 Jin-Pei Wang
Jin-Pei Wang Wen-Mao Zhang1
Wen-Mao Zhang1 Hong-Jun Chao
Hong-Jun Chao Ning-Yi Zhou
Ning-Yi Zhou- 1Wuhan Institute of Virology, Chinese Academy of Sciences, Wuhan, China
- 2University of Chinese Academy of Sciences, Beijing, China
- 3State Key Laboratory of Microbial Metabolism and School of Life Sciences & Biotechnology, Shanghai Jiao Tong University, Shanghai, China
A LysR-type transcriptional regulator (LTTR), PnpR, has previously been shown to activate the transcription of operons pnpA, pnpB, and pnpCDEFG for para-nitrophenol (PNP) degradation in Pseudomonas sp. strain WBC-3. Further preliminary evidence suggested the possible presence of an LTTR additional binding site in the promoter region of pnpCDEFG. In this study, an additional LTTR PnpM, which shows 44% homology to PnpR, was determined to activate the expression of pnpCDEFG. Interestingly, a pnpM-deleted WBC-3 strain was unable to grow on PNP but accumulating hydroquinone (HQ), which is the catabolic product from PNP degradation by PnpAB and the substrate for PnpCD. Through electrophoretic mobility shift assays (EMSAs) and promoter activity detection, only PnpR was involved in the activation of pnpA and pnpB, but both PnpR and PnpM were involved in the activation of pnpCDEFG. DNase I footprinting analysis suggested that PnpR and PnpM shared the same DNA-binding regions of 27 bp in the pnpCDEFG promoter. In the presence of PNP, the protection region increased to 39 bp by PnpR and to 38 bp by PnpM. Our data suggested that both PnpR and PnpM were involved in activating pnpCDEFG expression, in which PNP rather than the substrate hydroquinone for PnpCD is the inducer. Thus, during the PNP catabolism in Pseudomonas sp. strain WBC-3, pnpA and pnpB operons for the initial two reactions were controlled by PnpR, while the third operon (pnpCDEFG) for HQ degradation was activated by PnpM and PnpR. This study builds upon our previous findings and shows that two LTTRs PnpR and PnpM are involved in the transcriptional activation of these three catabolic operons. Specifically, our identification that an LTTR, PnpM, regulates pnpCDEFG expression provides new insights in an intriguing regulation system of PNP catabolism that is controlled by two regulators.
Introduction
It is well-known that versatile bacterial strains swiftly adapt and respond to polluted environments. One such adaptation is based on modulation of gene expression, particularly those encoding bacterial catabolism of pollutants. Various transcriptional regulation systems play an important role in adaptive responses (Cases and de Lorenzo, 2001; Shingler, 2003). Regulatory proteins are the key elements that control the transcription of catabolic operons to ensure the successful establishment of a catabolic pathway (Diaz and Prieto, 2000). The LysR-type transcriptional regulators (LTTRs) are the most abundant in prokaryotes, controlling diverse bacterial functions, including stress response, motility, antibiotic resistance, quorum sensing, aromatic compound degradation, and amino-acid biosynthesis (Schell, 1993; Maddocks and Oyston, 2008). Tetramers are the active form of LTTRs, and their interaction with the promoter regions generally occurs at two dissimilar sites: regulatory binding site (RBS) and activation binding site (ABS). The RBS contains an LTTR consensus binding motif (T-N11-A) and is usually centered near position −65 relative to the transcriptional start site of the activated promoter. While ABS, for which no conserved sequence motif has been identified thus far, is located near position −35. Although LTTRs are capable of binding DNA in the absence of inducer molecules, transcriptional activation of downstream genes requires inducers (Schell, 1993; Diaz and Prieto, 2000; Tropel and van der Meer, 2004; Maddocks and Oyston, 2008).
As a typical representative of mononitrophenols, para-nitrophenol (PNP) is toxic to humans and is widely utilized in the chemical syntheses of pharmaceuticals, dyes and pesticides (Karim and Gupta, 2002). So far, a number of bacterial strains capable of utilizing PNP have been isolated and their diverse catabolic pathways have been elucidated (Spain and Gibson, 1991; Jain et al., 1994; Roldan et al., 1998; Arora et al., 2014). Pseudomonas sp. strain WBC-3 utilizes PNP as its sole source of carbon, nitrogen, and energy. Strain WBC-3 metabolizes PNP via the hydroquinone (HQ) pathway and the genes involved in the PNP catabolism are located on three different operons: pnpA (encoding PNP 4-monoxygenase catalyzing monooxygenation of PNP to p-benzoquinone), pnpB [encoding p-benzoquinone (BQ) reductase converting p-benzoquinone to HQ], and pnpCDEFG (encoding the enzymes catalyzing the conversion of HQ to β-ketoadipate) (Zhang et al., 2009), as shown in Figure 1A. PnpR, encoded by a gene at least 16 kb away from the PNP catabolic operons, is a LTTR and found to be involved in the regulation of all three operons (Zhang et al., 2015). In other PNP utilizers, regulatory proteins were also proposed to be involved in PNP catabolism: (1) LTTR PnpR regulated HQ degradation based on the gene knock-out in Pseudomonas putida DLL-E4 (Shen et al., 2010); and (2) AraC-type regulator NphR regulated PNP oxidation based on the gene mutation and transcriptional activity analysis in Gram-positive Rhodococcus sp. strain PN1 (Takeo et al., 2008).
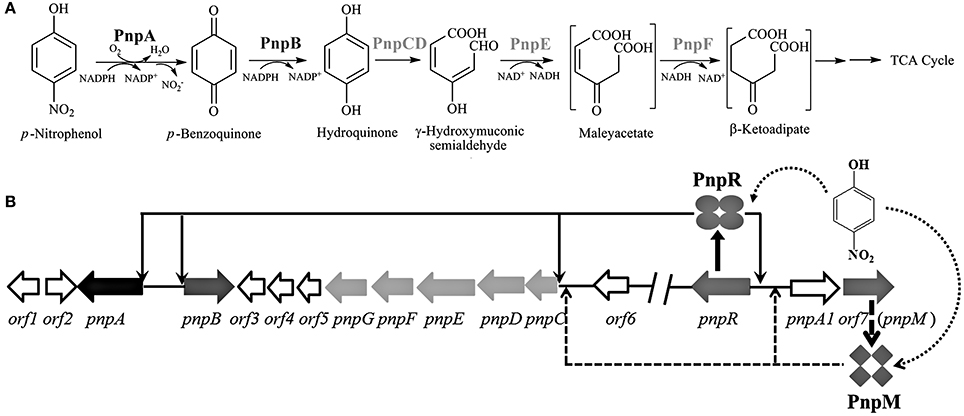
Figure 1. (A) Proposed pathway for PNP catabolism in Pseudomonas sp. strain WBC-3 (modified after Zhang et al., 2009) pnpA and pnpB are PNP 4-monooxygenase and p-benzoquinone reductase respectively; pnpCDEF encode α- and β-subunits of hydroquinone dioxygenase, γ-hydroxymuconic semialdehyde dehydrogenase and maleylacetate reductase, respectively. (B) Schematic of the regulatory circuit of the PNP catabolic cluster in Pseudomonas sp. strain WBC-3 (figure not drawn to scale, modified after Zhang et al., 2015). Four ellipses stand for PnpR tetramer, and four diamonds stand for PnpM tetramer. The curved and dotted arrow stands for binding of para-nitrophenol (PNP) with PnpR or PnpM. The down arrows stand for PnpR's activating four operons of pnpA, pnpB, pnpCDEFG, and pnpR. The up arrows stand for PnpM's activating two operons of pnpCDEFG and pnpA1M.PnpR regulates transcription initiation in four regions pnpA, pnpB, pnpCDEFG, and pnpR. PnpM regulates transcription initiation in two regions pnpCDEFG and pnpA1M.
Our previous work has shown that PnpR regulates all four operons of pnpR, pnpA, pnpB and pnpCDEFG in strain WBC-3 (Zhang et al., 2015). The promoter activities of pnpR, pnpA, and pnpB were completely lost after mutation in their corresponding RBSs. Surprisingly, no changes in promoter activity occurred between the mutant and wild type RBS of the promoter of the pnpCDEFG operon. This data suggested that the pnpCDEFG promoter possibly has evolved from a different origin, evidenced by a longer distance (20 nucleotides) between −35 box and RBS of pnpCDEFG promoter than those (9–10 nucleotides) of the other three promoters. Therefore, one of the reasons for this phenomenon was speculated to be an additional regulator involved in the activation of pnpCDEFG operon (Zhang et al., 2015). In this study, we confirmed that indeed an additional LTTR PnpM, encoded by a gene close to pnpR, was involved in the regulation of pnpCDEFG operon for HQ degradation in PNP degradation by strain WBC-3.
Materials and Methods
Bacterial Strains, Chemicals, Culture Media, and DNA Manipulations
The bacterial strains and their relevant genotypes are described in Table 1. Pseudomonas sp. strain WBC-3 was cultivated at 30°C in lysogeny broth (LB) or minimal medium (MM) with 0.5 mM of PNP as the sole source of carbon and nitrogen. Bacto agar was added to a final concentration of 15 g L−1 for the solid media. When necessary, antibiotics and other additions were used at the following concentrations: ampicillin (100 mg L−1), chloramphenicol (34 mg L−1), gentamicin (20 mg L−1), kanamycin (100 mg L−1), tetracycline hydrochloride (10 mg L−1) gentamicin (10 mg L−1), and ONPG (o-Nitrophenyl β-D-galactopyranoside, 4 mg L−1). Reagents were from Fluka Chemical Co. (Buchs, Switzerland) or Sigma Chemical Co. (St. Louis, MO).
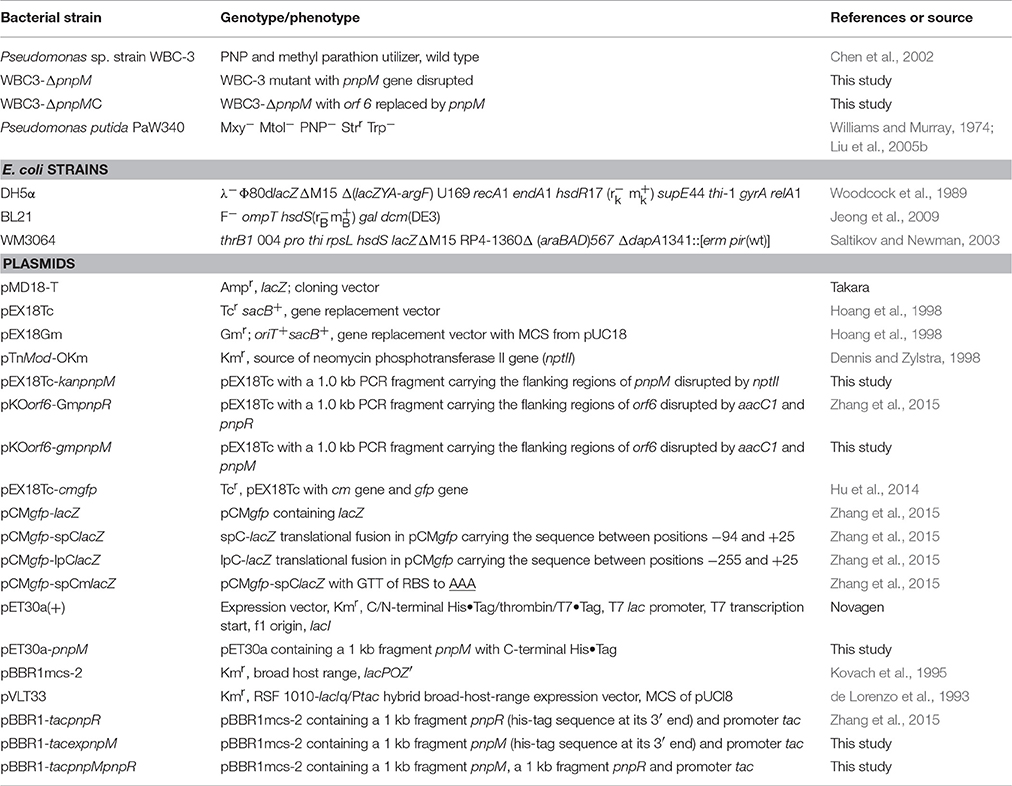
Table 1. Bacterial strains and plasmids in this study.
Plasmids used are summarized in Table 1, and the primers used are described in Table S1. All DNA manipulations were performed according to standard procedures (Sambrook and Maniatis, 1989). Restriction enzymes, DNA polymerases and T4 DNA ligase were used according to the manufacturers' specifications.
Gene Knockout and Complementation
The pnpM-knockout plasmid pEX18Tc-kanpnpM was constructed by fusion of the upstream and downstream fragments of pnpM that was amplified from genomic DNA of strain WBC-3 and the kanamycin resistance gene amplified from plasmid pTnMod-Okm (Dennis and Zylstra, 1998) to pEX18Tc (Hoang et al., 1998) with In-Fusion HD Cloning Kit (Clontech, Beijing, China). The construct pEX18Tc-kanpnpM was then transformed into E. coli WM3064 before it was conjugated with strain WBC-3 as previously described (Dehio and Meyer, 1997; Saltikov and Newman, 2003). The WBC-3ΔpnpM double-crossover recombinant was screened on LB plates with 15% (wt/vol) sucrose and 50 mg of L−1 kanamycin.
The entire pnpM together with its promoter region PpnpA1 and gentamycin resistant gene aacC1 was cloned into pKOorf6 (Zhang et al., 2015) using an In-Fusion kit, yielding construct pKOorf6-gmpnpM for pnpM complementation. The pnpM complemented strain WBC-3ΔpnpMC was obtained by the process of pnpM knockout, with the exception that 50 mg L−1 kanamycin was replaced by 10 mg L−1 gentamicin.
The growth of WT strain WBC-3 and its derivatives on 0.3 mM PNP or HQ, and their capability of utilizing the substrates was determined by monitoring their growth as well as the substrate consumption. The growth curves were fitted with modified Gompertz equation (Zwietering et al., 1990) with OriginPro 8 software.
Biotransformation
Biotransformation was performed as previously described (Chen et al., 2014) with slight modifications. Strain WBC-3ΔpnpM was grown in 100 ml MM containing 5% LB to an optical density at 600 nm (OD600) of 0.6 and then induced with 0.3 mM PNP for 6 h. The cultures were collected for high-performance liquid chromatography (HPLC) analyses, by a described method (Min et al., 2014). The specific activity is defined as the weight of cells required to convert 1 μM of substrate per minute at 30°C.
RNA Preparation and Real-Time Quantitative PCR (RT-qPCR)
Total RNA was purified with an RNAprep Pure Kit for Bacteria (Tiangen Biotech, Beijing, China), and reverse transcribed into cDNA with a PrimeScript RT Reagent kit (TaKaRa, Dalian, China). RT-qPCR was performed using the CFX96TM Real-Time PCR Detection System (Bio-Rad, Hercules, CA) following the iQ SYBR Green Supermix (Bio-Rad) manufacturer's recommendation. The expression levels of all of the genes were normalized to 16S rRNA gene expression as an internal standard and quantified according to a reported method (Livak and Schmittgen, 2001). All experiments were performed in triplicate.
5′ Rapid Amplification of cDNA Ends (5′-RACE)
The transcriptional start site (TSS) of the pnpA1M operon was determined using a SMARTer RACE cDNA amplification kit (Clontech, Mountain View, CA). First-strand cDNA was synthesized with a PrimeScript RT Reagent kit (TaKaRa, Dalian, China), and the cDNA was then performed with terminal transferase and dCTP for homopolymeric tailing. The tailed cDNA was then amplified using the abridged anchor primer (AAP) and pnpA1M-GSP2. Finally, this product was used as a template for nested PCR with AAP and primer pnpA1M-GSP3 and then cloned into pMD18-T (TaKaRa) for sequencing.
Expression and Purification of PnpM
pnpM was amplified from genomic DNA of strain WBC-3 and was cloned into pET-30a. The resulting plasmid pET30a-pnpM was transformed into E. coli BL21 (DE3) pLysS and purified as described (Liu and Zhou, 2012). PnpM purified from E. coli was demonstrated to be nonfunctional in subsequent experiments, and Pseudomonas putida strain PaW340, which is a mutant derivative of m-toluate/m-xylene utilizer Pseudomonas putida mt-2 and usually used as an expression host (Williams and Murray, 1974). It worthies to mention that Pseudomonas putida mt-2 and Pseudomonas putida PaW340 are not PNP utilizers (Liu et al., 2005b), and then the strain PaW340 was chosen as the alternative expression host.
Subsequently, the DNA fragment tac promoter (Ptac) amplified from plasmid pVLT33 (de Lorenzo et al., 1993) and the pnpM gene were cloned into pBBR1mcs-2 to construct the plasmid pBBR1-tacexpnpM. In addition, DNA fragments of Ptac, pnpM and pnpR were also cloned into pBBR1mcs-2 to construct the plasmid pBBR1-tacpnpMpnpR. The construct pBBR1mcs2-tacexpnpM was introduced into Pseudomonas putida strain PaW340. C-terminal His-tagged PnpM (PnpM-His6) was expressed and purified as previously described (Zhang et al., 2015). The concentration of purified PnpM-His6 was calculated using the Protein Assay Kit (Beyotime Co., Shanghai, China). The protein was stored in glycerol at −20°C.
Gel Filtration Chromatography
The sample containing PnpM-His6 was loaded onto a column (1.6 × 60 cm) of Hiload Superdex 200 pg (FPLC system, GE Healthcare, Little Chalfont, UK) that was equilibrated with 10 mM of Tris-HCl buffer (pH 7.5, with 0.1 M NaCl and 5% glycerol) for 2 h at a flow rate of 0.5 ml min−1. The target protein was then eluted with the same buffer. The native molecular mass of PnpM was estimated from a calibration curve plotted by using the standard proteins (Zhang et al., 2009).
Electrophoretic Mobility Shift Assays (EMSAs)
Electrophoretic Mobility Shift Assay (EMSA) was performed as previously described (Georgi et al., 2008) with minor modifications. DNA fragments containing respective promoters of pnpA, pnpB, pnpC, and pnpA1 were obtained by PCR with genomic DNA of strain WBC-3 as templates. Different amounts (0–2.0 μM) of purified PnpM-His6 were used in each reaction, respectively. Two DNA fragments (0.03 μM each), with approximately 400 bp respectively of gfp gene amplified from pEX18Tc-cmgfp (Hu et al., 2014), were used as the control DNA. After 30 min of incubation at room temperature, the samples were then loaded onto a 6% native polyacrylamide gel at 4°C and electrophoresed in Tris-borate buffer for 1.5 h at 130 V. Subsequently, the gels were stained with SYBR green I (BioTeKe Co., Beijing, China). DNA and DNA–protein complexes were visualized using an UltraBright LED Transilluminator (Maestrogen, Taiwan, China).
DNase I Footprinting Assay
Footprinting assays were performed as previously described (Zianni et al., 2006). A 270 bp DNA fragment of promoter pnpC was cloned into pUC18H (TOLO Biotech, Shanghai) in preparation of the probe. The correct construct was used as the template for further preparation of fluorescent 6-carboxyfluorescein (FAM)-labeled probes. The samples for DNase I footprinting were conducted as for the EMSAs. For each assay, the sample of binding reaction was treated as previously described (Zhang et al., 2015). The digested DNA was examined by a DNA Analyzer (Applied Biosystems, Waltham, MA, USA) and the GeneScan-LIZ500 standard size (Applied Biosystems) was used. The data analysis was the same as previously described (Wang et al., 2012).
Promoter Activity Analysis
β-Galactosidase assays were employed to analyze the expression of promoter-lacZ fusions in the surrogate host Pseudomonas putida PaW340, which is incapable of degrading PNP (Liu et al., 2005b). The activity was determined by SDS- and chloroform-permeabilized cells as described (Griffith and Wolf, 2002; Zhang et al., 2015).
Statistical Analysis
Statistical analysis was performed using Origin 8.0 software. Paired-samples tests were used to calculate probability values (p) of the transcription of pnpA, pnpC, and pnpM. Paired-samples tests and one-way analysis of variance (ANOVA) was used to calculate the probability values (P) for β-galactosidase activity analyses of pnpA, pnpC, and promoters. P-values of < 0.05 and < 0.01 were considered to be significant and highly significant, respectively.
Results
pnpM Is Essential in HQ Degradation
As hypothesized in the introduction, an additional regulator was probably involved in pnpCDEFG operon activation. The previously designated orf7 (Zhang et al., 2015) (orf WBC3-000012) from contig 001 (accession number KM019215) of strain WBC-3, right at the downstream of pnpA1 (orf WBC3-000011), was found to encode a LysR-type regulatory protein (accession number: AIV98012) (Figure 1B). The orf7 was re-designated pnpM and its product was 44% identical to PnpR that had previously been demonstrated to be involved in the four operons for PNP catabolism.
To investigate the possible regulatory role of PnpM in triggering catabolic operons for PNP degradation of strain WBC-3, pmpM was knocked out. The obtained mutant strain WBC3-ΔpnpM was no longer able to grow on PNP (Figure 2A) but could still transform PNP (yellow-green) to a brown substance without further degradation (test III in Figure 2B). This accumulated product was determined to be HQ by HPLC analysis. The insertion of plasmids in strain WBC-3 is technically challenging (Liu et al., 2005a), therefore, strain WBC3-ΔpnpM was complemented by inserting pnpM into the orf6 locus of mutant strain WBC3-ΔpnpM through a double cross-over. The complemented strain WBC3-ΔpnpMC, where pnpM replaced orf6, is able to grow on PNP (Figure 2A). Meanwhile, the wild-type strain WBC-3 was incapable of growing on HQ as the sole source of carbon (Figure 2A). However, PNP-induced cells of strain WBC-3 could degrade HQ with a specific activity of 0.78 ± 0.23 U mg−1. In a biotransformation experiment by strain WBC3-ΔpnpM, PNP consumption (249 μM) was approximately equivalent to the total accumulation of HQ (210 μM) and BQ (18 μM) (Figure 2B), indicating a nearly stoichiometric formation of HQ and BQ from PNP. These results suggested that PnpM was likely a positive regulator for the pnpCDEFG operon encoding enzymes for HQ degradation in PNP catabolism.
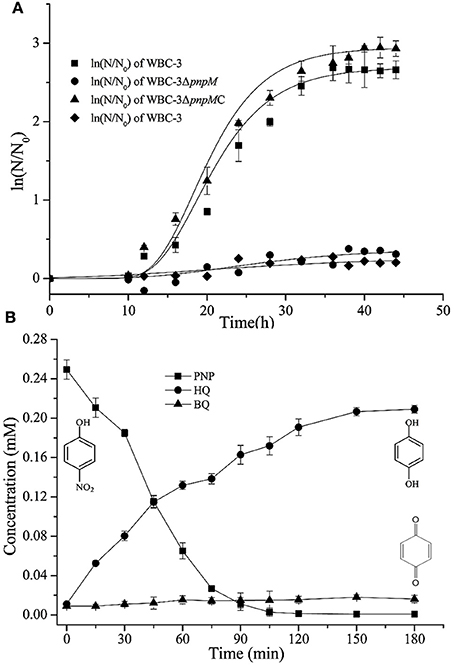
Figure 2. (A) Growth curve of strains WBC-3, WBC-3ΔpnpM, and WBC-3ΔpnpMC. Three strains were grown at 30°C for 44 h in MM containing 0.3 mM PNP, and strain WBC-3 was also grown in MM containing 0.3 mM HQ. The curves were fitted by the modified Gompertz model with Origin software. The values are averages of three independent experiments. Error bars indicate standard deviations. N, number of cells; N0, initial number of cells. Square (■) stands for strain WBC-3. Circle (•) stands for strain WBC-3ΔpnpM. Triangle (▴) stands for strain WBC-3ΔpnpMC that the gene pnpM is complemented into the genome of strain WBC3-ΔpnpM. Diamond (♦) stands for strain WBC-3 grown on MM containing 0.3 mM HQ. (B) Time course of PNP degradation by strain WBC-3ΔpnpM in whole-cell biotransformation. Samples were withdrawn at the time points indicated and treated immediately as described in the text. The disappearance of PNP and the appearance of the products (BQ and HQ) were quantified by HPLC. The experiments were performed in triplicate, the results shown are average values of three independent experiments, and error bars indicate standard deviations.
Transcriptional Analysis of PNP Catabolic Operons
To investigate the possible regulatory role on the transcriptional expression of the entire pnp gene cluster, the impact of PnpM was further analyzed by determining the transcriptional levels of all three PNP catabolic operons, as well as pnpA1M operon in both the WT strain and the mutant strain WBC-3ΔpnpM. When PNP is present, the transcriptional levels of four tested genes (each representing one of the four operons, pnpA, pnpB, pnpCDEFG, and pnpA1M as shown in Figure 1B) in WT strain WBC-3 were enhanced dramatically compared to the absence of PNP, with the transcriptional levels of pnpA, pnpB, pnpC, and pnpM being 3,465-, 2,490-, 1,292-, and 56-fold higher (Figure 3A), respectively, through qRT-PCR assay. The increased level of the three catabolic operons was at the same order of magnitude in WT strain WBC-3 from above figures, but the increased level of pnpA (130-fold) and pnpB (282-fold) in strain WBC-3ΔpnpM was about >10-fold that of pnpC (15-fold) (Figure 3B). In the pnpM-complemented strain WBC-3ΔpnpMC, the transcriptional levels of the three genes were similar to those of corresponding genes in the WT strain WBC-3 under the same conditions (Figure 3C). These results indicated that pnpM deletion had a greater impact on the transcription of pnpC than pnpA and pnpB.
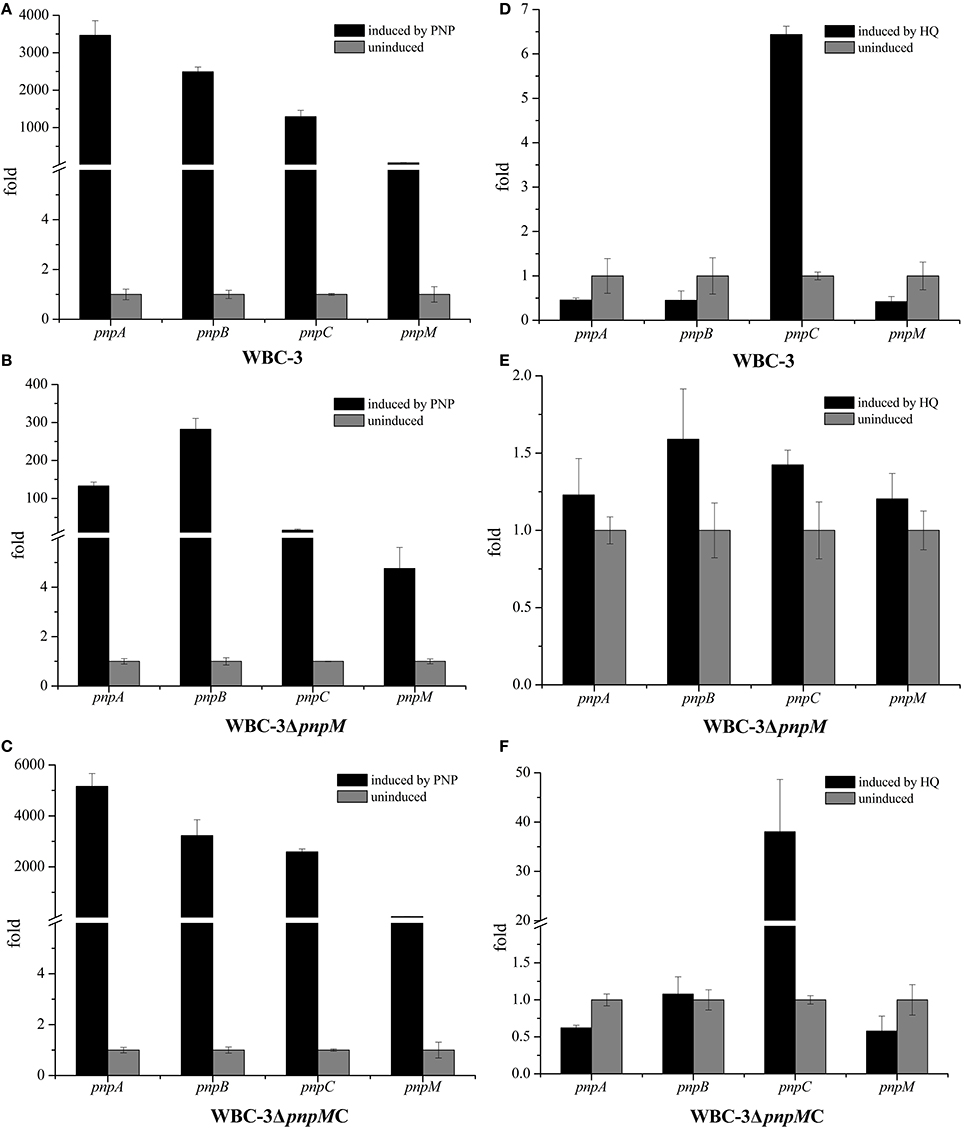
Figure 3. Transcriptional analysis of pnpA, pnpB, pnpCDEFG, and pnpA1M operons in wild type strain WBC-3 (A,D), pnpM knockout strain WBC-3ΔpnpM (B,E) and complemented pnpM-knockout strain WBC-3ΔpnpMC (C,F). The levels of gene expression in each sample were calculated as the fold expression ratio after normalization to 16S rRNA gene transcriptional levels. The values are averages of three independent qRT-PCR experiments. Error bars indicate standard deviations. There was a significant difference in the transcription of pnpA, pnpB, pnpC, and pnpM between strains WBC-3 and WBC-3ΔpnpM (p < 0.01, Paired-samples test), respectively. Black columns stand for the fold of gene expression level induced by PNP, and gray columns stand for the fold of gene expression level without PNP.
Similarly, the transcriptional levels of these genes were also analyzed under HQ induction conditions. In the presence of HQ, the transcriptional level of pnpC was increased in the WT strain and the pnpM-complemented strain (Figures 3D,F), but was significantly reduced in the presence of PNP (Figures 3A,C); while no change of the transcription level of pnpA, pnpB, and pnpM was observed between the induced and non-induced conditions (Figures 3D,F). In strain WBC3-ΔpnpM, the transcriptional levels of pnpA, pnpB, and pnpC were almost unaltered with or without HQ (Figure 3E). These results indicated that PnpM served as an activator for pnpC transcriptional expression under PNP induction.
PnpM Is a Tetramer
In order to obtain a functional regulator for in vitro assays, PnpM-His6 was overexpressed as a C-terminal His-tagged fusion protein from the pBBR1-tacexpnpM (a broad-host-range vector-based construct) in Pseudomonas putida PaW340, which is a mutant derivative of m-toluate/m-xylene utilizer Pseudomonas putida mt-2 and usually used as an expression host (Williams and Murray, 1974). It worthies to mention that Pseudomonas putida mt-2 and Pseudomonas putida PaW340 are not PNP utilizers (Liu et al., 2005b). The predicted size of the monomer of PnpM, 35 kDa, was confirmed by SDS-PAGE (Figure S1). By analytical gel filtration, the homologously produced PnpM-His6 was eluted as a single peak corresponding to 150 kDa (Figure S2) in the chromatography buffer with or without PNP. This was consistent with PnpM as a tetramer.
PnpM Binds with the Promoter Region of pnpC
The transcription start site (TSS) of pnpC was previously identified by 5′-RACE to be located to a T nucleotide, 23 nt upstream of the pnpC translation start codon (Zhang et al., 2015), as shown in Figure 5C. In this study, pnpA1 and pnpM were confirmed to be located in the same operon by RT-PCR. The TSS of pnpA1M operon was shown to be an A nucleotide, 54 nt upstream of the translation start codon of pnpA1. The putative −35 and −10 regions of the pnpC and pnpM promoters were proposed as shown in Figures 5C,D.
As previously reported, PnpR was capable of specifically binding the promoter of operons pnpA, pnpB, pnpCDEFG, and pnpR. It is worthy to note that, during a pre-experiment, PnpM in the range of 0–2μM was initially employed to bind with PpnpC (Figure 4A), where the DNA was found to be completely bound with 2.0 μM PnpM and this capacity was not changed with or without PNP. Here, EMSA was employed to assess the possible regulation of pnpA, pnpB, pnpC expression by PnpM. After, purified PnpM-His6 was incubated with the promoter regions of pnpA (PpnpA), pnpB (PpnpB), and pnpC (PpnpC) in the presence of PNP, PnpM was shown to specifically bind directly to the promoter region of pnpCDEFG, but not to the other promoters (Figure 4B). In addition, PnpM could bind to the promoter region of pnpA1M (Figure S3).
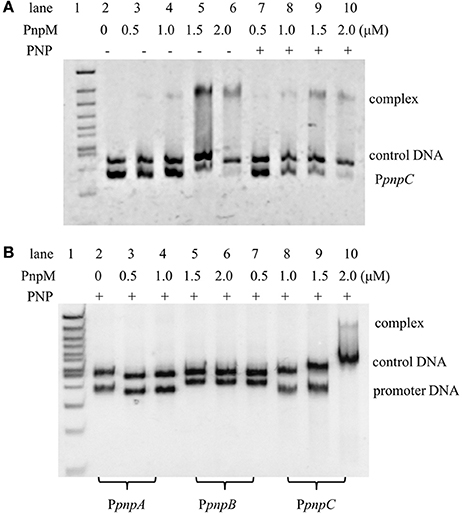
Figure 4. Electrophoretic mobility shift assays of PnpM binding with pnp promoters. (A) Electrophoretic mobility shift assays of PnpM binding with pnpC promoter (PpnpC). PnpM binding with PpnpC with PNP or without PNP. +, Stands for with PNP, −, stands for without PNP. The first lane was 100 bp ladder Marker, lanes 2–6 contain 0.03μM DNA, with 0μM, 0.5μM, 1.0μM, 1.5μM, and 2.0μM PnpM, respectively; lanes 7–10 contain 0.03μM DNA and 0.3 mM PNP, with 0.5μM, 1.0μM, 1.5μM, and 2.0μM PnpM, respectively. An approximately 400 bp DNA fragment gfp was used as a control DNA (0.03μM). Free probe is a fragment PpnpC of about 270 bp. (B) PnpM binding with pnp promoters in the present of PNP. The first lane was 100 bp ladder Marker, lanes 2–4 contain 0.03μM 283 bp DNA fragment of PpnpA and 0.3 mM PNP, with 0μM, 1.0μM, and 2.0μM PnpM, respectively; lanes 5–7 contain 0.03μM 334 bp DNA fragment of PpnpB and 0.3 mM PNP, with 0μM, 1.0μM, and 2.0μM PnpM, respectively; lanes 8–10 contain 0.03μM 270 bp DNA fragment of PpnpC and 0.3 mM PNP, with 0μM, 1.0μM, and 2.0μM PnpM, respectively. An approximately 400 bp DNA fragment gfp-1 was used as a control DNA (0.03μM).
Footprinting Analysis of PnpM and PnpR Binding Site to pnpC Promoter
DNase I footprinting was employed to determine the specific DNA motif within the pnpC promoter region that is bound by PnpR (Figure 5A) and PnpM (Figure 5B). After an approximately 270 bp 6-Fluorescein amidite (FAM)-labeled DNA fragment corresponding to the promoter region of pnpC was incubated with purified PnpM or PnpR, these complexes were treated with DNase I, followed by DNA fragment analysis by capillary electrophoresis. In the absence of PNP, PnpM protected a continuous 27 bp region that had also been determined to be from −88 to −62 relative to the pnpCDEFG TSS. The protection sequences contained an imperfect inverted repeat GTT-N11-AAC motif, which was similar to the T-N11-A motif of LTTRs consensus binding sequences, RBS, with positions −80 to −64 relative to the TSS of the pnpCDEFG operon. After analyzing the region of pnpC promoter, it was found that PnpM was bound to the sequence of putative RBS GTT-CGCGTTTCGCA-AAC in the promoter region of pnpCDEFG (Figure 5C). In the region of promoter pnpA1M, a similar putative sequence GTT-CGCGAAAGGCA-AAC was also present (Figure 5D). The protection region of PnpM was significantly increased from above a 27 bp sequence to 38 bp sequence by PnpM in the presence of PNP, and extended to the −51 region. This data tentatively suggested that the protection of extended 11 bp, from −61 to −51 relative to the TSS, and was the putative ABS (Figures 5B,C). The regulator PnpR shared the same 27 bp region DNA-binding regions of promoter pnpCDEFG with PnpM in the absence of the inducer (Figure 5A). The protection region was increased from 27 to 39 bp sequence by PnpR in the presence of PNP (Figure 5C).
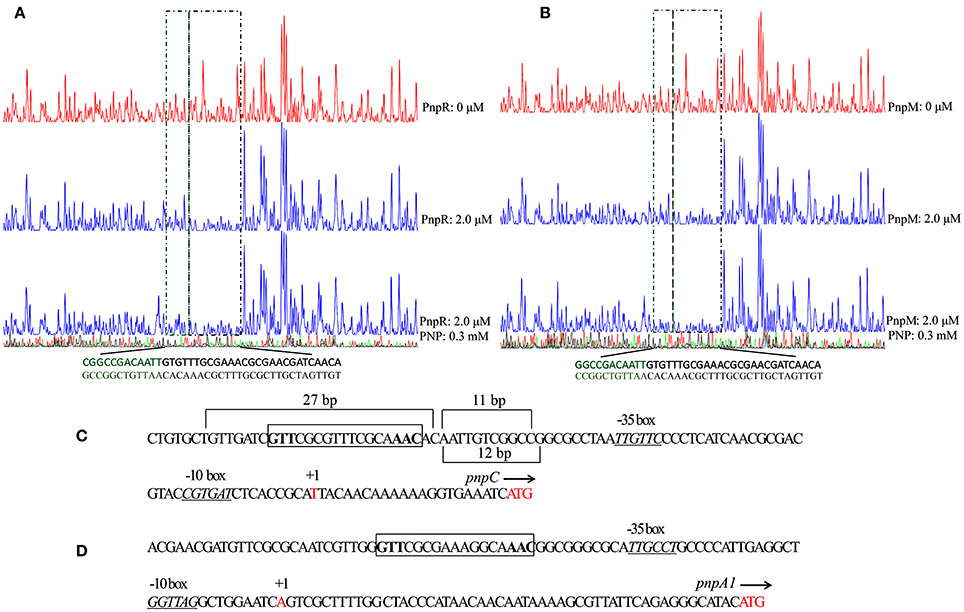
Figure 5. The DNase I footprinting analysis of PnpR (A) or PnpM (B) binding to PpnpC with PNP or without PNP. An amount of 0.03 μM probe PpnpC covering the entire intergenic region of pnpC was incubated with 2.0 μM PnpM in the EMSA buffer with or without PNP (0.3 mM). The intergenic fragment was labeled with 6-carboxyfluorescein (FAM) dye, incubated with 2.0 μM PnpM (blue line) or without PnpM (red line). The region protected without PNP by PnpM from DNase I cleavage are indicated with a black dotted box. The extended protected region with PNP are indicated with a green dotted box. PnpR with PpnpC and PNP was treated in the same way of PnpM. The organizations of upstream regions of pnpCDEFG (C) and pnpA1M (D) operons. The putative -10 boxes and -35 boxes are underlined and in italics. Start codon ATG and TSSs are in red. TSSs are denoted by arrow and +1, and the direction of arrow stands for the direction of gene transcription. The putative regulatory binding sites (RBSs) GTT-N11-AAC are boxed. The promoter regions of pnpCDEFG protected from DNase I digestion by PnpM without and with PNP. The protected region is 27 bp by PnpM or PnpR without PNP. When PNP was present, a region of 11 bp of the protection region was increased by PnpM, while a region of 12 bp by PnpR.
Effect of the Different Substrates on the Promoter Activity of pnpC Promoter
To further investigate pnpC promoter activity, the constructs pCMgfp-lpClacZ and pBBR1-tacexpnpM were transformed into strain PaW340 to measure pnpC promoter activities in response to various substrates. The expression of β-galactosidase activity in strain PaW340 was induced by following substrates: PNP, m-nitrophenol (MNP), o-nitrophenol (ONP), HQ, BQ and catechol, the final concentration of these six substrates was 0.3 mM for each one. No activity was detected except when PNP and HQ was used as inducers. The β-galactosidase activity of PNP-induced cells was 348.7- and HQ-induced cells was 35.8-fold higher (P < 0.05) than in non-induced cells, respectively (Figure 6A). These results were in agreement with in vivo pnpC expression measurements, further demonstrating that the major inducer for the promoter activity was PNP.
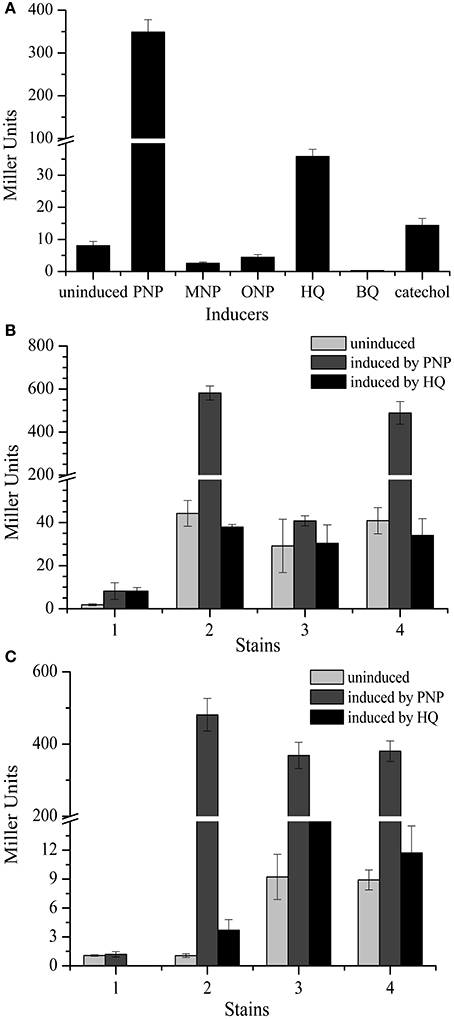
Figure 6. Determination of promoter activities by β-galactosidase. (A) Determination of promoter activities of pnpC operons induced by different substance. The β-galactosidase activity was determined in the strain PaW340 containing constructs pCMgfp-lpClacZ and pBBR1-tacexpnpM. The cells were induced by the following substrates: p-nitrophenol (PNP), m-nitrophenol (MNP), o-nitrophenol (ONP), hydroquinone (HQ), benzoquinone (BQ) and catechol, respectively. There was a significant difference in promoter activity between PNP and HQ as well as other substrates as inducers (p < 0.05). (B) Expression of promoter pnpA-lacZ translational fusions in strain PaW340. The β-galactosidase activities were determined in the strains: 1. PaW340 (pCMgfp-lpAlacZ+pBBR1-mcs2); 2. PaW340 (pCMgfp-lpAlacZ+pBBR1-tacpnpR); 3. PaW340 (pCMgfp-lpAlacZ+pBBR1-tacexpnpM); 4. PaW340 (pCMgfp-lpAlacZ+pBBR1-tacpnpMpnpR). There was a significant difference in the promoter activity of pnpA operon between regulator PnpR and PnpM with or without PNP (p < 0.05, Paired-samples test). (C) Expression of promoter pnpC-lacZ translational fusions in strain PaW340. The β-galactosidase activities were determined in the above strains: 1. PaW340 (pCMgfp-lpClacZ+pBBR1-mcs2); 2. PaW340 (pCMgfp-lpClacZ+pBBR1-tacpnpR); 3. PaW340 (pCMgfp-lpClacZ+pBBR1-tacexpnpM); 4. PaW340 (pCMgfp-lpClacZ+pBBR1-tacpnpMpnpR). There was a significant difference in the promoter activity of pnpC operon among different inducer (p < 0.05, Paired-samples test). Light gray columns stand for gene expression level without inducer. Gray columns stand for gene expression level induced by PNP. Black columns stand for gene expression level induced by HQ.
Determination of Promoter Activity of pnpC and pnpA Promoters
To further investigate pnpC promoter activity, pCMgfp-lpClacZ was used to see if cis-acting DNA sequences was required for the correct regulation of the expression of pnpR or pnpM or both in strain PaW340 carrying pBBR1-tacpnpR or pBBR1-tacexpnpM or pBBR1-tacpnpMpnpR. In addition, the above constructs with a promoter region were introduced in strain PaW340 carrying pBBR1mcs-2 as the negative controls. In the absence of PNP, the LacZ expression levels in the constructs with 270-bp corresponding pnpC promoter region were all evidently low. In the presence of PNP, the expression levels of LacZ were higher by 406-, 306-, or 316-fold in strain PaW340 carrying pCMgfp-lpClacZ plus pBBR1-tacpnpR or pBBR1-tacexpnpM or pBBR1-tacpnpMpnpR. In the presence of HQ, the expression levels of LacZ were higher by 3.6-, 88-, and 11-fold (Figure 6C), respectively. In order to test pnpA promoter activity with different regulators, strain PaW340 carrying pCMgfp-lpAlacZ (containing approximately 300-bp pnpA promoter-lacZ translational fusion) with above three individual constructs (pBBR1-tacpnpR or pBBR1-tacexpnpM or pBBR1-tacpnpMpnpR) was used. It can be concluded from Figure 6B that PNP-induced LacZ activity driven by the pnpA promoter were significantly increased only in the presence of pnpR but not pnpM. Therefore, these showed that only PnpR was involved in the transcriptional activation of pnpA, while PnpR and PnpM were both involved in the transcriptional activation of PpnpC.
Discussion
In our previous work, an LTTR PnpR was found to activate the transcription of four operons of pnpA, pnpB, pnpCDEFG, and pnpR involved in the PNP degradation by Pseudomonas sp. strain WBC-3 (Zhang et al., 2015). Mutation of the four promoters abolished the binding capability of purified PnpR. Moreover, the promoter activities were completely lost except for PpnpC. These observations led us to investigate the possible involvement of an additional regulator in the transcriptional activation of the pnpCDEFG operon. In the present work, an LTTR PnpM with 44% identity to PnpR was identified as an activator for the pnpCDEFG operon encoding HQ mineralization in the PNP catabolism, in addition to PnpR. This hypothesis was based on our previous study and has been addressed in the current study. Thus, we conclude that the transcriptional regulation of PNP catabolism in strain WBC-3 is triggered by both LTTRs PnpR and PnpM. PnpR activates pnpA and pnpB operons encoding the initial monooxygenation and reduction, and both PnpR and PnpM are involved in the transcriptional activation of the pnpCDEFG operon encoding the ring-cleavage of HQ and beyond. The principal inducer for all three catabolic operons is PNP and HQ might be a much weaker inducer and for the pnpCDEFG operon only.
With regard to other PNP utilizers, it was previously reported that an LTTR PnpRDLL−E4, with 85% identity to PnpM was found to be involved in the regulation of HQ degradation in Pseudomonas putida DLL-E4 (Shen et al., 2010; Chen et al., 2016). Unlike pnpM, pnpRDLL−E4 is adjacent to the HQ catabolic cluster pnpC1C2DECX1X2. However, no in vitro analysis of PnpRDLL−E was performed, such as EMSA, footprinting or promoter activity analysis. If the regulation system in strain DLL-E4 is similar to the one in strain WBC-3, genes homologous to pnpR from strain WBC-3 should be present in strain DLL-E4 but it has not been found so far. HQ is an important metabolite during aromatic catabolism and its pathway also exists as a downstream pathway in other aromatic degradations than PNP, such as alkylphenols degradation in strain Sphingomonas sp. strain TTNP3 (Kolvenbach et al., 2012); 4-hydroxyacetophenone degradation in Pseudomonas fluorescens ACB (Moonen et al., 2008); and 4-Fluorophenol degradation in Arthrobacter sp. strain IF1 (Ferreira et al., 2009). Of these bacterial strains, gene clusters similar to pnpCDEFG encoding HQ degradation are indeed present and putative regulators were also found to be nearby the HQ degradation gene clusters in some cases. For example, a putative AraC-type transcriptional regulator encoding-gene hqdR was next to the gene encoding hydroquinone-1,2-dioxygenase involved in the degradation of alkylphenols in Sphingomonas sp. strain TTNP3 (Kolvenbach et al., 2012). Nevertheless, no regulation study for HQ degradation has been reported in any bacterial strain.
In this study, two LysR transcriptional regulators PnpM and PnpR (44% identity) were shown to be involved in the activation of PNP catabolism in the strain WBC-3 in vitro, with both playing overlapping roles in the expression of pnpCDEFG as proposed in Figure 1B. One of well-studied examples for the involvement of multiple regulators is that of two LTTRs BenM and CatM which are complex regulatory circuits involved in benzoate consumption by Acinetobacter baylyi ADP1. Although both BenM and CatM activated the transcription of benABCDE operon, they responded to different inducers (Bundy et al., 2002; Ezezika et al., 2006, 2007; Craven et al., 2009). However, in this study, PnpR and PnpM both respond to the same inducer PNP (Figure 6C). In Gram-positive strain Corynebacterium glutamicum, two regulators GenR and GlxR are involved in the regulation of 3-hydroxybenzoate catabolism via gentisate. GenR is an IclR-Type specific regulator and GlxR is a CRP/FNR-type global regulator (Chao and Zhou, 2013, 2014). In contrast, PnpR and PnpM are both LysR-type specific regulators in this study. Although PnpM is a specific regulator for pnpCDEFG expression, it cannot singly function in the activation of all catabolic operons for PNP catabolism. Indeed, both PnpR and PnpM were involved in the intermediate HQ degradation and the starting compound PNP was the principal inducer rather than the substrate HQ for the enzymes pnpCDEFG encoded, since the latter was a much weaker inducer than the former in this system. This suggested that only HQ derived from PNP could then be further degraded by this system, and PNP degradation was sequentially activated by PnpR and PnpM in the presence of PNP. The observation of the incapability of strain WBC-3 growing on HQ is also clear and simple evidence for the above conclusion. However, it seems that the role of PnpR for pnpCDEFG operon can be replaced by PnpM in vitro, and it is unclear whether these two regulators may be in competition or act in a synergistic manner for pnpCDEFG promoter in vivo. This will require further efforts (such as chromatin immunoprecipitation assay) for a definite elucidation.
From the organization of pnp cluster in strain WBC-3, it can be tentatively concluded that the three operons may have evolved from different origins through patchwork assembly, each with their own promoters or regulators. Therefore, the complete degradation of PNP was regulated by two regulators, PnpR and PnpM, controlling three operons in total. Such complicated system for a single catabolic pathway may not be optimum and probably is an intermediate form during the adaptive evolution for PNP degradation. Indeed, most studied Gram-negative PNP utilizers were found to contain such three operons (Zhang et al., 2009, 2012; Shen et al., 2010; Wei et al., 2010), but with a recent, rare exception that the entire pnp genes are organized in a single operon in Gram-negative Burkholderia sp. strain SJ98, although they are very similar to their orthologous in strain WBC-3 (Min et al., 2014). Therefore, it can be reasonably considered that the transcriptional regulation of this single catabolic operon in strain SJ98 is less complex.
Usually, LTTRs bind to their regulated promoters at two sites regardless of the presence of relevant inducers: a strong RBS near position −65; and a weak ABS near position −35 (Schell, 1993; Porrua et al., 2007). In this study, the ABSs appeared from footprinting analyses (a 12-bp sequence binding with PnpR or a 11-bp sequence binding with PnpM only in the presence of the inducer PNP, as shown in Figure 5) may play a crucial role in activating the expression of pnpCDEFG. Although this observation is not common among LTTRs, several similar cases have been reported on the extended protection with relevant inducers. For instance, in the presence of an inducer, an additional 14 bp was protected by CatR, the regulator of catechol-degrading in Pseudomonas putida (Parsek et al., 1994, 1995). In Pseudomonas aeruginosa, the protection was extended 20 bp by Trpl for regulating tryptophan biosynthesis with an inducer (Chang and Crawford, 1990). The LigR-binding regions of the ligK promoter in Sphingobium sp. strain SYK-6 was extended by 16 bp (Kamimura et al., 2010). Interestingly, unlike the binding behavior of PnpR with the pnpC promoter, the binding region of promoters pnpA or pnpB by PnpR was unchanged with or without PNP (Zhang et al., 2015), indicating that the binding region was not only dependent on the regulator but also on its binding sequence. For most LysR proteins, the protein-DNA interaction region was shortened with the inducer. The mechanism of well-studied LTTRs, including AtzR, CbbR, OccR, OxyR, HadR, and QusR, was known as the sliding dimer model. These LTTRs cause the inducer-dependent shortening of the protected region from positions −80 to −20 to positions −80 to −30 and then a relaxation of the DNA bending. This was thought to be important in the release of the recognition site of RNA polymerase and the conformational change of regulator-DNA complex suitable for transcriptional activation (Toledano et al., 1994; Wang and Winans, 1995a,b; van Keulen et al., 2003; Porrua et al., 2007; Torii et al., 2013; Kubota et al., 2014). Nevertheless, in this study, the behavior of PnpM toward the pnpCDEFG promoter is somewhat different from the sliding dimer model for most LTTRs. The manner of DNA protection from PnpM binding to the pnpCDEFG promoter resembles the mentioned CatR, TrpI, or LigR, and the extended binding region in response to an inducer appears to be important for transcriptional activation.
Author Contributions
Conceived and designed the experiments: JW, WZ, and NZ. Performed the experiments: JW and WZ. Analyzed the data: JW, WZ, and HC. Wrote the paper: JW and NZ.
Funding
This work was funded by National Natural Science Foundation of China (NSFC) (31670107 and 31400068).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.01714/full#supplementary-material
Abbreviations
PNP, para-nitrophenol; HQ, hydroquinone; BQ, p-benzoquinone.
References
Arora, P. K., Srivastava, A., and Singh, V. P. (2014). Bacterial degradation of nitrophenols and their derivatives. J. Hazard. Mater. 266, 42–59. doi: 10.1016/j.jhazmat.2013.12.011
Bundy, B. M., Collier, L. S., Hoover, T. R., and Neidle, E. L. (2002). Synergistic transcriptional activation by one regulatory protein in response to two metabolites. Proc. Natl. Acad. Sci. U.S.A. 99, 7693–7698. doi: 10.1073/pnas.102605799
Cases, I., and de Lorenzo, V. (2001). The black cat/white cat principle of signal integration in bacterial promoters. EMBO J. 20, 1–11. doi: 10.1093/emboj/20.1.1
Chang, M., and Crawford, I. P. (1990). The roles of indoleglycerol phosphate and the TrpI protein in the expression of trpBA from Pseudomonas aeruginosa. Nucleic Acids Res. 18, 979–988. doi: 10.1093/nar/18.4.979
Chao, H., and Zhou, N. Y. (2013). GenR, an IclR-type regulator, activates and represses the transcription of gen genes involved in 3-hydroxybenzoate and gentisate catabolism in Corynebacterium glutamicum. J. Bacteriol. 195, 1598–1609. doi: 10.1128/JB.02216-12
Chao, H., and Zhou, N. Y. (2014). Involvement of the global regulator GlxR in 3-hydroxybenzoate and gentisate utilization by Corynebacterium glutamicum. Appl. Environ. Microbiol. 80, 4215–4225. doi: 10.1128/AEM.00290-14
Chen, Q. Z., Tu, H., Huang, F., Wang, Y. C., Dong, W. L., Wang, W. H., et al. (2016). Impact of pnpR, a LysR-type regulator-encoding gene, on the cellular processes of Pseudomonas putida DLL-E4. FEMS Microbiol. Lett. 363:fnw110. doi: 10.1093/femsle/fnw110
Chen, Y. F., Chao, H. J., and Zhou, N. Y. (2014). The catabolism of 2,4-xylenol and p-cresol share the enzymes for the oxidation of para-methyl group in Pseudomonas putida NCIMB 9866. Appl. Microbiol. Biotechnol. 98, 1349–1356. doi: 10.1007/s00253-013-5001-z
Chen, Y., Zhang, X., Liu, H., Wang, Y., and Xia, X. (2002). Study on Pseudomonas sp. WBC-3 capable of complete degradation of methylparathion. Wei Sheng Wu Xue Bao 42, 490–497. doi: 10.13343/j.cnki.wsxb.2002.04.017
Craven, S. H., Ezezika, O. C., Haddad, S., Hall, R. A., Momany, C., and Neidle, E. L. (2009). Inducer responses of BenM, a LysR-type transcriptional regulator from Acinetobacter baylyi ADP1. Mol. Microbiol. 72, 881–894. doi: 10.1111/j.1365-2958.2009.06686.x
Dehio, C., and Meyer, M. (1997). Maintenance of broad-host-range incompatibility group P and group Q plasmids and transposition of Tn5 in Bartonella henselae following conjugal plasmid transfer from Escherichia coli. J. Bacteriol. 179, 538–540. doi: 10.1128/jb.179.2.538-540.1997
de Lorenzo, V., Eltis, L., Kessler, B., and Timmis, K. N. (1993). Analysis of Pseudomonas gene products using lacIq/Ptrp-lac plasmids and transposons that confer conditional phenotypes. Gene 123, 17–24. doi: 10.1016/0378-1119(93)90533-9
Dennis, J. J., and Zylstra, G. J. (1998). Plasposons: modular self-cloning minitransposon derivatives for rapid genetic analysis of gram-negative bacterial genomes. Appl. Environ. Microbiol. 64, 2710–2715.
Diaz, E., and Prieto, M. A. (2000). Bacterial promoters triggering biodegradation of aromatic pollutants. Curr. Opin. Biotechnol. 11, 467–475. doi: 10.1016/S0958-1669(00)00126-9
Ezezika, O. C., Collier-Hyams, L. S., Dale, H. A., Burk, A. C., and Neidle, E. L. (2006). CatM regulation of the benABCDE operon: functional divergence of two LysR-type paralogs in Acinetobacter baylyi ADP1. Appl. Environ. Microbiol. 72, 1749–1758. doi: 10.1128/AEM.72.3.1749-1758.2006
Ezezika, O. C., Haddad, S., Clark, T. J., Neidle, E. L., and Momany, C. (2007). Distinct effector-binding sites enable synergistic transcriptional activation by BenM, a LysR-type regulator. J. Mol. Biol. 367, 616–629. doi: 10.1016/j.jmb.2006.09.090
Ferreira, M. I. M., Iida, T., Hasan, S. A., Nakamura, K., Fraaije, M. W., Janssen, D. B., et al. (2009). Analysis of two gene clusters involved in the degradation of 4-fluorophenol by Arthrobacter sp. strain IF1. Appl. Environ. Microbiol. 75, 7767–7773. doi: 10.1128/AEM.00171-09
Georgi, T., Engels, V., and Wendisch, V. F. (2008). Regulation of L-lactate utilization by the FadR-type regulator L1dR of Corynebacterium glutamicum. J. Bacteriol. 190, 963–971. doi: 10.1128/JB.01147-07
Griffith, K. L., and Wolf, R. E. (2002). Measuring beta-galactosidase activity in bacteria: cell growth, permeabilization, and enzyme assays in 96-well arrays. Biochem. Biophys. Res. Commun. 290, 397–402. doi: 10.1006/bbrc.2001.6152
Hoang, T. T., Karkhoff-Schweizer, R. R., Kutchma, A. J., and Schweizer, H. P. (1998). A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212, 77–86. doi: 10.1016/S0378-1119(98)00130-9
Hu, F., Jiang, X., Zhang, J. J., and Zhou, N. Y. (2014). Construction of an engineered strain capable of degrading two isomeric nitrophenols via a sacB- and gfp-based markerless integration system. Appl. Microbiol. Biotechnol. 98, 4749–4756. doi: 10.1007/s00253-014-5567-0
Jain, R. K., Dreisbach, J. H., and Spain, J. C. (1994). Biodegradation of p-nitrophenol via 1,2,4-benzenetriol by an Arthrobacter sp. Appl. Environ. Microbiol. 60, 3030–3032.
Jeong, H., Barbe, V., Lee, C. H., Vallenet, D., Yu, D. S., Choi, S. H., et al. (2009). Genome sequences of Escherichia coli B strains REL606 and BL21(DE3). J. Mol. Biol. 394, 644–652. doi: 10.1016/j.jmb.2009.09.052
Kamimura, N., Takamura, K., Hara, H., Kasai, D., Natsume, R., Senda, T., et al. (2010). Regulatory system of the protocatechuate 4,5-cleavage pathway genes essential for lignin downstream catabolism. J. Bacteriol. 192, 3394–3405. doi: 10.1128/JB.00215-10
Karim, K., and Gupta, S. K. (2002). Effects of alternative carbon sources on biological transformation of nitrophenols. Biodegradation 13, 353–360. doi: 10.1023/A:1022364616575
Kolvenbach, B. A., Dobrowinski, H., Fousek, J., Vlcek, C., Schaffer, A., Gabriel, F. L. P., et al. (2012). An unexpected gene cluster for downstream degradation of alkylphenols in Sphingomonas sp. strain TTNP3. Appl. Microbiol. Biotechnol. 93, 1315–1324. doi: 10.1007/s00253-011-3451-8
Kovach, M. E., Elzer, P. H., Hill, D. S., Robertson, G. T., Farris, M. A., Roop, R. M., et al. (1995). Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene 166, 175–176. doi: 10.1016/0378-1119(95)00584-1
Kubota, T., Tanaka, Y., Takemoto, N., Watanabe, A., Hiraga, K., Inui, M., et al. (2014). Chorismate-dependent transcriptional regulation of quinate/shikimate utilization genes by LysR-type transcriptional regulator QsuR in Corynebacterium glutamicum: carbon flow control at metabolic branch point. Mol. Microbiol. 92, 356–368. doi: 10.1111/mmi.12560
Liu, H., Wang, S. J., and Zhou, N. Y. (2005a). A new isolate of Pseudomonas stutzeri that degrades 2-chloronitrobenzene. Biotechnol. Lett. 27, 275–278. doi: 10.1007/s10529-004-8293-3
Liu, H., Zhang, J. J., Wang, S. J., Zhang, X. E., and Zhou, N. Y. (2005b). Plasmid-borne catabolism of methyl parathion and p-nitrophenol in Pseudomonas sp. strain WBC-3. Biochem. Biophys. Res. Commun. 334, 1107–1114. doi: 10.1016/j.bbrc.2005.07.006
Liu, T. T., and Zhou, N. Y. (2012). Novel L-cysteine-dependent maleylpyruvate isomerase in the gentisate pathway of Paenibacillus sp. strain NyZ101. J. Bacteriol. 194, 3987–3994. doi: 10.1128/JB.00050-12
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Maddocks, S. E., and Oyston, P. C. F. (2008). Structure and function of the LysR-type transcriptional regulator (LTTR) family proteins. Microbiology 154, 3609–3623. doi: 10.1099/mic.0.2008/022772-0
Min, J., Zhang, J. J., and Zhou, N. Y. (2014). The gene cluster for para-nitrophenol catabolism is responsible for 2-chloro-4-nitrophenol degradation in Burkholderia sp. strain SJ98. Appl. Environ. Microbiol. 80, 6212–6222. doi: 10.1128/AEM.02093-14
Moonen, M. J. H., Kamerbeek, N. M., Westphal, A. H., Boeren, S. A., Janssen, D. B., Fraaije, M. W., et al. (2008). Elucidation of the 4-hydroxyacetophenone catabolic pathway in Pseudomonas fluorescens ACB. J. Bacteriol. 190, 5190–5198. doi: 10.1128/JB.01944-07
Parsek, M. R., Kivisaar, M., and Chakrabarty, A. M. (1995). Differential DNA bending introduced by the Pseudomonas putida LysR-type regulator, CatR, at the plasmid-borne pheBA and chromosomal catBC promoters. Mol. Microbiol. 15, 819–828. doi: 10.1111/j.1365-2958.1995.tb02352.x
Parsek, M. R., Ye, R. W., Pun, P., and Chakrabarty, A. M. (1994). Critical nucleotides in the interaction of a LysR-type regulator with its target promoter region. catBC promoter activation by CatR. J. Biol. Chem. 269, 11279–11284.
Porrua, O., Garcia-Jaramillo, M., Santero, E., and Govantes, F. (2007). The LysR-type regulator AtzR binding site: DNA sequences involved in activation, repression and cyanuric acid-dependent repositioning. Mol. Microbiol. 66, 410–427. doi: 10.1111/j.1365-2958.2007.05927.x
Roldan, M. D., Blasco, R., Caballero, F. J., and Castillo, F. (1998). Degradation of p-nitrophenol by the phototrophic bacterium Rhodobacter capsulatus. Arch. Microbiol. 169, 36–42.
Saltikov, C. W., and Newman, D. K. (2003). Genetic identification of a respiratory arsenate reductase. Proc. Natl. Acad. Sci. U.S.A. 100, 10983–10988. doi: 10.1073/pnas.1834303100
Sambrook, J. F. E., and Maniatis, T. (1989). Molecular-Cloning - A Laboratory Manual, 2nd Edn. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
Schell, M. A. (1993). Molecular biology of the LysR family of transcriptional regulators. Annu. Rev. Microbiol. 47, 597–626. doi: 10.1146/annurev.mi.47.100193.003121
Shen, W. J., Liu, W. D., Zhang, J., Tao, J. A., Deng, H. H., Cao, H., et al. (2010). Cloning and characterization of a gene cluster involved in the catabolism of p-nitrophenol from Pseudomonas putida DLL-E4. Bioresour. Technol. 101, 7516–7522. doi: 10.1016/j.biortech.2010.04.052
Shingler, V. (2003). Integrated regulation in response to aromatic compounds: from signal sensing to attractive behaviour. Environ. Microbiol. 5, 1226–1241. doi: 10.1111/j.1462-2920.2003.00472.x
Spain, J. C., and Gibson, D. T. (1991). Pathway for biodegradation of para-nitrophenol in a Moraxella sp. Appl. Environ. Microbiol. 57, 812–819.
Takeo, M., Murakami, M., Niihara, S., Yamamoto, K., Nishimura, M., Kato, D., et al. (2008). Mechanism of 4-nitrophenol oxidation in Rhodococcus sp. strain PN1: characterization of the two-component 4-nitrophenol hydroxylase and regulation of its expression. J. Bacteriol. 190, 7367–7374. doi: 10.1128/JB.00742-08
Toledano, M. B., Kullik, I., Trinh, F., Baird, P. T., Schneider, T. D., and Storz, G. (1994). Redox-dependent shift of OxyR-DNA contacts along an extended DNA-binding site: a mechanism for differential promoter selection. Cell 78, 897–909. doi: 10.1016/S0092-8674(94)90702-1
Torii, H., Machida, A., Hara, H., Hatta, T., and Takizawa, N. (2013). The regulatory mechanism of 2,4,6-trichlorophenol catabolic operon expression by HadR in Ralstonia pickettii DTP0602. Microbiology 159, 665–677. doi: 10.1099/mic.0.063396-0
Tropel, D., and van der Meer, J. R. (2004). Bacterial transcriptional regulators for degradation pathways of aromatic compounds. Microbiol. Mol. Biol. Rev. 68, 474–500. doi: 10.1128/MMBR.68.3.474-500.2004
van Keulen, G., Ridder, A., Dijkhuizen, L., and Meijer, W. G. (2003). Analysis of DNA binding and transcriptional activation by the LysR-type transcriptional regulator CbbR of Xanthobacter flavus. J. Bacteriol. 185, 1245–1252. doi: 10.1128/JB.185.4.1245-1252.2003
Wang, L., and Winans, S. C. (1995a). High angle and ligand-induced low angle DNA bends incited by OccR lie in the same plane with OccR bound to the interior angle. J. Mol. Biol. 253, 32–38. doi: 10.1006/jmbi.1995.0533
Wang, L., and Winans, S. C. (1995b). The sixty nucleotide OccR operator contains a subsite essential and sufficient for OccR binding and a second subsite required for ligand-responsive DNA bending. J. Mol. Biol. 253, 691–702. doi: 10.1006/jmbi.1995.0583
Wang, Y., Cen, X. F., Zhao, G. P., and Wang, J. (2012). Characterization of a new GlnR binding box in the promoter of amtB in Streptomyces coelicolor Inferred a PhoP/GlnR competitive binding mechanism for transcriptional regulation of amtB. J. Bacteriol. 194, 5237–5244. doi: 10.1128/JB.00989-12
Wei, Q., Liu, H., Zhang, J. J., Wang, S. H., Xiao, Y., and Zhou, N. Y. (2010). Characterization of a para-nitrophenol catabolic cluster in Pseudomonas sp. strain NyZ402 and construction of an engineered strain capable of simultaneously mineralizing both para- and ortho-nitrophenols. Biodegradation 21, 575–584. doi: 10.1007/s10532-009-9325-4
Williams, P. A., and Murray, K. (1974). Metabolism of benzoate and the methylbenzoates by Pseudomonas putida (arvilla) mt-2: evidence for the existence of a TOL plasmid. J. Bacteriol. 120, 416–423.
Woodcock, D. M., Crowther, P. J., Doherty, J., Jefferson, S., Decruz, E., Noyerweidner, M., et al. (1989). Quantitative evaluation of Escherichia coli host strains for tolerance to cytosine methylation in plasmid and phage recombinants. Nucleic Acids Res. 17, 3469–3478. doi: 10.1093/nar/17.9.3469
Zhang, J. J., Liu, H., Xiao, Y., Zhang, X. E., and Zhou, N. Y. (2009). Identification and characterization of catabolic para-nitrophenol 4-monooxygenase and para-benzoquinone redctase from Pseudomonas sp. strain WBC-3. J. Bacteriol. 191, 2703–2710. doi: 10.1128/JB.01566-08
Zhang, S. Y., Sun, W., Xu, L., Zheng, X. M., Chu, X. Y., Tian, J., et al. (2012). Identification of the para-nitrophenol catabolic pathway, and characterization of three enzymes involved in the hydroquinone pathway, in Pseudomonas sp. 1-7. BMC Microbiol. 12:27. doi: 10.1186/1471-2180-12-27
Zhang, W. M., Zhang, J. J., Jiang, X., Chao, H. J., and Zhou, N. Y. (2015). Transcriptional activation of multiple operons involved in para-nitrophenol degradation by Pseudomonas sp. strain WBC-3. Appl. Environ. Microbiol. 81, 220–230. doi: 10.1128/AEM.02720-14
Zianni, M., Tessanne, K., Merighi, M., Laguna, R., and Tabita, F. R. (2006). Identification of the DNA bases of a DNase I footprint by the use of dye primer sequencing on an automated capillary DNA analysis instrument. J. Biomol. Tech. 17, 103–113.
Keywords: catabolism, hydroquinone pathway, LysR-type transcriptional regulator, para-nitrophenol, Pseudomonas sp. strain WBC-3, PnpM, PnpR
Citation: Wang J-P, Zhang W-M, Chao H-J and Zhou N-Y (2017) PnpM, a LysR-Type Transcriptional Regulator Activates the Hydroquinone Pathway in para-Nitrophenol Degradation in Pseudomonas sp. Strain WBC-3. Front. Microbiol. 8:1714. doi: 10.3389/fmicb.2017.01714
Received: 05 June 2017; Accepted: 24 August 2017;
Published: 14 September 2017.
Edited by:
Pankaj Kumar Arora, M. J. P. Rohilkhand University, IndiaReviewed by:
Jiandong Jiang, Nanjing Agricultural University, ChinaStefano Fedi, Università di Bologna, Italy
Copyright © 2017 Wang, Zhang, Chao and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ning-Yi Zhou, bmluZ3lpLnpob3VAc2p0dS5lZHUuY24=