 Peipei Zhang
Peipei Zhang Yan Liu
Yan Liu Wenwen Liu
Wenwen Liu Mengji Cao
Mengji Cao Sebastien Massart
Sebastien Massart Xifeng Wang
Xifeng Wang- 1State Key Laboratory for Biology of Plant Diseases and Insect Pests, Institute of Plant Protection, Chinese Academy of Agricultural Sciences, Beijing, China
- 2Laboratory of Phytopathology, University of Liège, Gembloux Agro-Bio Tech, Gembloux, Belgium
- 3National Citrus Engineering Research Center, Citrus Research Institute, Southwest University, Chongqing, China
To identify the pathogens responsible for leaf yellowing symptoms on wheat samples collected from Jinan, China, we tested for the presence of three known barley/wheat yellow dwarf viruses (BYDV-GAV, -PAV, WYDV-GPV) (most likely pathogens) using RT-PCR. A sample that tested negative for the three viruses was selected for small RNA sequencing. Twenty-five million sequences were generated, among which 5% were of viral origin. A novel polerovirus was discovered and temporarily named wheat leaf yellowing-associated virus (WLYaV). The full genome of WLYaV corresponds to 5,772 nucleotides (nt), with six AUG-initiated open reading frames, one non-AUG-initiated open reading frame, and three untranslated regions, showing typical features of the family Luteoviridae. Sequence comparison and phylogenetic analyses suggested that WLYaV had the closest relationship with sugarcane yellow leaf virus (ScYLV), but the identities of full genomic nucleotides and deduced amino acid sequence of coat protein (CP) were 64.9 and 86.2%, respectively, below the species demarcation thresholds (90%) in the family Luteoviridae. Furthermore, agroinoculation of Nicotiana benthamiana leaves with a cDNA clone of WLYaV caused yellowing symptoms on the plant. Our study adds a new polerovirus that is associated with wheat leaf yellowing disease, which would help to identify and control pathogens of wheat.
Introduction
In 43 countries, wheat (Triticum aestivum) is the most important food crop and the primary staple food, feeding at least one third of the world's population. However, wheat yield and quality are seriously impacted by diseases caused by fungi, bacteria, and viruses (Mehta, 2014). Nearly 50 viruses have been reported to infect wheat in the world, resulting in typical symptoms including mosaic, streak, yellowing, dwarfing, and rosette stunting (Lister and Ranieri, 1995; Wang et al., 2010; Rotenberg et al., 2016). Wheat yellow dwarf disease is a recurrent and costly problem throughout China, where it has caused serious epidemics seven times since the 1970 s. In early-planted winter wheat fields, when the densities of populations of aphids are high, yield losses on susceptible cultivars can reach to average 10~15% or even more than 50% in northwestern regions (Wang et al., 2010).
The causal agents of yellow dwarf disease are barley yellow dwarf viruses (BYDVs) and were previously classified into five strains based on epitope profile and aphid vector specificity (Rochow, 1969; Rochow and Muller, 1971). Now that the complete genomes of some of the BYDVs and their genome structures have been reported, the viruses are considered to be different species classified in the family Luteoviridae, that infect plants in the family Poaceae and are transmitted by aphids (Miller et al., 2002; Krueger et al., 2013). Luteoviridae comprise three genera: Enamovirus, Polerovirus, and Luteovirus, and seven species considered as “unassigned” members according to their genome characterization, nucleotide sequence identity and most efficient vector (Domier, 2012). Viruses in the Luteoviridae have linear, positive-sense, 5.5~6-kb RNA genomes with six recognized AUG-initiated open reading frames (ORFs), i.e., ORF 0–5 in poleroviruses and ORF 1–6 in luteoviruses and three or four untranslated regions (UTRs) (Miller et al., 1995). In addition, a small non-AUG-initiated ORF (ORF3a), required for long-distance movement, was predicted in poleroviruses, and luteoviruses through statistical analysis, and confirmed by functional analysis (Smirnova et al., 2015). The cereal-infecting members of Luteoviridae comprise BYDV-PAV, -MAV, -PAS, -kerII, and -kerIII species within the genus Luteovirus; cereal yellow dwarf virus-RPV (CYDV-RPV, formerly BYDV-RPV), CYDV-RPS and maize yellow dwarf virus-RMV (MYDV-RMV, formerly BYDV-RMV) in the genus Polerovirus as well as BYDV-SGV, and wheat yellow dwarf virus-GPV (WYDV-GPV, formerly BYDV-GPV) that have not yet been assigned to any genus (Miller and Rasochová, 1997; Hawkes and Jones, 2005; Luciozavaleta et al., 2007; Zhang et al., 2009; Domier, 2012; Krueger et al., 2013). In China, four species of Luteoviridae infect cereals according to Rochow's system of classification: BYDV-GAV, -PAV, MYDV-RMV, and WYDV-GPV (Zhou et al., 1987; Liu, F. et al., 2007; Wu et al., 2011). BYDV-GAV is very similar to BYDV-MAV (Jin et al., 2004; Zhang et al., 2009), WYDV-GPV is closely related to CYDV-RPV (Zhang et al., 2009), while BYDV-PAV-CN is highly diverged from the other BYDVs (Liu, F. et al., 2007; Wu et al., 2011).
Traditionally, viruses have been detected and identified using biological, electron microscopy, serological, and molecular biological methods, which depend on the development of antibodies or knowledge of sequences of potential pathogens. Next-generation sequencing (NGS, or deep sequencing), however, provides a powerful alternative for virus detection. The advantage of this method is that it no need prior knowledge of the host or viral information and can detect both RNA and DNA virus. When plants infected by any kinds of viruses, the small interfering RNAs (siRNAs) are generated and accumulated to respond to antiviral defense (Hamilton and Baulcombe, 1999; Mlotshwa et al., 2008). This defense is initiated by cleavage of viral dsRNA into viral derived siRNA (vsiRNA) by a Dicer family member (Voinnet, 2001; Baulcombe, 2004). Having noted that vsiRNA are often overlapping, it is possible to identify a virus through sRNA sequencing followed by assembly of sRNAs into a partial or even complete viral genome (Mierlo et al., 2010).
Since the early 1980s in China, a nationwide survey for wheat yellow dwarf disease has targeted multiple commercial field sites in the main wheat-growing regions where epidemics have been reported. RT-PCR, enzyme-linked immunosorbent assays (ELISA), and dot-blot hybridization were used to determine the occurrence of different luteoviruses in China (Liu, Y., et al., 2007). In the past, BYDV-GAV, -PAV or WYDV-GPV were detected in more than 80% of wheat samples that had yellowing and dwarfing symptoms, but this percentage dropped to 50% in more recent years (Zhao et al., 2010). Because we suspected that other novel viruses might be associated with wheat samples showing dull yellowing, dwarfing, and excessive tillering symptoms, next-generation sequencing was used to detect any viruses in a wheat sample collected from Jinan, in Shandong Province of China. As a result, we identified a new virus, temporarily named wheat leaf yellowing-associated virus (WLYaV).
Materials and Methods
Plant Material
During field surveys in April 2016, 12 wheat samples exhibiting flag leaf yellowing and little dwarfing were collected from Jinan, Shandong Province of China. Whole plants were put into plastic bags and their roots kept moist during transport to the laboratory. One hundred milligrams of fresh leaves were cut from the plants to extract total RNA; other leaves were stored at −70°C. Healthy wheat plants growing in an insect-proofed greenhouse served as controls.
RNA Extraction and Detection of Known Viruses
Total RNA of wheat leaves was extracted using TRIzol reagent according to the manufacturer's instruction (Invitrogen, USA). The quality of RNA was tested using a Nanodrop 2,000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA), and RNA integrity was verified by gel electrophoresis.
BYDV-GAV, -PAV and WYDV-GPV were detected by RT-PCR using specific primers detailed in Table S1, which also gives the sequences of all primers used in this study. B/W-YDV-infected plants that were used as positive controls were growing in the growth chamber (16 h light at 20°C/8 h dark at 18°C). Reverse transcription was performed in a total volume of 20 μL with 1 μg of RNA, 2 μL of reverse (R) primer (10 μM), 2 μL of dNTP Mix (each 2.5 mM; TaKaRa, China), 4 μL of 5× M-MLV buffer (Promega, USA), 1 μL of M-MLV reverse transcriptase (200 U/μL; Promega), 0.5 μL of Recombinant RNase Inhibitor (40 U/μL; TaKaRa) and DEPC water. The PCR contained 2 μL of cDNA, 2.5 μL of 10× buffer (Mg2+, 15 mM, TaKaRa), 2 μL of dNTP Mix (each 2.5 mM; TaKaRa), 0.5 μL of forward (F) and reverse (R) primer (10 μM), 0.2 μL of rTaq (5 U/μL; TaKaRa) and 17.3 μL of ddH2O. The PCR reaction was performed using thermal cycler (Bio RAD, Hercules, CA, USA) as follows: denaturation at 94°C for 3 min; 35 cycles at 94°C for 30 s, 58°C for 45 s and 72°C for 80 s; final extension at 72°C for 10 min (Zhao et al., 2010). PCR products were electrophoresed in 1% agarose gel and stained with ethidium bromide (EB).
Small RNA (sRNA) Sequencing
The quality of extracted RNA was tested using the Nanodrop 2000 spectrophotometer (Thermo Fisher Scientific) and Bio Analyzer 2100 (Agilent Technology, Santa Clara, CA, USA). An RNA sample with an RIN (RNA integrity number) value ≥6 was considered as high enough quality and used in the next steps. The small RNAs (sRNAs) were separated using PEG8000 and ligated to a 3′ adaptor. The 36~44-nt (ligated sRNAs) bands were purified using PAGE in a 15% denatured gel and linked to 5′ adaptor. The first-strand cDNA was synthesized using the pair-linked sRNAs, and 16 cycles of PCR amplification were performed. The 140~160-bp products, namely the sRNAs library, were purified by 3.5% agarose gel electrophoresis. The library was quantified by ECO (Illumina, San Diego, CA, USA) and submitted for sequencing on the Illumina 2,500 platform (Illumina).
Analysis and Assembly of sRNA Data
Raw reads from the Illumina platform were processed to trim adaptor sequences and low-quality reads. Unique sequences were then generated as clean reads by collapsing the identical sequences. The abundance of sRNAs was determined using the software Bowtie2 and default parameters (Langmead and Salzberg, 2012). Then the clean reads were assembled using the Velvet program with a k-mer of 17 as the minimal overlapping length required for joining sRNAs into larger contigs (Zerbino and Birney, 2008). Assembled contigs were screened against the GenBank nucleotide collection (nt) and non-redundant protein sequences (nr) databases using a BlastN and BlastX search using standard parameters respectively (https://blast.ncbi.nlm.nih.gov/Blast.cgi).
Validation and Completion of Viral Genome
Contigs and gaps were confirmed and filled by RT-PCR using specific primer pairs 1, 2, 3, 4, 1-2, 2-3, and 3-4 F/R (Table S1) designed according to the consensus sequences of the assembled viral contigs with more than 70-nt overlapping. The sugarcane yellow leaf virus (ScYLV, AF157029.1) genome sequence was used for the alignment and positioning of the contigs. The terminal sequences of the viral genome were obtained using 5′ and 3′ rapid amplification of cDNA ends (RACE) kits according to the manufacture (Invitrogen). The PCR products were collected using Wizard SV Gel and PCR Clean-Up System (Promega). The purified PCR products were then cloned into the pEASY-T5 vector (TransGen Biotech, China) and used to transformed Trans-T1 competent cells (TransGen Biotech) following the manufacturers' instructions. The clones harboring the transformed vector were identified by PCR and Sanger sequencing (Sangon Biotech (Shanghai) Co., China). The results of sequencing were assembled using DNAMAN (version 6) program (Lynnon Biosoft, San Ramon, CA, USA) with more than 70-nt overlapping regions to form the full-length viral genome.
Analysis of the Viral Genome and sRNA
After obtaining the whole viral genome, open reading frames (ORFs) were predicted using SnapGene software (Ian, 2004). The distribution and coverage of vsiRNA was determined using the software Bowtie2 under default parameters, and the results were exported to Excel (Microsoft, Redmond, WA, USA) for further analysis. Contigs were mapped to the WLYaV genome using the CLC Genomics Workbench (Qiagen, Valencia, CA, USA).
Identity Calculations and Phylogenetic Analyses
The identities of nucleotide sequences of the whole genome, untranslated regions and deduced amino acid sequences of seven ORFs of WLYaV with other viruses belonging to the family Luteoviridae (Table S2) that were retrieved from NCBI (http://www.ncbi.nlm.nih.gov/) were calculated by the Needle program (Liu et al., 2009). Sequences were aligned with the ClustalW method, and phylogenetic trees were constructed by the neighbor joining method using MEGA 6 software (Tamura et al., 2013). The reliability of each branch was evaluated with bootstrap (1000 repeats).
Agrobacterium Mediated Infectious cDNA Clone
The whole nucleotide sequence of WLYaV was divided into two overlapping parts, A (nt 1–3 059) and B (nt 3 035–5 772), a NcoI restriction enzyme site was within the overlapping region (nt 3 045–3 050). A and B were amplified by RT-PCR with specific primer pairs (A-Stu-F/A-Nco-R, B-Nco-F/B-Sal-R, respectively), adding restriction enzyme sites StuI and SalI, respectively. The amplification products of fragments A and B were purified and cloned into the pEASY-T5 vector and sequenced as above. pCB301 vector (kindly supplied by Prof. Xiaorong Tao, Nanjing Agricultural University), a binary vector with 2 × 35S promoter, ribozyme and NOS terminator (Shen et al., 2014; Wang et al., 2015), and plasmids containing fragment A and B without mutations were digested with restriction enzymes and ligated using T4 ligase (Promega, USA) to produce pCB301-WLYaV. Plasmid pCB301 (as a negative control) and pCB301-WLYaV were transferred to separate suspensions of Agrobacterium tumefaciens strain GV3101 (Wang et al., 2015). Then the abaxial surface of 2-week-old Nicotiana benthamiana leaves (3~4 leaves) was infiltrated with the bacterial cultures (Chen et al., 2016). At 14 days post inoculation (dpi), systemic virus dissemination was evaluated in non-inoculated leaves by RT-PCR using specific primer pair CP-F/R (Table S1).
Results
Detection of Known Luteoviruses in Wheat Samples
In 12 wheat samples from Jinan, China that had yellow dwarf symptoms, three known B/W-YDVs (-GAV, -PAV and -GPV) in China were detected. Eight samples were infected with one of the BYDVs, and none had a mixed infection (Table S3). None of the above three viruses were detected in sample JN-U3, which exhibited dull yellowing on the leaf margins and little dwarfing (Figures 1B,C) comparing with the healthy sample (Figure 1A), while bright yellowing was found on samples infected by B/W-YDVs. Therefore, sample JN-U3 was used for the next experiment.

Figure 1. Symptoms on wheat samples collected from Jinan, China. (A) Symptomless, apparently healthy sample. (B,C) Sample JN-U3 infected with virus newly discovered, with dull yellowing and little dwarfing.
Analysis and Assemble of sRNA Library
The sRNA library from sample JN-U3 was constructed and sequenced using Illumina Hiseq 2,500 platform. In total, 25,605,387 clean reads that had passed quality control were produced after removing adaptor sequences and low-quality reads. The length of the clean reads ranged from 18 to 25 nt (Figure 2); 21-nt reads were the most abundant, followed by 24 and 22-nt reads. After assembly using the Velvet program, 8,798 contigs were generated.
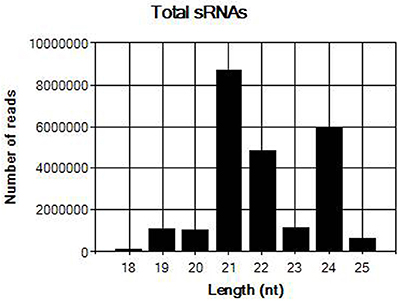
Figure 2. Length distribution of total sRNAs in sample JN-U3.
Identification and Sequencing of Unknown Viral Genome
After blast analysis of the assembled contigs, four contigs of 1,151, 701, 718, and 1,714 nucleotides, respectively, had high identities with several viruses in the family Luteoviridae, and may be associated with the yellowing symptom. The higher identities were observed with ScYLV, with 83% identity on 99% coverage of the sequence for contig 3, for example. Four contigs were aligned to different positions of the ScYLV genome, and their relative positions and gaps were predicted. RT-PCR and RACE-PCR were performed to confirm the contig sequences, fill gaps and obtain the terminal sequences (Figure S1). The RACE-PCR was repeated three times with the similar results. Finally, the assembled genome of this virus was constituted by 5,772 nt, and deposited in GenBank as accession KY605226. The other 11 samples from Jinan were negative for WLYaV (Table S3) using RT-PCR and primer CP-F/R (Table S1).
Characterization of the Viral Genome and sRNAs
Because the newly discovered virus was found in a wheat sample that had dull yellowing and little dwarfing symptoms, the virus has been temporarily named wheat leaf yellowing-associated virus (WLYaV). Six ORFs (ORF0, ORF1, ORF2, ORF3, ORF4, ORF5) were predicted by SnapGene software, and 5′ and 3′ UTRs, one intergenic-UTR (I-UTR) and ORF3a were also found in the viral genome (Figure 3A), suggesting the genome organization is similar to that of viruses in the genus Polerovirus (Miller et al., 1995). The 5′ UTR consists of 58 nt starting with ACUAAA, while the conserved sequence that start the 5′ end of poleroviruses is ACAAAA (Guilley et al., 1994; Moonan et al., 2000; Mo et al., 2010). ORF0 (nt 59~877) is the first ORF encoding a 31.4 kD protein (P0), which is a putative RNA-silencing suppressor (RSS) (Pfeffer et al., 2002, 2017; Kozlowska-Makulska et al., 2010). A putative F-box like motif (IPIIL) conserved among poleroviruses (LPxxL/I) has been found (Figure S2), which is required for RSS function (Pazhouhandeh et al., 2006). ORF1 (nt 228~2,099) encodes a 69.1 kD putative polyprotein (P1) with a proteinase motif that can generate three proteins including the viral genome-linked protein (VPg) (Figure S2) (Miller et al., 2002; Nickel et al., 2008). ORF2 (nt 1,658~3,364) and ORF1 compose a putative fusion protein of RdRp (P1-P2 fusion) by a -1 ribosomal frameshift at nt 1,658, which is regulated by a GGGAAAC sequence at nt 1,658~1,664 within the overlapping region of ORF1 and ORF2 and is involved in the conserved GDD amino acid core (Figure S2) (Prüfer et al., 1992; Nixon et al., 2002). ORF2 is followed by a 188 nt I-UTR (nt 3,365~3,552). This sequence contains the ORF3a starting with a putative CUG initiation codon (nt 3,435~3,569; 4.91 kD), which encodes a protein that involved in the viral long-distance movement (Smirnova et al., 2015). ORF3a, ORF3~ORF5 might be expressed through the 3′ co-terminal sgRNAs, yielding the putative capsid, CP-RTD (RTP) and movement proteins (Hwang et al., 2013). ORF3 (nt 3,553~4,143) encodes a 21.7 kD protein of putative coat protein (P3). ORF4 (nt 3,584~4,036) present within ORF3 and encodes a 17.0 kD putative movement protein (P4). ORF5 (nt 4,144~5,577) and ORF3 encode a fusion protein (P3–P5 fusion), a putative readthrough protein that may be involved in virus transmission (Brault et al., 1995). The 3′ UTR consists of 195 nt (nt 5,578~5,772) without a polyA tail.
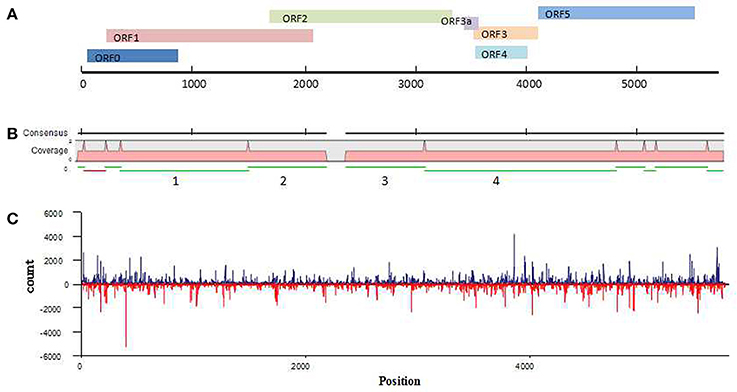
Figure 3. Analysis of WLYaV genome. (A) Characteristics of WLYaV genome, full length: 5,772 nt; 5′UTR: nt 1~58, with ACUAAA start; ORF0: nt 59~877, encodes a 31.4 kD protein (P0), a putative RNA-silencing suppressor (RSS); ORF1: nt 228~2,099, encodes a 69.1 kD putative polyprotein (P1); ORF2: nt 1,658~3,364, encodes a putative fusion protein of RdRp (P1–P2 fusion) by a −1 ribosomal frameshift with ORF1; ORF3a: nt 3,435~3,569, encodes a protein that may be involved in long-distance movement; ORF3: nt 3,553~4,143, encodes a 21.7 kD protein, a putative coat protein (P3); ORF4: nt 3,584~4,036, within ORF3, encodes a 17.0 kD protein, a putative movement protein (P4); ORF5: nt 4,144~5,577, encodes a putative fusion protein, a read-through protein (P3–P5 fusion) with ORF3 by suppression of termination; 3′UTR: nt 5,578~5,772, without a polyA tail. (B) Contigs mapped to the WLYaV genome: 11 of 8,798 contigs were remapped to the WLYaV genome, which covered 99% of the genome; the four numbered contigs were positioned using a blast search of known viruses. (C) Hotspots along the WLYaV genome, blue: positive strand, red: reverse strand. 5.25% of total sRNAs reads were derived from WLYaV, which were well-scattered on the viral positive and reverse strands except for several peaks at ORF0 (RSS), ORF3 (CP), and ORF5 (RTD).
After the complete viral genome was obtained, total contigs were remapped to the viral genome (Figure 3B). This time, we obtained 11 mapped contigs, which nearly covered the entire genome, while initially only 4 of the 11 contigs had homologies with Luteoviridae family. This difference may be due to the low identities of the seven unrecognized contigs with other known viruses.
Analysis of vsiRNAs
To identify the hotspots of viral siRNAs (vsiRNAs), we analyzed the genome region targeted by RNAi by mapping the sRNAs on the viral genome (Figure 3C). A total of 1 301,797 reads (5.25% of total sRNAs reads) were mapped. They were well-scattered on the viral genome and anti-genome strands except for several peaks at ORF0 (RSS), ORF3 (CP) and ORF5 (RTD). The higher number of siRNAs corresponding to CP and RTD would correspond to a higher representativity of the RNA targets that correspond to both genomic and subgenomic RNA species.
To comprehensively understand vsiRNAs, we analyzed the size, polar distribution and base bias of vsiRNAs (Figure 4). The vsiRNAs were mostly 19~24-nt long; the 22- and 21-nt sRNA species had the highest percentages, together making up more than 90% of the total (Figure 4A). We also found that the number of vsiRNAs reads from the anti-genome strand was slightly higher than from the genome strand. It was also observed that the vsiRNAs were evenly distributed in the positive and negative strands of the genome, indicating that the vsiRNAs were formed from the replication intermediate. In addition, a bias toward base A and U especially in the anti-genome was observed (52.1% in the genome, 67.3% in the anti-genome) (Figure 4B).
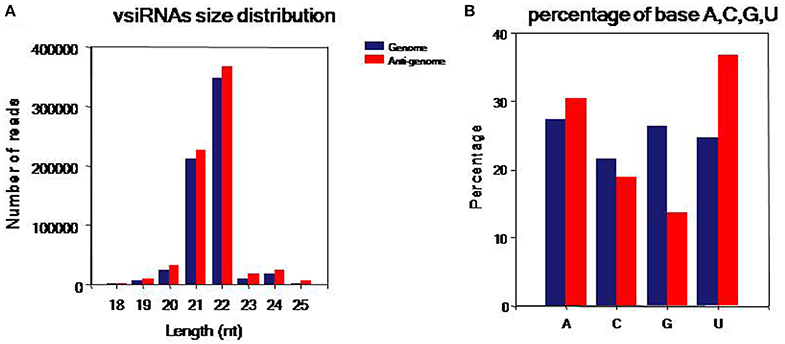
Figure 4. Characteristics of the vsiRNAs. (A) Size distribution of vsiRNAs, 18~25 nt long, with 22- and 21-nt sRNAs predominated. (B) Percentage of A/U/G/C bases in vsiRNAs, a bias toward bases A and U especially in the anti-genome was found (52.06% in genome, 67.32% in anti-genome). Blue: genome, red: anti-genome.
Identity Calculation and Phylogenetic Analyses with Other Viruses in Family Luteoviridae
The full-length genome sequence and the predicted UTR and ORFs of WLYaV were compared with those of 26 other viruses in the family Luteoviridae (Table 1). The nucleotide identities ranged from 42.3 to 64.9% for the full-length genome, 17.8 to 50.0% for the 5′UTR, 33.0–60.3% for the I-UTR, and 11.1–48.4% for the 3′UTR, respectively. Moreover, the deduced amino acid sequence identities were 2.1–25.4% for ORF0, 4.5–48.9% for ORF1, 10.8–62.3% for ORF1-2 fusion, 31.1–72.7% for ORF3a, 29.7–86.2% for ORF3, 20.2–74.0% for ORF4, 29.9–66.0% for ORF3-5 fusion, respectively. It is worth emphasizing that the CP produced by ORF3 shared the highest identities among the viruses, and ORF0 had the lowest identities (Table 1). WLYaV had the closest relationship with ScYLV (64.9% for full length nucleotide and 86.2% for deduced amino acid sequences of ORF3). When the full-length genome and ORF3 deduced amino acid sequences of 10 representative ScYLV isolates (Elsayed et al., 2017) were compared with WLYaV (Table S4), the identities were 63.7~65.3% and 85.7~86.2%, respectively, below the species demarcation thresholds (90%) in the family Luteoviridae (Domier, 2012; Simmonds et al., 2017).
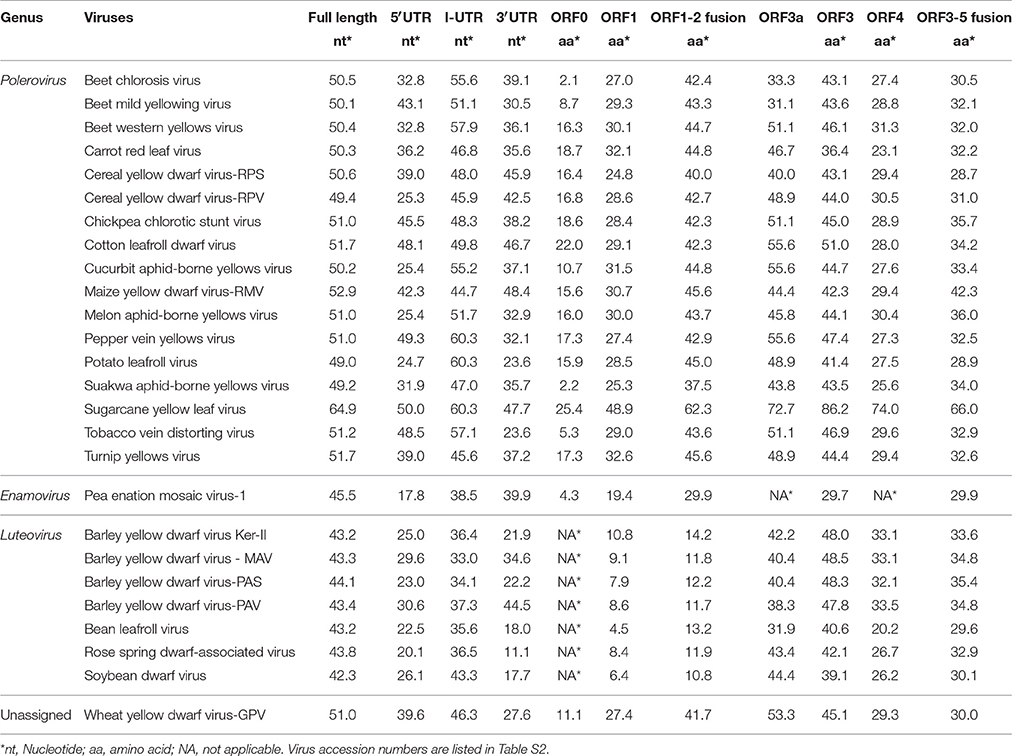
Table 1. Percent nucleotide and amino acid sequence identities of WLYaV and other members in Luteoviridae.
The neighbor-joining phylogenetic analyses based on the nucleotide sequences of the full-length genome and deduced amino acid sequences of RdRp were performed with a bootstrap of 1,000 replications (Figure 5), which are widely used as important criteria for assigning virus species to a genus in the family Luteoviridae. Besides, the phylogenetic relationship of CP sequences was analyzed due to a possible recombinant site had been identified between RdRp and CP of ScYLV (Moonan et al., 2000; Smith et al., 2000). The three phylogenetic trees had similar topologies and showed that WLYaV was most closely related to ScYLV, genus Polerovirus. It's worth noted that in the CP tree, WLYaV, and ScYLV clustered with the members of genus Luteovirus, in contrast, two known luteoviruses, soybean dwarf virus (SbDV) and bean leafroll virus (BLRV) clustered with poleroviruses (Figure 5C). Therefore, a similar recombination event has probably occurred in the ancestral WLYaV genome. It could be speculated that this event may have occurred in the ancestral WLYaV and ScYLV, as similar to a drawing scenario by Domier et al. (2002) with the two luteoviruses, SbDV and BLRV. Furthermore, when the phylogenetic trees were constructed using the 10 representative isolates of ScYLV (Figure S3), WLYaV and ScYLVs were on different branches, giving the same result and indicating WLYaV and ScYLV were different. When the identities and phylogenetic analyses are considered together, this new virus could be defined as a new member of genus Polerovirus, family Luteoviridae according to criterion of the International Committee on Taxonomy of Viruses (ICTV).
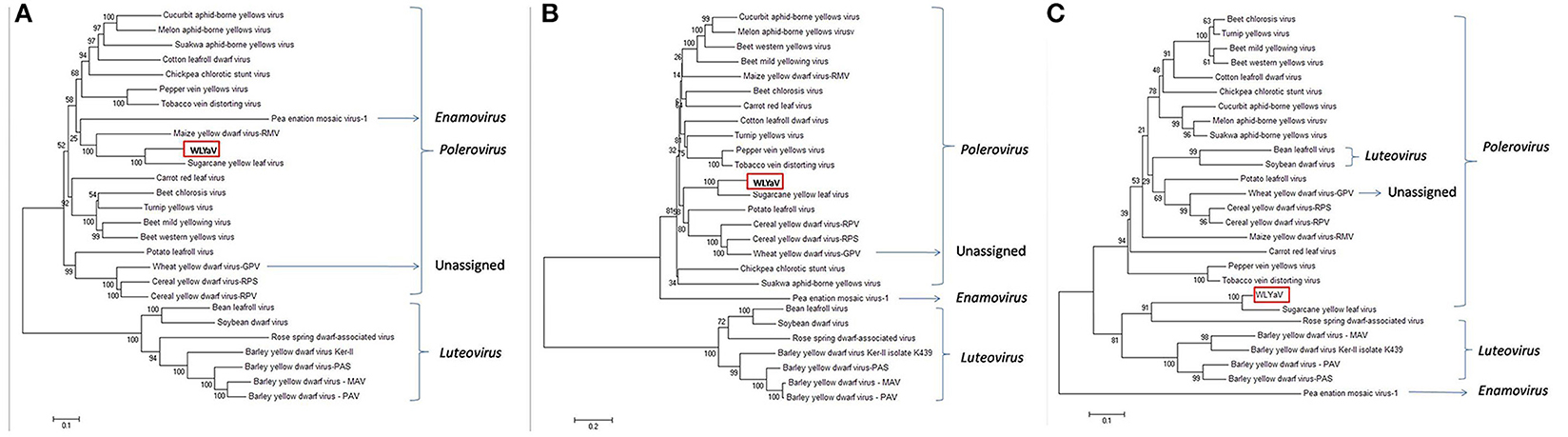
Figure 5. Phylogenetic analyses of WLYaV with other viruses in the family Luteoviridae. The phylogenetic trees were generated using the neighbor-joining method by MEGA 6 software. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1,000 replicates) is shown next to the branches. Phylogenetic tree based on (A) complete genome nucleotide sequences, deduced (B) RdRp and (C) CP amino acid sequences. The three phylogenetic trees had similar topologies, and both phylogenetic trees showed that WLYaV was most closely related to ScYLV and belonged to genus Polerovirus. Virus accession numbers are listed in Table S2.
Agroinoculation Infectivity Assay
The full-length cDNA clone of WLYaV was constructed using vector pCB301, namely pCB301-WLYaV (Figure 6A), and confirmed by colony PCR using primers A-Stu-F and B-Sal-R (Figure 6B) and Sanger sequencing. The positive clone was used to agroinoculate 2-week-old leaves of N. benthamiana plants. Typical systemic symptom of leaf yellowing was observed 2 weeks later and more obvious 4 weeks later (Figure 6C). Leaves were confirmed as systemically infected using RT-PCR and subsequent sequencing of the amplification product (Figure 6D). This test was repeated three times; each time about 60–70% plants were systemically infected.
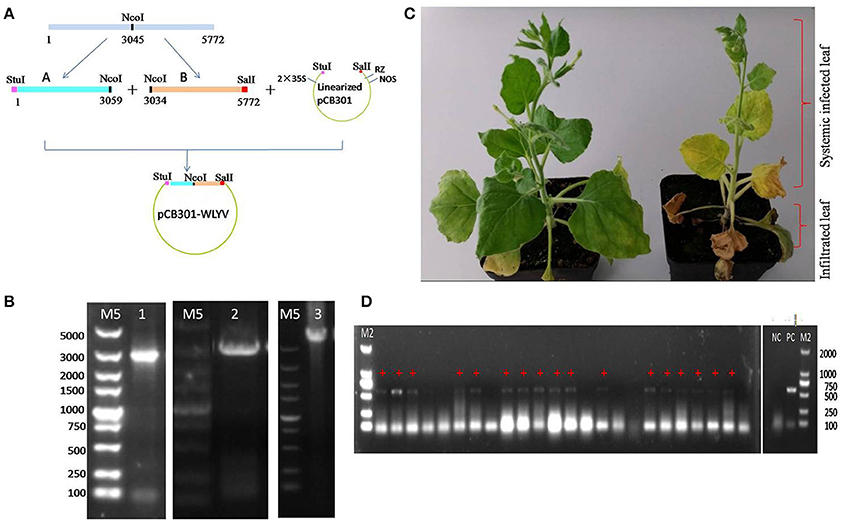
Figure 6. Agroinoculation infectivity assay of cDNA clone on N. benthamiana. (A) Construction of infectious cDNA clone. Whole viral genome was cloned using fragments A and B digested with StuI, NcoI and SalI and ligated with StuI-SalI digested pCB301 vector, 2 × 35S: promoter, RZ: ribozyme, NOS: teminator. (B) Confirmation of the constructs obtained by PCR, M5: DL 5 000 DNA marker, 1: pEASYT5-A, 2: pEASYT5-B, 3: pCB301-WLYaV. (C) Symptoms on infected N. benthamiana 4 weeks after infiltration of lower 3~4 leaves; upper leaves are systemically infected. Left: inoculated with pCB301 as negative control, no obvious symptoms; right: inoculated with pCB301-WLYaV, stunted plants have leaf yellowing. (D) Detection of WLYaV in N. benthamiana by RT-PCR 14 days post-inoculation, M2: DL 2 000 DNA marker, 17/24 plants were positive, NC: negative control (pCB301-inoculated N. benthamiana), PC: positive control (JN-U3), +: exhibiting yellowing symptom.
Discussion
The recent advent of NGS, combined with bioinformatics, has accelerated the discovery and identification of virus sequences in organisms including plant, fungi and insects because it does not depend on previous information and can detect both known and unknown viruses (Kreuze et al., 2009; Wu et al., 2010; Li et al., 2012; Massart et al., 2014; Chen et al., 2015; Wang et al., 2016). More than 100 novel DNA and RNA plant viruses from different genera and families have been identified in recent years (Roossinck et al., 2015; Wu et al., 2015; Xin et al., 2017a,b). This technology also has been used to identify viruses in family Luteoviridae, resulting in the discovery of novel viruses such as pepper yellow leaf curl virus, citrus vein enation virus, and maize yellow mosaic virus (MaYMV) (Dombrovsky et al., 2013; Vives et al., 2013; Chen et al., 2016). Importantly, the discovery of a new viral species must be followed by its biological characterization to evaluate its impact at biosecurity, commercial, regulatory, and scientific levels (Massart et al., 2017). Our work is an example for identification of a novel virus from winter wheat sample showing dull yellowing and little dwarfing symptoms by means of sRNA sequencing technology, combined with RT-PCR and RACE-PCR to obtain the full-length genome sequence, which is the first step of its biological characterization. This virus, WLYaV, has a positive-polarity ssRNA genome with seven ORFs encoding at least seven proteins. BLAST analysis showed that WLYaV has similarities to members of the family Luteoviridae, and it has the highest identity with ScYLV.
Although the multiple sequence alignments of the coding and non-coding regions of the genome indicated that WLYaV had lower identities with other members in family Luteoviridae (described above), it has many features similar to those of members of the genus Polerovirus, either in genome organization or conserved sequences. Similar to other poleroviruses, WLYaV has an extra ORF0 at the 5′ terminal that is absent in luteoviruses and lacks the 3′ proximal ORF6 present in luteoviruses. The Luteoviridae block (ORFs 3, 4, and 5) was also found in its genome. The WLYaV and ScYLV F-box like motifs appear similar (IPIIL and VPILL, respectively) and both slightly differ from the conserved motif LPxxL/I found in other poleroviruses. The putative RdRp of WLYaV is encoded by ORF2 and it might be expressed by −1 ribosomal frameshift driven by the −1 frameshift signals at the overlap of ORF1 and 2 (nt 1,658~2,099). These include a slippery site (GGGAAAC in WLYaV, nt 1,658~1,664), which is a common feature of the viruses in family Luteoviridae (Krueger et al., 2013). ScYLV has the polerovirus conserved sequence ACAAAA at both of the 5′ extremity and the ends of ORF2, while WLYaV does not have this conserved sequence. Instead, there is a sequence ACUAAA respectively at the beginning of 5′ terminal and the end of ORF2 (nt 3,350–3,355) of WLYaV. Similarly, CYDV-RPS and PLRV have a conserved sequence ACAAAA at the end of ORF2, but having CAAAAC and CUUUAU respectively at the beginning of the 5′ terminal. According to the identities and phylogenetic relationships, WLYaV has the closest relationship with ScYLV, and an inter-species recombination may have occurred independently during the course of evolution and might contribute to genetic diversity of the members of family Luteoviridae. All the features suggested that WLYaV belongs to genus Polerovirus.
ScYLV, a member of the genus Polerovirus, family Luteoviridae, causes severe leaf symptoms in sugarcane and exhibits significant genetic diversity with two phylogroups (Elsayed et al., 2017). Sequences comparison and phylogenetic analyses of WLYaV with 10 representative isolates of ScYLV (Table S4, Figure S3) indicated that WLYaV and ScYLV are distinct. Considering all the results, we propose WLYaV as a new species according to the criteria in the 9th Report of the ICTV for species demarcation in the family Luteoviridae (Domier, 2012).
Analysis of WLYaV vsiRNAs revealed similar trend with MaYMV vsiRNAs (Chen et al., 2016), i.e., hotspots within 3′ half of the genome probably due to the production of sgRNA which is required for CP and RTD expression. The hotspots within 5′ terminal suggested the structural elements, such as stem-loops, at the 5′ terminus, and coding regions may be preferentially targeted by the host Dicer (Jaag et al., 2003; Miller et al., 2015). In Arabidopsis thaliana, 21-, 22- and 24-nt long siRNAs were produced by DCL4, DCL2 and DCL3, respectively (Mlotshwa et al., 2008). Most WLYaV siRNAs were 22 and 21 nt sRNAs while the total sRNAs peaked at 21 and 24 nt. Therefore, it is assumed that DCL2 and DCL4 likely play important role in generating WLYaV siRNAs whereas in most viruses and viroids, the 21 nt species is the predominant class (Donaire et al., 2009; Ma et al., 2011; Li et al., 2012; Zhang et al., 2014). For poleroviruses such as cotton leafroll dwarf virus, MaYMV, and brassica yellows virus, the abundant vsiRNA class is 22 nt (Silva et al., 2011; Chen et al., 2016; Zhou et al., 2017). This suggests that these poleroviruses and WLYaV may affect DCL4 activity which is then compensated by DCL2.
To complete the Koch's postulates for new pathogen identification, we constructed infectious cDNA clone of WLYaV, which was then used to inoculate N. benthamiana, similar to the method used to identify other viruses. A cDNA clone of MaYMV (genus Polerovirus, family Luteoviridae) caused systemic, symptomless infection of N. benthamiana, but not in maize and oat plants (Chen et al., 2016). Citrus tristeza virus (a closterovirus), which failed to agroinfect citrus plants and lacked an experimental herbaceous host, was also successfully used to agroinoculate N. benthamiana (Ambrós et al., 2011). In our study, at 14 d after inoculation WLYaV was detected by RT-PCR in non-inoculated leaves of the plant with symptom of yellowing. This result suggested that WLYaV can be transmitted to another host and also cause disease. For the future, the important characterization steps to evaluate the risks for the wheat production will be identifying WLYaV vector species and evaluating virus prevalence and symptomatology in wheat fields on a large scale (Massart et al., 2017). More specifically, we will collect potential vector insect species in the wheat field where the samples were collected for transmission assays in the laboratory. We will give closer attention to Melanaphis sacchari (sugarcane aphid) and Rhopalosiphum maidis (corn leaf aphid), which reportedly transmit the closest relative of WLYaV, ScYLV (Chinnaraja and Viswanathan, 2015).
In conclusion, we identified a new wheat virus that is associated with wheat leaf yellowing disease, sequenced, and analyzed its whole genome sequence and organization. Additionally, we constructed an infectious cDNA clone of WLYaV that can systemically infect plants of N. benthamiana and cause leaf yellowing symptoms, thus adding a step to fulfill Koch's postulates. There are still many questions about WLYaV to be answered: what are its natural vector insects, host ranges, distribution and damage in China? However, we anticipate that findings from this study will lead to a better understanding of the incidence and distribution of different wheat viruses in China, and then determine the best control measures.
Author Contributions
XW, Conceived and designed the experiments. PZ, YL, WL, and MC, Performed the experiments. PZ, WL, MC, and SM, Analyzed the data. XW, PZ, and SM, Wrote the manuscript. All authors read and approved the final manuscript.
Funding
This research was supported by the Inter-Governmental S&T Cooperation Proposal (2016YFE0131000) and the Special Fund for Agro-scientific Research in the Public Interest (201303021).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was supported by the Inter-Governmental S&T Cooperation Proposal (2016YFE0131000) and the Special Fund for Agro-scientific Research in the Public Interest (201303021). We also thank Prof. Xiaorong Tao, Nanjing Agricultural University for providing the pCB301 vector.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.01689/full#supplementary-material
References
Ambrós, S., El-Mohtar, C., Ruiz-Ruiz, S., Peña, L., Guerri, J., Dawson, W. O., et al. (2011). Agroinoculation of citrus tristeza virus causes systemic infection and symptoms in the presumed nonhost Nicotiana benthamiana. Mol. Plant Microbe Interact. 24:1119. doi: 10.1094/MPMI-05-11-0110
Brault, V., van den Heuvel, J. F., Verbeek, M., Ziegler-graff, V., Reutenauer, A., Herrbach, E., et al. (1995). Aphid transmission of beet western yellows luteovirus requires the minor capsid read-through protein P74. EMBO J. 14, 650–659.
Chen, S., Huang, Q., Wu, L., and Qian, Y. (2015). Identification and characterization of a maize-associated mastrevirus in China by deep sequencing small RNA populations. Virol. J. 12:156. doi: 10.1186/s12985-015-0384-3
Chen, S., Jiang, G., Wu, J., Liu, Y., Qian, Y., and Zhou, X. (2016). Characterization of a novel polerovirus infecting maize in China. Viruses 8:120. doi: 10.3390/v8050120
Chinnaraja, C., and Viswanathan, R. (2015). Quantification of sugarcane yellow leaf virus in sugarcane following transmission through aphid vector, Melanaphis sacchari. Virus Dis. 26, 237–242. doi: 10.1007/s13337-015-0267-7
Dombrovsky, A., Glanz, E., Lachman, O., Sela, N., Doron-Faigenboim, A., and Antignus, Y. (2013). The complete genomic sequence of pepper yellow leaf curl virus (PYLCV) and its implications for our understanding of evolution dynamics in the genus polerovirus. PLoS ONE 8:e70722. doi: 10.1371/journal.pone.0070722
Domier, L. L. (2012). “Family luteoviridae,” in Virus Taxonomy: Ninth Report of the International Committee on Taxonomy of Viruses, eds M. Q. K. Andrew, L. Elliot, J. A. Michael, and E. B. Carstens (San Diego, CA: Elsevier Academic Press), 1045–1053.
Domier, L. L., McCoppin, N. K., Larsen, R. C., and D'Arcy, C. J. (2002). Nucleotide sequence shows that bean leafroll virus has a luteovirus-like genome organization. J. Gen. Virol. 83, 1791–1798. doi: 10.1099/0022-1317-83-7-1791
Donaire, L., Wang, Y., Gonzalezibeas, D., Mayer, K. F., Aranda, M. A., and Llave, C. (2009). Deep-sequencing of plant viral small RNAs reveals effective and widespread targeting of viral genomes. Virology 392, 203–214. doi: 10.1016/j.virol.2009.07.005
Elsayed, A. I., Boulila, M., Odero, D. C., and Komor, E. (2017). Phylogenetic and recombination analysis of sorghum isolates of sugarcane yellow leaf virus. Plant Pathol. doi: 10.1111/ppa.12708. [Epub ahead of print].
Guilley, H., Wipf-Scheibel, C., Richards, K., Lecoq, H., and Jonard, G. (1994). Nucleotide sequence of cucurbit aphid-borne yellows luteovirus. Virology 202, 1012–1017. doi: 10.1006/viro.1994.1429
Hamilton, A. J., and Baulcombe, D. C. (1999). A species of small antisense RNA in posttranscriptional gene silencing in plants. Science 286, 950–952. doi: 10.1126/science.286.5441.950
Hawkes, J. R., and Jones, R. A. C. (2005). Incidence and distribution of Barley yellow dwarf virus and Cereal yellow dwarf virus in over-summering grasses in a mediterranean-type environment. Aust. J. Agric. Res. 56, 257–270. doi: 10.1071/AR04259
Hwang, Y. T., Kalischuk, M., Fusaro, A. F., Waterhouse, P. M., and Kawchuk, L. (2013). Small RNA sequencing of Potato leafroll virus-infected plants reveals an additional subgenomic RNA encoding a sequence-specific RNA-binding protein. Virology 438, 61–69. doi: 10.1016/j.virol.2012.12.012
Jaag, H. M., Kawchuk, L., Rohde, W., Fischer, R., Emans, N., and Prüfer, D. (2003). An unusual internal ribosomal entry site of inverted symmetry directs expression of a potato leafroll polerovirus replication-associated protein. Proc. Natl. Acad. Sci. U.S.A. 100, 8939–8944. doi: 10.1073/pnas.1332697100
Jin, Z., Wang, X., Chang, S., and Zhou, G. (2004). The complete nucleotide sequence and its organization of the genome of Barley yellow dwarf virus-GAV. SCI. China Ser. C Life Sci. 47, 175–182. doi: 10.1360/03yc0076
Kozlowska-Makulska, A., Guilley, H., Szyndel, M. S., Beuve, M., Lemaire, O., Herrbach, E., et al. (2010). P0 proteins of European beet-infecting poleroviruses display variable RNA silencing suppression activity. J. Gen. Virol. 91, 1082–1091. doi: 10.1099/vir.0.016360-0
Kreuze, J. F., Perez, A., Untiveros, M., Quispe, D., Fuentes, S., Barker, I., et al. (2009). Complete viral genome sequence and discovery of novel viruses by deep sequencing of small RNAs: a generic method for diagnosis, discovery and sequencing of viruses. Virology 388, 1–7. doi: 10.1016/j.virol.2009.03.024
Krueger, E., Beckett, R., Gray, S., and Miller, W. A. (2013). The complete nucleotide sequence of the genome of Barley yellow dwarf virus-RMV reveals it to be a new Polerovirus distantly related to other yellow dwarf viruses. Front. Microbiol. 4:205. doi: 10.3389/fmicb.2013.00205
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Li, R., Gao, S., Hernandez, A. G., Wechter, W. P., Fei, Z., and Li, K. (2012). Deep sequencing of small RNAs in tomato for virus and viroid identification and strain differentiation. PLoS ONE 7:e37127. doi: 10.1371/journal.pone.0037127
Lister, R., and Ranieri, R. (1995). “Distribution and economic importance of Barley yellow dwarf,” in Barley Yellow Dwarf: 40 Years of Progress, eds C. J. D'Arcy and P. A. Burnett (St. Paul, MN: APS Press), 29–53.
Liu, F., Wang, X., Liu, Y., Xie, J., Gray, S. M., Zhou, G., et al. (2007). A Chinese isolate of barley yellow dwarf virus-PAV represents a third distinct species within the PAV serotype. Arch. Virol. 152, 1365–1373. doi: 10.1007/s00705-007-0947-8
Liu, X., Wu, J., Wang, J., Liu, X., Zhao, S., Li, Z., et al. (2009). WebLab: a data-centric, knowledge-sharing bioinformatic platform. Nucleic Acids Res. 37, W33–W39. doi: 10.1093/nar/gkp428
Liu, Y., Sun, B. X., Zheng, C., and Zhou, G. (2007). Three digoxigenin-labeled cDNA probes for specific detection of the natural population of Barley yellow dwarf viruses in China by dot-blot hybridization. J. Virol. Methods 145, 22–29. doi: 10.1016/j.jviromet.2007.05.006
Luciozavaleta, E., Smith, D. M., and Gray, S. M. (2007). Variation in transmission efficiency among Barley yellow dwarf virus-RMV isolates and clones of the normally inefficient aphid vector, Rhopalosiphum padi. Phytopathology 91, 792–796. doi: 10.1094/PHYTO.2001.91.8.792
Ma, M., Huang, Y., Gong, Z., Zhuang, L., Li, C., Yang, H., et al. (2011). Discovery of DNA viruses in wild-caught mosquitoes using small RNA high throughput sequencing. PLoS ONE 6:e24758. doi: 10.1371/journal.pone.0024758
Massart, S., Candresse, T., Gil, J., Lacomme, C., Predjana, L., Ravnikar, M., et al. (2017). Framework for the evaluation of biosecurity, commercial, regulatory and scientific impacts of plant viruses and viroids identified by NGS technologies. Front. Microbiol. 8:45. doi: 10.3389/fmicb.2017.00045
Massart, S., Olmos, A., Jijakli, H., and Candresse, T. (2014). Current impact and future directions of high throughput sequencing in plant virus diagnostics. Virus Res. 188, 90–96. doi: 10.1016/j.virusres.2014.03.029
Mehta, Y. R. (ed.). (2014). “Wheat and wheat production constraints,” in Wheat Diseases and their Management (Cham: Springer Press), 1–16.
Mierlo, J. T. V., Cleef, K. W. R. V., and Rij, R. P. V. (2010). Small silencing RNAs: piecing together a viral genome. Cell Host Microbe 7, 87–89. doi: 10.1016/j.chom.2010.02.001
Miller, W. A., and Rasochová, L. (1997). Barley yellow dwarf viruses. Annu. Rev. Phytopathol. 35, 167–190. doi: 10.1146/annurev.phyto.35.1.167
Miller, W. A., Dinesh-Kumar, S. P., and Paul, C. P. (1995). Luteovirus gene expression. Crit. Rev. Plant Sci. 14, 179–211. doi: 10.1080/07352689509701926
Miller, W. A., Jackson, J., and Feng, Y. (2015). Cis- and trans-regulation of luteovirus gene expression by the 3' end of the viral genome. Virus Res. 206, 37–45. doi: 10.1016/j.virusres.2015.03.009
Miller, W. A., Liu, S., and Beckett, R. (2002). Barley yellow dwarf virus: Luteoviridae or Tombusviridae? Mol. Plant Pathol. 3, 177–183. doi: 10.1046/j.1364-3703.2002.00112.x
Mlotshwa, S., Pruss, G. J., and Vance, V. (2008). Small RNAs in viral infection and host defense. Trends Plant Sci. 13, 375–382. doi: 10.1016/j.tplants.2008.04.009
Mo, X., Chen, Z., and Chen, J. (2010). Complete nucleotide sequence and genome organization of a Chinese isolate of Tobacco vein distorting virus. Virus Genes 41, 425–431. doi: 10.1007/s11262-010-0524-1
Moonan, F., Molina, J., and Mirkov, T. E. (2000). Sugarcane yellow leaf virus: an emerging virus that has evolved by recombination between luteoviral and poleroviral ancestors. Virology 269, 156–171. doi: 10.1006/viro.1999.0162
Nickel, H., Kawchuk, L., Twyman, R. M., Zimmermann, S., Junghans, H., Winter, S., et al. (2008). Plantibody-mediated inhibition of the Potato leafroll virus P1 protein reduces virus accumulation. Virus Res. 136, 140–145. doi: 10.1016/j.virusres.2008.05.001
Nixon, P. L., Rangan, A., Kim, Y. G., Rich, A., Hoffman, D. W., Hennig, M., et al. (2002). Solution structure of a Luteoviral P1-P2 frameshifting mRNA pseudoknot. J. Mol. Biol. 322, 621–633. doi: 10.1016/S0022-2836(02)00779-9
Pazhouhandeh, M., Dieterle, M., Marrocco, K., Lechner, E., Berry, B., Brault, V., et al. (2006). F-box-like domain in the polerovirus protein P0 is required for silencing suppressor function. Proc. Natl. Acad. Sci. U.S.A. 103, 1994–1999. doi: 10.1073/pnas.0510784103
Pfeffer, S., Dunoyer, P., Heim, F., Richards, K. E., Jonard, G., and Ziegler-Graff, V. (2002). P0 of beet western yellows virus is a suppressor of posttranscriptional gene silencing. J. Virol. 76, 6815–6824. doi: 10.1128/JVI.76.13.6815-6824.2002
Pfeffer, S., Dunoyer, P., Heim, F., Richards, K. E., Jonard, G., and Zieglergraff, V. (2017). Retraction for pfeffer et al. P0 of beet western yellows virus is a suppressor of posttranscriptional gene silencing. J. Virol. 91:e00022–17. doi: 10.1128/JVI.00022-17
Prüfer, D., Tacke, E., Schmitz, J., Kull, B., Kaufmann, A., and Rohde, W. (1992). Ribosomal frameshifting in plants: a novel signal directs the -1 frameshift in the synthesis of the putative viral replicase of potato leafroll luteovirus. EMBO J. 11, 1111–1117.
Rochow, W. F. (1969). Biological properties of four isolates of barley yellow dwarf virus. Phytopathology 59, 1580–1589.
Rochow, W. F., and Muller, I. (1971). A fifth variant of barley yellow dwarf virus in New York. Plant Dis. 55, 874–877.
Roossinck, M. J., Martin, D. P., and Roumagnac, P. (2015). Plant virus metagenomics: advances in virus discovery. Phytopathology 105, 716–727. doi: 10.1094/PHYTO-12-14-0356-RVW
Rotenberg, D., Bockus, W. W., Whitfield, A. E., Hervey, K., Baker, K. D., Ou, Z., et al. (2016). Occurrence of viruses and associated grain yields of paired symptomatic and nonsymptomatic tillers in Kansas winter wheat fields. Phytopathology 106, 202–210. doi: 10.1094/PHYTO-04-15-0089-R
Shen, Y., Zhao, X., Yao, M., Li, C., Miriam, K., Xue, Z., et al. (2014). A versatile complementation assay for cell-to-cell and long distance movements by cucumber mosaic virus based agro-infiltration. Virus Res. 190, 25–33. doi: 10.1016/j.virusres.2014.06.013
Silva, T. F., Romanel, E. A., Andrade, R. R., Farinelli, L., Østerås, M., Deluen, C., et al. (2011). Profile of small interfering RNAs from cotton plants infected with the polerovirus cotton leafroll dwarf virus. BMC Mol. Biol. 12:40. doi: 10.1186/1471-2199-12-40
Simmonds, P., Adams, M. J., Benko, M., Breitbart, M., Brister, J. R., Carstens, E. B., et al. (2017). Consensus statement: virus taxonomy in the age of metagenomics. Nat. Rev. Microbiol. 15, 161–168. doi: 10.1038/nrmicro.2016.177
Smirnova, E., Firth, A. E., Miller, W. A., Scheidecker, D., Brault, V., Reinbold, C., et al. (2015). Discovery of a small non-AUG-initiated ORF in poleroviruses and luteoviruses that is required for long-distance movement. PLoS Pathog. 11:e1004868. doi: 10.1371/journal.ppat.1004868
Smith, G. R., Borg, Z., Lockhart, B. E., Braithwaite, K. S., and Gibbs, M. J. (2000). Sugarcane yellow leaf virus: a novel member of the Luteoviridae that probably arose by inter-species recombination. J. Gen. Virol. 81, 1865–1869. doi: 10.1099/0022-1317-81-7-1865
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Vives, M. C., Velázquez, K., Pina, J. A., Moreno, P., Guerri, J., and Navarro, L. (2013). Identification of a new enamovirus associated with citrus vein enation disease by deep sequencing of small RNAs. Phytopathology 103, 1077–1086. doi: 10.1094/PHYTO-03-13-0068-R
Voinnet, O. (2001). RNA silencing as a plant immune system against viruses. Trends Genet. 17, 449–459. doi: 10.1016/S0168-9525(01)02367-8
Wang, Q., Ma, X., Qian, S., Zhou, X., Sun, K., Chen, X., et al. (2015). Rescue of a plant negative-strand RNA virus from cloned cDNA: insights into enveloped plant virus movement and morphogenesis. PLoS Pathog. 11:e1005223. doi: 10.1371/journal.ppat.1005223
Wang, S., Zhang, C., Cheng, R., Yu, X., and Lu, J. (2016). A Cripavirus in the brown planthopper, Nilaparvata lugens. J. Gen. Virol. 97, 706–714. doi: 10.1099/jgv.0.000394
Wang, X., Liu, Y., Han, C., Wu, Y., and Zhao, Z. (2010). Present situation and developing strategies for the research and control of wheat viral diseases. Plant Prot. 36, 13–19. doi: 10.3969/j.issn.0529-1542.2010.03.004
Wu, B., Blanchardletort, A., Liu, Y., Zhou, G., Wang, X., and Elena, S. F. (2011). Dynamics of molecular evolution and phylogeography of Barley yellow dwarf virus-PAV. PLoS ONE 6:e16896. doi: 10.1371/journal.pone.0016896
Wu, Q., Ding, S., Zhang, Y., and Zhu, S. (2015). Identification of viruses and viroids by next-generation sequencing and homology-dependent and homology-independent algorithms. Ann. Rev. Phytopathol. 53, 425–444. doi: 10.1146/annurev-phyto-080614-120030
Wu, Q., Luo, Y., Lu, R., Lau, N., Lai, E. C., Li, W. X., et al. (2010). Virus discovery by deep sequencing and assembly of virus-derived small silencing RNAs. Proc. Natl. Acad. Sci. U.S.A. 107, 1606–1611. doi: 10.1073/pnas.0911353107
Xin, M., Cao, M., Liu, W., Ren, Y., Lu, C., and Wang, X. (2017a). The genomic and biological characterization of Citrullus lanatus cryptic virus infecting watermelon in China. Virus Res. 232, 106–112. doi: 10.1016/j.virusres.2017.02.009
Xin, M., Cao, M., Liu, W., Ren, Y., Zhou, X., and Wang, X. (2017b). Two negative-strand RNA viruses identified in watermelon represent a novel clade in the order Bunyavirales. Front. Microbiol. 8:1514. doi: 10.3389/fmicb.2017.01514
Zerbino, D. R., and Birney, E. (2008). Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18, 821–829. doi: 10.1101/gr.074492.107
Zhang, W., Cheng, Z., Xu, L., Wu, M., Waterhouse, P., Zhou, G., et al. (2009). The complete nucleotide sequence of the barley yellow dwarf GPV isolate from China shows that it is a new member of the genus Polerovirus. Arch. Virol. 154, 1125–1128. doi: 10.1007/s00705-009-0415-8
Zhang, Z., Qi, S., Tang, N., Zhang, X., Chen, S., Zhu, P., et al. (2014). Discovery of replicating circular RNAs by RNA-seq and computational algorithms. PLoS Pathog. 10:e1004553. doi: 10.1371/journal.ppat.1004553
Zhao, K., Liu, Y., and Wang, X. (2010). Reverse transcription loop-mediated isothermal amplification of DNA for detection of barley yellow dwarf viruses in China. J. Virol. Methods 169, 211–214. doi: 10.1016/j.jviromet.2010.06.020
Zhou, C., Zhang, X., Liu, S., Wang, Y., Li, D., Yu, J. L., et al. (2017). Synergistic infection of BrYV and PEMV 2 increases the accumulations of both BrYV and BrYV-derived siRNAs in Nicotiana benthamiana. Sci. Rep. 7:45132. doi: 10.1038/srep45132
Keywords: deep sequencing, wheat, leaf yellowing, Luteoviridae, Polerovirus, infectious cDNA clone
Citation: Zhang P, Liu Y, Liu W, Cao M, Massart S and Wang X (2017) Identification, Characterization and Full-Length Sequence Analysis of a Novel Polerovirus Associated with Wheat Leaf Yellowing Disease. Front. Microbiol. 8:1689. doi: 10.3389/fmicb.2017.01689
Received: 10 May 2017; Accepted: 21 August 2017;
Published: 06 September 2017.
Edited by:
David Gilmer, University of Strasbourg, FranceReviewed by:
Eugene I. Savenkov, Swedish University of Agricultural Sciences, SwedenHideki Kondo, Okayama University, Japan
W. Allen Miller, Iowa State University, United States
Copyright © 2017 Zhang, Liu, Liu, Cao, Massart and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sebastien Massart, c2ViYXN0aWVuLm1hc3NhcnRAdWxnLmFjLmJl
Xifeng Wang, eGZ3YW5nQGlwcGNhYXMuY24=