of ochratoxin A by Bacillus amyloliquefaciens ASAG1. Food Addit. Contam. A Chem. Anal. Control Expo. Risk Assess. 32, 564–571. doi: 10.1080/19440049.2014.991948
Chaves-Lopez, C., Serio, A., Gianotti, A., Sacchetti, G., Ndagijimana, M., Ciccarone, C., et al. (2015). Diversity of food-borne Bacillus volatile compounds and influence on fungal growth. J. Appl. Microbiol. 119, 487–499. doi: 10.1111/jam.12847
Chen, X. H., Koumoutsi, A., Scholz, R., Eisenreich, A., Schneider, K., Heinemeyer, I., et al. (2007). Comparative analysis of the complete genome sequence of the plant growth-promoting bacterium Bacillus Amyloliquefaciens Fzb42. Nat. Biotechnol. 25, 1007–1014. doi: 10.1038/nbt1325
Chen, X. Y., Zhang, Y. Y., Fu, X. C., Li, Y., and Wang, Q. (2016). Isolation and characterization of Bacillus amyloliquefaciens PG12 for the biological control of apple ring rot. Postharvest Biol. Technol. 115, 113–121. doi: 10.1016/j.postharvbio.2015.12.021
Chowdhury, S. P., Uhl, J., Grosch, R., Alquéres, S., Pittroff, S., Dietel, K., et al. (2015). Cyclic lipopeptides of Bacillus amyloliquefaciens FZB42 subsp. plantarum colonizing the lettuce rhizosphere enhance plant defense responses towards the bottom rot pathogen Rhizoctonia solani. Mol. Plant Microbe Interact. 28, 984–995. doi: 10.1094/MPMI-03-15-0066-R
Cimermancic, P., Medema, M. H., Claesen, J., Kurita, K., Wieland Brown, L. C., Mavrommatis, K., et al. (2014). Insights into secondary metabolism from a global analysis of prokaryotic biosynthetic gene clusters. Cell 158, 412–421. doi: 10.1016/j.cell.2014.06.034
Cochrane, R. V. K., and Vederas, J. C. (2014). Highly selective but multifunctional oxygenases in secondary metabolism. Acc. Chem. Res. 47, 3148–3161. doi: 10.1021/ar500242c
Doublet, B., Schwarz, S., Kehrenberg, C., and Cloeckaert, A. (2005). Florfenicol resistance gene floR is part of a novel transposon. Antimicrob. Agents Chemother. 49, 2106–2108. doi: 10.1128/AAC.49.5.2106-2108.2005
Duan, J., Jiang, W., Cheng, Z., Heikkila, J. J., and Glick, B. R. (2013). The complete genome sequence of the plant growth-promoting bacterium Pseudomonas sp. UW4. PLoS ONE 8:e58640. doi: 10.1371/journal.pone.0058640
Dunlap, C. A., Kim, S.-J., Kwon, S.-W., and Rooney, A. P. (2015). Phylogenomic analysis shows that Bacillus amyloliquefaciens subsp. plantarum is a later heterotypic synonym of Bacillus methylotrophicus. Int. J. Syst. Evol. Microbiol. 65, 2104–2109. doi: 10.1099/ijs.0.000226
Dunlap, C. A., Kim, S.-J., Kwon, S.-W., and Rooney, A. P. (2016). Bacillus velezensis is not a later heterotypic synonym of Bacillus amyloliquefaciens; Bacillus methylotrophicus, Bacillus amyloliquefaciens subsp. plantarum and “Bacillus oryzicola” are later heterotypic synonyms of Bacillus velezensis based on phylogenomics. Int. J. Syst. Evol. Microbiol. 66, 1212–1217. doi: 10.1099/ijsem.0.000858
Eom, J. S., and Choi, H. S. (2016). Inhibition of Bacillus cereus growth and toxin production by Bacillus amyloliquefaciens RD7-7 in fermented soybean products. J. Microbiol. Biotechnol. 26, 44–55. doi: 10.4014/jmb.1509.09090
Felsenstein, J. (1981). Evolutionary trees from DNA sequences: a maximum likelihood approach. J. Mol. Evol. 17, 368–376. doi: 10.1007/BF01734359
Franks, S. E., Ebrahimi, C., Hollands, A., Okumura, C. Y., Aroian, R. V., Nizet, V., et al. (2014). Novel role for the yceGH tellurite resistance genes in the pathogenesis of Bacillus anthracis. Infect Immun. 82, 1132–1140. doi: 10.1128/IAI.01614-13
Fu, Z., Liu, Y., Chen, C., Guo, Y., Ma, Y., Yang, Y., et al. (2016). Characterization of fosfomycin resistance gene, fosB, in methicillin-resistant Staphylococcus aureus isolates. PLoS ONE 11:e0154829. doi: 10.1371/journal.pone.0154829
Gadhave, K. R., and Gange, A. C. (2016). Plant-associated Bacillus spp. alter life-history traits of the specialist insect Brevicoryne brassicae L. Agric. For. Entomol. 18, 35–42. doi: 10.1111/afe.12131
Ghelardi, E., Salvetti, S., Ceragioli, M., Gueye, S. A., Celandroni, F., and Senesi, S. (2012). Contribution of surfactin and SwrA to flagellin expression, swimming, and surface motility in Bacillus subtilis. Appl. Environ. Microbiol. 78, 6540–6544. doi: 10.1128/AEM.01341-12
Glick, B. R., Jacobson, C. B., Schwarze, M. M. K., and Pasternak, J. J. (1994). 1-Aminocyclopropane-1-carboxylic acid deaminase mutants of the plant growth promoting rhizobacterium Pseudomonas putida GR12-2 do not stimulate canola root elongation. Can. J. Microbiol. 40, 911–915. doi: 10.1139/m94-146
Goris, J., Konstantinidis, K. T., Klappenbach, J. A., Coenye, T., Vandamme, P., and Tiedje, J. M. (2007). DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 57, 81–91. doi: 10.1099/ijs.0.64483-0
Gowrishankar, S., Sivaranjani, M., Kamaladevi, A., Ravi, A. V., Balamurugan, K., and Karutha Pandian, S. (2016). Cyclic dipeptide cyclo(l-leucyl-l-prolyl) from marine Bacillus amyloliquefaciens mitigates biofilm formation and virulence in Listeria monocytogenes. Pathog. Dis. 74:ftw017. doi: 10.1093/femspd/ftw017
Graf, N., Wenzel, M., and Altenbuchner, J. (2016). Identification and characterization of the vanillin dehydrogenase YfmT in Bacillus subtilis 3NA. Appl. Microbiol. Biotechnol. 100, 3511–3521. doi: 10.1007/s00253-015-7197-6
Gupta, A., Gopal, M., Thomas, G. V., Manikandan, V., Gajewski, J., Thomas, G., et al. (2014). Whole genome sequencing and analysis of plant growth promoting bacteria isolated from the rhizosphere of plantation crops coconut, cocoa and arecanut. PLoS ONE 9:e104259. doi: 10.1371/journal.pone.0104259
Gurr, G. M., and You, M. (2016). Conservation biological control of pests in the molecular era: new opportunities to address old constraints. Front. Plant Sci. 6:1255. doi: 10.3389/fpls.2015.01255
Hahnke, R. L., Meier-Kolthoff, J. P., García-López, M., Mukherjee, S., Huntemann, M., Ivanova, N. N., et al. (2016). Genome-based taxonomic classification of bacteroidetes. Front. Microbiol. 7:2003. doi: 10.3389/fmicb.2016.02003
Hosoya, S., Yamane, K., Takeuchi, M., and Sato, T. (2002). Identification and characterization of the Bacillus subtilis D-glucarate/galactarate utilization operon ycbCDEFGHJ. FEMS Microbiol. Lett. 210, 193–199. doi: 10.1016/s0378-1097(02)00612-2
Hu, K. H., Liu, E., Dean, K., Gingras, M., DeGraff, W., and Trun, N. J. (1996). Overproduction of three genes leads to camphor resistance and chromosome condensation in Escherichia coli. Genetics 143, 1521–1532.
Idris, E. E. S., Iglesias, D. J., Talon, M., and Borriss, R. (2007). Tryptophan-dependent production of indole-3-acetic acid (IAA) affects level of plant growth promotion by Bacillus amyloliquefaciens FZB42. Mol. Plant Microb. Interact. 20, 619–626. doi: 10.1094/MPMI-20-6-0619
Islam, S., Akanda, A. M., Prova, A., Islam, M. T., and Hossain, M. M. (2016). Isolation and identification of plant growth promoting rhizobacteria from cucumber rhizosphere and their effect on plant growth promotion and disease suppression. Front. Microbiol. 6:1360. doi: 10.3389/fmicb.2015.01360
Jacob, J., Evers, S., Bischoff, K., Carlier, C., and Courvalin, P. (1994). Characterization of the sat4 gene encoding a streptothricin acetyltransferase in Campylobacter coli BE/G4. FEMS Microbiol. Lett. 120, 13–17. doi: 10.1016/0378-1097(94)00168-5
Jeske, O., Jogler, M., Petersen, J., Sikorski, J., and Jogler, C. (2013). From genome mining to phenotypic microarrays: planctomycetes as source for novel bioactive molecules. Anton. Leeuw. 104, 551–567. doi: 10.1007/s10482-013-0007-1
Kim, J. H., Ji, C. J., Ju, S. Y., Yang, Y. M., Ryu, S. H., Kwon, Y., et al. (2003). Bacillus licheniformis contains two more PerR-like proteins in addition to PerR, Fur, and Zur orthologues. PLoS ONE 11:e0155539. doi: 10.1371/journal.pone.0155539
Kimura, M. (1980). A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 16, 111–120. doi: 10.1007/BF01731581
Lagerlof, J., Ayuke, F., Bejai, S., Jorge, G., Lagerqvis, E., Meijer, J., et al. (2015). Potential side effects of biocontrol and plant-growth promoting Bacillus amyloliquefaciens bacteria on earthworms. Appl. Soil Ecol. 96, 159–164. doi: 10.1016/j.apsoil.2015.08.014
Lara, E., and Belbahri, L. (2011). SSU rRNA reveals major trends in oomycete evolution. Fungal Divers 49, 93–100. doi: 10.1007/s13225-011-0098-9
Li, M. H. T., Ung, P. M. U., Zajkowski, J., Garneau-Tsodikova, S., and Sherman, D. H. (2009). Automated genome mining for natural products. BMC Bioinf. 10:185. doi: 10.1186/1471-2105-10-185
Liu, S., Lee, L., Tai, C., Hung, C., Chang, Y., Wolfram, J., et al. (1992). Cloning of an Erwinia herbicola gene necessary for gluconic acid production and enhanced mineral phosphate solubilization in Escherichia coli HB101: nucleotide sequence and probable involvement in biosynthesis of the coenzyme pyrroloquinoline quinone. J. Bacteriol. 174, 5814–5819. doi: 10.1128/jb.174.18.5814-5819.1992
Liu, Y. P., Chen, L., Zhang, N., Li, Z. F., Zhang, G. S., Xu, Y., et al. (2016). Plant-microbe communication enhances auxin biosynthesis by a root-associated bacterium, Bacillus amyloliquefaciens SQR9. Mol. Plant Microbe Interact. 29, 324–330. doi: 10.1094/MPMI-10-15-0239-R
Loper, J. E., Hassan, K. A., Mavrodi, D. V., Davis, E. W., Lim, C. K., Shaffer, B. T., et al. (2012). Comparative genomics of plant-associated Pseudomonas spp.: insights into diversity and inheritance of traits involved in multitrophic interactions. PLoS Genet. 8:e1002784. doi: 10.1371/journal.pgen.1002784
Luchi, N., Ghelardini, L., Belbahri, L., Quartier, M., and Santini, A. (2013). Rapid detection of Ceratocystis platani inoculum by quantitative real-time PCR assay. Appl. Environ. Microbiol. 79, 5394–5404. doi: 10.1128/AEM.01484-13
Machado, H., Sonnenschein, E. C., Melchiorsen, J., and Gram, L. (2015). Genome mining reveals unlocked bioactive potential of marine gram-negative bacteria. BMC Genomics. 16:158. doi: 10.1186/s12864-015-1365-z
Magno-Perez-Bryan, M. C., Martinez-Garcia, P. M., Hierrezuelo, J., Rodriguez-Palenzuela, P., Arrebola, E., Ramos, C., et al. (2015). Comparative genomics within the Bacillus genus reveal the singularities of two robust Bacillus amyloliquefaciens biocontrol strains. Mol. Plant Microbe Interact. 28, 1102–1116. doi: 10.1094/MPMI-02-15-0023-R
Mazzola, M., Cook, R. J., Thomashow, L. S., Weller, D. M., and Pierson, L. S. (1992). Contribution of phenazine antibiotic biosynthesis to the ecological competence of fluorescent pseudomonads in soil habitats. Appl. Environ. Microbiol. 58, 2616–2624.
Mefteh, F., Daoud, A., Chenari Bouket, A., Alenezi, F. N., Luptakova, L., Rateb, M. E., et al. (2017). Fungal root microbiome from healthy and brittle leaf diseased date palm trees (Phoenix dactylifera L.) reveals a hidden untapped arsenal of antibacterial and broad spectrum antifungal secondary metabolites. Front. Microbiol. 8:307. doi: 10.3389/fmicb.2017.00307
Meier-Kolthoff, J. P., Auch, A. F., Klenk, H. P., and Göker, M. (2013). Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinf. 14:60. doi: 10.1186/1471-2105-14-60
Miller, S. H., Browne, P., Prigent-Combaret, C., Combes-Meynet, E., Morrissey, J. P., and O'Gara, F. (2010). Biochemical and genomic comparison of inorganic phosphate solubilization in Pseudomonas species. Environ. Microbiol. Rep. 2, 403–411. doi: 10.1111/j.1758-2229.2009.00105.x
Mlaik, N., Bakonyi, J., Borsodi, A., Woodward, S., Belbahri, L., and Mechichi, T. (2015). Microbial diversity in tanning wastewaters treatment reactors. Environ. Prog. Sustain. Energy 34, 401–410. doi: 10.1002/ep.12000
Moore, C. M., Gaballa, A., Hui, M., Ye, R. W., and Helmann, J. D. (2005). Genetic and physiological responses of Bacillus subtilis to metal ion stress. Mol. Microbiol. 57, 27–40. doi: 10.1111/j.1365-2958.2005.04642.x
Neyfakh, A. A., Borsch, C. M., and Kaatz, G. W. (1993). Fluoroquinolone resistance protein NorA of Staphylococcus aureus is a multidrug efflux transporter. Antimicrob. Agents Chemother. 37, 128–129. doi: 10.1128/AAC.37.1.128
Ng, L. C., Sariah, M., Sariam, O., Radziah, O., and Abidin, M. A. Z. (2016). PGPM-induced defense-related enzymes in aerobic rice against rice leaf blast caused by Pyricularia oryzae. Eur. J. Plant Pathol. 145:167. doi: 10.1007/s10658-015-0826-1
Nguyen, V. D., Wolf, C., Mäder, U., Lalk, M., Langer, P., Lindequist, U., et al. (2007). Transcriptome and proteome analyses in response to 2-methylhydroquinone and 6-brom-2-vinyl-chroman-4-on reveal different degradation systems involved in the catabolism of aromatic compounds in Bacillus subtilis. Proteomics 7, 1391–1408. doi: 10.1002/pmic.200700008
Niazi, A., Manzoor, S., Asari, S., Bejai, S., Meijer, J., and Bongcam-Rudloff, E. (2014). Genome analysis of Bacillus amyloliquefaciens subsp. plantarum UCMB5113: A rhizobacterium that improves plant growth and stress management. PLoS ONE 9:e104651. doi: 10.1371/journal.pone.0104651
Noguchi, N., Sasatsu, M., and Kono, M. (1993). Genetic mapping in Bacillus subtilis 168 of the aadK gene which encodes aminoglycoside 6-adenylyltransferase. FEMS Microbiol. Lett. 114, 47–52. doi: 10.1111/j.1574-6968.1993.tb06549.x
Ochsner, U. A., Vasil, M. L., Alsabbagh, E., Parvatiyar, K., and Hassett, D. J. (2000). Role of the Pseudomonas aeruginosa oxyR-recG operon in oxidative stress defense and DNA repair: OxyR-dependent regulation of katB-ankB, ahpB, and ahpC-ahpF. J. Bacteriol. 182, 4533–4544. doi: 10.1128/JB.182.16.4533-4544.2000
Ohki, R., Tateno, K., Takizawa, T., Aiso, T., and Murata, M. (2005). Transcriptional termination control of a novel ABC transporter gene involved in antibiotic resistance in Bacillus subtilis. J. Bacteriol. 187, 5946–5954. doi: 10.1128/JB.187.17.5946-5954.2005
Olson, A., Aerts, A., Asiegbu, F., Belbahri, L., Bouzid, O., Broberg, A., et al. (2012). Insight into trade-off between wood decay and parasitism from the genome of a fungal forest pathogen. New Phytol. 194, 1001–1013. doi: 10.1111/j.1469-8137.2012.04128.x
Osman, D., and Cavet, J. S. (2010). Bacterial metal-sensing proteins exemplified by ArsR-SmtB family repressors. Nat. Prod. Rep. 27, 668–680. doi: 10.1039/b906682a
Ozer, E. A., Allen, J. P., and Hauser, A. R. (2014). Characterization of the core and accessory genomes of Pseudomonas aeruginosa using bioinformatic tools Spine and AGEnt. BMC Genomics 15:737. doi: 10.1186/1471-2164-15-737
Perez, K. J., Viana, J. D. S., Lopes, F. C., Pereira, J. Q., dos Santos, D. M., Oliveira, J. S., et al. (2017). Bacillus spp. isolated from puba as a source of biosurfactants and antimicrobial lipopeptides. Front. Microbiol. 8:61. doi: 10.3389/fmicb.2017.00061
Petersen, T. N., Brunak, S., von Heijne, G., and Nielsen, H. (2011). SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8, 785–786. doi: 10.1038/nmeth.1701
Piddington, C. S., Kovacevich, B. R., and Rambosek, J. (1995). Sequence and molecular characterization of a DNA region encoding the dibenzothiophene desulfurization operon of Rhodococcus sp. strain IGTS8. Appl. Environ. Microbiol. 61, 468–475.
Prazdnova, E. V., Chistyakov, V. A., Churilov, M. N., Mazanko, M. S., Bren, A. B., Volski, A., et al. (2015). DNA-protection and antioxidant properties of fermentates from Bacillus amyloliquefaciens B-1895 and Bacillus subtilis KATMIRA1933. Lett. Appl. Microbiol. 61, 549–554. doi: 10.1111/lam.12491
Prospero, S., Vercauteren, A., Heungens, K., Belbahri, L., and Rigling, D. (2013). Phytophthora diversity and the population structure of Phytophthora ramorum in Swiss ornamental nurseries. Plant Pathol. 62, 1063–1071. doi: 10.1111/ppa.12027
Rademacher, C., and Masepohl, B. (2012). Copper-responsive gene regulation in bacteria. Microbiology 158(Pt 10), 2451–2464. doi: 10.1099/mic.0.058487-0
Ravari, S. B., and Heidarzadeh, N. (2014). Isolation and characterization of rhizosphere auxin producing Bacilli and evaluation of their potency on wheat growth improvement. Arch. Agron. Soil Sci. 60, 895–905. doi: 10.1080/03650340.2013.856003
Rekik, I., Chaabane, Z., Missaoui, A., Chenari Bouket, A., Luptakova, L., Elleuch, A., et al. (2017). Effects of untreated and treated wastewater at the morphological, physiological and biochemical levels on seed germination and development of sorghum (Sorghum bicolor (L) Moench), alfalfa (Medicago sativa L) and fescue (Festuca arundinacea Schreb). J. Hazard. Mater. 326, 165–176. doi: 10.1016/j.jhazmat.2016.12.033
Rey, M. W., Ramaiya, P., Nelson, B. A., Brody-Karpin, S. D., Zaretsky, E. J., Tang, M., et al. (2004). Complete genome sequence of the industrial bacterium Bacillus licheniformis and comparisons with closely related Bacillus species. Genome Biol. 5:R77. doi: 10.1186/gb-2004-5-10-r77
Richter, M., and Rosselló-Móra, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. U.S.A. 106, 19126–19131. doi: 10.1073/pnas.0906412106
Saengsanga, T., Siripornadulsil, W., and Siripornadulsil, S. (2016). Molecular and enzymatic characterization of alkaline lipase from Bacillus amyloliquefaciens E1PA isolated from lipid-rich food waste. Enzyme Microb. Technol. 82, 23–33. doi: 10.1016/j.enzmictec.2015.08.005
Sakaguchi, R., Amano, H., and Shishido, K. (1988). Nucleotide sequence homology of the tetracycline-resistance determinant naturally maintained in Bacillus subtilis Marburg 168 chromosome and the tetracycline-resistance gene of B. subtilis plasmid pNS1981. Biochim. Biophys. Acta 950, 441–444. doi: 10.1016/0167-4781(88)90142-X
Sellami, M., Khlifi, A., Frikha, F., Miled, N., Belbahri, L., and Ben Rebah, F. (2016). Agro-industrial waste based growth media optimization for biosurfactant production by Aneurinibacillus migulanus. J. Microbiol. Biotechnol Food Sci. 5, 578–583. doi: 10.15414/jmbfs.2016.5.6.578-583
Shakeel, M., Rais, A., Hassan, M. N., and Yusuf Hafeez, F. (2015). Root associated Bacillus sp. improves growth, yield and zinc translocation for Basmati rice (Oryza sativa) varieties. Front. Microbiol. 6:1286. doi: 10.3389/fmicb.2015.01286
Shao, J. H., Li, S. Q., Zhang, N., Cui, X. S., Zhou, X., Zhang, G. S., et al. (2015). Analysis and cloning of the synthetic pathway of the phytohormone indole-3-acetic acid in the plant-beneficial Bacillus amyloliquefaciens SQR9. Microb. Cell Fact. 14:130. doi: 10.1186/s12934-015-0323-4
Shen, X., Hu, H., Peng, H., Wang, W., and Zhang, X. (2013). Comparative genomic analysis of four representative plant growth-promoting rhizobacteria in Pseudomonas. BMC Genomics. 14:271. doi: 10.1186/1471-2164-14-271
Skinnider, M. A., Dejong, C. A., Rees, P. N., Johnston, C. W., Li, H., Webster, A. L. H., et al. (2015). Genomes to natural products prediction informatics for secondary metabolomes (PRISM). Nucleic Acids Res. 43, 9645–9662. doi: 10.1093/nar/gkv1012
Somers, E., Ptacek, D., Gysegom, P., Srinivasan, M., and Vanderleyden, J. (2005). Azospirillum brasilense produces the auxin-like phenylacetic acid by using the key enzyme for indole-3-acid biosynthesis. Appl. Environ. Microbiol. 71, 1803–1810. doi: 10.1128/AEM.71.4.1803-1810.2005
Someya, Y., Yamaguchi, A., and Sawai, T. (1995). A novel glycylcycline, 9-(N,N-dimethylglycylamido)-6-demethyl-6-deoxytetracycline, is neither transported nor recognized by the transposon Tn10-encoded metal-tetracycline/H+ antiporter. Antimicrob. Agents Chemother. 39, 247–249. doi: 10.1128/AAC.39.1.247
Srivastava, S., Bist, V., Srivastava, S., Singh, P. C., Trivedi, P. K., Asif, M. H., et al. (2016). Unraveling aspects of Bacillus amyloliquefaciens mediated enhanced production of rice under biotic stress of Rhizoctonia solani. Front. Plant Sci. 7:587. doi: 10.3389/fpls.2016.00587
Sun, P., Hui, C., Wang, S., Wan, L., Zhang, X., and Zhao, Y. (2016). Bacillus amyloliquefaciens biofilm as a novel biosorbent for the removal of crystal violet from solution. Colloids Surf. B. Biointerfaces. 139, 164–170. doi: 10.1016/j.colsurfb.2015.12.014
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Tan, S. Y., Gu, Y., Yang, C. L., Dong, Y., Mei, X. L., Shen, Q. R., et al. (2016). Bacillus amyloliquefaciens T-5 may prevent Ralstonia solanacearum infection through competitive exclusion. Biol. Fertil. Soils 52, 341–351. doi: 10.1007/s00374-015-1079-z
Vacheron, J., Desbrosses, G., Bouffaud, M.-L., Touraine, B., Moënne-Loccoz, Y., Muller, D., et al. (2013). Plant growth-promoting rhizobacteria and root system functioning. Front. Plant Sci. 4:356. doi: 10.3389/fpls.2013.00356
Van Heel, A. J., de Jong, A., Montalbán-López, M., Kok, J., and Kuipers, O. P. (2013). BAGEL3: automated identification of genes encoding bacteriocins and (non-)bactericidal post translationally modified peptides. Nucleic Acids Res. 41, W448–W453. doi: 10.1093/nar/gkt391
Verma, A., Singh, V. K., and Gaur, S. (2016). Computational based functional analysis of Bacillus phytases. Comput. Biol. Chem. 60, 53–58. doi: 10.1016/j.compbiolchem.2015.11.001
Vlamakis, H., Chai, Y., Beauregard, P., Losick, R., and Kolter, R. (2013). Sticking together: building a biofilm the Bacillus subtilis way. Nat. Rev. Microbiol. 11, 157–168. doi: 10.1038/nrmicro2960
Wang, H., Yang, L., Ping, Y. H., Bai, Y. G., Luo, H. Y., Huang, H. Q., et al. (2016). Engineering of a Bacillus amyloliquefaciens strain with high neutral protease producing capacity and optimization of its fermentation conditions. PLoS ONE 11:e0146373. doi: 10.1371/journal.pone.0146373
Wang, J., Zhao, D., Liu, Y., Ao, X., Fan, R., Duan, Z., et al. (2014). Antagonism against Beauveria bassiana by lipopeptide metabolites produced by entophyte Bacillus amyloliquefaciens strain SWB16. Wei Sheng Wu Xue Bao 54, 778–785.
Wang, X. M., Bai, Y. J., Cai, Y. J., and Zheng, X. H. (2017). Biochemical characteristics of three feruloyl esterases with a broad substrate spectrum from Bacillus amyloliquefaciens H47. Process Biochem. 53, 109–115. doi: 10.1016/j.procbio.2016.12.012
Weber, T., Blin, K., Duddela, S., Krug, D., Kim, H. U., Bruccoleri, R., et al. (2015). antiSMASH 3.0 - a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 43, W237–W243. doi: 10.1093/nar/gkv437
Wei, Z., Huang, J. F., Yang, C. L., Xu, Y. C., Shen, Q. R., and Chen, W. (2015). Screening of suitable carriers for Bacillus amyloliquefaciens strain QL-18 to enhance the biocontrol of tomato bacterial wilt. Crop Prot. 75, 96–103. doi: 10.1016/j.cropro.2015.05.010
Whistler, C. A., Corbell, N. A., Sarniguet, A., Ream, W., and Loper, J. E. (1998). The two-component regulators GacS and GacA influence accumulation of the stationary-phase sigma factor sigmaS and the stress response in Pseudomonas fluorescens Pf-5. J. Bacteriol. 180, 6635–6641.
Wu, B., Wang, X., Yang, L., Yang, H., Zeng, H., Qiu, Y. M., et al. (2016). Effects of Bacillus amyloliquefaciens ZM9 on bacterial wilt and rhizosphere microbial communities of tobacco. Appl. Soil Ecol. 103, 1–12. doi: 10.1016/j.apsoil.2016.03.002
Wu, L., Wu, H.-J., Qiao, J., Gao, X., and Borriss, R. (2015). Novel routes for improving biocontrol activity of Bacillus based bioinoculants. Front. Microbiol. 6:1395. doi: 10.3389/fmicb.2015.01395
Wu, X., Wang, X., Drlica, K., and Zhao, X. (2011). A Toxin-antitoxin module in Bacillus subtilis can both mitigate and amplify effects of lethal stress. PLoS ONE. 6:e23909. doi: 10.1371/journal.pone.0023909
Xie, S.-S., Wu, H.-J., Zang, H.-Y., Wu, L.-M., Zhu, Q.-Q., and Gao, X.-W. (2014). Plant growth promotion by spermidine-producing Bacillus subtilis OKB105. Mol. Plant Microbe Interact. 27, 655–663. doi: 10.1094/MPMI-01-14-0010-R
Yang, L., Wang, H., Lv, Y., Bai, Y. G., Luo, H. Y., Shi, P. J., et al. (2016). Construction of a rapid feather-degrading bacterium by overexpression of a highly efficient alkaline keratinase in its parent strain Bacillus amyloliquefaciens K11. J. Agric. Food Chem. 64, 78–84. doi: 10.1021/acs.jafc.5b04747
Yoon, S. H., Ha, S. M., Kwon, S., Lim, J., Kim, Y., Seo, H., et al. (2017). Introducing EzBioCloud: a taxonomically united database of 16S rRNA and whole genome assemblies. Int. J. Syst. Evol. Microbiol. 67, 1613–1617. doi: 10.1099/ijsem.0.001755
Zhang, J. H., Xue, Q. H., Gao, H., Lai, H. X., and Wang, P. (2016). Bacterial degradation of crude oil using solid formulations of Bacillus strains isolated from oil-contaminated soil towards microbial enhanced oil recovery application. RSC Adv. 6, 5566–5574. doi: 10.1039/C5RA23772F
Zhang, N., Yang, D. Q., Kendall, J. R. A., Borriss, R., Druzhinina, I. S., Kubicek, C. P., et al. (2016). Comparative genomic analysis of Bacillus amyloliquefaciens and Bacillus subtilis reveals evolutional traits for adaptation to plant-associated habitats. Front. Microbiol. 7:2039. doi: 10.3389/fmicb.2016.02039
Zhang, S., Wang, D., Wang, Y., Hasman, H., Aarestrup, F. M., Alwathnani, H. A., et al. (2015). Genome sequences of copper resistant and sensitive Enterococcus faecalis strains isolated from copper-fed pigs in Denmark. Stand. Genomic Sci. 2015, 10:35. doi: 10.1186/s40793-015-0021-1
Zhi, Y., Wu, Q., and Xu, Y. (2017). Genome and transcriptome analysis of surfactin biosynthesis in Bacillus amyloliquefaciens MT45. Sci. Rep. 7:40976. doi: 10.1038/srep40976
Ziemert, N., Podell, S., Penn, K., Badger, J. H., Allen, E., and Jensen, P. R. (2012). The natural product domain seeker NaPDoS: a phylogeny based bioinformatic tool to classify secondary metabolite gene diversity. PLoS ONE 7:e34064. doi: 10.1371/journal.pone.0034064
Zimmer, W., Aparicio, C., and Elmerich, C. (1991). Relationship between tryptophane biosynthesis and indole-3-acetic acid production in Azospirillum: identification and sequencing of a trpGDC cluster. Mol. Gen. Genet. 229, 41–51. doi: 10.1007/BF00264211
Zuhlke, M. K., Schluter, R., Henning, A. K., Lipka, M., Mikolasch, A., Schumann, P., et al. (2016). A novel mechanism of conjugate formation of bisphenol a and its analogues by Bacillus amyloliquefaciens: detoxification and reduction of estrogenicity of bisphenols. Int. Biodeterior. Biodegrad. 109, 165–173. doi: 10.1016/j.ibiod.2016.01.019
Keywords: bioinformatics, genome mining, Bacillus amyloliquefaciens, biocontrol bacteria, secondary metabolism
Citation: Belbahri L, Chenari Bouket A, Rekik I, Alenezi FN, Vallat A, Luptakova L, Petrovova E, Oszako T, Cherrad S, Vacher S and Rateb ME (2017) Comparative Genomics of Bacillus amyloliquefaciens Strains Reveals a Core Genome with Traits for Habitat Adaptation and a Secondary Metabolites Rich Accessory Genome. Front. Microbiol. 8:1438. doi: 10.3389/fmicb.2017.01438
Received: 20 May 2017; Accepted: 17 July 2017;
Published: 03 August 2017.
Edited by:
Carlos Alberto Moreira-Filho, Faculdade de Medicina da Universidade de São Paulo, BrazilReviewed by:
Gaurav Sharma, University of California, Davis, United StatesChiachi Hwang, Montana State University, United States
Copyright © 2017 Belbahri, Chenari Bouket, Rekik, Alenezi, Vallat, Luptakova, Petrovova, Oszako, Cherrad, Vacher and Rateb. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lassaad Belbahri, bGFzc2FhZC5iZWxiYWhyaUB1bmluZS5jaA==