 Pauline Chaignaud1,2
Pauline Chaignaud1,2 Bruno Maucourt1
Bruno Maucourt1 Marion Weiman3Adriana Alberti3
Marion Weiman3Adriana Alberti3 Steffen Kolb2,4
Steffen Kolb2,4 Stéphane Cruveiller3
Stéphane Cruveiller3 Stéphane Vuilleumier1
Stéphane Vuilleumier1 Françoise Bringel1*
Françoise Bringel1*- 1Department of Molecular Genetics, Genomics, and Microbiology, UMR 7156 Université de Strasbourg (UNISTRA)-Centre National de la Recherche Scientifique, Strasbourg, France
- 2Department of Ecological Microbiology, University of Bayreuth, Bayreuth, Germany
- 3UMR 8030 Centre National de la Recherche Scientifique-CEA, DSV/IG/Genoscope, LABGeM, Evry, France
- 4Institute of Landscape Biogeochemistry-Leibniz Centre for Agricultural Landscape Research (ZALF), Müncheberg, Germany
Bacterial adaptation to growth with toxic halogenated chemicals was explored in the context of methylotrophic metabolism of Methylobacterium extorquens, by comparing strains CM4 and DM4, which show robust growth with chloromethane and dichloromethane, respectively. Dehalogenation of chlorinated methanes initiates growth-supporting degradation, with intracellular release of protons and chloride ions in both cases. The core, variable and strain-specific genomes of strains CM4 and DM4 were defined by comparison with genomes of non-dechlorinating strains. In terms of gene content, adaptation toward dehalogenation appears limited, strains CM4 and DM4 sharing between 75 and 85% of their genome with other strains of M. extorquens. Transcript abundance in cultures of strain CM4 grown with chloromethane and of strain DM4 grown with dichloromethane was compared to growth with methanol as a reference C1 growth substrate. Previously identified strain-specific dehalogenase-encoding genes were the most transcribed with chlorinated methanes, alongside other genes encoded by genomic islands (GEIs) and plasmids involved in growth with chlorinated compounds as carbon and energy source. None of the 163 genes shared by strains CM4 and DM4 but not by other strains of M. extorquens showed higher transcript abundance in cells grown with chlorinated methanes. Among the several thousand genes of the M. extorquens core genome, 12 genes were only differentially abundant in either strain CM4 or strain DM4. Of these, 2 genes of known function were detected, for the membrane-bound proton translocating pyrophosphatase HppA and the housekeeping molecular chaperone protein DegP. This indicates that the adaptive response common to chloromethane and dichloromethane is limited at the transcriptional level, and involves aspects of the general stress response as well as of a dehalogenation-specific response to intracellular hydrochloric acid production. Core genes only differentially abundant in either strain CM4 or strain DM4 total 13 and 58 CDS, respectively. Taken together, the obtained results suggest different transcriptional responses of chloromethane- and dichloromethane-degrading M. extorquens strains to dehalogenative metabolism, and substrate- and pathway-specific modes of growth optimization with chlorinated methanes.
Introduction
Chlorinated one-carbon compounds chloromethane (CM, CH3Cl) and dichloromethane (DCM, CH2Cl2) are toxic chemicals that occur in both pristine and polluted environments. CM is the most abundant halogenated trace gas in the atmosphere, where it contributes to ozone destruction (Carpenter et al., 2014). It is mainly produced by vegetation (Derendorp et al., 2012; Hardacre and Heal, 2013; Rhew et al., 2014; Bringel and Couée, 2015). DCM is one of the most commonly manufactured chlorinated chemicals. It is used for its solvent properties, and is a frequently found contaminant at polluted sites. Some methylotrophic microorganisms are able to utilize chlorinated methanes (such as CM and DCM) as their sole carbon and energy source for growth (Muller et al., 2011a; Nadalig et al., 2014). Bacterial CM and DCM utilization starts with dehalogenation, causing diverse physiological stresses such as production of intracellular HCl, which lowers pH and increases ionic strength, and formation of DNA adducts (Kayser and Vuilleumier, 2001; Torgonskaya et al., 2011; Michener et al., 2014b, 2016). The mechanisms allowing methylotrophic bacteria to cope with dehalogenation-induced stress are still poorly understood, despite potential use of such bacteria for bioremediation.
The aerobic alphaproteobacterium Methylobacterium extorquens is the most extensively studied methylotroph. It is found in a wide variety of habitats, including plants, soil, wastewater, and clouds (Amato et al., 2007; Kolb, 2009; Bringel and Couée, 2015). The well-studied M. extorquens strains CM4 and DM4, whose genomes have been sequenced (Vuilleumier et al., 2009; Marx et al., 2012), utilize the chlorinated compounds CM and DCM, respectively, as their sole source of carbon and energy. Degradation pathways for CM and DCM have been characterized in these strains, and are also found in many other species (Muller et al., 2011a; Nadalig et al., 2014). The ability to grow on chlorinated methanes has been demonstrated by genetic, biochemical and recent experimental evolution studies to require the expression of essential dehalogenation-associated genes that differ for CM and DCM. To our knowledge no strains able to metabolize both CM and DCM has been isolated so far. Genes cmuA and cmuB are essential for CM dehalogenation by the cmu (CM-utilization) pathway (Vannelli et al., 1999). The two-domain methyltransferase/corrinoid-binding CmuA protein catalyzes methyl transfer from CM to a cobalt-corrin cofactor (Studer et al., 2001), and the methylcobalamin:tetrahydrofolate methyltransferase CmuB transfers the resulting corrinoid-bound methyl group to tetrahydrofolate (H4F) (Studer et al., 1999). For each molecule of CM, one methyl-H4F and one HCl are produced. For DCM degradation, a glutathione-dependent dehalogenase of the glutathione S-transferase family encoded by dcmA converts DCM into formaldehyde and two molecules of HCl (Vuilleumier and Leisinger, 1996; Kayser et al., 2002).
Although protons and chloride ions are produced in both cases, the processing of carbon from chlorinated methanes for production of biomass and energy proceeds differently in CM- and DCM-utilizing M. extorquens strains (Michener et al., 2016). Thus, toxic chlorinated methanes CM and DCM may generate both similar and compound- and pathway-specific adaptive responses. In this work, we analyzed these responses in terms of global gene expression, by sequencing cDNA libraries of M. extorquens strains grown either with CM or with DCM.
Materials and Methods
Bacterial Cultivation and RNA Purification
Strains CM4 and DM4 were grown aerobically in 1.2L Erlenmeyer flasks closed with gas-tight screw caps with Mininert® valves (Supelco) in Methylobacterium mineral medium (M3) (modified as described in Roselli et al., 2013), with shaking (120 rpm) at 30°C. For 220-mL cultures, one-carbon growth substrates were supplied at 10 mM final concentration, by adding either aqueous solutions of 2.75 mL of filter-sterilized aqueous stock solution of methanol (800 mM), 141 μL of neat dichloromethane, or 40 mL of gaseous chloromethane (assuming a Henry constant of 0.0106 m3⋅atm⋅mol−1 at 30°C) (Chen et al., 2012). Upon reaching mid-exponential phase (OD600 ~0.15), growth was stopped by addition of 27.5 mL stabilization buffer. This buffer was prepared by mixing 5 mL of phenol and 5 mL of 1 M sodium acetate pH 5.5, then after centrifugation at 1,800 g for 3 min, 5 mL of the lower phase was mixed with 95 mL of absolute ethanol. Resulting cell suspensions were centrifuged at 5,000 rpm for 5 min at 4°C, and suspended in 10 mL TE containing 2 mg⋅mL−1 lysozyme (Euromedex). After 15 min incubation at 37°C, each cell suspension was centrifuged 10 min at 4°C, the obtained pellet resuspended in 10 mL of Trizol (Invitrogen), and 2.5 mL of chloroform was added. RNA was precipitated with isopropanol and washed with ethanol (70%), then resuspended in DEPC-water and treated with DNAse (Turbo DNAse, Invitrogen). DNA depletion was checked by PCR (see Table S1 for primers). RNA quality was checked with the RNA 6000 Nano kit on a Bioanalyzer 2100 (Agilent Technologies), and quantified with the Qubit RNA kit (Invitrogen). Depletion of rRNA was obtained by treating 5 μg of total RNA with the Gram-negative RiboZero Magnetic kit (Tebu-Bio) according to the manufacturer's protocol.
cDNA Library Preparation, Sequencing and Data Normalization
rRNA-depleted RNA (50–60 ng in 5 μL) were fragmented by adding 13 μL of the “Fragment, prime, finish mix” of the “Purify and fragment mRNA” kit (Illumina). Then, cDNA libraries were constructed with the TruSeq stranded mRNA LT kit (Illumina) following the manufacturer's protocol. Obtained cDNA libraries were quality checked (Bioanalyzer, DNA 1000 kit, Agilent Technologies) prior to HiSeq2000 sequencing. Paired-end sequence mapping was used to limit mapping artifacts and remove mapping ambiguities between gene paralogs. Factor size normalization of the raw counts was performed using a set of 55 reference “housekeeping” genes instead of the standard method using the complete set of CDS (Anders et al., 2013), as a few genes had very high read numbers in growth with chlorinated methanes only. The 55 reference genes cover a wide range of expression levels but had unchanged transcript abundance in the tested biological replicates (log2fc value between −0.9 and 1.2) (Table S2). Normalization was validated by comparison with the complete set of CDS for the methanol dataset (Figure S1). Gene transcripts were defined as differentially abundant when the log2 of fold-change values (log2fc) between cultures grown with chlorinated methanes and methanol were ≥2.0 or ≤ −2 (Yang et al., 2015). Each condition was analyzed in duplicate, with the average of read sense and antisense for each condition. Data were considered significant when False Discovery Rate (i.e., FDR) was ≤0.1 as previously described (Benjamini and Hochberg, 1995). The complete RNA-Seq dataset is accessible online (https://www.genoscope.cns.fr/agc/microscope/transcriptomic/NGSProjectRNAseq.php?projType=RNAseq).
RT-qPCR
Retrotranscription was performed from 2.0 μg of DNA-depleted RNA extracted using the Nucleospin RNA plus kit (Macherey Nagel) combined with Turbo DNase and Turbo DNA free kit (Invitrogen) using Superscript III (Invitrogen) and random hexanucleotide primers (Invitrogen) following manufacturers' protocols, except that RNaseOUT™ was replaced by RNasin™ (40 U⋅μL−1; Invitrogen), with inclusion of appropriate controls (reactions without RNA template or Superscript III enzyme). Amplification was performed in qPCR 96 wells plates (Agilent Technologies) covered with Microseal B adhesive seals (Bio-Rad) using the Thermocycler Stratagene Mx3005P (MxPro software v4.10; Agilent Technologies). Master mix containing 7.5 μL Brilliant III SYBR™ Green low ROX qPCR master mix (Agilent Technologies), 0.225 μL of each primer (Eurofins) at 20 μM and 3.3 μL molecular biology grade water was mixed with 3.75 μL template cDNA. After 3 min pre-denaturation at 95°C, 40 cycles of 20 s at 95°C and 20 s at 60°C, a final one-cycle step of 1 min at 95°C with ramping from 60 to 95°C was applied to obtain dissociation curves for quality control of PCR products. For each biological triplicate, technical duplicates were analyzed and compared to standard curves with gDNA (0.1–1⋅10−5 ng⋅μL−1) and a no template control. SYBR™ green fluorescence data normalized with Rox fluorescence were analyzed using the package qpcR (v1.40) within R for Ct determination (Ritz and Spiess, 2008). Fold change values were calculated by the 2−ΔΔCt method (Schmittgen and Livak, 2008), with rrsA as internal control and dcmA in DM4 cultures grown with methanol as the reference condition.
Comparative Genomics Analysis
Comparative analysis of M. extorquens genomes was done in MaGe on the Genoscope MicroScope online platform (Vallenet et al., 2017), with the sequenced genomes of 5 strains of M. extorquens, i.e., the 2 dehalogenating strains CM4 (Genbank accession numbers CP001298, CP001299, CP001300) and DM4 (FP103042, FP103043, and FP103044), and 3 other strains AM1 (CP001511, CP001512, CP001513, and CP001514), PA1 (CP000908), and BJ001 (CP001029, CP001030, and CP001031) (Vuilleumier et al., 2009; Marx et al., 2012). All studied strains contain at least one plasmid, except for PA1. Proteins responsible for dehalogenation of CM or DCM are exclusively encoded by M. extorquens CM4 and DM4 genomes, respectively. The common genome called hereafter “core” was operationally identified using the MaGe Pan/Core-genome tool (https://www.genoscope.cns.fr/agc/microscope/compgenomics/pancoreTool.php?), by defining shared CDS as encoding proteins displaying at least 80% amino acid identity over 80% of CDS length in all considered genomes. Other genes were assigned either to the variable genome when present in at least 2 genomes, to the dehalogenation-associated genome when shared only by strains CM4 and DM4, and to the strain-specific genome when found only in one genome, respectively. Genomic islands of at least 5 kb were defined using the MaGe “Regions of Genomic Plasticity” tool (https://www.genoscope.cns.fr/agc/microscope/compgenomics/genomicIsland.php?) with the genomes of the 5 strains above, and applying a specificity score cutoff of 40 for all compared genomes.
Results
Acquisition of specific dehalogenase genes by M. extorquens does not necessarily lead to growth with chlorinated methanes (Kayser et al., 2002; Michener et al., 2014a,b, 2016). Adaptation may require other specific genes associated with dehalogenative pathways, as well as modulation of expression of common “household” genes to optimize metabolic flux and responses to dehalogenation-associated stresses. In this study, the relative contribution of core, variable and strain-specific genomes in M. extorquens strains growing with halogenated methanes was investigated using a combination of comparative genomics and transcriptomics.
The Potentially Dehalogenation-Associated Genome of M. extorquens Is Limited
Beyond specific genes associated with dehalogenation [at least 6 genes of the cmu pathway for strain CM4 (Michener et al., 2016), and 4 genes of the dcm islet for strain DM4 (Muller et al., 2011a, respectively)], few strain-specific genes shared by CM- and DCM-dehalogenating strains were identified through comparison with 3 other high quality assembled genomes from M. extorquens (Figure 1A). The strains AM1, PA1, and BJ001 were experimentally checked for their inability to grow on chloromethane or dichloromethane (data not shown). The gene content in the 5 M. extorquens genomes totals 12,273 unique CDS, representing 12.3 Mb. The core genome shared by all 5 M. extorquens genomes is extensive (3,489 CDS) ranging from 55 to 68% (55% for CM4; 61% for DM4). M. extorquens strains CM4 and DM4 share most of their genome content (75%, 4,424 CDS) and extensive gene synteny (Figure 1B). The specific genome for strain CM4 comprises 1,512 CDS (24% of total), and that of strain DM4 952 CDS (17%), respectively. In contrast, the variable genome shared only by the 2 dehalogenating strains was only 163 CDS, representing less than 3% of their total genome size, and in the range of the shared variable genome for any pair of the 5 strains considered (Figure 1A). A majority (97 CDS) had no predicted function and 42 genes were found in synteny on plasmids pCMU01 in CM4 and plasmid p1METDI in DM4 (Table S3).
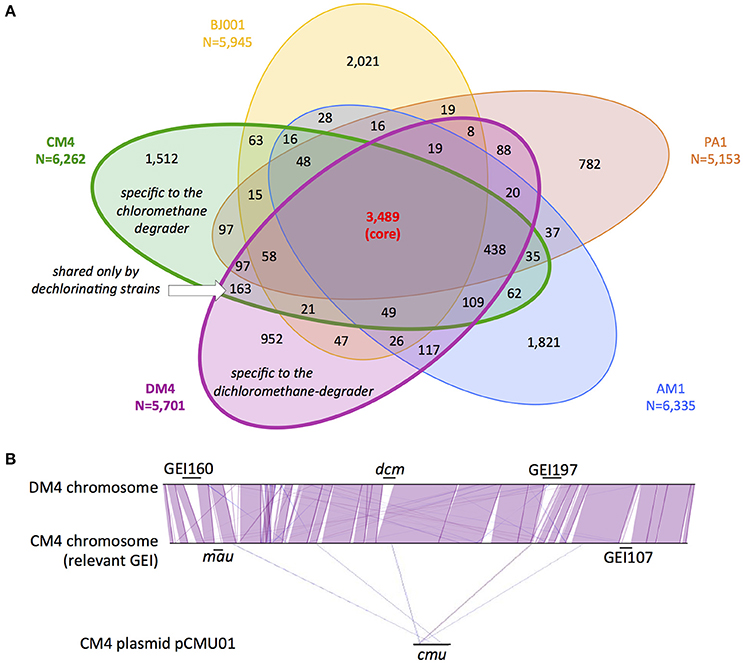
Figure 1. Gene content and synteny in genomes of M. extorquens CM4 and DM4. Comparative genome analyses were performed on the sequenced and assembled genomes of M. extorquens dehalogenating strains CM4 and DM4, as well as strains AM1, PA1, and BJ001 (Vuilleumier et al., 2009; Marx et al., 2012), using MaGe (Vallenet et al., 2017) on the MicroScope online platform (see Materials and Methods). (A) Common homologous genes were defined and assigned to either core (found in all 5 strains), variable (found in 2–4 strains), shared (found only in CM4 and DM4) or specific (found in one strain only) genomes. (B) Synteny of CM4 and DM4 genomes indicating genomic islands (GEI) relevant to C1 metabolism, i.e., dcm for dichloromethane utilization and mau for methylamine utilization (Vuilleumier et al., 2009), and cmu for CM utilization (380 kb plasmid pCMU01, Marx et al., 2012) (see Table 3 for GEI details). Synteny involved groups of at least 6 homologous genes (purple lines, strand conservation; blue lines, strand inversion).
Common Transcriptomic Response of M. extorquens Strains to Chlorinated Methanes as Growth Substrates
Methylobacterium extorquens strains CM4 and DM4 were grown with CM and with DCM, respectively. Longer generation times were observed for growth with CM and DCM compared to with methanol, as observed previously (Muller et al., 2011b; Roselli et al., 2013). Global profiles of gene expression were assessed by RNA-Seq with sequencing coverage exceeding 600X (Table 1). Only 29 CDS of strain CM4 and 36 CDS of strain DM4 showed no reads. Obtained data were normalized (Table S1; Figure S1) and verified by qPCR for a set of 16 genes spanning 2 orders of magnitude of gene expression (Table S2; Figure S2). Only genes with satisfactory false discovery rates (p-value < 0.1) were further analyzed and included 1,245 and 301 genes for the genomes of strain CM4 and strain DM4, respectively.
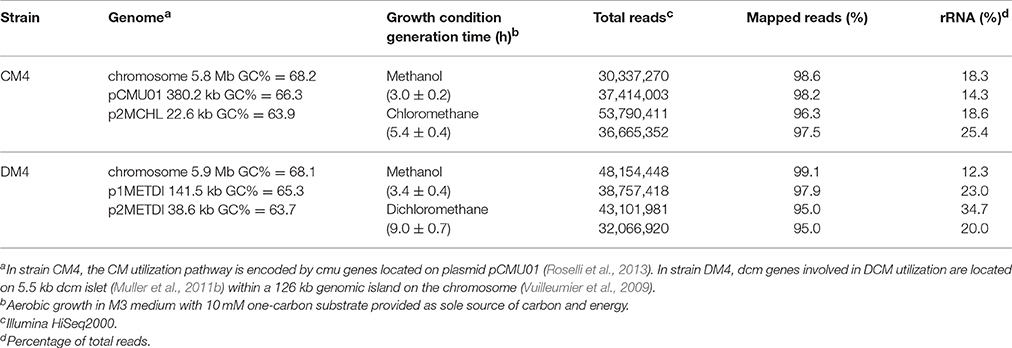
Table 1. Overview of genomic properties of M. extorquens CM4 and DM4 and obtained RNA-Seq data.
Transcript abundance of strains grown with chlorinated methanes CM or DCM and with methanol, the reference methylotrophic growth substrate for M. extorquens, were then compared. A total number of 150 genes for CM4 and 190 genes for DM4 were detected as differentially abundant genes (Table 2). Overall gene expression of common genes during growth with methanol was similar in strains CM4 and DM4, as expected (Figure S1). More pronounced differences were observed between patterns of gene expression for common genes of M. extorquens strains CM4 and DM4 grown with CM or with DCM (Table S4), with only few genes showing the same trend with CM and DCM relative to methanol (Figure 2). Strikingly, essentially all 163 CDS only shared by the 2 dehalogenating strains and not found in any of the 3 other non-dehalogenating M. extorquens strains (Figure 1A) lacked differential expression between chlorinated methanes and methanol (with the exception of METDI4814, less abundant with DCM) (Table S3; Figure S3B). Among those, 42 genes were plasmid-borne and co-localized on the largest plasmids in both strains (Table S3), with some displaying high transcript abundance (Figure S3).
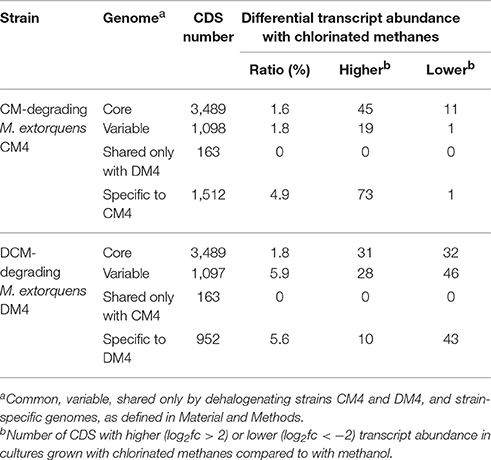
Table 2. Differential expression of core, variable, shared and strain-specific CDS during growth with chlorinated methanes.
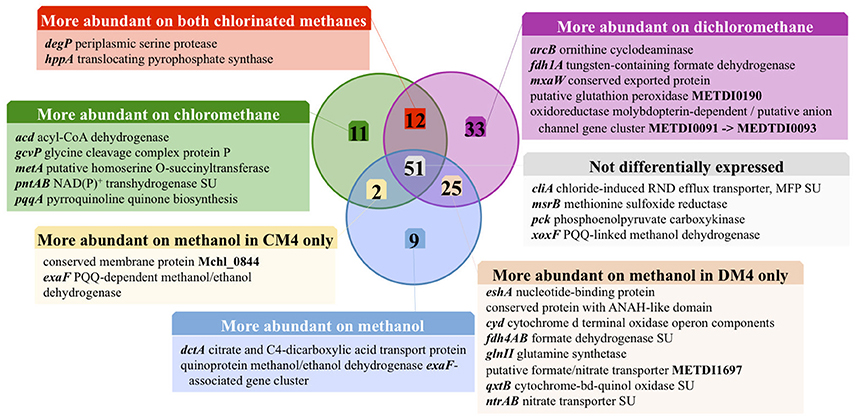
Figure 2. Carbon source-dependent transcript abundance of common core genes in M. extorquens CM4 and DM4. Different categories were defined on the basis of the log2 fold-change (log2fc) values of RNA-Seq reads for M. extorquens CM4 grown with CM (green circle) and for M. extorquens DM4 grown with DCM (purple circle) versus methanol (blue cercle). Complete gene names, log2fc values for each category are detailed in Table S4.
Of the 12 common genes encoded by the core genome more highly expressed during growth with either CM and DCM than with methanol (Table S4; Figure 2), only 2, degP and hppA, have predicted functions. Gene degP encodes a putative periplasmic serine protease whose Escherichia coli homolog HtrA (39% amino acid identity) is a central housekeeping molecular chaperone protein controlling the production of functional, properly folded outer-membrane proteins (Ge et al., 2014). In Carboxydothermus hydrogenoformans, the membrane-bound proton translocating pyrophosphatase HppA (48% amino acid identity to M. extorquens homologs) uses energy from pyrophosphate hydrolysis to build up a proton motive force by proton extrusion (Belogurov and Lahti, 2002). In Rhodospirullum rubrum, the closely homologous HppA (72% amino acid identity) is involved in stress bioenergetics and in particular salt stress (Lopez-Marques et al., 2004).
The number of genes of the core genome with significantly lower expression on chlorinated methanes is also low (9 genes, Figure 2). These may rather be methanol-induced rather than genes repressed by chlorinated methanes, since 7 of these genes belong to predicted operons associated with the alternative alcohol dehydrogenase ExaF to the paradigmatic methanol dehydrogenase encoded by mxa genes (Good et al., 2016; Table S4).
The 10 most highly transcribed genes during growth with chlorinated methanes in strains CM4 and DM4 (Figure 3) included 3 common to both strains, albeit with values of log2fc <2 compared to growth with methanol. Of the other 7 genes highly transcribed on chlorinated methanes, only strain-specific genes directly associated with dehalogenation (5 for strain CM4, 2 for strain DM4), and not common genes, showed log2fc values >2. Transcription responses for growth with CM and DCM were analyzed in more detail.
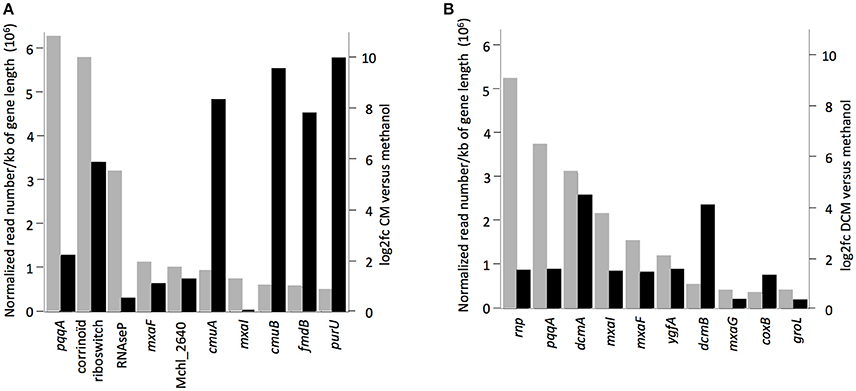
Figure 3. Genes with highest transcript abundance in cultures grown with chlorinated methanes compared to with methanol. Black rectangles indicate values of log2fc (values higher than 2.0 or lower than −2.0 mean normalized reads more abundant in cultures grown with chlorinated methanes or with methanol, respectively). Gray rectangles represent normalized read numbers divided by gene length in kb. RNA-Seq data from cultures of (A) M. extorquens CM4, (B) M. extorquens DM4.
The Chloromethane Transcriptome of Strain CM4
Under the conditions tested, a total of 137 genes (2% of the CM4 transcriptome) had higher transcript abundance with CM than with methanol. Only 43 of these belong to the core genome defined here for M. extorquens (log2fc ≥ 2; Table S5; Figure 4A). Among these, 11 core genes only differentially expressed in strain CM4 had not previously been associated with M. extorquens growth with CM. They include the pnt gene cluster encoding a NADH/NADPH transhydrogenase with cross-membrane proton translocation activity (Chou et al., 2015); two consecutive paralogs of pqqA, a precursor of the redox active dehydrogenase cofactor PQQ (Ochsner et al., 2015); ykuD, encoding a transpeptidase of a large enzyme family associated with cell wall biology (Bielnicki et al., 2005); ada encoding a bifunctional transcriptional activator which acts in response to alkylation damage (51% amino acid identity with the well-characterized E. coli homolog) (McCarthy and Lindahl, 1985); ibpA encoding a small heat shock protein (sHSP) with 57% amino acid identity to E. coli IbpA, which protects enzymes against oxidative stress (Kitagawa et al., 2002).
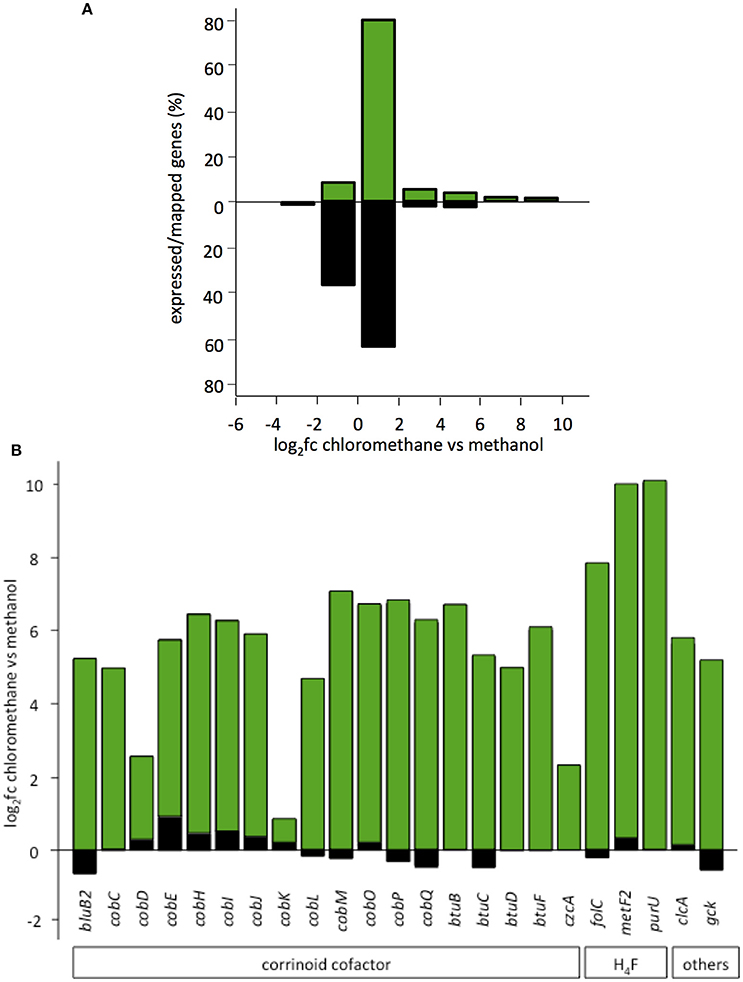
Figure 4. Involvement of plasmid pCMU01 in the chloromethane transcriptome. (A) Expressed percentage of total genes per log2fc range. Genes encoded by plasmid pCMU01 (in green) and by the M. extorquens CM4 chromosome (in black) were grouped according to their log2fc values. Chromosome and plasmid pCMU01 encode 6,262 and 361 genes, and the total percentage of expressed genes is 1.4 and 17.5%, respectively. (B) Differential expression of pCMU01- and chromosome-encoded paralogs.
The major contribution in the CM transcriptome involves strain-specific genes either directly or indirectly associated with dehalogenation (Figure 5). First, 40 genes known from previous work to be associated with cmu gene clusters in CM-degrading strains, and found on the pCMU01 plasmid in strain CM4, showed highest relative expression (log2fc values 7.7–10) during growth with CM (Table S5; also see Figure 4A). Most of these genes are involved in metabolism and transport of the corrinoid and H4F cofactors essential for CM dehalogenation by the cmu pathway (Studer et al., 2001) (Table S5; Figure 4B). A gene cluster specifically shared between sequenced genomes of CM degrading isolates [acxABC, Roselli et al., 2013 >81% amino acid identity with the characterized aerobic bacterium Xanthobacter autotrophicus acetone carboxylase components (Sluis et al., 2002)] was also more expressed in cells grown with CM (see Figure S3A for log2fc values and expression level) although its functional connection with the cmu pathway remains to be characterized. Second, half of the plasmid pCMU01-borne genes more highly expressed on CM had homologous copies located on the chromosome (31 genes out of 62 plasmid-borne genes with higher transcript abundance; Table S5; Figure 3B). All the corresponding chromosomal paralogs were, in contrast, not differentially expressed. Again, a majority of plasmid-borne homologs were associated with the essential corrinoid and tetrahydrofolate cofactors of CM dehalogenation by the cmu pathway (Figure 3B). Similarly, the pCMU01 plasmid homolog of the clc H+/Cl− antiporter gene was more highly expressed on CM (log2fc value of 5.5), unlike its chromosomal homolog (68% identity at the protein level; Figure S3A). Taken together, this argues strongly for a key role of plasmid pCMU01-borne genes in adaptation to growth with CM in strain CM4.
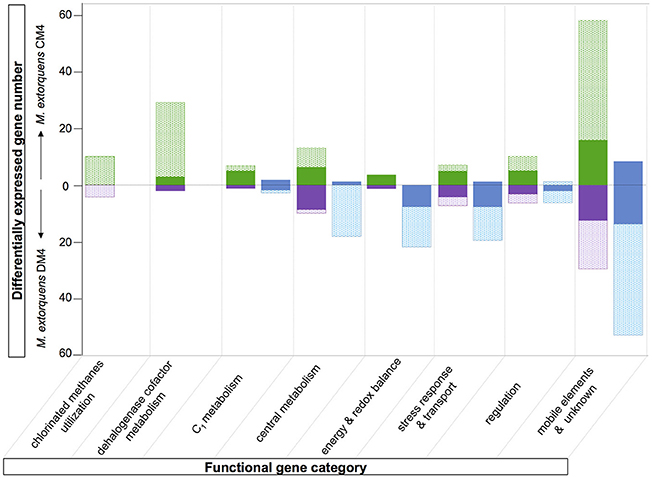
Figure 5. Overview comparison of the chloromethane and dichloromethane transcriptomes. Differentially abundant genes with predicted function are clustered in different functional categories (see Tables S6, S7). The fractions of genes of the core and variable genomes are indicated in plain and dotted rectangles, respectively. Transcript with higher abundance in chloromethane, dichloromethane or methanol are in green, purple or blue, respectively.
The Dichloromethane Transcriptome of Strain DM4
Under the conditions tested, only 3% of the DM4 transcriptome was altered in growth with DCM relative to methanol. Among these, 69 genes (1% of the transcriptome) showed higher transcript abundance in cultures grown with DCM (Table 2). These included the 4 genes of the dcm islet, i.e., dcmA coding for the DCM dehalogenase essential for growth with DCM; dcmR involved in its own transcription as well as that of dcmA (Leisinger et al., 1994; Kayser et al., 2002); and dcmB and dcmC genes of unknown function (Table S6). This is the first reported evidence for transcription of dcmC and its modulation by DCM, and confirms previous results for dcmB (Muller et al., 2011b). Transcripts of dcmA and dcmB were among the most abundant with DCM (Figure 3B).
The majority of genes showing differential abundance with DCM only (33 genes) belong to the core genome of M. extorquens, and significantly exceeds the number of M. extorquens genes of the core genome also more highly abundant during growth with CM (12 genes), or those only more highly expressed during growth with CM but not DCM (11 genes) (Figure 2). The specific functions of most of these genes remain elusive, although an association with redox status is suggested, with a putative glutathione peroxidase (METDI0190) and 2 sets of genes associated with uncharacterized molybdenum-dependent enzymes (METDI2693, METDI0091- METDI0093) showing increased transcript abundance (Table S6). Of those, the gene encoding the glutathione peroxidase was highly expressed (Figure S3B).
The contribution of the 2 plasmids of DCM-degrading strain DM4 (Table 1) in the DCM transcriptome is limited (2 uncharacterized genes among 186 CDS were differentially expressed). This suggests that unlike for the CM-degrading strain, DM4 plasmids do not play a role in adaptation to DCM in strain DM4.
The set of genes from strain DM4 with higher expression on methanol is also larger than that of strain CM4 (Figure 5). The majority of these genes is found in gene clusters with redundant gene content (Figure S4), and often associated with genomic islands (GEIs) (Table 3, and see next subsection).
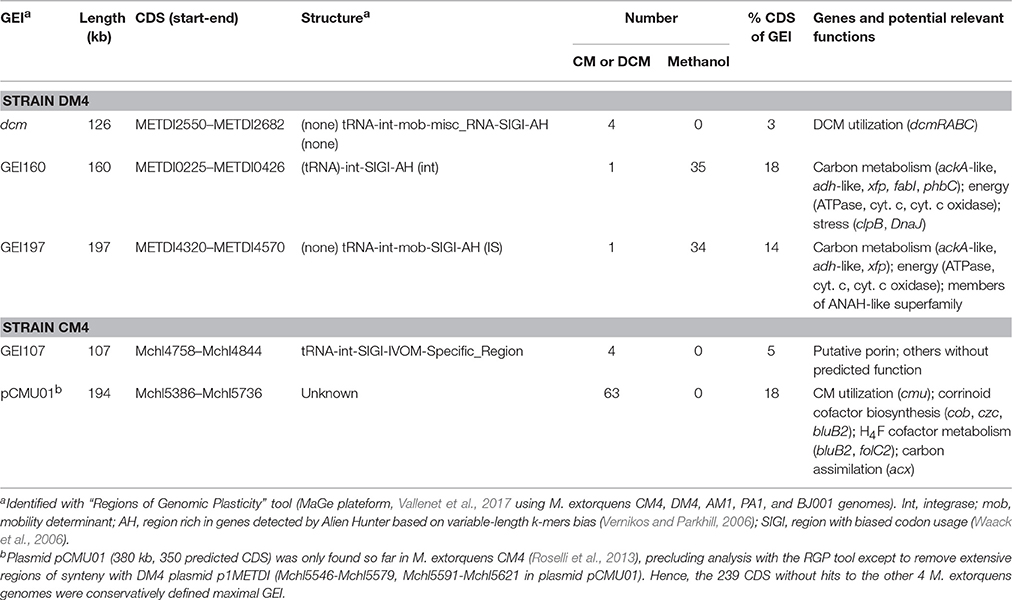
Table 3. GEIs containing genes with differential transcript abundance.
Expression of Genomic Island-Borne Genes in Dehalogenating M. extorquens Strains
As plasmids, GEIs are major agents of rapid genome evolution and adaptation of catabolic pathways in bacteria, and are often found integrated in the chromosome (van der Meer and Sentchilo, 2003). The potential role of GEIs in adaptation of M. extorquens to growth with chlorinated methanes was investigated. Out of the 14 and 11 specific GEIs detected in strains CM4 and DM4, respectively, only 3 GEIs contained genes with differential expression (Table 3). Such genes only represented a minor part of GEIs, and their roles beyond that of genes associated with dehalogenation remain unknown. It is striking that no other genes of the 126 kb dcm GEI of strain DM4 were differentially transcribed beyond those of the 5.5 kb dcm islet, unlike many genes associated with cmu gene clusters on plasmid pCMU01 (Table 3).
Discussion
Dehalogenation-Specific Gene Complement
The number of strain-specific genes, as such potentially associated with dehalogenative metabolism, was relatively small (Figure 1A) and genes with an annotation suggestive of a role in this context were not detected (Table S3). Moreover, apart from dehalogenase genes, such genes were not especially prone to differential expression with chlorinated methanes (Tables S5, S6; Figures 3–5). Further, none of the few genes shared only by CM- and DCM-degrading strains among a small group of 5 M. extorquens strains were differentially expressed during growth with chlorinated methanes. Nevertheless looking for upregulated genes in CM/DCM-grown cultures vs. methanol-grown cultures will not detect constitutively expressed genes that support dehalogenation growth, and other complementary approaches need to be assessed (Ochsner et al., 2017). Among highly transcribed genes, a few had predicted functions in plasmid transfer and replication (genes icmL, repA, repB, traD; Figure S3). Thus, the part of the transcriptional response common to both chlorinated methanes was limited (Table S3). Adaptation to growth with chlorinated methanes seems to be largely a function of fine-tuning gene content/expression rather than large-scale changes as discussed in the following.
Adaptive Stress Response
Indications for a transcriptional adaptive stress response to the utilization of chlorinated methanes in M. extorquens obtained here confirm previous suggestions from mutant growth phenotypes in the case of DCM (Muller et al., 2011b) and proteomic data in the case of CM (Roselli et al., 2013). Whereas expression of several genes associated with the general stress response was modulated (Tables S3, S5, S6; Figure 5), genes potentially associated with dehalogenation-specific stress were of particular interest. Since bacteria that grow with chlorinated methanes have to cope with production of intracellular hydrochloric acid production, one key question to address was which of intracellular proton or chloride buildup represents a larger stress for M. extorquens.
Intracellular generation of protons during growth with chlorinated methanes is expected to be detrimental for growth through intracellular acidification and also through dissipation of the proton-motive force and associated ATP production. It thus seems significant that the membrane-bound proton translocating pyrophosphatase hppA was one of only 12 genes of the core genome with higher expression on both chlorinated methanes. HppA-driven proton extrusion and restoration of the proton-motive force involves an additional energy expense through pyrophosphatase hydrolysis (Belogurov and Lahti, 2002).
Similarly, the H+/Cl− antiporter ClcA involved in adaptation to chloride stress affords chloride efflux at a cost for the proton-motive force. The corresponding gene was expressed at high constitutive levels in strain DM4 (Figure S3B), as shown recently for DCM-degrading isolates including strain DM4 (Michener et al., 2014b). In case of strain CM4, clcA displayed moderate constitutive expression, but a pCMU01 plasmid-borne paralog clcA2 (69% amino acid identity) was significantly more highly expressed in cultures on CM (Table S5; Figure S3A). This suggests that clcA-driven chloride extrusion is crucial for growth with both chlorinated methanes, and may involve different paralogs of this large gene family. Recent work showed that clcA, when transcribed from its native promoter cloned from strain DM4, confers higher fitness for growth with DCM but not with CM in Methylobacterium strains not previously exposed to chlorinated methanes (Michener et al., 2014a, 2016). It is noteworthy that the uncharacterized RND efflux transporter CliABC previously identified as chloride-induced in strain DM4 (Muller et al., 2011b) was found here to be even more expressed than gene clcA (Figure S3B). Taken together, the obtained data suggest that an increase in intracellular chloride levels represents more of the problem for growing strains of M. extorquens than intracellular proton production.
Regulation under Dehalogenative Methylotrophic Conditions
Strain-specific genes were among the most transcribed genes, especially those within gene clusters involved in chlorinated methane dehalogenation, i.e., dcmA and cmuAB genes (Figure 3). Here, CM-dependent transcription involved much larger changes in expression compared to DCM-dependent transcription (log2fc of 10 vs. 4, respectively). This confirms previous studies with RT-qPCR and transcriptional fusions of the cmuA promoter (Farhan Ul Haque et al., 2013). Among genes with increased expression levels with chlorinated methanes identified in this work, a significant number are annotated as putative regulatory genes (9 for CM and 6 for DCM respectively; Tables S5, S6). The transcriptional regulator of dcmA has been preliminarily characterized (La Roche and Leisinger, 1991; Muller et al., 2011b). In contrast, the genetic determinants involved in regulating the expression of the cmu pathway remain to be identified (Roselli et al., 2013).
The contribution of genes less expressed during growth with CM than with methanol was limited in the case of the core genome (11 genes) and even more so for the CM4 strain-specific genes (2 genes; Figure 5). This suggests that gene downregulation mediated by CM in strain CM4 is limited. On the other hand, a striking finding was that many plasmid pCMU01-borne paralogs but not their chromosomal homologs had higher transcript abundance in CM cultures (Figure 4B). This suggests the existence of as yet uncharacterized regulation mechanisms favoring expression of plasmid-encoded over chromosomally encoded gene paralogs in response to growth with CM.
Effects of Dehalogenation Metabolism on Transcription of Methylotrophy Genes
High levels of transcripts of genes mxaFI for subunits of the canonical methanol dehydrogenase (MDH) of M. extorquens (Amaratunga et al., 1997) were observed in methanol cultures, as expected, but also in cultures grown with chlorinated methanes (Figure 3). Since dehalogenation of DCM leads to formaldehyde, one explanation might be that transcription of methanol dehydrogenase MxaFI, which also converts formaldehyde (Nunn and Lidstrom, 1986), is also induced by formaldehyde. However, this does not strictly apply to growth with CM, as formaldehyde is not a direct product of CM dehalogenation by the cmu pathway (Vannelli et al., 1999; Figure 6) and chemical equilibrium between free formaldehyde and methylene-tetrahydrofolate (CH2 = H4F) does not favor free formaldehyde (Kallen and Jencks, 1966). This suggests that methanol dehydrogenase expression may be induced by other downstream metabolites common to CM and methanol catabolism, such as other folate-bound C1 compounds or formate, or that mxaFI expression is not switched off in strain CM4. On the other hand, other recently identified dehydrogenases active with methanol did not show the same transcriptional profile. In particular, the expression of genes for the XoxF-type enzymes was not modulated by chlorinated methanes. As for the PQQ-dependent ExaF-type dehydrogenase active with ethanol, methanol and formaldehyde (Good et al., 2016), seven genes directly upstream of gene exaF showed higher transcript levels in methanol-grown cultures in both CM4 and DM4 strains, including for gene exaF in strain CM4 (Table S5). Our results suggest complex modes of cross-regulation of key enzymes of methylotrophic metabolism in response to different C1 substrates that mostly involve genes that belong to the core genome (Figure 5).
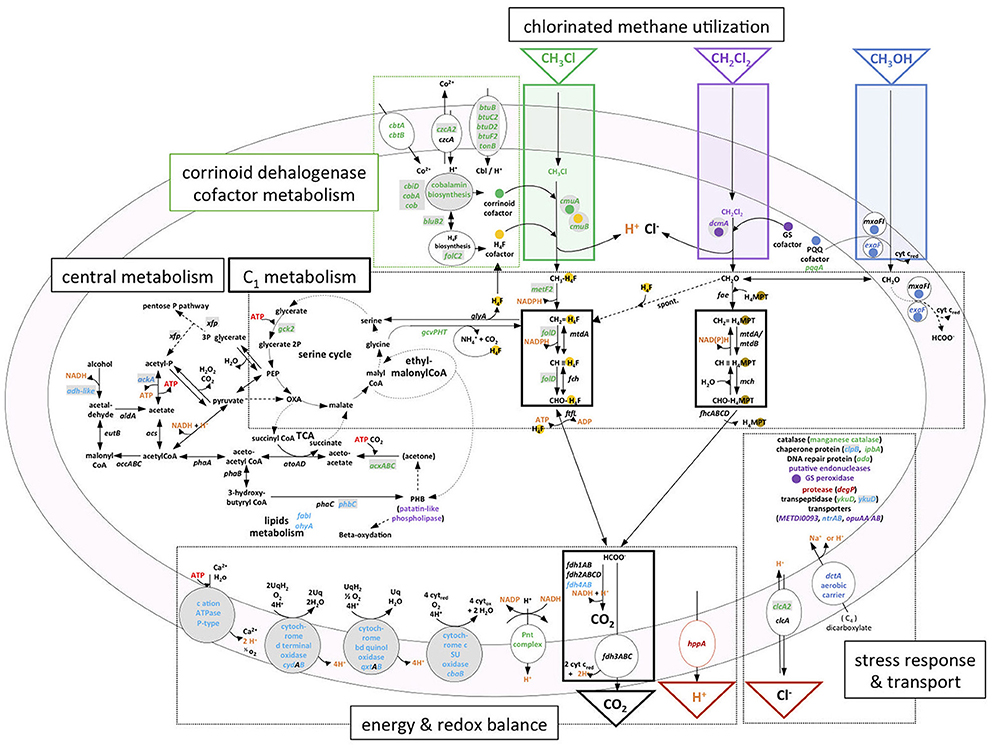
Figure 6. Transcriptional response and metabolism of chlorinated C1 compounds chloromethane and dichloromethane in M. extorquens. Chloromethane dehalogenation and methyl transfer to tetrahydrofolate (H4F) reactions are boxed in green; dichloromethane dehalogenation to formaldehyde in purple; methanol oxidation to formaldehyde in blue. Within C1 metabolism, black boxes delimit common methylotrophic modules (Chistoserdova et al., 2009): H4F and tetrahydromethanopterin (H4MPT)-dependent C1 transfer reactions, and formate oxidation for C1 dissimilation with C02 production. Serine and ethylmalonyl-CoA cycles for C1 assimilation (Ochsner et al., 2015) are schematized with two connected circles. Reactions related to essential co-factors for chlorinated methane utilization are highlighted with colored spots; H4F (yellow), corrinoid cofactor (green), glutathione (purple), and pyrroquinoline quinone (blue). GEI-located genes in M. extorquens DM4 or in M. extorquens CM4 (plasmid pCMU01) are shown in gray-shaded boxes. RNA-Seq data are summarized using gene color-coding for more abundant transcripts on CM (green), DCM (purple), both CM and DCM (dark red), methanol [both M. extorquens CM4 and DM4 (dark blue), DM4 only (light blue)]. Compounds associated with energy-dependent transformations are highlighted in orange. Cbi, cobinamide; Cbl, cobalamin; CoA, coenzyme A; PEP, phosphoenolpyruvate; OXA, oxaloacetate; TCA, tricarboxylic acid cycle.
Metabolic Fine-Tuning for Dehalogenative Methylotrophic Growth
Several genes of M. extorquens strains CM4 and DM4 that show significant modulation of expression with chlorinated methanes have predicted functions in energy homeostasis and redox processes. However, both the types of genes involved and the differential transcription patterns often differed between CM- and DCM-degrading strains. In the case of the DCM-degrading strain DM4, several gene clusters containing cytochrome oxidase genes displayed lower transcript counts in DCM cultures (Table S6). In particular, a substantial set of GEI-associated genes (Table 3), belonging to the variable genome of M. extorquens and potentially linked to cytochrome electron transport and thereby the transmembrane H+ gradient, had enhanced transcript abundance in cultures grown with methanol (Figure S4). Other potentially energy-associated adjustments of M. extorquens DM4 include very high transcript levels for various genes involved in metabolism of fatty acids and polyhydroxyalkanoates (Table S6), which are a storage form of carbon and energy (Escapa et al., 2012), suggesting that growth on DCM vs. methanol may involved different carbon and energy spillage control processes.
For the CM-degrading strain CM4, in contrast, obtained data suggest that energy balance adjustments as a function of growth with CM or methanol involve NAD(P)-linked metabolism. Whereas only one enzymatic step from methanol to formate involves one molecule of NAD(P)+ (MtdA/MtdB), for each molecule of CM transformed to formate, two molecules of NAD(P)H are potentially generated from NAD(P)+ in two successive reactions specifically catalyzed by MetF2 and FolD (Figure 6), These enzymes can use both NAD+ and NADP+ as cofactors, although M. extorquens CM4 FolD activity had only been assessed so far with NADP+ (Marx and Lidstrom, 2004). The corresponding genes metF2 and folD are two of the most differentially expressed genes in the CM transcriptome (Table S5). Importantly, the cmu pathway specifically features FolD rather than MtdA and Fch for oxidation of C1 carbon to formate (Studer et al., 2002) unlike growth on methanol (Marx and Lidstrom, 2004). MetF2 and FolD may thus have different preferences than MtdA/MtdB for using NAD+ or NADP+ as cofactors. Should this be the case, significant altering of the intracellular pools of NAD+ and NADP+ during growth with CM compared to methanol could become growth limiting if levels of oxidized cofactors are limiting. A candidate for metabolic fine-tuning in this context is the membrane-bound transhydrogenase encoded by the chromosomal pnt gene cluster. All three pnt genes were significantly more expressed on CM at both transcription (Table S5) and protein (Roselli et al., 2013) levels. Transhydrogenase catalyzes the reversible reaction NADPH + NAD+ + <=> NADP+ + NADH + (Carroll et al., 2015). In addition to NADP+ regeneration via the membrane-bound transhydrogenase, concomitant proton efflux could thus potentially also help maintain internal cellular pH during CM dehalogenation.
During growth with CM, the high demand for oxidized NAD+/NADP+ cofactors may alter CH2 = H4F flux toward the serine cycle by limiting the carbon flux toward formate production. A compensatory metabolic rerouting of CH2 = H4F toward formate formation could be needed and may be achieved by the components of the so-called glycine cleavage complex (Figure 6), which were more highly abundant at both the transcript (GcvPHT, Table S5) and protein levels (GcvT; Roselli et al., 2013).
In conclusion, utilization of horizontally transferred genes for growth of M. extorquens with CM and DCM presumably involved required several post-transfer adjustments, as shown in recent experimental evolution experiments (Michener et al., 2014a, 2016). The new data obtained in the present study highlight potential global adjustments at the transcriptional level and more generally, the importance of substrate- and pathway-dependent genome adaptation following acquisition of new growth-supporting abilities such as degradation of toxic halogenated compounds. Clearly, M. extorquens represents a model of choice to address these issues in the future.
Author Contributions
PC, BM, and FB prepared RNA. PC and AA constructed cDNA libraries. PC and FB performed RNA-Seq analysis. BM carried out RT-qPCR assays and data processing. FB designed the study. PC, MW, SV, FB, and SC participated in bioinformatic analysis. PC, BM, SK, SV, SC, and FB were involved in data analysis and interpretation. PC, SV, and FB wrote the manuscript. All authors read and approved the final manuscript.
Funding
This work was partially funded by a Ph.D grant to PC from Région Alsace (France) and DFG grant Ko2912/5-1 (Germany), the German Academic Exchange Service (DAAD), the University of Bayreuth, and French-German mobility funds PROCOPE (Deutscher Akademischer Austausch Dienst), the French Agence Nationale de la Recherche (ANR grant 14CE35-005-01), as well as support for RNA-Seq analysis by a GIS IBiSA grant-in-kind to FB. The funding bodies had no impact on the design of the study, data collection, analysis, interpretation of data, nor in writing the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We gratefully acknowledge Beatrice Segurens (CEA CNRS-UMR 8030, France) for RNA extraction protocol optimization, David A. C. Beck (University of Washington, Seattle, USA) and Anne Francez-Charlot (ETH Zurich, Switzerland) for checking proposed reference genes in their unpublished M. extorquens RNA-Seq libraries.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.01600/full#supplementary-material
Abbreviations
CM, chloromethane; DCM, dichloromethane; GEI, genomic island; H4F, tetrahydrofolate.
References
Amaratunga, K., Goodwin, P. M., O'Connor, D., and Anthony, C. (1997). The methanol oxidation genes mxaFJGIR(S)ACKLD in Methylobacterium extorquens. J. Bacteriol. 146, 31–38. doi: 10.1016/S0378-1097(96)00399-0
Amato, P., Parazols, M., Sancelme, M., Laj, P., Mailhot, G., and Delort, A.-M. (2007). Microorganisms isolated from the water phase of tropospheric clouds at the Puy de Dôme: major groups and growth abilities at low temperatures: microorganisms from the water phase of tropospheric clouds. FEMS Microbiol. Ecol. 59, 242–254. doi: 10.1111/j.1574-6941.2006.00199.x
Anders, S., McCarthy, D. J., Chen, Y., Okoniewski, M., Smyth, G. K., Huber, W., et al. (2013). Count-based differential expression analysis of RNA sequencing data using R and Bioconductor. Nature protocols 8, 1765–1786. doi: 10.1038/nprot.2013.099
Belogurov, G. A., and Lahti, R. (2002). A lysine substitute for K+. A460K mutation eliminates K+ dependence in H+-pyrophosphatase of Carboxydothermus hydrogenoformans. J. Biol. Chem. 277, 49651–49654. doi: 10.1074/jbc.M210341200
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Statist. Soc. B. 57, 289–300.
Bielnicki, J., Devedjiev, Y., Derewenda, U., Dauter, Z., Joachimiak, A., and Derewenda, Z. S. (2005). Bacillus subtilis YkuD protein at 2.0 Å resolution: insights into the structure and function of a novel, ubiquitous family of bacterial enzymes. Proteins 62, 144–151. doi: 10.1002/prot.20702
Bringel, F., and Couée, I. (2015). Pivotal roles of phyllosphere microorganisms at the interface between plant functioning and atmospheric trace gas dynamics. Front. Microbiol. 6:486. doi: 10.3389/fmicb.2015.00486
Carpenter, L. J., Reimann, S., Burkholder, J. B., Clerbaux, C., Hall, B. D., Hossaini, R., et al. (2014). “Ozone-depleting substances (ODSs) and other gases of interest to the Montreal protocol,” in Scientific Assessment of Ozone Depletion: 2014, Global Ozone Research and Monitoring Project, eds A. Engel and S. A. Montzka (Geneva: World Meteorological Organization), 1.1–1.101.
Carroll, S., Chubiz, L., Agashe, D., and Marx, C. (2015). Parallel and divergent evolutionary solutions for the optimization of an engineered central metabolism in Methylobacterium extorquens AM1. Microorganisms 3, 152–174. doi: 10.3390/microorganisms3020152
Chen, F., Freedman, D. L., Falta, R. W., and Murdoch, L. C. (2012). Henry's law constants of chlorinated solvents at elevated temperatures. Chemosphere 86, 156–165. doi: 10.1016/j.chemosphere.2011.10.004
Chistoserdova, L., Kalyuzhnaya, M. G., and Lidstrom, M. E. (2009). The expanding world of methylotrophic metabolism. Annu. Rev. Microbiol. 63, 477–499. doi: 10.1146/annurev.micro.091208.073600
Chou, H. H., Marx, C. J., and Sauer, U. (2015). Transhydrogenase promotes the robustness and evolvability of Escherichia coli deficient in NADPH production. PLoS Genet. 11:e1005007. doi: 10.1371/journal.pgen.1005007
Derendorp, L., Wishkerman, A., Keppler, F., McRoberts, C., Holzinger, R., and Rockmann, T. (2012). Methyl chloride emissions from halophyte leaf litter: dependence on temperature and chloride content. Chemosphere 87, 483–489. doi: 10.1016/j.chemosphere.2011.12.035
Escapa, I. F., García, J. L., Bühler, B., Blank, L. M., and Prieto, M. A. (2012). The polyhydroxyalkanoate metabolism controls carbon and energy spillage in Pseudomonas putida: PHA metabolism in Pseudomonas putida. Environ. Microbiol. 14, 1049–1063. doi: 10.1111/j.1462-2920.2011.02684.x
Farhan Ul Haque, M., Nadalig, T., Bringel, F., Schaller, H., and Vuilleumier, S. (2013). Fluorescence-based bacterial bioreporter for specific detection of methyl halide emissions in the environment. Appl. Environ. Microbiol. 79, 6561–6567. doi: 10.1128/aem.01738-13
Ge, X., Wang, R., Ma, J., Liu, Y., Ezemaduka, A. N., Chen, P. R., et al. (2014). DegP primarily functions as a protease for the biogenesis of β-barrel outer membrane proteins in the Gram-negative bacterium Escherichia coli. FEBS J. 281, 1226–1240. doi: 10.1111/febs.12701
Good, N. M., Vu, H. N., Suriano, C. J., Subuyuj, G. A., Skovran, E., and Martinez-Gomez, N. C. (2016). Pyrroloquinoline quinone-containing ethanol dehydrogenase in Methylobacterium extorquens AM1 extends lanthanide-dependent metabolism to multi-carbon substrates. J. Bacteriol. 198, 3109–3118. doi: 10.1128/JB.00478-16
Hardacre, C. J., and Heal, M. R. (2013). Characterization of methyl bromide and methyl chloride fluxes at temperate freshwater wetlands. J. Geophys. Res. Atmos. 118, 977–991. doi: 10.1029/2012jd018424
Kallen, R. G., and Jencks, W. P. (1966). The mechanism of the condensation of formaldehyde with tetrahydrofolic acid. J. Biol. Chem. 241, 5851–5863.
Kayser, M. F., Ucurum, Z., and Vuilleumier, S. (2002). Dichloromethane metabolism and C1 utilization genes in Methylobacterium strains. Microbiology 148, 1915–1922. doi: 10.1099/00221287-148-6-1915
Kayser, M. F., and Vuilleumier, S. (2001). Dehalogenation of dichloromethane by dichloromethane dehalogenase/glutathione S-transferase leads to the formation of DNA adducts. J. Bacteriol. 183, 5209–5212. doi: 10.1128/JB.183.17.5209-5212.2001
Kitagawa, M., Miyakawa, M., Matsumura, Y., and Tsuchido, T. (2002). Escherichia coli small heat shock proteins, IbpA and IbpB, protect enzymes from inactivation by heat and oxidants: E. coli small heat shock proteins. Eur. J. Biochem. 269, 2907–2917. doi: 10.1046/j.1432-1033.2002.02958.x
Kolb, S. (2009). Aerobic methanol-oxidizing bacteria in soil. FEMS Microbiol. Lett. 300, 1–10. doi: 10.1111/j.1574-6968.2009.01681.x
La Roche, S. D., and Leisinger, T. (1991). Identification of dcmR, the regulatory gene governing expression of dichloromethane dehalogenase in Methylobacterium sp. strain DM4. J. Bacteriol. 173, 6714–6721. doi: 10.1128/jb.173.21.6714-67
Leisinger, T., Bader, R., Hermann, R., Schmid-Appert, M., and Vuilleumier, S. (1994). Microbes, enzymes and genes involved in dichloromethane utilization. Biodegradation 5, 237–248. doi: 10.1007/BF00696462
Lopez-Marques, R. L., Perez-Castineira, J. R., Losada, M., and Serrano, A. (2004). Differential regulation of soluble and membrane-bound inorganic pyrophosphatases in the photosynthetic bacterium Rhodospirillum rubrum provides insights into pyrophosphate-based stress bioenergetics. J. Bacteriol. 186, 5418–5426. doi: 10.1128/JB.186.16.5418-5426.2004
Marx, C. J., Bringel, F., Chistoserdova, L., Moulin, L., Farhan Ul Haque, M., Fleischman, D. E., et al. (2012). Complete genome sequences of six strains of the genus Methylobacterium. J. Bacteriol. 194, 4746–4748. doi: 10.1128/jb.01009-12
Marx, C. J., and Lidstrom, M. E. (2004). Development of an insertional expression vector system for Methylobacterium extorquens AM1 and generation of null mutants lacking mtdA and/or fch. Microbiology 150, 9–19. doi: 10.1099/mic.0.26587-0
McCarthy, T. V., and Lindahl, T. (1985). Methyl phosphotriesters in alkylated DNA are repaired by the Ada regulatory protein of Escherichia coli. Nucl. Acids Res. 13, 2683–2698. doi: 10.1093/nar/13.8.2683
Michener, J. K., Camargo Neves, A. A., Vuilleumier, S., Bringel, F., and Marx, C. J. (2014a). Effective use of a horizontally-transferred pathway for dichloromethane catabolism requires post-transfer refinement. eLife 3:4279. doi: 10.7554/eLife.04279
Michener, J. K., Vuilleumier, S., Bringel, F., and Marx, C. J. (2014b). Phylogeny poorly predicts the utility of a challenging horizontally transferred gene in Methylobacterium strains. J. Bacteriol. 196, 2101–2107. doi: 10.1128/jb.00034-14
Michener, J. K., Vuilleumier, S., Bringel, F., and Marx, C. J. (2016). Transfer of a catabolic pathway for chloromethane in Methylobacterium strains highlights different limitations for growth with chloromethane or with dichloromethane. Front. Microbiol. 7:1116. doi: 10.3389/fmicb.2016.01116
Muller, E. E. L., Bringel, F., and Vuilleumier, S. (2011a). Dichloromethane-degrading bacteria in the genomic age. Res. Microbiol. 162, 869–876. doi: 10.1016/j.resmic.2011.01.008
Muller, E. E. L., Hourcade, E., Louhichi-Jelail, Y., Hammann, P., Vuilleumier, S., and Bringel, F. (2011b). Functional genomics of dichloromethane utilization in Methylobacterium extorquens DM4. Environ. Microbiol. 13, 2518–2534. doi: 10.1111/j.1462-2920.2011.02524.x
Nadalig, T., Greule, M., Bringel, F., Keppler, F., and Vuilleumier, S. (2014). Probing the diversity of chloromethane-degrading bacteria by comparative genomics and isotopic fractionation. Front. Microbiol 5:523. doi: 10.3389/fmicb.2014.00523
Nunn, D. N., and Lidstrom, M. E. (1986). Phenotypic characterization of 10 methanol oxidation mutant classes in Methylobacterium sp. strain AM1. J. Bacteriol. 166, 591–597. doi: 10.1128/jb.166.2.591-597.1986
Ochsner, A. M., Christen, M., Hemmerle, L., Peyraud, R., Christen, B., and Vorholt, J. A. (2017). Transposon sequencing uncovers an essential regulatory function of phosphoribulokinase for methylotrophy. Curr. Biol. 27, 1–10. doi: 10.1016/j.cub.2017.07.025
Ochsner, A. M., Sonntag, F., Buchhaupt, M., Schrader, J., and Vorholt, J. A. (2015). Methylobacterium extorquens: methylotrophy and biotechnological applications. Appl. Microbiol. Biotechnol. 99, 517–534. doi: 10.1007/s00253-014-6240-3
Rhew, R. C., Whelan, M. E., and Min, D. H. (2014). Large methyl halide emissions from south Texas salt marshes. Biogeoscience 11, 6427–6434. doi: 10.5194/bg-11-6427-2014
Ritz, C., and Spiess, A. N. (2008). qpcR: an R package for sigmoidal model selection in quantitative real-time polymerase chain reaction analysis. Bioinformatics 24, 1549–1551. doi: 10.1093/bioinformatics/btn227
Roselli, S., Nadalig, T., Vuilleumier, S., and Bringel, F. (2013). The 380 kb pCMU01 plasmid encodes chloromethane utilization genes and redundant genes for vitamin B12- and tetrahydrofolate-dependent chloromethane metabolism in Methylobacterium extorquens CM4: a proteomic and bioinformatics study. PLoS ONE 8:e56598. doi: 10.1371/journal.pone.0056598
Schmittgen, T. D., and Livak, K. J. (2008). Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 3, 1101–1108. doi: 10.1038/nprot.2008.73
Sluis, M. K., Larsen, A. R., Krum, J. G., Anderson, R., Metcalf, W. W., and Ensign, S. A. (2002). Biochemical, molecular, and genetic analyses of the acetone carboxylases from Xanthobacter autotrophicus strain Py2 and Rhodobacter capsulatus strain B10. J. Bacteriol. 184, 2969–2977. doi: 10.1128/JB.184.11.2002
Studer, A., McAnulla, C., Büchele, R., Leisinger, T., and Vuilleumier, S. (2002). Chloromethane induced genes define a third C1 utilization pathway in Methylobacterium chloromethanicum CM4. J. Bacteriol. 184, 3476–3484. doi: 10.1128/JB.184.13.3476-34
Studer, A., Stupperich, E., Vuilleumier, S., and Leisinger, T. (2001). Chloromethane:tetrahydrofolate methyl transfer by two proteins from Methylobacterium chloromethanicum strain CM4. Eur. J. Biochem. 268, 2931–2938. doi: 10.1046/j.1432-1327.2001.02182.x
Studer, A., Vuilleumier, S., and Leisinger, T. (1999). Properties of the methylcobalamin: H4 folate methyltransferase involved in chloromethane utilization by Methylobacterium sp. strain CM4. Eur. J. Biochem. 264, 242–249. doi: 10.1046/j.1432-1327.1999.00629.x
Torgonskaya, M. L., Doronina, N. V., Hourcade, E., Trotsenko, Y. A., and Vuilleumier, S. (2011). Chloride-associated adaptive response in aerobic methylotrophic dichloromethane-utilising bacteria. J. Basic Microbiol. 51, 296–303. doi: 10.1002/jobm.201000280
Vallenet, D., Calteau, A., Cruveiller, S., Gachet, M., Lajus, A., Josso, A., et al. (2017). MicroScope in 2017: an expanding and evolving integrated resource for community expertise of microbial genomes. Nucl. Acids Res. 45, D517–D528. doi: 10.1093/nar/gkw1101
van der Meer, J. R., and Sentchilo, V. (2003). Genomic islands and the evolution of catabolic pathways in bacteria. Curr. Opin. Biotechnol. 14, 248–254. doi: 10.1016/S0958-1669(03)00058-2
Vannelli, T., Messmer, M., Studer, A., Vuilleumier, S., and Leisinger, T. (1999). A corrinoid-dependent catabolic pathway for growth of a Methylobacterium strain with chloromethane. Proc. Natl. Acad. Sci. U.S.A. 96, 4615–4620. doi: 10.1073/pnas.96.8.4615
Vernikos, G. S., and Parkhill, J. (2006). Interpolated variable order motifs for identification of horizontally acquired DNA: revisiting the Salmonella pathogenicity islands. Bioinformatics 22, 2196–2203. doi: 10.1093/bioinformatics/btl369
Vuilleumier, S., Chistoserdova, L., Lee, M.-C., Bringel, F., Lajus, A., Zhou, Y., et al. (2009). Methylobacterium genome sequences: a reference blueprint to investigate microbial metabolism of C1 compounds from natural and industrial sources. PLoS ONE 4:e5584. doi: 10.1371/journal.pone.0005584
Vuilleumier, S., and Leisinger, T. (1996). Protein engineering studies of dichloromethane dehalogenase /glutathione S-transferase from Methylophilus sp. strain DM11 Ser12 but not Tyr6 is required for enzyme activity. Eur. J. Biochem. 239, 410–417. doi: 10.1111/j.1432-1033.1996.0410u.x
Waack, S., Keller, O., Asper, R., Brodag, T., Damm, C., Fricke, W. F., et al. (2006). Score-based prediction of genomic islands in prokaryotic genomes using hidden Markov models. BMC Bioinform. 7:142. doi: 10.1186/1471-2105-7-142
Keywords: dehalogenation, chloromethane, dichloromethane, GEI, genomic island, genome adaptation, methylotrophy
Citation: Chaignaud P, Maucourt B, Weiman M, Alberti A, Kolb S, Cruveiller S, Vuilleumier S and Bringel F (2017) Genomic and Transcriptomic Analysis of Growth-Supporting Dehalogenation of Chlorinated Methanes in Methylobacterium. Front. Microbiol. 8:1600. doi: 10.3389/fmicb.2017.01600
Received: 25 June 2017; Accepted: 07 August 2017;
Published: 01 September 2017.
Edited by:
Ivan Berg, Universität Münster, GermanyReviewed by:
Nicholas Coleman, University of Sydney, AustraliaJeremy Semrau, University of Michigan, United States
Copyright © 2017 Chaignaud, Maucourt, Weiman, Alberti, Kolb, Cruveiller, Vuilleumier and Bringel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Françoise Bringel, ZnJhbmNvaXNlLmJyaW5nZWxAdW5pc3RyYS5mcg==