 Ilaria Cavallari1†
Ilaria Cavallari1† Gloria Scattolin2†
Gloria Scattolin2† Micol Silic-Benussi1
Micol Silic-Benussi1 Vittoria Raimondi1
Vittoria Raimondi1 Donna M. D'Agostino3*
Donna M. D'Agostino3* Vincenzo Ciminale1,2*
Vincenzo Ciminale1,2*- 1Veneto Institute of Oncology IOV-IRRCS, Padova, Italy
- 2Department of Surgery, Oncology, and Gastroenterology, University of Padova, Padova, Italy
- 3Department of Biomedical Sciences, University of Padova, Padova, Italy
Viruses must exploit the cellular biosynthetic machinery and evade cellular defense systems to complete their life cycles. Due to their crucial roles in cellular bioenergetics, apoptosis, innate immunity and redox balance, mitochondria are important functional targets of many viruses, including tumor viruses. The present review describes the interactions between mitochondria and proteins coded by the human tumor viruses human T-cell leukemia virus type 1, Epstein-Barr virus, Kaposi's sarcoma-associated herpesvirus, human hepatitis viruses B and C, and human papillomavirus, and highlights how these interactions contribute to viral replication, persistence and transformation.
Introduction
It is estimated that about 20% of human cancer cases world-wide are caused by infection with bacteria, parasites, or viruses (de Martel et al., 2012; IARC Monograph, 2012). Among these infectious agents, viruses have the greatest reliance on cellular pathways for completion of their life cycles, and have evolved complex mechanisms to manipulate the anabolic and proliferative capacity of the host cell while minimizing effects on cell death and destruction by the immune system.
Viruses that are linked to human cancer include human-T-cell leukemia virus type 1 (HTLV-1), Epstein-Barr virus (EBV), Kaposi's sarcoma-associated herpesvirus (KSHV), hepatitis viruses B and C (HBV and HCV, respectively), high-risk genotypes of human papillomavirus (HPV, e.g., HPV-16, HPV-18), and Merkel cell polyomavirus (MCPyV). In addition, the AIDS-causing retrovirus human immunodeficiency virus is indirectly linked to cancer through its immunosuppressive effects, which favor transformation by other tumor viruses, especially EBV and KSHV (IARC Monograph, 2012).
Many of the strategies used by viruses to replicate and persist in host cells intersect at the level of mitochondria (reviewed by Claus and Liebert, 2014). This is not surprising, given the range of essential roles of these organelles in multiple cellular processes. (i) Energy supply: as the sites of pyruvate- and fatty acid oxidation, the citric acid cycle and the electron transport chain, mitochondria are the main source of ATP in most differentiated eukaryotic cells. (ii) Ca2+ signaling: through their ability to take up Ca2+ from the cytoplasm, mitochondria act as key regulators of intracellular calcium homeostasis. (iii) ROS homeostasis: the mitochondrial electron transport chain is a major source of reactive oxygen species (ROS) that influence cell turnover. (iv) Apoptosis: proteins present within mitochondria (e.g., cytochrome c) and on the outer mitochondrial membrane (e.g., Bax, Bak) play a key role in the triggering of intrinsic apoptosis. (v) Innate immunity: MAVS (Mitochondrial antiviral signaling protein), a component of RIG-1 (retinoic acid-inducible gene I) signaling, is located on the outer mitochondrial membrane.
All of the human tumor viruses except MCPyV (the most recently identified human tumor virus, associated with a rare form of skin cancer Feng et al., 2008b) code for one or more proteins that affect mitochondrial function.
The present review summarizes current knowledge on the interplay between mitochondria and proteins produced by HTLV-1, EBV, KSHV, HBV, HCV, and HPV (Table 1), and how these proteins influence the replication strategies and oncogenic properties of these viruses (Table 2).
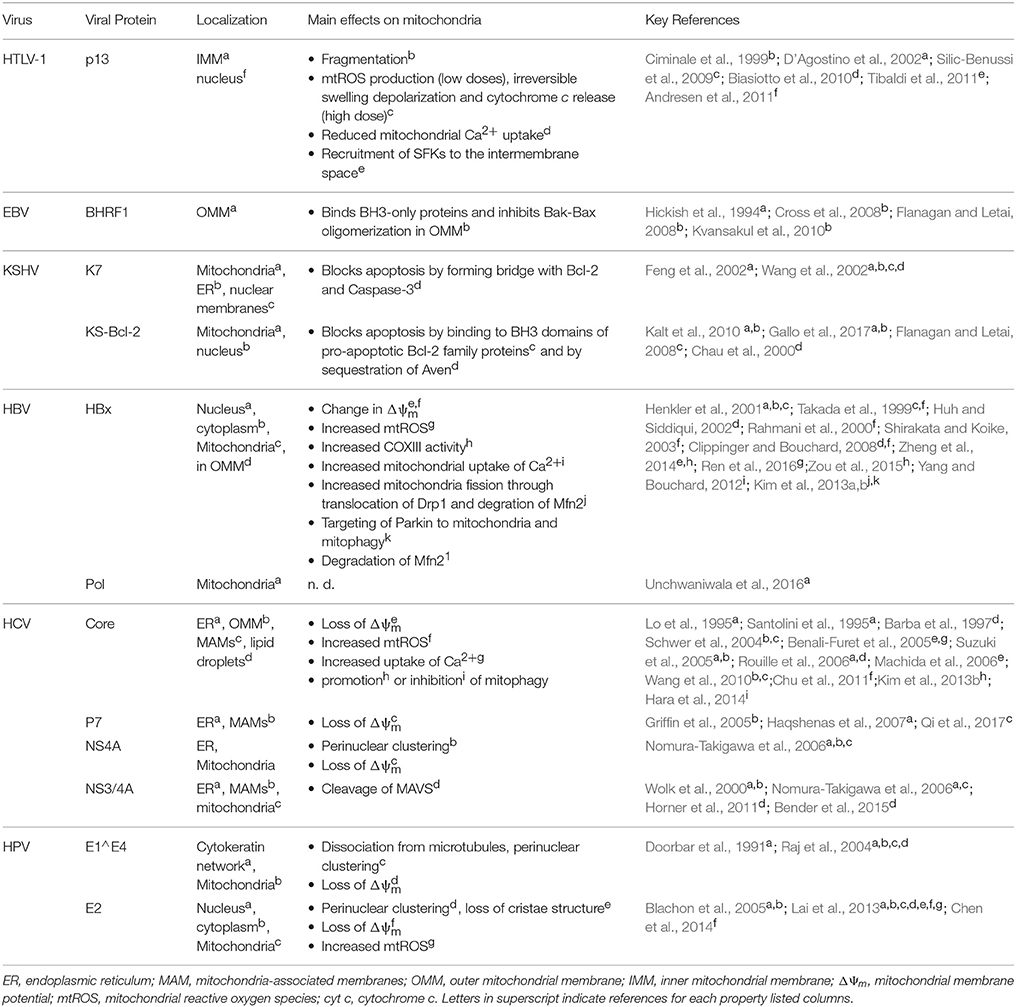
Table 1. Localization and main mitochondrial effects of viral proteins.
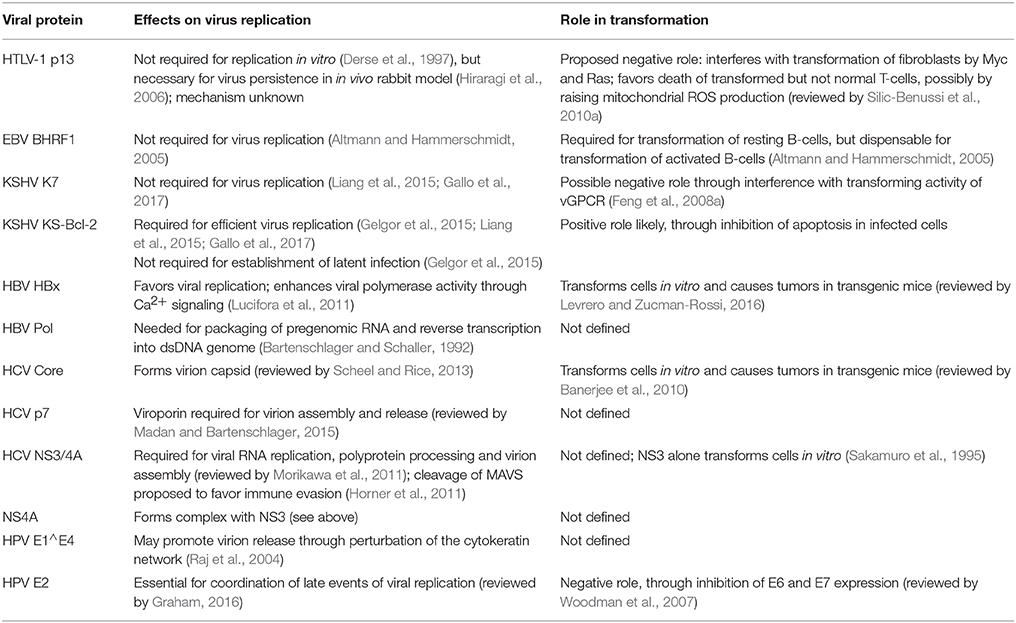
Table 2. Role of viral mitochondrial proteins in replication and transformation.
HTLV-1
HTLV-1 is a retrovirus that infects at least 10 million people worldwide, with most cases identified in southwestern Japan, the Caribbean basin, sub-Saharan Africa, and Brazil (reviewed by Gessain and Cassar, 2012). HTLV-1 is transmitted through transfer of infected cells during breast-feeding, sexual contact, and exposure to blood and results in persistent infection, mainly in CD4+ T-cells. About 3–5% of infected patients develop an aggressive neoplasm of mature CD4+ T-cells named adult T-cell leukemia/lymphoma (ATLL) or a neurological disease named tropical spastic paraparesis/HTLV-associated myelopathy (TSP/HAM) after a latency period of decades (ATLL) or years (TSP/HAM). Other diseases associated with HTLV-1 infection include uveitis, infective dermatitis, myositis and other pathologies with an important inflammatory component (reviewed by Goncalves et al., 2010). Almost 40 years after the discovery of HTLV-1, we still lack an accurate measure of the global burden of infection, there is no HTLV-1 vaccine, and biomarkers to predict clinical outcome remain to be identified (reviewed by Willems et al., 2017).
In addition to the gag, pol, pro and env gene products produced by all retroviruses, the genome of HTLV-1 codes for nonstructural proteins named Tax, Rex, p12, p13, p21Rex, p30/Tof, and HBZ (reviewed by Journo et al., 2009; Lairmore et al., 2012). Tax and Rex are essential for completion of the replication cycle, with Tax driving transcription from the viral 5′-LTR (long terminal repeat) promoter, and Rex promoting expression of incompletely spliced transcripts, including those coding for the virion proteins. Tax and HBZ are considered to be the principal viral factors that drive development of ATLL (reviewed by Panfil et al., 2016).
p13
p13 is an 87-amino acid protein that accumulates primarily in the inner mitochondrial membrane (Table 1; D'Agostino et al., 2002). p13 is considered to be an accessory protein, as its deletion from the viral genome does not abolish viral replication in vitro (Derse et al., 1997). However, studies carried out in a rabbit model of HTLV-1 infection indicated that p13 is important for establishing a persistent infection in vivo (Hiraragi et al., 2006).
Studies of transfected HeLa cells demonstrated that mitochondrial accumulation of p13 is directed by a 10-residue mitochondrial targeting signal near its amino terminus (MTS; amino acids 22-31) which, unlike most MTS, is not cleaved during mitochondrial import (Ciminale et al., 1999). The MTS contains 4 arginines and folds into an amphipathic α-helix (D'Agostino et al., 2002). The carboxy-terminal half of p13 contains a putative hinge (amino acids 42-48) and β-sheet (amino acids 65-75) (Silic-Benussi et al., 2010a) and a cluster of prolines that mediate binding to proteins containing SH3 domains (Ghorbel et al., 2006; Tibaldi et al., 2011).
Expression of p13 in HeLa cells alters mitochondrial morphology and produces isolated clusters of round-shaped, fragmented mitochondria (Ciminale et al., 1999). These effects depend on the presence of the 4 arginines that constitute the charged face of the amphipathic α-helix (Silic-Benussi et al., 2004). When added to isolated rat liver mitochondria, synthetic p13 protein induces an inward K+ current, mitochondrial swelling and loss of mitochondrial membrane potential (ΔΨm). However, this depolarization triggers a compensatory increase in electron transport chain activity, which restores ΔΨm but raises levels of mitochondrial reactive oxygen species (ROS) and lowers the threshold for opening of the permeability transition pore (PTP), a channel that regulates apoptosis (reviewed by Bernardi et al., 2015). These effects are dose-dependent, as low concentrations of p13 induce mitochondrial swelling and increased mitochondrial ROS, but not mitochondrial depolarization, while high levels of p13 cause irreversible swelling, depolarization and cytochrome c release (Silic-Benussi et al., 2009).
p13 corresponds to the carboxy-terminal portion of p30/Tof, a nucleolar/nuclear accessory protein whose activities include suppression of Tax and Rex expression and modulation of cellular transcription (reviewed by Anupam et al., 2013). Despite this sequence overlap, p13 and p30/Tof are expressed from distinct alternatively spliced mRNAs. Studies of the temporal regulation of HTLV-1 gene expression in infected cells demonstrated that the mRNA coding for p13 is expressed as a late gene (Cavallari et al., 2011, 2013; Rende et al., 2011) and is Rex-dependent (Cavallari et al., 2015).
The structural and functional properties of p13 suggest that it may act as a viroporin (reviewed by D'Agostino et al., 2005a,b). The viroporins are a family of small, hydrophobic viral proteins with one or more membrane-spanning domains that, upon oligomerization in membranes, alter membrane permeability to ions and small molecules through the formation of channels or pores, and change the trafficking, processing and lifespan of membrane-associated proteins. Although these alterations have diverse effects on infected cells, the principal role of viroporins is to promote the assembly and egress of virus particles (reviewed by Nieva et al., 2012; Scott and Griffin, 2015).
The activity of Ca2+ as a second messenger is crucial for the metabolism and function of all cells, including T-cells (reviewed by Fracchia et al., 2013), the main targets of HTLV-1 infection in vivo. Ca2+ messages are delivered in the form of transient elevations in cytosolic Ca2+ concentration upon its release from organelles, especially the ER, and upon entry from the extracellular environment; mitochondria participate in this process through their ability to take up Ca2+. Experiments carried out in HeLa cells expressing organelle-targeted aequorins revealed that p13 specifically reduces mitochondrial Ca2+ uptake (Biasiotto et al., 2010).
The impact of p13 on mitochondrial Ca2+ flux might intersect with that of p12, a 99-residue viral protein that accumulates in the endoplasmic reticulum (ER) and cis-Golgi (reviewed by Van Prooyen et al., 2010). p12 increases Ca2+ release from the ER, thus promoting activation of NFAT (nuclear factor of activated T-cells), a key mitogenic transcription factor whose activity is controlled by the calcium-dependent phosphatase calcineurin (Kim et al., 2003). p12-mediated release of ER calcium stores and p13-mediated interference with mitochondrial calcium uptake could be predicted to amplify Ca2+ signals in response to T-cell receptor activation with consequent enhancement of NFAT activation (Silic-Benussi et al., 2010a). In alternative, this combination of effects could convert a Ca2+ signal to a prolonged increase in cytosolic Ca2+, which might trigger apoptosis.
Efforts to understand the function of p13 through identification of its binding partners indicated that it interacts with farnesyl pyrophosphate synthetase (FPPS) (Lefebvre et al., 2002); with members of the Src family of protein kinases (Tibaldi et al., 2011), and with the HTLV-1 regulatory protein Tax (Andresen et al., 2011). Yeast 2-hybrid screening assays also indicated binding of p13 to a protein of the nucleoside monophosphate kinase superfamily and to actin-binding protein 280 (Hou et al., 2000), but these interactions were not explored in detail.
Binding of p13 to Src family kinases (SFKs) involves p13's proline-rich C-terminal domain and the kinases' SH3 domains (Tibaldi et al., 2011). This interaction promotes the accumulation of SFKs in the mitochondrial intermembrane space and enhances their tyrosine kinase activity, while interfering with targeting of p13 to the inner membrane and attenuating the effects of p13 on ΔΨm (Tibaldi et al., 2011).
Experiments carried out in 293T cells and HeLa cells showed that when co-expressed with Tax, p13 undergoes ubiquitination, becomes more stable, and is partially rerouted to nuclear speckles containing Tax (Andresen et al., 2011). These effects are mediated by direct interaction of Tax with p13 through a disulphide bond involving cysteine 27 in p13. Results of co-immunoprecipitation and LTR-reporter assays showed that the p13-Tax interaction interferes with binding of Tax to the transcription cofactor CBP/p300 and leads to a decrease in Tax-mediated viral gene transcription, a crucial step in viral replication. The finding that substitution of the p13 start codon with isoleucine in an HTLV-1 proviral clone resulted in an increase in viral gene expression supports the proposal that p13 might acts as a negative regulator of HTLV-1 replication (Andresen et al., 2011).
The effect of ablation of p13 expression on the ability of the virus to transform primary T-cells needs to be investigated. However, results of studies of primary cells and cell lines forced to express p13 indicate that it may limit the oncogenic potential of HTLV-1. p13 interfered with the ability of Myc and Ras to transform rat embryo fibroblasts, and HeLa cells expressing p13 exhibited a proliferation defect in vitro and were less tumorigenic in nude mice compared to parental cells (Silic-Benussi et al., 2004). Subsequent comparisons of the effects of p13 in the T-cell line Jurkat and normal primary T-cells showed that the protein induced ROS production in both cell types, and slowed proliferation and increased apoptotic death in Jurkat cells, but activated normal primary T-cells (Silic-Benussi et al., 2010a,b). These observations suggest that p13 might expand the pool of untransformed infected cells and on the other hand favor the elimination of transformed cells, thus increasing HTLV-1's adaptation to the host and lifelong persistence of the infection. It will be interesting to determine whether the effects of p13 on T-cell activation are connected to its impact on Ca2+ homeostasis.
EBV and KSHV
EBV and KSHV are members of the γ-herpesvirus subfamily of the Herpesviridae, a family of large, enveloped viruses with a linear double-stranded DNA genome. EBV targets mainly B-cells and epithelial cells, while KSHV targets mainly B-cells and endothelial cells. Like all herpesviruses, the life cycle of EBV and KSHV comprises latent states that favor viral persistence and a lytic phase that produces virus particles. EBV infection is present in more than 90% of adults in a uniform distribution worldwide. Although studies of global KSHV epidemiology are incomplete, it appears to be more restricted geographically to parts of sub-Saharan Africa, the Mediterranean basin, Brazil and China, and in certain subpopulations such as men who have sex with men (Morrison et al., 2015). Primary infection with EBV and KSHV occurs mainly through contact with saliva during early childhood and usually does not cause overt clinical symptoms, except for a self-limiting polyclonal B-lymphoproliferative disease termed infectious mononucleosis that arises in at least 25% of patients who become infected with EBV in adolescence or early adulthood. Neoplasias associated with EBV include an endemic form of Burkitt's lymphoma, post-transplant lymphoproliferative disorders, some cases of Hodgkin's lymphoma, and nasopharyngeal carcinoma (reviewed by Young et al., 2016). KSHV is linked to 3 types of tumors, with immunosuppression representing a risk factor: the endothelial-derived tumor Kaposi's sarcoma (KS, always KSHV-positive), a B-cell malignancy named primary effusion lymphoma (PEL; always KSHV-positive, sometimes also EBV-positive) and a plasmablastic form of the B-lymphoproliferative disorder multicentric Castleman disease (MCD, KSHV-positive in about half of cases) (reviewed by Bhutani et al., 2015). EBV and KSHV share 59 gene homologs whose similarity ranges from 41 to 75% (Russo et al., 1996); both viruses possess genes that closely resemble cellular genes. Proteins of EBV and KSHV with an impact on mitochondria include EBV BHRF1, BZLF1, and LMP2A, and KSHV K7 and KS-Bcl-2 (Table 1).
EBV: BHRF1, BZLF1 BALF1, LMP2A
BHRF1 is a 191-amino acid, early lytic-phase EBV protein with similarity to cellular Bcl-2 family proteins (Pearson et al., 1987) that blocks the mitochondrial arm of apoptosis mediated by pro-apoptotic Bcl-2 proteins (Henderson et al., 1993). In this manner, BHRF1 promotes the survival of EBV-infected cells, thus promoting viral persistence/replication. The production of viral Bcl-2 homologs (v-Bcl-2) is shared by several other viruses, including KSHV, which produces KS-Bcl-2 (see below; reviewed by Kvansakul et al., 2017) (Figures 1A,B).
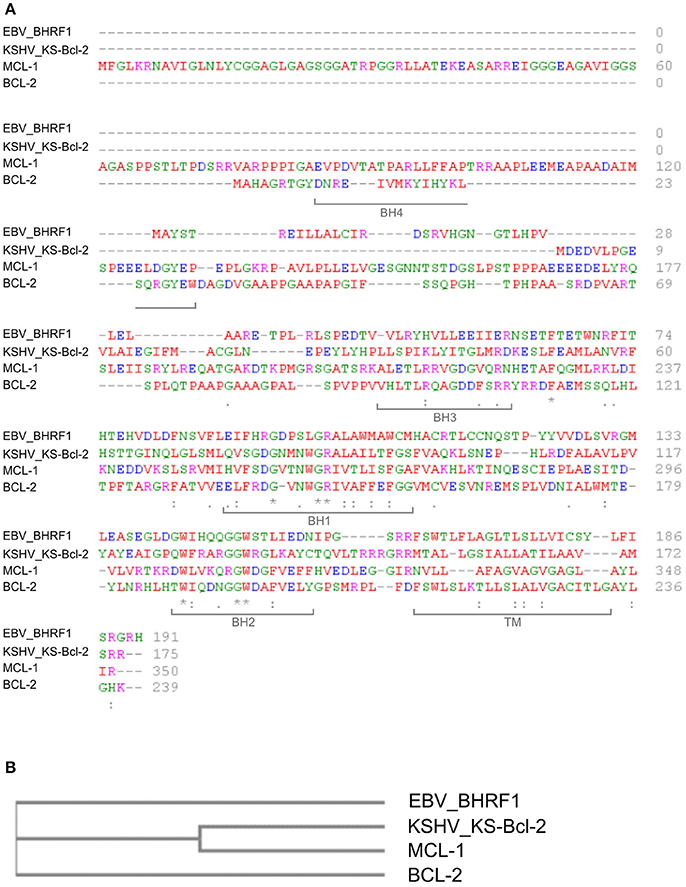
Figure 1. Sequence comparison of human Bcl-2 family proteins and viral homologues. (A) Multiple sequence alignment. Protein sequences were obtained from the UniProt database (http://www.uniprot.org/) and analyzed with the online software Clustal Omega (http://www.ebi.ac.uk/Tools/msa/clustalo/). Amino acids are labeled in different colors according to their biochemical properties (red: small/hydrophobic; blue: acidic; magenta: basic; green: hydroxyl/sulfhydryl/amine/Glycine). Asterisks indicate single conserved residues; periods indicate residues with similar properties, and brackets indicate locations of the BH and TM domains in Bcl-2 (UniProtKB–P10415 (BCL2_HUMAN). Accession IDs: KS-Bcl-2 (>sp|F5HGJ3|ARBH_HHV8P); BHRF1 (>sp|P03182|EAR_EBVB9); Bcl-2 (>sp|P10415|BCL2_HUMAN)); Mcl-1 (>sp|Q07820|MCL1_HUMAN). (B) Cladogram representing the similarity between human Bcl-2 and Mcl-1 and the viral orthologs of EBV (EBV_BHRF1) and KSHV (KSHV_KS-Bcl-2). The cladrogram was generated with the online software Clustal Omega.
BHRF1 accumulates in the outer mitochondrial membrane (OMM) of B-cells, a distribution similar to that of Bcl-2 (Hickish et al., 1994). Mitochondrial targeting of BHRF1 is directed by a C-terminal sequence that shares homology with the transmembrane domain (TM) present in some cellular Bcl-2 family members (Bcl-2 38%, Bcl-xl 32%, Bax 34%) (Hickish et al., 1994). As shown in Figure 1A, BHRF1 also has all of the Bcl-2 homology (BH) domains except for BH4, which is in general poorly conserved among Bcl-2 family members.
The anti-apoptotic effects of BHRF1 are mainly due to its ability to bind to BH3-only pro-apoptotic Bcl-2 proteins and to inhibit the formation of Bax/Bak oligomers in the OMM (Cross et al., 2008; Flanagan and Letai, 2008; Kvansakul et al., 2010; Milian et al., 2015). BHRF1 binds to the BH3-only proteins Bim, Bak, Bid and PUMA (Kvansakul et al., 2010). Structural studies of the BHRF1:Bim/Bak complexes indicated that an aspartic acid at position 100 of BHRF1 is essential for its association with the BH3 domains of Bim and Bak, in interactions similar to those occurring between Bim and the cellular anti-apoptotic protein Bcl-xL (Kvansakul et al., 2010). EBV also codes for BALF1, a cytoplasmic protein with homology to Bcl-2 family members that was shown by one group to interfere with the anti-apoptotic activity of BHRF1 (Bellows et al., 2002) but by others to inhibit apoptosis (Marshall et al., 1999) and promote transformation (Hsu et al., 2012).
BZLF1 (also known as ZEBRA, Zta) is a 245-amino acid EBV protein of the basic leucine zipper (b-zip) family that is essential for transcription of EBV lytic-phase genes and replication of viral genomes (reviewed by McKenzie and El-Guindy, 2015). Experiments carried out in EBV-positive B-cell lines indicated that BZLF1 can interact with the mitochondrial protein mtSSB, a single-stranded DNA-binding protein that is needed for replication of the mitochondrial genome. Cells in the lytic phase showed reduced mtDNA synthesis and contained fewer mitochondrial genomes, while silencing of mtSSB interfered with BZLF1-dependent entry into the lytic phase. Imaging analyses of cells expressing FLAG-tagged BZLF1 indicated that BZLF1 partially redirects mtSSB from mitochondria to the nucleus, which is the main site of BZLF1 accumulation (Wiedmer et al., 2008). Expression of BZLF1 in HeLa cells resulted in fused masses of mitochondria, some of which were localized near the nucleus (LaJeunesse et al., 2005); it is however unclear how BZLF1 induces these changes in mitochondrial morphology.
EBV LMP2A (latent membrane protein 2A) is a 497-amino acid protein with 12 transmembrane domains that accumulates mainly in plasma membrane rafts, where it influences signal transduction pathways affecting cell activation, proliferation, survival and migration (reviewed by Cen and Longnecker, 2015). Pal et al. (2014) showed that LMP2A has indirect effects on mitochondria. Expression of LMP2A in EBV-negative gastric- and breast cancer cell lines resulted in increased mitochondrial fission accompanied by an increase in migration and induction of the epithelial-mesenchymal transition. These effects were attributed to LMP2A's ability to stimulate the Notch pathway, which in turn upregulates Drp1 (dynamin-related protein 1), a protein that induces mitochondrial fission (Pal et al., 2014).
KSHV: K7, KS-Bcl-2
K7 is a 126-amino acid protein that shows homology to the cellular protein Survivin, a member of the family of inhibitor of apoptosis proteins (IAPs). K7 is detected in the endoplasmic reticulum, nucleus and in mitochondria (Feng et al., 2002; Wang et al., 2002) and is expressed early after induction of the lytic cycle (Wang et al., 2002). It contains an amino-terminal, atypical MTS, a putative transmembrane domain partially overlapping the MTS, a baculovirus IAP repeat (BIR), and a carboxy-terminal BH2 domain (Wang et al., 2002).
K7's BH2 and BIR domains mediate its direct interaction with Bcl-2 and activated caspase-3, respectively; the resulting bridge between K7, Bcl-2, and activated caspase-3 inhibits the apoptotic caspase cascade (Wang et al., 2002). Another anti-apoptotic function of K7 involves its association with CAML (calcium-modulating cyclophilin ligand), an ER protein that controls intracellular Ca2+ homeostasis (Feng et al., 2002). The CAML-K7 interaction alters the changes in cytosolic Ca2+ induced by thapsigargin, an inhibitor of the sarcoplasmic/endoplasmic Ca2+-ATPase (SERCA), whose role is to transport Ca2+ from the cytosol to the ER. In this manner, K7 protects thapsigargin-treated cells from Ca2+-induced loss of ΔΨm and apoptosis (Feng et al., 2002).
K7 also interacts with a helix-coiled region (HC) of Rubicon, an inhibitor of autophagosomal maturation that forms a complex with Beclin 1, UVRAG and Vps34 (Liang et al., 2013). K7 increases the expression of Rubicon and enhances its interaction with the autophagy machinery, resulting in a block in the autophagy process. Cells expressing a virus knocked out for K7 were able to produce virus, but expression of the viral proteins K3 and K8 was reduced (Liang et al., 2013). K7-mediated regulation of K3 and K8 expression would have an important effect on viral persistence, as K3 induces internalization and degradation of MHC-I complexes, thus favoring the escape of virus-infected cells from the control of the immune system, while K8 is part of the viral lytic DNA replication complex (Liang et al., 2013).
Another binding partner of K7 is vGPCR, a KSHV homolog of the human interleukin-8 receptor that induces tumors in nude mice. K7 induces the degradation of vGPCR through the ER-associated degradation pathway, resulting in a decrease in tumorigenicity (Feng et al., 2008a).
KS-Bcl-2 is a 175-amino acid protein that is expressed from KSHV ORF16. KS-Bcl-2 shares about 60% overall sequence homology with cellular proteins of the Bcl-2 family, with high conservation of the BH1, BH2 and TM domains (Figure 1A; Sarid et al., 1997). KS-Bcl-2 appears to be more closely related to MCL-1 than to Bcl-2 in terms of sequence homology (Figure 1B) and functional activity (Flanagan and Letai, 2008).
The first study of KS-Bcl-2 described a punctate cytoplasmic distribution, indicative of accumulation in organelle membranes, but did not identify these organelles (Sarid et al., 1997). More recent studies of KS-Bcl-2 indicated its partial localization in mitochondria and in the nucleus (Gallo et al., 2017) or nucleoli (Kalt et al., 2010); these studies employed tagged versions of KS-Bcl-2 and a fluorescent marker to distinguish the mitochondrial compartment in different cell lines. The combination of tagging and cell lines used in these studies likely influenced KS-Bcl-2's localization: in the study by Gallo et al., when expressed in 293A cells, FLAG-tagged KS-Bcl-2 colocalized with the fluorescent mitochondrial marker in compact crescent-shaped structures lying against the nucleus, while in HUVEC cells the protein colocalized with the marker in structures resembling the web-like mitochondrial network (Gallo et al., 2017).
The KS-Bcl-2 mRNA is detected in KS lesions and in PEL cells upon induction of the lytic cycle and is required for lytic reactivation and replication (Gelgor et al., 2015; Liang et al., 2015). The best-defined roles of KS-Bcl-2 are to inhibit both apoptosis and autophagy. KS-Bcl-2 impedes apoptosis through interactions with the BH3 domains of pro-apoptotic proteins of the Bcl-2 family (Bim, Bid, PUMA, Bik, NOXA, Bmf) (Flanagan and Letai, 2008) and through its association with Aven, a protein that interferes with the ability of Apaf-1 to activate caspase 9 (Chau et al., 2000). Inhibition of autophagy by KS-Bcl-2 is mediated by its binding to Beclin 1, a key component of the autophagy pathway (Pattingre et al., 2005). Interestingly, the role of KS-Bcl-2 in promoting viral reactivation and replication is apparently independent from its effects on apoptosis and autophagy (Gelgor et al., 2015; Liang et al., 2015).
The nuclear targeting of KS-Bcl-2 was mapped to its amino-terminal 17 amino acids and was found to be important for the protein's effects of viral replication (Gallo et al., 2017). Nucleolar accumulation of KS-Bcl-2 depended on its association with the cellular nucleolar protein GLTSCR2/PICT-1, and interfered with KS-Bcl-2's anti-apoptotic properties (Kalt et al., 2010).
HBV
HBV is a small, enveloped DNA virus of the Hepadnaviridae family that is transmitted parenterally through blood and other body fluids. Most HBV-infected patients are asymptomatic or present signs of acute liver disease and inflammation. Active viral replication and insufficient immune clearance may result in chronic hepatitis, with tissue injury, inflammation and regeneration that can progress to cirrhosis, liver failure and hepatocellular carcinoma (HCC), an aggressive neoplasm with a dismal prognosis. Despite the availability of a prophylactic vaccine since the 1980s, approximately 257 million persons are chronically infected with HBV worldwide, with high-prevalence areas in the African and Western Pacific regions (statistics for 2015, World Health Organization, 2017).
HBV produces two proteins that interact with mitochondria: HBx and Pol (Table 1).
HBx
The 154-amino acid HBx protein favors HBV replication, influences cellular transcription, signal transduction, cell proliferation and survival, and is considered to be a key contributor to the oncogenic potential of HBV (reviewed by Benhenda et al., 2009; Motavaf et al., 2013). Takada et al. provided the first evidence for mitochondrial accumulation of HBx, which was accompanied by aggregation of mitochondria near the nucleus, accumulation of p53 in mitochondria, loss of ΔΨm and increased apoptotic death (Takada et al., 1999). A subsequent study revised HBx's targeting properties to include the nucleus and cytoplasm (Henkler et al., 2001). The mitochondrially-associated fraction of HBx was detected in the outer mitochondrial membrane in the hepatoma cell line Huh7, in the hepatocarcinoma cell line HepG2, and in primary rat hepatocytes (Huh and Siddiqui, 2002; Clippinger and Bouchard, 2008). Experiments carried out with GFP fusion proteins in Huh7 cells indicated that residues 68-117 of HBx confer mitochondrial targeting (Shirakata and Koike, 2003).
HBx was shown to induce loss of ΔΨm when expressed in Huh7 cells (Takada et al., 1999; Rahmani et al., 2000; Shirakata and Koike, 2003) or HepG2 cells (Clippinger and Bouchard, 2008). However, primary rat hepatocytes expressing HBx were protected from loss of ΔΨm induced by treatment with TNF-α, a property attributed to HBx's ability to induce the NF-κB pathway (Clippinger and Bouchard, 2008). More recently, Zheng et al. (2014) showed that HBx increases ΔΨm in HepG2 cells, an effect that was correlated with increased levels of cytochrome c oxidase III (COXIII) and increased COX activity. HBx-induced upregulation of COXIII expression and activity were also observed in the immortalized hepatocyte cell line HL-7702 (Zou et al., 2015). Although no changes in ΔΨm were observed in this system, HBx increased cellular ROS production (Zou et al., 2015).
An HBx-mediated increase in cellular ROS was associated with activation of the STAT3 and NF-κB pathways (Waris et al., 2001), both of which are important in inflammation and transformation. HBx-induced cellular ROS was also accompanied by induction of cyclooxygenase 2 (COX-2), an enzyme that catalyzes the production of mediators of the inflammatory response (Lim et al., 2010). The observation that ROS-enhancing agents such as the chemotherapeutic drug adriamycin increase the stability of HBx protein suggests the existence of a ROS-HBx positive feedback loop (Wang et al., 2003). Such a positive role for ROS would fit in with results of a recent study which indicated that HBV infection induces oxidative stress, in part through the ability of HBx to increase mitochondrial ROS, and that ROS favor replication of the virus (Ren et al., 2016).
Binding partners of HBx in mitochondria include the voltage-dependent anion channel 3 (VDAC3), which resides in the OMM (Rahmani et al., 2000), and heat shock protein 60 (HSP60), a multifunctional chaperone that is located mainly in the mitochondrial matrix (Tanaka et al., 2004). Studies carried out in Huh7 cells showed that HBx-induced ROS promotes the translocation of the serine/threonine kinase Raf-1 (C-Raf) from the cytoplasm to mitochondria, where it forms complexes with HBx (Chen and Siddiqui, 2007).
HBx-induced alterations in intracellular calcium signaling play an important role in HBV replication (Bouchard et al., 2001). HBx increases the basal levels of cytosolic Ca2+ in HepG2 cells (McClain et al., 2007) and augments the spike in cytosolic Ca2+ levels provoked by ATP (Chami et al., 2003). The HBx-mediated increase in cytosolic Ca2+ reflects influx of Ca2+ into cells due to store-operated calcium entry and is associated to greater uptake of Ca2+ by mitochondria (Yang and Bouchard, 2012).
HBx-expressing cells also show changes in mitochondrial dynamics with increased fission, which results both from phosphorylation and mitochondrial translocation of the fission protein Drp1 and from ubiquitination/degradation of Mitofusin 2 (Mfn2), a protein that mediates mitochondrial fusion (Kim et al., 2013a). The study by Kim et al. also showed that HBx promotes mitophagy, a specialized form of autophagy that eliminates dysfunctional mitochondria. This effect was attributed to increased expression and mitochondrial targeting of Parkin, a ubiquitin ligase whose substrates provide a signal for sequestration and degradation of damaged mitochondria (reviewed by Bernardini et al., 2017). Thus, by enhancing mitophagy, HBV could promote cell survival and possibly viral persistence. On the other hand, induction of mitochondrial fission may lead to mitochondrial injury, which might play a role in the pathogenesis of HBV-related liver disease (Kim et al., 2013a).
HBx can either directly affect apoptosis or modify the response of cells to apoptotic stimuli such as TNF-α (reviewed by Rawat et al., 2012). The varied effects of HBx on cell death likely depend on its expression levels and on the cell context.
Pol
HBV Pol is an 832-residue protein whose principal role is to direct packaging of viral pregenomic RNA (pgRNA) molecules into capsids and to reverse-transcribe the RNA into the dsDNA genome. In a study of the sites of HBV replication, Pol was observed to accumulate mainly in mitochondria (Unchwaniwala et al., 2016). A segment of Pol spanning residues 141-160 functioned as a mitochondrial targeting sequence (MTS) when attached to GFP and was important for pgRNA packaging, but could be deleted from Pol without abolishing its mitochondrial targeting, indicating that additional amino-terminal sequences contribute MTS properties. Based on the finding that neither the pgRNA nor the viral Core protein (Cp) localized to the mitochondria during replication, Unchwaniwala et al. (2016) suggested that the binding of pgRNA to the MTS may block mitochondrial localization, so that Pol bound to pgRNA would be incorporated into in capsids, while unbound Pol would accumulate in mitochondria. Although the role of Pol in mitochondria remains to be understood, the protein is known to have diverse activities besides HBV genome packaging and reverse transcription, including interference with the antiviral interferon response (Wu et al., 2007; Wang and Ryu, 2010), an effect that would favor viral persistence in chronically infected patients.
HCV
HCV belongs to the Flaviviridae family of small, enveloped viruses with a single-stranded RNA genome. About 80–85% of HCV-infected persons fail to eliminate the virus due to the ability of HCV to evade innate and adaptive immune surveillance. Persistent HCV infection is associated with liver pathologies (chronic hepatitis, hepatic steatosis, hepatic fibrosis, cirrhosis, and hepatocellular carcinoma) and lymphoproliferative disorders. Although new treatment regimens with direct-acting antivirals (DAAs) are capable of eliminating the infection, HCV remains an important clinical and social problem due to the high cost of DAAs, susceptibility of cured patients to reinfection, and the lack of a prophylactic vaccine (reviewed by Webster et al., 2015; Dustin et al., 2016). Approximately 71 million persons are chronically infected with HCV, in a heterogeneous world-wide distribution with higher prevalence in the Eastern Mediterranean and European regions (statistics for 2015, World Health Organization, 2017).
The 9.6 kb HCV genome codes for a polyprotein precursor that is cleaved by viral and cellular proteases into the structural proteins Core, E1 and E2, which make up the virion, and nonstructural proteins p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B, which are involved in polyprotein processing, genome replication and virion assembly (reviewed by Scheel and Rice, 2013). The viral life cycle takes place in the ER and an ER-derived membranous web associated with lipid droplets, and results in ER stress (Dash et al., 2016). Core, p7, NS3/4, NS4A, and NS5A show partial accumulation in mitochondria or MAM (Table 1).
Core
Core is a 177-amino acid protein that is derived from the amino-terminal end of the polyprotein precursor by two proteolytic cleavage events (Okamoto et al., 2008). In addition to forming the virion capsid, Core has multiple effects on viral and cellular pathways, and can protect against or sensitize cells to apoptotic stimuli (reviewed by Kao et al., 2016). Studies of cell lines ectopically expressing Core indicated its accumulation mainly in the ER (Lo et al., 1995; Santolini et al., 1995), in both the ER and outer mitochondrial membrane (Suzuki et al., 2005), or in MAMs and the OMM (Schwer et al., 2004). The association of Core with the ER/MAMs/OMM is mediated by an amphipathic alpha helical sequence near its carboxy terminus (Schwer et al., 2004; Suzuki et al., 2005).
The targeting properties of Core are influenced by its interactions with other viral proteins. When expressed with E1, Core was targeted mainly to lipid droplets (Barba et al., 1997). Analyses of HCV-infected Huh7 cells indicated accumulation of Core mainly in lipid droplets and associated membranes (Rouille et al., 2006) or mainly in the ER and to a lesser extent in mitochondria (Schwer et al., 2004). The fraction of Core associated with mitochondria in infected cells is restricted to the outer mitochondrial membrane (OMM) or possibly MAMs (Wang et al., 2010).
The phenotype of Core-transgenic mice includes hepatic steatosis and hepatocellular carcinoma (Moriya et al., 1997, 1998). The hepatocytes of these mice showed age-dependent accumulation of Core in the nucleus and in morphologically altered, dysfunctional mitochondria (Moriya et al., 1998), and signs of oxidative stress (Moriya et al., 2001), suggesting a direct role for Core in the oxidative stress and impaired mitochondrial function observed in patients with HCV-induced hepatitis (Farinati et al., 1995; Barbaro et al., 1999). Core was also detected in mitochondria of hepatocytes from mice co-expressing Core, E1, E2, and p7; the mitochondria contained reduced levels of glutathione and NADPH, indicating oxidative stress, and showed impaired Complex I activity, reduced respiration, and increased ROS (Korenaga et al., 2005).
Further studies of Core-expressing cell lines confirmed that it induces mitochondrial ROS (Okuda et al., 2002; Chu et al., 2011), as well as loss of ΔΨm (Benali-Furet et al., 2005; Machida et al., 2006), and release of cytochrome c into the cytoplasm (Okuda et al., 2002; Benali-Furet et al., 2005). Other effects of Core include increased lipid peroxidation (Okuda et al., 2002; Machida et al., 2006), accumulation of lipid droplets (Chu et al., 2011), increased autophagy (Chu et al., 2011), and apoptosis (Benali-Furet et al., 2005). Studies in Huh7 cells and the B-cell line Raji linked the effects of Core on ΔΨm and cellular ROS to activation of the STAT3 pathway and induction of DNA damage (Machida et al., 2006).
Core also induces an accumulation of Ca2+ in the cytoplasm due to a defect in SERCA function (Benali-Furet et al., 2005). The consequent loading of mitochondria with Ca2+ favors mitochondrial ROS production and triggers the mitochondrial permeability transition (Li et al., 2007). Augmented mitochondrial uptake of Ca2+ as well as inhibition of Complex I activity and increased ROS and glutathione oxidation are also observed when Core is added to isolated mitochondria (Korenaga et al., 2005). The effects of Core on intracellular Ca2+ likely contribute to the signs of ER stress observed in an osteosarcoma cell line expressing HCV (Piccoli et al., 2006, 2007, 2009). In Huh7 cells, HCV was found to favor ROS induction, loss of ΔΨm and apoptosis triggered by treatment with the pro-oxidant t-BOOH; interestingly, these effects that were lost in the absence of Core, E1, E2, and p7 (Wang et al., 2010).
Analyses of Core deletion mutants indicated that its first 75 amino acids mediate binding to the mitochondrial matrix protein HSP60, and are required for Core-mediated induction of intracellular ROS (measured using DCF-DA) and sensitization to TNF-α-induced apoptosis (Kang et al., 2009); the location in which the Core-HSP60 interaction influences ROS production was not identified.
Core also interacts with Prohibitin, a multifunctional protein that influences mitochondrial function and apoptosis (reviewed by Peng et al., 2015). Core-expressing HepG2 cells expressed increased levels of Prohibitin, but reduced levels of COX subunits I and II, suggesting that the Core-Prohibitin interaction interferes with proper assembly and activity of respiratory chain complexes, which would be expected to result in increased mitochondrial ROS production (Tsutsumi et al., 2009).
Studies of the impact of HCV on mitophagy indicated either a promoting (Kim et al., 2013b) or blocking (Hara et al., 2014) effect. The inhibitory effect was attributed to the ability of Core to interact with and inhibit mitochondrial accumulation of Parkin (Hara et al., 2014). Persistence of damaged mitochondria due to the Core-Parkin interaction may thus contribute to HCV-associated liver damage and HCC.
Alcohol consumption worsens the clinical course of chronic hepatitis C. A study of Huh7 cells provided evidence that Core may contribute to oxidative stress that arises during the metabolism of ethanol by cytochrome p4502E1 (CYP2E1) (Otani et al., 2005).
Hepatitis C patients and Core-transgenic mice show hepatic accumulation of iron (Farinati et al., 1995; Moriya et al., 2010). In iron-overload experiments, hepatocytes from Core-transgenic mice and Core-expressing HepG2 cells showed impaired upregulation of the antioxidant heme-oxygenase-1, which is normally induced by excess iron (Moriya et al., 2010). Such a defect in the iron-overload response could contribute to deregulated ROS production and scavenging (Fujinaga et al., 2011).
p7, NS3/4A, and NS5A
There is also evidence that the HCV nonstructural proteins p7, NS3/4A, and NS5A influence mitochondrial function. p7 is a 63-amino acid hydrophobic protein that forms hexameric ion channels in membranes and is classified as a viroporin (reviewed by Nieva et al., 2012). p7 is required for the assembly and release of infectious virions, thus making it a potential target for antiviral therapy (reviewed by Madan and Bartenschlager, 2015). p7 accumulates mainly in the ER (Carrere-Kremer et al., 2002; Isherwood and Patel, 2005; Haqshenas et al., 2007; Vieyres et al., 2013) and in MAMs (Griffin et al., 2004, 2005). A recent study of the impact of HCV on the interferon response indicated that p7 induces mitochondrial membrane depolarization and binds to IFI6-16, an interferon response protein that stabilizes ΔΨm, an effect that is counteracted by p7 (Qi et al., 2017).
NS3 is a 631-amino acid protein with serine protease- and RNA helicase activities that, in association with the 54-amino acid accessory protein NS4A, forms a membrane-bound complex that is required for HCV RNA replication, polyprotein processing and virion assembly (reviewed by Morikawa et al., 2011), and is a target of DAAs. NS3/4A also cleaves several cellular proteins, including MAVS, a component of the innate immune response signaling pathway triggered by binding of pathogen-associated molecular patterns (PAMPs) to RIG-1 (Meylan et al., 2005; Belgnaoui et al., 2011). In Huh7 cells, MAVS is detected in peroxisomes, mitochondria and MAMs (Seth et al., 2005; Horner et al., 2011) and is susceptible to cleavage by NS3/4A in these compartments (Horner et al., 2011; Bender et al., 2015). Consistent with these observations, there is evidence that NS3/4A is partially targeted to mitochondria (Wolk et al., 2000; Nomura-Takigawa et al., 2006). Huh7 cells expressing both NS3 and NS4A showed increased sensitivity to an apoptosis-inducing drug, suggesting that NS3/4A may contribute to HCV-associated hepatic injury by priming cells to death stimuli (Nomura-Takigawa et al., 2006). Huh7 cells expressing NS4A alone showed accumulation of the protein in the ER and in mitochondria that were perinuclear, doughnut-shaped, and depolarized. NS4A-expressing cells released cytochrome c in the cytoplasm and showed increased spontaneous death compared to control cells or cells coexpressing NS3 and NS4A (Nomura-Takigawa et al., 2006).
NS5A is a 467-amino acid ER/membranous web-associated phosphoprotein that regulates viral replication and assembly, and is a target of DAA (reviewed by Ross-Thriepland and Harris, 2015). When expressed in Huh7 cells, NS5A activates the NF-κB and STAT3 pathways through Ca2+-mediated induction of oxidative stress (Gong et al., 2001). Studies of a GFP-NS5A fusion protein in HEK293 cells indicated a role for NS5A in tethering the ER to mitochondria through a mechanism that involves binding of NS5A to phosphatidylinositol 4-kinase PI4KA III-α (PI4KA) (Siu et al., 2016). GFP-NS5A-expressing cells contained fragmented mitochondria but were more resistant to apoptosis induced by hydrogen peroxide compared to control cells, suggesting that NS5A may favor survival of HCV-infected cells under conditions of oxidative stress (Siu et al., 2016).
HPV
Human papillomaviruses (HPVs), members of the Papillomaviridae family, are small non-enveloped viruses that possess a double-stranded circular DNA genome and show a tropism for epithelial cells (reviewed by Doorbar et al., 2015). The HPV genome codes for 6 early genes named E1, E2, E4, E5, E6, and E7, which regulate virus replication, and 2 late genes named L1 and L2, which make up the virion capsid. The pattern of HPV gene expression is tightly controlled by the differentiation status of the infected cell: after penetration into the mucosal or cutaneous epithelium through microlesions, replication initiates with expression of early genes in the basal layer, while expression of late proteins making up the virus particles is restricted to the upper layers (Tomaic, 2016).
HPV is classified into more than 200 genotypes based on the sequence of the L1 (major capsid) gene. The genotypes are grouped into 5 genera, the 2 most numerous of which are named alpha and beta. Alpha-HPVs are further classified as low-risk or high-risk based on their connection with cancer (reviewed by Egawa et al., 2015). The low-risk viruses (e.g., HPV-6 and HPV-11) cause warts and other benign proliferative lesions of the skin and mucosa, while high-risk viruses (e.g., HPV-16 and HPV-18) are associated with cervical carcinoma and cancers of the anogenital and head-and neck regions. Beta-HPVs infect cutaneous epithelia and may contribute to the initiation of non-melanoma skin cancers, with immunosuppression representing a risk factor (Tomaic, 2016). HPV-mediated transformation is driven by the viral proteins E6 and E7 (reviewed by Doorbar et al., 2015; Tomaic, 2016). The frequency of HPV-associated pathologies should be reduced with the availability, since 2006, of vaccines based on virus-like particles containing the L1 protein of selected high-risk or high- plus low-risk genotypes (reviewed by Harper and DeMars, 2017).
E1∧E4
Two HPV proteins, E1∧E4 and E2, partially localize to mitochondria (Table 1, Figure 2). E1∧E4 is produced by a spliced mRNA that joins the first few codons from the E1 ORF in frame to the E4 ORF, resulting in a fusion protein of about 90-120 amino acids, depending on the genotype. E1∧E4 is the most abundantly expressed viral protein in the mid-upper epithelial layers of productively infected lesions (reviewed by Doorbar, 2013). Doorbar et al. (1991) showed that E1∧E4 binds to and collapses the cytokeratin network in mature human keratinocytes. Collapse of the cytokeratin network leads to accumulation of E1∧E4 in mitochondria, mediated by an N-terminal leucine-rich sequence (Raj et al., 2004). E1∧E4-containing mitochondria dissociate from microtubules, are clustered in the perinuclear region and exhibit a severe reduction in ΔΨm; these changes are associated with increased apoptosis (Raj et al., 2004).
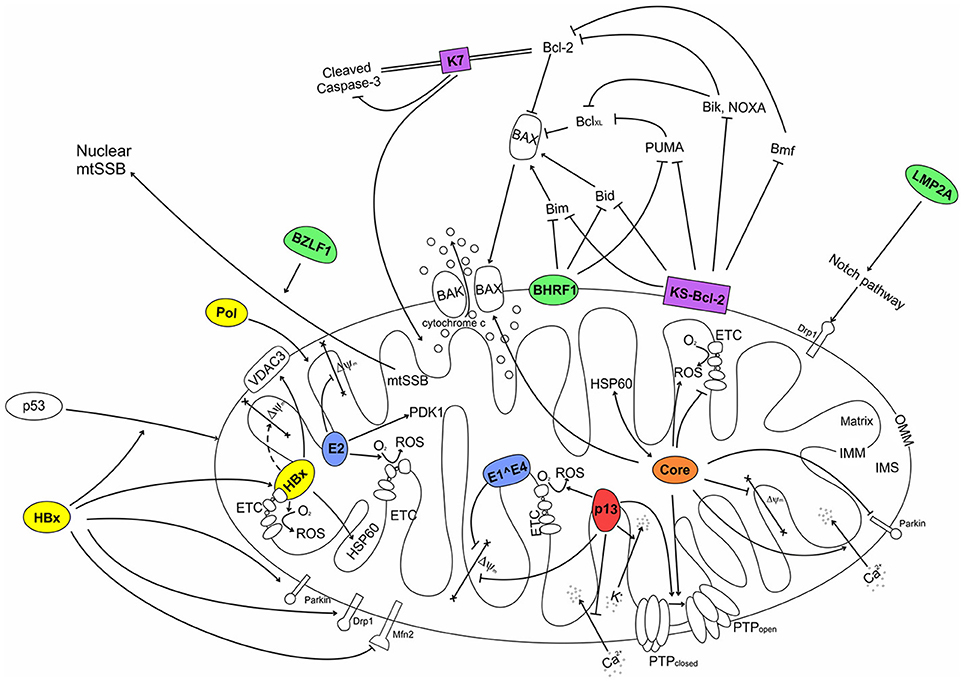
Figure 2. Interactions of human tumor virus proteins with mitochondria. HTLV-1 (red): p13 causes an inward K+ current that leads to mitochondrial swelling, depolarization and increased ROS production that lowers the PTP opening threshold. p13 also reduces mitochondrial Ca2+ uptake. EBV (green): The Bcl-2 homolog BHRF1 localizes at the OMM and binds to Bim, Bid and PUMA, resulting in the inhibition of Bax translocation to the OMM. BZLF1 interacts with mtSSB. LMP2A increases expression of Drp1 (dynamin-related protein 1) through stimulation of the Notch pathway. KSHV (purple): The Mcl-1 homolog KS-Bcl-2 localizes at the OMM and can bind and inhibit a variety of BH3-only proteins, resulting in the inhibition of Bax-Bak oligomerization at the OMM. The K7 protein forms a bridge between cellular Bcl-2 and cleaved Caspase-3, resulting in inhibition of Caspase-3 activity. HBV (yellow): HBx interacts with the Complex IV subunit COXIII and increases ROS generation by the ETC; HBx can interact with VDAC3 and HSP60. Furthermore, HBx can induce p53 translocation to mitochondria. HBx was also shown to influence mitochondrial dynamics through its interaction with Drp1 and Mnf2. Polymerase (Pol) contains an amino-terminal MTS that determines its mitochondrial targeting; it impact on mitochondria remains to be understood. HCV (orange): Core increases mitochondrial respiration, ROS generation, and uptake of Ca2+, which sensitizes PTP opening. Core also inhibits translocation of Parkin to mitochondria, favors/facilitates/promotes Bax-Bak oligomerization, and interacts with the matrix chaperone HSP60. HPV (blue): The E2 protein interacts with IMM proteins and induce expression of the matrix protein PDK1 (pyruvate dehydrogenase kinase 1); E2 also increases ROS generation in mitochondria. The E1∧E4 protein causes loss of ΔΨm. OMM, outer mitochondrial membrane; IMM, inner mitochondrial membrane; IMS, inter-membrane space; ROS, reactive oxygen species; MTS, mitochondrial targeting sequence; ETC, electron transport chain; PTP, permeability transition pore; not determined.
E2
E2 is a protein of about 365 amino acids (HPV genotype 16; its length varies among genotypes) whose expression gradually increases from the mid- to upper layers of infected lesions. This pattern of expression reflects the key role of E2 in the coordination of late events of viral replication through its influence on transcription, viral genome replication and partitioning in dividing cells, and RNA processing (reviewed by Graham, 2016).
A comparison of the properties of E2 from low-risk (genotypes 6, 11) and high-risk (genotypes 16, 18) viruses revealed stable nuclear compartmentalization of low-risk E2, and shuttling of high-risk E2 between the nucleus and cytoplasm, a property that was connected to the ability of high-risk E2 to induce apoptosis (Blachon et al., 2005). A more recent analysis of E2 trafficking provided evidence that the cytoplasmic fraction of E2 eventually accumulates in mitochondria. Lai et al. studied GFP-tagged E2 proteins from HPV-18 and HPV-6 (high- and low-risk genotypes, respectively) in a keratinocyte cell line (Lai et al., 2013). Time-lapse studies showed that HPV-18 GFP-E2 was gradually relocalized from the nucleus to mitochondria, whereas HPV-6 GFP-E2 remained predominantly nuclear and was rerouted to mitochondria only when expressed at high levels. Mass spectrometry analysis of proteins co-immunoprecipitated with HPV-18 E2 revealed the association of E2 with many mitochondrial proteins, mostly of the IMM, including subunits of Complexes III, IV, and V. Cells expressing HPV-18 GFP-E2 exhibited perinuclear clustering of the mitochondria and loss of the cristae structure. HPV-18 GFP-E2-expressing keratinocytes showed higher levels of mitochondrial ROS however, this effect did not induce cytochrome c release or apoptosis.
Lai et al. (2013) also showed that HPV-18 E2 stabilizes HIF-1α and induces the expression of known HIF-1α target genes, including PDK1 (pyruvate dehydrogenase kinase 1) and CAIX (carbonic anhydrase IX). These changes were accompanied by a modest but statistically significant increase in lactate production, an indication of a shift toward glycolytic metabolism. These effects were not observed in cells expressing low-risk HPV-6 E2. Although mitochondrial localization of HPV-18 E2 was evident Lai et al.'s transfection assays, their results of immunohistochemistry assays revealed cytoplasmic staining of HPV-18 E2 in a CINII lesion (grade II cervical intraepithelial neoplasia) and mainly nuclear accumulation of HPV-6 E2 in a benign condyloma.
A study by Chen et al. (2014) indicated that HPV-induced mitochondrial alterations also involve interplay between E2 and C1QBP (complement C1q-binding protein, also named gC1qR, receptor of the globular heads of complement C1q), a predominantly mitochondrial protein that has many roles, including regulation of mitochondrial and ER morphology and cell metabolism (Hu et al., 2013). Both HPV-16 E2 and C1QBP were less abundant in cervical carcinoma samples compared to non-neoplastic cervical tissue, suggesting a negative role for these proteins in the growth of cervical epithelial cells (Chen et al., 2014). Ectopic expression of E2 in keratinocytes led to upregulation of C1QBP and signs of mitochondrial dysfunction, including increased cellular ROS, augmented levels of cytosolic Ca2+, loss of ΔΨm, and increased apoptosis; similar effects were observed in cells overexpressing C1QBP. These properties described for E2 contrast with previous observations made for the HPV oncoproteins E6 and E7, which downregulate C1QBP expression and protect cells from apoptosis (Gao et al., 2011; Chen et al., 2014). It is noteworthy that HCV Core protein also binds to C1QBP, resulting in impaired cytokine production (Song et al., 2016).
Concluding Remarks
Mitochondria play a central role in key biological processes, including energy conservation, cell death and Ca2+ signaling. These processes are involved both in the control of physiological tissue homeostasis and in the process of neoplastic transformation. It is thus not surprising that tumor viruses have developed common strategies to impinge on mitochondrial function. The experimental evidence collected so far suggests five major mechanisms by which proteins coded by tumor viruses interact with mitochondria (see Figure 2): (i) direct inhibition of apoptosis by viral Bcl-2 homologs; (ii) deregulation of cellular bioenergetics through the interaction with ETC components and inner mitochondrial membrane complexes; (iii) changes in the mitochondrial production of ROS, which indirectly influence cell turnover; (iv) changes in mitochondrial Ca2+ uptake, which influence Ca2+ homeostasis and Ca2+-dependent signal transduction pathways; (v) interaction with OMM proteins that control innate immunity. Mitochondrial viral proteins can also alter mitochondrial morphology through interactions with the fission/fusion machinery.
Although much experimental information supports these points of interaction between tumor virus-encoded proteins and mitochondria, it must be emphasized that in most cases these results were obtained from experimental systems based on the ectopic overexpression of the viral protein of interest, in the absence of other viral proteins, and in cell types that are not the targets of natural infection by the viruses, conditions that may not recapitulate the situation in naturally infected cells. Further investigation is thus needed in order to validate and clarify the effects of viral-encoded mitochondrial proteins in the context of the viruses' life cycle and in connection to their pathogenic properties in the natural host.
In addition to providing information about the life cycle and pathogenesis of human tumor viruses, the examination of virus-mitochondrial interactions using more refined experimental systems will enhance our understanding of basic cellular processes controlled by mitochondria, thus paving the road to the identification of new strategies to clear viral infections and treat cancer patients.
Author Contributions
IC prepared the paragraph on HPV and Table 1. GS prepared the paragraph on EBV and Figures 1, 2. MS-B prepared the paragraph on HSHV. VR prepared the paragraph on HTLV-1. VC prepared the paragraph on HBV and revised the final version of the paper. DD prepared the paragraph on HCV, Table 2 and revised the final version of the paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Luigi Chieco-Bianchi for discussions. Research in VC laboratory is supported by an Investigator Grant from the Associazione Italiana per la Ricerca sul Cancro (AIRC IG # 17794), a University of Padova Ateneo grant, and a University of Verona-Veneto Institute of Oncology Joint Project grant. DD research is supported by the University of Padova. IC, VR, and MS-B are supported by fellowships from the Veneto Institute of Oncology and GS is a recipient of a Ph.D. fellowship from the Fondazione Cassa di Risparmio di Padova e Rovigo.
References
Altmann, M., and Hammerschmidt, W. (2005). Epstein-Barr virus provides a new paradigm: a requirement for the immediate inhibition of apoptosis. PLoS Biol. 3:e404. doi: 10.1371/journal.pbio.0030404
Andresen, V., Pise-Masison, C. A., Sinha-Datta, U., Bellon, M., Valeri, V., Washington Parks, R., et al. (2011). Suppression of HTLV-1 replication by Tax-mediated re-routing of the p13 viral protein to nuclear speckles. Blood 11, 1549–1559. doi: 10.1182/blood-2010-06-293340
Anupam, R., Doueiri, R., and Green, P. L. (2013). The need to accessorize: molecular roles of HTLV-1 p30 and HTLV-2 p28 accessory proteins in the viral life cycle. Front. Microbiol. 4:275. doi: 10.3389/fmicb.2013.00275
Banerjee, A., Ray, R. B., and Ray, R. (2010). Oncogenic potential of hepatitis C virus proteins. Viruses 2, 2108–2133. doi: 10.3390/v2092108
Barba, G., Harper, F., Harada, T., Kohara, M., Goulinet, S., Matsuura, Y., et al. (1997). Hepatitis C virus core protein shows a cytoplasmic localization and associates to cellular lipid storage droplets. Proc. Natl. Acad. Sci. U.S.A. 94, 1200–1205.
Barbaro, G., Di Lorenzo, G., Asti, A., Ribersani, M., Belloni, G., Grisorio, B., et al. (1999). Hepatocellular mitochondrial alterations in patients with chronic hepatitis C: ultrastructural and biochemical findings. Am. J. Gastroenterol. 94, 2198–2205. doi: 10.1111/j.1572-0241.1999.01294.x
Bartenschlager, R., and Schaller, H. (1992). Hepadnaviral assembly is initiated by polymerase binding to the encapsidation signal in the viral RNA genome. EMBO J 11, 3413–3420.
Belgnaoui, S. M., Paz, S., and Hiscott, J. (2011). Orchestrating the interferon antiviral response through the mitochondrial antiviral signaling (MAVS) adapter. Curr. Opin. Immunol. 23, 564–572. doi: 10.1016/j.coi.2011.08.001
Bellows, D. S., Howell, M., Pearson, C., Hazlewood, S. A., and Hardwick, J. M. (2002). Epstein-Barr virus BALF1 is a BCL-2-like antagonist of the herpesvirus antiapoptotic BCL-2 proteins. J. Virol. 76, 2469–2479. doi: 10.1128/jvi.76.5.2469-2479.2002
Benali-Furet, N. L., Chami, M., Houel, L., De Giorgi, F., Vernejoul, F., Lagorce, D., et al. (2005). Hepatitis C virus core triggers apoptosis in liver cells by inducing ER stress and ER calcium depletion. Oncogene 24, 4921–4933. doi: 10.1038/sj.onc.1208673
Bender, S., Reuter, A., Eberle, F., Einhorn, E., Binder, M., and Bartenschlager, R. (2015). Activation of Type I and III interferon response by mitochondrial and peroxisomal MAVS and inhibition by Hepatitis C Virus. PLoS Pathog. 11:e1005264. doi: 10.1371/journal.ppat.1005264
Benhenda, S., Cougot, D., Buendia, M. A., and Neuveut, C. (2009). Hepatitis B virus X protein molecular functions and its role in virus life cycle and pathogenesis. Adv. Cancer Res. 103, 75–109. doi: 10.1016/S0065-230X(09)03004-8
Bernardi, P., Rasola, A., Forte, M., and Lippe, G. (2015). The mitochondrial permeability transition pore: channel formation by F-ATP synthase, integration in signal transduction, and role in pathophysiology. Physiol. Rev. 95, 1111–1155. doi: 10.1152/physrev.00001.2015
Bernardini, J. P., Lazarou, M., and Dewson, G. (2017). Parkin and mitophagy in cancer. Oncogene 36, 1315–1327. doi: 10.1038/onc.2016.302
Bhutani, M., Polizzotto, M. N., Uldrick, T. S., and Yarchoan, R. (2015). Kaposi sarcoma-associated herpesvirus-associated malignancies: epidemiology, pathogenesis, and advances in treatment. Semin. Oncol. 42, 223–246. doi: 10.1053/j.seminoncol.2014.12.027
Biasiotto, R., Aguiari, P., Rizzuto, R., Pinton, P., D'Agostino, D. M., and Ciminale, V. (2010). The p13 protein of human T cell leukemia virus type 1 (HTLV-1) modulates mitochondrial membrane potential and calcium uptake. Biochim. Biophys. Acta 1797, 945–951. doi: 10.1016/j.bbabio.2010.02.023
Blachon, S., Bellanger, S., Demeret, C., and Thierry, F. (2005). Nucleo-cytoplasmic shuttling of high risk human Papillomavirus E2 proteins induces apoptosis. J. Biol. Chem. 280, 36088–36098. doi: 10.1074/jbc.M505138200
Bouchard, M. J., Wang, L. H., and Schneider, R. J. (2001). Calcium signaling by HBx protein in hepatitis B virus DNA replication. Science 294, 2376–2378. doi: 10.1126/science.294.5550.2376
Carrere-Kremer, S., Montpellier-Pala, C., Cocquerel, L., Wychowski, C., Penin, F., and Dubuisson, J. (2002). Subcellular localization and topology of the p7 polypeptide of hepatitis C virus. J. Virol. 76, 3720–3730. doi: 10.1128/JVI.76.8.3720-3730.2002
Cavallari, I., Rende, F., Bender, C., Romanelli, M. G., D'Agostino, D. M., and Ciminale, V. (2013). Fine tuning of the temporal expression of HTLV-1 and HTLV-2. Front. Microbiol. 4:235. doi: 10.3389/fmicb.2013.00235
Cavallari, I., Rende, F., Bona, M. K., Sztuba-Solinska, J., Silic-Benussi, M., Tognon, M., et al. (2015). Expression of alternatively spliced human T-Cell Leukemia Virus Type 1 mRNAs is influenced by mitosis and by a novel cis-acting regulatory sequence. J. Virol. 90, 1486–1498. doi: 10.1128/JVI.02298-15
Cavallari, I., Rende, F., D'Agostino, D. M., and Ciminale, V. (2011). Converging strategies in expression of human complex retroviruses. Viruses 3, 1395–1414. doi: 10.3390/v3081395
Cen, O., and Longnecker, R. (2015). Latent Membrane Protein 2 (LMP2). Curr. Top. Microbiol. Immunol. 391, 151–180. doi: 10.1007/978-3-319-22834-1_5
Chami, M., Ferrari, D., Nicotera, P., Paterlini-Brechot, P., and Rizzuto, R. (2003). Caspase-dependent alterations of Ca2+ signaling in the induction of apoptosis by hepatitis B virus X protein. J. Biol. Chem. 278, 31745–31755. doi: 10.1074/jbc.M304202200
Chau, B. N., Cheng, E. H., Kerr, D. A., and Hardwick, J. M. (2000). Aven, a novel inhibitor of caspase activation, binds Bcl-xL and Apaf-1. Mol. Cell 6, 31–40. doi: 10.1016/S1097-2765(05)00021-3
Chen, J., and Siddiqui, A. (2007). Hepatitis B virus X protein stimulates the mitochondrial translocation of Raf-1 via oxidative stress. J. Virol. 81, 6757–6760. doi: 10.1128/JVI.00172-07
Chen, Z. L., Su, Y. J., Zhang, H. L., Gu, P. Q., and Gao, L. J. (2014). The role of the globular heads of the C1q receptor in HPV-16 E2-induced human cervical squamous carcinoma cell apoptosis via a mitochondria-dependent pathway. J. Transl. Med. 12:286. doi: 10.1186/s12967-014-0286-y
Chu, V. C., Bhattacharya, S., Nomoto, A., Lin, J., Zaidi, S. K., Oberley, T. D., et al. (2011). Persistent expression of hepatitis C virus non-structural proteins leads to increased autophagy and mitochondrial injury in human hepatoma cells. PLoS ONE 6:e28551. doi: 10.1371/journal.pone.0028551
Ciminale, V., Zotti, L., D'Agostino, D. M., Ferro, T., Casareto, L., Franchini, G., et al. (1999). Mitochondrial targeting of the p13II protein coded by the x-II ORF of human T-cell leukemia/lymphotropic virus type I (HTLV-I). Oncogene 18, 4505–4514.
Claus, C., and Liebert, U. G. (2014). A renewed focus on the interplay between viruses and mitochondrial metabolism. Arch. Virol. 159, 1267–1277. doi: 10.1007/s00705-013-1841-1
Clippinger, A. J., and Bouchard, M. J. (2008). Hepatitis B virus HBx protein localizes to mitochondria in primary rat hepatocytes and modulates mitochondrial membrane potential. J. Virol. 82, 6798–6811. doi: 10.1128/JVI.00154-08
Cross, J. R., Postigo, A., Blight, K., and Downward, J. (2008). Viral pro-survival proteins block separate stages in Bax activation but changes in mitochondrial ultrastructure still occur. Cell Death Differ 15, 997–1008. doi: 10.1038/cdd.2008.14
D'Agostino, D. M., Bernardi, P., Chieco-Bianchi, L., and Ciminale, V. (2005a). Mitochondria as Functional Targets of Proteins Coded by Human Tumor Viruses. Adv. Cancer Res. 94, 87–142. doi: 10.1016/S0065-230X(05)94003-7
D'Agostino, D. M., Ranzato, L., Arrigoni, G., Cavallari, I., Belleudi, F., Torrisi, M. R., et al. (2002). Mitochondrial alterations induced by the p13II protein of human T-cell leukemia virus type 1. Critical role of arginine residues. J. Biol. Chem. 277, 34424–34433. doi: 10.1074/jbc.M203023200
D'Agostino, D. M., Silic-Benussi, M., Hiraragi, H., Lairmore, M. D., and Ciminale, V. (2005b). The human T-cell leukemia virus type 1 p13II protein: effects on mitochondrial function and cell growth. Cell Death Differ 12(Suppl. 1), 905–915. doi: 10.1038/sj.cdd.4401576
Dash, S., Chava, S., Aydin, Y., Chandra, P. K., Ferraris, P., Chen, W., et al. (2016). Hepatitis C virus infection induces autophagy as a prosurvival mechanism to alleviate hepatic ER-stress response. Viruses 8:150. doi: 10.3390/v8050150
de Martel, C., Ferlay, J., Franceschi, S., Vignat, J., Bray, F., Forman, D., et al. (2012). Global burden of cancers attributable to infections in 2008: a review and synthetic analysis. Lancet Oncol. 13, 607–615. doi: 10.1016/S1470-2045(12)70137-7
Derse, D., Mikovits, J., and Ruscetti, F. (1997). X-I and X-II open reading frames of HTLV-I are not required for virus replication or for immortalization of primary T-cells in vitro. Virology 237, 123–128.
Doorbar, J. (2013). The E4 protein; structure, function and patterns of expression. Virology 445, 80–98. doi: 10.1016/j.virol.2013.07.008
Doorbar, J., Egawa, N., Griffin, H., Kranjec, C., and Murakami, I. (2015). Human papillomavirus molecular biology and disease association. Rev. Med. Virol. 25(Suppl. 1), 2–23. doi: 10.1002/rmv.1822
Doorbar, J., Ely, S., Sterling, J., McLean, C., and Crawford, L. (1991). Specific interaction between HPV-16 E1-E4 and cytokeratins results in collapse of the epithelial cell intermediate filament network. Nature 352, 824–827.
Dustin, L. B., Bartolini, B., Capobianchi, M. R., and Pistello, M. (2016). Hepatitis C virus: life cycle in cells, infection and host response, and analysis of molecular markers influencing the outcome of infection and response to therapy. Clin. Microbiol. Infect. 22, 826–832. doi: 10.1016/j.cmi.2016.08.025
Egawa, N., Egawa, K., Griffin, H., and Doorbar, J. (2015). Human Papillomaviruses; Epithelial Tropisms, and the Development of Neoplasia. Viruses 7, 3863–3890. doi: 10.3390/v7072802
Farinati, F., Cardin, R., De Maria, N., Della Libera, G., Marafin, C., Lecis, E., et al. (1995). Iron storage, lipid peroxidation and glutathione turnover in chronic anti-HCV positive hepatitis. J. Hepatol. 22, 449–456.
Feng, H., Dong, X., Negaard, A., and Feng, P. (2008a). Kaposi's sarcoma-associated herpesvirus K7 induces viral G protein-coupled receptor degradation and reduces its tumorigenicity. PLoS Pathog 4:e1000157. doi: 10.1371/journal.ppat.1000157
Feng, H., Shuda, M., Chang, Y., and Moore, P. S. (2008b). Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 319, 1096–1100. doi: 10.1126/science.1152586
Feng, P., Park, J., Lee, B. S., Lee, S. H., Bram, R. J., and Jung, J. U. (2002). Kaposi's sarcoma-associated herpesvirus mitochondrial K7 protein targets a cellular calcium-modulating cyclophilin ligand to modulate intracellular calcium concentration and inhibit apoptosis. J. Virol. 76, 11491–11504. doi: 10.1128/JVI.76.22.11491-11504.2002
Flanagan, A. M., and Letai, A. (2008). BH3 domains define selective inhibitory interactions with BHRF-1 and KSHV BCL-2. Cell Death Differ 15, 580–588. doi: 10.1038/sj.cdd.4402292
Fracchia, K. M., Pai, C. Y., and Walsh, C. M. (2013). Modulation of T cell metabolism and function through calcium signaling. Front. Immunol. 4:324. doi: 10.3389/fimmu.2013.00324
Fujinaga, H., Tsutsumi, T., Yotsuyanagi, H., Moriya, K., and Koike, K. (2011). Hepatocarcinogenesis in hepatitis C: HCV shrewdly exacerbates oxidative stress by modulating both production and scavenging of reactive oxygen species. Oncol. 81(Suppl. 1), 11–17. doi: 10.1159/000333253
Gallo, A., Lampe, M., Gunther, T., and Brune, W. (2017). The viral Bcl-2 homologs of kaposi's sarcoma-associated herpesvirus and rhesus rhadinovirus share an essential role for viral replication. J. Virol. 91:e01875-16. doi: 10.1128/JVI.01875-16
Gao, L. J., Gu, P. Q., Fan, W. M., Liu, Z., Qiu, F., Peng, Y. Z., et al. (2011). The role of gC1qR in regulating survival of human papillomavirus 16 oncogene-transfected cervical cancer cells. Int. J. Oncol. 39, 1265–1272. doi: 10.3892/ijo.2011.1108
Gelgor, A., Kalt, I., Bergson, S., Brulois, K. F., Jung, J. U., and Sarid, R. (2015). Viral Bcl-2 Encoded by the Kaposi's Sarcoma-Associated Herpesvirus Is Vital for Virus Reactivation. J. Virol. 89, 5298–5307. doi: 10.1128/JVI.00098-15
Gessain, A., and Cassar, O. (2012). Epidemiological Aspects and World Distribution of HTLV-1 Infection. Front. Microbiol. 3:388. doi: 10.3389/fmicb.2012.00388
Ghorbel, S., Sinha-Datta, U., Dundr, M., Brown, M., Franchini, G., and Nicot, C. (2006). Human T-cell leukemia virus type I p30 nuclear/nucleolar retention is mediated through interactions with RNA and a constituent of the 60 S ribosomal subunit. J. Biol. Chem. 281, 37150–37158. doi: 10.1074/jbc.M603981200
Goncalves, D. U., Proietti, F. A., Ribas, J. G., Araujo, M. G., Pinheiro, S. R., Guedes, A. C., et al. (2010). Epidemiology, treatment, and prevention of human T-cell leukemia virus type 1-associated diseases. Clin. Microbiol. Rev. 23, 577–589. doi: 10.1128/CMR.00063-09
Gong, G., Waris, G., Tanveer, R., and Siddiqui, A. (2001). Human hepatitis C virus NS5A protein alters intracellular calcium levels, induces oxidative stress, and activates STAT-3 and NF-kappa B. Proc. Natl. Acad. Sci. U.S.A. 98, 9599–9604. doi: 10.1073/pnas.171311298
Graham, S. V. (2016). Human Papillomavirus E2 protein: linking replication, transcription, and RNA processing. J. Virol. 90, 8384–8388. doi: 10.1128/JVI.00502-16
Griffin, S., Clarke, D., McCormick, C., Rowlands, D., and Harris, M. (2005). Signal peptide cleavage and internal targeting signals direct the hepatitis C virus p7 protein to distinct intracellular membranes. J. Virol. 79, 15525–15536. doi: 10.1128/JVI.79.24.15525-15536.2005
Griffin, S. D., Harvey, R., Clarke, D. S., Barclay, W. S., Harris, M., and Rowlands, D. J. (2004). A conserved basic loop in hepatitis C virus p7 protein is required for amantadine-sensitive ion channel activity in mammalian cells but is dispensable for localization to mitochondria. J. Gen. Virol. 85(Pt 2), 451–461. doi: 10.1099/vir.0.19634-0
Haqshenas, G., Mackenzie, J. M., Dong, X., and Gowans, E. J. (2007). Hepatitis C virus p7 protein is localized in the endoplasmic reticulum when it is encoded by a replication-competent genome. J. Gen. Virol. 88(Pt 1), 134–142. doi: 10.1099/vir.0.82049-0
Hara, Y., Yanatori, I., Ikeda, M., Kiyokage, E., Nishina, S., Tomiyama, Y., et al. (2014). Hepatitis C virus core protein suppresses mitophagy by interacting with parkin in the context of mitochondrial depolarization. Am. J. Pathol. 184, 3026–3039. doi: 10.1016/j.ajpath.2014.07.024
Harper, D. M., and DeMars, L. R. (2017). HPV vaccines - A review of the first decade. Gynecol. Oncol. 146, 196–204. doi: 10.1016/j.ygyno.2017.04.004
Henderson, S., Huen, D., Rowe, M., Dawson, C., Johnson, G., and Rickinson, A. (1993). Epstein-Barr virus-coded BHRF1 protein, a viral homologue of Bcl-2, protects human B cells from programmed cell death. Proc. Natl. Acad. Sci. U.S.A. 90, 8479–8483.
Henkler, F., Hoare, J., Waseem, N., Goldin, R. D., McGarvey, M. J., Koshy, R., et al. (2001). Intracellular localization of the hepatitis B virus HBx protein. J. Gen. Virol. 82(Pt 4), 871–882. doi: 10.1099/0022-1317-82-4-871
Hickish, T., Robertson, D., Clarke, P., Hill, M., di Stefano, F., Clarke, C., et al. (1994). Ultrastructural localization of BHRF1: an Epstein-Barr virus gene product which has homology with bcl-2. Cancer Res. 54, 2808–2811.
Hiraragi, H., Kim, S. J., Phipps, A. J., Silic-Benussi, M., Ciminale, V., Ratner, L., et al. (2006). Human T-lymphotropic virus type 1 mitochondrion-localizing protein p13(II) is required for viral infectivity in vivo. J. Virol. 80, 3469–3476. doi: 10.1128/JVI.80.7.3469-3476.2006
Horner, S. M., Liu, H. M., Park, H. S., Briley, J., and Gale, M. Jr. (2011). Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc. Natl. Acad. Sci. U.S.A. 108, 14590–14595. doi: 10.1073/pnas.1110133108
Hou, X., Foley, S., Cueto, M., and Robinson, M. A. (2000). The human T-cell leukemia virus type I (HTLV-I) X region encoded protein p13(II) interacts with cellular proteins. Virology 277, 127–135. doi: 10.1006/viro.2000.0604
Hsu, W. L., Chung, P. J., Tsai, M. H., Chang, C. L., and Liang, C. L. (2012). A role for Epstein-Barr viral BALF1 in facilitating tumor formation and metastasis potential. Virus Res. 163, 617–627. doi: 10.1016/j.virusres.2011.12.017
Hu, M., Crawford, S. A., Henstridge, D. C., Ng, I. H., Boey, E. J., Xu, Y., et al. (2013). p32 protein levels are integral to mitochondrial and endoplasmic reticulum morphology, cell metabolism and survival. Biochem. J. 453, 381–391. doi: 10.1042/BJ20121829
Huh, K. W., and Siddiqui, A. (2002). Characterization of the mitochondrial association of hepatitis B virus X protein, HBx. Mitochondrion 1, 349–359. doi: 10.1016/S1567-7249(01)00040-X
IARC Monograph (2012). Biological agents. Volume 100 B. A review of human carcinogens. IARC Monogr. Eval. Carcinog. Risks Hum. 100(Pt B), 1–441.
Isherwood, B. J., and Patel, A. H. (2005). Analysis of the processing and transmembrane topology of the E2p7 protein of hepatitis C virus. J. Gen. Virol. 86(Pt 3), 667–676. doi: 10.1099/vir.0.80737-0
Journo, C., Douceron, E., and Mahieux, R. (2009). HTLV gene regulation: because size matters, transcription is not enough. Future Microbiol. 4, 425–440. doi: 10.2217/fmb.09.13
Kalt, I., Borodianskiy-Shteinberg, T., Schachor, A., and Sarid, R. (2010). GLTSCR2/PICT-1, a putative tumor suppressor gene product, induces the nucleolar targeting of the Kaposi's sarcoma-associated herpesvirus KS-Bcl-2 protein. J. Virol. 84, 2935–2945. doi: 10.1128/JVI.00757-09
Kang, S. M., Kim, S. J., Kim, J. H., Lee, W., Kim, G. W., Lee, K. H., et al. (2009). Interaction of hepatitis C virus core protein with Hsp60 triggers the production of reactive oxygen species and enhances TNF-alpha-mediated apoptosis. Cancer Lett. 279, 230–237. doi: 10.1016/j.canlet.2009.02.003
Kao, C. C., Yi, G., and Huang, H. C. (2016). The core of hepatitis C virus pathogenesis. Curr. Opin. Virol. 17, 66–73. doi: 10.1016/j.coviro.2016.01.009
Kim, S. J., Ding, W., Albrecht, B., Green, P. L., and Lairmore, M. D. (2003). A conserved calcineurin-binding motif in human T lymphotropic virus type 1 p12I functions to modulate nuclear factor of activated T cell activation. J. Biol. Chem. 278, 15550–15557. doi: 10.1074/jbc.M210210200
Kim, S. J., Khan, M., Quan, J., Till, A., Subramani, S., and Siddiqui, A. (2013a). Hepatitis B virus disrupts mitochondrial dynamics: induces fission and mitophagy to attenuate apoptosis. PLoS Pathog. 9:e1003722. doi: 10.1371/journal.ppat.1003722
Kim, S. J., Syed, G. H., and Siddiqui, A. (2013b). Hepatitis C virus induces the mitochondrial translocation of Parkin and subsequent mitophagy. PLoS Pathog. 9:e1003285. doi: 10.1371/journal.ppat.1003285
Korenaga, M., Wang, T., Li, Y., Showalter, L. A., Chan, T., Sun, J., et al. (2005). Hepatitis C virus core protein inhibits mitochondrial electron transport and increases reactive oxygen species (ROS) production. J. Biol. Chem. 280, 37481–37488. doi: 10.1074/jbc.M506412200
Kvansakul, M., Caria, S., and Hinds, M. G. (2017). The Bcl-2 family in host-virus interactions. Viruses 9:pii: E290. doi: 10.3390/v9100290
Kvansakul, M., Wei, A. H., Fletcher, J. I., Willis, S. N., Chen, L., Roberts, A. W., et al. (2010). Structural basis for apoptosis inhibition by Epstein-Barr virus BHRF1. PLoS Pathog. 6:e1001236. doi: 10.1371/journal.ppat.1001236
Lai, D., Tan, C. L., Gunaratne, J., Quek, L. S., Nei, W., Thierry, F., et al. (2013). Localization of HPV-18 E2 at mitochondrial membranes induces ROS release and modulates host cell metabolism. PLoS ONE 8:e75625. doi: 10.1371/journal.pone.0075625
Lairmore, M. D., Haines, R., and Anupam, R. (2012). Mechanisms of human T-lymphotropic virus type 1 transmission and disease. Curr. Opin. Virol. 2, 474–481. doi: 10.1016/j.coviro.2012.06.007
LaJeunesse, D. R., Brooks, K., and Adamson, A. L. (2005). Epstein-Barr virus immediate-early proteins BZLF1 and BRLF1 alter mitochondrial morphology during lytic replication. Biochem. Biophys. Res. Commun. 333, 438–442. doi: 10.1016/j.bbrc.2005.05.120
Lefebvre, L., Vanderplasschen, A., Ciminale, V., Heremans, H., Dangoisse, O., Jauniaux, J. C., et al. (2002). Oncoviral bovine leukemia virus G4 and human T-cell leukemia virus type 1 p13(II) accessory proteins interact with farnesyl pyrophosphate synthetase. J. Virol. 76, 1400–1414. doi: 10.1128/JVI.76.3.1400-1414.2002
Levrero, M., and Zucman-Rossi, J. (2016). Mechanisms of HBV-induced hepatocellular carcinoma. J. Hepatol. 64, S84–S101. doi: 10.1016/j.jhep.2016.02.021
Li, Y., Boehning, D. F., Qian, T., Popov, V. L., and Weinman, S. A. (2007). Hepatitis C virus core protein increases mitochondrial ROS production by stimulation of Ca2+ uniporter activity. FASEB J. 21, 2474–2485. doi: 10.1096/fj.06-7345com
Liang, Q., Chang, B., Brulois, K. F., Castro, K., Min, C. K., Rodgers, M. A., et al. (2013). Kaposi's sarcoma-associated herpesvirus K7 modulates Rubicon-mediated inhibition of autophagosome maturation. J. Virol. 87, 12499–12503. doi: 10.1128/JVI.01898-13
Liang, Q., Chang, B., Lee, P., Brulois, K. F., Ge, J., Shi, M., et al. (2015). Identification of the essential role of viral Bcl-2 for Kaposi's Sarcoma-Associated Herpesvirus Lytic Replication. J. Virol. 89, 5308–5317. doi: 10.1128/JVI.00102-15
Lim, W., Kwon, S. H., Cho, H., Kim, S., Lee, S., and Ryu, W. S. (2010). HBx targeting to mitochondria and ROS generation are necessary but insufficient for HBV-induced cyclooxygenase-2 expression. J. Mol. Med. 88, 359–369. doi: 10.1007/s00109-009-0563-z
Lo, S. Y., Masiarz, F., Hwang, S. B., Lai, M. M., and Ou, J. H. (1995). Differential subcellular localization of hepatitis C virus core gene products. Virology 213, 455–461. doi: 10.1006/viro.1995.0018
Lucifora, J., Arzberger, S., Durantel, D., Belloni, L., Strubin, M., Levrero, M., et al. (2011). Hepatitis B virus X protein is essential to initiate and maintain virus replication after infection. J. Hepatol. 55, 996–1003. doi: 10.1016/j.jhep.2011.02.015
Machida, K., Cheng, K. T., Lai, C. K., Jeng, K. S., Sung, V. M., and Lai, M. M. (2006). Hepatitis C virus triggers mitochondrial permeability transition with production of reactive oxygen species, leading to DNA damage and STAT3 activation. J. Virol. 80, 7199–7207. doi: 10.1128/JVI.00321-06
Madan, V., and Bartenschlager, R. (2015). Structural and Functional Properties of the Hepatitis C Virus p7 Viroporin. Viruses 7, 4461–4481. doi: 10.3390/v7082826
Marshall, W. L., Yim, C., Gustafson, E., Graf, T., Sage, D. R., Hanify, K., et al. (1999). Epstein-Barr virus encodes a novel homolog of the bcl-2 oncogene that inhibits apoptosis and associates with Bax and Bak. J. Virol. 73, 5181–5185.
McClain, S. L., Clippinger, A. J., Lizzano, R., and Bouchard, M. J. (2007). Hepatitis B virus replication is associated with an HBx-dependent mitochondrion-regulated increase in cytosolic calcium levels. J. Virol. 81, 12061–12065. doi: 10.1128/JVI.00740-07
McKenzie, J., and El-Guindy, A. (2015). Epstein-Barr Virus Lytic Cycle Reactivation. Curr. Top. Microbiol. Immunol. 391, 237–261. doi: 10.1007/978-3-319-22834-1_8
Meylan, E., Curran, J., Hofmann, K., Moradpour, D., Binder, M., Bartenschlager, R., et al. (2005). Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437, 1167–1172. doi: 10.1038/nature04193
Milian, E., Prats, E., Cairo, J. J., Godia, F., and Vives, J. (2015). BHRF1 exerts an antiapoptotic effect and cell cycle arrest via Bcl-2 in murine hybridomas. J. Biotechnol. 209, 58–67. doi: 10.1016/j.jbiotec.2015.06.379
Morikawa, K., Lange, C. M., Gouttenoire, J., Meylan, E., Brass, V., Penin, F., et al. (2011). Nonstructural protein 3-4A: the Swiss army knife of hepatitis C virus. J. Viral Hepat. 18, 305–315. doi: 10.1111/j.1365-2893.2011.01451.x
Moriya, K., Fujie, H., Shintani, Y., Yotsuyanagi, H., Tsutsumi, T., Ishibashi, K., et al. (1998). The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat. Med. 4, 1065–1067. doi: 10.1038/2053
Moriya, K., Miyoshi, H., Shinzawa, S., Tsutsumi, T., Fujie, H., Goto, K., et al. (2010). Hepatitis C virus core protein compromises iron-induced activation of antioxidants in mice and HepG2 cells. J. Med. Virol. 82, 776–792. doi: 10.1002/jmv.21661
Moriya, K., Nakagawa, K., Santa, T., Shintani, Y., Fujie, H., Miyoshi, H., et al. (2001). Oxidative stress in the absence of inflammation in a mouse model for hepatitis C virus-associated hepatocarcinogenesis. Cancer Res. 61, 4365–4370.
Moriya, K., Yotsuyanagi, H., Shintani, Y., Fujie, H., Ishibashi, K., Matsuura, Y., et al. (1997). Hepatitis C virus core protein induces hepatic steatosis in transgenic mice. J. Gen. Virol. 78(Pt 7), 1527–1531. doi: 10.1099/0022-1317-78-7-1527
Morrison, B. J., Labo, N., Miley, W. J., and Whitby, D. (2015). Serodiagnosis for tumor viruses. Semin. Oncol. 42, 191–206. doi: 10.1053/j.seminoncol.2014.12.024
Motavaf, M., Safari, S., Saffari Jourshari, M., and Alavian, S. M. (2013). Hepatitis B virus-induced hepatocellular carcinoma: the role of the virus x protein. Acta Virol. 57, 389–396. doi: 10.4149/av_2013_04_389
Nieva, J. L., Madan, V., and Carrasco, L. (2012). Viroporins: structure and biological functions. Nat. Rev. Microbiol. 10, 563–574. doi: 10.1038/nrmicro2820
Nomura-Takigawa, Y., Nagano-Fujii, M., Deng, L., Kitazawa, S., Ishido, S., Sada, K., et al. (2006). Non-structural protein 4A of Hepatitis C virus accumulates on mitochondria and renders the cells prone to undergoing mitochondria-mediated apoptosis. J. Gen. Virol. 87(Pt 7), 1935–1945. doi: 10.1099/vir.0.81701-0
Okamoto, T., Omori, H., Kaname, Y., Abe, T., Nishimura, Y., Suzuki, T., et al. (2008). A single-amino-acid mutation in hepatitis C virus NS5A disrupting FKBP8 interaction impairs viral replication. J. Virol. 82, 3480–3489. doi: 10.1128/JVI.02253-07
Okuda, M., Li, K., Beard, M. R., Showalter, L. A., Scholle, F., Lemon, S. M., et al. (2002). Mitochondrial injury, oxidative stress, and antioxidant gene expression are induced by hepatitis C virus core protein. Gastroenterology 122, 366–375. doi: 10.1053/gast.2002.30983
Otani, K., Korenaga, M., Beard, M. R., Li, K., Qian, T., Showalter, L. A., et al. (2005). Hepatitis C virus core protein, cytochrome P450 2E1, and alcohol produce combined mitochondrial injury and cytotoxicity in hepatoma cells. Gastroenterology 128, 96–107. doi: 10.1053/j.gastro.2004.10.045
Pal, A. D., Basak, N. P., Banerjee, A. S., and Banerjee, S. (2014). Epstein-Barr virus latent membrane protein-2A alters mitochondrial dynamics promoting cellular migration mediated by Notch signaling pathway. Carcinogenesis 35, 1592–1601. doi: 10.1093/carcin/bgu069
Panfil, A. R., Martinez, M. P., Ratner, L., and Green, P. L. (2016). Human T-cell leukemia virus-associated malignancy. Curr. Opin. Virol. 20, 40–46. doi: 10.1016/j.coviro.2016.08.009
Pattingre, S., Tassa, A., Qu, X., Garuti, R., Liang, X. H., Mizushima, N., et al. (2005). Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122, 927–939. doi: 10.1016/j.cell.2005.07.002
Pearson, G. R., Luka, J., Petti, L., Sample, J., Birkenbach, M., Braun, D., et al. (1987). Identification of an Epstein-Barr virus early gene encoding a second component of the restricted early antigen complex. Virology 160, 151–161.
Peng, Y. T., Chen, P., Ouyang, R. Y., and Song, L. (2015). Multifaceted role of prohibitin in cell survival and apoptosis. Apoptosis 20, 1135–1149. doi: 10.1007/s10495-015-1143-z
Piccoli, C., Quarato, G., Ripoli, M., D'Aprile, A., Scrima, R., Cela, O., et al. (2009). HCV infection induces mitochondrial bioenergetic unbalance: causes and effects. Biochim. Biophys. Acta 1787, 539–546. doi: 10.1016/j.bbabio.2008.11.008
Piccoli, C., Scrima, R., D'Aprile, A., Ripoli, M., Lecce, L., Boffoli, D., et al. (2006). Mitochondrial dysfunction in hepatitis C virus infection. Biochim. Biophys. Acta 1757, 1429–1437. doi: 10.1016/j.bbabio.2006.05.018
Piccoli, C., Scrima, R., Quarato, G., D'Aprile, A., Ripoli, M., Lecce, L., et al. (2007). Hepatitis C virus protein expression causes calcium-mediated mitochondrial bioenergetic dysfunction and nitro-oxidative stress. Hepatology 46, 58–65. doi: 10.1002/hep.21679
Qi, H., Chu, V., Wu, N. C., Chen, Z., Truong, S., Brar, G., et al. (2017). Systematic identification of anti-interferon function on hepatitis C virus genome reveals p7 as an immune evasion protein. Proc. Natl. Acad. Sci. U.S.A. 114, 2018–2023. doi: 10.1073/pnas.1614623114
Rahmani, Z., Huh, K. W., Lasher, R., and Siddiqui, A. (2000). Hepatitis B virus X protein colocalizes to mitochondria with a human voltage-dependent anion channel, HVDAC3, and alters its transmembrane potential. J. Virol. 74, 2840–2846. doi: 10.1128/JVI.74.6.2840-2846.2000
Raj, K., Berguerand, S., Southern, S., Doorbar, J., and Beard, P. (2004). E1 empty set E4 protein of human papillomavirus type 16 associates with mitochondria. J. Virol. 78, 7199–7207. doi: 10.1128/JVI.78.13.7199-7207.2004
Rawat, S., Clippinger, A. J., and Bouchard, M. J. (2012). Modulation of apoptotic signaling by the hepatitis B virus X protein. Viruses 4, 2945–2972. doi: 10.3390/v4112945
Ren, J. H., Chen, X., Zhou, L., Tao, N. N., Zhou, H. Z., Liu, B., et al. (2016). Protective Role of Sirtuin3 (SIRT3) in Oxidative Stress Mediated by Hepatitis B Virus X Protein Expression. PLoS ONE 11:e0150961. doi: 10.1371/journal.pone.0150961
Rende, F., Cavallari, I., Corradin, A., Silic-Benussi, M., Toulza, F., Toffolo, G. M., et al. (2011). Kinetics and intracellular compartmentalization of HTLV-1 gene expression: nuclear retention of HBZ mRNA. Blood 117, 4855–4859. doi: 10.1182/blood-2010-11-316463
Ross-Thriepland, D., and Harris, M. (2015). Hepatitis C virus NS5A: enigmatic but still promiscuous 10 years on! J. Gen. Virol. 96(Pt 4), 727–738. doi: 10.1099/jgv.0.000009
Rouille, Y., Helle, F., Delgrange, D., Roingeard, P., Voisset, C., Blanchard, E., et al. (2006). Subcellular localization of hepatitis C virus structural proteins in a cell culture system that efficiently replicates the virus. J. Virol. 80, 2832–2841. doi: 10.1128/JVI.80.6.2832-2841.2006
Russo, J. J., Bohenzky, R. A., Chien, M. C., Chen, J., Yan, M., Maddalena, D., et al. (1996). Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc. Natl. Acad. Sci. U.S.A. 93, 14862–14867.
Sakamuro, D., Furukawa, T., and Takegami, T. (1995). Hepatitis C virus nonstructural protein NS3 transforms NIH 3T3 cells. J. Virol. 69, 3893–3896.
Santolini, E., Pacini, L., Fipaldini, C., Migliaccio, G., and Monica, N. (1995). The NS2 protein of hepatitis C virus is a transmembrane polypeptide. J. Virol. 69, 7461–7471.
Sarid, R., Sato, T., Bohenzky, R. A., Russo, J. J., and Chang, Y. (1997). Kaposi's sarcoma-associated herpesvirus encodes a functional bcl-2 homologue. Nat. Med. 3, 293–298.
Scheel, T. K., and Rice, C. M. (2013). Understanding the hepatitis C virus life cycle paves the way for highly effective therapies. Nat. Med. 19, 837–849. doi: 10.1038/nm.3248
Schwer, B., Ren, S., Pietschmann, T., Kartenbeck, J., Kaehlcke, K., Bartenschlager, R., et al. (2004). Targeting of hepatitis C virus core protein to mitochondria through a novel C-terminal localization motif. J. Virol. 78, 7958–7968. doi: 10.1128/JVI.78.15.7958-7968.2004
Scott, C., and Griffin, S. (2015). Viroporins: structure, function and potential as antiviral targets. J. Gen. Virol. 96, 2000–2027. doi: 10.1099/vir.0.000201
Seth, R. B., Sun, L., Ea, C. K., and Chen, Z. J. (2005). Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122, 669–682. doi: 10.1016/j.cell.2005.08.012
Shirakata, Y., and Koike, K. (2003). Hepatitis B virus X protein induces cell death by causing loss of mitochondrial membrane potential. J. Biol. Chem. 278, 22071–22078. doi: 10.1074/jbc.M301606200
Silic-Benussi, M., Biasiotto, R., Andresen, V., Franchini, G., D'Agostino, D. M., and Ciminale, V. (2010a). HTLV-1 p13, a small protein with a busy agenda. Mol. Aspects Med. 31, 350–358. doi: 10.1016/j.mam.2010.03.001
Silic-Benussi, M., Cannizzaro, E., Venerando, A., Cavallari, I., Petronilli, V., La Rocca, N., et al. (2009). Modulation of mitochondrial K+ permeability and reactive oxygen species production by the p13 protein of human T-cell leukemia virus type 1. Biochim. Biophys. Acta 1787, 947–954. doi: 10.1016/j.bbabio.2009.02.001
Silic-Benussi, M., Cavallari, I., Zorzan, T., Rossi, E., Hiraragi, H., Rosato, A., et al. (2004). Suppression of tumor growth and cell proliferation by p13II, a mitochondrial protein of human T cell leukemia virus type 1. Proc. Natl. Acad. Sci. U.S.A. 101, 6629–6634. doi: 10.1073/pnas.0305502101
Silic-Benussi, M., Marin, O., Biasiotto, R., D'Agostino, D. M., and Ciminale, V. (2010b). Effects of human T-cell leukemia virus type 1 (HTLV-1) p13 on mitochondrial K+ permeability: a new member of the viroporin family? FEBS Lett. 584, 2070–2075. doi: 10.1016/j.febslet.2010.02.030
Siu, G. K., Zhou, F., Yu, M. K., Zhang, L., Wang, T., Liang, Y., et al. (2016). Hepatitis C virus NS5A protein cooperates with phosphatidylinositol 4-kinase IIIalpha to induce mitochondrial fragmentation. Sci. Rep. 6:23464. doi: 10.1038/srep23464
Song, X., Yao, Z., Yang, J., Zhang, Z., Deng, Y., Li, M., et al. (2016). HCV core protein binds to gC1qR to induce A20 expression and inhibit cytokine production through MAPKs and NF-kappaB signaling pathways. Oncotarget 7, 33796–33808. doi: 10.18632/oncotarget.9304
Suzuki, R., Sakamoto, S., Tsutsumi, T., Rikimaru, A., Tanaka, K., Shimoike, T., et al. (2005). Molecular determinants for subcellular localization of hepatitis C virus core protein. J. Virol. 79, 1271–1281. doi: 10.1128/JVI.79.2.1271-1281.2005
Takada, S., Shirakata, Y., Kaneniwa, N., and Koike, K. (1999). Association of hepatitis B virus X protein with mitochondria causes mitochondrial aggregation at the nuclear periphery, leading to cell death. Oncogene 18, 6965–6973.
Tanaka, Y., Kanai, F., Kawakami, T., Tateishi, K., Ijichi, H., Kawabe, T., et al. (2004). Interaction of the hepatitis B virus X protein (HBx) with heat shock protein 60 enhances HBx-mediated apoptosis. Biochem. Biophys. Res. Commun. 318, 461–469. doi: 10.1016/j.bbrc.2004.04.046
Tibaldi, E., Venerando, A., Zonta, F., Bidoia, C., Magrin, E., Marin, O., et al. (2011). Interaction between the SH3 domain of Src family kinases and the proline-rich motif of HTLV-1 p13: a novel mechanism underlying delivery of Src family kinases to mitochondria. Biochem. J. 439, 505–516. doi: 10.1042/BJ20101650
Tomaic, V. (2016). Functional Roles of E6 and E7 Oncoproteins in HPV-Induced Malignancies at Diverse Anatomical Sites. Cancers (Basel). 8:95. doi: 10.3390/cancers8100095
Tsutsumi, T., Matsuda, M., Aizaki, H., Moriya, K., Miyoshi, H., Fujie, H., et al. (2009). Proteomics analysis of mitochondrial proteins reveals overexpression of a mitochondrial protein chaperon, prohibitin, in cells expressing hepatitis C virus core protein. Hepatology 50, 378–386. doi: 10.1002/hep.22998
Unchwaniwala, N., Sherer, N. M., and Loeb, D. D. (2016). Hepatitis B Virus Polymerase Localizes to the Mitochondria, and Its Terminal Protein Domain Contains the Mitochondrial Targeting Signal. J. Virol. 90, 8705–8719. doi: 10.1128/JVI.01229-16
Van Prooyen, N., Andresen, V., Gold, H., Bialuk, I., Pise-Masison, C., and Franchini, G. (2010). Hijacking the T-cell communication network by the human T-cell leukemia/lymphoma virus type 1 (HTLV-1) p12 and p8 proteins. Mol. Aspects Med. 31, 333–343. doi: 10.1016/j.mam.2010.07.001
Vieyres, G., Brohm, C., Friesland, M., Gentzsch, J., Wolk, B., Roingeard, P., et al. (2013). Subcellular localization and function of an epitope-tagged p7 viroporin in hepatitis C virus-producing cells. J. Virol. 87, 1664–1678. doi: 10.1128/JVI.02782-12
Wang, H., and Ryu, W. S. (2010). Hepatitis B virus polymerase blocks pattern recognition receptor signaling via interaction with DDX3: implications for immune evasion. PLoS Pathog. 6:e1000986. doi: 10.1371/journal.ppat.1000986
Wang, H. W., Sharp, T. V., Koumi, A., Koentges, G., and Boshoff, C. (2002). Characterization of an anti-apoptotic glycoprotein encoded by Kaposi's sarcoma-associated herpesvirus which resembles a spliced variant of human survivin. EMBO J. 21, 2602–2615. doi: 10.1093/emboj/21.11.2602
Wang, J. H., Yun, C., Kim, S., Lee, J. H., Yoon, G., Lee, M. O., et al. (2003). Reactive oxygen species modulates the intracellular level of HBx viral oncoprotein. Biochem. Biophys. Res. Commun. 310, 32–39. doi: 10.1016/j.bbrc.2003.08.113
Wang, T., Campbell, R. V., Yi, M. K., Lemon, S. M., and Weinman, S. A. (2010). Role of Hepatitis C virus core protein in viral-induced mitochondrial dysfunction. J. Viral Hepat. 17, 784–793. doi: 10.1111/j.1365-2893.2009.01238.x
Waris, G., Huh, K. W., and Siddiqui, A. (2001). Mitochondrially associated hepatitis B virus X protein constitutively activates transcription factors STAT-3 and NF-kappa B via oxidative stress. Mol. Cell. Biol. 21, 7721–7730. doi: 10.1128/MCB.21.22.7721-7730.2001
Webster, D. P., Klenerman, P., and Dusheiko, G. M. (2015). Hepatitis C. Lancet 385, 1124–1135. doi: 10.1016/S0140-6736(14)62401-6
Wiedmer, A., Wang, P., Zhou, J., Rennekamp, A. J., Tiranti, V., Zeviani, M., et al. (2008). Epstein-Barr virus immediate-early protein Zta co-opts mitochondrial single-stranded DNA binding protein to promote viral and inhibit mitochondrial DNA replication. J. Virol. 82, 4647–4655. doi: 10.1128/JVI.02198-07
Willems, L., Hasegawa, H., Accolla, R., Bangham, C., Bazarbachi, A., Bertazzoni, U., et al. (2017). Reducing the global burden of HTLV-1 infection: an agenda for research and action. Antiviral Res. 137, 41–48. doi: 10.1016/j.antiviral.2016.10.015
Wolk, B., Sansonno, D., Krausslich, H. G., Dammacco, F., Rice, C. M., Blum, H. E., et al. (2000). Subcellular localization, stability, and trans-cleavage competence of the hepatitis C virus NS3-NS4A complex expressed in tetracycline-regulated cell lines. J. Virol. 74, 2293–2304. doi: 10.1128/JVI.74.5.2293-2304.2000
Woodman, C. B., Collins, S. I., and Young, L. S. (2007). The natural history of cervical HPV infection: unresolved issues. Nat. Rev. Cancer 7, 11–22. doi: 10.1038/nrc2050
Wu, M., Xu, Y., Lin, S., Zhang, X., Xiang, L., and Yuan, Z. (2007). Hepatitis B virus polymerase inhibits the interferon-inducible MyD88 promoter by blocking nuclear translocation of Stat1. J. Gen. Virol. 88(Pt 12), 3260–3269. doi: 10.1099/vir.0.82959-0
Yang, B., and Bouchard, M. J. (2012). The hepatitis B virus X protein elevates cytosolic calcium signals by modulating mitochondrial calcium uptake. J. Virol. 86, 313–327. doi: 10.1128/JVI.06442-11
Young, L. S., Yap, L. F., and Murray, P. G. (2016). Epstein-Barr virus: more than 50 years old and still providing surprises. Nat. Rev. Cancer 16, 789–802. doi: 10.1038/nrc.2016.92
Zheng, B. Y., Fang, X. F., Zou, L. Y., Huang, Y. H., Chen, Z. X., Li, D., et al. (2014). The co-localization of HBx and COXIII upregulates COX-2 promoting HepG2 cell growth. Int. J. Oncol. 45, 1143–1150. doi: 10.3892/ijo.2014.2499
Keywords: Mitochondria, EBV, HTLV-1, HPV, HBV, HCV, KSHV
Citation: Cavallari I, Scattolin G, Silic-Benussi M, Raimondi V, D'Agostino DM and Ciminale V (2018) Mitochondrial Proteins Coded by Human Tumor Viruses. Front. Microbiol. 9:81. doi: 10.3389/fmicb.2018.00081
Received: 30 October 2017; Accepted: 12 January 2018;
Published: 06 February 2018.
Edited by:
Akio Adachi, Tokushima University, JapanReviewed by:
Renaud Mahieux, École normale supérieure de Lyon, FranceMasao Matsuoka, Kumamoto University, Japan
Mari Kannagi, Tokyo Medical and Dental University, Japan
Copyright © 2018 Cavallari, Scattolin, Silic-Benussi, Raimondi, D'Agostino and Ciminale. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Donna M. D'Agostino, ZG0uZGFnb3N0aW5vQHVuaXBkLml0
Vincenzo Ciminale, di5jaW1pbmFsZUB1bmlwZC5pdA==
†These authors have contributed equally to this work.