 María J. Torres‡
María J. Torres‡ Emilio Bueno†‡
Emilio Bueno†‡ Andrea Jiménez-Leiva
Andrea Jiménez-Leiva Juan J. Cabrera
Juan J. Cabrera Eulogio J. Bedmar
Eulogio J. Bedmar Socorro Mesa*
Socorro Mesa* María J. Delgado*
María J. Delgado*- Department of Soil Microbiology and Symbiotic Systems, Estación Experimental del Zaidín, Consejo Superior de Investigaciones Científicas, Granada, Spain
The powerful greenhouse gas, nitrous oxide (N2O) has a strong potential to drive climate change. Soils are the major source of N2O and microbial nitrification and denitrification the main processes involved. The soybean endosymbiont Bradyrhizobium diazoefficiens is considered a model to study rhizobial denitrification, which depends on the napEDABC, nirK, norCBQD, and nosRZDYFLX genes. In this bacterium, the role of the regulatory cascade FixLJ-FixK2-NnrR in the expression of napEDABC, nirK, and norCBQD genes involved in N2O synthesis has been previously unraveled. However, much remains to be discovered regarding the regulation of the respiratory N2O reductase (N2OR), the key enzyme that mitigates N2O emissions. In this work, we have demonstrated that nosRZDYFLX genes constitute an operon which is transcribed from a major promoter located upstream of the nosR gene. Low oxygen was shown to be the main inducer of expression of nosRZDYFLX genes and N2OR activity, FixK2 being the regulatory protein involved in such control. Further, by using an in vitro transcription assay with purified FixK2 protein and B. diazoefficiens RNA polymerase we were able to show that the nosRZDYFLX genes are direct targets of FixK2.
Introduction
Nitrous oxide (N2O) is a powerful greenhouse gas (GHG) and a major cause of ozone layer depletion with an atmospheric lifetime of 114 years and, based on its radiative capacity, an estimated 300-fold greater potential for global warming compared with that of carbon dioxide (CO2). Hence, N2O accounts for approximately 10% of total emissions with respect to the impact of each individual GHGs on global warming (Intergovernmental Panel on Climate Change [IPCC], 2014). Due to its environmental impact, a better understanding of the pathways implicated in the generation and consumption of N2O has received great interest (Thomson et al., 2012).
Despite the existence of multiple pathways for N2O generation in soils such as nitrifier denitrification, nitrite oxidation, heterotrophic denitrification, ammonia oxidation, anaerobic ammonium oxidation (anammox) and dissimilatory nitrate reduction to ammonium (DNRA), it is generally assumed that nitrification and denitrification are the principal processes that contribute to the emissions of N2O from terrestrial ecosystems (for a review see Stein, 2011; Schreiber et al., 2012; Butterbach-Bahl et al., 2014). Denitrification is widespread within the domain of Bacteria being dominant within Proteobacteria (Shapleigh, 2006). However, it has been evinced that some archaea (Treusch et al., 2005) and fungi (Takaya, 2002; Prendergast-Miller et al., 2011) may also denitrify. Most of the studies about denitrification have been focused on Gram-negative bacteria that occupy terrestrial niches, using the alpha-proteobacterium Paracoccus (Pa.) denitrificans as well as the gamma-proteobacteria Pseudomonas (Ps.) stutzeri and Ps. aeruginosa as model organisms (Zumft, 1997). The reactions of denitrification are catalyzed by periplasmic (Nap) or membrane-bound (Nar) nitrate reductase, nitrite reductases (NirK/NirS), nitric oxide (NO) reductases (cNor, qNor, or CuANor) and nitrous oxide reductase (N2OR) encoded by nap/nar, nirK/nirS, nor, and nos genes, respectively. The physiological, biochemical and molecular aspects of denitrification have been covered by a collection of reviews published elsewhere (Zumft, 1997; van Spanning et al., 2005, 2007; Kraft et al., 2011; Richardson, 2011; Bueno et al., 2012).
In contrast to the numerous sources of N2O, nitrous oxide reductase (NosZ) is the only known biological enzyme involved in its removal by reduction to N2 (reviewed by Thomson et al., 2012). A new cluster of atypical nosZ genes, designated clade II, have been recently identified (Sanford et al., 2012; Jones et al., 2013) which are also present in genomes lacking the nirS and/or nirK gene. This suggests that non-denitrifiers also contribute to N2O removal (Jones et al., 2013).
Nitrous oxide reductase is a homodimer with molecular weight of 120-160 kDa, a copper content of ∼12 Cu atoms, and a sulfide content of ∼2 S2- ions per dimer (Rasmussen et al., 2000). The enzyme contains two copper sites: CuA, and CuZ, a tetranuclear μ4-sulfide-bridged cluster liganded by seven histidine residues, which has been proposed to be the active center for N2O reduction. The expression, maturation, and maintenance of the NosZ catalytic subunit require several other auxiliary proteins (Zumft, 2005) being all encoded together by a typical gene cluster that contains six genes (nosRZDFYL). This core cluster is, in some cases, associated with an additional gene, nosX (reviewed by Zumft and Kroneck, 2007). Mutation analyses demonstrated that NosDFY or NosL are involved in the maturation of the NosZ CuZ, but not in the biogenesis of the CuA site (reviewed by Zumft and Kroneck, 2007; van Spanning, 2011). NosR and NosX do not participate in CuZ biogenesis but do play a role in N2O reduction in vivo altering the state of the CuZ site during turnover and supporting the catalytic activity of NosZ (Wunsch and Zumft, 2005). NosR, apart from its putative role as electron donor to NosZ, might also act as a regulator, since it is needed for Ps. stutzeri nosZ and nosD transcription (Honisch and Zumft, 2003).
Low O2 conditions and NO have been suggested as the main signal molecules for induction of nos genes expression (reviewed by Zumft and Kroneck, 2007). Both signals are perceived and transduced via transcriptional regulators belonging to the cyclic AMP receptor protein (CRP)/fumarate and nitrate reductase (FNR) superfamily. This family carries diverse mnemonics, such as ANR, DNR, NNR, NnrR, FNR or FixK but all refer to the same type of regulatory protein with similar domain structure. Proteins that form part of the DNR clade such as DNR/DnrD/NNR from Ps. aeruginosa, Ps. stutzeri, and Pa. denitrificans, respectively (van Spanning et al., 1999; Vollack and Zumft, 2001; Zumft and Kroneck, 2007; Arai et al., 2013), control nos genes expression in response to NO, while low oxygen is perceived by [4Fe-4S]2+ cluster-containing FNR- and FnrP-type proteins such as Pa. denitrificans FnrP (Bergaust et al., 2012) or Ps. aeruginosa ANR (Trunk et al., 2010).
Bradyrhizobium diazoefficiens (Delamuta et al., 2013; formerly B. japonicum), the endosymbiont of soybeans, possesses the ability to denitrify under both free-living and symbiotic lifestyles. In B. diazoefficiens the denitrification process depends on the napEDABC, nirK, norCBQD, and nosRZDYFLX genes, coding for Nap, copper-containing NirK, c-type Nor and the N2OR, respectively (Velasco et al., 2001, 2004; Mesa et al., 2002; Delgado et al., 2003; Bedmar et al., 2005).
Expression of B. diazoefficiens denitrification genes required low oxygen tension and in the case of norCBQD genes the presence of NO is also needed (Bueno et al., 2017). In this bacterium, perception and transduction of the ‘low-oxygen’ signal are mediated by a complex network comprising two interconnected regulatory cascades, the FixLJ–FixK2–NnrR and the RegSR–NifA (Sciotti et al., 2003). In the latter cascade, an oxygen concentration at or below 0.5% is required for activation of the oxygen-sensitive NifA protein and subsequent induction of essential nitrogen fixation genes (Sciotti et al., 2003). Under anoxic conditions in the presence of NO3-, NifA is also necessary for the maximal expression of napE-lacZ, nirK-lacZ, and norC-lacZ fusions (Bueno et al., 2010). Moreover, global transcription analyses of a regR mutant in comparison to the wild-type (WT), both grown in anoxic denitrifying conditions showed that RegR is also involved in the regulation of B. diazoefficiens norCBQD and nosRZDYFLX genes (Torres et al., 2014).
In contrast as reported for the RegSR-NifA cascade, activation of expression of the FixLJ-FixK2-NnrR-dependent targets requires a moderate decrease in the oxygen concentration in the gas phase (≤5%), where the haem-based sensory kinase FixL senses the ‘low-oxygen’ signal, phosphorylates itself and transfers the phosphoryl group to the FixJ response regulator. Then, FixJ activates transcription of the fixK2 gene, encoding the FixK2 protein, a CRP/FNR-like transcriptional regulator. FixK2 induces, in turn, expression of the napEDABC, nirK, and norCBQD denitrification genes involved in N2O production (Velasco et al., 2001; Mesa et al., 2002; Robles et al., 2006) as well as other regulatory genes [e.g., rpoN1, fixK1, and nnrR; (Nellen-Anthamatten et al., 1998; Mesa et al., 2003, 2008)]. The latter, the CRP/FNR-type NnrR protein adds an additional control level to the FixLJ-FixK2 cascade integrating the NOx signal necessary for induction of norCBQD genes expression (Mesa et al., 2003; Bueno et al., 2017). Within the CRP/FNR family, FixK2 belongs to the FixK subgroup, whose members, in contrast to the O2-sensitive proteins Ps. aeruginosa ANR and Pa. denitrificans FnrP, lack the cysteine motif required to bind an [4Fe-4S]2+ cluster (reviewed in Korner et al., 2003; Mesa et al., 2006). Particularly, FixK2 activity is subjected to posttranslational control by oxidation of its singular cysteine residue at position 183 (Mesa et al., 2009). B. diazoefficiens NnrR forms part of the NnrR clade, proteins that cover a similar function to the one defined for DNR-type proteins on the control of denitrification genes expression in response to NO (Bueno et al., 2017). Recently, we observed that B. diazoefficiens napEDABC, nirK, and norCBQD promoters exhibited differences with regard to their dependence on low oxygen (microoxia), NOx, and the regulatory proteins FixK2 and NnrR. While microoxic conditions were sufficient to induce expression of napEDABC and nirK genes and this control directly depends on FixK2, norCBQD genes expression depends on NO, NnrR being the candidate that directly interacts with norCBQD promoter (Bueno et al., 2017).
As described for other CRP/FNR members, FixK2 acts as a dimeric form which binds to a twofold symmetric DNA sequence present at distinct distances within the promoter region of regulated genes (Browning and Busby, 2004). Specifically, the FixK2 box corresponds to TTG(A/C)-N6-(T/G)CAA (Bonnet et al., 2013), which matches reasonably well with the previously described consensus binding site for FixK-type proteins (TTGA-N6-TCAA) (Fischer, 1994; Dufour et al., 2010).
While substantial progress has been made on the external signals (microxia and NO) and the manner by which the FixK2 and NnrR proteins control the expression of B. diazoefficiens napEDABC, nirK, and norCBQD genes involved in N2O synthesis, the regulation of nosRZDYFLX genes involved in N2O reduction to N2, the key step to N2O mitigation, has been very poorly explored in this bacterium. In the present work, we show the transcriptional arrangement of the nosRZDYFLX genes in B. diazoefficiens. We also expanded the knowledge on nosRZDYFLX regulation by studying the involvement of low oxygen, and NOx in nos expression as well as the role of FixK2 and NnrR regulatory proteins in this control. By using in vitro transcription (IVT) activation assays we demonstrated, for first time, that the nosRZDYFLX genes are direct targets of FixK2.
Materials and Methods
Bacterial Strains, Media, and Growth Conditions
Bacterial strains used in this work are compiled in Table 1. Escherichia coli cells were cultivated in Luria Bertani medium (Miller, 1972) at 37°C. When needed, antibiotics were used at the following concentrations (in μg/ml): ampicillin, 200; kanamycin, 30; spectinomycin, 25; streptomycin, 25; tetracycline, 10.
TABLE 1. Bacterial strains and plasmids used in this study.
Bradyrhizobium diazoefficiens cells were cultured oxically and microoxically basically as described earlier (Bueno et al., 2017). While Peptone-Salts-Yeast extract (PSY) medium (Regensburger and Hennecke, 1983; Mesa et al., 2008) was employed in routine oxic cultures, Yeast Extract-Mannitol (YEM) medium (Daniel and Appleby, 1972) was used as standard medium in our experiments. After growth under oxic conditions in PSY medium, cells were collected by centrifugation (8.000 g for 10 min at 4°C), and washed twice with YEM medium. Next, washed cells were used to inoculate, at a 600 nm optical density (OD600) of 0.2, 17 ml or 500 ml rubber stoppered tubes or Erlenmeyer flasks containing 3 ml or 150 ml of YEM medium amended or not with 10 mM KNO3, respectively. Next, cells were incubated for 24 h under low oxygen conditions, either at initial 0.5% O2 or at 2% O2 (in this case the headspace was exchanged every 8–16 h). The latter conditions were chosen to study the specific control of the FixK2 and NnrR regulatory proteins. To analyze the effect of the different NOx, microoxically incubated cells were subsequently exposed for 5 h to 10 mM KNO3, 500 μM NaNO2, 50 μM NO (from a saturated NO solution [1.91 mM at 20°C]), and 0.15% (30 mM) N2O. 10 μM or 100 μM of the NO-scavenger cPTIO [2-(4-Carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide; carboxy-PTIO potassium salt; Sigma] was added from the beginning to the WT and ΔnnrR strain cultures grown microoxically (2% O2) in the presence of 10 mM KNO3 for 24 h, in order to analyze the effect of removing the excess of NO on the expression of nosR-lacZ or N2OR activity, respectively. Antibiotics were added to the B. diazoefficiens cultures at the following concentrations (μg/ml); chloramphenicol, 20; streptomycin, 200; kanamycin, 200; tetracycline, 100 (solid cultures), 25 (liquid cultures); spectinomycin, 200.
Plasmids and Bacterial Strains Construction
Plasmids used in this study are listed in Table 1. Primer sequences in this work are compiled in Supplementary Table S1. For construction of transcriptional reporter fusion plasmids, 5′ DNA fragments for the nosR (558; 132; 128 and 75 bp), nosZ (1024 bp) and nosD (875 pb) promoter regions were amplified using primers’ pair a1/PnosR.r, PnosRfull.f/PnosR.r, PnosRhalf.f/PnosR.r, PnosRno.f/PnosR.r, PnosZ.f/PnosZ.r and c1/c2, respectively (Supplementary Table S1). The PCR products were then individually ligated into the pGEM®-T vector (Promega), digested with EcoRI or EcoRI-PstI and cloned into the lacZ fusion suicide vector pSUP3535 (Mesa et al., 2003), to yield plasmids pBG0301, pBG0304, pBG0305, pBG0306, pBG0302, and pBG0303, respectively (see Table 1 for details). The correct orientation of the inserts was verified by sequencing. Plasmids pBG0301, pBG0302, pBG0303, pBG0304, pBG0305, and pBG0306 were integrated by homologous recombination into the chromosome of WT B. diazoefficiens 110spc4, yielding strains 110spc4-BG0301, 110spc4-BG0302, 110spc4-BG0303, 110spc4-BG0304, 110spc4-BG0305, 110spc4-BG0306. Plasmid pBG0301 was also integrated into the chromosome of napA (GRAP1), nirK (GRK308), fixK2 (9043), and nnrR (8678) mutants, yielding strains GRPA1-BG0301, GRK308-BG0301, 9043-BG0301, and 8678-BG0301, respectively (Table 1). Correct recombination into the chromosome of the corresponding recipient strain was checked by PCR analyses.
The plasmid used as transcription template was based on the plasmid pRJ9519 which contains a B. diazoefficiens rrn transcriptional terminator (Beck et al., 1997). The nosRZDFYLX promoter was PCR-amplified with nosR_For_Transc and nosR_Rev_Transc primers, subsequently restricted with XbaI and EcoRI, and finally cloned as a 486-bp fragment into pRJ9519, yielding plasmid pDB4020. The correct nucleotide sequence was confirmed by sequencing.
Analysis of nosRZDFYLX Genes Co-transcription by RT-PCR
End-point reverse transcription-polymerase chain reaction (RT-PCR) was performed to investigate the transcriptional architecture of nosRZDFYLX genes. First, B. diazoefficiens cells were grown under 0.5% initial O2 concentration to an OD600 of ∼0.4 in YEM medium supplemented with 10 mM KNO3. Cell harvest and isolation of total RNA were done as described previously (Hauser et al., 2007; Lindemann et al., 2007; Mesa et al., 2008). First strand cDNA synthesis was performed with the SuperScript II reverse transcriptase (Invitrogen) according to the supplier’s guidelines, using 1 μg of total RNA and primers c2 and g2 that hybridize in the complementary sequence of nosD and nosX genes. The obtained cDNA was next used for amplification of putative intergenic regions between nosR and nosX (blr0314-blr0320) using primers’ pairs labeled as b1/b2-to-g1/g2 and flanking regions using primers’ pair labeled as a1/a2 and h1/h2 (Supplementary Table S1), essentially as described by Sambrook and Russell (2001). In negative controls, reverse transcriptase was omitted in the reaction. Positive control PCR reactions were performed with B. diazoefficiens genomic DNA as template.
5′ RACE of B. diazoefficiens nosRZDFLYX Genes
The transcription start sites of nos genes were determined with the RACE (Rapid Amplification of cDNA Ends) method as described by Sambrook and Russell (2001). Cell cultivation and harvest as well as total RNA isolation were carried out as described above for the RT-PCR experiments. First strand cDNA synthesis was performed with the SuperScript II reverse transcriptase (Invitrogen) according to the supplier’s guidelines, using 0.8 μg of total RNA and primer SP1_nosR. After the reaction, dNTPs and primers were removed with the GeneJET PCR Purification Kit (Thermo Fisher Scientific) and products were eluted in 15 μl of 10 mM Tris-HCl, pH 8.5. Poli-A tails were added to 5′ end of cDNAs with the terminal deoxynucleotidyl transferase (Thermo Fisher Scientific) and final products were diluted with purified water to final volume of 1 ml. Amplification reactions were carried out with primers (dT)17-adaptor-primer, adaptor-primer and SP2_ nosR primers using the following PCR program: 95°C for 5 min; (95°C for 30 s; 48°C for 30 s; 72°C for 45 s) × 5 cycles; (95°C for 30 s; 55°C for 30 s; 72°C for 45 s) × 30 cycles; 72°C for 10 min and hold at 4°C. DNA libraries were constructed by cloning the PCR products into pGEM-T easy vector (Promega). Plasmid DNA of individual clones was purified with QIAprep Spin Miniprep Kit (Qiagen) and Sanger sequenced using SP6 as primer. Transcription start sites were identified as the first nucleotide sequenced after the poly-A sequence.
Analysis of nosRZDFYLX Gene Expression by qRT-PCR
Expression of nosR was also analyzed by qRT-PCR using an iQTM5 Optical System (Bio-Rad, Foster City, CA, United States). B. diazoefficiens WT and napA, nirK, fixK2, and nnrR mutant strains were grown in YEM medium amended with 10 mM NO3- under initial 0.5% O2 (WT, napA and nirK mutant strains) or 2% O2 (WT, fixK2 and nnrR mutant strains) for 24 h. Cell harvest, isolation of total RNA and cDNA synthesis were done as described previously (Hauser et al., 2007; Lindemann et al., 2007; Mesa et al., 2008). Primers for the PCR reactions (nosR_qRT_PCR_F/ nosR_qRT_PCR_R; Supplementary Table S1) were designed with the Clone Manager Suite 9 software to have melting temperatures between 57 and 62°C and generate PCR products of 50–100 bp. Each PCR reaction contained 9.5 μl of iQTM SYBR Green Supermix (Bio-Rad), 2 μM (final concentration) of individual primers and appropriate dilutions of different cDNA samples in a total volume of 19 μl. Reactions were run in triplicate. Melting curves were generated to verify the specificity of the amplification. Relative changes in gene expression were calculated as described by Pfaffl (2001). Expression of the 16S rrn gene was used as reference for normalization (primers 16S_qRT_For and 16S_qRT_Rev; Supplementary Table S1).
β-Galactosidase Activity Determination
β-galactosidase activity was determined by using permeabilised cells from at least three independently grown cultures assayed in triplicate essentially as previously described (Cabrera et al., 2016). Specific activities were calculated in Miller units (Miller, 1972).
N2OR Activity
B. diazoefficiens cells were incubated microoxically (2% O2) for 24 h in YEM medium supplemented or not with 10 mM NO3-. In the latter conditions, parallel replicates were also exposed to 100 μM of the NO-scavenger cPTIO. Next, cells were washed three times with YEM medium and 30 μl gaseous aliquots of 2% N2O in 98% N2 (0.15% N2O final concentration in the headspace) were injected into the rubber stoppered Erlenmeyer flasks. After 5 h of incubation at 30°C at 185 rpm, gas-liquid phase equilibration was reached and 500-μl gaseous aliquots were taken from the headspace to analyze N2O consumption by gas chromatography as described previously (Tortosa et al., 2015).
The protein concentration was estimated using the Bradford method (Bio-Rad Laboratories) with a standard curve constructed with varying bovine serum albumin (BSA) concentrations. N2OR activity was determined by using cells from at least three independently biological grown cultures.
Immunoblot Analyses
B. diazoefficiens cells incubated micooxically (2% O2) in YEM medium in the presence or absence of 10 mM NO3- for 24 h, were harvested and the soluble fraction of the cells was obtained by following the protocol previously described by Delgado et al. (2003). The resulting membrane pellet was discarded and the supernatant, containing the soluble fraction, was concentrated to about 100 μl by using AmiconR Ultra-2 centrifugal filter devices (Millipore) and stored at -20°C until their use. Protein concentration was estimated as described above.
For immunodetection of NosZ, protein samples (10 μg of the soluble fraction) were separated by 12% SDS-polyacrylamide gel electrophoresis (PAGE) as described by Laemmli (1970). Then, proteins were transferred to nylon or PVDF membranes (Millipore). The membrane was then incubated in blocking buffer [5% non-fat dry milk in TTBS buffer containing 50 mM Tris-HCl pH 7.5, 0.15 mM NaCl and 0.1% Tween 20], with overnight shaking at 4°C. Afterward, the membrane was then washed with TTBS buffer (four times for 10 min each), before being incubated in 10 ml of blocking buffer containing 1/1000 (v/v) antibody dilution (anti-NosZ of Pa. denitrificans; Felgate et al., 2012). The membrane was subsequently incubated by shaking gently for 1 h at room temperature (RT). Further, the membrane was then washed with TTBS and incubated for 1 h at RT with a 1/3500 (v/v) dilution of the secondary antibody (sheep anti-IgG: peroxidase antibody produced in donkeys; A3415 Sigma–Aldrich) in blocking buffer. Next the membrane was washed four times with TTBS before adding 500 μl of ECL Select western-blotting detection reagent (GE Healthcare, Amersham) followed by Chemiluminescent signal detection in a Chemidoc XRS (Universal Hood II, Bio-Rad). The Quantity One software (Bio-Rad) was used for image analyses.
Purification of B. diazoefficiens RNA Polymerase
Purification of the B. diazoefficiens holoenzyme was carried by using a modified protocol similar to the one described by Beck et al. (1997). 25 g (wet weight) of B. diazoefficiens 110spc4 cells grown oxically in PSY supplemented with 0.1% arabinose until late exponential phase were used for each purification batch. All purification steps were performed at 4°C. Cells were resuspended in 70 ml of TGED buffer (10 mM Tris-HCl [pH 8.0], 10% glycerol, 1 mM EDTA, 0.1 mM dithiothreitol [DTT]) containing 0.02 M NaCl and 1 mM ABSF and disrupted in a French pressure cell (three passes at 1000 psi). The crude extract was treated with polyethyleneimine to a final concentration of 0.3%. The pellet obtained after centrifugation (15 min; 27,000 × g) was washed with TGED buffer (0.2 M NaCl), and protein containing RNAP was washed three times in TGED buffer (0.8 M NaCl). In all recovery steps, the supernatant was collected and precipitated again by adding solid (NH4)2SO4 to 65% final saturation (43 g per 100 ml). The precipitate was collect by centrifugation (30 min; 27,000 × g), dissolved in 30 ml of TGED buffer (0.02 M NaCl) and, dialyzed against 1 liter of TGED buffer (0.02 M NaCl). The dialyzed sample was loaded onto an HiTrap Q FF column (GE Healthcare), from which it was eluted by a linear 0.02–1.2 M NaCl gradient. Fractions containing RNAP (as judged by standard transcription assays) were pooled and loaded onto a heparin agarose column (HiTrap Heparin HP; GE Healthcare). Equilibration and elution buffers were similar to those used in the HiTrap Q FF chromatography. Peak fractions contained the RNAP (indicated by general IVT assays performed according Beck et al., 1997) were pooled, concentrated by ultrafiltration (YM30 membrane, Amicon), and dialyzed and stored in TGED buffer (0.02 M NaCl) containing 50% glycerol at -20° or -80°. The purity of the active fractions was tested by SDS-PAGE. Protein concentrations were determined with Bio-Rad assay solution, with BSA as the standard.
IVT Activation Assay
Multiple-round in vitro transcription (IVT) assays were carried out as described previously (Beck et al., 1997; Mesa et al., 2008). Plasmid pDB4020 was used as template to study the capacity of the FixK2 protein to initiate transcription from the nosRZDFYLX promoter. Expression and purification of an oxidation-insensitive C-terminal Histidine-tagged C183S FixK2 protein variant (C183S-FixK2-His6; Bonnet et al., 2013) were carried out as described in (Mesa et al., 2005). Purified FixK2 protein was used at concentrations of 1.25 or 2.5 μM dimer.
Runoff transcripts of 286 and 180 nucleotides produced in vitro following the procedure used by Mesa et al. (2005) were used as RNA size markers. Transcripts were visualized with a PhosphorImager and signal intensities were determined with the Bio-Rad Quantity One software (Bio-Rad).
Results
Transcriptional Organization of the B. diazoefficiens nosRZDFYLX Genes
Analysis of the nosRZDFYLX sequence did not reveal any predicted transcriptional termination signals1 which is an indication that they might be transcribed as an operon. Overlapping coding regions between nosR and nosZ, as well as between nosD, F, Y, and L stop and start codons, suggest translational couplings between nosRZ and nosDFYL. However, unlike these translational couplings, there is a short intergenic region of 14 nucleotides between nosZ and nosD and 11 nucleotides between nosL and nosX.
In order to investigate the transcriptional architecture of nosRZDFYLX genes, end-point RT-PCR was performed to detect intergenic regions between each pair of correlative genes. To ensure that the amplified RT-PCR product was from the template mRNA, each RT-PCR reaction had a negative control (without reverse transcriptase) and a positive control (genomic DNA). First, total RNA was isolated from B. diazoefficiens WT cells cultured with initial 0.5% O2 concentration in the presence of NO3- and subsequently reverse transcribed to cDNA. As shown in Figure 1A, specific cDNA products were obtained for intergenic regions designed as b-to-g, but not from those labeled as “a” and “h” corresponding to flanking regions of the nosRZDFYLX genes. These findings reveal that B. diazoefficiens nosRZDFYLX genes constitute a transcriptional unit, although we cannot discard the presence of additional internal promoters.
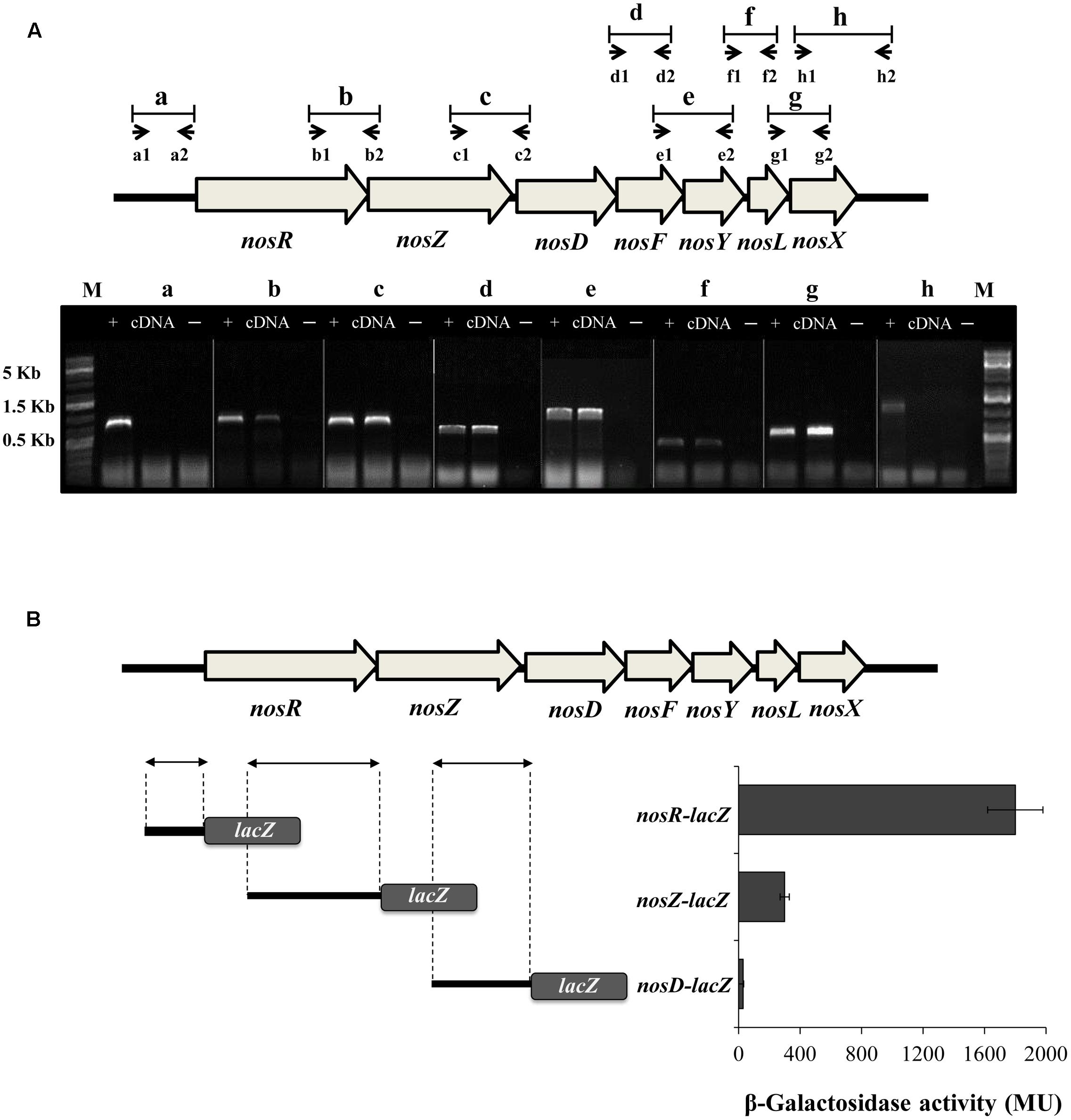
FIGURE 1. Transcriptional organization of the B. diazoefficiens nosRZDFYLX genes. (A) Putative intergenic regions probed by RT-PCR are labeled as “b-to-g” and the small arrows below depict positions and orientation of the PCR primers used for each intergenic region. Combinations of primers a1/a2 and h1/h2 were used to amplify “a” and “h” flanking regions. Primer sequences are shown in Supplementary Table S1. RT-PCR products of each primers’ pair combination were loaded on an agarose gel. Total RNA served as the template for cDNA synthesis by using gene specific primers that hybridize in the complementary sequence of nosD and nosX genes. PCR amplifications using genomic DNA as template (+) or without reverse transcriptase (–) served as positive and negative controls, respectively. The sizes of the marker (M) bands are labeled on the left side. (B) β-galactosidase activity from the DNA regions upstream of nosR, nosZ, and nosD genes fused to the lacZ reporter gene (on the right). On the left, the DNA regions fused to lacZ are depicted by arrows. In (A,B), B. diazoefficiens wild-type (WT) cells were grown for 24 h under low oxygen conditions (initial 0.5% O2) with 10 mM KNO3. Data expressed as Miller units (MU) represent mean values and error bars from triplicate samples from at least two independent cultures.
To test any potential transcription from the DNA regions upstream of the nosR, nosZ, and nosD genes, we determined β-Galactosidase activity of chromosomally integrated transcriptional fusions between the DNA regions preceding the annotated nosR, nosZ, nosD genes and the reporter gene lacZ (Figure 1B). After growing B. diazoefficiens cells under an initial O2 concentration of 0.5% O2 in the presence of NO3-, the highest transcriptional expression was driven from the nosR-lacZ fusion compared to the nosZ-lacZ and nosD-lacZ fusions (Figure 1B). These results strongly suggest that transcription of nosRZDFYLX mainly depends on a promoter present in the DNA region upstream of nosR. However, although β-galactosidase activity from the nosZ-lacZ fusion was sixfold lower to that observed from the nosR-lacZ fusion, we cannot exclude the possibility that another internal promoter upstream of nosZ might exist.
In order to map transcription initiation within the nosR promoter region, we identified their Transcriptional Start Sites (TSS) by using 5′-RACE. As shown in Figure 2A, we identify two TSS (TSS1 and TSS2) that initiate at a G and T, 84 and 57 bp upstream of the putative translational start codon, respectively. Analysis of the 5′ region of nosR revealed the presence of a purine-rich Shine-Dalgarno-like sequence (GAGG) four bases in front of the nosR putative translational start codon. Exhaustive inspection of the nosR promoter region failed to identify any putative conserved -35/-10- or -24/-12-type elements associated to σ70-dependent or σ54-dependent promoters. However, we noticed the presence of an imperfect palindromic sequence (TTGATCCAGCGCAA) positioned at 40.5 and 67.5 bp from TSS1 and TSS2, respectively (Figure 2A). This sequence resembles reasonably well the consensus sequence of the binding site for FixK-type proteins, 5′-TTGA-N6-TCAA-3′ (Fischer, 1994; Dufour et al., 2010) and specifically the consensus FixK2 binding site [TTG(A/C)-N6-(T/G)CAA] recently reported by Bonnet et al. (2013) based on the solved FixK2-DNA complex structure.
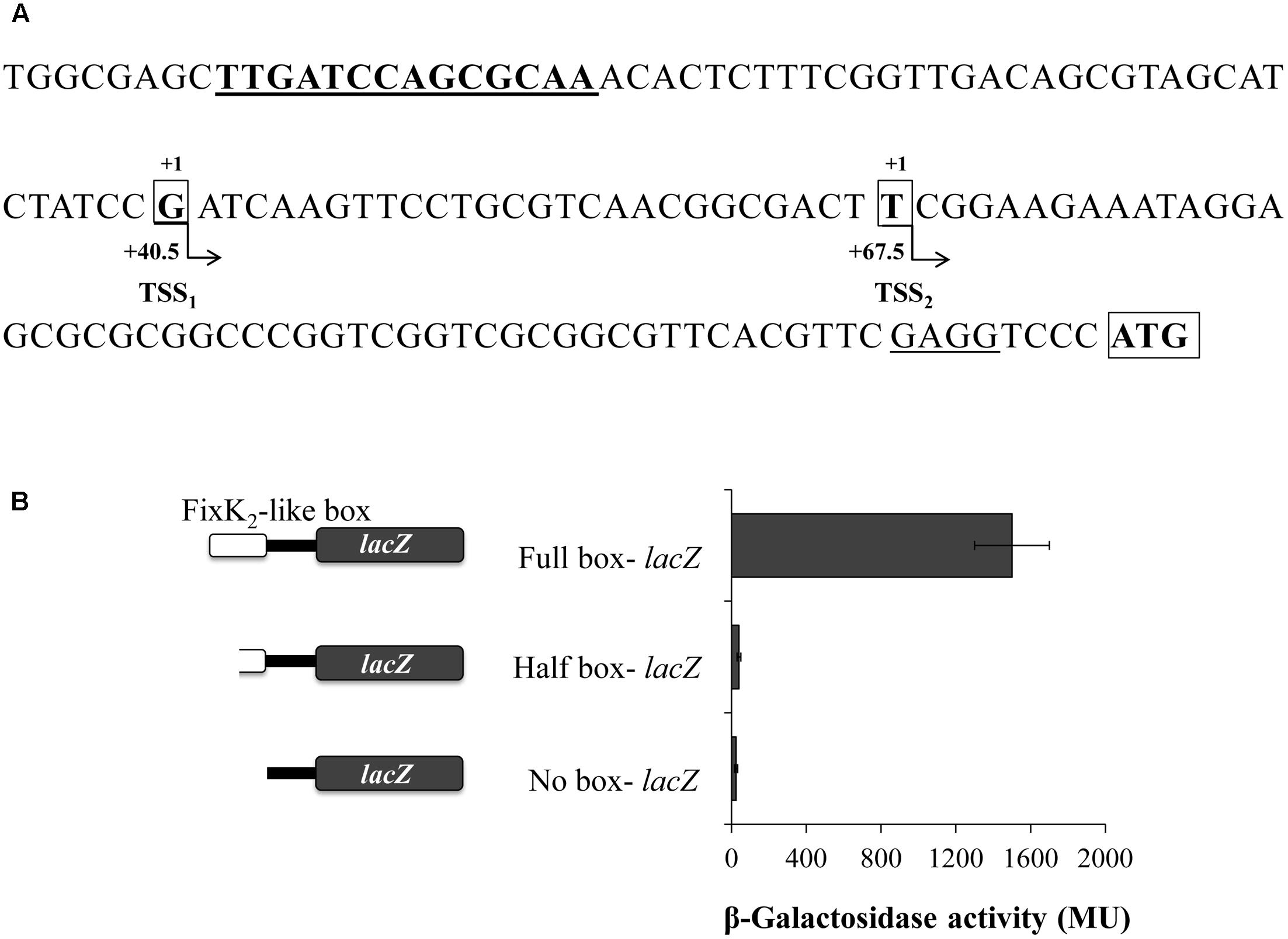
FIGURE 2. Structure of the B. diazoefficiens nosR promoter. (A) Sequence and schematic representation of the nosR promoter region. Nucleotides corresponding to the transcription start sites (TSS) located at +40.5 and +67.5 bp from the axis of symmetry of the putative FixK2-like box (bold and underlined) identified by RACE are shown in bold, marked with “+1” above and highlighted with an open black box. The annotated nosR translation start codon (ATG; http://genome.annotation.jp/RhizoBase) is shown in bold with an open black box. A putative ribosome binding site of nosR is underlined. (B) β-Galactosidase activity from nosR-lacZ fusions containing different lengths of the FixK2-like box is indicated on the left side of the figure. Cells were cultured for 24 h under low oxygen conditions (0.5% O2) with 10 mM KNO3. Data expressed as Miller units (MU) represent mean values and error bars from triplicate samples from at least two independent cultures.
In order to examine the importance of the FixK2-like box identified within the nosR promoter region in its transcription, we studied the transcriptional expression derived from a battery of nosR-lacZ fusions harboring the full or half FixK2-like box, or a deletion of this box (plasmids pBG0304, pBG0305, and pBG0306, respectively) (Figure 2B and Table 1). These plasmids were integrated into B. diazoefficiens WT and β-galactosidase activity was measured in cells cultured under initial 0.5% O2 with NO3-. In contrast to the significant induction of the nosR-lacZ transcriptional fusion containing the full FixK2-like site, expression of nosR-lacZ constructs carrying half or deleted FixK2-like site was basal, which showed the importance of the presence of this FixK2-like binding site in the induction of nosR (Figure 2B).
Low Oxygen Is the Main Signal Which Induces Expression of the nosRZDFYLX Operon
To address the effect of low oxygen and NOx in the expression of the nosRZDFYLX operon, we analyzed β-galactosidase activity of the nosR-lacZ transcriptional fusion in WT cells cultured oxically or under initial 0.5% O2, both for 24 h, and later exposed to different NOx (NO3-, NO2-, NO, or N2O) for additional 5 h-period. As shown in Figure 3A, β-galactosidase activity values were basal in cells incubated under oxic conditions. Similar basal levels were observed under oxic conditions in the presence of NO3- (data not shown). However, when cells were cultured under 0.5% O2, expression of the nosR-lacZ fusion significantly increased (about fourfold) as compared to oxic conditions (Figure 3A). The presence of NO3-, but not of NO2-, NO, or N2O, slightly increased nosR-lacZ expression (about 1.5-fold) compared to that observed in cells incubated microoxically in the absence of any NOx (Figure 3A).
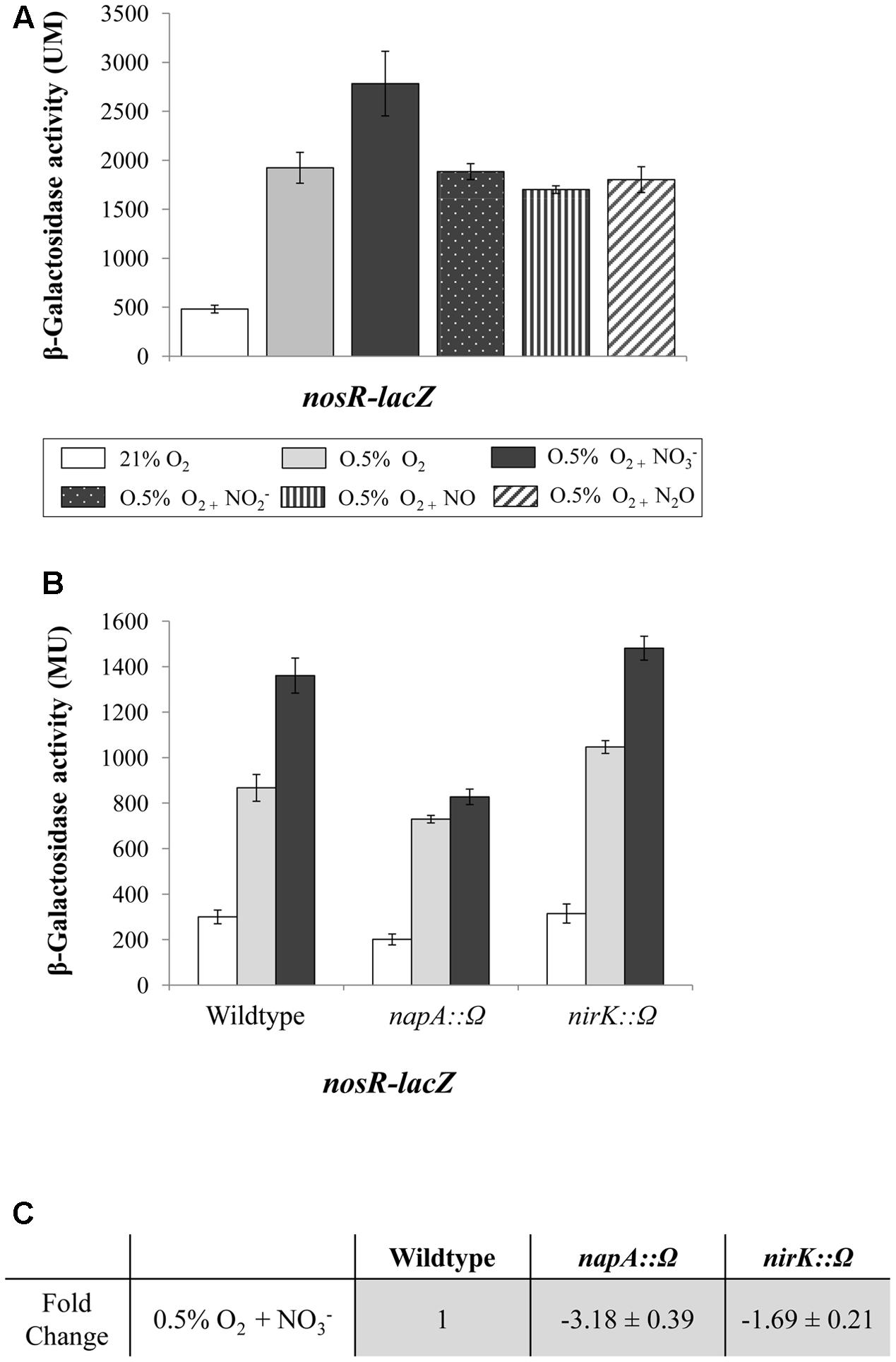
FIGURE 3. Low-oxygen is the main inducer of nosRZDFYLX expression. (A) β-Galactosidase activity derived from a nosR-lacZ fusion in B. diazoefficiens cells grown oxically or under 0.5% O2 for 24 h. Then, cells were incubated for another 5 h with or without 10 mM KNO3, 500 μM NaNO2, 50 μM NO, and 30 mM N2O. (B) β-Galactosidase activity from the nosR-lacZ fusion in the B. diazoefficiens WT, and mutant strains napA and nirK. Cells were grown oxically (white bars) or under 0.5% O2 in the absence (gray bars) or in the presence of 10 mM KNO3 (black bars) during 24 h. (C) Expression of nosR measured by qRT-PCR. After RNA isolation from cells grown under 0.5% O2 in the presence of 10 mM KNO3, qRT-PCR reactions were performed with cDNA synthesized from three independent RNA samples assayed in three parallel reactions. Fold-change values refer to differences of expression in the napA and nirK mutants relative to the WT. In (A,B) data expressed as Miller units (MU) are means with standard error bars from at least two independent cultures, assayed in triplicate.
Next, we were interested to confirm that the lack of NO3- reduction products does not affect nosR-lacZ expression. Therefore, β-galactosidase activity from the nosR-lacZ fusion was individually analyzed in napA or nirK mutant strains which are unable to reduce NO3- or NO2-, respectively (Velasco et al., 2001; Delgado et al., 2003). Again, a slight induction of the nosR-lacZ fusion in the WT cells cultured under 0.5% O2 in the presence of NO3- was observed (Figure 3B), however, no change was detected in the napA mutant cultured under the same conditions, suggesting a requirement of NO3- reduction on nosR-lacZ expression. By contrary, induction by NO3- of the nosR-lacZ fusion was retained in the nirK mutant indicating that NO2- reduction products (NO or N2O) are not required for activating the expression of nosRZDFYLX genes. These results were validated by qRT-PCR analyses (Figure 3C). Similarly as we observed by using the nosR-lacZ fusion, nosR expression was reduced in the napA mutant (3.18-fold) compared to WT cells, while it was not significantly affected in the nirK mutant (1.69-fold), all cultured in the presence of NO3-. However, we cannot conclude that the lack of NO3--mediated induction of nos genes observed in the napA mutant (Figures 3B,C) is due to the absence of NO2-, since the addition of NO2- to the medium did not increase nosR-lacZ expression (Figure 3A). Taken together, results from Figures 3A–C suggest that microoxia is the main signal that induces expression of B. diazoefficiens nosRZDFYLX genes.
Selective Regulation of nosRZDFYLX Genes by FixK2 But Not by NnrR
In B. diazoefficiens, sensing and transduction of the decrease in O2 concentration are mediated by two interlinked O2-responsive regulatory cascades, the FixLJ-FixK2-NnrR and the RegSR-NifA (Sciotti et al., 2003). A mild decrease in the O2 concentration in the gas phase (≤5%) is sufficient to activate expression of FixLJ-FixK2-dependent targets, however, a 10-fold lower O2 concentration (≤0.5%) is necessary for NifA-mediated activation. In order to investigate how FixK2 and NnrR control the microoxic expression of nosRZDFYLX genes, we analyzed β-Galactosidase activity from the nosR-lacZ fusion in the WT and ΔfixK2 and ΔnnrR strains, incubated for 24 h oxically, and microoxically (2% O2) in the absence or the presence of 10 mM of KNO3. In these experiments, 2% O2 concentration was chosen as a middle concentration between 5% (needed for FixLJ-FixK2 cascade activation) and 0.5% (required for the activation of the low O2-responsive NifA protein), in order to circumvent any possible influence by NifA regulation in our assays.
As observed in Figure 4A, microoxic induction of nosR-lacZ was completely abolished in the absence of a functional fixK2 gene, however, it was retained in the ΔnnrR strain, suggesting that microoxic expression of nosRZDFYLX genes depends on FixK2 but not on NnrR. When cells were cultured microoxically in the presence of NO3-, expression of the nosR-lacZ fusion was significantly reduced in the fixK2 mutant (about threefold) compared to that observed in the WT cells (Figure 4A). However, β-galactosidase activity of the nosR-lacZ fusion was slightly reduced in the nnrR mutant (about 1.75-fold) compared to the WT (Figure 4A). This slight reduction of the expression of the nosR gene in the nnrR mutant is probably due to the toxic effect of NO that is accumulated in nnrR cells as previously reported by Bueno et al. (2017). To check this hypothesis, a NO scavenger (cPTIO) was added during growth of WT and ΔnnrR cells under microoxic conditions with NO3-. As shown in Figure 4A, while no effect of cPTIO was observed in WT cells, nosR-lacZ expression in ΔnnrR cells increased about 40% to that observed in the absence of cPTIO (right panel), which almost corresponds to the expression pattern of the WT. Thus, this indicates that nos expression could be partially recovered in the ΔnnrR mutant when NO was sequestered by cPTIO.
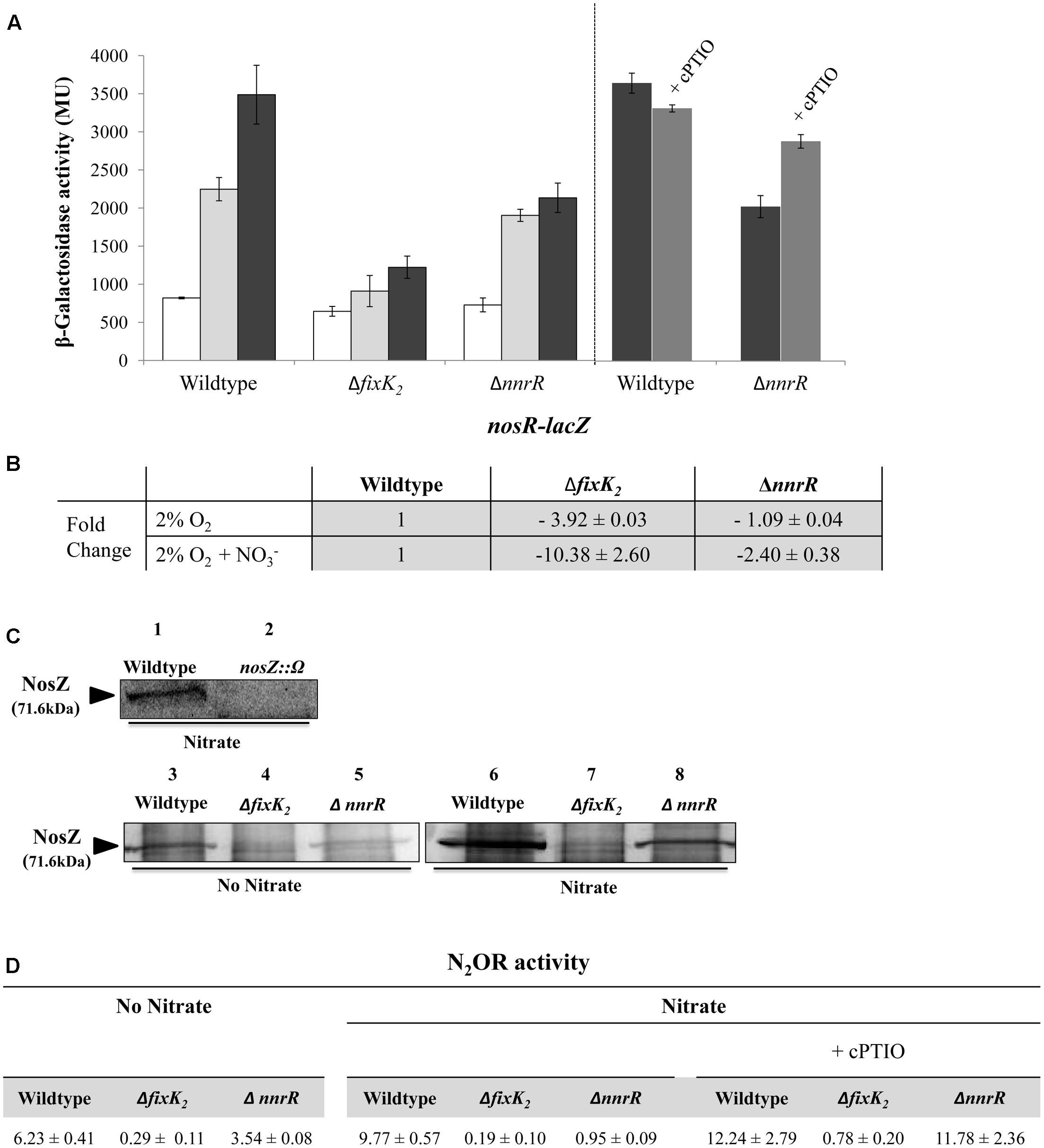
FIGURE 4. Control of nosRZDFYLX expression by the regulatory proteins FixK2 and NnrR. (A) β-Galactosidase activity expressed as Miller units (MU) from the nosR-lacZ transcriptional fusion chromosomally integrated in the B. diazoefficiens WT strain, and ΔnnrR, and ΔfixK2 strains grown oxically (white bars), under 2% O2 in the absence (light gray bars) or in the presence of 10 mM KNO3 (black bars) for 24 h. In the right panel, 10 μM of the NO-scavenger cPTIO was added to a series of cultures containing NO3- (dark gray bars). (B) Expression of nosR by qRT-PCR in the WT, and ΔnnrR, and ΔfixK2 strains. qRT-PCR reactions were performed with cDNA synthesized from three independent RNA samples assayed in triplicate. Fold-change values refer to differences of expression in the ΔnnrR, and ΔfixK2 mutants relative to the WT. (C) Western-blotted SDS-PAGE gels of the soluble fraction from the WT and ΔnnrR, and ΔfixK2 strains probed with anti-NosZ antibody from Pa. denitrificans. As control, a B. diazoefficens nosZ mutant was used. The size of B. diazoefficiens NosZ is labeled on the left side. (D) Nitrous oxide reductase (N2OR) activity in the WT and ΔnnrR, and ΔfixK2 strains expressed as nmol N2O consumed × (mg prot-1) h-1. In (B–D), cells were grown under 2% O2 in the absence or in the presence of 10 mM KNO3 during 24 h. 100 μM of cPTIO was added to some of the cultures containing NO3- in (D). In (A,B,D), data shown as means with standard errors from at least two independent cultures, assayed in triplicate.
The different control of nosR expression by FixK2 or NnrR was also confirmed by qRT-PCR analyses. When cells were cultured microoxically in the absence of NO3-, expression of nosR was reduced in the fixK2 mutant (3.92-fold) compared to that observed in the WT cells (Figure 4B), however, it was almost not affected in the nnrR mutant (Figure 4B). When NO3- was added to medium, a significant reduction of nosR expression (10.38-fold) was observed in the fixK2 mutant but only a slight decrease (2.4-fold) was detected in the nnrR mutant, both compared to the WT cultured in the same conditions (Figure 4B). Taken together, these results suggest FixK2 as the transcriptional activator of nos genes in response to microoxic conditions.
The differential dependency of nosRZDFYLX expression on FixK2 and NnrR was also confirmed at protein level by immunoblot analyses using antibodies raised against purified Pa. denitrificans NosZ (Felgate et al., 2012). Firstly, we were able to identify NosZ protein in the soluble fraction of B. diazoefficiens cells cultured under microoxic conditions (2% O2) with NO3-, since a prominent band of about 70 kDa found in the WT was readily undetectable in the nosZ mutant (Figure 4C, lanes 1 and 2). The size of this band corresponds to the predicted molecular mass of B. diazoefficiens NosZ subunit (71.6 kDa; ProtParam tool2). NosZ was already detected in the WT cells cultured microoxically (Figure 4C, lane 3) but the presence of NO3- slightly increased NosZ steady-state levels (Figure 4C, lane 6). This is in line with the observed NO3--mediated induction of the nosR-lacZ fusion (Figures 3A,B, 4A). Similarly as the expression pattern observed for the nosR-lacZ fusion, NosZ was present in the soluble fraction of ΔnnrR cells cultured microoxically either in the absence or in the presence of nitrate (Figure 4C, lanes 5 and 8), although at a slightly lower concentration than in the WT cells. As expected, the band of about 70 kDa corresponding to NosZ was absent in the soluble fractions of the ΔfixK2, independently of the presence or absence of NO3- in the incubation medium (Figure 4C, lanes 4 and 7).
Finally, we determined N2O reductase (N2OR) activity in B. diazoefficiens WT and fixK2 and nnrR mutant strains as the capacity to reduce a defined initial N2O concentration. As shown in Figure 4D, values of N2OR activity in WT cells correlated with NosZ steady-state levels in B. diazoefficiens cells (Figure 4C), where a slight induction (about 1.6-fold) of activity was observed in the WT cells in the presence of NO3- (Figure 4D) compared to that observed in exclusively microoxic conditions. In line with the expression pattern of the nosR-lacZ fusion (Figure 4A), nosR expression (Figure 4B) and NosZ detection (Figure 4C, lanes 4 and 7), N2OR activity was severely impaired in the ΔfixK2 strain cultivated microoxically independently of the presence of NO3- (Figure 4D). Under microoxic conditions, cells of the ΔnnrR strain showed a milder decrease of N2OR activity (about 1.75-fold) compared to that observed in WT cells (Figure 4D), which was significantly diminished further (about 10-fold) in the presence of NO3- (Figure 4D). As we have mentioned above, this strong decrease is probably due to the higher NO accumulation capacity of ΔnnrR cells grown microoxically with nitrate compared to WT cells grown under the same conditions (Bueno et al., 2017). In fact, when cPTIO was added during growth, ΔnnrR cells restored its ability to reduce N2O reaching WT N2OR activity values (Figure 4D). These data discard the involvement of NnrR as direct regulator of nos expression and suggest that the incapacity of ΔnnrR to reduce N2O under microoxic conditions with NO3- is probably due to the accumulation of NO. Taken together, these results pointed out that FixK2 is the key transcriptional regulator involved in nosRZDFYLX expression.
The nosRZDFYLX Operon Is a Novel Direct Target of FixK2
In order to investigate whether FixK2 could have a direct role on nosRZDFYLX activation, we monitored RNA synthesis by multiple-round IVT. The nosR promoter region was cloned into the template plasmid pRJ9519 (Beck et al., 1997), which carries an rrn terminator, yielding plasmid pDB4020. In these experiments, purified C183S-FixK2-His6 (Bonnet et al., 2013), hereafter referred as FixK2, and RNA polymerase (RNAP) holoenzyme from B. diazoefficiens that was purified in this work (see Material and Methods) were used. In the absence of FixK2, B. diazoefficiens RNAP was unable to transcribe the nosR promoter efficiently (Figure 5, lane 3), whereas it produced a vector-encoded transcript that served as an internal reference. In the presence of FixK2 (1.25 and 2.5 μM dimer), B. diazoefficiens RNAP transcribed the nosRZDFYLX promoter producing a single specific transcript larger than 286 nucleotides (Figure 5, lanes 4 and 5, respectively), which probably initiate at TSS1. This suggested that the nosR promoter is directly activated by FixK2 and that transcription from TSS1 depends on FixK2, at least, in in vitro conditions.
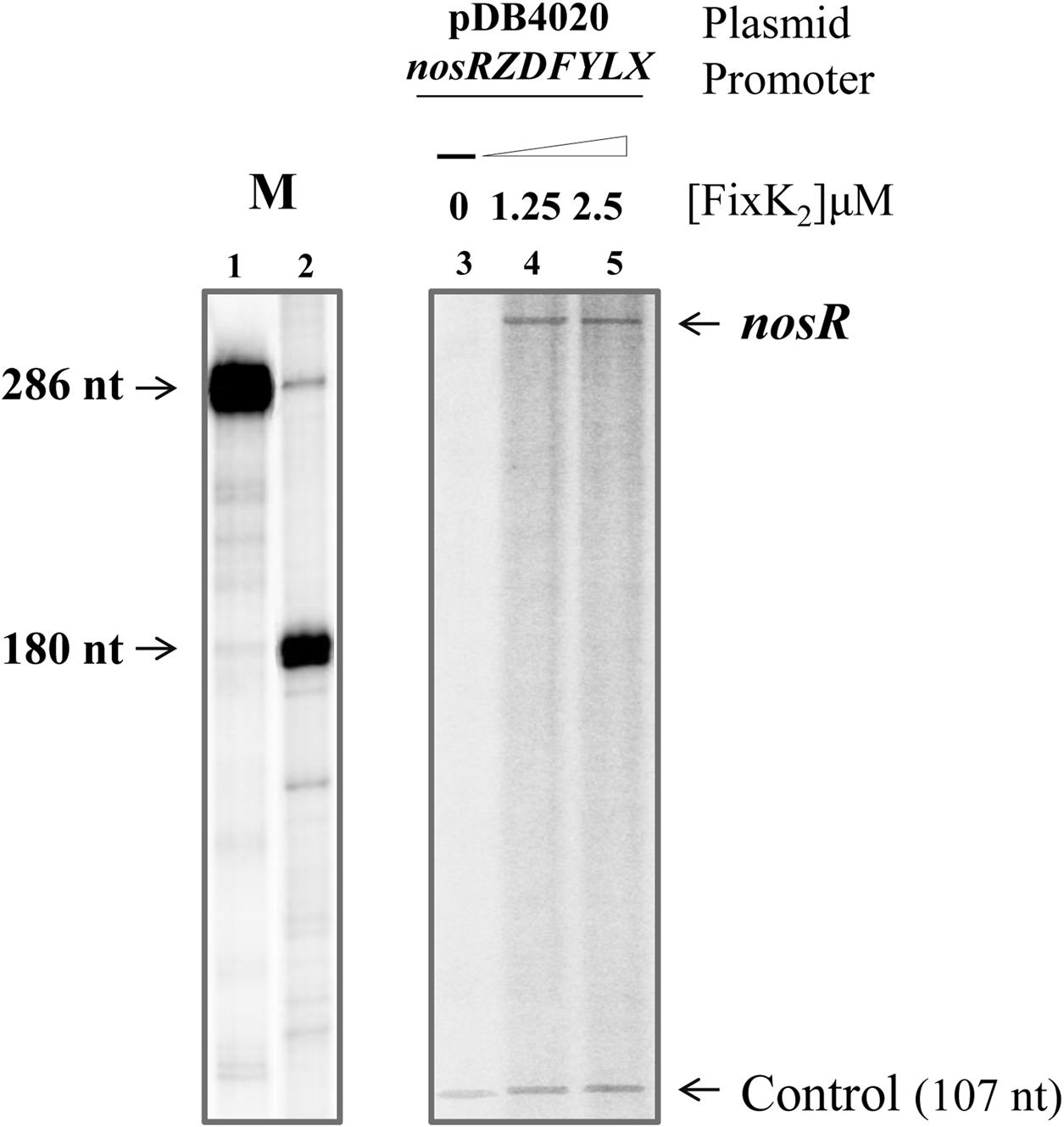
FIGURE 5. FixK2-mediated IVT activation from the nosRZDFYLX promoter. pDB4020 plasmid containing the nosR promoter cloned upstream of the rrrn terminator was used for multiple-round IVT assays with different amounts of purified FixK2 protein and RNAP from B. diazoefficiens. FixK2 concentrations were as follows: no protein, 1.25 and 2.5 μM (lanes 3–5). Transcripts synthesized in vitro in the presence of [α-32P]UTP were separated on a 6% denaturing polyacrylamide gel and visualized by phosphorimager analysis of the dried gel. Markers transcripts (M) of 286 and 180 nucleotides loaded in lanes 1 and 2 were produced as described by Mesa et al. (2005). The 107-nucleotide transcript present in all lanes originates from a promoter located on the plasmid vector and serves as internal reference. Shown are the results from a transcription experiment that was repeated at least once. Both panels correspond to the same gel. nt, nucleotides.
Discussion
Given the damaging effect on climate change of the powerful GHG N2O, strategies to mitigate their emissions have to be developed in order to increase agricultural efficiency and decrease current levels of N2O production, to satisfy the demands of continuing population growth (Richardson et al., 2009; Thomson et al., 2012). These strategies should include a better understanding of the environmental and molecular factors that contribute to the biological generation and consumption of N2O. B. diazoefficiens, the endosymbiont of soybeans, contributes to N2O emissions given its capacity to carry out the denitrification process under both free-living and symbiotic conditions. Despite the significant knowledge available in this rhizobial species on the regulation of the three first enzymes of denitrification (Nap, NirK, and cNor) involved in N2O production (Bueno et al., 2017), the regulatory mechanisms involved in the control of the key step in N2O mitigation (the reduction of N2O to N2) in response to low oxygen and NOx has not been covered in detail. Previous studies have demonstrated that expression of a nosZ-lacZ fusion depends on low O2, the presence of NO3- and the FixLJ, FixK2 and NosR regulatory proteins (Velasco et al., 2004). The capacity of B. diazoefficiens to couple N2O reduction to growth as well as a role for the NasST regulatory system on modulation of nosZ gene transcription has also been reported (Sánchez et al., 2013, 2014). Furthermore, recent studies have demonstrated the capacity of NasT to interact with B. diazoefficiens nosR 5′-leader RNA (Sánchez et al., 2017).
In this work, we have dissected, for the first time, the transcriptional organization of the nosRZDFYLX genes in B. diazoefficiens. By using RT-PCR we found that the nosRZDFYLX genes are transcribed as a single polycistronic mRNA and thus, they are organized as an operon. The transcriptional arrangement of the nos genes in other denitrifiers indicate the existence of a diversity of transcriptionally active promoters detected across the nos genes between different bacterial species (Zumft and Kroneck, 2007). Supporting our findings, the Ps. aeruginosa nos genes are arranged in a single hexacistronic nosRZDFYL operon (Arai et al., 2013). A single nosZ transcript was identified in Ps. fluorescens as well (Philippot et al., 2001). However, in Ps. stutzeri three units of monocistronic nosR and nosZ, and the nosDFYLtatE operon (Cuypers et al., 1992; Vollack and Zumft, 2001; Honisch and Zumft, 2003) have been proposed. Similarly, the transcriptional organization of the nos cluster of both Ensifer meliloti and Pa. denitrificans comprises three transcripts: nosR, nosZ, and nosDF(Y), and nosCR, nosZ, and nosDFYLX, respectively (Holloway et al., 1996; van Spanning, 2011). In order to confirm the results obtained by RT-PCR, we looked for transcriptionally active promoters within B. diazoefficiens nosRZDFYLX operon analyzing the transcriptional strength driven by the DNA regions upstream to the nosR, nosZ, and nosD genes. Interestingly, the highest transcriptional activity was derived from the DNA region upstream of the nosR gene compared to that detected from the nosZ gene, and no transcription was observed from the 5′ DNA region of the nosD gene. The presence of a transcriptionally active promoter upstream of the nosZ gene was previously demonstrated by using a nosZ-lacZ transcriptional fusion (Velasco et al., 2004) and by performing 5′-RACE (Sánchez et al., 2017). However, since a binding motif for FixK-type regulators was only present within the promoter region of nosR, we suggest that this promoter plays the major role in B. diazoefficiens nosRZDFYLX regulation.
In this work, we have identified two nosR TSS, i.e., TSS1 and TSS2, positioned at +40.5 and +67.5 bp, respectively, from the axis of symmetry of the FixK-like binding site (TTGATCCAGCGCAA). Similarly, a TSS at +40.5 from the axis of symmetry of the FixK box has been recently identified by Sánchez et al. (2017). In contrast to our results, the TSS at +67.5 bp was not identified in the latter studies. This discrepancy could be due to the different growth conditions used by Sánchez et al. (2017) where cells were cultured in HMM medium (Sameshima-Saito et al., 2006) under anoxic conditions (replacement of O2 by N2 in the gas phase). FixK2-like boxes are present within the promoters of the B. diazoefficiens napEDABC (TTGATCCAGATCAA), nirK (TTGTTGCAGCGCAA), and norCBDQD (TTGCGCCCTGACAA) genes (Velasco et al., 2001; Mesa et al., 2002; Delgado et al., 2003; Supplementary Figure S1). Interestingly, only the napEDABC-associated FixK2 box as well as the nosR-box identified in this work, matches quite well with the consensus FixK2 box, TTG(A/C)-N6-(T/G)CAA (Mesa et al., 2008, 2009; Bonnet et al., 2013; Supplementary Figure S1). Deletion of this FixK2-like box resulted in the complete shutdown of nosR-lacZ expression, indicating its essential role in the transcription of the nosRZDFYLX operon.
Cells of B. diazoefficiens grown oxically showed a basal expression of the nosR-lacZ fusion. In this regard, previous observations showed that the Ps. stutzeri nosZ gene can also be expressed at high O2 concentrations (Miyahara et al., 2010). Supporting these findings, it was recently demonstrated the capacity of both Ps. stutzeri and Pa. denitrificans to reduce N2O under oxic conditions (Desloover et al., 2014; Qu et al., 2015).
Similarly as described for napEDABC genes (Bueno et al., 2017), we found that microoxia is sufficient to induce expression of the nosR-lacZ fusion, NosZ levels as well as N2OR activity. In contrast to that observed for nosR/NosZ expression and activity, previous results reported that microoxic expression of B. diazoefficiens norCBQD genes required the presence of either NO3-, NO2-, or NO, the latter being the signal molecule involved in such control (Bueno et al., 2017). The slight induction of the nosR-lacZ fusion in WT cells cultured in the presence of NO3- was not observed in cells of a napA mutant which does not reduce NO3-. However, results from Figure 3A suggest that any of the NOx derived from NO3- reduction (NO2-, NO, or N2O) are not inducers of nosR-lacZ expression. Furthermore, NO is not required for nosR-lacZ induction, since WT levels of nosR expression were observed in a nirK mutant which does not reduce NO2- to NO. Likewise as we found in this work, previous studies suggested N2O as a weak inducer of nosZ genes in several bacteria (Kroneck et al., 1989; Richardson et al., 1991; Sabaty et al., 1999). Taken together, these observations suggest a very mild effect of NOx in the expression of nos genes. Therefore, it might be possible that a change in the cellular redox state derived from NO3- reduction by Nap is involved in nosR-lacZ induction. In fact, our own previous results demonstrated the involvement of the B. diazoefficiens redox-responsive regulatory protein RegR on the expression of nos genes (Torres et al., 2014). Alternatively, the NasST system might be involved in the NO3--mediated response of nos genes expression (Sánchez et al., 2014).
Microoxic induction of the nosRZDFYLX genes as well as NosZ expression in B. diazoefficiens depends on FixK2, but not on NnrR. The dependency of nosRZDFYLX transcription on FixK2 was demonstrated by IVT transcription experiments carried out with oxically purified protein in collaboration with B. diazoefficiens RNAP. In the same manner, microoxic induction of the B. diazoefficiens napEDABC genes depends on FixK2, but not on NnrR, probably due to its NOx-independent expression (Bueno et al., 2017). In fact, FixK2 also activates transcription of napEDABC genes (Bueno et al., 2017).
In contrast to our results, NO has been proposed as the signal that upregulates the nosR, nosZ, and nosD promoters in Ps. aeruginosa, Ps. stutzeri, and Pa. denitrificans (reviewed by Zumft and Kroneck, 2007). In Rhodobacter sphaeroides IL106 nosZ expression depends on one of the reduction products of NO3-, suggesting NO as the signal molecule, too (Sabaty et al., 1999). Further, global gene expression analysis carried out with E. meliloti showed induction of nos genes in response to NO (Meilhoc et al., 2010). NO-dependent induction of nos genes in Ps. aeruginosa, Ps. Stutzeri, or Pa. denitrificans is processed via the regulatory proteins DNR/DnrD/NNR, respectively (van Spanning et al., 1999; Vollack and Zumft, 2001; Arai et al., 2013). While Ps. aeruginosa DNR is under the control of the low O2-sensing protein ANR (Trunk et al., 2010), transcription of dnrD in Ps. stutzeri is activated in cells grown under O2 limitation conditions, being particularly strong in denitrifying cells, but not under the control of the low-O2 sensor FnrA (Vollack et al., 1999). A particular case constitutes Pa. denitrificans, where N2O reduction is subjected to a robust regulation by FnrP and NNR in response to low oxygen (via FnrP) or NO (via NNR) (Bergaust et al., 2012).
The reduced induction of nosR/NosZ expression observed in ΔnnrR cells cultured with NO3- that has also been described previously for napEDABC genes expression (Bueno et al., 2017), might be a consequence of the higher capacity to accumulate NO by the nnrR mutant strain compared to the WT strain (Bueno et al., 2017). Supporting this hypothesis, when NO was removed by adding the NO-scavenger cPTIO to the ΔnnrR cultures with NO3-, nosR-lacZ expression as well as N2OR activity restored to WT levels. It might be possible that the NosZ catalytic center Cuz which remains in a redox-inert, paramagnetic state Cuz∗ (Wunsch and Zumft, 2005), is inactivated in the presence of NO accumulated by the nnrR mutant (Dell’Acqua et al., 2011). However, the precise mechanism involved in NosZ inactivation by NO is still unknown.
This work performed with the model rhizobial denitrifier B. diazoefficiens expands the understanding of the environmental and regulatory factors involved in the reduction of N2O, the key step that mitigates N2O emissions. We hope that our results would help to establish action plans for the development of practical strategies for mitigation of N2O emissions from legume crops.
Author Contributions
MT, EB, MD, and SM conceived and designed the study. MT, EB, AJ-L, and JC performed the experiments. MT, EB, AJ-L, JC, MD, and SM analyzed the results. MT, EB, MD, and SM wrote the manuscript. EB critically revised the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by Fondo Europeo de Desarrollo Regional (FEDER)-co-financed grants (AGL2013-45087-R and AGL2015-63651-P) from the Ministerio de Economía y Competitividad (Spain). Grant P12-AGR-1968 and support from the Junta de Andalucía to Group BIO-275 are also acknowledged. MT was supported by a contract funded by Grant AGL2013-45087-R. AJ-L was financed by a Ph.D. contract associated to Grant P12-AGR-1968. EB was supported by the Consejo Superior de Investigaciones Cientificas JAE-DOC Programme co-financed by European Social Fund (ESF).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Germán Tortosa (EEZ, CSIC, Granada, Spain) for the excellent technical assistance. Juan J. Lázaro and Alfonso Lázaro (EEZ, CSIC, Granada, Spain) are acknowledged for their help in B. diazoefficiens RNAP purification. We also thank D. Richardson (UEA, Norwich, United Kingdom) for the gift of the Pa. denitrificans NosZ polyclonal antibodies. We acknowledge support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.01621/full#supplementary-material
Footnotes
References
Arai, H., Roh, J. H., Eraso, J. M., and Kaplan, S. (2013). Transcriptome response to nitrosative stress in Rhodobacter sphaeroides 2.4.1. Biosci. Biotechnol. Biochem. 77, 111–118. doi: 10.1271/bbb.120601
Beck, C., Marty, R., Klausli, S., Hennecke, H., and Gottfert, M. (1997). Dissection of the transcription machinery for housekeeping genes of Bradyrhizobium japonicum. J. Bacteriol. 179, 364–369. doi: 10.1128/jb.179.2.364-369.1997
Bedmar, E. J., Robles, E. F., and Delgado, M. J. (2005). The complete denitrification pathway of the symbiotic, nitrogen-fixing bacterium Bradyrhizobium japonicum. Biochem. Soc. Trans. 33, 141–144. doi: 10.1042/BST0330141
Bergaust, L., van Spanning, R. J., Frostegard, A., and Bakken, L. R. (2012). Expression of nitrous oxide reductase in Paracoccus denitrificans is regulated by oxygen and nitric oxide through FnrP and NNR. Microbiology 158, 826–834. doi: 10.1099/mic.0.054148-0
Bonnet, M., Kurz, M., Mesa, S., Briand, C., Hennecke, H., and Grutter, M. G. (2013). The structure of Bradyrhizobium japonicum transcription factor FixK2 unveils sites of DNA binding and oxidation. J. Biol. Chem. 288, 14238–14246. doi: 10.1074/jbc.M113.465484
Browning, D. F., and Busby, S. J. (2004). The regulation of bacterial transcription initiation. Nat. Rev. Microbiol. 2, 57–65. doi: 10.1038/nrmicro787
Bueno, E., Mesa, S., Bedmar, E. J., Richardson, D. J., and Delgado, M. J. (2012). Bacterial adaptation of respiration from oxic to microoxic and anoxic conditions: redox control. Antioxid. Redox. Signal. 16, 819–852. doi: 10.1089/ars.2011.4051
Bueno, E., Mesa, S., Sánchez, C., Bedmar, E. J., and Delgado, M. J. (2010). NifA is required for maximal expression of denitrification genes in Bradyrhizobium japonicum. Environ. Microbiol. 12, 393–400. doi: 10.1111/j.1462-2920.2009.02076.x
Bueno, E., Robles, E. F., Torres, M. J., Krell, T., Bedmar, E. J., Delgado, M. J., et al. (2017). Disparate response to microoxia and nitrogen oxides of the Bradyrhizobium japonicum napEDABC, nirK and norCBQD denitrification genes. Nitric Oxide 68, 137–149. doi: 10.1016/j.niox.2017.02.002
Butterbach-Bahl, K., Baggs, E. M., Dannenmann, M., Kiese, R., and Zechmeister-Boltenstern, S. (2014). Nitrous oxide emissions from soils: How well do we understand the processes and their controls? Philos. Trans. R. Soc. Lond. B Biol. Sci. 368:20130122. doi: 10.1098/rstb.2013.0122
Cabrera, J. J., Salas, A., Torres, M. J., Bedmar, E. J., Richardson, D. J., Gates, A. J., et al. (2016). An integrated biochemical system for nitrate assimilation and nitric oxide detoxification in Bradyrhizobium japonicum. Biochem. J. 473, 297–309. doi: 10.1042/bj20150880
Cuypers, H., Viebrock-Sambale, A., and Zumft, W. G. (1992). NosR, a membrane-bound regulatory component necessary for expression of nitrous oxide reductase in denitrifying Pseudomonas stutzeri. J. Bacteriol. 174, 5332–5339. doi: 10.1128/jb.174.16.5332-5339.1992
Daniel, R. M., and Appleby, C. A. (1972). Anaerobic-nitrate, symbiotic and aerobic growth of Rhizobium japonicum: effects on cytochrome P450, other haemoproteins, nitrate and nitrite reductases. Biochim. Biophys. Acta 275, 347–354. doi: 10.1016/0005-2728(72)90215-0
Delamuta, J. R., Ribeiro, R. A., Ormeno-Orrillo, E., Melo, I. S., Martinez-Romero, E., and Hungria, M. (2013). Polyphasic evidence supporting the reclassification of Bradyrhizobium japonicum group Ia strains as Bradyrhizobium diazoefficiens sp. nov. Int. J. Syst. Evol. Microbiol. 63, 3342–3351. doi: 10.1099/ijs.0.049130-0
Delgado, M. J., Bonnard, N., Tresierra-Ayala, A., Bedmar, E. J., and Muller, P. (2003). The Bradyrhizobium japonicum napEDABC genes encoding the periplasmic nitrate reductase are essential for nitrate respiration. Microbiology 149, 3395–3403. doi: 10.1099/mic.0.26620-0
Dell’Acqua, S., Pauleta, S. R., Moura, I., and Moura, J. J. (2011). The tetranuclear copper active site of nitrous oxide reductase: the CuZ center. J. Biol. Inorg. Chem. 16, 183–194. doi: 10.1007/s00775-011-0753-3
Desloover, J., Roobroeck, D., Heylen, K., Puig, S., Boeckx, P., Verstraete, W., et al. (2014). Pathway of nitrous oxide consumption in isolated Pseudomonas stutzeri strains under anoxic and oxic conditions. Environ. Microbiol. 16, 3143–3152. doi: 10.1111/1462-2920.12404
Dufour, Y. S., Kiley, P. J., and Donohue, T. J. (2010). Reconstruction of the core and extended regulons of global transcription factors. PLoS Genet. 6:e1001027. doi: 10.1371/journal.pgen.1001027
Felgate, H., Giannopoulos, G., Sullivan, M. J., Gates, A. J., Clarke, T. A., Baggs, E., et al. (2012). The impact of copper, nitrate and carbon status on the emission of nitrous oxide by two species of bacteria with biochemically distinct denitrification pathways. Environ. Microbiol. 14, 1788–1800. doi: 10.1111/j.1462-2920.2012.02789.x
Fischer, H. M. (1994). Genetic regulation of nitrogen fixation in rhizobia. Microbiol. Rev. 58, 352–386.
Hauser, F., Pessi, G., Friberg, M., Weber, C., Rusca, N., Lindemann, A., et al. (2007). Dissection of the Bradyrhizobium japonicum NifA+σ54 regulon, and identification of a ferredoxin gene (fdxN) for symbiotic nitrogen fixation. Mol. Genet. Genomics 278, 255–271. doi: 10.1007/s00438-007-0246-9
Holloway, P., McCormick, W., Watson, R. J., and Chan, Y. K. (1996). Identification and analysis of the dissimilatory nitrous oxide reduction genes, nosRZDFY, of Rhizobium meliloti. J. Bacteriol. 178, 1505–1514. doi: 10.1128/jb.178.6.1505-1514.1996
Honisch, U., and Zumft, W. G. (2003). Operon structure and regulation of the nos gene region of Pseudomonas stutzeri, encoding an ABC-Type ATPase for maturation of nitrous oxide reductase. J. Bacteriol. 185, 1895–1902. doi: 10.1128/JB.185.6.1895-1902.2003
Intergovernmental Panel on Climate Change [IPCC] (2014). Climate change 2014: Synthesis Report. Contribution of Working Groups I, II and III to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change. Geneva: IPCC. doi: 10.1017/CBO9781107415416
Jones, C. M., Graf, D. R., Bru, D., Philippot, L., and Hallin, S. (2013). The unaccounted yet abundant nitrous oxide-reducing microbial community: a potential nitrous oxide sink. ISME J. 7, 417–426. doi: 10.1038/ismej.2012.125
Korner, H., Sofia, H. J., and Zumft, W. G. (2003). Phylogeny of the bacterial superfamily of Crp-Fnr transcription regulators: exploiting the metabolic spectrum by controlling alternative gene programs. FEMS Microbiol. Rev. 27, 559–592. doi: 10.1016/S0168-6445(03)00066-4
Kraft, B., Strous, M., and Tegetmeyer, H. E. (2011). Microbial nitrate respiration–genes, enzymes and environmental distribution. J. Biotechnol. 155, 104–117. doi: 10.1016/j.jbiotec.2010.12.025
Kroneck, P. M., Antholine, W. A., Riester, J., and Zumft, W. G. (1989). The nature of the cupric site in nitrous oxide reductase and of CuA in cytochrome c oxidase. FEBS Lett. 248, 212–213. doi: 10.1016/0014-5793(89)80464-8
Laemmli, U. K. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685. doi: 10.1038/227680a0
Lindemann, A., Moser, A., Pessi, G., Hauser, F., Friberg, M., Hennecke, H., et al. (2007). New target genes controlled by the Bradyrhizobium japonicum two-component regulatory system RegSR. J. Bacteriol. 189, 8928–8943. doi: 10.1128/JB.01088-07
Meilhoc, E., Cam, Y., Skapski, A., and Bruand, C. (2010). The response to nitric oxide of the nitrogen-fixing symbiont Sinorhizobium meliloti. Mol. Plant Microbe Interact. 23, 748–759. doi: 10.1094/MPMI-23-6-0748
Mesa, S., Bedmar, E. J., Chanfon, A., Hennecke, H., and Fischer, H. M. (2003). Bradyrhizobium japonicum NnrR, a denitrification regulator, expands the FixLJ-FixK2 regulatory cascade. J. Bacteriol. 185, 3978–3982. doi: 10.1128/JB.185.13.3978-3982.2003
Mesa, S., Hauser, F., Friberg, M., Malaguti, E., Fischer, H. M., and Hennecke, H. (2008). Comprehensive assessment of the regulons controlled by the FixLJ-FixK2-FixK1 cascade in Bradyrhizobium japonicum. J. Bacteriol. 190, 6568–6579. doi: 10.1128/JB.00748-08
Mesa, S., Hennecke, H., and Fischer, H. M. (2006). A multitude of CRP/FNR-like transcription proteins in Bradyrhizobium japonicum. Biochem. Soc. Trans. 34, 156–159. doi: 10.1042/BST0340156
Mesa, S., Reutimann, L., Fischer, H. M., and Hennecke, H. (2009). Posttranslational control of transcription factor FixK2, a key regulator for the Bradyrhizobium japonicum-soybean symbiosis. Proc. Natl. Acad. Sci. U.S.A. 106, 21860–21865. doi: 10.1073/pnas.0908097106
Mesa, S., Ucurum, Z., Hennecke, H., and Fischer, H. M. (2005). Transcription activation in vitro by the Bradyrhizobium japonicum regulatory protein FixK2. J. Bacteriol. 187, 3329–3338. doi: 10.1128/JB.187.10.3329-3338.2005
Mesa, S., Velasco, L., Manzanera, M. E., Delgado, M. J., and Bedmar, E. J. (2002). Characterization of the norCBQD genes, encoding nitric oxide reductase, in the nitrogen fixing bacterium Bradyrhizobium japonicum. Microbiology 148, 3553–3560. doi: 10.1099/00221287-148-11-3553
Miller, J. H. (1972). Experiments in Molecular Genetics. New York, NY: Cold Spring Harbor Laboratory.
Miyahara, M., Kim, S. W., Fushinobu, S., Takaki, K., Yamada, T., Watanabe, A., et al. (2010). Potential of aerobic denitrification by Pseudomonas stutzeri TR2 to reduce nitrous oxide emissions from wastewater treatment plants. Appl. Environ. Microbiol. 76, 4619–4625. doi: 10.1128/AEM.01983-09
Nellen-Anthamatten, D., Rossi, P., Preisig, O., Kullik, I., Babst, M., Fischer, H. M., et al. (1998). Bradyrhizobium japonicum FixK2, a crucial distributor in the FixLJ-dependent regulatory cascade for control of genes inducible by low oxygen levels. J. Bacteriol. 180, 5251–5255.
Pfaffl, M. W. (2001). A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29:e45. doi: 10.1093/nar/29.9.e45
Philippot, L., Mirleau, P., Mazurier, S., Siblot, S., Hartmann, A., Lemanceau, P., et al. (2001). Characterization and transcriptional analysis of Pseudomonas fluorescens denitrifying clusters containing the nar, nir, nor and nos genes. Biochim. Biophys. Acta 1517, 436–440. doi: 10.1016/S0167-4781(00)00286-4
Prendergast-Miller, M. T., Baggs, E. M., and Johnson, D. (2011). Nitrous oxide production by the ectomycorrhizal fungi Paxillus involutus and Tylospora fibrillosa. FEMS Microbiol. Lett. 316, 31–35. doi: 10.1111/j.1574-6968.2010.02187.x
Qu, Z., Bakken, L. R., Molstad, L., Frostegard, A., and Bergaust, L. (2015). Transcriptional and metabolic regulation of denitrification in Paracoccus denitrificans allows low but significant activity of nitrous oxide reductase under oxic conditions. Environ. Microbiol. 18, 2951–2963. doi: 10.1111/1462-2920.13128
Rasmussen, T., Berks, B. C., Sanders-Loehr, J., Dooley, D. M., Zumft, W. G., and Thomson, A. J. (2000). The catalytic center in nitrous oxide reductase, CuZ, is a copper-sulfide cluster. Biochemistry 39, 12753–12756. doi: 10.1021/bi001811i
Regensburger, B., and Hennecke, H. (1983). RNA polymerase from Rhizobium japonicum. Arch. Microbiol. 135, 103–109. doi: 10.1007/BF00408017
Richardson, D., Felgate, H., Watmough, N., Thomson, A., and Baggs, E. (2009). Mitigating release of the potent greenhouse gas N2O from the nitrogen cycle - could enzymic regulation hold the key? Trends Biotechnol. 27, 388–397. doi: 10.1016/j.tibtech.2009.03.009
Richardson, D. J. (2011). “Redox complexes of the nitrogen cycle,” in Nitrogen Cycling in Bacteria, ed. J. W. B. Moir (Norfolk: Caister Academic Press), 23–39.
Richardson, D. J., Bell, L. C., McEwan, A. G., Jackson, J. B., and Ferguson, S. J. (1991). Cytochrome c2 is essential for electron transfer to nitrous oxide reductase from physiological substrates in Rhodobacter capsulatus and can act as an electron donor to the reductase in vitro. Correlation with photoinhibition studies. Eur. J. Biochem. 199, 677–683. doi: 10.1111/j.1432-1033.1991.tb16170.x
Robles, E. F., Sánchez, C., Bonnard, N., Delgado, M. J., and Bedmar, E. J. (2006). The Bradyrhizobium japonicum napEDABC genes are controlled by the FixLJ-FixK2-NnrR regulatory cascade. Biochem. Soc. Trans. 34, 108–110. doi: 10.1042/BST0340108
Sabaty, M., Schwintner, C., Cahors, S., Richaud, P., and Vermeglio, A. (1999). Nitrite and nitrous oxide reductase regulation by nitrogen oxides in Rhodobacter sphaeroides f. sp. denitrificans IL106. J. Bacteriol. 181, 6028–6032.
Sambrook, J., and Russell, D. W. (2001). Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory.
Sameshima-Saito, R., Chiba, K., Hirayama, J., Itakura, M., Mitsui, H., Eda, S., et al. (2006). Symbiotic Bradyrhizobium japonicum reduces N2O surrounding the soybean root system via nitrous oxide reductase. Appl. Environ. Microbiol. 72, 2526–2532. doi: 10.1128/AEM.72.4.2526-2532.2006
Sánchez, C., Itakura, M., Mitsui, H., and Minamisawa, K. (2013). Linked expressions of nap and nos genes in a Bradyrhizobium japonicum mutant with increased N2O reductase activity. Appl. Environ. Microbiol. 79, 4178–4180. doi: 10.1128/AEM.00703-13
Sánchez, C., Itakura, M., Okubo, T., Matsumoto, T., Yoshikawa, H., Gotoh, A., et al. (2014). The nitrate-sensing NasST system regulates nitrous oxide reductase and periplasmic nitrate reductase in Bradyrhizobium japonicum. Environ. Microbiol. 16, 3263–3274. doi: 10.1111/1462-2920.12546
Sánchez, C., Mitsui, H., and Minamisawa, K. (2017). Regulation of nitrous oxide reductase genes by NasT-mediated transcription antitermination in Bradyrhizobium diazoefficiens. Environ. Microbiol. Rep. 9, 389–396. doi: 10.1111/1758-2229.12543
Sanford, R. A., Wagner, D. D., Wu, Q., Chee-Sanford, J. C., Thomas, S. H., Cruz-Garcia, C., et al. (2012). Unexpected nondenitrifier nitrous oxide reductase gene diversity and abundance in soils. Proc. Natl. Acad. Sci. U.S.A. 109, 19709–19714. doi: 10.1073/pnas.1211238109
Schreiber, F., Wunderlin, P., Udert, K. M., and Wells, G. F. (2012). Nitric oxide and nitrous oxide turnover in natural and engineered microbial communities: biological pathways, chemical reactions, and novel technologies. Front. Microbiol. 3:372. doi: 10.3389/fmicb.2012.00372
Sciotti, M. A., Chanfon, A., Hennecke, H., and Fischer, H. M. (2003). Disparate oxygen responsiveness of two regulatory cascades that control expression of symbiotic genes in Bradyrhizobium japonicum. J. Bacteriol. 185, 5639–5642. doi: 10.1128/JB.185.18.5639-5642.2003
Shapleigh, J. P. (2006). “The denitrifying prokaryotes,” in The Prokaryotes: A Handbook on the Biology of Bacteria, eds M. Dworkin, S. Falkow, E. Rosenberg, K. H. Schleifer, and E. Stackebrandt (New York, NY: Springer Science+Business Media), 769–792.
Simon, R., Priefer, U., and Pühler, A. (1983). “Vector plasmids for in vivo and in vitro manipulation of gram-negative bacteria”, in Molecular Genetics of the Bacteria-Plant Interaction, ed A. Pühler (Heidelberg: Springer), 98–106.
Stein, L. Y. (2011). Surveying N2O-producing pathways in bacteria. Methods Enzymol. 486, 131–152. doi: 10.1016/B978-0-12-381294-0.00006-7
Takaya, N. (2002). Dissimilatory nitrate reduction metabolisms and their control in fungi. J. Biosci. Bioeng. 94, 506–510. doi: 10.1016/S1389-1723(02)80187-6
Thomson, A. J., Giannopoulos, G., Pretty, J., Baggs, E. M., and Richardson, D. J. (2012). Biological sources and sinks of nitrous oxide and strategies to mitigate emissions. Philos. Trans. R. Soc. Lond. B Biol. Sci. 367, 1157–1168. doi: 10.1098/rstb.2011.0415
Torres, M. J., Argandona, M., Vargas, C., Bedmar, E. J., Fischer, H. M., Mesa, S., et al. (2014). The global response regulator RegR controls expression of denitrification genes in Bradyrhizobium japonicum. PLoS ONE 9:e99011. doi: 10.1371/journal.pone.0099011
Tortosa, G., Hidalgo, A., Salas, A., Bedmar, E. J., Mesa, S., and Delgado, M. J. (2015). Nitrate and flooding induce N2O emissions from soybean nodules. Symbiosis 67, 125–133. doi: 10.1007/s13199-015-0341-3
Treusch, A. H., Leininger, S., Kletzin, A., Schuster, S. C., Klenk, H. P., and Schleper, C. (2005). Novel genes for nitrite reductase and Amo-related proteins indicate a role of uncultivated mesophilic crenarchaeota in nitrogen cycling. Environ. Microbiol. 7, 1985–1995. doi: 10.1111/j.1462-2920.2005.00906.x
Trunk, K., Benkert, B., Quack, N., Munch, R., Scheer, M., Garbe, J., et al. (2010). Anaerobic adaptation in Pseudomonas aeruginosa: definition of the Anr and Dnr regulons. Environ. Microbiol. 12, 1719–1733. doi: 10.1111/j.1462-2920.2010.02252.x
van Spanning, R. J. (2011). “Structure, function, regulation and evolution of the nitrite and nitrous oxide reductase: denitrification enzymes with a b-propeller fold,” in Nitrogen Cycling in Bacteria, ed. J. W. B. Moir (Norfolk: Caister Academic Press), 135–161.
van Spanning, R. J., Delgado, M. J., and Richardson, D. J. (2005). “The nitrogen cycle: denitrification and its relationship to N2 fixation,” in Nitrogen Fixation in Agriculture, Forestry, Ecology and the Environment, eds D. Werner and W. E. Newton (Dordrecht: Springer), 277–342. doi: 10.1007/1-4020-3544-6_13
van Spanning, R. J., Houben, E., Reijnders, W. N., Spiro, S., Westerhoff, H. V., and Saunders, N. (1999). Nitric oxide is a signal for NNR-mediated transcription activation in Paracoccus denitrificans. J. Bacteriol. 181, 4129–4132.
van Spanning, R. J., Richardson, D. J., and Ferguson, S. J. (2007). “Introduction to the biochemistry and molecular biology of denitrification,” in Biology of the Nitrogen Cycle, eds H. Bothe, S. J. Ferguson, and W. E. Newton (Amsterdam: Elsevier Science), 3–20.
Velasco, L., Mesa, S., Delgado, M. J., and Bedmar, E. J. (2001). Characterization of the nirK gene encoding the respiratory, Cu-containing nitrite reductase of Bradyrhizobium japonicum. Biochim. Biophys. Acta 1521, 130–134. doi: 10.1016/S0167-4781(01)00279-2
Velasco, L., Mesa, S., Xu, C. A., Delgado, M. J., and Bedmar, E. J. (2004). Molecular characterization of nosRZDFYLX genes coding for denitrifying nitrous oxide reductase of Bradyrhizobium japonicum. Antonie Van Leeuwenhoek 85, 229–235. doi: 10.1023/B:ANTO.0000020156.334242470.db
Vollack, K. U., Hartig, E., Korner, H., and Zumft, W. G. (1999). Multiple transcription factors of the FNR family in denitrifying Pseudomonas stutzeri: characterization of four fnr-like genes, regulatory responses and cognate metabolic processes. Mol. Microbiol. 31, 1681–1694. doi: 10.1046/j.1365-2958.1999.01302.x
Vollack, K. U., and Zumft, W. G. (2001). Nitric oxide signaling and transcriptional control of denitrification genes in Pseudomonas stutzeri. J. Bacteriol. 183, 2516–2526. doi: 10.1128/JB.183.8.2516-2526.2001
Wunsch, P., and Zumft, W. G. (2005). Functional domains of NosR, a novel transmembrane iron-sulfur flavoprotein necessary for nitrous oxide respiration. J. Bacteriol. 187, 1992–2001. doi: 10.1128/JB.187.6.1992-2001.2005
Zumft, W. G. (1997). Cell biology and molecular basis of denitrification. Microbiol. Mol. Biol. Rev. 61, 533–616.
Zumft, W. G. (2005). Biogenesis of the bacterial respiratory CuA, Cu-S enzyme nitrous oxide reductase. J. Mol. Microbiol. Biotechnol. 10, 154–166. doi: 10.1159/000091562
Keywords: climate change, denitrification, greenhouse gas, nitrous oxide, nitrous oxide reductase, regulation
Citation: Torres MJ, Bueno E, Jiménez-Leiva A, Cabrera JJ, Bedmar EJ, Mesa S and Delgado MJ (2017) FixK2 Is the Main Transcriptional Activator of Bradyrhizobium diazoefficiens nosRZDYFLX Genes in Response to Low Oxygen. Front. Microbiol. 8:1621. doi: 10.3389/fmicb.2017.01621
Received: 07 June 2017; Accepted: 09 August 2017;
Published: 30 August 2017.
Edited by:
Diana Elizabeth Marco, Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), ArgentinaReviewed by:
Stephen Spiro, University of Texas at Dallas, United StatesRosa María Martínez-Espinosa, University of Alicante, Spain
James Moir, University of York, United Kingdom
Copyright © 2017 Torres, Bueno, Jiménez-Leiva, Cabrera, Bedmar, Mesa and Delgado. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: María J. Delgado, bWRlbGdhZG9AZWV6LmNzaWMuZXM= Socorro Mesa, c29jb3Jyby5tZXNhQGVlei5jc2ljLmVz
†Present address: Emilio Bueno, Laboratory for Molecular Infection Medicine Sweden (MIMS), Department of Molecular Biology, Umeå University, Umeå, Sweden
‡These authors have contributed equally to this work.