 Heike M. Freese
Heike M. Freese Anika Methner
Anika Methner Jörg Overmann
Jörg Overmann- Leibniz-Institut DSMZ-Deutsche Sammlung von Mikroorganismen und Zellkulturen, Braunschweig, Germany
The marine Roseobacter group encompasses numerous species which occupy a large variety of ecological niches. However, members of the genus Phaeobacter are specifically adapted to a surface-associated lifestyle and have so far been found nearly exclusively in disjunct, man-made environments including shellfish and fish aquacultures, as well as harbors. Therefore, the possible natural habitats, dispersal and evolution of Phaeobacter spp. have largely remained obscure. Applying a high-throughput cultivation strategy along a longitudinal Pacific transect, the present study revealed for the first time a widespread natural occurrence of Phaeobacter in the marine pelagial. These bacteria were found to be specifically associated to mesoplankton where they constitute a small but detectable proportion of the bacterial community. The 16S rRNA gene sequences of 18 isolated strains were identical to that of Phaeobacter gallaeciensis DSM26640T but sequences of internal transcribed spacer and selected genomes revealed that the strains form a distinct clade within P. gallaeciensis. The genomes of the Pacific and the aquaculture strains were highly conserved and had a fraction of the core genome of 89.6%, 80 synteny breakpoints, and differed 2.2% in their nucleotide sequences. Diversification likely occurred through neutral mutations. However, the Pacific strains exclusively contained two active Type I restriction modification systems which is commensurate with a reduced acquisition of mobile elements in the Pacific clade. The Pacific clade of P. gallaeciensis also acquired a second, homolog phosphonate transport system compared to all other P. gallaeciensis. Our data indicate that a previously unknown, distinct clade of P. gallaeciensis acquired a limited number of clade-specific genes that were relevant for its association with mesozooplankton and for colonization of the marine pelagial. The divergence of the Pacific clade most likely was driven by the adaptation to this novel ecological niche rather than by geographic isolation.
Introduction
Bacteria of the genus Phaeobacter belong to the widespread and often abundant marine Roseobacter group (Buchan et al., 2005; Wagner-Döbler and Biebl, 2006; Simon et al., 2017). They are adapted to a surface associated lifestyle and are characterized by a high versatility of metabolic pathways (Martens et al., 2006; Dickschat et al., 2010; Newton et al., 2010; Thole et al., 2012). The cells attach tightly to abiotic and biotic surfaces, outcompeting other bacteria and can even invade established epiphytic communities (Rao et al., 2005, 2010; Frank et al., 2015). Colonization is accompanied by the production of the antibiotic tropodithietic acid (TDA) which inhibits a variety of bacteria including pathogenic vibrios (Brinkhoff et al., 2004; Porsby et al., 2008; Prado et al., 2009). The inhibitory activity may also involve antibacterial peptides encoded by hybrid non-ribosomal peptide synthetase/polyketide synthases (Ruiz-Ponte et al., 1999; Martens et al., 2007). Due to their antibiotic activity, members of the genus Phaeobacter exert a beneficial effect on different fish and shellfish and may act as probiotic in aquacultures (D’Alvise et al., 2012; Kesarcodi-Watson et al., 2012; Karim et al., 2013). However, Phaeobacter cells associated with senescent algae switch from a mutualistic to a parasitic lifestyle and produce algaecides (“roseobacticide”) leading to the lysis of algal cells (Seyedsayamdost et al., 2011b).
Despite their high metabolic versatility and contrary to other representatives of the Roseobacter group (Billerbeck et al., 2016; Sonnenschein et al., 2017), Phaeobacter gallaeciensis, P. inhibens, and P. porticola have so far been detected exclusively in different aquacultures (Porsby et al., 2008; Prado et al., 2009; Balcazar et al., 2010; Xue et al., 2016) or on sessile invertebrates and abiotic surface in harbors (Gram et al., 2015; Breider et al., 2017). In addition, only a few solitary isolates from macroalgae, seaweed, and intertidal mudflats at coastal shores of Australia, France and Germany have been reported (Rao et al., 2005; Martens et al., 2006; Penesyan et al., 2009; Doghri et al., 2015).
Two strains of P. inhibens which had been isolated from an aquaculture at the Atlantic Coast of north western Spain (strain DSM 17395) and from a macroalga near Sydney, Australia (strain 2.10, DSM 24588), respectively, had identical 16S rRNA sequences, a genome sequence identity of 97%, a very high percentage of shared genes (88–93%), and a high synteny of genomes and plasmids (Thole et al., 2012). Considering that Phaeobacter spp. were mostly detected on surfaces in man-made environments, the presence of highly similar genotypes in locations as far as 18,000 km apart suggest that rapid means of dispersal exist for these bacteria. Also, populations of Phaeobacter spp. might exist in other marine environments which so far have escaped detection by established molecular or cultivation-based approaches. Accordingly, Phaeobacter may also occur associated to phytoplankton or zooplankton in the open ocean. We hypothesize that attachment to and transport by planktonic eukaryotes link the Phaeobacter populations in distant coastal environments.
In metagenomics databases from the oceans like the Global Ocean Sampling, Phaeobacter spp. are not detectable. Although, short 16S rRNA gene sequence fragments related to Phaeobacter spp. are present in the Tara Ocean 16S rRNA sequence database1, they could not be unambiguously classified, as these short read sequences are also identical to several other Roseobacter group genera like Sulfitobacter and Ruegeria which are known to be globally distributed (Sonnenschein et al., 2017). Different Phaeobacter species and related genera have very low 16S rRNA gene sequence differences. In combination with their low abundance this currently precludes a reliable detection of these bacteria by culture-independent methods suitable for large sample sets. We therefore conducted a systematic study to detect Phaeobacter in the open Pacific Ocean along a longitudinal transect from New Zealand to Alaska employing advanced high-throughput enrichment methods with improved growth media. Subsequent comparative genomics of selected Phaeobacter isolates revealed a distinct subpopulation of P. gallaeciensis with particular adaptations. These findings contribute to our understanding of the evolution and dispersal of marine surface-associated bacteria.
Materials and Methods
Sampling and Sample Preparation
Plankton samples were collected during cruise SO248 of the RV Sonne in May 2016 at 12 open water stations in the Pacific along a longitudinal transect between Auckland (New Zealand) and Dutch Harbor (Alaska) (Table 1). For sampling, a 3.2 m long Bongo net with an opening diameter of 61 cm and mesh sizes of 100 or 300 μm was employed. Vertical tows were conducted from 150 m water depth to the surface with a constant velocity of 0.2 m s-1 and horizontal hauls were done at the water surface for 45 min at a speed of 1.5–2 knots. Two ml biovolume of concentrated plankton was homogenized with a glass tissue grinder (Kimble Chase Gerresheimer, United States) and diluted with artificial sea water (ASW; Bruns et al., 2003) supplemented with 10 mM HEPES, pH 7.6. Samples from the supernatant of the plankton sample were used for parallel inoculations to investigate the bacteria which were not firmly attached to the zooplankton. A workflow scheme of the combined methods is depicted in Supplementary Figure S1.
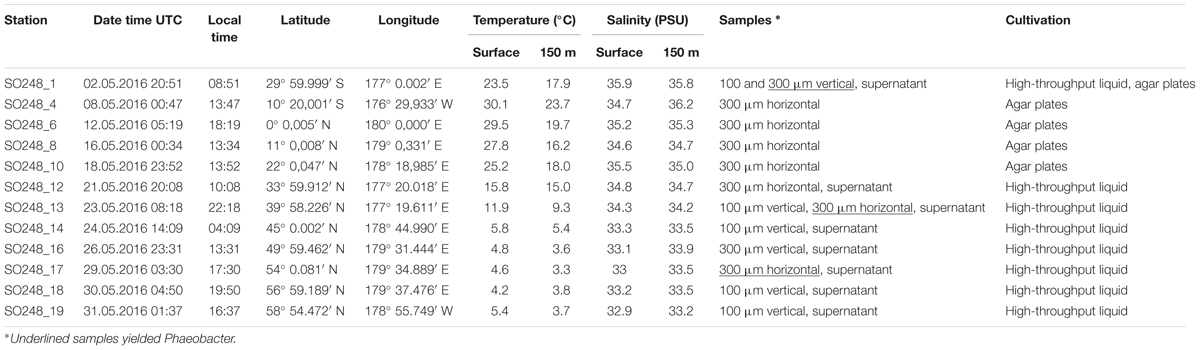
TABLE 1. Characteristics of samples and sampling depths. Temperature and salinity were recorded by a CTD system and taken from Badewien et al. (2016).
Enrichment, Isolation, and Screening Strategies
High-throughput liquid serial dilutions were performed on board in 96 deepwell plates (2 ml MASTERBLOCK®, Greiner Bio-One GmbH) with 48–128 inoculated wells per dilution (10-4 to 10-10) and sample. Two different media were successfully employed to grow Phaeobacter. Medium HD contained some complex organic substrates at low concentrations and based on ASW supplemented with 0.6 mM glucose, 0.25 g l-1 yeast extract, 0.5 g l-1 peptone, 20 mg l-1 cycloheximide, 0.2 mM ferric citrate, 1 ml l-1 vitamin solution (Karsten and Drake, 1995), and 1 ml l-1 trace element solution SL 10 (Widdel et al., 1983). Medium AM was more alkaline (pH 9) and based on ASW after Martens et al. (2006) but without silicate and with 10 mM CHES, 0.2 mM ferric citrate, 1 ml l-1 selenite-tungstate solution (Tschech and Pfennig, 1984), supplemented with 0.5 mM glucose, 0.5 mM D-alanine, and 0.2 mM Tween® 80. To test isolation success on solidified media, plankton homogenate was streaked onto Difco marine broth Bacto agar (BD) in dilutions from 10-4 to 10-8 in duplicates. Deepwell plates and agar plates were incubated at 15°C while one parallel of each dilution of solid medium was incubated at in situ temperature for at least 10 days to test for temperature effects.
To detect Phaeobacter and closely related genera, cultures were screened first on board and later in our home lab via PCR using the specific forward primer PHA-16S-129f (5′-AAC GTG CCC TTC TCT AAG G-3′; Gram et al., 2015) and the universal reverse primer 907r (5′-CCG TCA ATT CMT TTG AGT TT-3′; Lane, 1991) (details see Supplementary Material). Positive cultures were sequenced and identified according to the NCBI type material database (Federhen, 2015). Most probable numbers (MPNs) of Phaeobacter were calculated after Jarvis et al. (2010). Strains of P. gallaeciensis were isolated from positive enrichments by streaking on solid media.
Sequencing of the 16S rRNA Gene and the Internal Transcribed Spacer Region
To sequence the complete 16S rRNA gene and the downstream internal transcribed spacer (ITS) region, isolates were grown in marine broth, harvested by centrifugation and DNA was extracted by the Qiagen Blood & Tissue Kit. Amplification products were generated with primers 27f (5′-AGA GTT TGA TCM TGG CTC AG-3′; Lane, 1991) and 23S-130r (5′-GGG TTB CCC CAT TCR G-3′; Fisher and Triplett, 1999) and sequenced by Sanger sequencing. ITS sequences were aligned using ClustalW implemented in MEGA6 (Tamura et al., 2013) and manually curated. The phylogenetic tree was calculated employing the best fit option of the Maximum Likelihood method (Kimura 2-parameter model with gamma distribution, complete gap deletion) in MEGA6.
Genome Sequencing, Assembly, and Annotation
Based on the results of the phylogenetic analysis, two strains originating from distant sampling locations were chosen for genome analysis. Genomic DNA was extracted with the JETFLEX Genomic DNA Purification Kit (Genomed). SMRT sequencing was carried out on the PacBio RSII (Pacific Biosciences, Menlo Park, CA, United States) using the P6 chemistry (details see Supplementary Material). PacBio reads were assembled de novo in the SMRT Portal 2.3.0 and were corrected by paired-end Illumina reads, which were sequenced on the MiSeq (PE150), using the Burrows-Wheeler Aligner (Li and Durbin, 2009) and the CLC Genomics Workbench 7.0.1. The final assembly was circularized and adjusted to the replication system as start point2. Genome sequences were automatically annotated using Prokka 1.8 (Seemann, 2014) and were deposited in the NCBI GenBank (accession numbers: CP021040–CP021052). Information for all other Phaeobacter strains which were used for genomic comparisons are given in Supplementary Table S1.
Genome Comparisons
Polymorphic genome sites in the Phaeobacter genus were extracted from a core genome alignment using Parsnp and Gingr (Treangen et al., 2014). A phylogenetic network was calculated from the resulting matrix which contained 68,858 characters with the NeighborNet algorithm in SplitsTree 4.13.1 (Huson and Bryant, 2006). Phylogenetic distances of P. gallaeciensis were inferred from pairwise comparisons of complete genome sequences via the GGDC 2.1 web service (formula 2; Meier-Kolthoff et al., 2013). A BIONJ tree was calculated with the R package ape and rooted at midpoint (Gascuel, 1997; Paradis et al., 2004). Whole chromosome alignments of P. gallaeciensis were obtained with Mauve (Darling et al., 2010), sequence similarity between genomes estimated by BLAST (Altschul et al., 1990) and plotted together with the R package genoPlotR (Guy et al., 2010). Nucleotide diversity between Pacific and the other strains was calculated for all aligned ortholog genes along the genome with the R package PopGenome (Pfeifer et al., 2014). Prophages were identified with PHASTER (Arndt et al., 2016) and classified by Virfam (Lopes et al., 2014). Genomic islands were predicted with the IslandViewer web server (Dhillon et al., 2013). Mobile elements of the other P. gallaeciensis had been identified previously (Freese et al., submitted) and were included in the analysis. Orthologs of P. gallaeciensis were inferred via proteinortho (Lechner et al., 2011) and the core genome was defined as orthologs present in all strains. Subsequently, clade-specific orthologs were identified and their KEGG orthologies (ko identifiers) were determined using KAAS (Moriya et al., 2007) using a threshold of 30%. Methylation motifs were detected using the SMRT Portal with the RS Modification and Motif Analysis and were identified using REBASE (Roberts et al., 2015). Methylase sequence homologs for the hits were identified using BLAST.
Results
For the first time, Phaeobacter spp. were successfully enriched from the open ocean by employing our high-throughput liquid cultivation. Enrichments were obtained from very distantly located samples taken in the South Pacific (30°S), North Pacific (40°N), and in the Bering Sea (54°N) (Figure 1) where surface water temperatures differed between 23.5 and 4.6°C (Table 1). The observations that representatives of the phylogenetically related genera Pseudophaeobacter and Leisingera were also enriched and that total MPNs exceeded those of Phaeobacter by 1–2 orders of magnitude (Table 2) indicate that both types of liquid media were not highly selective for Phaeobacter. Cultivation attempts on marine broth agar failed for samples from the open Pacific (Table 1) even when liquid enrichments from the same station (SO248_1) yielded Phaeobacter. Most notably, Phaeobacter were exclusively enriched from size fractions >300 μm which consists mostly of larger zooplankton organisms. In contrast neither the supernatant nor the size fraction >100 μm yielded enrichments of Phaeobacter even at stations SO248_1 and SO248_13 where both size fractions were tested in parallel.
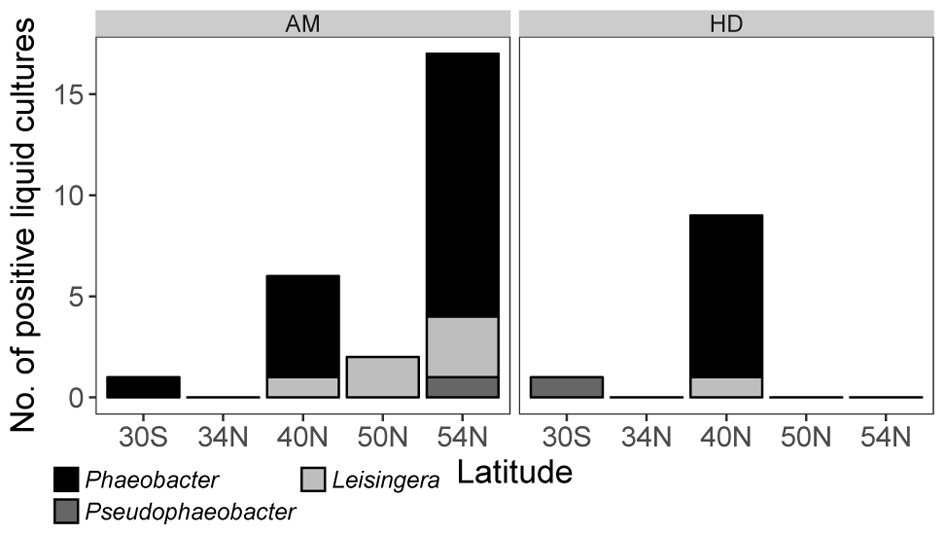
FIGURE 1. Number of positive enrichments of Phaeobacter and phylogenetically related genera obtained from zooplankton > 300 μm collected along a longitudinal transect in the Pacific (respective Latitudes of sampling sites are given) in defined medium AM or complex medium HD.
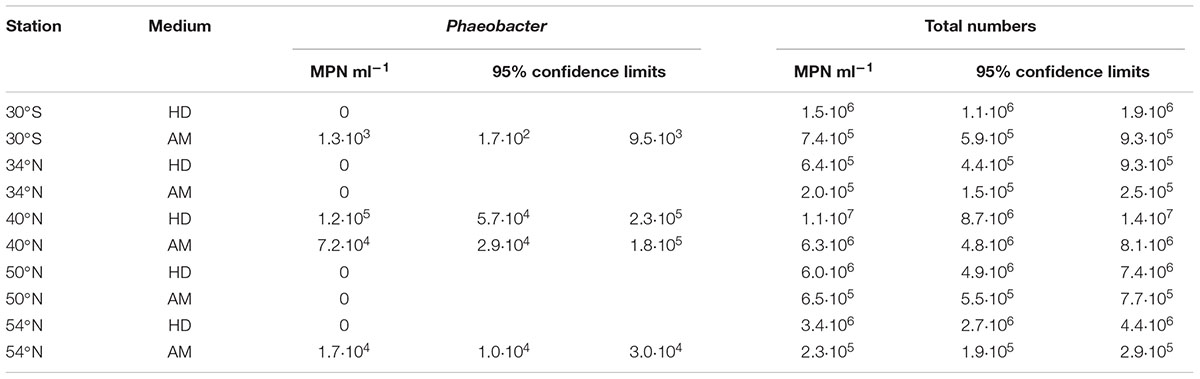
TABLE 2. Most probable numbers (MPNs) of Phaeobacter per ml plankton biovolume.
The specific MPNs calculated from the results of the liquid dilution series revealed highest Phaeobacter abundances of 7.2⋅104 to 1.2⋅105 cells (ml plankton biovolume)-1 in the North Pacific (Table 2). In the warmer Southern Pacific, the Phaeobacter abundance was 2 orders of magnitude lower since only one well of the liquid dilution series proved to be positive (Figure 1).
A total of 18 strains were isolated from the samples collected in the North Pacific and the Bering Sea. The 16S rRNA gene sequences of all isolates were identical to the sequence of the P. gallaeciensis type strain DSM 26640T and to all other isolates of P. gallaeciensis which are available to date. The ITS sequences of all Pacific strains were also identical but clustered separately from the other P. gallaeciensis sequences due to five polymorphic sites (Figure 2). Based on the results of the ITS analysis, two strains isolated from locations 1500 km apart in the North Pacific (strain P128) and the Bering Sea (strain P129) and which had been enriched in two different media were chosen for genome sequencing. Their high quality, closed genome sequences were generated and compared to the five existing genome sequences of P. gallaeciensis strains originating from aquacultures in Spain and France (Supplementary Table S1).
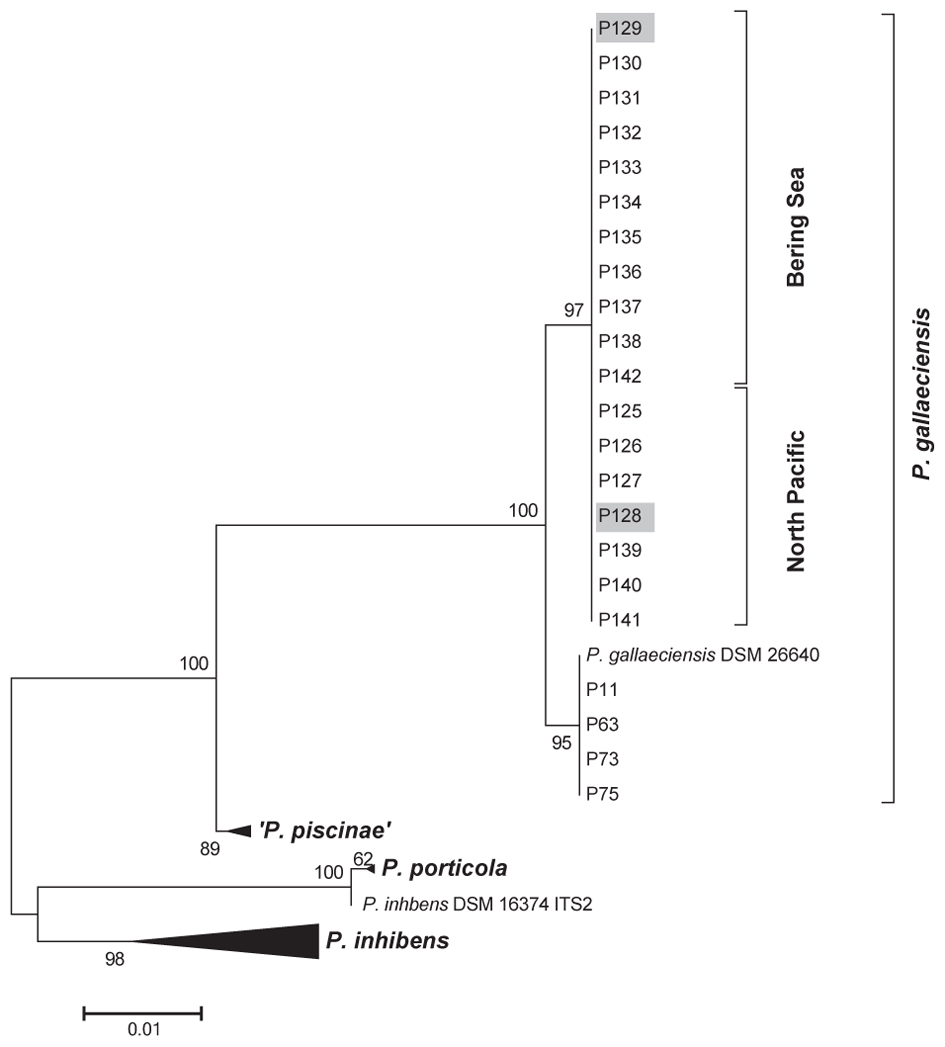
FIGURE 2. Maximum likelihood phylogenetic tree of Phaeobacter strains based on ITS sequences. For comparison, Phaeobacter strains representing different ITS lineages identified previously (Breider et al., 2017; Freese et al., submitted) were chosen. The tree based on 767 aligned nucleotide positions. Numbers adjacent to branches give percentage of support from 1000 bootstrap replicates. Strains chosen for genome sequencing are marked.
The characteristics of the P. gallaeciensis genomes are summarized in Table 3. They share a large core genome (median, 89.6%), a high nucleotide similarity (median whole genome nucleotide similarity estimated as digital DNA:DNA hybridisation, 100%) and are highly syntenous with not more than 80 breakpoints (Figure 3). A considerable number of synteny breaks (31.5%) resulted from small sequence length variations (median 36 bp) in intergenic regions and 39.7% of the breaks occurred due to differences in gene content. The two isolates from the open Pacific were nearly identical in their genome sequence and differed by just 17 SNPs. Of these SNPs, 6 were located in intergenic regions whereas 11 were located in single core orthologs (nine non-synonymous, two synonymous). The two genomes of the Pacific strains differed by only 15 genes which are located on a small, 15-kb plasmid present in P129 but not P128 (labeled f in Figure 3). This plasmid carried a replication system and also a restriction system. Among the total of seven P. gallaeciensis strains, 84,374 SNPs were detected which were nearly homogenously distributed over the whole genome (Supplementary Figure S2). The Pacific strains differed by 83,953 SNPs from the five aquaculture P. gallaeciensis strains whereas much fewer differences were detected among the latter (405 SNPs). Phylogenetic network analysis confirmed that the Pacific isolates constitute a separate clade within P. gallaeciensis with no indication of recombination between the clades (Figure 4). Between the two clades, the average nucleotide similarity of homolog blocks still amounted to 97.8%. A higher sequence divergence was only detected for the RepABC-5 plasmid which also differed in gene content (Figure 3; plasmid labeled ‘b’ or ‘x’ have only 94.7% sequence similarity).
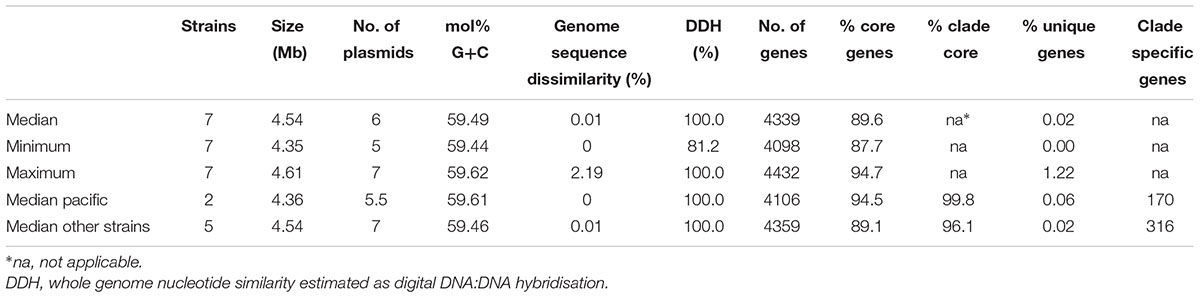
TABLE 3. Characteristics of Phaeobacter gallaeciensis genomes.
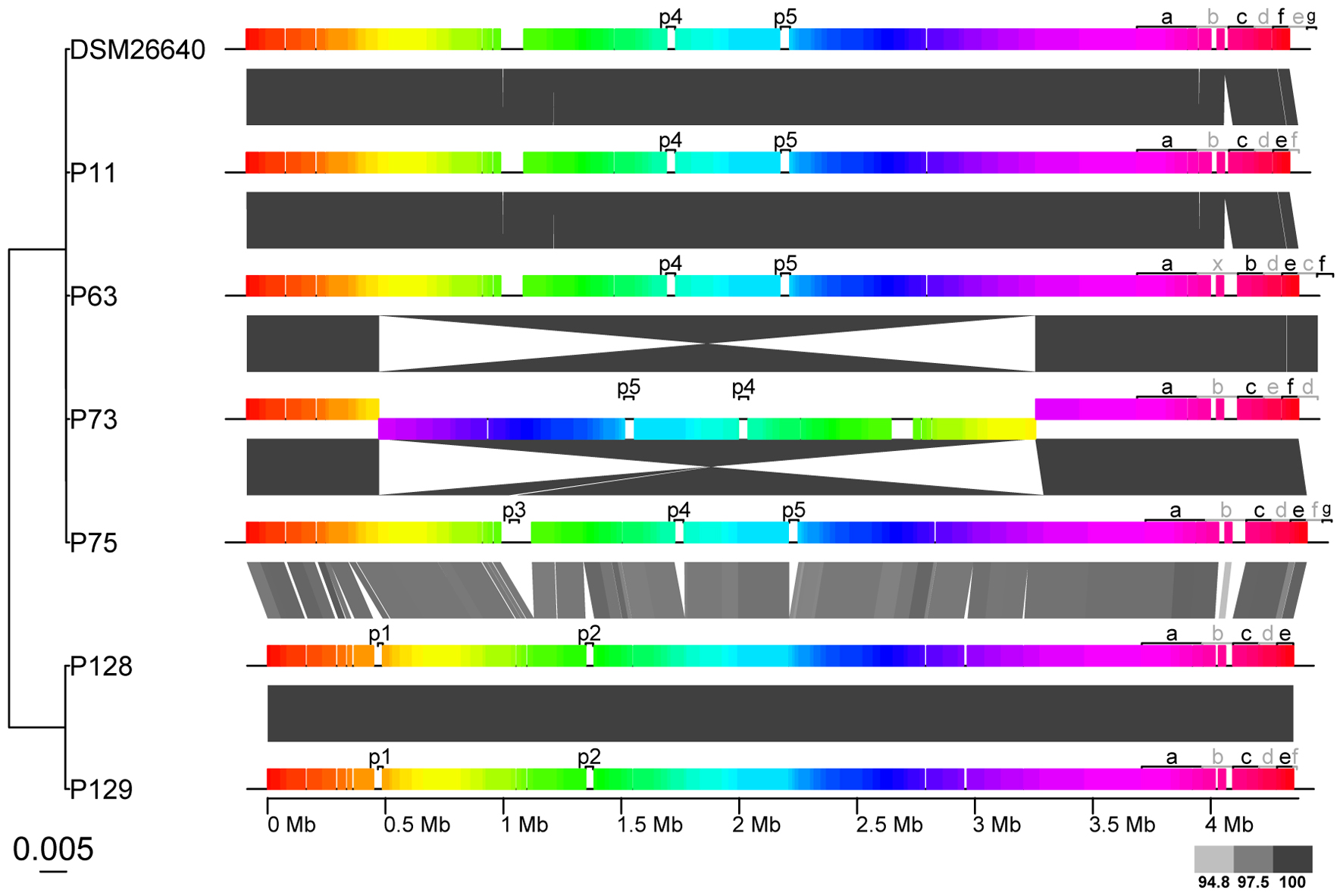
FIGURE 3. Synteny and sequence similarity of the 7 Phaeobacter gallaeciensis strains. The Mauve alignment depicts large collinear, and presumably homologous blocks as colored rectangles. Collinear blocks cover 87.2–92.4% of the whole genomes. Gray scale of wedges between the genomes indicates the level of sequence identity (%) between genomic blocks. The genome phylogeny on the left was inferred from pairwise genome to genome nucleotide sequence distances (GGDC) using BIONJ and the scale gives the genomic distance. Prophages (labeled p), and plasmids (labeled according to their names, for instance “a” corresponding to pP11_a or pP128_a) are also depicted.
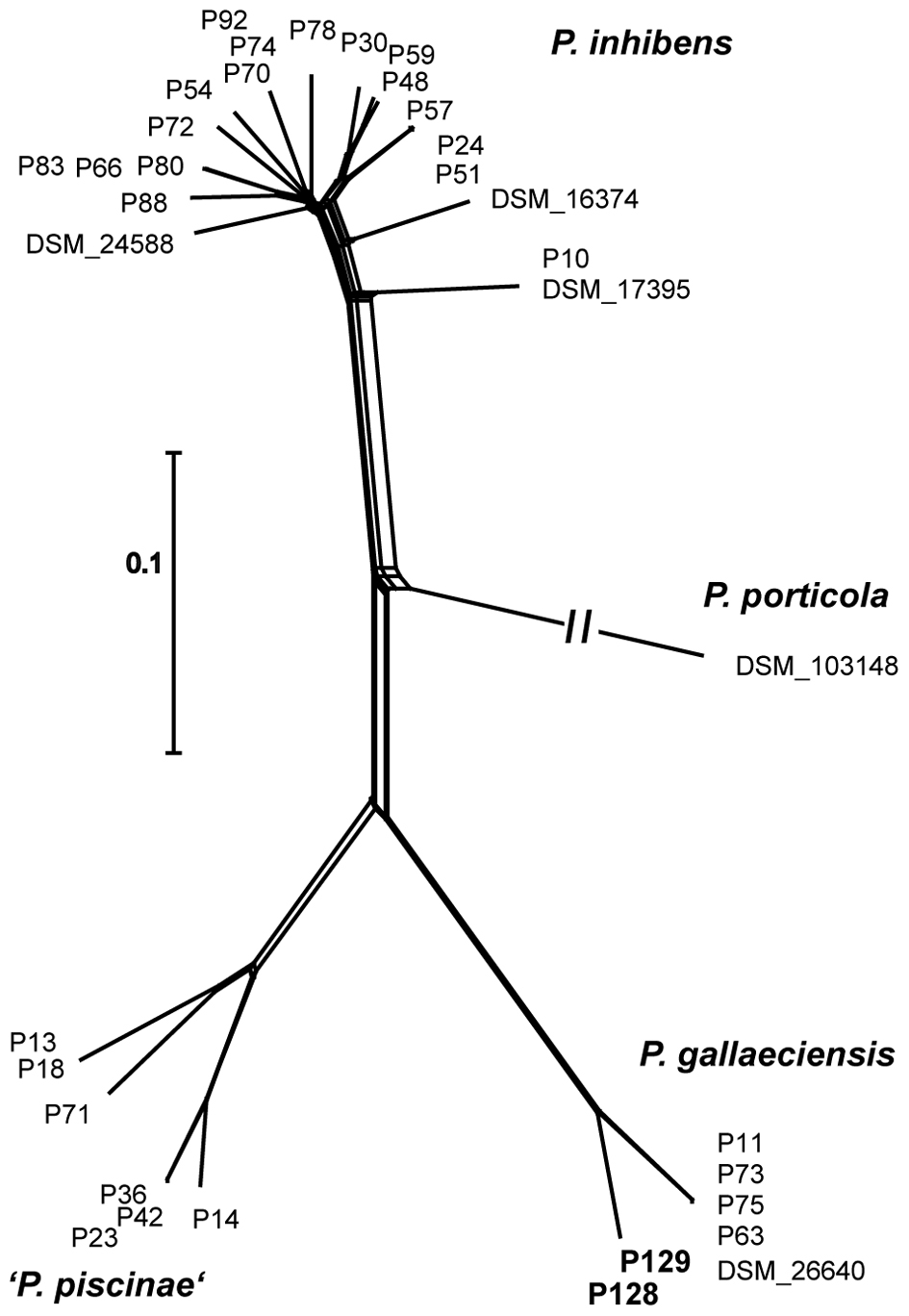
FIGURE 4. Phylogenetic network of 35 Phaeobacter strains inferred by NeighborNet analysis from 68858 core-genome SNPs estimated by Parsnp. Scale bar, 0.1 changes per polymorphic site. The long branch to P. porticola is depicted incompletely and Pacific strains are highlighted in bold.
Phaeobacter gallaeciensis strains from the Pacific contained significantly fewer mobile genetic elements than the five strains from aquacultures (t-test p < 0.002). Plasmids, prophages, and genomic islands together amounted 10.6% (464 kb) and 14.4% (653 kb) of the genomes, respectively (Figure 5). Significant differences were also apparent when only the mobile elements located in the chromosomes are considered (216 and 292 kb, t-test p < 0.001). While the overall number of prophages did not differ between the two P. gallaeciensis clades, different taxa of prophages were present. Both Pacific strains contained two Myoviridae (labeled p1 and p2; Figure 3) whereas all members of the aquaculture clade harbored a representative each of the Siphoviridae (p4) and of the Podoviridae (p5). One aquaculture strain, P75, in addition contained a member of the Myoviridae (p3), which was totally dissimilar on nucleotide and protein level to the Myoviridae detected in the Pacific P. gallaeciensis. Interestingly, this latter prophage occurred in a large 100 kb-region which contains several transferred genetic elements but is completely lacking in the Pacific clade (Figure 3). At this particular position, other Phaeobacter species also contained different mobile elements (Freese et al., submitted) which may indicate a selective loss in the Pacific clade after its divergence.
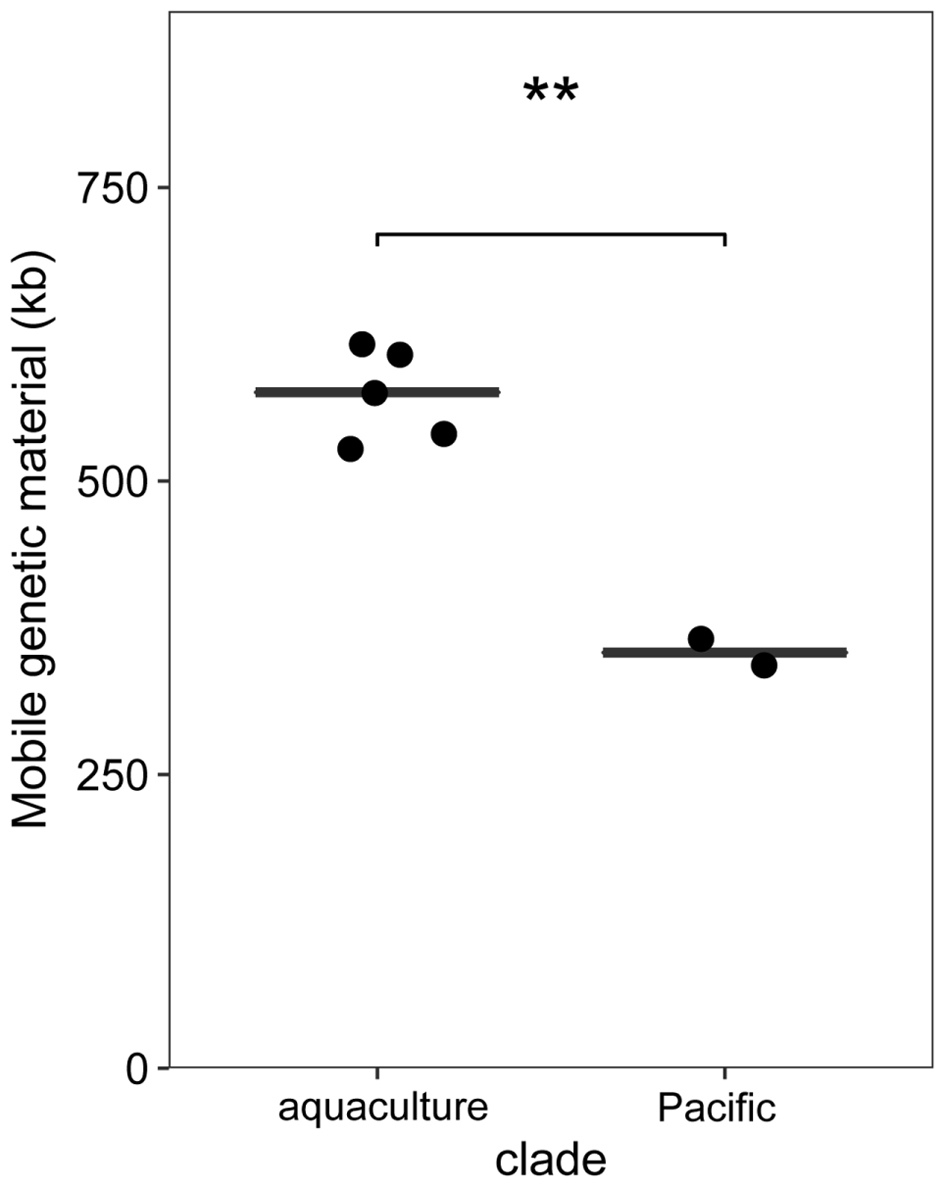
FIGURE 5. Differences in the summed sequences of mobile elements (plasmids, phages, genomic islands) between P. gallaeciensis clades. Horizontal lines show median and ∗∗ denotes t-test with p < 0.002.
All P. gallaeciensis strains contained the TDA gene cluster and genes involved in surface attachment and biofilm formation (like motility, chemotaxis, polysaccharide biosynthesis and transport, rfb genes). On the other hand, comparative analysis of the functional gene content identified 170 and 316 clade-specific orthologs in the Pacific and aquaculture strains of P. gallaeciensis, respectively (Table 3). Functional assignments were possible for a limited number of these orthologs (Table 4). In the aquaculture strains these innovative orthologs often represented functional orphans, i.e., they occur isolated in metabolic pathways like in the terpenoid metabolism, or as part of xenobiotics biodegradation or host infections. The aquaculture strains may also contain few clade-specific genes coding for alternative reactions in carbohydrate or amino acid metabolism like a glutamate dehydrogenase (K00262), which may be involved in fixation or release of ammonia.
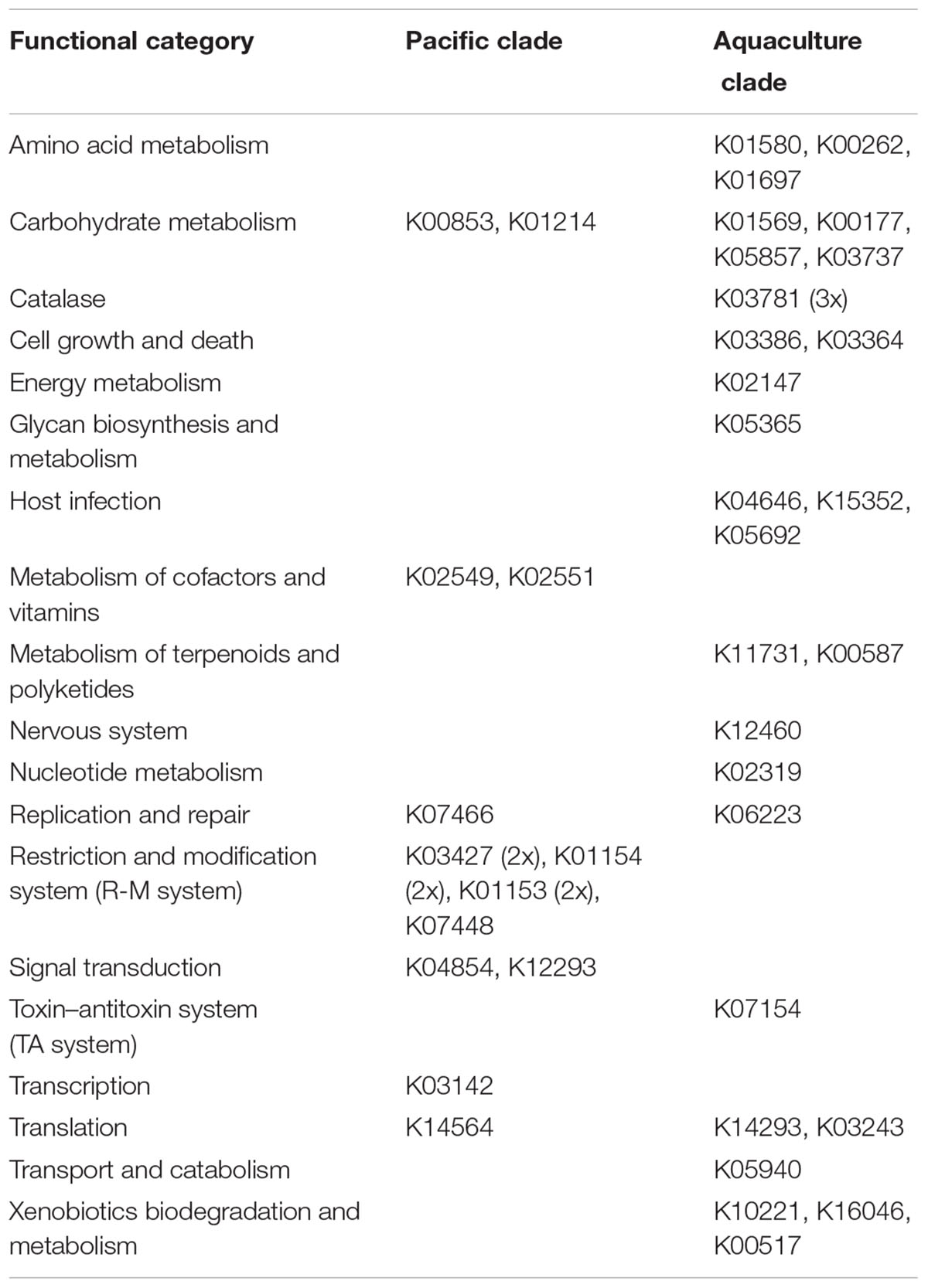
TABLE 4. Specific functional orthologs of Phaeobacter gallaeciensis clades.
In contrast to the aquaculture strains, the major innovative function of the Pacific strains is a complete Type I restriction/modification system (K03427, K01154, K01153) which occurred twice (i.e., PhaeoP128_00141 to 00144 and PhaeoP128_00349 to 00351). A detailed analysis of methylation patterns revealed two previously unknown Type I restriction motifs which occur exclusively in the Pacific clade (AGCN6GTCY and AGCAN8TTYG; Table 5). On average, 99.1% (98.2–99.7%) of these two motifs were found to be methylated in the genomes of the Pacific strains, indicating that the Type I restriction modification systems are active. All P. gallaeciensis strains encode an identical, active Type II restriction system (modification methylase BabI, ribonuclease HII). Some further methylated motifs of restrictions systems were detected but the corresponding genes for methylases could not be identified. Remarkably, the Pacific clade of P. gallaeciensis also acquired a second, homolog phosphonate transport system (PhnCDE) in addition to the phosphonate transport system/C–P lyase enzyme complex (PhnGHIJKLM) present in all other P. gallaeciensis.
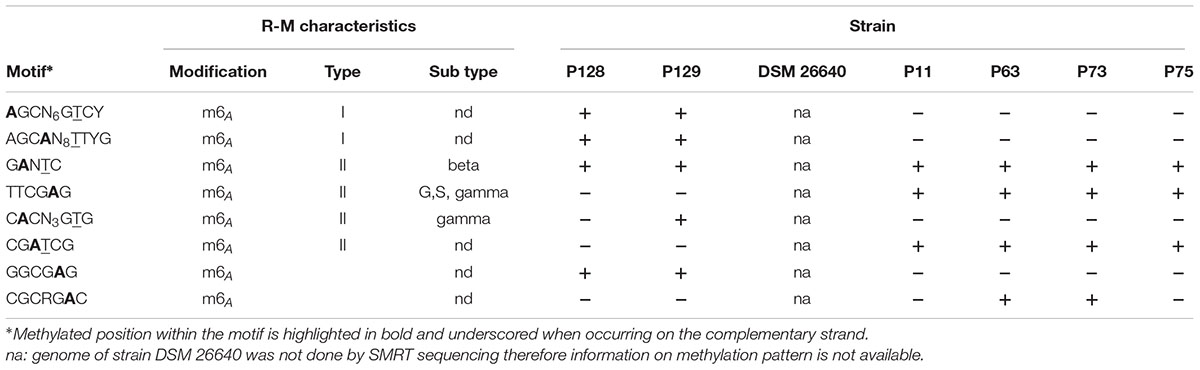
TABLE 5. Presence of restriction system motifs in Phaeobacter gallaeciensis strains.
Discussion
Phaeobacter spp. were detected in the >300 μm size plankton fraction which contained mostly mesozooplankton except for station SO248_12 at 34°N where samples were dominated by radiolarian macrocolonies. In contrast, Phaeobacter spp. was neither detected in the free-living bacterioplankton nor in the plankton fraction >100 μm. Due to the overall larger biovolume of the smaller plankton, the mesozooplankton (>300 μm) was highly diluted in the size fraction >100 μm which likely is the reason for the lack of Phaeobacter in these samples. Our results reveal that Phaeobacter occurs associated with zooplankton in the open ocean but in contrast to our initial expectations we found no evidence for their association with phytoplankton. Although Phaeobacter was previously shown to colonize macroalgae and laboratory cultures of dinoflagellates (Rao et al., 2005; Frank et al., 2015) a symbiotic interaction with algae was so far only demonstrated for the coccolithophore E. huxleyi (Seyedsayamdost et al., 2011a,b). In addition to zooplankton, Phaeobacter spp. may therefore also be specifically associated with some phytoplankton taxa which were not prevalent during the time of our cruise (e.g., Alvain et al., 2008).
Whereas, little information is available on the bacterial colonization of mesozooplankton in the open ocean, the total abundance of bacteria associated with estuarine or North Sea zooplankton was found to range between 1.2⋅108 and 3.6⋅1012 (ml zooplankton biovolume)-1 (Møller et al., 2007; Bickel and Tang, 2014). Based on these total bacterial cell numbers, the abundance of Phaeobacter spp. detected by our cultivation-based approach would amount to ≤0.1% of all zooplankton-associated bacteria. By comparison, marine vibrios constitute between 1 and 26% of the associated bacterial community (Heidelberg et al., 2002). The low abundance of Phaeobacter spp. on Pacific mesozooplankton resembles the low percentage of P. inhibens sequence reads determined in biofilms colonizing inert surface or marine animals in harbors (Gram et al., 2015) and thus may constitute a typical feature of the genus. As Phaeobacter can exert antibacterial activities against marine pathogens and prevent biofouling at low cell densities (Rao et al., 2007) they are likely of ecological relevance for their hosts even at the low abundances deduced in the present study.
So far, Phaeobacter spp. have neither been found associated with zooplankton in coastal and shelf areas (Møller et al., 2007; Tang et al., 2009) or in oligotrophic open ocean (Shoemaker and Moisander, 2015). Elevated temperatures (above ∼18°C) were considered as a main factor determining the occurrence of Phaeobacter in harbors (Gram et al., 2015) but we could not confirm this for the open ocean. It remains to be investigated if Phaeobacter is preferentially associated with specific zooplankton taxa and changes in abundance according to the seasonal abundance of their hosts as was described for other bacterial groups (Turner et al., 2009; Tang et al., 2010). However, considering that zooplankton-associated bacteria typically constitute only <0.1–0.3% of all water column bacteria in the marine environment (Møller et al., 2007; Bickel and Tang, 2014), Phaeobacter spp. must represent a very rare bacterial group in marine water samples with abundances lower (<10-3 %) than the detection limit of many studies employing next generation sequencing (0.64%; Pochon et al., 2013). Commensurate with these results the metagenomes of a large number of oceanic samples typically do not contain any 16S rRNA gene sequence specific for Phaeobacter spp. In contrast, our cultivation-based high-throughput approach in optimized media enabled us to recover representative genotypes despite their low in situ abundance and to study their specific features.
Even by cultivation-based methods, Phaeobacter spp. have so far only been detected in anthropogenic habitats like aquacultures and harbors (Hjelm et al., 2004; Prado et al., 2009; Gram et al., 2015; Xue et al., 2016) or were sporadically isolated from shoreline samples (Rao et al., 2005; Martens et al., 2006; Penesyan et al., 2009). In general solid marine broth agar media have been employed to isolate Phaeobacter. Although we used Pacific samples for which in situ temperatures were close to the temperature optimum of growth of P. gallaeciensis DSM 26640T and P. inhibens DSMZ 16374T (23–27 and 27–29°C, respectively; Ruiz-Ponte et al., 1998; Martens et al., 2006), our attempts to isolate Phaeobacter on solid media were not successful. This suggests that agar media might be less suitable for recovering Phaeobacter from the open ocean. The Phaeobacter strains obtained in the present study employing optimized liquid enrichment strategies represent the first isolates from the open ocean environment. These genotypes constitute a phylogenomically separate subclade and hence do not represent known variants that link the distant coastal populations across the globe. Therefore their genomes were investigated for potential mechanisms of adaption to the open ocean habitat and to the inferred association to mesozooplankton.
All known genetic elements characterizing the surface- and host-associated lifestyle of Phaeobacter (Thole et al., 2012; Frank et al., 2015) are generally present in both P. gallaeciensis clades. The most prominent genomic features of the Pacific strains are their two complete, exclusive, and active Type I restriction modification systems which act as phage defense mechanism (Stern and Sorek, 2011; Loenen et al., 2014). Unlike their aquaculture counterparts, the Pacific strains only contained one type of prophage despite the high abundance and diversity of bacteriophages present in the ocean (Breitbart, 2012), suggesting an effective protection of the host against a broader range of bacteriophages. These restriction systems also are likely to prevent the horizontal transfer of foreign DNA through gene transfer agents (Stern and Sorek, 2011; Lang et al., 2012) which represent a major mechanism of gene acquisition of the genus Phaeobacter (Freese et al. submitted). This may explain the lower number and functional diversity of mobile elements observed in the Pacific strains of P. gallaeciensis compared to their aquaculture relatives. In addition, the slight reduction of the genome size of the Pacific strains by 0.18 Mb maybe related to an incipient genome streamlining as a consequence of nutrient limitation (Giovannoni et al., 2014; Luo and Moran, 2014). In this context it is remarkable that the genotypes of the Pacific P. gallaeciensis clade not only encode the complete phosphonate transport and degradation complex (cf. Villarreal-Chiu et al., 2012) like the aquaculture strains but in addition acquired and maintain a second set of phosphonate transporter (PhnCDE) despite their genome reduction. Phosphonates are particular prominent among marine invertebrate (Quin, 2000). Phosphonates also constitute a third of the dissolved organic phosphorus in oceanic waters and marine bacteria from the Roseobacter group have been shown to be capable of utilizing these compounds (Martinez et al., 2010). Pacific genotypes of P. gallaeciensis may therefore be particularly adapted to this alternative source of phosphorus.
Zooplankton surfaces are colonized by distinct bacterial communities which vary between zooplankton species and body regions (Tang et al., 2010). It has yet to be determined whether P. gallaeciensis can preferentially colonize the body surface or the intestinal tract of mesozooplankton or is also present on other organisms and particles >300 μm. Zooplankton functions as microbial hotspots since it provides a nutrient-richer environment than the surrounding water (Tang et al., 2010). A possible reciprocal advantage of P. gallaeciensis for the host could be related to the formation of TDA and other antibiotics that might affect the success of other bacteria to colonize zooplankton.
The genomes of the two Pacific strains were highly similar (97.8% nucleotide similarity; fraction of core genome, 89.6%) to those of all other P. gallaeciensis strains from aquacultures. A genome conservation such as that observed in Phaeobacter is rare and even exceeds that of species with specialized ecological niches like the oligotrophic pelagic SAR11 subclade 1a (77.7% fraction of the core genome; Grote et al., 2012) or symbiotic Vibrio fischeri (80.4% core genome; Bongrand et al., 2016). Only some small obligate intracellular bacteria like the Chlamydia psittaci group (89.5% core genome; Voigt et al., 2012) and Rickettsia (84–93% core genome; Fuxelius et al., 2008) reach a similarly high genome conservation, but have a much smaller genome size. This raises the question which mechanisms underlie the unexpected high similarity of the P. gallaeciensis genomes.
Based on the homogenous random distribution of SNPs over the genome, recombination events and gene-specific sweeps through the population of P. gallaeciensis are unlikely (Marttinen et al., 2012; Shapiro et al., 2012). Instead, the different lineages must have diversified by neutral mutations. The two Pacific P. gallaeciensis strains detected in the mesozooplankton fraction differed by only 17 SNPs and were isolated from water samples 1500 km apart. Water currents have been shown to transport Pacific copepods over large distances up to 5000 km (Tatebe et al., 2010). The coastal strains of P. gallaeciensis for which genomes are available were isolated from aquacultures in Spain and France (Supplementary Table S1) which are located at a distance of 8,600–10,200 km to the habitats of the Pacific P. gallaeciensis isolates. These aquaculture strains differed by 83,953 SNPs from the Pacific strains. Our data allow to infer hypotheses regarding the mechanism of genomic differentiation within the species P. gallaeciensis. The rate of spontaneous mutation has recently been determined for Ruegeria pomeroyi, another member of the Roseobacter group, as 1.39⋅10-10 base-1 generation-1 (Sun et al., 2017). Accordingly, P. gallaeciensis with a genome size of 4.54 Mb would take 1585 generations to acquire a SNP. R. pomeroyi reaches 45 generations per year in its marine environment (Sun et al., 2017). Due to the higher substrate supply, bacteria associated with marine copepods grow at rates which are 3–18 times higher than free-living bacteria (Tang et al., 2010). They can attain growth rates between 0.7 and 1.2 d-1, corresponding to generation times of 0.58–0.99 days, depending on the feeding status of the zooplankton (Tang, 2005; Møller et al., 2007). Based on these faster growth rates, the 1585 generations would take between 919 and 1569 days or 2.5 to 4.3 years. Correspondingly, the 17 SNPs that distinguish the two genomes of the Pacific strains may have accumulated over 20 to 38.7 years. By a similar calculation, it would take between 164,940 and 180,501 years to acquire the larger number of SNPs that distinguish the Pacific from the phylogenetically related aquaculture strains. This time frame is substantially larger than the time required for the global overturn of ocean waters by surface currents and thermohaline circulation (1000–2000 years; Döös et al., 2012) which renders geographic isolation in different water bodies a highly unlikely mechanism of genomic divergence in P. gallaeciensis.
Conclusion
Our data indicate that a previously unknown, distinct clade of P. gallaeciensis acquired a limited number of clade-specific genes which may be relevant for its association with mesozooplankton and the colonization of the marine pelagial. The divergence of the Pacific clade was most likely driven by the adaptation to this novel ecological niche rather than by geographic isolation. The distribution pattern observed in the present study also provides first indications for possible biotic interactions between the pelagic lineage of P. gallaeciensis and marine mesozooplankton.
Author Contributions
HF and AM performed the sampling and enrichments. AM conducted the screening and isolation. HF performed the analysis of genome sequences and other data. HF and JO designed the study and wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The work was supported by the Deutsche Forschungsgemeinschaft (DFG) within the Transregional Collaborative Research Centre “Roseobacter” (TRR 51/2 TA07). We thank the crew of RV Sonne for their great support during the cruise SO248 which was funded by the German Federal Ministry of Education and Research (BMBF) within the BacGeoPac project (03G0248A), and Maria Pinto Gomes Ribeiro Teixeira who pre-processed samples of the horizontal haul. We are grateful for the fruitful discussions with Boyke Bunk and Johannes Sikorski who also helped with Supplementary Figure S2. We thank Cathrin Spröer for genome sequencing, Franziska Klann, Nicole Heyer and Simone Severitt for excellent technical assistance, and Isabel Schober for bioinformatics support and submission of genomes to NCBI.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.01659/full#supplementary-material
FIGURE S1 | Flow scheme of combined methods applied in the current study.
FIGURE S2 | Distribution of nucleotide diversity (Pi) per gene between P. gallaeciensis clades ordered along the genome of P75 by means of the locus_tag number.
Footnotes
References
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Alvain, S., Moulin, C., Dandonneau, Y., and Loisel, H. (2008). Seasonal distribution and succession of dominant phytoplankton groups in the global ocean: a satellite view. Glob. Biogeochem. Cycles 22:GB3001. doi: 10.1029/2007GB003154
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Badewien, T. H., Winkler, H., Arndt, K. L., and Simon, M. (2016). Physical Oceanography during SONNE Cruise SO248 (BacGeoPac). Oldenburg: University of Oldenburg. doi: 10.1594/PANGAEA.864673
Balcazar, J. L., Lee, N. M., Pintado, J., and Planas, M. (2010). Phylogenetic characterization and in situ detection of bacterial communities associated with seahorses (Hippocampus guttulatus) in captivity. Syst. Appl. Microbiol. 33, 71–77. doi: 10.1016/j.syapm.2009.11.005
Bickel, S. L., and Tang, K. W. (2014). Zooplankton-associated and free-living bacteria in the York River, Chesapeake Bay: comparison of seasonal variations and controlling factors. Hydrobiologia 722, 305–318. doi: 10.1007/s10750-013-1725-0
Billerbeck, S., Wemheuer, B., Voget, S., Poehlein, A., Giebel, H. A., Brinkhoff, T., et al. (2016). Biogeography and environmental genomics of the Roseobacter-affiliated pelagic CHAB-I-5 lineage. Nat. Microbiol. 1:16063. doi: 10.1038/nmicrobiol.2016.63
Bongrand, C., Koch, E. J., Moriano-Gutierrez, S., Cordero, O. X., McFall-Ngai, M., Polz, M. F., et al. (2016). A genomic comparison of 13 symbiotic Vibrio fischeri isolates from the perspective of their host source and colonization behavior. ISME J. 10, 2907–2917. doi: 10.1038/ismej.2016.69
Breider, S., Freese, H. M., Spröer, C., Simon, M., Overmann, J., and Brinkhoff, T. (2017). Phaeobacter porticola sp. nov., an antibiotic-producing bacterium isolated from a sea harbour. Int. J. Syst. Evol. Microbiol. doi: 10.1099/ijsem.0.001879 [Epub ahead of print].
Breitbart, M. (2012). Marine viruses: truth or dare. Annu. Rev. Mar. Sci. 4, 425–448. doi: 10.1146/annurev-marine-120709-142805
Brinkhoff, T., Bach, G., Heidorn, T., Liang, L., Schlingloff, A., and Simon, M. (2004). Antibiotic production by a Roseobacter clade-affiliated species from the German Wadden Sea and its antagonistic effects on indigenous isolates. Appl. Environ. Microbiol. 70, 2560–2565. doi: 10.1128/AEM.70.4.2560-2565.2003
Bruns, A., Phillip, H., Cypionka, H., and Brinkhoff, T. (2003). Aeromicrobium marinum sp. nov., an abundant pelagic bacterium isolated from the German Wadden Sea. Int. J. Syst. Evol. Microbiol. 53, 1917–1923. doi: 10.1099/ijs.0.02735-0
Buchan, A., González, J. M., and Moran, M. A. (2005). Overview of the marine Roseobacter lineage. Appl. Environ. Microbiol. 71, 5665–5677. doi: 10.1128/AEM.71.10.5665-5677.2005
D’Alvise, P. W., Lillebø, S., Prol-García, M. J., Wergeland, H. I., Nielsen, K. F., Bergh, O., et al. (2012). Phaeobacter gallaeciensis reduces Vibrio anguillarum in cultures of microalgae and rotifers, and prevents vibriosis in cod larvae. PLoS ONE 7:e43996. doi: 10.1371/journal.pone.0043996
Darling, A. E., Mau, B., and Perna, N. T. (2010). progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 5:e11147. doi: 10.1371/journal.pone.0011147
Dhillon, B. K., Chiu, T. A., Laird, M. R., Langille, M. G., and Brinkman, F. S. (2013). IslandViewer update: improved genomic island discovery and visualization. Nucleic Acids Res. 41, W129–W132. doi: 10.1093/nar/gkt394
Dickschat, J. S., Zell, C., and Brock, N. L. (2010). Pathways and substrate specificity of DMSP catabolism in marine bacteria of the Roseobacter clade. Chembiochem 11, 417–425. doi: 10.1002/cbic.200900668
Doghri, I., Rodrigues, S., Bazire, A., Dufour, A., Akbar, D., Sopena, V., et al. (2015). Marine bacteria from the French Atlantic coast displaying high forming-biofilm abilities and different biofilm 3D architectures. BMC Microbiol. 15:231. doi: 10.1186/s12866-015-0568-4
Döös, K., Nilsson, J., Nycander, J., Brodeau, L., and Ballarotta, M. (2012). The World Ocean Thermohaline Circulation. J. Phys. Oceanogr. 42, 1445–1460. doi: 10.1175/JPO-D-11-0163.1
Federhen, S. (2015). Type material in the NCBI taxonomy database. Nucleic Acids Res. 43, D1086–D1098. doi: 10.1093/nar/gku1127
Fisher, M. M., and Triplett, E. W. (1999). Automated approach for ribosomal intergenic spacer analysis of microbial diversity and its application to freshwater bacterial communities. Appl. Environ. Microbiol. 65, 4630–4636.
Frank, O., Michael, V., Pauker, O., Boedeker, C., Jogler, C., Rohde, M., et al. (2015). Plasmid curing and the loss of grip - The 65-kb replicon of Phaeobacter inhibens DSM 17395 is required for biofilm formation, motility and the colonization of marine algae. Syst. Appl. Microbiol. 38, 120–127. doi: 10.1016/j.syapm.2014.12.001
Fuxelius, H. H., Darby, A. C., Cho, N. H., and Andersson, S. G. (2008). Visualization of pseudogenes in intracellular bacteria reveals the different tracks to gene destruction. Genome Biol. 9:R42. doi: 10.1186/gb-2008-9-2-r42
Gascuel, O. (1997). BIONJ: an improved version of the NJ algorithm based on a simple model of sequence data. Mol. Biol. Evol. 14, 685–695. doi: 10.1093/oxfordjournals.molbev.a025808
Giovannoni, S. J., Thrash, J., and Temperton, B. (2014). Implications of streamlining theory for microbial ecology. ISME J. 8, 1553–1565. doi: 10.1038/ismej.2014.60
Gram, L., Rasmussen, B. B., Wemheuer, B., Bernbom, N., Ng, Y. Y., Porsby, C. H., et al. (2015). Phaeobacter inhibens from the Roseobacter clade has an environmental niche as a surface colonizer in harbors. Syst. Appl. Microbiol. 38, 483–493. doi: 10.1016/j.syapm.2015.07.006
Grote, J., Thrash, J., Huggett, M. J., Landry, Z. C., Carini, P., Giovannoni, S. J., et al. (2012). Streamlining and core genome conservation among highly divergent members of the SAR11 clade. mBio 3:e00252-12. doi: 10.1128/mBio.00252-12
Guy, L., Roat Kultima, J., and Andersson, S. G. E. (2010). genoPlotR: comparative gene and genome visualization in R. Bioinformatics 26, 2334–2335. doi: 10.1093/bioinformatics/btq413
Heidelberg, J. F., Heidelberg, K. B., and Colwell, R. R. (2002). Bacteria of the gamma-subclass Proteobacteria associated with zooplankton in Chesapeake Bay. Appl. Environ. Microbiol. 68, 5498–5507. doi: 10.1128/AEM.68.11.5498-5507.2002
Hjelm, M., Riaza, A., Formoso, F., Melchiorsen, J., and Gram, L. (2004). Seasonal incidence of autochthonous antagonistic Roseobacter spp. and Vibrionaceae strains in a turbot larva (Scophthalmus maximus) rearing system. Appl. Environ. Microbiol. 70, 7288–7294. doi: 10.1128/AEM.70.12.7288-7294.2004
Huson, D. H., and Bryant, D. (2006). Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23, 254–267. doi: 10.1093/molbev/msj030
Jarvis, B., Wilrich, C., and Wilrich, P. T. (2010). Reconsideration of the derivation of Most Probable Numbers, their standard deviations, confidence bounds and rarity values. J. Appl. Microbiol. 109, 1660–1667. doi: 10.1111/j.1365-2672.2010.04792.x
Karim, M., Zhao, W., Rowley, D., Nelson, D., and Gomez-Chiarri, M. (2013). Probiotic strains for shellfish aquaculture: protection of eastern oyster, Crassostrea virginica, larvae and juveniles againsl bacterial challenge. J. Shellfish Res. 32, 401–408. doi: 10.2983/035.032.0220
Karsten, G. R., and Drake, H. L. (1995). Comparative assessment of the aerobic and anaerobic microfloras of earthworm guts and forest soils. Appl. Environ. Microbiol. 61, 1039–1044.
Kesarcodi-Watson, A., Miner, P., Nicolas, J. L., and Robert, R. (2012). Protective effect of four potential probiotics against pathogen-challenge of the larvae of three bivalves: pacific oyster (Crassostrea gigas), flat oyster (Ostrea edulis) and scallop (Pecten maximus). Aquaculture 34, 29–34. doi: 10.1016/j.aquaculture.2012.02.029
Lane, D. J. (1991). “16S/23S rRNA sequencing,” in Nucleic Acid Techniques in Bacterial Systematics, eds E. Stackebrandt and M. Goodfellow (Chichester: John Wiley & Sons), 115–175.
Lang, A. S., Zhaxybayeva, O., and Beatty, J. T. (2012). Gene transfer agents: phage-like elements of genetic exchange. Nat. Rev. Microbiol. 10, 472–482. doi: 10.1038/nrmicro2802
Lechner, M., Findeiß, S., Steiner, L., Marz, M., Stadler, P. F., and Prohaska, S. J. (2011). Proteinortho: detection of (Co-)orthologs in large-scale analysis. BMC Bioinformatics 12:124. doi: 10.1186/1471-2105-12-124
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Loenen, W. A. M., Dryden, D. T. F., Raleigh, E. A., and Wilson, G. G. (2014). Type I restriction enzymes and their relatives. Nucleic Acids Res. 42, 20–44. doi: 10.1093/nar/gkt847
Lopes, A., Tavares, P., Petit, M. A., Guerois, R., and Zinn-Justin, S. (2014). Automated classification of tailed bacteriophages according to their neck organization. BMC Genomics 15:1027. doi: 10.1186/1471-2164-15-1027
Luo, H., and Moran, M. A. (2014). Evolutionary ecology of the marine Roseobacter clade. Microbiol. Mol. Biol. Rev. 78, 573–587. doi: 10.1128/MMBR.00020-14
Martens, T., Gram, L., Grossart, H. P., Kessler, D., Muller, R., Simon, M., et al. (2007). Bacteria of the Roseobacter clade show potential for secondary metabolite production. Microb. Ecol. 54, 31–42. doi: 10.1007/s00248-006-9165-2
Martens, T., Heidorn, T., Pukall, R., Simon, M., Tindall, B. J., and Brinkhoff, T. (2006). Reclassification of Roseobacter gallaeciensis Ruiz-Ponte et al. 1998 as Phaeobacter gallaeciensis gen. nov., comb. nov., description of Phaeobacter inhibens sp. nov., reclassification of Ruegeria algicola (Lafay et al. 1995) Uchino et al. 1999 as Marinovum algicola gen. nov., comb. nov., and emended descriptions of the genera Roseobacter, Ruegeria and Leisingera. Int. J. Syst. Evol. Microbiol. 56, 1293–1304. doi: 10.1099/ijs.0.63724-0
Martinez, A., Tyson, G. W., and DeLong, E. F. (2010). Widespread known and novel phosphonate utilization pathways in marine bacteria revealed by functional screening and metagenomic analyses. Environ. Microbiol. 12, 222–238. doi: 10.1111/j.1462-2920.2009.02062.x
Marttinen, P., Hanage, W. P., Croucher, N. J., Connor, T. R., Harris, S. R., Bentley, S. D., et al. (2012). Detection of recombination events in bacterial genomes from large population samples. Nucleic Acids Res. 40:e6. doi: 10.1093/nar/gkr928
Meier-Kolthoff, J. P., Auch, A. F., Klenk, H. P., and Göker, M. (2013). Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics 14:60. doi: 10.1186/1471-2105-14-60
Møller, E. F., Riemann, L., and Søndergaard, M. (2007). Bacteria associated with copepods: abundance, activity and community composition. Aquat. Microb. Ecol. 47, 99–106. doi: 10.3354/ame047099
Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A. C., and Kanehisa, M. (2007). KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 35, W182–W185. doi: 10.1093/nar/gkm321
Newton, R. J., Griffin, L. E., Bowles, K. M., Meile, C., Gifford, S., Givens, C. E., et al. (2010). Genome characteristics of a generalist marine bacterial lineage. ISME J. 4, 784–798. doi: 10.1038/ismej.2009.150
Paradis, E., Claude, J., and Strimmer, K. (2004). APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20, 289–290. doi: 10.1093/bioinformatics/btg412
Penesyan, A., Marshall-Jones, Z., Holmstrom, C., Kjelleberg, S., and Egan, S. (2009). Antimicrobial activity observed among cultured marine epiphytic bacteria reflects their potential as a source of new drugs. FEMS Microbiol. Ecol. 69, 113–124. doi: 10.1111/j.1574-6941.2009.00688.x
Pfeifer, B., Wittelsbürger, U., Ramos-Onsins, S. E., and Lercher, M. J. (2014). PopGenome: an efficient swiss army knife for population genomic analyses in R. Mol. Biol. Evol. 31, 1929–1936. doi: 10.1093/molbev/msu136
Pochon, X., Bott, N. J., Smith, K. F., and Wood, S. A. (2013). Evaluating detection limits of next-generation sequencing for the surveillance and monitoring of international marine pests. PLoS ONE 8:e73935. doi: 10.1371/journal.pone.0073935
Porsby, C. H., Nielsen, K. F., and Gram, L. (2008). Phaeobacter and Ruegeria species of the Roseobacter clade colonize separate niches in a Danish turbot (Scophthalmus maximus)-rearing farm and antagonize Vibrio anguillarum under different growth conditions. Appl. Environ. Microbiol. 74, 7356–7364. doi: 10.1128/AEM.01738-08
Prado, S., Montes, J., Romalde, J. L., and Barja, J. L. (2009). Inhibitory activity of Phaeobacter strains against aquaculture pathogenic bacteria. Int. Microbiol. 12, 107–114.
Rao, D., Skovhus, T., Tujula, N., Holmstrom, C., Dahllof, I., Webb, J. S., et al. (2010). Ability of Pseudoalteromonas tunicata to colonize natural biofilms and its effect on microbial community structure. FEMS Microbiol. Ecol. 73, 450–457. doi: 10.1111/j.1574-6941.2010.00917.x
Rao, D., Webb, J. S., Holmstrom, C., Case, R., Low, A., Steinberg, P., et al. (2007). Low densities of epiphytic bacteria from the marine alga Ulva australis inhibit settlement of fouling organisms. Appl. Environ. Microbiol. 73, 7844–7852. doi: 10.1128/AEM.01543-07
Rao, D., Webb, J. S., and Kjelleberg, S. (2005). Competitive interactions in mixed-species biofilms containing the marine bacterium Pseudoalteromonas tunicata. Appl. Environ. Microbiol. 71, 1729–1736. doi: 10.1128/AEM.71.4.1729-1736.2005
Roberts, R. J., Vincze, T., Posfai, J., and Macelis, D. (2015). REBASE–a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res. 43, D298–D299. doi: 10.1093/nar/gku1046
Ruiz-Ponte, C., Cilia, V. Lambert, C., and Nicolas, J. L. (1998). Roseobacter gallaeciensis sp. nov., a new marine bacterium isolated from rearings and collectors of the scallop Pecten maximus. Int. J. Syst. Microbiol. 48, 537–542.
Ruiz-Ponte, C., Samain, J. F., Sánchez, J. L., and Nicolas, J. L. (1999). The benefit of a Roseobacter species on the survival of scallop larvae. Mar. Biotechnol. 1, 52–59. doi: 10.1007/PL00011751
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Seyedsayamdost, M. R., Carr, G., Kolter, R., and Clardy, J. (2011a). Roseobacticides: small molecule modulators of an algal-bacterial symbiosis. J. Am. Chem. Soc. 133, 18343–18349. doi: 10.1021/ja207172s
Seyedsayamdost, M. R., Case, R. J., Kolter, R., and Clardy, J. (2011b). The Jekyll-and-Hyde chemistry of Phaeobacter gallaeciensis. Nat. Chem. 3, 331–335. doi: 10.1038/nchem.1002
Shapiro, B. J., Friedman, J., Cordero, O. X., Preheim, S. P., Timberlake, S. C., Szabó, G., et al. (2012). Population genomics of early events in the ecological differentiation of bacteria. Science 336, 48–51. doi: 10.1126/science.1218198
Shoemaker, K. M., and Moisander, P. H. (2015). Microbial diversity associated with copepods in the North Atlantic subtropical gyre. FEMS Microbiol. Ecol. 91:fiv064. doi: 10.1093/femsec/fiv064
Simon, M., Scheuner, C., Meier-Kolthoff, J. P., Brinkhoff, T., Wagner-Dobler, I., Ulbrich, M., et al. (2017). Phylogenomics of Rhodobacteraceae reveals evolutionary adaptation to marine and non-marine habitats. ISME J. 11, 1483–1499. doi: 10.1038/ismej.2016.198
Sonnenschein, E. C., Nielsen, K. F., D’Alvise, P., Porsby, C. H., Melchiorsen, J., Heilmann, J., et al. (2017). Global occurrence and heterogeneity of the Roseobacter-clade species Ruegeria mobilis. ISME J. 11, 569–583. doi: 10.1038/ismej.2016.111
Stern, A., and Sorek, R. (2011). The phage-host arms-race: shaping the evolution of microbes. Bioessays 33, 43–51. doi: 10.1002/bies.201000071
Sun, Y., Powell, K. E., Sung, W., Lynch, M., Moran, M. A., and Luo, H. (2017). Spontaneous mutations of a model heterotrophic marine bacterium. ISME J. 11, 1713–1718. doi: 10.1038/ismej.2017.20
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Tang, K., Dziallas, C., Hutalle-Schmelzer, K., and Grossart, H. P. (2009). Effects of food on bacterial community composition associated with the copepod Acartia tonsa Dana. Biol. Lett. 5, 549–553. doi: 10.1098/rsbl.2009.0076
Tang, K. W. (2005). Copepods as microbial hotspots in the ocean: effects of host feeding activities on attached bacteria. Aquat. Microb. Ecol. 38, 31–40. doi: 10.3354/ame038031
Tang, K. W., Turk, V., and Grossart, H. P. (2010). Linkage between crustacean zooplankton and aquatic bacteria. Aquat. Microb. Ecol. 61, 261–277. doi: 10.3354/ame01424
Tatebe, H., Yasuda, I., Saito, H., and Shimizu, Y. (2010). Horizontal transport of the calanoid copepod Neocalanus in the North Pacific: the influences of the current system and the life history. Deep Sea Res. I 57, 409–419. doi: 10.1016/j.dsr.2009.11.009
Thole, S., Kalhoefer, D., Voget, S., Berger, M., Engelhardt, T., Liesegang, H., et al. (2012). Phaeobacter gallaeciensis genomes from globally opposite locations reveal high similarity of adaptation to surface life. ISME J. 6, 2229–2244. doi: 10.1038/ismej.2012.62
Treangen, T. J., Ondov, B. D., Koren, S., and Phillippy, A. M. (2014). The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 15, 524. doi: 10.1186/s13059-014-0524-x
Tschech, A., and Pfennig, N. (1984). Growth yield increase linked to caffeate reduction in Acetobacterium woodii. Arch. Microbiol. 137, 163–167. doi: 10.1007/BF00414460
Turner, J. W., Good, B., Cole, D., and Lipp, E. K. (2009). Plankton composition and environmental factors contribute to Vibrio seasonality. ISME J. 3, 1082–1092. doi: 10.1038/ismej.2009.50
Villarreal-Chiu, J. F., Quinn, J. P., and McGrath, J. W. (2012). The genes and enzymes of phosphonate metabolism by bacteria, and their distribution in the marine environment. Front. Microbiol. 3:19. doi: 10.3389/fmicb.2012.00019
Voigt, A., Schöfl, G., and Saluz, H. P. (2012). The Chlamydia psittaci genome: a comparative analysis of intracellular pathogens. PLoS ONE 7:e35097. doi: 10.1371/journal.pone.0035097
Wagner-Döbler, I., and Biebl, H. (2006). Environmental biology of the marine Roseobacter lineage. Annu. Rev. Microbiol. 60, 255–280. doi: 10.1146/annurev.micro.60.080805.142115
Widdel, F., Kohring, G. W., and Mayer, F. (1983). Studies on dissimilatory sulfate-reducing bacteria that decompose fatty acids. Arch. Microbiol. 134, 286–294. doi: 10.1007/BF00407804
Keywords: Phaeobacter gallaeciensis, zooplankton, attached bacteria, bacterial adaptation, genome evolution, high-throughput cultivation
Citation: Freese HM, Methner A and Overmann J (2017) Adaptation of Surface-Associated Bacteria to the Open Ocean: A Genomically Distinct Subpopulation of Phaeobacter gallaeciensis Colonizes Pacific Mesozooplankton. Front. Microbiol. 8:1659. doi: 10.3389/fmicb.2017.01659
Received: 28 April 2017; Accepted: 16 August 2017;
Published: 31 August 2017.
Edited by:
Bernd Wemheuer, University of New South Wales, AustraliaReviewed by:
James T. Hollibaugh, University of Georgia, United StatesJulia Grosse, GEOMAR Helmholtz Centre for Ocean Research Kiel (HZ), Germany
Copyright © 2017 Freese, Methner and Overmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Heike M. Freese, aGVpa2UuZnJlZXNlQGRzbXouZGU=